

Abstract
Doxorubicin is an anthracycline antibiotic that is widely used to treat different types of malignancy. Here we studied whether doxorubicin could be used to render tumor cells susceptible to apoptosis by NK and T cells. Pretreatment with sub-apoptotic doses of doxorubicin sensitized tumor cell lines of various histotypes to both NK and T cells resulting in a 3.7 to 32.7 % increase in lysis (2.5 mean fold increase, p<0.0001) and 2.9 to 14.2 % increase in lysis (3.0 mean fold increase, p<0.05) respectively. The sensitizing effect of the drug was primarily dependent on TRAIL/TRAILR signaling, but not on Fas-ligand (FasL), perforin, NKG2D or DNAM-1. The central role of the TRAIL signaling pathway was further supported by an increased expression of TRAIL-R2 on doxorubicin-treated tumor cells and by down-regulation of cFLIP, the inhibitors of death receptor-mediated apoptosis. Compared to untreated cells, pretreatment of tumor cells with doxorubicin showed increased processing and activation of caspase-8 upon co-culture with NK or T cells. The significance of this treatment strategy was confirmed using a xenogeneic tumor-bearing mouse model. Tumor progression was delayed in mice that received either NK cells (p<0.05) or T cells (p<0.0001) following doxorubicin treatment compared to mice receiving either cell type alone. Moreover, combined infusion of both NK and T cells following doxorubicin treatment not only delayed tumor progression, but significantly improved the long-term survival (p<0.01). Based on these findings we propose that doxorubicin can be used to improve the efficacy of adoptive cell therapy in patients with cancer.
Keywords: Doxorubicin, Adoptive cell therapy, Natural killer cells, Tumor-infiltrating lymphocytes
Introduction
Several studies have demonstrated that intratumoral infiltration of T and NK cells correlates with favorable prognosis in patients with cancer 1–3. Although of clinical benefit, tumor-infiltrating T cells can exert an immunological pressure leading to tumor escape variants 4–5. Two relatively frequent immune escape events include down-regulation of tumor antigens and MHC class I 6–8. If such events can be offset, clinical responses of adoptive cell therapy may be improved. Previous studies have demonstrated that several chemotherapy agents have immune-sensitizing effects on tumors through up-regulation of death receptors 9–11. The death ligand TNF-related apoptosis-inducing ligand (TRAIL) binds to TRAIL-R1 and -R2 which triggers an apoptotic signal initiated by cleavage of caspase-8 12–13. In contrast to tumor cells, non-neoplastic cells tend to express the TRAIL decoy receptors TRAIL-R3 and -R4, which render them less sensitive to TRAIL-mediated killing 14–16. We recently demonstrated that exposure to the proteasome inhibitor bortezomib induces increased expression of TRAIL-R2 in a variety of tumor cells leading to increased susceptibility to TRAIL-mediated killing 17. These findings, in combination with reports from studies evaluating the safety and clinical efficacy of recombinant TRAIL (rTRAIL) or monoclonal antibodies directed against TRAIL-R1 and -R2 18–21, have warranted additional studies on the synergistic potential of combining chemotherapy with TRAIL targeted therapy. The topoisomerase II inhibitor doxorubicin have previously been demonstrated to sensitize tumors to killing by rTRAIL 16, 22–25. Upon activation, both NK and T cells express high levels of TRAIL 26–27. In order to increase the tumor immunogenicity to improve targeting of immune escape variants with differential expression of MHC class I, we studied whether conventional chemotherapeutic drugs sensitize tumors to combined NK and T cell therapy. Our data demonstrate that sub-therapeutic doses of doxorubicin sensitize human tumor cells to killing by NK and T cells via augmented TRAIL signaling. Furthermore, treatment with doxorubicin in combination with infusion of both NK and T cells significantly delayed tumor progression and prolonged survival in tumor-bearing mice. Our findings present a novel concept that administration of doxorubicin prior to adoptive transfer of ex vivo expanded NK cells and/or T cells augment the anti-tumor effect of the infused cells and that the sensitization of tumor cells to NK and T cell-mediated lysis is a result of increased signaling via the TRAIL/TRAIL-R pathway.
Materials and Methods
Isolation of NK cells and tumor specific T cells
NK cells were isolated from PBMC from healthy donors (ethical permission # 20010305, 01-50) or melanoma patients (ethical permission # 2009/848-3) by negative selection using immunomagnetic beads (Miltenyi Biotech, Bergisch Gladbach, Germany). Before expansion, isolated NK cells were assessed for purity by flow cytometry using human anti-CD3 and CD56 monoclonal antibodies (Biolegend, San Diego, CA, USA). Purified NK cells (range 95.3 ± 6.1 % purity with no T cell contamination) were co-cultured with either irradiated autologous PBMC (25Gy) or allogeneic EBV transformed B cells (100Gy) at a 10:1 ratio (feeder cells:NK cells) in X-vivo 20 medium (Lonza, Verviers, Belgium) supplemented with 5% AB serum (Karolinska Hospital, Stockholm, Sweden) and 1000 U/ml IL-2 (Novartis Pharma GmbH, Nurnberg, Germany) at 37°C. Medium was replenished with 500 U/ml IL-2 on days 5 and 8 after expansion start. NK cells were tested for their cytotoxic activity on days 11–14. Biopsies from HLA-A2 positive melanoma patients (ethical permission # 2009/848-3) were dissected into small pieces and transferred to 24-well plates in X-vivo 20 medium supplemented with 5% AB serum and 6000 U/ml IL-2. Tumor infiltrating lymphocytes (TILs) were stained with monoclonal antibodies directed against CD56, CD4, and CD8 (Biolegend, San Diego, CA, USA) and screened for antigen-specificity by intracellular staining with antibodies against interferon-gamma (IFNγ) and IL-2 (Biolegend, San Diego, CA, USA), after co-culture with T2 cells pulsed with HLA-A2 binding peptides; gp100, MAGE-3, Survivin and Mart-1 (Genecust, Dudelang, Luxembourg). 8.4 % of the cells from the TIL culture stained positive for IFNγ and IL-2 after co-culture with Mart-1(ELAGIGILTV) peptide pulsed T2 cells. None of the other epitopes tested were recognized by the TILs. Thereafter, TILs were rapidly expanded for two weeks in X-vivo 20 media supplemented with 50 ng/ml anti-CD3 antibody (Janssen-Cilag, Sollentuna, Sweden) and irradiated (40Gy) allogeneic PBMC at 37°C. IL-2 (500U/ml) was added to the culture medium on days 1, 5 and 8. TIL cultures were on average 97.1 ± 2.8 % CD3 positive with no NK cell contamination. Killing by NK and T cells was confirmed against K562 cells (supplemental figure 1A). The reason for using our Mart-1 specific tumor cells from a melanoma patient to use as effector T-cells against the Mart-1 positive J82 cell line. There are not any known methods to obtain TILs from bladder biopsies and that type of material is not accessible to us. Therefore, we used TILs from a melanoma patient that was HLA-matched with J82 and that recognized the Mart-1 antigen which is expressed on J82.
Cell lines
The melanoma cell line GEWA-MEL was established from tumor biopsy and was passaged not more than 8 times when tested for functional and phenotypical properties. Cells were grown at 37°C in 20% RPMI1640 medium (Gibco, Paisley, UK), 20% F12 HAM medium (Biological Industries, Beit Ha’emek, Israel), 60 % DMEM medium (Gibco, Paisley, UK), 25mM Hepes (Gibco, Paisley, UK), 200 mM L-Glutamine (Gibco, Paisley, UK), 100μg/ml Streptamycin (Gibco, Paisley, UK), 100 U/ml Penicillin (Gibco, Paisley, UK) supplemented with 20% FBS (Gibco, Paisley, UK). Cell lines tested include; melanoma: EST074, EST115, EST149, EST025, EST066, EST112, JUSO; bladder carcinoma: J82, renal cell carcinoma: A498, MAR, JOHW; colorectal carcinoma: HCT116; breast carcinoma: T47D; prostate carcinoma: DU145; fibroblast cell lines: BJ1, AG08164. The JOHW and MAR cell lines were established in NHLBI/NCI laboratories from surgically resected tumor specimens. All the cell lines were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, 25 mM Hepes, 100 μg/ml streptomycin, and 100 U/ml penicillin. STR identifier (Applied biosystems, Warrington, UK) was used to verify the origin of tumor cell lines EST112, EST074, EST149, EST025, EST066, EST115 and J82. Tumor cell lines were stained with monoclonal antibodies against TRAIL-R1, TRAIL-R2, MICA, MICB, ULBP (pan-antibody), MHC class I, HLA-E, FAS and acquired by LSR II flow cytometer (BD Biosciences, San Jose, CA, USA). All flow cytometry data was analyzed using the FlowJo software (Tree Star, Ashland, OR, USA). The J82 cell line was chosen for our mechanistic studies due to its resistance to killing by NK cells and T cells.
Killing and degranulation assay
Target cells were treated for 16 hours with 150–500 ng/ml doxorubicin (TEVA, Helsingborg, Sweden). For each cell line, the dose of doxorubicin was titrated to levels where sensitization of tumor cells was achieved without affecting the viability of the cells. (Supplemental figure 1B). Subsequently, cells were washed and labeled with radioactive 51Cr (30μCi) (PerkinElmer, Groningen, The Netherlands) for 1 hour at 37 °C. Expanded NK and T cells were seeded in a 96-well V-bottom plate (Corning lifesciences, New York, NY, USA) together with target cells (5,000 cells/well) and incubated for 4–18 hours at 37°C at Effector-to-Target (E:T) ratios ranging between 0.1:1–10:1. Supernatants were transferred to 96 well LUMA-plates (Perkin Elmer, Groningen, Netherlands) and analyzed for chromium release in a Microbeta scintillation counter (TRILUX 1450, Wallac, Perkin Elmer, Groningen, The Netherlands). Specific lysis percentage was calculated according to the formula [(test sample release - spontaneous release) / (maximum release - spontaneous release)] * 100. Spontaneous release represents 51Cr release from target cells in medium alone and maximum release represents the 51Cr release from target cells lysed in medium plus 5% Triton X-100 (Sigma-Aldrich, Saint Louis, Missouri, USA). In blocking experiments, prior to co-culture with target cells, effector cells were pre-treated with blocking antibodies against TRAIL (RIK-2, 10μg/ml), Fas-ligand (NOK-1, 2.5μg/ml), NKG2D (1D11, 10μg/ml), DNAM-1 (5 μg/ml) or NKp44 (P44-8, 10μg/ml) (LEAF purified anti-human antibodies, Biolegend, San Diego, CA, USA). Concanamycin A (CMA, 100nM, Sigma-Aldrich, Saint Louis, Missouri, USA) was used to degrade perforin in killing assays. For degranulation assay, tumor cells were co-cultured with NK cells for 1 hour and stained for CD3, CD56, Annexin-V and Live/Dead cell marker and degranulation was measured by staining for CD107a (Biolegend, San Diego, CA, USA) and acquired on a Fortessa LSRII (Becton Dickinson, CA, USA). Statistical analysis of cytotoxicity data was performed with Student’s t-test.
RNA interference
Cells were seeded in 6-well plates 24 hours before transfection. Non-targeting siRNA (D-001810-10, Dharmacon, Chicago, IL, USA) and caspase-8 siRNA (L-003466-00, Dharmacon, Chicago, IL, USA) were diluted in 50 μl of OPTI-MEM I medium (Gibco, Paisley, UK) and mixed with 4 μl of INTERFERin siRNA Transfection Reagent (Polyplus-transfection, 409-10, Illkirch, France). After a 10 min incubation at room temperature the complexes were added to the cells. The final concentration of siRNA in the medium was 50 nM. Cells were tested in cytotoxicity assay 24 hours after transfection.
Immunoblotting
Cells were lysed in CelLytic M Cell lysis reagent (Sigma, St Louis, MO, USA), supplemented with protease inhibitor cocktail (Sigma St Louis, MO, USA). The protein concentration was determined using the BCA protein assay (Pierce, Rockford, IL, USA). The samples were mixed with Laemmli’s buffer, boiled for 5 min, subjected to SDS-PAGE and blotted onto nitrocellulose membrane (Bio-Rad Laboratories, Germany), which was blocked for 1 hour with 5% non-fat milk in PBS and probed with primary antibodies diluted in PBS containing 2 % BSA (A3059, Sigma, St Louis, MO, USA) and 0.05 % Tween-20 (P1379, Sigma, St Louis, MO, USA). The following antibodies were used: mouse anti-caspase-8 (kindly provided by Drs P. Krammer and I. Lavrik, German Cancer Research Center (DKFZ), Heidelberg, Germany), anti-c-FLIP (Cell signaling, Danvers, MA, USA), anti-b-actin (Sigma St Louis, MO, USA) and anti-GAPDH (Trevigen, Gaithersburg, MD, USA). The recognized proteins were detected using horseradish peroxidase-labeled secondary antibodies: anti-mouse IgG (Pierce, Rockford, IL, USA), anti-rabbit IgG (Pierce, Rockford, IL, USA), and an enhanced chemiluminescence kit (Amersham, Buckinghamshire, UK).
Xenograft mouse model
All animal studies were approved by the ethical review board at Karolinska Institutet (N42/10). SCID/Beige mice (Charles River, Sulzfeld, Germany) were injected with 3 × 106 J82 tumor cells subcutaneously in the right flank. There are no known orthotopic xenograft mouse models present for the J82 cell line, why subcutaneous implantation was chosen, also facilitating the monitoring of tumor size by palpation. On day 7 (when tumors became palpable) and on days 14 and 21 after tumor inoculation, mice were injected intravenously (I.V. 100 μl) with 1 mg/kg of doxorubicin or with 100 μl PBS. Mice were euthanized when tumors reached a volume of 800 mm3. Twenty-four hours following doxorubicin injection (on days 8, 15 and 22 after tumor inoculation), mice were injected I.V (100 μl) with 1.0 – 2.0 × 106 NK cells, 2.0 – 4.6 × 106 TILs (both isolated from the same melanoma patient) or PBS. In cytotoxicity assays, killing of J82 tumors was approximately 2-fold higher by NK cells compared to TILs; therefore a 1:2 ratio of infused NK cells and TILs was used. Mice treated with the combination of both NK and TILs received the same number of total cells compared to mice receiving NK or TILs alone. Statistical data on tumor progression and survival was analyzed by two-way ANOVA and Log-rank test respectively.
Results
Doxorubicin sensitizes human tumor cells to NK cell-mediated killing
To test whether doxorubicin could sensitize tumor cells to killing by NK cells, we first exposed the J82 bladder cancer cell line and the GEWA-MEL melanoma cell line to sub-apoptotic doses of doxorubicin for 24 hours followed by co-culture with allogeneic or autologous NK cells, respectively. Pretreatment with doxorubicin augmented NK cell-mediated killing of J82 cells by 2.5 ± 0.6 fold (p<0.01, n=6) and GEWA-MEL cells by 4.0 ± 1.8 fold (p<0.05, n=4) (Figure 1). To expand these findings, experiments were performed on a panel of tumor cell lines of various origins. We found that these experiments corroborated our initial finding showing a sensitizing effect on the cell lines tested ranging from 3.7 to 32.7 % increase in lysis of doxorubicin treated compared to untreated tumor cells (2.5 mean fold increase, p < 0.0001, n = 14,) (Table 1). Importantly, doxorubicin treatment did not sensitize the fibroblast cell lines AG08164 and BJ1 to NK cell-mediated killing, suggesting a tumor antigen-specific effect (Table 1). Due to its relatively high resistance to NK and T cell lysis, the J82 cell line was later used for mechanistic studies of doxorubicin-induced sensitization.
Figure 1. Increased NK cell-mediated killing of doxorubicin-treated allogeneic and autologous tumor cells.
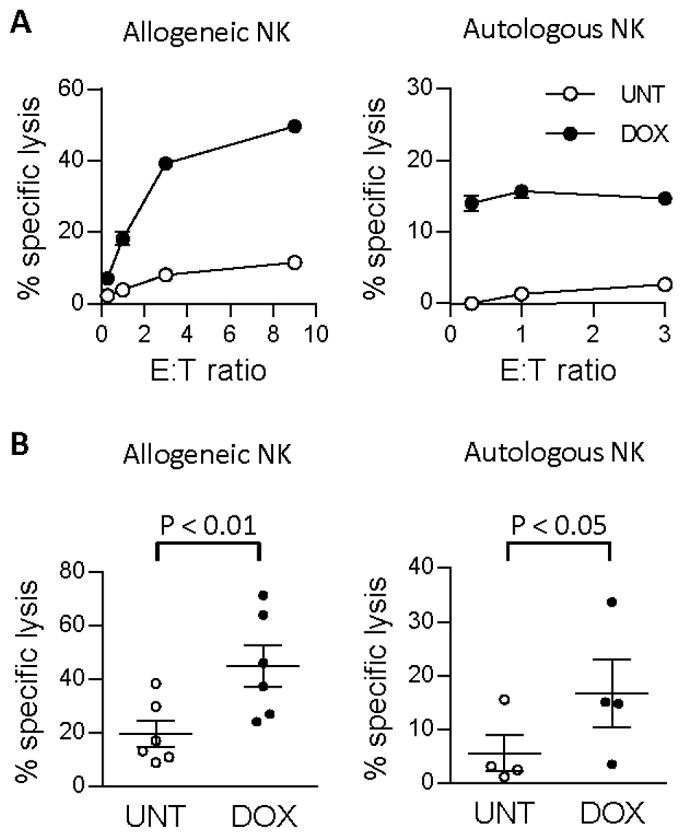
(A) J82 tumors (allogeneic) and GEWA-MEL tumors (autologous) were treated with 200 ng/ml doxorubicin and tested for susceptibility to NK cell-mediated killing in an 18h 51Cr-release assay. (B) Summary of 51Cr-release assays of allogeneic (n=6, p<0.01) and autologous (n=4, p< 0.05) NK cell killing of J82 and GEWA-MEL cell lines respectively. Effector-target (E:T) ratios ranged from 0.1:1–3:1, UNT = untreated, DOX = doxorubicin-treated.
Table 1. Doxorubicin sensitizes tumor cell lines of various histiotypes to NK and T cell-mediated killing.
Cell lines of different tissue origin were pre-treated with doxorubicin (DOX) at concentrations varying between 200–500 ng/ml and tested for susceptibility to killing by NK cells or HLA-A2 matched tumor-antigen specific TILs cells in 5–18 hour 51Cr-release assays at E:T ratios ranging from 0.3–3:1. All tumor cell lines tested for lysis by TILs were Mart-1 positive.
| Cell line | DOX(ng/ml) | NK cell lysis (%)
|
E:T ratio | Co-culture | T cell lysis (%)
|
E:T ratio | Co-culture | ||
|---|---|---|---|---|---|---|---|---|---|
| UNT | DOX | UNT | DOX | ||||||
|
|
|
|
|||||||
| GEWA-MEL | 200 | 3.2 ± 0.9 | 15.1 ± 1.5 | 1:1 | 18 h | 1.8 ± 0.2 | 12.6 ± 0.7 | 3:1 | 18 h |
| EST074 | 200 | 19.8 ± 2.7 | 23.5 ± 2.2 | 2,5:1 | 5h | 5.1 ± 3.5 | 8.0 ± 2.2 | 2,5:1 | 5h |
| EST149 | 200 | ND | ND | ND | ND | 4.1 ± 0.1 | 7.5 ± 6.1 | 0,3:1 | 18 h |
| EST025 | 200 | 14.4 ± 2.3 | 22.3 ± 1.8 | 1:1 | 18h | 20.8 ± 1.4 | 35.0 ± 1.1 | 2,5:1 | 18h |
| EST066 | 200 | 8.1 ± 2.0 | 16.2 ± 2.2 | 1:1 | 5h | 6.6 ± 1.4 | 9.6 ± 2.0 | 1:1 | 5h |
| J82 | 200 | 13.3 ± 2.1 | 46.0 ± 1.7 | 1:1 | 18h | 3.8 ± 0.8 | 16.7± 4.1 | 1:1 | 18h |
| EST115 | 200 | 14.9 ± 1.4 | 27.7 ± 3.8 | 1:1 | 18h | ND | ND | ||
| JUSO | 200 | 5.1 ± 0.3 | 11.3 ± 1.9 | 1:1 | 5h | ND | ND | ||
| EST112 | 250 | 12.0 ± 0.6 | 23.2 ± 0.9 | 1:1 | 5h | ND | ND | ||
| A498 | 200 | 14.0 ± 2.4 | 38.5 ± 0.6 | 1:1 | 18 h | ND | ND | ||
| MAR | 250 | 9.0 ± 0.3 | 18.1 ± 1.0 | 1:1 | 5h | ND | ND | ||
| JOHW | 500 | 8.1 ± 0.8 | 39.2 ± 0.8 | 3:1 | 5h | ND | ND | ||
| HCT116 | 250 | 30.1 ± 7.0 | 53.7 ± 8.3 | 0,3:1 | 18h | ND | ND | ||
| T47D | 200 | 6.9 ± 1.5 | 13.4 ± 2.3 | 0,3:1 | 18h | ND | ND | ||
| DU145 | 200 | 11.0 ± 1.2 | 26.6 ± 1.6 | 1:1 | 18h | ND | ND | ||
| BJ1 | 250 | 5.2 ± 0.7 | 3.4 ± 1.3 | 1:1 | 18 | ND | ND | ||
| AG08164 | 250 | 9.7 ± 1.0 | 9.9 ± 0.6 | 3:1 | 18 | 3.5 ± 2.2 | 2.3 ± 2.8 | ||
Improved NK cell-targeting of doxorubicin-treated tumor cells is mediated by up-regulation of TRAIL-receptors and down-regulation of cFLIP
To understand the mechanism behind the sensitization we assessed changes in NK cell receptor ligand expression on the tumor cell lines following exposure to doxorubicin. This experiment revealed an increased expression of TRAIL-R2 on the tumor cell lines studied following treatment with sub-apoptotic doses of doxorubicin for 12 hours (Figure 2A and B). In contrast, no changes in the expression of MHC class I, Fas, MICA/B, ULBP-1/2 and HLA-E were observed in any of the tumor cell lines studied (data not shown). As even low concentrations of doxorubicin (200 ng/ml) sensitized the J82 bladder cancer cell line to killing by recombinant TRAIL (Figure 2C) despite the relatively minor up-regulation of TRAIL-R2 expression (Figure 2B), we next assessed whether this was related to a downregulation of the caspase-8 inhibitory protein cFLIP as reported by Vucolova and colleagues 25. Indeed, Western blot analysis showed downregulation of cFLIP in the J82 cell line following exposure to doxorubicin (Figure 2D), which highlighted the role for TRAIL-R signaling in addition to upregulation of TRAIL-R2. Downregulation of cFLIP was also observed in the GEWA and JOHW cell lines after incubation with doxorubicin (Supplemental figure 1E).
Figure 2. Doxorubicin up-regulates TRAIL-R2 and reduces cFLIP expression and sensitizes tumors to TRAIL-mediated killing.
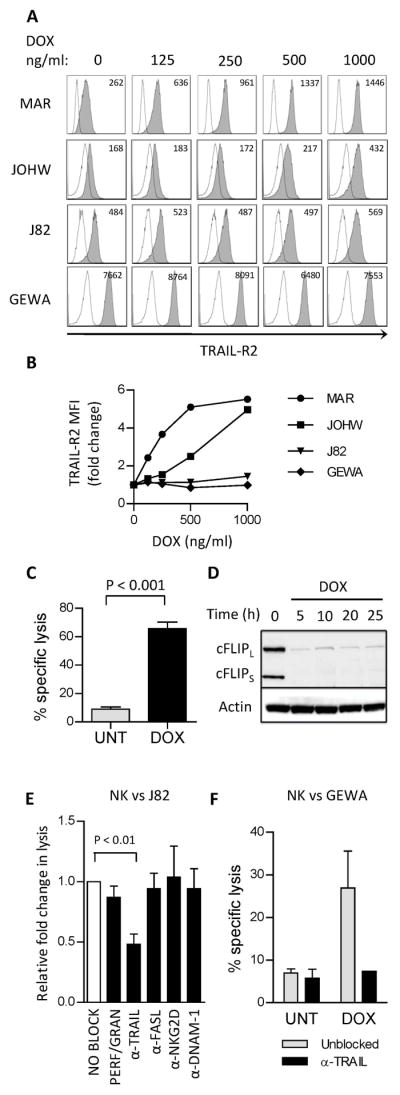
(A) Surface expression of TRAIL-R2 on tumor cells after treatment with DOX (18 hours). Open histograms represent isotype control. Values in histograms represent mean fluorescence intensity (MFI) of TRAIL-R2 expression. (B) Fold change in MFI of TRAIL-R2 expression in tumor cells after DOX-treatment. Similar results were obtained in the repeated experiment. (C) Killing of J82 cells that were either untreated (UNT) or treated with 200 ng/ml doxorubicin (DOX) by rTRAIL (200 ng/ml) measured in an 18h 51Cr-release assay. The experiment was repeated twice with similar results. (D) Expression of the short form (cFLIPS) and the long form (cFLIPL) of cellular FLICE inhibitory protein (cFLIP) in J82 cells after treatment with DOX (200 ng/ml). (E) NK cell-mediated killing of DOX-treated J82 tumors. The figure illustrates fold change in cytotoxicity between untreated and doxorubicin-treated tumors in presence of blocking agents. (unblocked is set to 1.0, white bar). Blocking agents used were concanamycin A (CMA, n=4) or neutralizing antibodies against TRAIL (n=4), FasL (n=4), NKG2D (n=3) and DNAM-1 (n=2). (F) Killing of GEWA tumor cells that were either untreated or treated with 200 ng/ml doxorubicin by expanded NK cells, measured in an 18h 51Cr-release assay. NK cells were incubated with blocking antibody against TRAIL (10 μg/ml) 30 min prior to co-culture with tumor cells. (E:T ratio = 1:1). UNT = untreated, DOX = doxorubicin treated.
To verify the impact of TRAIL/TRAIL-R signaling in doxorubicin induced sensitization of tumor cells to NK cells, we next performed cytotoxicity assays following blockade of TRAIL, FasL or the perforin pathway. The relative reduction of NK cell-mediated killing of doxorubicin-treated J82 tumor cells was 52.0 ± 19.3 % (p<0.01) in presence of neutralizing antibodies to TRAIL. Conversely, the relative fold change in lysis of doxorubicin-treated tumor cells compared to untreated tumor cells was not significantly altered in presence of CMA targeting the perforin pathway or neutralizing antibodies to FasL, NKG2D or DNAM-1 (Figure 2E). An abrogation of DOX-induced sensitization to NK cell mediated killing was also observed in the GEWA cell line in presence of neutralizing antibodies to TRAIL (Figure 2F).
NK cell-mediated killing of doxorubicin-treated tumor cells is dependent on caspase-8 activity
To further support that killing of doxorubicin-treated tumors is dependent on death receptor signaling, activity of caspase-8 in J82 tumor cells was analyzed by intracellular staining following co-culture with NK cells. These experiments showed a 2-fold increase in cells expressing active caspase-8 in doxorubicin-treated compared to untreated tumor cells (Figure 3A). Silencing of caspase-8 in J82 tumor cells resulted in significantly lower NK cell-mediated killing than in mock-transfected cells (p<0.05) and doxorubicin treatment did not induce sensitization of tumor cells with silenced caspase-8 (Figure 3B–C). In addition to caspase-8 activation, enhanced cleavage of Bid was observed in doxorubicin-treated tumor cells following co-culture with NK cells (Supplemental Figure 1C).
Figure 3. Increase in cleavage of caspase-8 in doxorubicin-treated tumor cells following culture with NK cells.

(A) Activation of caspase-8 in J82 tumor cells after treatment (18 hours) with 200 ng/ml DOX and 4 hour co-culture with NK cells. Effector : Target ratio = 1:1. Experiment was repeated once with similar results. (B) Expression of pro- and cleaved form of caspase-8 in untreated or DOX-treated (200 ng/ml, 16 hours) J82 cells after 2 hour co-culture with NK cells. Effector : Target ratio = 1:1 Graph is representative of three independent experiments. (C) NK cell-mediated killing of J82 tumor cells transfected with non-targeting or caspase-8-targeting siRNA with or without treatment with 200 ng/ml DOX. Graph is representative of three independent experiments. UNT = untreated, DOX = doxorubicin-treated.
Doxorubicin sensitizes tumor cells to lysis by tumor specific T cells via augmented TRAIL signaling
Compared to resting peripheral blood T cells, the expression of TRAIL is up-regulated on activated CTLs and TILs 27–28. To investigate whether T cells would also exhibit an increased ability to kill doxorubicin-treated tumor cells, TILs from a melanoma biopsy were expanded and tested for killing of HLA-A2-matched Mart-1 positive tumor cells. Compared to untreated tumor cells, an 8.5 ± 3.7 fold increase in T cell-mediated killing of doxorubicin-treated J82 tumor cells, expressing low levels of Mart-1 antigen, was observed (p=0.057, n=3) and a 2.0 ± 0.2 fold increased killing (p<0.05, n=4) was observed for the autologous melanoma tumor cell line (GEWA-MEL) (Figure 4A–B). The role for doxorubicin-mediated sensitization of T cell-mediated killing was also verified using a panel of HLA-A2 positive, Mart-1 positive tumor cell lines, where we observed a 2.9 to 14.2 % increase in killing (3.0 mean fold increase, p<0.05, n=6) in the analyzed cell lines after doxorubicin treatment (Table 1). Similar to when co-cultured with NK cells, co-culture of tumor antigen-specific T cells with tumor cells increased activity of caspase-8 in doxorubicin-treated compared to untreated tumor cells (data not shown). In presence of TRAIL neutralizing antibody, but not FasL, NKG2D, DNAM-1 or CMA neutralizing antibodies, the relative change in T cell-mediated killing of doxorubicin-treated J82 tumor cells was reduced by 42.6 ± 18.3 % (p<0.05) (Figure 4C). A reduction of T cell-mediated killing was also observed in doxorubicin treated GEWA cells, in presence of neutralizing antibodies to TRAIL (Figure 4D).
Figure 4. Doxorubicin sensitizes tumors to lysis by tumor specific T cells via augmented TRAIL signaling.
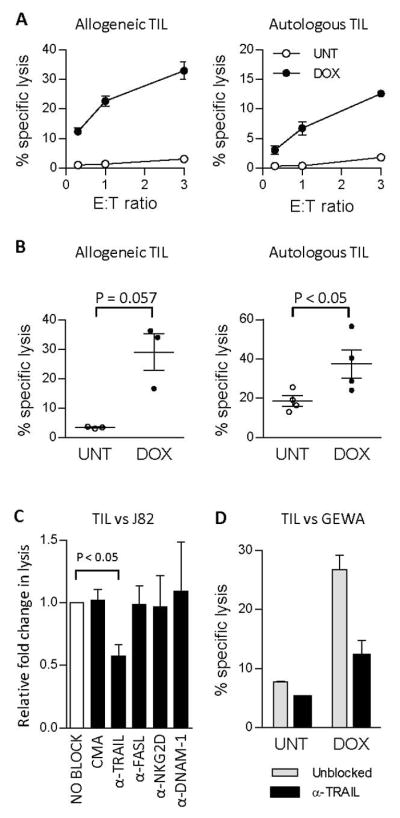
(A) TIL killing against untreated or DOX-treated (200 ng/ml) allogeneic J82 tumors or autologous GEWA-MEL tumors in an 18 h 51Cr release assay. (B). Summary of 51Cr-release assays of allogeneic (n=3, p=0.057) and autologous (n=4, p<0.05) TIL killing of J82 and GEWA-MEL cell lines respectively. Effector-target (E:T) ratios ranged from 0.1:1–3:1 (C) Summary of TIL-mediated killing of DOX-treated J82 tumors. The figure illustrates fold change in cytotoxicity between untreated and doxorubicin-treated tumors in presence of blocking agents (unblocked is set to 1.0, white bar). Blocking agents used were concanamycin A (CMA, n=4) or neutralizing antibodies against TRAIL (n=4), Fas-ligand (n=4), NKG2D (n=3) and DNAM-1 (n=2). (D) TIL-mediated killing of GEWA tumor cells that were either untreated or treated with 200 ng/ml doxorubicin, measured in an 18h 51Cr-release assay. TILs were incubated with blocking antibody against TRAIL (10 μg/ml) 30 min prior to co-culture with tumor cells (E:T ratio = 3:1). UNT = untreated, DOX = doxorubicin-treated.
Combined doxorubicin treatment and adoptive NK and T cell infusion result in significantly delayed tumor progression and prolonged survival in vivo
Tumor-bearing SCID/Beige mice were treated with doxorubicin followed by infusion of NK cells and/or TILs. Compared to untreated mice, treatment with doxorubicin alone did not result in any delay of tumor growth (Figure 5A). Treatment with doxorubicin resulted in a reduced expression of cFLIP in tumors (data not shown). In mice treated with doxorubicin, a significantly delayed tumor growth was observed after infusions of NK cells (p<0.05, Figure 5B) or TILs (p<0.0001, Figure 5C) compared to mice treated with doxorubicin alone. Following doxorubicin treatment, a further delay in tumor progression (p<0.001) and prolonged survival (median survival 72.4 ± 10.0 days) was observed in mice receiving a combination of NK cells and T cells compared to mice receiving this combination without doxorubicin (median survival 53.8 ± 6.9 days, p<0.01) (Figure 5D–E). Furthermore, in all mice receiving doxorubicin, we observed an initial delay in tumor progression (from start of treatment until day 35 after treatment start) in mice receiving combined infusion of NK cells and TILs compared to mice receiving infusion of NK cells (p<0.05, Supplemental figure 2A) or TILs (p<0.05, Supplemental figure 2B). However, after 35 days a rapid tumor progression was observed in all treatment groups and no difference in survival was observed.
Figure 5. Anti-tumor effect of adoptively transferred NK and T cells is augmented by doxorubicin pretreatment.
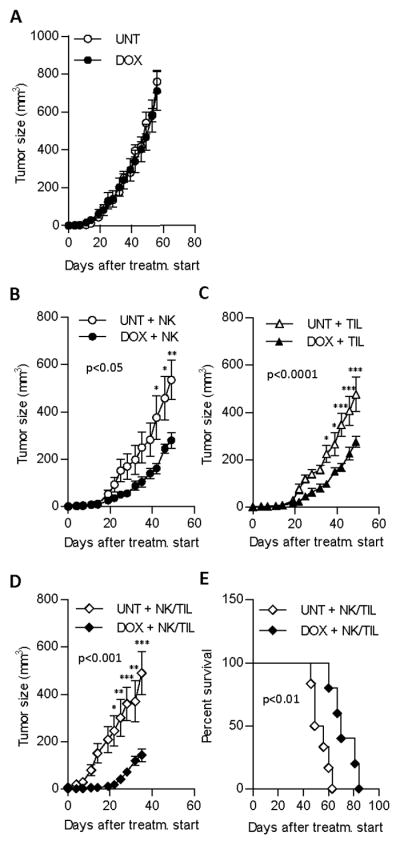
(A) Tumor progression in mice receiving either injections of DOX or PBS (UNT). Progression of J82 tumors in SCID/Beige mice receiving cell therapy alone or in combination with doxorubicin (DOX) from day of treatment start until sacrifice of mice. Comparison of (B) NK cell therapy (p<0.05, tumor progression), (C) TIL therapy (p<0.0001, tumor progression) or (D) NK and TIL combination therapy (p<0.001, tumor progression). (E) Log-rank test of survival of SCID/Beige mice receiving adoptive transfer of NK cell and TILs without pre-treatment (median survival 72.4 ± 10.0 days) or with pre-treatment with 1 mg/kg DOX (median survival 53.8 ± 6.9 days) (p<0.01). At least 5 mice were included in each group. Statistical data on tumor progression and survival was analyzed by two-way ANOVA and Log-rank test respectively and Student’s t-test was used to calculate differences in tumor size at the indicated time points. The experiment was performed twice with similar results. * p < 0.05, ** p < 0.01, *** p < 0.001
Discussion
Here we demonstrate that doxorubicin sensitizes tumors to NK and T cell-mediated killing via augmented TRAIL signaling. Several different chemotherapy agents have recently been shown to sensitize tumors to TRAIL-mediated killing 9–11, 16, 22–25, 29–31. We and others have demonstrated that the proteasome inhibitor bortezomib can sensitize tumors to NK cell-mediated killing via augmented TRAIL signaling 17, 23. In an ongoing clinical phase I trial of bortezomib treatment followed by infusion of autologous NK cells, clinical responses include 10/20 patients with stable disease including two patients with metastatic tumors who had more than a 30% decline in serum tumor markers and four patients with a minor response 32. Although durable clinical responses may be achieved, more patients need to be evaluated to determine the efficacy of adoptive NK cell therapy. Adoptive T cell therapy based on transfer of tumor infiltrating lymphocytes has produced objective clinical responses in patients with melanoma 33–34. In addition, with the advent of adoptive transfer of genetically engineered T cells, response rates have improved in the past few years 35–36. However, randomized clinical trials of adoptive T cell transfer are warranted in order to interpret the clinical response of this individualized treatment strategy. Moreover, the frequency of patients with a complete response is relatively low. Thus, further improvements in cell-based therapies are needed.
A relatively frequent event used by tumors to escape T cell mediated recognition is down-regulation of MHC class I 6–8. Such tumors instead become sensitive to killing by NK cells. Thus, targeting tumors by both T and NK cells may result in improved clinical responses. In the present study, we sought to identify an agent with the ability to sensitize tumors to killing by both NK and tumor specific T cells. Instead of sensitizing tumors to NK cell-mediated killing, proteasome inhibitors render tumors resistant to killing by tumor specific T cells possibly due to disrupted presentation of tumor antigens. Therefore, the use of bortezomib is precluded as pre-treatment to adoptive T cell therapy 37. Mizutani et. al, demonstrated that doxorubicin can sensitize fresh bladder cancer cells to lysis by lymphokine activated killer cells, as well as resting purified natural killer cells and T lymphocytes in vitro 38. In our study, we show that doxorubicin sensitizes tumors of several different origins to TRAIL-mediated lysis by NK cells and TILs that have been expanded for adoptive transfer ex vivo. In addition, in tumor-bearing mice receiving doxorubicin pretreatment prior to adoptive transfer, a significantly delayed tumor progression was observed in mice receiving infusions of both NK cells and TILs compared to mice receiving either NK cells or TILs. Possibly, the combined use of NK and T cells increased the targeting of tumor cells with different immune escape phenotypes, which may have contributed to the reduced tumor progression. However, in mice that did not receive doxorubicin pretreatment, we observed that combined transfer of NK and T cells resulted in a faster tumor progression than in mice receiving NK cells or T cells alone. Possibly, in the absence of doxorubicin, the infused cells may compete for growth factors or the NK cells have killed the T cells. It has previously been demonstrated that infused NK cells can kill stressed T cells via NKG2D39. It has also been shown that NK cells can inhibit other NK cells via the 2B4 receptor which can also induce fratricide among NK cells40
In our phenotypic analysis of doxorubicin-treated tumor cells, we confirmed other reports demonstrating upregulation of TRAIL-R2 in several cell lines after treatment with doxorubicin 22, 24. In our experiments sensitization of J82 cells to rTRAIL was observed even at low doses of doxorubicin and only minor changes in TRAIL-R2 but no changes in TRAIL-R1 expression after exposure to doxorubicin in ranges between 125–250 ng/ml. However, the anti-apoptotic cFLIPs were rapidly degraded in these tumor cells following exposure to doxorubicin. Several studies have investigated the role of doxorubicin in modulating the expression of cFLIP in tumor cells 41–42. As a consequence, tumors treated with doxorubicin were more sensitive to killing by activated NK cells expressing high levels of TRAIL. Furthermore, doxorubicin treatment of the J82 tumor cell line which has a very low expression of melanoma antigen was sufficient to sensitize this tumor to killing by TILs. Importantly, not all cell lines were sensitized to NK cell and TIL killing by low dose doxorubicin. It is possible that a higher dose of doxorubicin may have sensitized these tumors. Furthermore, tumor cell lines not responding to doxorubicin treatment may lack expression of essential molecules in the TRAIL apoptotic signaling pathway. Ramakrishnan and colleagues demonstrated that murine T cells raised against tumor antigens were able to kill tumor cells that lacked expression of the corresponding tumor antigen following treatment with doxorubicin. A doxorubicin-mediated increase in the tumor cells permeability to granzyme B was reported to create a bystander effect that augmented the anti-tumor response also against tumor cells that lacked antigen expression 43. Our studies demonstrate that selective blockade of the perforin/granzyme pathway had only a minor effect on the ability of TILs to kill doxorubicin-treated tumor cells. However, in presence of neutralizing TRAIL antibodies, the level of both NK and T cell mediated cytotoxicity was significantly reduced. Furthermore, the NK cell-mediated apoptosis in tumor cells was dependent on caspase-8 signaling.
Several studies have evaluated targeting TRAIL receptors in patients with advanced solid tumors. Although the maximum tolerated dose was not reached, in the majority of these trials, the common consensus is that TRAIL receptor targeting should be combined with chemotherapy. In three recent phase I clinical studies, TRAIL targeting in combination with chemotherapy has demonstrated objective clinical responses 44–46. In combination with paclitaxel and carboplatin, administration of dulanermin (recombinant human TRAIL) or mapatumumab have generated overall response rates of 58% and 19% respectively in patients with non-small-cell lung cancer 44–45. Although the results from these studies are promising, combined therapy with doxorubicin and cell-based adoptive therapy may have additional clinical benefits for several reasons; the terminal half-life of dulanermin, lexatumumab and mapatumumab is less than one hour, 16 and 19 days respectively 20, 47. The anti-tumor effect of adoptively transferred immune cells, which can be detected several months after cell transfer 48, is potentially greater. Moreover, immune cells in the tumor micro-environment produce cytokines that can recruit other immune cells that could further contribute to tumor eradication. At therapeutic doses, paclitaxel as well as other commonly used chemotherapy agents, can significantly inhibit NK cell mediated killing while doxorubicin has a minimal impact on the viability and cytotoxicity of NK cells 49. Our results indicate that following exposure to doxorubicin, tumor cells remained sensitive to NK cell killing for 72 hours in vitro, thus providing a relatively long therapeutic window for NK cell therapy (supplemental figure 1D).
Our findings present a novel use of doxorubicin as a pre-treatment of adoptive infusion of expanded NK cells and T cells. Since a combination approach increases the chances of targeting immune escape variants of tumor cells, with variable expression of MHC class I and low expression of tumor antigens, doxorubicin pre-treatment coupled with infusion of autologous NK and T cells may be a therapeutic option for the treatment of cancer.
Supplementary Material
Supplemental figure 1. (A) NK and T cell specific lysis of K562 cells in a 51Cr-release assay. (B) Viability of renal cell carcinoma cell line JOHW after 18 hour incubation with 1 μg/ml doxorubicin (DOX). Similar data was obtained with the MAR cell line after 18 hour incubation with 500 ng/ml DOX (C) Expression of truncated BID (tBID) in J82 tumor cells after pretreatment with DOX (200 ng/ml, 18 hours) with or without co-culture with NK cells (Effector : Target ratio = 1:1, 4 hours) (D) Duration of NK cell-meditated cytotoxicity against untreated and DOX-treated J82 tumors. Tumor cells were treated for 18 hour and tested either directly (DOX 0 hours) after wash or after incubation in DOX-free medium for 24, 48 or 72 hours. (E) Expression of cFLIP in JOHW and GEWA cells after treatment with 500 ng/ml and 200 ng/ml doxorubicin (18 h). (F) NK cell-mediated killing of MAR tumor cells that were either untreated or treated with 250 ng/ml doxorubicin measured in an 18h 51Cr-release assay. NK cells were incubated with blocking antibody against TRAIL (10 μg/ml) 30 min prior to co-culture with tumor cells (E:T ratio = 3:1)
Supplemental figure 2. (A) Tumor progression in DOX-treated (1 mg/kg) SCID/Beige mice receiving infusions with a combination of NK cells and TILs or NK cells alone. (p<0.05, tumor progression from start of treatment to day 35 after treatment start) or (B) a combination of NK cells and TILs or TILs alone (p<0.05, tumor progression from start of treatment to day 35 after treatment start). At least 5 mice were included in each group. Statistical data on tumor progression was analyzed by two-way ANOVA and Student’s t-test was used to calculate differences in tumor size at the indicated time points. The experiment was performed twice with similar results. * p < 0.05, ** p < 0.01, *** p < 0.001.
Brief description.
We show for the first time that doxorubicin sensitizes tumor cells to killing by ex vivo expanded NK cells and tumor-specific T cells due to increased TRAIL-receptor signaling. In addition, we demonstrate that pre-treatment with doxorubicin augments the anti-tumor effect of adoptively transferred NK and T cells in tumor-bearing mice. Our findings will have an impact on the advancement of novel treatment strategies in the field of adoptive cell therapy against cancer.
Acknowledgments
Grant Number for this article is NIH HL002345-13
We would like to acknowledge Professor Rolf Kiessling for providing us with research material as well as the staff at the animal facility at MTC, Karolinska Institutet. This work was supported by funding from The Swedish Research Council, The Swedish Cancer Society, The European Research Council Society, Karolinska Institutet, Jeanssons Stiftelser, Åke Wibergs Stiftelse, Magnus Bergvalls Stiftelse, Fredrik och Ingrid Thurings Stiftelse, Stiftelsen Clas Groschinskys Minnesfond, and The Swedish Society of Medicine.
Abbreviations
- TRAIL
TNF-related apoptosis-inducing ligand
- TIL
Tumor infiltrating lymphocyte
- cFLIP
cellular FLICE inhibitory protein
Footnotes
Conflict of interest
None of the authors have any conflict of interest to declare
References
- 1.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 2.Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, Aridome K, Hokita S, Aikou T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. 2000;88:577–83. [PubMed] [Google Scholar]
- 3.Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, Martos JA, Moreno M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–8. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 5.Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, Balsamo M, Conte R, Benelli R, Minghelli S, Solari N, Gualco M, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 2012;72:1407–15. doi: 10.1158/0008-5472.CAN-11-2544. [DOI] [PubMed] [Google Scholar]
- 6.Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, Real LM, Garrido F, Cabrera T. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics. 2008;60:439–47. doi: 10.1007/s00251-008-0303-5. [DOI] [PubMed] [Google Scholar]
- 7.Vitale M, Rezzani R, Rodella L, Zauli G, Grigolato P, Cadei M, Hicklin DJ, Ferrone S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998;58:737–42. [PubMed] [Google Scholar]
- 8.Rongcun Y, Salazar-Onfray F, Charo J, Malmberg KJ, Evrin K, Maes H, Kono K, Hising C, Petersson M, Larsson O, Lan L, Appella E, et al. Identification of new HER2/neu-derived peptide epitopes that can elicit specific CTL against autologous and allogeneic carcinomas and melanomas. J Immunol. 1999;163:1037–44. [PubMed] [Google Scholar]
- 9.He Q, Huang Y, Sheikh MS. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene. 2004;23:2554–8. doi: 10.1038/sj.onc.1207351. [DOI] [PubMed] [Google Scholar]
- 10.Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–71. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 11.Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene. 2005;24:4609–23. doi: 10.1038/sj.onc.1208585. [DOI] [PubMed] [Google Scholar]
- 12.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–90. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 13.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 14.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–20. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 15.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 16.Evdokiou A, Bouralexis S, Atkins GJ, Chai F, Hay S, Clayer M, Findlay DM. Chemotherapeutic agents sensitize osteogenic sarcoma cells, but not normal human bone cells, to Apo2L/TRAIL-induced apoptosis. Int J Cancer. 2002;99:491–504. doi: 10.1002/ijc.10376. [DOI] [PubMed] [Google Scholar]
- 17.Lundqvist A, Abrams SI, Schrump DS, Alvarez G, Suffredini D, Berg M, Childs R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66:7317–25. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 18.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O’Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, Ashkenazi A, Jubb AM, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28:2839–46. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 19.Trarbach T, Moehler M, Heinemann V, Kohne CH, Przyborek M, Schulz C, Sneller V, Gallant G, Kanzler S. Phase II trial of mapatumumab, a fully human agonistic monoclonal antibody that targets and activates the tumour necrosis factor apoptosis-inducing ligand receptor-1 (TRAIL-R1), in patients with refractory colorectal cancer. Br J Cancer. 2010;102:506–12. doi: 10.1038/sj.bjc.6605507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, Barrett M, Judson I, Kaye S, Fox NL, Halpern W, Corey A, et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res. 2007;13:6187–94. doi: 10.1158/1078-0432.CCR-07-0950. [DOI] [PubMed] [Google Scholar]
- 21.Wakelee HA, Patnaik A, Sikic BI, Mita M, Fox NL, Miceli R, Ullrich SJ, Fisher GA, Tolcher AW. Phase I and pharmacokinetic study of lexatumumab (HGS-ETR2) given every 2 weeks in patients with advanced solid tumors. Ann Oncol. 2010;21:376–81. doi: 10.1093/annonc/mdp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu XX, Kakehi Y, Mizutani Y, Nishiyama H, Kamoto T, Megumi Y, Ito N, Ogawa O. Enhancement of TRAIL/Apo2L-mediated apoptosis by adriamycin through inducing DR4 and DR5 in renal cell carcinoma cells. Int J Cancer. 2003;104:409–17. doi: 10.1002/ijc.10948. [DOI] [PubMed] [Google Scholar]
- 23.Sayers TJ, Brooks AD, Koh CY, Ma W, Seki N, Raziuddin A, Blazar BR, Zhang X, Elliott PJ, Murphy WJ. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003;102:303–10. doi: 10.1182/blood-2002-09-2975. [DOI] [PubMed] [Google Scholar]
- 24.Wu XX, Jin XH, Zeng Y, El Hamed AM, Kakehi Y. Low concentrations of doxorubicin sensitizes human solid cancer cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-receptor (R) 2-mediated apoptosis by inducing TRAIL-R2 expression. Cancer Sci. 2007;98:1969–76. doi: 10.1111/j.1349-7006.2007.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaculova A, Kaminskyy V, Jalalvand E, Surova O, Zhivotovsky B. Doxorubicin and etoposide sensitize small cell lung carcinoma cells expressing caspase-8 to TRAIL. Mol Cancer. 2010;9:87. doi: 10.1186/1476-4598-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berg M, Lundqvist A, McCoy P, Jr, Samsel L, Fan Y, Tawab A, Childs R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11:341–55. doi: 10.1080/14653240902807034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirandola P, Ponti C, Gobbi G, Sponzilli I, Vaccarezza M, Cocco L, Zauli G, Secchiero P, Manzoli FA, Vitale M. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood. 2004;104:2418–24. doi: 10.1182/blood-2004-04-1294. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Hu X, Zimmerman M, Waller JL, Wu P, Hayes-Jordan A, Lev D, Liu K. TNFalpha cooperates with IFN-gamma to repress Bcl-xL expression to sensitize metastatic colon carcinoma cells to TRAIL-mediated apoptosis. PLoS One. 2011;6:e16241. doi: 10.1371/journal.pone.0016241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seitz SJ, Schleithoff ES, Koch A, Schuster A, Teufel A, Staib F, Stremmel W, Melino G, Krammer PH, Schilling T, Muller M. Chemotherapy-induced apoptosis in hepatocellular carcinoma involves the p53 family and is mediated via the extrinsic and the intrinsic pathway. Int J Cancer. 2010;126:2049–66. doi: 10.1002/ijc.24861. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Liu R, Qiu J, Yin P, Luo F, Su J, Li W, Chen C, Fan X, Zhang J, Zhuang G. Combination of human Fas (CD95/Apo-1) ligand with adriamycin significantly enhances the efficacy of antitumor response. Cell Mol Immunol. 2009;6:167–74. doi: 10.1038/cmi.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacFarlane M, Ahmad M, Srinivasula SM, Fernandes-Alnemri T, Cohen GM, Alnemri ES. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J Biol Chem. 1997;272:25417–20. doi: 10.1074/jbc.272.41.25417. [DOI] [PubMed] [Google Scholar]
- 32.Lundqvist A, Berg M, Smith A, Childs RW. Bortezomib Treatment to Potentiate the Anti-tumor Immunity of Ex-vivo Expanded Adoptively Infused Autologous Natural Killer Cells. J Cancer. 2011;2:383–5. doi: 10.7150/jca.2.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA, Rogers-Freezer L, Morton KE, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–51. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, Kammula US, Hughes MS, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundqvist A, Su S, Rao S, Childs R. Cutting edge: bortezomib-treated tumors sensitized to NK cell apoptosis paradoxically acquire resistance to antigen-specific T cells. J Immunol. 2010;184:1139–42. doi: 10.4049/jimmunol.0902856. [DOI] [PubMed] [Google Scholar]
- 38.Mizutani Y, Yoshida O, Miki T. Adriamycin-mediated potentiation of cytotoxicity against freshly isolated bladder cancer cells by autologous non-activated peripheral blood lymphocytes and tumor infiltrating lymphocytes. J Urol. 1999;162:2170–5. doi: 10.1016/S0022-5347(05)68154-2. [DOI] [PubMed] [Google Scholar]
- 39.Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115:4293–301. doi: 10.1182/blood-2009-05-222190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taniguchi RT, Guzior D, Kumar V. 2B4 inhibits NK-cell fratricide. Blood. 2007;110:2020–3. doi: 10.1182/blood-2007-02-076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White SJ, Kasman LM, Kelly MM, Lu P, Spruill L, McDermott PJ, Voelkel-Johnson C. Doxorubicin generates a proapoptotic phenotype by phosphorylation of elongation factor 2. Free Radic Biol Med. 2007;43:1313–21. doi: 10.1016/j.freeradbiomed.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.White-Gilbertson SJ, Kasman L, McKillop J, Tirodkar T, Lu P, Voelkel-Johnson C. Oxidative stress sensitizes bladder cancer cells to TRAIL mediated apoptosis by down-regulating anti-apoptotic proteins. J Urol. 2009;182:1178–85. doi: 10.1016/j.juro.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, Altiok S, Celis E, Gabrilovich DI. Chemotherapy enhances tumor cell susceptibility to CTL-mediated killing during cancer immunotherapy in mice. J Clin Invest. 2010;120:1111–24. doi: 10.1172/JCI40269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soria JC, Smit E, Khayat D, Besse B, Yang X, Hsu CP, Reese D, Wiezorek J, Blackhall F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J Clin Oncol. 2010;28:1527–33. doi: 10.1200/JCO.2009.25.4847. [DOI] [PubMed] [Google Scholar]
- 45.Leong S, Cohen RB, Gustafson DL, Langer CJ, Camidge DR, Padavic K, Gore L, Smith M, Chow LQ, von Mehren M, O’Bryant C, Hariharan S, et al. Mapatumumab, an antibody targeting TRAIL-R1, in combination with paclitaxel and carboplatin in patients with advanced solid malignancies: results of a phase I and pharmacokinetic study. J Clin Oncol. 2009;27:4413–21. doi: 10.1200/JCO.2008.21.7422. [DOI] [PubMed] [Google Scholar]
- 46.Mom CH, Verweij J, Oldenhuis CN, Gietema JA, Fox NL, Miceli R, Eskens FA, Loos WJ, de Vries EG, Sleijfer S. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res. 2009;15:5584–90. doi: 10.1158/1078-0432.CCR-09-0996. [DOI] [PubMed] [Google Scholar]
- 47.Tolcher AW, Mita M, Meropol NJ, von Mehren M, Patnaik A, Padavic K, Hill M, Mays T, McCoy T, Fox NL, Halpern W, Corey A, et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol. 2007;25:1390–5. doi: 10.1200/JCO.2006.08.8898. [DOI] [PubMed] [Google Scholar]
- 48.Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17:6287–97. doi: 10.1158/1078-0432.CCR-11-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Markasz L, Stuber G, Vanherberghen B, Flaberg E, Olah E, Carbone E, Eksborg S, Klein E, Skribek H, Szekely L. Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol Cancer Ther. 2007;6:644–54. doi: 10.1158/1535-7163.MCT-06-0358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. (A) NK and T cell specific lysis of K562 cells in a 51Cr-release assay. (B) Viability of renal cell carcinoma cell line JOHW after 18 hour incubation with 1 μg/ml doxorubicin (DOX). Similar data was obtained with the MAR cell line after 18 hour incubation with 500 ng/ml DOX (C) Expression of truncated BID (tBID) in J82 tumor cells after pretreatment with DOX (200 ng/ml, 18 hours) with or without co-culture with NK cells (Effector : Target ratio = 1:1, 4 hours) (D) Duration of NK cell-meditated cytotoxicity against untreated and DOX-treated J82 tumors. Tumor cells were treated for 18 hour and tested either directly (DOX 0 hours) after wash or after incubation in DOX-free medium for 24, 48 or 72 hours. (E) Expression of cFLIP in JOHW and GEWA cells after treatment with 500 ng/ml and 200 ng/ml doxorubicin (18 h). (F) NK cell-mediated killing of MAR tumor cells that were either untreated or treated with 250 ng/ml doxorubicin measured in an 18h 51Cr-release assay. NK cells were incubated with blocking antibody against TRAIL (10 μg/ml) 30 min prior to co-culture with tumor cells (E:T ratio = 3:1)
Supplemental figure 2. (A) Tumor progression in DOX-treated (1 mg/kg) SCID/Beige mice receiving infusions with a combination of NK cells and TILs or NK cells alone. (p<0.05, tumor progression from start of treatment to day 35 after treatment start) or (B) a combination of NK cells and TILs or TILs alone (p<0.05, tumor progression from start of treatment to day 35 after treatment start). At least 5 mice were included in each group. Statistical data on tumor progression was analyzed by two-way ANOVA and Student’s t-test was used to calculate differences in tumor size at the indicated time points. The experiment was performed twice with similar results. * p < 0.05, ** p < 0.01, *** p < 0.001.