

Abstract
The incorporation of omics approaches into symptom science research can provide researchers with information about the molecular mechanisms that underlie symptoms. Most of the omics analyses in symptom science have used a single omics approach. Therefore, these analyses are limited by the information contained within a specific omics domain (e.g., genomics and inherited variations, transcriptomics and gene function). A multi-staged data-integrated multi-omics (MS-DIMO) analysis integrates multiple types of omics data in a single study. With this integration, a MS-DIMO analysis can provide a more comprehensive picture of the complex biological mechanisms that underlie symptoms. The results of a MS-DIMO analysis can be used to refine mechanistic hypotheses and/or discover therapeutic targets for specific symptoms. The purposes of this paper are to: (1) describe a MS-DIMO analysis using “Symptom X” as an example; (2) discuss a number of challenges associated with specific omics analyses and how a MS-DIMO analysis can address them; (3) describe the various orders of omics data that can be used in a MS-DIMO analysis; (4) describe omics analysis tools; and (5) review case exemplars of MS-DIMO analyses in symptom science. This paper provides information on how a MS-DIMO analysis can strengthen symptom science research through the prioritization of functional genes and biological processes associated with a specific symptom.
Keywords: symptom, multi-omics, genomics, gene expression, methylation, cross-validation
Symptom science research is focused on the discovery of the underlying mechanisms for symptoms and the development of interventions targeting these mechanisms (National Institute of Nursing Research, 2016). However, the most common symptoms reported by patients with chronic conditions (e.g., fatigue, pain, depression, sleep disturbance; Miaskowski et al., 2017) occur as a result of complex interactions among a patient’s demographic and molecular characteristics, environmental influences, and disease and treatment states. Because of the complex and multifactorial nature of most symptoms, the mechanisms that underlie them remain poorly understood.
Given this complexity, the use of a variety of omics approaches (e.g., genomics, epigenomics, transcriptomics, proteomics, metabolomics) is needed to increase our understanding of the molecular mechanisms that underlie these symptoms (Tully & Grady, 2015). To date, most of the studies of associations between symptoms and molecular mechanisms have used a single type of omics data in their analysis. For example, one study (Koleck et al., 2017) evaluated for associations between cognitive dysfunction and breast cancer-related candidate genes in survivors of breast cancer. In another study (Dorsey et al., 2019), an evaluation was done of differentially expressed genes (DEGs) and pathways in patients with acute low back pain. Other studies have focused on associations between symptoms and changes in proteomic (Goo et al., 2012) and metabolomic (Chou et al., 2020) profiles.
While the use of a single type of omics analysis provides some information on mechanisms, several limitations warrant consideration. A single omics analysis is limited to a specific biologic domain. For example, while a genetic association study discovers gene variants associated with a symptom, this type of analysis does not provide information on gene expression (i.e., gene function). Given that symptoms occur as a result of interactions among multiple levels of biology, the use of a single omics analysis does not allow for an examination of these complex processes.
A multi-omics analysis can address some of these limitations. Systems biology (i.e., the study of complex biological systems) is an analytical framework that can be used to integrate omics data with symptom data (S. Founds, 2018). Systems biology is an interdisciplinary science that uses knowledge from biology, bioinformatics, computer science, mathematics, medicine, and nursing to study each level of human biology, their interactions, and the system’s responses to genetic and environmental changes (S. A. Founds, 2009; Kirschner, 2005; Weston & Hood, 2004).
While many types of systems biology analyses exist, an integrated omics analysis offers numerous advantages for symptom science research. A symptom is a complex phenotype, that is the result of individual, environmental, and genetic factors. Compared to an analysis that uses only a single source of omics data, an integrated omics analysis combines data from multiple levels of biology and provides an improved identification and interpretation of the omics-phenotype relationship (Hasin et al., 2017; Misra et al., 2018; Sun & Hu, 2016). A data-integrated omics analysis can be classified as either meta-dimensional or multi-staged (Ritchie et al., 2015). A meta-dimensional analysis is one in which all sources of data are combined simultaneously. In contrast, a multi-staged analysis follows a “stepwise” approach (Ritchie et al., 2015, p. 86). The purposes of this paper are to: 1) describe a multi-staged data-integrated multi-omics (MS-DIMO) analysis using a transcriptome analysis first design; 2) discuss challenges with specific omics analyses and how a MS-DIMO analysis can address them; 3) describe other orders of omics data that can be used in a MS-DIMO analysis; 4) describe omics analysis tools; and 5) provide case exemplars of MS-DIMO analyses in symptom science.
A MS-DIMO Analysis Starting With Functional Gene Products
A MS-DIMO analysis is divided into multiple stages using a hierarchical approach. Namely, at each stage of the process a new type of data is analyzed (Ritchie et al., 2015). For example, transcriptomic (e.g., gene expression) data can be analyzed in Stage 1 and genomic (e.g., genetic association) data can be analyzed in Stage 2. Loci (e.g., genes) that have significant associations with a characteristic of interest (e.g., symptom) are advanced for subsequent analysis. With these steps, the identification of a causal molecular signal is strengthened. Through the ordering of the stages of a MS-DIMO analysis, genes and biological processes associated with a symptom are prioritized. A major strength of a MS-DIMO analysis is its ability to filter and refine data guided by biological information to obtain more meaningful and biologically relevant results. These results can be used to refine mechanistic hypotheses and/or discover potential therapeutic targets.
While the stages of a MS-DIMO analysis can be ordered in numerous ways, the first stage is important because it has a direct impact on the type of omics data that will be used in subsequent stages. In this paper, we describe a MS-DIMO analysis that uses transcriptomic data in Stage 1 and highlight the strengths of this approach. Alternative approaches to ordering omics data are discussed in subsequent sections of this manuscript.
A transcriptomic analysis provides valuable insights into gene function. Stage 1 of a MS-DIMO analysis can take advantage of this type of measurement to evaluate the relationship between a symptom and a functional gene product. The evaluation of genes using gene expression data in Stage 1 of a MS-DIMO analysis reduces the genome “search space” from millions or billions (e.g., genetic or methylation data) to hundreds or thousands of loci. This reduction in the number of statistical tests increases the power to detect significant associations between genes and a symptom of interest. The following section describes and Figure 1 illustrates an example of using this approach with “Symptom X.”
Figure 1.
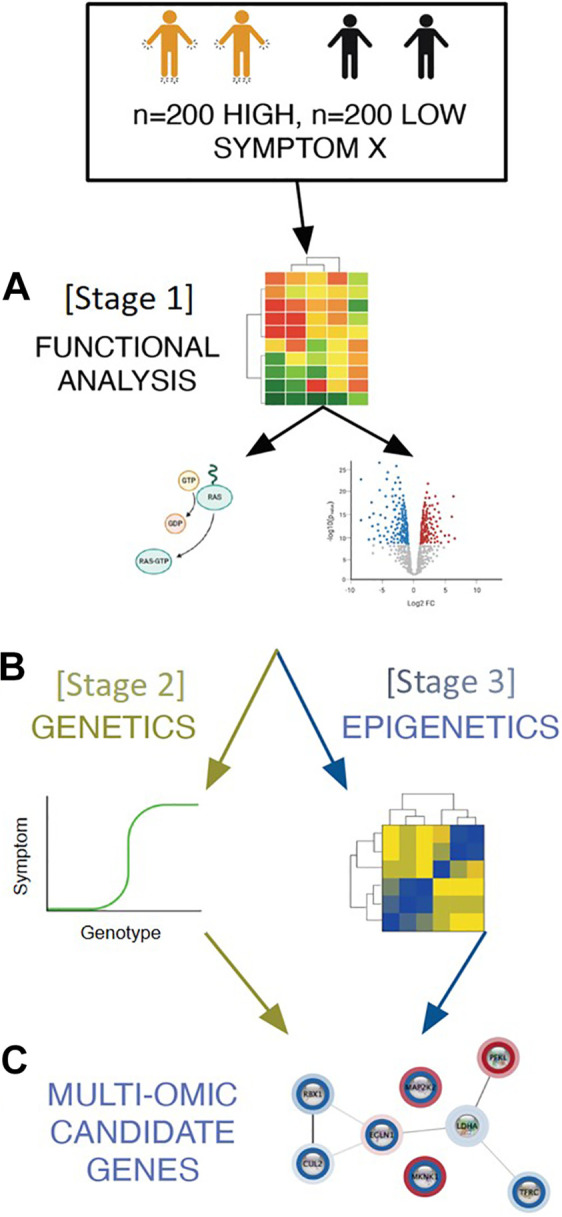
Multi-staged data-integrated multi-omics analysis: Symptom X exemplar. Patients with a chronic condition are recruited to participate in a study that is evaluating for associations between multiple levels of omics data and Symptom X. Patients are divided into groups based on their report of the severity of Symptom X. (A) In Stage 1, candidate genes are identified that are differentially expressed between patients in the Low and High Symptom X groups. (B) In Stage 2, these candidate genes are evaluated for genetic associations between the Low and High Symptom X groups. In Stage 3, the candidate genes are evaluated for differential methylation state between the Low and High Symptom X groups. (C) A list of multi-omic candidate genes are identified.
A MS-DIMO analysis is designed to discover molecular mechanisms underlying “Symptom X” in patients with an acute or chronic condition. In this study, 400 patients completed a valid and reliable instrument to assess “Symptom X.” Of the 400 patients, the extreme phenotypes for “Symptom X” are identified (i.e., 200 with low, 200 with high symptom severity scores). In Stage 1 of the MS-DIMO analysis, DEGs between the Low and High “Symptom X” groups are identified (Figure 1A). These DEGs are used as candidates in the next two stages of the MS-DIMO analysis (Figure 1B). In Stage 2, single nucleotide polymorphisms (SNPs) in these genes are selected using specific criteria (e.g., genomic location, functional evidence) and are evaluated for associations with membership in the Low versus the High “Symptom X” groups (Figure 1B). For Stage 3, the DEGs are used to evaluate for differential methylation between the Low and High “Symptom X” groups. This MS-DIMO analysis results in a list of candidate genes (Figure 1C) that were found to have significant associations in Stages 2 and/or 3. These candidate genes can be validated in subsequent samples and/or be evaluated in intervention studies that target the underlying mechanism(s) of “Symptom X.” Alternatively, these genes are good candidates to evaluate their functional relationship to each other (e.g., expression quantitative trait loci (eQTLs; e.g. genetic variants associated with levels of gene expression (Kukurba & Montgomery, 2015)), methylation quantitative trait loci (meQTL; e.g. genetic variants associated with levels of DNA methylation (Ritchie et al., 2015))).
Challenges and Benefits of a MS-DIMO Analysis
While this exemplar appears straightforward, omics data are inherently complex and large in scale (Dreisbach & Koleck, 2020). This complexity and magnitude present multiple problems that need to be considered when ordering omics analyses, as well as when collecting, analyzing, and integrating these data (Jagadish et al., 2014). The following sections describe common challenges with various types of omics data and methods that can be used to address these challenges.
Functional Information
Next-generation sequencing technologies have made the collection and evaluation of large amounts of omics data cost effective and time efficient. While the number of studies that have examined associations between symptoms and omics data have increased (Cashion et al., 2016; Fu et al., 2020), the interpretation of the results of molecular association studies are limited by a lack of functional information. When genetic or epigenetic associations are identified, their functional impact is difficult to interpret because linked variants are identified rather than a putative causal variant. By testing only those loci that had evidence of functional effects (i.e., gene expression) in Stage 1 of the MS-DIMO analysis, a clearer picture of the mechanism(s) that underlie a symptom begins to emerge in subsequent stages. These subsequent evaluations of additional omics data (e.g., genetic, epigenetic) are guided by specific hypotheses (e.g., inherited variation, genetic regulation).
Multiple Hypothesis Testing Burden
The multiple hypothesis testing burden is another challenge associated with high throughput omics data because millions of independent tests are run simultaneously (e.g., a genome-wide association study with 1 million SNPs). When a large number of tests are evaluated with an uncorrected alpha of 0.05, many of the positive results must be attributed to chance. To uncover true positive associations, the Type I error rate needs to be controlled.
Two approaches can be used to control the Type I error rate, namely: the family-wise error rate (FWER) and the false discovery rate (FDR). The FWER is the probability of making at least one Type I error. Two methods to control the FWER are the Bonferroni correction (Bonferroni, 1936) and Šidák’s procedure (Šidák, 1967). The FDR is the proportion of results that are incorrectly rejected. Procedures to control the FDR include the Benjamini-Hochberg step-up procedure (Benjamini & Hochberg, 1995) and Storey’s q-value approach (Storey, 2002).
The procedures that are used to control the FWER are extremely strict because they control for the probability of any Type I error. This conservative adjustment severely reduces the power to detect true positives by falsely rejecting a proportion of true positives (Shaffer, 1995). In contrast, the FDR has increased power to detect true positive associations while maintaining the number of Type I errors at a pre-specified alpha (Benjamini & Hochberg, 1995). For more details on various FDR procedures see the report by Korthauer and colleagues (2019).
Another approach to decrease the multiple hypothesis testing burden is to simply reduce the total number of tests. Evaluation of larger omics datasets (e.g., genetic, methylation) are often limited by the large number of statistical tests (e.g., one million loci) and small sample sizes (e.g., <1,000). A MS-DIMO analysis, with gene expression data in Stage 1, allows for the analysis of omics data with the fewest number of tests. This approach reduces the absolute number of tests performed and significantly reduces the multiple hypothesis testing burden. In addition, in gene expression analyses, each individual test may have increased power to identify differences because the effect sizes (i.e., log fold change; Harrison et al., 2019; McCarthy & Smyth, 2009) are expected to be larger. Because of this increased power and reduced number of tests, smaller samples can be evaluated. More detailed descriptions of power and sample size estimation are beyond the scope of this manuscript. Studies discussing power calculation with genetic (Skol et al., 2006), gene expression (Hart et al., 2013), and methylation (Tsai & Bell, 2015) data are described elsewhere.
Signal-to-Noise Ratio
Another statistical consideration associated with the analysis of large amounts of multi-omics data is the ability to detect a true biological signal. Assessment of the signal-to-noise ratio provides information on the accuracy of an analysis in identifying a true biological signal that is associated with a symptom versus noise (i.e., non-contributory biological data; Ideker et al., 2011). Methods that can be used to improve the signal-to-noise ratio include the addition of filters and/or integrators into the analysis (Ideker et al., 2011). In addition, the MS-DIMO analysis assists in filtering out unrelated biological data by reducing the number of tests conducted in subsequent stages of analysis.
Filters reduce noise by removing unrelated biological data (e.g., elimination of variants in distant regions of the suspected genes; Ideker et al., 2011). One type of filter uses previously published information to reduce the number of loci within the analysis. For example, in a study designed to evaluate for associations between pain and a regulatory mechanism, one can limit this evaluation to sites within the promoter region of “pain” genes. By focusing on loci in putative regulatory regions, a regulatory hypothesis is evaluated and the number of loci that warrant evaluation is reduced.
Integrators are methods that piece together individual units of data or different types of data to increase an effect size (e.g., identifying genes within shared biological pathways; Ideker et al., 2011). One example would be to select specific genes based on higher orders of biological knowledge. A number of integrators are available that contain a variety of molecular data organized by higher orders of biology (e.g., Reactome (Joshi-Tope et al., 2005), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Aoki-Kinoshita & Kanehisa, 2007), WikiPathways (Pico et al., 2008)) that provide a priori hypotheses for testing. Higher orders of biology are structured into biological pathways that represent relationships between genes, proteins, molecules, cells, other organisms, and the environment. As genes do not function in isolation, the selection of genes within biological pathways that have known associations with a symptom of interest for a MS-DIMO analysis provides a context for the interpretation of a potential mechanism for that symptom. While information within these databases can help to reduce the number of loci in a biologically meaningful way, they are limited by the data that are available. For example, novel gene-gene interactions that are not defined in these databases will be missed.
An example of an investigator-generated integrator that can be used to identify biological interactions is co-expression network analysis. Through an evaluation of gene co-expression patterns, unique networks of genes (i.e., modules) are identified (van Dam et al., 2018). Genes within these networks tend to be functionally related and co-regulated (Singer et al., 2005; van Dam et al., 2018). For example, a weighted gene co-expression network constructs networks using gene expression data and assigns weighted values to the strength of the co-expression between each gene (Zhang & Horvath, 2005). Using these co-expressed genes in a MS-DIMO analysis can improve the signal-to-noise ratio by focusing on genes that have the potential to function in shared biological processes.
Validation of Multi-Omics Results
While the integration of some or all of the methods described above can improve the signal-to-noise ratio in omics analyses, the risk of detecting noise rather than a true biological signal remains high. Therefore, the validation of results is critical for all omics studies (Ioannidis & Khoury, 2011). A MS-DIMO analysis is a type of biological validation (i.e., conceptual replication; Picho et al., 2016) because the findings from the Stage 1 analysis may be validated in Stage 2 (Ideker et al., 2011).
While validation in an independent sample is ideal (Loscalzo, 2012), it may not be an option because of limitations in available data and costs. In this situation, internal validation procedures are an acceptable alternative. While a number of internal validation procedures can be used (Perng & Aslibekyan, 2020; Sung et al., 2012), the most common methods, described below, are: cross-validation, meta-analysis, and bootstrap. The selection of the most appropriate validation procedure should be guided by a power analysis within each stage of the MS-DIMO analysis.
Internal twofold cross-validation
Cross-validation procedures involve dividing the sample into two or more subsets; conducting independent omics analyses on each subset; and conducting reciprocal analyses on all of the subsets to determine if the findings are validated in the independent subsets (Sung et al., 2012). As illustrated in Figure 2, with an internal twofold cross-validation study, the sample is split into two subsets. At each stage of this MS-DIMO analysis, the findings are validated across the two subsets (Figure 2A). For the first fold, Subset A is used to identify candidates (e.g., DEGs), that will be tested in Subset B for other associations (e.g., differential methylation or genetic association). For the second fold, the reciprocal analysis is performed (e.g., differential expression using Subset B, followed by differential methylation or genetic association in Subset A). Then, the overall significance of the loci discovered from each fold of the analysis are evaluated by combining the p-values of each independent test utilizing a meta-analytic approach (see below) using Fisher’s combined probability test (Figure 2B; Fisher, 1925, 1948).
Figure 2.

Internal twofold cross-validation design. (A) For the first fold (F1), a differential gene expression analysis will be conducted on Subset A. The differentially expressed genes (DEGs) identified in Step 1 will be tested in Subset B as candidate genes for genetic association (GA) and differential methylation (DM). For the second fold (F2), a reciprocal analysis will be conducted: a differential gene expression analysis will be conducted on Subset B with the resulting DEGs used as candidates for the GA and DM analyses in Step 2 for Subset A. (B) The overall significance of the loci from each fold are evaluated with Fisher’s combined probability test.
Cross-validation procedures are particularly useful when the relationships between various types of biological data are not clear and potentially confounded (e.g., gene expression is regulated by DNA methylation), because each type of data is used as a candidate for the other data type. In addition, cross-validation procedures may offer a solution for issues associated with sample batch effects (e.g., sample processing on different days or at different sites) or for situations where sample characteristics exhibit more variation between than within groups (e.g., merging sample data from different study sites). A major limitation of cross-validation in a single sample is the large variability in the data that are generated because the sample is divided into smaller groups. In addition, as the number of subsets or folds in the cross-validation increases, the analyses become more computationally challenging. Another limitation of cross-validation procedures is that the sample size relative to power will decrease when the sample is divided into smaller groups.
Meta-analytic approach to combine omics results
Meta-analysis procedures integrate omics data from two or more analyses to increase the ability to identify a true biological signal and to validate findings (Gao, 2016; Tseng et al., 2012). In a MS-DIMO analysis, this procedure can be used to determine the candidates that progress to the next stage of the analysis (Figure 3). As with cross-validation, the sample is split into two or more smaller samples. In Stage 1, a single omics analysis (e.g., gene expression) is completed separately for each group. Using a meta-analysis procedure (e.g., Fisher’s combined probability test; Fisher, 1925, 1948), the results of each independent analysis (e.g., DEGs) from Stage 1 are combined to produce an overall p-value. Then, based on the results of the combined tests, candidates are selected for Stage 2. This procedure is repeated in Stage 2 with a different type of omics data (e.g., genetic association). Similar to the cross-validation procedure, meta-analytic methods may not be appropriate for small samples because splitting the sample into smaller subsets will result in insufficient power.
Figure 3.

Internal validation with meta-analysis. A sample of 400 patients is evaluated for a symptom of interest. This sample is split into two smaller groups of equal size (n = 200). In Stage 1, independent differential gene expression analyses are performed on both Group 1 and Group 2. The differentially expressed genes (DEGs) from both Groups in Stage 1 are combined using uncorrected p-values with Fisher’s combined probability test. DEGs that were present in both samples are advanced as candidate genes in the independent genetic association (GA) analyses in Stage 2 for Groups 1 and 2. Genes with genetic variants that are significantly associated with the symptom of interest from both groups are combined using Fisher’s combined probability test.
Cross-validation approach for small sample sizes
In situations where sample sizes prohibit the use of the twofold cross-validation design (Fu et al., 2005) or meta-analytic methods, differential gene expression, methylation, and genetic association analyses can be cross-validated using a bootstrap procedure. Bootstrap procedures utilize a resampling technique whereby a subset of patients from the larger sample are selected and evaluated followed by the selection and evaluation of another subset. This procedure is repeated n times (Efron, 1979). One limitation is the large amount of computation time required to perform the procedure (Braga-Neto & Dougherty, 2004). Efron’s enhanced bootstrap procedure (Braga-Neto & Dougherty, 2004; Fu et al., 2005) as described by Harrell can be used to perform this analysis (Harrel, 2015).
Other Options for Ordering Omics Data in a MS-DIMO Analysis
Depending on the research question or hypothesis, other molecular starting points warrant consideration when ordering the omics data for a MS-DIMO analysis (Buescher & Driggers, 2016). For example, the central dogma of molecular biology (Crick, 1970) can be used to determine the order of the molecular data based on the flow of genetic information. Alternatively, the order can be based on the results of previous research. For example, previously identified biological pathways (e.g., mitophagy, oxidative stress) can provide the initial starting point to select candidate genes for Stage 1.
Researchers must weigh the costs associated with sample collection, processing, and storage against available resources (e.g., patient participation, funding) when designing the stages of a MS-DIMO analysis. While resource limitations may impact the types of omics data that are selected for the various stages of the analysis, a MS-DIMO analysis provides some economic efficiency. A MS-DIMO analysis can reduce the overall number of analyses and produce results that have increased power to detect true biological signals. Other common molecular starting points are discussed below.
Omics analysis guided by inherited variation
The use of genetic data in Stage 1 is one of the most common starting points. For example, the triangle method (Holzinger & Ritchie, 2012) or a three-staged approach uses genetic data in Stage 1. The goal of this analysis is to discover functional SNPs. In Stage 1, SNPs are evaluated for their associations with a symptom either using an exploratory approach (i.e., genome-wide) or with pre-specified candidate genes. SNPs that meet a pre-specified level of significance are advanced to the next stage of the analysis.
In Stage 2, these SNPs are used to test for associations with another type of omics data (e.g., gene expression, methylation, proteins; Ritchie et al., 2015). The SNPs that are associated with levels of gene expression are termed eQTLs (Kukurba & Montgomery, 2015). These eQTLs provide valuable information on the associations between gene variants and a symptom (Nica & Dermitzakis, 2013). Stage 3 of the triangle method involves an evaluation of the correlations between remaining genes and the symptom. A strength of evaluating for genetic variation in Stage 1 is that genetic data are easily accessible and more affordable to collect, process, and store than other types of omics data.
One limitation of this approach is that SNPs with large effects drive the subsequent associations between gene expression and the symptom (Holzinger & Ritchie, 2012). For example, while SNPs with small effects may contribute to the development of a symptom through their interaction with other SNPs, they would not be identified in this approach (Holzinger & Ritchie, 2012). Another limitation is the multiple hypothesis testing burden that results from the large number of association tests that are done. For example, in genome-wide association studies, significance of the identified SNPs are evaluated based on a genome-wide significance threshold (i.e., p < 5 × 10−8; Pe’er et al., 2008). For many symptom studies, this extremely small significance threshold cannot be met because of small sample sizes. However, this limitation can be overcome in a MS-DIMO analysis because, in Stage 1, the significance threshold for genomic variation can be relaxed to an exploratory threshold (e.g., p < 0.10). Then, the identified genes are evaluated in subsequent stages of the analysis using a stricter significance threshold (i.e., p < 0.05). Alternatively, the number of tests can be decreased by reducing the genome search space to only genes that were associated with the symptom.
Omics analysis guided by an epigenetic signal (microRNA, methylation)
The use of epigenetic data in Stage 1 of a MS-DIMO analysis prioritizes the identification of mechanisms involved in gene regulation. For example, research questions that are designed to explore the effects of a treatment (e.g., chemotherapy) or environmental stimuli (e.g., stress) on gene regulation and symptom severity may benefit from the use of epigenetic data in Stage 1. As with a genome-wide analysis of genetic variation, one limitation for starting with an epigenome analysis is the multiple hypothesis testing burden that results from the large number of association tests that are run. To address this limitation, a filter can be used to reduce the total number of tests. For example, meQTL that were previously identified can be used as candidates in Stage 1. Alternatively, CpG sites that have methylation levels that are associated with expression changes (i.e., expression-associated CpGs (eCpG)) can be evaluated in Stage 1 (Kennedy et al., 2018).
The use of epigenetic data in Stage 1 can be guided by information in the Encyclopedia of DNA Elements (ENCODE), a growing catalogue of functional DNA elements within the human genome. The purpose of this database is to improve our understanding of how gene expression is regulated by determining the locations and functions of regulatory elements (e.g., promoters, transcriptional regulatory elements, histone modifications; Luo et al., 2020). ENCODE employs multiple data-processing pipelines, including chromatin immunoprecipitation next-generation sequencing (ChIP-seq) technologies and whole-genome bisulfite sequencing, to discover functional elements. These technologies aid in the discovery of new regulatory regions; increase our knowledge of how interactions between proteins and DNA influence gene expression; and are valuable resources for MS-DIMO analyses (Encode Project Consortium, 2012).
Omics analysis guided by metabolites
Metabolites are derived from a variety of sources (e.g., host, microorganisms, diet). An evaluation of metabolomic data in Stage 1 of a MS-DIMO analysis can be used to identify biomarkers and system-level effects of metabolites (Johnson et al., 2016). Given that metabolomic panels are smaller in scale than genomic or epigenomic data, one advantage of using metabolomic data in Stage 1 is that it greatly reduces the scope of biological data that is explored. In contrast, this approach may be limited by the evaluation of a predefined set of metabolites (e.g., targeted metabolomics) or by what is known in metabolite databases (e.g., untargeted metabolomics; Johnson et al., 2016). These limitations may reduce the ability to detect important mechanistic factors (e.g., changes in regulation, transport) and limit the interpretation of the study findings (Pinu et al., 2019).
Omics Analysis Tools
The expertise of the research team and access to the necessary analytic tools need to be considered when designing a MS-DIMO analysis. For example, omics data are usually analyzed using complex computational methods with software packages like Bioconductor for R (Sepulveda, 2020). These software programs require basic levels of programing and genomics knowledge to use. However, researchers may lack this level of expertise or not have access to bioinformaticians. Therefore, programs that facilitate data analysis and interpretation are needed.
Numerous online resources are available to assist researchers to interpret omics analyses through visual exploration of the data (e.g., University of California Santa Cruz Genome Browser (Kent et al., 2002), Ensembl (Yates et al., 2020), Broad Integrative Genomics Viewer (Thorvaldsdottir et al., 2013)). These user friendly genomic resources are excellent sources for annotation information. For example, they provide updated information on gene and mRNA alignment, expression, and function, as well as information on gene and disease association study results.
In addition, user-friendly resources are available to analyze multiple types of omics data individually (e.g., shinyGAStool for genetic data (Hoffmann & Kober, 2020), MetaboAnalyst (Chong et al., 2018) for metabolomics, OpenMS (Rost et al., 2016) for metabolomics or proteomics) or collectively (e.g., Galaxy (Jalili et al., 2020)). A non-exhaustive list of publicly available software packages for different omics analyses are listed in Supplemental Table 1 (https://doi.org/10.5281/zenodo.4558052). Additional resources are provided in two reviews (Misra et al., 2018; Spicer et al., 2017). For more customized analyses, multiple software tools and resources are available (e.g., Bioconductor in R (Sepulveda, 2020), packages in Python like Biopython (Cock et al., 2009), Genome Analysis Toolkit (GATK) (McKenna et al., 2010)). Because these resources are open-source and maintained by individuals or small research teams, they vary in terms of online support and program maintenance.
Case Exemplars of MS-DIMO Analyses in Symptom Science
Work has already begun in symptoms science research to utilize a MS-DIMO analysis. In Box 1, we present three case exemplars that used a MS-DIMO analysis and discuss how this approach strengthened the study findings (Hong et al., 2018; Kober et al., 2020; Saligan et al., 2018).
Conclusion
The use of a MS-DIMO analysis in symptom science research has the potential to increase our understanding of molecular mechanisms that underly acute and chronic symptoms. The staging, filter, and refinement of multiple types of omics data that occur with an MS-DIMO analysis strengthens the causal molecular signal, accumulates biological evidence (Picho et al., 2016), and provides a biologically guided integration of multiple sources of data. In addition, a MS-DIMO analysis facilitates exploratory analyses (Kimmelman et al., 2014). Furthermore, the integration of multiple types of omics data into an analysis assists with the prioritization of functional or predictive genes and biological processes associated with a specific symptom. These results can be used to explore additional mechanisms that underlie symptom occurrence, severity, and distress.
Box 1.
Case Exemplars of MS-DIMO Analyses in Symptom Science.
Exemplar 1—Our research team conducted a MS-DIMO analysis that built on a previous study that found that the hypoxia inducible factor 1 signaling pathway (HIF-1 SP) was perturbed in breast cancer survivors with and without paclitaxel-induced peripheral neuropathy (PIPN) (Kober et al., 2018). For this analysis (Kober et al., 2020), we investigated genes within the HIF-1 SP that were both differentially methylated and expressed between the two survivor groups (n = 50). In Stage 1, we evaluated for differential methylation in the promoter regions of the genes within the HIF-1 SP. In Stage 2, we evaluated for differential ribonucleic acid (RNA) expression of these genes between the two survivor groups. Twelve loci across eight genes were both differentially methylated and expressed between survivors with and without PIPN. In Stage 3, we evaluated the functional role of these genes using protein-protein interaction (PPI) network connectivity and functional enrichment tests. All eight genes were significantly enriched for PPIs. In addition, seven Kyoto Encyclopedia of Genes and Genomes (KEGG) and three Reactome pathways were enriched for these eight differentially methylated and expressed genes. Then, these eight candidate genes were evaluated in animal models of neuropathic pain. The mitogen-activated protein kinase 1 interacting serine/threonine kinase 1 (MKNK1) gene had RNA expression and methylation differences associated with neuropathic pain in both breast cancer survivors and animal models. The strength of these findings supports the need for validation of these genes as potential targets for therapeutic intervention.
These findings were strengthened by multiple design decisions that reduced our search space of relevant genes associated with PIPN. By utilizing our previous research findings and domain specific information from the KEGG database, we were able to reduce our initial search space in Stage 1 to 100 genes within a biologically relevant pathway. Furthermore, the evaluation of only the promoter regions of these genes reduced the search space to functional gene regulatory regions. These steps, coupled with the integration of multiple levels of omics data, strengthened our analyses by reducing the number of statistical tests and directing our evaluation of more biologically relevant mechanisms.
Exemplar 2—Saligan and colleagues (2018) used a MS-DIMO analysis to explore the relationships between fatigue severity and gene expression (RNA and proteins) associated with T lymphocyte proliferation in men (n = 30) receiving radiation therapy for prostate cancer. In Stage 1, RNA levels of 327 genes were found to be differentially expressed between the initiation and middle of radiation therapy. Of these genes, arginase type 1 (ARG1) was significantly upregulated between the initiation and middle of radiation therapy and was found to be negatively correlated with the change in absolute lymphocyte count in patients with high fatigue. In Stage 2, differences in protein levels of ARG1 and arginine in plasma were evaluated. While differences in protein levels did not meet statistical significance, a trend in the levels of ARG1 and arginine was observed for patients with high fatigue. While limited, these findings support the role of ARG1 and arginine in T lymphocyte suppression and the development of fatigue in men with prostate cancer and warrant further study.
Exemplar 3—Hong and colleagues (2018) conducted a MS-DIMO analysis using epigenetic and genetic data to evaluate the occurrence of pre-term spontaneous birth in Black mothers (n = 300). In Stage 1, differentially methylated sites were compared between mothers with spontaneous pre-term birth and mothers with full-term births. In subsequent stages, genome-wide data from regions near these differentially methylated sites were evaluated. Two sites on the cytohesin 1 interacting protein (CYTIP) and long intergenic non-protein coding RNA 114 (LINC00114) genes were found to be associated with spontaneous pre-term birth. Of note, the differentially methylated sites were not associated with between group differences in genetic variation. These results suggest that epigenetic changes associated with spontaneous pre-term birth may not be driven by genetic variations and future research should evaluate for functional changes in CYTIP and LINC00114. The authors suggest that these genes may be used as early predictive markers of pre-term birth and may serve as targets for prevention or drug development.
References
Hong, X., Sherwood, B., Ladd-Acosta, C., Peng, S., Ji, H., Hao, K., Burd, I., Bartell, T. R., Wang, G., Tsai, H. J., Liu, X., Ji, Y., Wahl, A., Caruso, D., Lee-Parritz, A., Zuckerman, B., & Wang, X. (2018). Genome-wide DNA methylation associations with spontaneous preterm birth in US blacks: Findings in maternal and cord blood samples. Epigenetics, 13(2), 163–172. https://doi.org/10.1080/15592294.2017.1287654
Kober, K. M., Lee, M. C., Olshen, A., Conley, Y. P., Sirota, M., Keiser, M., Hammer, M. J., Abrams, G., Schumacher, M., Levine, J. D., & Miaskowski, C. (2020). Differential methylation and expression of genes in the hypoxia-inducible factor 1 signaling pathway are associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors and with preclinical models of chemotherapy-induced neuropathic pain. Molecular Pain, 16, 1–15. https://doi.org/10.1177/1744806920936502
Kober, K. M., Olshen, A., Conley, Y. P., Schumacher, M., Topp, K., Smoot, B., Mazor, M., Chesney, M., Hammer, M., Paul, S. M., Levine, J. D., & Miaskowski, C. (2018). Expression of mitochondrial dysfunction-related genes and pathways in paclitaxel-induced peripheral neuropathy in breast cancer survivors. Molecular Pain, 14, 1–16. https://doi.org/10.1177/1744806918816462
Saligan, L. N., Lukkahatai, N., Zhang, Z., Cheung, C. W., & Wang, X. (2018). Altered Cd8+ T lymphocyte response triggered by arginase 1: Implication for fatigue intensification during localized radiation therapy in prostate cancer patients. Neuropsychiatry (London), 8(4), 1249–1262. https://doi.org/10.4172/Neuropsychiatry.1000454
Supplemental Material
Supplemental Material, sj-pdf-1-brn-10.1177_10998004211003980 for Multi-Staged Data-Integrated Multi-Omics Analysis for Symptom Science Research by Carolyn S. Harris, Christine A. Miaskowski, Anand A. Dhruva, Janine Cataldo and Kord M. Kober in Biological Research For Nursing
Footnotes
Author Contributions: Carolyn S. Harris contributed to conception and design, contributed to acquisition and interpretation, drafted manuscript, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Christine Miaskowski contributed to conception and design, contributed to interpretation, drafted manuscript, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Anand A. Dhruva contributed to interpretation, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Janine Cataldo contributed to interpretation, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Kord M. Kober contributed to conception and design, contributed to acquisition and interpretation, drafted manuscript, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This manuscript was supported by grants from the National Cancer Institute (NCI, CA107091, CA118658, CA233774), the American Cancer Society (#IRG-97-150-13), and the National Institute of Nursing Research of the National Institutes of Health (T32NR016920). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Dr. Miaskowski is an American Cancer Society Clinical Research Professor. Carolyn Harris is supported by a grant from the American Cancer Society (134336-DSCN-20-073-01-SCN).
ORCID iDs: Carolyn S. Harris  https://orcid.org/0000-0002-7080-4990
https://orcid.org/0000-0002-7080-4990
Kord M. Kober
https://orcid.org/0000-0001-9732-3321
Supplemental Material: Supplemental material for this article is available online.
References
- Aoki-Kinoshita K. F., Kanehisa M. (2007). Gene annotation and pathway mapping in KEGG. In Bergman N. H. (Ed.), Comparative genomics (Vol. 396, pp. 71–91). Humana Press. https://doi-org.ucsf.idm.oclc.org/10.1007/978-1-59745-515-2_6 [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. Journal of the Royal Statistical Society, 57(1), 289–399. [Google Scholar]
- Bonferroni C. E. (1936). Teoria statistica delle classi e calcolo delle probabilita [Statistical theory of classes and calculation of probabilities]. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze, 8, 3–62. [Google Scholar]
- Braga-Neto U. M., Dougherty E. R. (2004). Is cross-validation valid for small-sample microarray classification? Bioinformatics, 20(3), 374–380. 10.1093/bioinformatics/btg419 [DOI] [PubMed] [Google Scholar]
- Buescher J. M., Driggers E. M. (2016). Integration of omics: More than the sum of its parts. Cancer & Metababolism, 4, 4. 10.1186/s40170-016-0143-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashion A. K., Gill J., Hawes R., Henderson W. A., Saligan L. (2016). National Institutes of Health Symptom Science Model sheds light on patient symptoms. Nursing Outlook, 64(5), 499–506. 10.1016/j.outlook.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D. S., Xia J. (2018). MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Research, 46(W1), W486–W494. 10.1093/nar/gky310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou Y. J., Kober K. M., Kuo C. H., Yeh K. H., Kuo T. C., Tseng Y. J., Miaskowski C., Liang J. T., Shun S. C. (2020). A pilot study of metabolomic pathways associated with fatigue in survivors of colorectal cancer. Biological Research for Nursing, 23(1), 42–49. 10.1177/1099800420942586 [DOI] [PubMed] [Google Scholar]
- Cock P. J., Antao T., Chang J. T., Chapman B. A., Cox C. J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B., de Hoon M. J. (2009). Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422–1423. 10.1093/bioinformatics/btp163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. (1970). Central dogma of molecular biology. Nature, 227, 561–563. [DOI] [PubMed] [Google Scholar]
- Dorsey S. G., Renn C. L., Griffioen M., Lassiter C. B., Zhu S., Huot-Creasy H., McCracken C., Mahurkar A., Shetty A. C., Jackson-Cook C. K., Kim H., Henderson W. A., Saligan L., Gill J., Colloca L., Lyon D. E., Starkweather A. R. (2019). Whole blood transcriptomic profiles can differentiate vulnerability to chronic low back pain. PLoS One, 14(5), e0216539. 10.1371/journal.pone.0216539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreisbach C., Koleck T. A. (2020). The state of data science in genomic nursing. Biological Research for Nursing, 22(3), 309–318. 10.1177/1099800420915991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B. (1979). Bootstrap methods: Another look at the jacknife. The Annals of Statistics, 7, 1–26. 10.1214/aos/1176344552 [DOI] [Google Scholar]
- Encode Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R. A. (1925). Statistical methods for research workers. Oliver and Boyd. [Google Scholar]
- Fisher R. A. (1948). Questions and answers #14. The American Statistician, 2(5), 30–31. [Google Scholar]
- Founds S. (2018). Systems biology for nursing in the era of big data and precision health. Nursing Outlook, 66(3), 283–292. 10.1016/j.outlook.2017.11.006 [DOI] [PubMed] [Google Scholar]
- Founds S. A. (2009). Introducing systems biology for nursing science. Biological Research for Nursing, 11(1), 73–80. 10.1177/1099800409331893 [DOI] [PubMed] [Google Scholar]
- Fu M. R., Kurnat-Thoma E., Starkweather A., Henderson W. A., Cashion A. K., Williams J. K., Katapodi M. C., Reuter-Rice K., Hickey K. T., Barcelona de Mendoza V., Calzone K., Conley Y. P., Anderson C. M., Lyon D. E., Weaver M. T., Shiao P. K., Constantino R. E., Wung S. F., Hammer M. J., Voss J. G., Coleman B. (2020). Precision health: A nursing perspective. International Journal of Nursing Sciences, 7(1), 5–12. 10.1016/j.ijnss.2019.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W. J., Carroll R. J., Wang S. (2005). Estimating misclassification error with small samples via bootstrap cross-validation. Bioinformatics, 21(9), 1979–1986. 10.1093/bioinformatics/bti294 [DOI] [PubMed] [Google Scholar]
- Gao X. (2016). Statistical method for integrative platform analysis: Application to integration of proteomic and microarray data. In Jung K. (Ed.), Methods in molecular biology (Vol. 1362, pp. 199–207). Humana Press. 10.1007/978-1-4939-3106-4_13 [DOI] [PubMed] [Google Scholar]
- Goo Y. A., Cain K., Jarrett M., Smith L., Voss J., Tolentino E., Tsuji J., Tsai Y. S., Panchaud A., Goodlett D. R., Shulman R. J., Heitkemper M. (2012). Urinary proteome analysis of irritable bowel syndrome (IBS) symptom subgroups. Journal of Proteome Research, 11(12), 5650–5662. 10.1021/pr3004437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrel F. E. (2015). Multivariable modeling strategies. In Regression modeling strategies: With applications to linear models, logistic and ordinal regression, and survival analysis (2nd ed., pp. 63–102). Springer. [Google Scholar]
- Harrison P. F., Pattison A. D., Powell D. R., Beilharz T. H. (2019). Topconfects: A package for confident effect sizes in differential expression analysis provides a more biologically useful ranked gene list. Genome Biology, 20(1), 67. 10.1186/s13059-019-1674-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S. N., Therneau T. M., Zhang Y., Poland G. A., Kocher J. P. (2013). Calculating sample size estimates for RNA sequencing data. Journal of Computational Biology: A Journal of Computational Molecular Cell Biology, 20(12), 970–978. 10.1089/cmb.2012.0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y., Seldin M., Lusis A. (2017). Multi-omics approaches to disease. Genome Biology, 18(1), 1–15. 10.1186/s13059-017-1215-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T., Kober K. M. (2020). shinyGAStool. https://github.com/kordk/shinyGAStool
- Holzinger E. R., Ritchie M. D. (2012). Integrating heterogeneous high-throughput data for meta-dimensional pharmacogenomics and disease-related studies. Pharmacogenomics, 13(2), 213–222. 10.2217/PGS.11.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong X., Sherwood B., Ladd-Acosta C., Peng S., Ji H., Hao K., Burd I., Bartell T. R., Wang G., Tsai H. J., Liu X., Ji Y., Wahl A., Caruso D., Lee-Parritz A., Zuckerman B., Wang X. (2018). Genome-wide DNA methylation associations with spontaneous preterm birth in US blacks: Findings in maternal and cord blood samples. Epigenetics, 13(2), 163–172. 10.1080/15592294.2017.1287654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T., Dutkowski J., Hood L. (2011). Boosting signal-to-noise in complex biology: Prior knowledge is power. Cell, 144(6), 860–863. 10.1016/j.cell.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis J. P., Khoury M. J. (2011). Improving validation practices in “omics” research. Science, 334(6060), 1230–1232. 10.1126/science.1211811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadish H. V., Gehrke J., Labrinidis A., Papakonstantinou Y., Patel J. M., Ramakrishnan R., Shahabi C. (2014). Big data and its technical challenges. Communications of the ACM, 57(7), 86–94. 10.1145/2611567 [DOI] [Google Scholar]
- Jalili V., Afgan E., Gu Q., Clements D., Blankenberg D., Goecks J., Taylor J., Nekrutenko A. (2020). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Research, 48(W1), W395–W402. 10.1093/nar/gkaa434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. H., Ivanisevic J., Siuzdak G. (2016). Metabolomics: Beyond biomarkers and towards mechanisms. Nature Reviews Molecular Cell BIology, 17, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G., Gillespie M., Vastrik I., D’Eustachio P., Schmidt E., de Bono B., Jassal B., Gopinath G. R., Wu G. R., Matthews L., Lewis S., Birney E., Stein L. (2005). Reactome: A knowledgebase of biological pathways. Nucleic Acids Research, 33(Database issue), D428–432. 10.1093/nar/gki072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy E. M., Goehring G. N., Nichols M. H., Robins C., Mehta D., Klengel T., Eskin E., Smith A. K., Conneely K. N. (2018). An integrated-omics analysis of the epigenetic landscape of gene expression in human blood cells. BMC Genomics, 19(1), 476. 10.1186/s12864-018-4842-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D. (2002). The human genome browser at UCSC. Genome Research, 12(6), 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman J., Mogil J. S., Dirnagl U. (2014). Distinguishing between exploratory and confirmatory preclinical research will improve translation. PLoS Biology, 12(5), 1–4. 10.1371/journal.pbio.1001863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner M. W. (2005). The meaning of systems biology. Cell, 121(4), 503–504. 10.1016/j.cell.2005.05.005 [DOI] [PubMed] [Google Scholar]
- Kober K. M., Lee M. C., Olshen A., Conley Y. P., Sirota M., Keiser M., Hammer M. J., Abrams G., Schumacher M., Levine J. D., Miaskowski C. (2020). Differential methylation and expression of genes in the hypoxia-inducible factor 1 signaling pathway are associated with paclitaxel-induced peripheral neuropathy in breast cancer survivors and with preclinical models of chemotherapy-induced neuropathic pain. Molecular Pain, 16, 1–15. 10.1177/1744806920936502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleck T. A., Bender C. M., Clark B. Z., Ryan C. M., Ghotkar P., Brufsky A., McAuliffe P. F., Rastogi P., Sereika S. M., Conley Y. P. (2017). An exploratory study of host polymorphisms in genes that clinically characterize breast cancer tumors and pretreatment cognitive performance in breast cancer survivors. Breast Cancer—Targets and Therapy, 9, 95–110. 10.2147/BCTT.S123785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthauer K., Kimes P. K., Duvallet C., Reyes A., Subramanian A., Teng M., Shukla C., Alm E. J., Hicks S. C. (2019). A practical guide to methods controlling false discoveries in computational biology. Genome Biology, 20(1), 1–21. 10.1186/s13059-019-1716-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukurba K. R., Montgomery S. B. (2015). RNA sequencing and analysis. Cold Spring Harbor Protocols, 2015(11), 951–969. 10.1101/pdb.top084970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscalzo J. (2012). Irreproducible experimental results: Causes, (mis)interpretations, and consequences. Circulation, 125(10), 1211–1214. 10.1161/CIRCULATIONAHA.112.098244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Hitz B. C., Gabdank I., Hilton J. A., Kagda M. S., Lam B., Myers Z., Sud P., Jou J., Lin K., Baymuradov U. K., Graham K., Litton C., Miyasato S. R., Strattan J. S., Jolanki O., Lee J. W., Tanaka F. Y., Adenekan P., O’Neill E., Cherry J. M. (2020). New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Research, 48(D1), D882–D889. 10.1093/nar/gkz1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy D. J., Smyth G. K. (2009). Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics, 25(6), 765–771. 10.1093/bioinformatics/btp053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A. (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaskowski C., Barsevick A., Berger A., Casagrande R., Grady P. A., Jacobsen P., Kutner J., Patrick D., Zimmerman L., Xiao C., Matocha M., Marden S. (2017). Advancing symptom science through symptom cluster research: Expert panel proceedings and recommendations. Journal of the National Cancer Institute, 109(4). 10.1093/jnci/djw253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra B. B., Langefeld C. D., Olivier M., Cox L. A. (2018). Integrated omics: Tools, advances, and future approaches. Journal of Molecular Endocrinology, 62(1), R21–R45. 10.1530/JME-18-0055 [DOI] [PubMed] [Google Scholar]
- National Institute of Nursing Research. (2016). The NINR strategic plan: Advancing science, improving lives. https://www.ninr.nih.gov/sites/files/docs/NINR_StratPlan2016_reduced.pdf
- Nica A. C., Dermitzakis E. T. (2013). Expression quantitative trait loci: Present and future. Philosophical Transactions of the Royal Society B, 368(1620), 1–6. 10.1098/rstb.2012.0362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pe’er I., Yelensky R., Altshuler D., Daly M. J. (2008). Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genetic Epidemiology, 32(4), 381–385. 10.1002/gepi.20303 [DOI] [PubMed] [Google Scholar]
- Perng W., Aslibekyan S. (2020). Find the needle in the haystack, then find it again: Replication and validation in the ‘omics era. Metabolites, 10(7), 286. 10.3390/metabo10070286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picho K., Maggio L. A., Artino A. R., Jr. (2016). Science: The slow march of accumulating evidence. Perspectives on Medical Education, 5(6), 350–353. 10.1007/s40037-016-0305-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico A. R., Kelder T., van Iersel M. P., Hanspers K., Conklin B. R., Evelo C. (2008). WikiPathways: Pathway editing for the people. PLoS Biology, 6(7), 1403–1407. 10.1371/journal.pbio.0060184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinu F. R., Beale D. J., Paten A. M., Kouremenos K., Swarup S., Schirra H. J., Wishart D. (2019). Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites, 9(4), 1–31. 10.3390/metabo9040076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. D., Holzinger E. R., Li R., Pendergrass S. A., Kim D. (2015). Methods of integrating data to uncover genotype-phenotype interactions. Nature Reviews Genetics, 16(2), 85–97. 10.1038/nrg3868 [DOI] [PubMed] [Google Scholar]
- Rost H. L., Sachsenberg T., Aiche S., Bielow C., Weisser H., Aicheler F., Andreotti S., Ehrlich H. C., Gutenbrunner P., Kenar E., Liang X., Nahnsen S., Nilse L., Pfeuffer J., Rosenberger G., Rurik M., Schmitt U., Veit J., Walzer M., Wojnar D., Wolski W. E., Schilling O., Choudhary J. S., Malmstrom L., Aebersold R., Reinert K., Kohlbacher O. (2016). OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nature Methods, 13(9), 741–748. 10.1038/nmeth.3959 [DOI] [PubMed] [Google Scholar]
- Saligan L. N., Lukkahatai N., Zhang Z., Cheung C. W., Wang X. (2018). Altered Cd8+ T lymphocyte response triggered by arginase 1: Implication for fatigue intensification during localized radiation therapy in prostate cancer patients. Neuropsychiatry (London), 8(4), 1249–1262. 10.4172/Neuropsychiatry.1000454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda J. L. (2020). Using R and Bioconductor in clinical genomics and transcriptomics. Journal of Molecular Diagnostics, 22(1), 3–20. 10.1016/j.jmoldx.2019.08.006 [DOI] [PubMed] [Google Scholar]
- Shaffer J. P. (1995). Multiple hypothesis testing. Annual Reviews in Psychology, 46, 561–584. 10.1146/annurev.ps.46.020195.003021 [DOI] [Google Scholar]
- Šidák Z. (1967). Rectangular confidence regions for the means of multivariate normal distributions. Journal of the American Statistical Association, 62(318), 626–633. 10.1080/01621459.1967.10482935 [DOI] [Google Scholar]
- Singer G. A., Lloyd A. T., Huminiecki L. B., Wolfe K. H. (2005). Clusters of co-expressed genes in mammalian genomes are conserved by natural selection. Molecular Biology and Evolution, 22(3), 767–775. 10.1093/molbev/msi062 [DOI] [PubMed] [Google Scholar]
- Skol A. D., Scott L. J., Abecasis G. R., Boehnke M. (2006). Joint analysis is more efficient than replication-based analysis for 2-stage genome-wide association studies. Nature Genetics, 38(2), 209–213. 10.1038/ng1706 [DOI] [PubMed] [Google Scholar]
- Spicer R., Salek R. M., Moreno P., Canueto D., Steinbeck C. (2017). Navigating freely-available software tools for metabolomics analysis. Metabolomics, 13(9), 106. 10.1007/s11306-017-1242-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. D. (2002). A Direct approach to false discovery rates. Journal of the Royal Statistical Society, 64(3), 479–498. [Google Scholar]
- Sun Y. V., Hu Y. J. (2016). Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases. In Kumar D. (Ed.), Advances in genetics (Vol. 93, pp. 147–190). 10.1016/bs.adgen.2015.11.004 [DOI] [PMC free article] [PubMed]
- Sung J., Wang Y., Chandrasekaran S., Witten D. M., Price N. D. (2012). Molecular signatures from omics data: From chaos to consensus. Biotechnology Journal, 7(8), 946–957. 10.1002/biot.201100305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H., Robinson J. T., Mesirov J. P. (2013). Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P. C., Bell J. T. (2015). Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. International Journal of Epidemiology, 44(4), 1429–1441. 10.1093/ije/dyv041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng G. C., Ghosh D., Feingold E. (2012). Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Research, 40(9), 3785–3799. 10.1093/nar/gkr1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully L. A., Grady P. A. (2015). A path forward for genomic nursing research. Research in Nursing and Health, 38(3), 177–179. 10.1002/nur.21659 [DOI] [PubMed] [Google Scholar]
- van Dam S., Vosa U., van der Graaf A., Franke L., de Magalhaes J. P. (2018). Gene co-expression analysis for functional classification and gene-disease predictions. Briefings in Bioinformatics, 19(4), 575-592. 10.1093/bib/bbw139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston A. D., Hood L. (2004). Systems biology, proteomics, and the future of health care: Toward predictive, preventative, and personalized medicine. Journal of Proteome Research, 3, 179–196. 10.1021/pr0499693 [DOI] [PubMed] [Google Scholar]
- Yates A. D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M. R., Armean I. M., Azov A. G., Bennett R., Bhai J., Billis K., Boddu S., Marugan J. C., Cummins C., Davidson C., Dodiya K., Fatima R., Gall A.…Flicek P. (2020). Ensembl 2020. Nucleic Acids Research, 48(D1), D682–D688. 10.1093/nar/gkz966 [DOI] [PMC free article] [PubMed]
- Zhang B., Horvath S. (2005). A general framework for weighted gene co-expression network analysis. Statistical Applications in Genetics and Molecular Biology, 4(17). 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material, sj-pdf-1-brn-10.1177_10998004211003980 for Multi-Staged Data-Integrated Multi-Omics Analysis for Symptom Science Research by Carolyn S. Harris, Christine A. Miaskowski, Anand A. Dhruva, Janine Cataldo and Kord M. Kober in Biological Research For Nursing