

Hydrothermally treated Al- and Ga-stabilized stuffed Li garnets, nominally Li7La3Zr2O12, have been investigated by single-crystal X-ray diffraction to determine structural and site-occupation alterations due to LiI/HI exchange.
Keywords: solid-state electrolyte, LLZO, hydrothermal degradation, crystal structure, structure analysis, garnet, lattice expansion
Abstract
Single crystals of an Li-stuffed, Al- and Ga-stabilized garnet-type solid-state electrolyte material, Li7La3Zr2O12 (LLZO), have been analysed using single-crystal X-ray diffraction to determine the pristine structural state immediately after synthesis via ceramic sintering techniques. Hydrothermal treatment at 150 °C for 28 d induces a phase transition in the Al-stabilized compound from the commonly observed cubic Ia
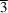 d structure to the acentric I
d structure to the acentric I
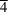 3d subtype. LiI ions at the interstitial octahedrally (4 + 2-fold) coordinated 48e site are most easily extracted and AlIII ions order onto the tetrahedral 12a site. Deep hydration induces a distinct depletion of LiI at this site, while the second tetrahedral site, 12b, suffers only minor LiI loss. Charge balance is maintained by the incorporation of HI, which is bonded to an O atom. Hydration of Ga-stabilized LLZO induces similar effects, with complete depletion of LiI at the 48e site. The LiI/HI exchange not only leads to a distinct increase in the unit-cell size, but also alters some bonding topology, which is discussed here.
3d subtype. LiI ions at the interstitial octahedrally (4 + 2-fold) coordinated 48e site are most easily extracted and AlIII ions order onto the tetrahedral 12a site. Deep hydration induces a distinct depletion of LiI at this site, while the second tetrahedral site, 12b, suffers only minor LiI loss. Charge balance is maintained by the incorporation of HI, which is bonded to an O atom. Hydration of Ga-stabilized LLZO induces similar effects, with complete depletion of LiI at the 48e site. The LiI/HI exchange not only leads to a distinct increase in the unit-cell size, but also alters some bonding topology, which is discussed here.
Introduction
The garnet family, X
3
Y
2
Z
3O12, has been well described mineralogically and crystallographically in recent decades (Novak & Gibbs, 1971 ▸), and is of interest to a range of scientists from the fields of geoscience and technology, due to its thermodynamic stability in a variety of geological environments and its flexible structure, which can host ∼60 different chemical elements as major and minor components (Geiger, 2013 ▸; Baxter et al., 2013 ▸). Furthermore, the so-called Li-stuffed garnets, e.g. Li4La3Zr2Li3O12, or as sum formula, Li7La3Zr2O12 (LLZO), have raised particular interest as promising materials for use as solid-state electrolytes in all solid-state Li batteries due to their superior Li-ion conductivity (Cussen, 2010 ▸; Wang et al., 2020 ▸; Murugan et al., 2007 ▸; Samson et al., 2019 ▸). Pure end-member LLZO is tetragonal, has the space group I41/acd (Awaka et al., 2009 ▸) and has distinctly lower Li-ion conductivities. The high Li-ion conductivity is associated with the ‘standard’ cubic garnet structure with Ia
d symmetry. The latter can be stabilized by various aliovalent substitutions, e.g. by small amounts of AlIII, which – in the first experiments – entered the structure as a contaminant from the corundum crucibles during synthesis (Geiger et al., 2011 ▸; Buschmann et al., 2011 ▸). The incorporation of GaIII into LLZO also increases the Li-ion conductivity, but induces a reduction of the symmetry to I
3d (Rettenwander et al., 2016 ▸; Wagner et al., 2016a
▸; Robben et al., 2016 ▸), i.e. the space group of hydrogarnet Ca3Al2(O4H4)3 (Lager et al., 1987 ▸). A stabilization of the cubic structure for nominally pure LLZO is also possible by the uptake of HI, which raises a question about LLZO stability in hydrous environments (Larraz et al., 2013 ▸). Several studies have investigated the role of LiI/HI exchange under different environmental conditions and found that LLZO-type materials are distinctly unstable in the presence of moisture (Ma et al., 2015 ▸; Galven et al., 2011 ▸, 2012 ▸, 2013 ▸; Larraz et al., 2015 ▸; Orera et al., 2016 ▸; Liu et al., 2019 ▸). Surfaces quickly degrade with the formation of LiOH and Li2CO3, and an increase in unit-cell parameters is observed as LiI/HI exchange progresses. Until now, only a few studies have systematically investigated the mechanisms behind this structural degradation. In recent studies, we have shown, using diffraction methods, that significant LiI is especially lost from the interstitial sites of the structure in AlIII-, GaIII- and TaV-substituted LLZOs during ageing at room temperature under high humidity (Hiebl et al., 2019 ▸; Redhammer et al., 2021a
▸,b
▸). In this article, we report on the deep hydration of Al- and Ga-substituted LLZO using hydrothermal treatment of single-crystalline material.
Experimental methodology
Synthesis and aging of material
Single crystals of Al- and Ga-stabilized LLZO were obtained using a solid-state ceramic sintering method, which has been described in detail elsewhere (Rettenwander et al., 2016 ▸; Wagner et al., 2016a ▸). In brief, Li2CO3 (with an excess of 10%), La2O3, ZrO2 and Al2O3 or Ga2O3 were carefully mixed in the required stoichiometric proportions for the nominal compositions Li6.55La3Zr2Al0.15O12 (Al15-LLZO) and Li5.8La3Zr2Ga0.4O12 (Ga40-LLZO). The mixtures were pressed into pellets and preheated at 850 °C for 4 h for decarbonatization. The samples were subsequently milled in a high-energy ball mill under alcohol using ZrO2 balls, then dried, pelletized and sintered at 1230 °C for 6 h. This yielded a dense ceramic consisting of large individual crystallites of up to 150 µm. Structural refinements were carried out on the fresh LLZOs within 24 h of synthesis to assess the unaltered structural state. Parts of the pellets were crushed carefully and aged in a 45 ml Teflon-lined autoclave (25 mg sample and 250 ml distilled water) at 150 °C for a period of 28 d. The pH value of the liquid, in which the crystals were submerged, was measured after the experiment; the pH value was ∼13 for both compositions. The hydrated single LLZO crystallites were then filtered off and the remaining liquid was left to evaporate. The resulting precipitate was identified as Li2CO3 using powder diffraction (PXRD), i.e. proving that LiI was extracted from the LLZO material.
Refinement
Information on data collection and refinement results is given in Table 1 ▸. Indexing the diffraction data for pristine Al-LLZO yields the space-group symmetry Ia
d. Refining the structure with framework cations and O atoms results in only two strong residual electron-density peaks at the 24d and 96h positions. Assuming that Li atoms occupy only these two positions, then there must be a distinct overpopulation at the 24d site. Thus, AlIII is assigned to the 24d site and its content was fixed using the chemical composition calculated from energy dispersive X-ray (EDX) analysis on a similar material synthesized using an identical experimental setup (Rettenwander et al., 2016 ▸). The Li content was allowed to refine freely.
Table 1. Experimental details.
For all structures: Z = 8. Experiments were carried out at 298 K with Mo Kα radiation using a Bruker SMART APEX diffractometer. Absorption was corrected for by multi-scan methods (APEX2; Bruker, 2012 ▸). LLZO-Al15-pristine = fresh sample of Al-doped LLZO, measured directly after the end of the synthesis, LLZO-Al15-hydro-150C = Al-doped LLZO aged hydrothermally at 150 °C, LLZO-Ga40-pristine = fresh sample of Ga-doped LLZO, measured directly after the end of the synthesis and LLZO-Ga40-hydro-150C = Ga-doped LLZO aged hydrothermally at 150 °C.
| LLZO-Al15-pristine | LLZO-Al15-hydro-150C | LLZO-Ga40-pristine | LLZO-Ga40-hydro-150C | |
|---|---|---|---|---|
| Crystal data | ||||
| Chemical formula | Al0.15La2.95Li5.73O12Zr2 | Al0.15H5.52La2.88Li1.64O12Zr1.95 | Ga0.28La2.94Li6.44O12.00Zr2.00 | Ga0.26H3.90La2.96Li1.99O12Zr2 |
| M r | 827.89 | 790.95 | 847.67 | 821.46 |
| Crystal system, space group | Cubic, I a\overline{3}d | Cubic, I\overline{4}3d | Cubic, I\overline{4}3d | Cubic, I\overline{4}3d |
| a (Å) | 12.9637 (2) | 13.0738 (2) | 12.9669 (2) | 13.06720 (12) |
| V (Å3) | 2178.65 (10) | 2234.63 (10) | 2180.26 (10) | 2231.25 (6) |
| μ (mm−1) | 13.24 | 12.61 | 13.88 | 13.57 |
| Crystal size (mm) | 0.12 × 0.11 × 0.07 | 0.13 × 0.12 × 0.08 | 0.13 × 0.13 × 0.10 | 0.12 × 0.11 × 0.09 |
| Data collection | ||||
| T min, T max | 0.22, 0.39 | 0.21, 0.36 | 0.19, 0.25 | 0.21, 0.36 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 33130, 455, 441 | 34201, 819, 819 | 35033, 1054, 1045 | 35657, 918, 915 |
| R int | 0.029 | 0.026 | 0.038 | 0.028 |
| (sin θ/λ)max (Å−1) | 0.840 | 0.804 | 0.884 | 0.838 |
| Refinement | ||||
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.016, 0.032, 1.51 | 0.013, 0.029, 1.27 | 0.013, 0.026, 1.29 | 0.018, 0.037, 1.12 |
| No. of reflections | 455 | 819 | 1054 | 918 |
| No. of parameters | 25 | 44 | 48 | 43 |
| No. of restraints | 0 | 1 | 2 | 3 |
| H-atom treatment | – | Only H-atom coordinates refined | – | Only H-atom coordinates refined |
| Δρmax, Δρmin (e Å−3) | 0.71, −0.44 | 0.58, −0.51 | 0.53, −0.65 | 0.54, −0.52 |
| Absolute structure | – | Refined as an inversion twin | Refined as an inversion twin | Refined as an inversion twin |
| Absolute structure parameter | – | 0.50 (3) | 0.50 (4) | 0.99944 (15) |
For pristine Ga-stabilized LLZO, indexing of the diffraction data yields the space-group symmetry I
3d. Three different sites are identified from residual electron-density maps for the LiI ions: at Wykoff positions 12a, 12b and 48e. It is evident that GaIII must be located at the 12a position, as the 12a site becomes distinctly overpopulated when refined with only LiI. In subsequent refinements, both the Li1 and Li2 sites are assumed to be fully occupied and the electron density is modelled with Li + Ga = 1. The result is that GaIII almost exclusively resides at the 12a position. Refinement of the anisotropic atomic displacement parameters (adps) is possible for all atoms using the same strategy as that applied by Wagner et al. (2016a
▸,b
▸). The data of the hydrothermally treated Ga-LLZO sample can also be indexed using I
3d symmetry when the model for untreated material is used as the starting point. The Li1 site is again assumed to be fully occupied and its electron density was modelled with Li + Ga = 1. This approach is considered valid as the resultant GaIII content is similar (albeit slightly higher) to that obtained for the untreated sample. The Li2 and, in particular, the Li3 sites are distinctly depleted in Li and no anisotropic atomic displacement refinement is possible. Thus, the isotropic adps are adjusted and fixed to the U
eq value refined for the Li1 site; anisotropic adps could be obtained for Li1. Protons are located close to the O1 atom using residual electron-density maps. Fixing the U
eq value of hydrogen yields reliable occupation factors for this site and an almost charge-balanced chemical formula (with a slight surplus of 0.35 negative charges).
For deeply hydrated Al-LLZO, indexing of data yields a change in symmetry from Ia
d to acentric I
3d. The structure of this compound is refined using the model of the Ga-stabilized LLZO, as described above for the La-, Zr- and the two O-atom positions. The electron densities at Li1 and Li2 were modelled first with only Li+ ions. In this case, the Li1 position is distinctly overpopulated, while the occupation of the Li2 position is low, and there is no indication that the Li3 position is occupied at all. Consequently, all AlIII is assigned to the Li1 site and its occupation is fixed to the value used in the unaltered sample, consistent with the assumption that no Al left the structure during hydration. Refinement of the anisotropic adps is not possible for the Li1 site, and the isotropic adp is very small, so the isotropic adp of Li1 was adjusted in such a way that it has a similar value to the U
eq value of the Li2 site (where anisotropic adp refinements were possible) and fixed as such in subsequent refinements. Two distinct residual electron-density peaks are identified in residual electron-density maps: one high (2.5 e Å−3), very close to the Zr-atom position, and another at ∼0.8 Å from the O1 atom. This latter (x, y, z) position is close to the proposed H-atom positions given by Larraz et al. (2013 ▸) and Orera et al. (2016 ▸). Hence, this residual density is assigned to the H atom, which is bonded to the O1 atom. Independent refinement of the x, y and z positions, and the occupation of the H atom is possible, whereas the isotropic adp had to be fixed. A residual density close to Zr can be explained by positional disorder at this site, similar to that reported for deeply hydrated Ta-substituted LLZO, which also transformed to the I
3d structure. However, there is a marked decrease in the reliability factors associated with the refinements when ZrIV disorder is applied, e.g. there is no sign of another site close to O2 that would allow for another H atom to be bonded to the O2 atom. Furthermore, no reliable residual electron-density peaks can be detected.
Two additional crystals were hydrated in the same way and both were then analysed; the results are consistent with those reported in the tables and below.
Results and discussion
In the pristine state, Al-LLZO shows the typical garnet structure with Ia
d symmetry. LaIII occupies the eightfold-coordinated 24c site with two symmetrically independent La—O bond lengths (see Table 2 ▸). A slight deficit in the LaIII ion site occupation is observed, which is in line with previous studies (Hiebl et al., 2019 ▸; Rettenwander et al., 2016 ▸; Wagner et al., 2016a
▸,b
▸). The ZrIV ions are located at the 16a position with a regular sixfold oxygen coordination and a bond length of 2.076 (16) Å. As depicted in Fig. 1 ▸, the garnet structure comprises an integrated framework constructed of edge-sharing octahedral and dodecahedral sites, in which the Li atoms are located at both the regular 24d tetrahedral site (Li1) and at the interstitial 96h position (Li2), often denoted to have a distorted octahedral coordination [Li2—O distances range between 1.854 (15) and 2.646 (14) Å, with the average of the four smaller bond lengths being 2.085 Å]. AlIII substitutes into the 24d position and the four equivalent Li—O lengths are 1.9044 (17) Å; both the 24d and the 96h positions show distinct vacancies.
Table 2. Selected geometric parameters (Å).
| LLZO-Al15-pristine | |||
| La1—O1i | 2.5110 (17) | Li2—O1 | 1.854 (15) |
| La1—O1 | 2.5951 (17) | Li2—O1vii | 2.085 (14) |
| Zr1—O1ii | 2.1076 (16) | Li2—O1vi | 2.159 (15) |
| Li1—Li2iii | 1.616 (14) | Li2—O1viii | 2.242 (14) |
| Li1—O1iv | 1.9044 (17) | Li2—Li2viii | 2.46 (2) |
| Li1—Li2v | 2.367 (14) | Li2—O1ix | 2.646 (14) |
| Li2—Li2vi | 0.79 (3) | ||
| LLZO-Al15-hydro-150C | |||
| La1—O1i | 2.506 (3) | Zr1B—O2xii | 2.06 (3) |
| La1—O1 | 2.518 (3) | Zr1B—O2iv | 2.10 (3) |
| La1—O2x | 2.544 (3) | Zr1B—O1ii | 2.14 (3) |
| La1—O2i | 2.607 (3) | Zr1B—O1xiv | 2.19 (3) |
| Zr1A—O1xi | 2.007 (4) | Zr1B—O1xi | 2.36 (3) |
| Zr1A—O2xii | 2.217 (4) | Li1—O1xv | 1.998 (3) |
| Zr1B—O2xiii | 1.85 (3) | Li2—O2xvi | 1.976 (3) |
| LLZO-Ga40-pristine | |||
| La1—O2xiii | 2.4935 (19) | Li2—O2xvi | 1.9246 (19) |
| La1—O1i | 2.5264 (19) | Li2—Li3xviii | 2.340 (9) |
| La1—O2i | 2.587 (2) | Li3—O1 | 1.878 (14) |
| La1—O1xvii | 2.5975 (19) | Li3—O1vii | 2.075 (8) |
| Zr1—O2xiii | 2.0823 (18) | Li3—O2i | 2.108 (9) |
| Zr1—O1ii | 2.1346 (18) | Li3—O1viii | 2.229 (8) |
| Li1—Li3iii | 1.645 (8) | Li3—Li3vii | 2.513 (13) |
| Li1—O1xv | 1.8941 (18) | Li3—O2vii | 2.639 (8) |
| LLZO-Ga40-hydro-150C | |||
| La1—O1i | 2.514 (3) | Li1—Li3iii | 1.5 (2) |
| La1—O1xvii | 2.525 (3) | Li1—O1xv | 1.987 (4) |
| La1—O2xiii | 2.539 (4) | Li2—O2xvi | 1.982 (4) |
| La1—O2i | 2.603 (4) | Li2—Li3xviii | 3.2 (2) |
| Zr1—O1ii | 2.031 (3) | Li3—O1vii | 2.4 (2) |
| Zr1—O2xiii | 2.187 (4) | Li3—Li3viii | 2.6 (4) |
Symmetry codes: (i) z, x, y; (ii) x − {1\over 4}, z − {1\over 4}, y − {1\over 4}; (iii) −z + {3\over 4}, y − {1\over 4}, −x + {1\over 4}; (iv) z, −x, −y + {1\over 2}; (v) −y + {1\over 2}, z − {1\over 2}, x; (vi) −x + {1\over 4}, z − {1\over 4}, y + {1\over 4}; (vii) y − {1\over 4}, −x + {1\over 4}, −z + {3\over 4}; (viii) −y + {1\over 4}, x + {1\over 4}, −z + {3\over 4}; (ix) y, −z + {1\over 2}, x + {1\over 2}; (x) −x, y − {1\over 2}, −z + {1\over 2}; (xi) z − {1\over 4}, y − {1\over 4}, x − {1\over 4}; (xii) −y + {1\over 2}, z, −x; (xiii) −x, −y + {1\over 2}, z; (xiv) y − {1\over 4}, x − {1\over 4}, z − {1\over 4}; (xv) −z + {3\over 4}, −y + {1\over 4}, x + {1\over 4}; (xvi) x + {3\over 4}, −z + {1\over 4}, −y + {3\over 4}; (xvii) x, −y, −z + {1\over 2}; (xviii) y + {3\over 4}, −x + {1\over 4}, −z + {3\over 4}.
Figure 1.
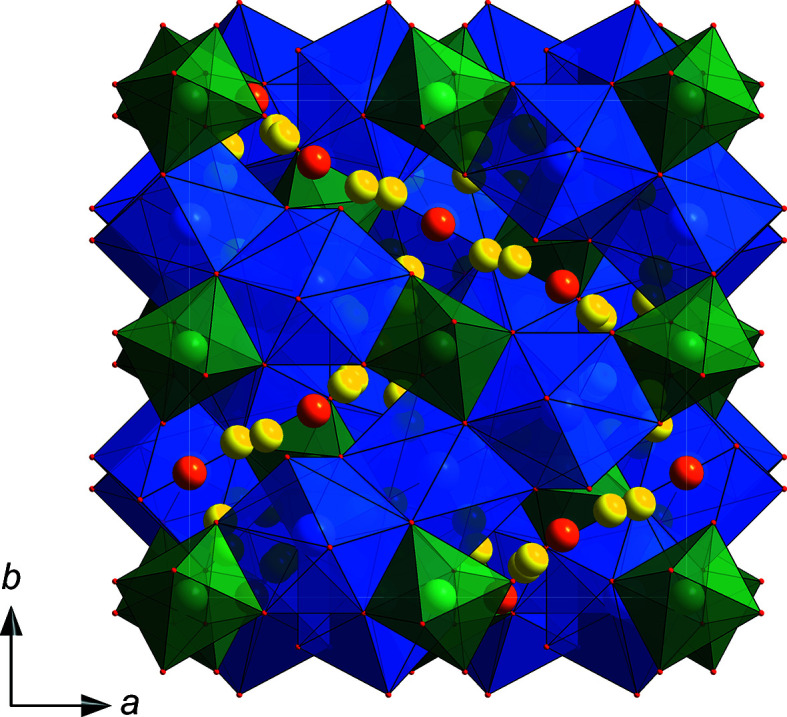
The crystal structure of Al-stabilized LLZO (space group Ia
d) in a polyhedral format. LaO8 sites are in light blue and ZrO6 octahedra in sea green, whereas the Li1 (orange) and Li2 sites (yellow) are shown as spheres only for clarity and to highlight their diffusion pathway.
The hydrothermally altered sample of Al-LLZO shows additional Bragg peaks of type k = odd and l = odd that obey Ia
d symmetry. Calculated precession images of the hk0 plane for unaltered and altered Al-LLZO are compared in Fig. 2 ▸, where some Bragg peaks that obey Ia
d symmetry are marked. Indexing of the observed data definitively yield the space-group symmetry I
3d, in accordance with the findings of Larraz et al. (2013 ▸) and Orera et al. (2016 ▸) for LiI/HI-exchanged LLZO. The symmetry reduction is associated with several rearrangements in the structural architecture. The oxygen site at 96h in Ia
d splits into two different 48e positions, i.e. O1 and O2, in I
3d, thus allowing for two different sets of Zr—O and four different La—O bond lengths. The ZrIV ion shifts from (0, 0, 0) with site symmetry
(Ia
d) to the 16c position (x, x, x) with site symmetry 3 (I
3d). Some positional disorder is observed at the Zr position, where around 20% of the ZrIV is displaced to a general 48e position, with a Zr—Zr offset of ∼0.39 (3) Å. Free refinement of the site occupancies of these two Zr positions total 1.96 ZrIV atoms per formula unit, i.e. very close to the expected value of 2.0. Whereas in Ia
d, the six Zr—O bond lengths are equivalent to a value of 2.1076 (16) Å. In the hydrothermally altered structure, ZrIV is in a very distorted octahedral environment at 16c, with Zr1A—O1xi bond lengths of 2.007 (4) Å (×3), and Zr1A—O2xii of 2.217 (4) Å (×3) (see Table 2 ▸ for symmetry codes). For the general position, the Zr1B—O bonds range between 1.85 (3) and 2.36 (3) Å (Table 2 ▸). It is interesting to note that a similar behaviour was found during hydrothermal alteration of Li6La3ZrTaO12 (LLZTO). Pristine LLZTO shows Ia
d space-group symmetry but deep hydration induces a symmetry reduction to I
3d. A disorder at the Zr/Ta site is also observed in hydrated LLZTO similar to that in hydrated Al-stabilized LLZO in this study. It would appear that the symmetry reduction, induced by LiI/HI exchange, causes a large distortion of the O-atom environment around the 16c position and induces some positional disorder.
Figure 2.
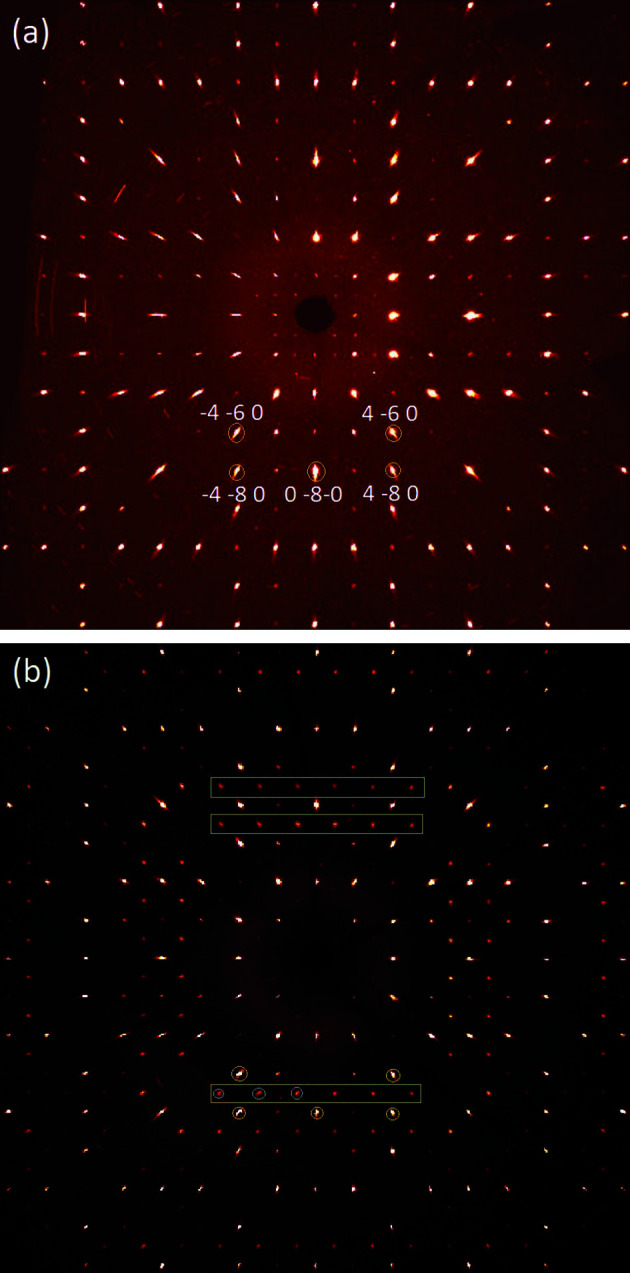
Reconstructed precession images of the hk0 plane of (a) a single crystal of Al-stabilized LLZO directly after synthesis with some selected Bragg reflections indexed and (b) a single crystal after hydrothermal alteration at 150 °C for 28 d. Note the presence of sharp superstructure reflections, which obey Ia
d symmetry. In part (b), the yellow encircled Bragg reflections are the same as in part (a), while the blue encircled reflections in the rectangular box correspond (from left to right) to the
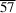 0,
0,
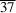 0,
0,
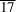 0,… Bragg reflections.
0,… Bragg reflections.
The regular 24d tetrahedral site of the Ia
d garnet structure splits into two different sites, 12a (Li1) and 12b (Li2), upon symmetry reduction to I
3d. AlIII is ordered onto the 12a (Li1) site but a distinct number of vacancies are observed on both sites. While ∼4.5 apfu LiI occupy the interstitial 96h position in the pristine state, this position (Li3 at 48e) is completely unoccupied in the deeply hydrated form, i.e. all the LiI ions have vacated the interstitial octahedral site.
Recently, Redhammer et al. (2021b
▸) observed a progressive increase of the tetrahedral site (24d and 12a) occupation in Al-stabilized LLZO during continuous LiI/HI exchange in a humid atmosphere and under mild hydrothermal conditions. The shift and ordering of LiI from the interstitial site to the regular tetrahedral site, and the preference of LiI and AlIII for the 12a position, are described as triggers for the symmetry reduction from Ia
d to I
3d (Redhammer et al., 2021b
▸). A study of the structure of the deeply hydrated samples here shows that, after complete recovery of LiI from the interstitial site, the tetrahedral 12a and 12b sites also take part in the LiI/HI exchange. When compared to the data of Redhammer et al. (2021b
▸), it is obvious that, in this study, more LiI is extracted from the 12a site [∼1.4 to 0.64 (2) apfu], but there is only a moderate change in site occupation at 12b [∼1.1 to 0.82 (2) apfu]. As LiI is extracted from the structure, HI is incorporated and bonds with the O1 atom. The LiI ions in the 12a tetrahedron are thus coordinated by four OH groups, with the O—H vector pointing towards the empty Li3 site (compare with Fig. 3 ▸). The proposed position of HI is in line with that found by Orera et al. (2016 ▸) for pure undoped LLZO based on neutron diffraction on polycrystalline powders. Refinements indicate that the deeply hydrated Al-LLZO has a composition of La2.88Zr1.95Al0.15Li1.64H5.52O12. It is also worth noting that a significantly lower number of LaIII ions are found at the 24d site, so it would appear that LaIII also leaves the structure under hydrothermal conditions. This is in line with the observations of Redhammer et al. (2021a
▸,b
▸), who observed an instability of LLZO powders in highly humid air, with decomposition of LLZO, leading to the formation of lanthanite La2(CO3)3·8H2O within ∼30 d of exposure.
Figure 3.
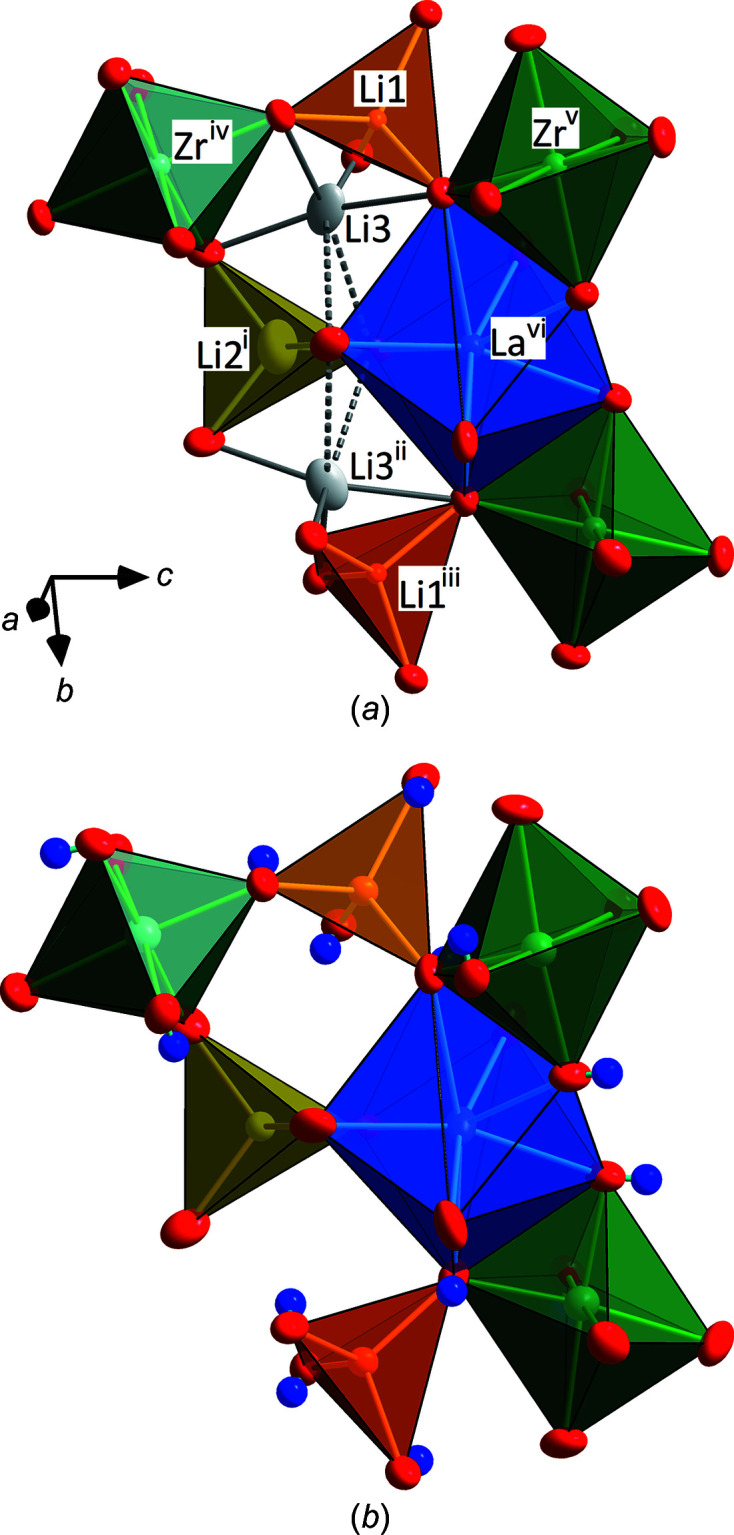
Polyhedral representations of part of the crystal structure of Ga-stabilized LLZO (space group I
3d) in the (a) pristine and (b) hydrothermally altered state viewed on the (12,
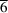 ,3) plane. LaO8 sites are in light blue, ZrO6 octahedra in green and Li1, Li2 and Li3 sites in orange, yellow and grey, respectively. The dashed grey bonds around the Li3 site are the two most distant. [Symmetry codes: (i) −z +
,3) plane. LaO8 sites are in light blue, ZrO6 octahedra in green and Li1, Li2 and Li3 sites in orange, yellow and grey, respectively. The dashed grey bonds around the Li3 site are the two most distant. [Symmetry codes: (i) −z +
 , −y +
, −y +
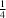 , x −
; (ii) −z + 1, −x +
, x −
; (ii) −z + 1, −x +
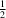 , y; (iii) −x + 1, −y +
, z; (iv) −x +
, y, −z; (v) −x +
, −y, z +
; (vi) −z +
, −y +
, x +
.]
, y; (iii) −x + 1, −y +
, z; (iv) −x +
, y, −z; (v) −x +
, −y, z +
; (vi) −z +
, −y +
, x +
.]
The LiI/HI exchange is accompanied by a large increase in the a unit-cell parameter from 12.9637 (2) to 13.0738 (2) Å, which is among the largest values yet recorded for hydrated LLZO, cf. 13.06245 (4) Å at 77 K for hydrated LLZO with an Li2.3H4.7La3Zr2O12 composition (Orera et al., 2016 ▸) or 13.0530 (8) Å for Li3.08H3.52La3Zr2O12Ta0.4 (Yow et al., 2016 ▸). A replacement of stronger Li—O bonds by weaker O—H bonds and the creation of a large number of vacant sites, especially around the empty 48e (Li3) site, both require more space and are considered responsible for the lattice expansion.
Pristine unaltered Ga-stabilized LLZO shows I
3d symmetry. A section of this crystal structure is illustrated in Fig. 3 ▸, together with that of the hydrothermally treated sample. Both the tetrahedrally coordinated 12a and 12b positions appear to be fully occupied, with the GaIII ions ordered onto the 12a position. In contrast to the Li-stuffed garnets with Ia
d symmetry, the decreased number of vacancies at the regular tetrahedral sites are considered to be a characteristic feature of the I
3d garnet structure. The 48e interstitial site [Li3 in Fig. 3 ▸(a)] is occupied by 3.71 LiI apfu, equivalent to being ∼62% full. All of the tetrahedral faces of the fourfold oxygen coordination around the Li1 and Li2 sites are shared with neighbouring Li3 sites, with interatomic contacts of 1.645 (8) (Li1—Li3) and 2.340 (9) Å (Li2—Li3), thereby forming a three-dimensional network that is responsible for the good Li-ion conductivity in this compound. The dodecahedral site has a small number of vacancies, while the regular octahedral positions are fully occupied with ZrIV, resulting in a composition of La2.94Zr2.00Ga0.28Li6.43O12 for the unaltered Ga-stabilized LLZO material.
A smaller and larger set of La—O1/O2 bonds are observed at the dodecahedral site [2.4935 (19)–2.5264 (19) and 2.587 (2)–2.5975 (19) Å] and the octahedron is regular with two independent Zr—O bonds ranging between 2.0823 (18) and 2.1346 (18) Å. Therefore, the octahedra are much less distorted than those in the altered Al-LLZO with the same symmetry (Table 2 ▸). The Li1 (12a) site, which hosts GaIII, is slightly smaller than the Li2 (12b) site, whereas the Li3 site, as in Al-LLZO, shows a very distorted 4 + 2-fold coordination, with bond lengths between 1.878 (4) and 2.639 (8) Å; the average of the four shorter bonds is 2.073 Å, i.e. slightly smaller than in the pristine Al-LLZO [see Fig. 3 ▸(a)].
As in Al-LLZO, hydrothermal treatment of Ga-LLZO induces LiI/HI exchange. Again, a large increase of the lattice parameter to 13.06720 (12) Å is close to the value in hydrated Al-LLZO, suggesting that values around 13.07 Å represent an upper limit for lattice-parameter increase due to hydration. From site-occupation refinements, very minor LiI ions are found at the interstitial 48e position, i.e. this site is almost completely depleted. One key difference with Al-LLZO is that no LiI is lost from the 12a position. It seems that GaIII ions pin LiI at this site. However, LiI ions are extracted from the 12b site where the amount of LiI is reduced from 1.49 apfu in the pristine to 0.50 apfu in the altered sample. Protons are again located close to the O1 atom [Fig. 3 ▸(b)], giving rise to a fully hydrated Li1(OH)4 coordination around the 12a site. Some differences to Al-LLZO are, however, evident: there is no significant reduction in the La-site occupation and no residual electron density is observed close to the ZrIV ions, i.e. there is no indication of disorder at the 16c position in altered Ga-LLZO. Nevertheless, the ZrO6 octahedron is much more distorted in the altered sample, with Zr—O bond lengths ranging between 2.03 (3) and 2.187 (4) Å; the difference between the two independent Zr—O bonds increases with prolonged LiI/HI exchange. This was outlined by Redhammer et al. (2021b ▸) and the data of this study fit the extrapolated trends observed there. A distinct alteration is also observed within the coordination sphere of the 24d site due to HI incorporation. The most prominent effects include the reduction of the longer La1—O1xvii distance by ∼0.073 Å, as well as changes in the three other La—O bond lengths. In addition, Li1—O1xv and Li2—O2xvi are extended in the altered sample (see Table 2 ▸).
Supplementary Material
Crystal structure: contains datablock(s) global, LLZO-Al15-pristine, LLZO-Al15-hydro-150C, LLZO-Ga40-pristine, LLZO-Ga40-hydro-150C. DOI: 10.1107/S2053229621012250/ep3020sup1.cif
Structure factors: contains datablock(s) LLZO-Al15-pristine. DOI: 10.1107/S2053229621012250/ep3020LLZO-Al15-pristinesup2.hkl
Structure factors: contains datablock(s) LLZO-Al15-hydro-150C. DOI: 10.1107/S2053229621012250/ep3020LLZO-Al15-hydro-150Csup3.hkl
Structure factors: contains datablock(s) LLZO-Ga40-pristine. DOI: 10.1107/S2053229621012250/ep3020LLZO-Ga40-pristinesup4.hkl
Structure factors: contains datablock(s) LLZO-Ga40-hydro-150C. DOI: 10.1107/S2053229621012250/ep3020LLZO-Ga40-hydro-150Csup5.hkl
References
- Awaka, J., Kijima, N., Hayakawa, H. & Akimoto, J. (2009). J. Solid State Chem. 182, 2046–2052.
- Baxter, E. F., Caddick, M. J. & Ague, J. J. (2013). Elements, 9, 415–419.
- Bruker (2012). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., Giacovazzo, C., Mallamo, M., Mazzone, A., Polidori, G. & Spagna, R. (2012). J. Appl. Cryst. 45, 357–361.
- Buschmann, H., Dölle, J., Berendts, S., Kuhn, A., Bottke, P., Wilkening, M., Heitjans, P., Senyshyn, A., Ehrenberg, H., Lotnyk, A., Duppel, V., Kienle, L. & Janek, J. (2011). Phys. Chem. Chem. Phys. 13, 19378–19392. [DOI] [PubMed]
- Cussen, E. (2010). Functional Oxides, edited by D. W. Bruce, D. O’Hare & R. I. Walton, pp. 119–202: London: John Wiley & Sons Ltd.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Galven, C., Dittmer, J., Suard, E., Le Berre, F. & Crosnier-Lopez, M.-P. (2012). Chem. Mater. 24, 3335–3345.
- Galven, C., Fourquet, J.-L., Crosnier-Lopez, M.-P. & Le Berre, F. (2011). Chem. Mater. 23, 1892–1900.
- Galven, C., Suard, E., Mounier, D., Crosnier-Lopez, M.-P. & Le Berre, F. (2013). J. Mater. Res. 28, 2147–2153.
- Geiger, C. A. (2013). Elements, 9, 447–452.
- Geiger, C. A., Alekseev, E., Lazic, B., Fisch, M., Armbruster, T., Langner, R., Fechtelkord, M., Kim, N., Pettke, T. & Weppner, W. (2011). Inorg. Chem. 50, 1089–1097. [DOI] [PubMed]
- Hiebl, C., Young, D., Wagner, R., Wilkening, H. M. R., Redhammer, G. J. & Rettenwander, D. (2019). J. Phys. Chem. C, 123, 1094–1098.
- Lager, G. A., Armbruster, T. & Faber, J. (1987). Am. Mineral. 72, 756–765.
- Larraz, G., Orera, A. & Sanjuán, M. L. (2013). J. Mater. Chem. A, 1, 11419–11428.
- Larraz, G., Orera, A., Sanz, J., Sobrados, I., Diez-Gómez, V. & Sanjuán, M. L. (2015). J. Mater. Chem. A, 3, 5683–5691.
- Liu, X., Chen, Y., Hood, Z. D., Ma, C., Yu, S., Sharafi, A., Wang, H., An, K., Sakamoto, J., Siegel, D. J., Cheng, Y., Jalarvo, N. H. & Chi, M. (2019). Energy Environ. Sci. 12, 945–951.
- Ma, C., Rangasamy, E., Liang, C., Sakamoto, J., More, K. L. & Chi, M. (2015). Angew. Chem. Int. Ed. 54, 129–133. [DOI] [PubMed]
- Murugan, R., Thangadurai, V. & Weppner, W. (2007). Angew. Chem. Int. Ed. 46, 7778–7781. [DOI] [PubMed]
- Novak, G. A. & Gibbs, G. V. (1971). Am. Mineral. 56, 791–825.
- Orera, A., Larraz, G., Rodríguez-Velamazán, J. A., Campo, J. & Sanjuán, M. L. (2016). Inorg. Chem. 55, 1324–1332. [DOI] [PubMed]
- Redhammer, G. J., Badami, P., Meven, M., Ganschow, S., Berendts, S., Tippelt, G. & Rettenwander, D. (2021a). Appl. Mater. Interfaces, 13, 350–359. [DOI] [PubMed]
- Redhammer, G. J., Tippelt, G., Portenkirchner, A. & Rettenwander, D. (2021b). Crystals, 11, 721.
- Rettenwander, D., Redhammer, G., Preishuber-Pflügl, F., Cheng, L., Miara, L., Wagner, R., Welzl, A., Suard, E., Doeff, M. M., Wilkening, M., Fleig, J. & Amthauer, G. (2016). Chem. Mater. 28, 2384–2392. [DOI] [PMC free article] [PubMed]
- Robben, L., Merzlyakova, E., Heitjans, P. & Gesing, T. M. (2016). Acta Cryst. E72, 287–289. [DOI] [PMC free article] [PubMed]
- Samson, A. J., Hofstetter, K., Bag, S. & Thangadurai, V. (2019). Energy Environ. Sci. 12, 2957–2975.
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Wagner, R., Redhammer, G. J., Rettenwander, D., Senyshyn, A., Schmidt, W., Wilkening, M. & Amthauer, G. (2016a). Chem. Mater. 28, 1861–1871. [DOI] [PMC free article] [PubMed]
- Wagner, R., Redhammer, G. J., Rettenwander, D., Tippelt, G., Welzl, A., Taibl, S., Fleig, J., Franz, A., Lottermoser, W. & Amthauer, G. (2016b). Chem. Mater. 28, 5943–5951. [DOI] [PMC free article] [PubMed]
- Wang, C., Fu, K., Kammampata, S. P., McOwen, D. W., Samson, A. J., Zhang, L., Hitz, G. T., Nolan, A. M., Wachsman, E. D., Mo, Y., Thangadurai, V. & Hu, L. (2020). Chem. Rev. 120, 4257–4300. [DOI] [PubMed]
- Yow, Z. F., Oh, Y. L., Gu, W. Y., Rao, R. P. & Adams, S. (2016). Solid State Ionics, 292, 122–129.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, LLZO-Al15-pristine, LLZO-Al15-hydro-150C, LLZO-Ga40-pristine, LLZO-Ga40-hydro-150C. DOI: 10.1107/S2053229621012250/ep3020sup1.cif
Structure factors: contains datablock(s) LLZO-Al15-pristine. DOI: 10.1107/S2053229621012250/ep3020LLZO-Al15-pristinesup2.hkl
Structure factors: contains datablock(s) LLZO-Al15-hydro-150C. DOI: 10.1107/S2053229621012250/ep3020LLZO-Al15-hydro-150Csup3.hkl
Structure factors: contains datablock(s) LLZO-Ga40-pristine. DOI: 10.1107/S2053229621012250/ep3020LLZO-Ga40-pristinesup4.hkl
Structure factors: contains datablock(s) LLZO-Ga40-hydro-150C. DOI: 10.1107/S2053229621012250/ep3020LLZO-Ga40-hydro-150Csup5.hkl