

ABSTRACT
We propose that beyond its role in WNT secretion, WLS/GPR177 (wntless, WNT ligand secretion mediator) acts as an essential regulator controlling protein glycosylation, endoplasmic reticulum (ER) homeostasis, and dendritic cell (DC)-mediated immunity. WLS deficiency in bone marrow-derived DCs (BMDCs) resulted in poor growth and an inability to mount cytokine and T-cell responses in vitro, phenotypes that were irreversible by the addition of exogenous WNTs. In fact, WLS was discovered to integrate a protein complex in N-glycan-dependent and WLS domain-selective manners, comprising ER stress sensors and lectin chaperones. WLS deficiency in BMDCs led to increased ER stress response and macroautophagy/autophagy, decreased calcium efflux from the ER, and the loss of CALR (calreticulin)-CANX (calnexin) cycle, and hence protein hypo-glycosylation. Consequently, DC-specific wls-null mice were unable to develop both Th1-, Th2- and Th17-associated responses in the respective autoimmune and allergic disease models. These results suggest that WLS is a critical chaperone in maintaining ER homeostasis, glycoprotein quality control and calcium dynamics in DCs.
Abbreviations: ATF6: activating transcription factor 6; ATG5: autophagy related 5; ATG12: autophagy related 12; ATG16L1: autophagy related 16 like 1; ATP2A1/SERCA1: ATPase sarcoplasmic/endoplasmic reticulum Ca2+ transporting 1; BALF: bronchoalveolar lavage fluid; BFA: brefeldin A; BMDC: bone marrow-derived dendritic cell; CALR: calreticulin; CANX: calnexin; CCL2/MCP-1: C-C motif chemokine ligand 2; CNS: central nervous system; CT: C-terminal domain; DTT: dithiothreitol; DNAJB9/ERDJ4: DnaJ heat shock protein family (Hsp40) member B9; EAE: experimental autoimmune encephalomyelitis; EIF2A/eIF2α: eukaryotic translation initiation factor 2A; EIF2AK3/PERK: eukaryotic translation initiation factor 2 alpha kinase 3; ERN1/IRE1: endoplasmic reticulum (ER) to nucleus signaling 1; GFP: green fluorescent protein; HSPA5/GRP78/BiP: heat shock protein A5; IFNA: interferon alpha; IFNAR1: interferon alpha and beta receptor subunit 1; IFNB: interferon beta; IFNG/INFγ: interferon gamma; IFNGR2: interferon gamma receptor 2; IL6: interleukin 6; IL10: interleukin 10; IL12A: interleukin 12A; IL23A: interleukin 23 subunit alpha; ITGAX/CD11c: integrin subunit alpha X; ITPR1/InsP3R1: inositol 1,4,5-trisphosphate receptor type 1; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; OVA: ovalbumin; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PLF: predicted lipocalin fold; PPP1R15A/GADD34: protein phosphatase 1 regulatory subunit 15A; RYR1/RyanR1: ryanodine receptor 1, skeletal muscle; SD: signal domain; TGFB/TGF-β: transforming growth factor beta family; Th1: T helper cell type 1; Th17: T helper cell type 17; TM: tunicamycin; TNF/TNF-α: tumor necrosis factor; UPR: unfolded protein response; WLS/wntless: WNT ligand secretion mediator.
KEYWORDS: Dendritic cells, er stress, gpr177, protein glycosylation, unfold protein response
Introduction
WLS/GPR177 (wntless, WNT ligand secretion mediator) is a conserved, multi-pass endoplasmic reticulum (ER) transmembrane protein known to control the transport and secretion of WNT molecules to initiate WNT signaling [1,2]. Emerging studies have highlighted the importance of WNT signaling pathway in regulating immunity, which has been clearly exemplified in dendritic cells (DCs) [3], a critical cell type in initiating and regulating adaptive immune responses [4,5]. Loss of Wls function results in retrograde Golgi–ER transport and ER stress [6], which is associated with the pathogenesis of many diseases [7–9]; also, loss or defect of Wls function causes early embryonic death [10] and multiple developmental defects in lymphopoiesis, neurogenesis, and osteogenesis in adults [11–13]. These deficiencies, by inference, have all been attributed to the loss of WNT secretion and signaling, but one intriguing question remains as to whether there exists additional role of WLS within the ER beyond its classically defined function as a WNT cargo receptor.
ER is involved in virtually all cellular function in protein folding, posttranslational modification, including N-linked glycosylation, Ca2+ homeostasis, lipid synthesis and metabolism. The majority of proteins synthesized in the ER are decorated with N-linked glycans critical for protein maturation and as a quality control signal. Within the ER, folding and modification of nascent proteins are facilitated through the actions of a series of molecular chaperones, lectins, and glycosidases [14,15]. When misfolded, proteins are usually subjected to ER-associated degradation (ERAD). The ER stress can be caused by a number of mechanisms, including impaired ER glycoprotein quality control, protein glycosylation and folding, or blockade of calcium channels, which ultimately determine cell fate through a series of adaptive responses, such as the unfolded protein response (UPR) consisting of complex cytoplasmic and nuclear signaling pathways, or triggering of autophagy and apoptosis [14,16,17].
The UPR is a highly conserved pathway that ensures sensitivity of the ER to the stresses of synthesis, folding, modification, and secretory demands of proteins associated with environmental stimuli [18,19], in which HSPA5/GRP78/BiP (heat shock protein family A [Hspa70] member 5) is a key component and dissociates from three types of ER-localized transmembrane signal transducers to lead to their activation. These transducers include protein kinases ERN1/IRE1 (endoplasmic reticulum [ER] to nucleus signaling 1), EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3), and the transcription factor ATF6 (activating transcription factor 6) [18]. Also, as a part of the ER quality control machinery, CANX (calnexin) and CALR (calreticulin) are key components of the CANX-CALR cycle responsible for the folding of newly synthesized proteins and for glycoprotein quality control pathways in the ER. These actions, collectively, promote correct folding, eliminate faulty protein, and restore ER homeostasis, wherein well-balanced coordination and activity are critical. While the importance of ER quality control has been well appreciated, the mechanisms of action and coordination in the ER remain to be fully defined. Herein, we present evidence supporting a novel role of WLS in mediating the assembly of a molecular chaperone complex involving the CANX-CALR cycle, UPR sensors (ERN1, EIF2AK3, ATF6, HSPA5) and calcium regulators, while its deficiency and dysregulation significantly alter dendritic cell function both in vitro and in vivo.
Results
Multiple functional deficiencies are noted in wls-null DCs
To explore the functions of WLS in DCs, we generated DC-specific wls-null mice (wlsfx/fx; Fig. S1A–C) which were viable and showed similar percentages of BMDC subsets (ITGAX/CD11c+ CD40+ and ITGAX+ CD80+; Fig. S1D and S1E) as those seen in WT and Wls-heterozygous mice (Wlsfx/+). However, wls-null BMDCs formed fewer colonies and differentiated poorly in the presence of CSF2/GM-CSF (colony stimulating factor 2), as compared to those of wild-type BMDCs (Figure 1A). In fact, wls-null BMDCs exhibited significantly higher percentages of apoptosis and autophagy (Figures 1B and 1C), as evidenced by the increased expression of autophagic markers, MAP1LC3B/LC3B, ATG5, ATG12, ATG16 L1, and PIK3C3/VSP34 (Figure 1D, S4A and S4B) as compared to those seen in wild-type BMDCs. Notably, this effect could not be reversed by treatment of the cells with WNT1 or WNT3A (Figures 1E and 1F). Further, analysis of DC-derived cytokines showed that at baseline, the percentages of the cells expressing TNF/TNF-α, TGFB/TGF-β, IL6, IL10, IL12A, and IL23A were lower in BMDCs lacking Wls than those in wild-type BMDCs (Figure 1G). Following stimulation with lipopolysaccharide (LPS), lower levels of expression for major histocompatibility complex II (MHC class II), CD80, CD86, IL6, IL10 and IL12A were observed in wls-null BMDCs as compared to those seen in WT cells, while the expression of MHC class I appeared to be unchanged (Figure 1H and S1F-J); also, when BMDCs were co-cultured with autologous T cells, significantly lower levels of IFNG/IFN-γ and TNF were found (Figure 1I).
Figure 1.
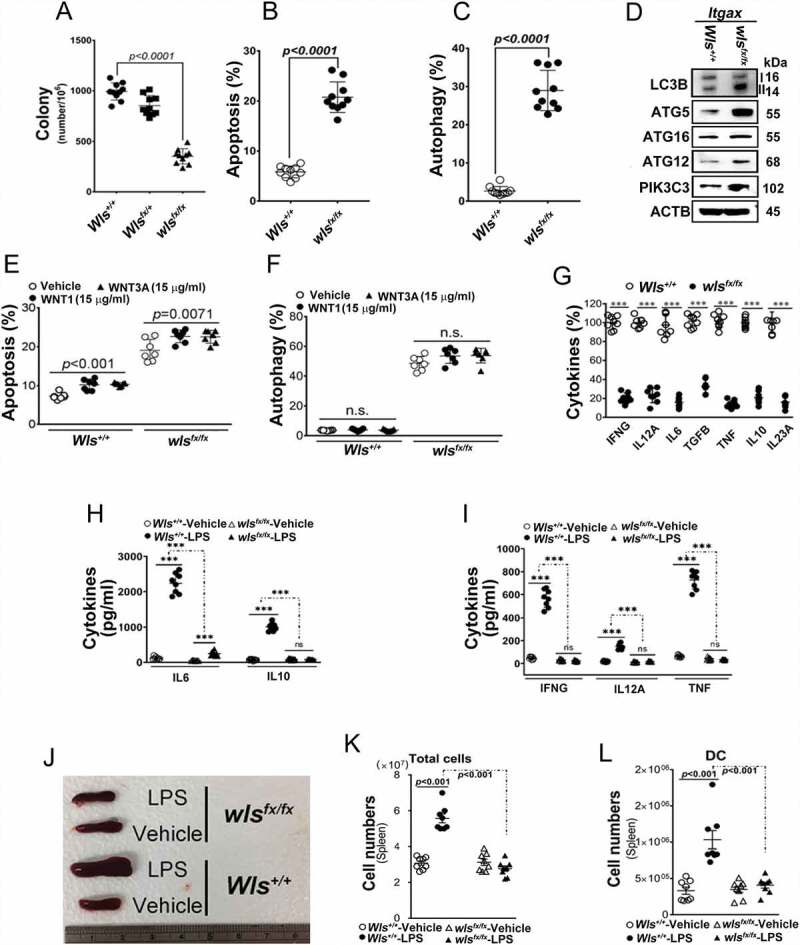
Effect of WLS deficiency on BMDCs. (A) Colony formation of BMDCs with wild-type, heterozygous, or wls-null (wlsfx/fx) genotypes (n = 8). (B and C) Percentage of apoptotic and autophagic BMDCs with wild-type, heterozygous, or wls-null genotypes (n = 8). (D) Western blots of autophagic markers (LC3B, PIK3C3, ATG5, ATG12 and ATG16L1) in lysates of wild-type and wls-null BMDCs (n = 3; p< 0.001). (E) Percentage of apoptotic BMDCs with wild-type and wls-null genotypes by WNT1 and WNT3A treatment. (F) Percentage of autophagic BMDCs with wild-type and wls-null genotypes after WNT1 and WNT3A treatment. (G) Level of cytokine-expressing cells in wild-type or wls-null (wlsfx/fx) BMDCs. (H) Levels of IL12A, IL6, and IL10 secreted by wild-type and wls-null BMDCs, or (I) IFNG and TNF secreted by cocultured CD4+ T cells in the presence or absence of LPS. (J) Spleen size in wild-type and DC-specific wls-null mice with or without LPS treatment. Flow cytometric analysis of the total number of (K) splenocytes and (L) DCs in wild-type and DC-specific wls-null mice after LPS treatment (n = 8; p< 0.001)
Moreover, after stimulation with M. Tuberculosis, significantly lower levels of TNF, IL10, IL6 and IFNG were found in a co-culture system of wls-deficient BMDCs and CD4+ T cells (Fig. S1K and S1L). Consequently, BMDCs with wls deficiency failed to prime CD4+ T-cell proliferation in the co-culture system (Fig. S1M). Also, after LPS challenge, the spleens of wild-type, but not DC-specific wls-null mice, increased in size (Figure 1J), concomitant with significantly increased total number of spleen cells and DCs (Figures 1K and 1L). These functional effects of lipopolysaccharide (LPS) challenge could not be observed in wls-null BMDCs.
WLS deficiency results in ER stress and the loss of ER quality control
As WLS is an ER membrane protein, WLS deficiency might result in ER stress response and its quality control [20,21]. Indeed, wls-null BMDCs showed significantly increased ER stress response, as evidenced by enhanced EIF2AK3-EIF2A activity (Figure 2A and S4C), and interestingly, reduction in the levels of CANX, ITPR1/InsP3R1 (inositol trisphosphate receptor isoform 1) and RYR1/RyanR1 were noted, while, to a lesser degree, the levels of CALR and HSPA5, in wls-null BMDCs (Figure 2A and S4C). In contrast, no apparent changes in the levels of EIF2AK3, ATP2A1/SERCA1, WNT1 and WNT5A (Figure 2A and S4C). The effects of EIF2AK3-EIF2A signaling on protein expression were investigated in wls-null BMDCs by analyzing ER-associated ribosomal complexes using transmission electron microscopy (TEM). As shown in Figure 2B, ribosomal complexes (red arrows) and WLS in the ER were present in greater numbers in wild-type BMDCs than those seen in wls-deficient BMDCs. Further, as a consequence of ER stress, the level of intracellular calcium in wls-null BMDCs was lower both at resting and after stimulation with a chemokine, CCL2/MCP-1 (C-C motif chemokine ligand 2) (Figure 2C) than those seen in WT BMDCs.
Figure 2.
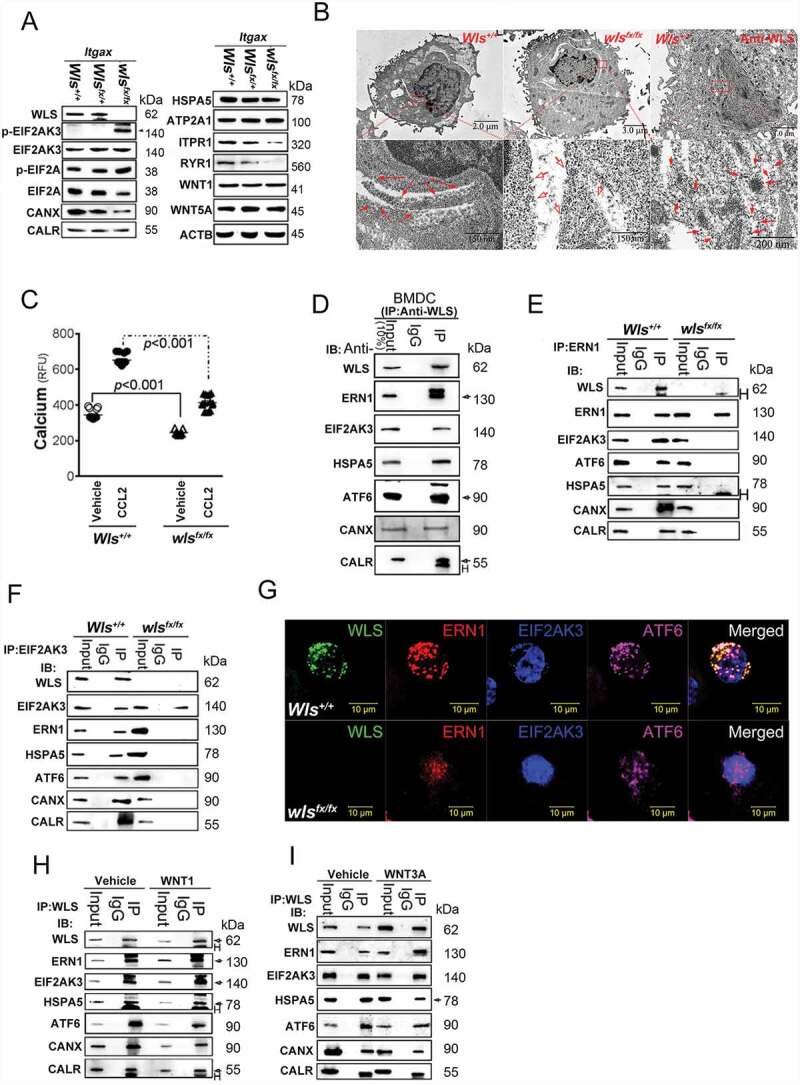
WLS deficiency results in ER stress and the loss of ER quality control. (A) Western blot analysis of UPR sensors, EIF2AK3-ElF2A signals, and calcium regulators in wild-type (+/+), heterozygous (fx/+), and wls-null (fx/fx) Itgax-Cre BMDCs. (B) TEM analysis of the ultrastructure and translational ribosome complex in wild-type and wls-null BMDCs. Red arrow, translational ribosomal complex. (C) intracellular calcium levels in wild-type and wls-null BMDCs following treatment with CCL2 (0.7 nM). (D-F) Western blotting analysis of WLS, ERN1, EIF2AK3, ATF6, HSPA5 CANX, and CALR in anti-WLS, anti-ERN1, and anti-EIF2AK3 immunoprecipitates probed with the respective antibodies in wild-type or wls-deficient BMDCs. Data were generated from three independent experiments. (G) Confocal immunofluorescent imaging of WLS, ERN1, EIF2AK3, and ATF6 in wild-type or wls-null BMDCs. (H-I), Western blot analysis of WLS, ERN1, EIF2AK3, ATF6, HSPA5, CANX, and CALR in anti-WLS immunoprecipitates of BMDCs treated with WNT1 and WNT3A, respectively
To identify the molecular mechanism underlying the functional defect in wls-null DCs, WLS-interacting proteins in anti-WLS Ab immune precipitates were determined using matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry. Results showed that as an ER membrane protein, WLS was found to interact with UPR sensor proteins, ERN1, HSPA5, EIF2AK3, and ATF6, as detected in anti-WLS immune precipitates of BMDCs (Figure 2D) and confirmed by reciprocal immune precipitations with the respective protein-specific Abs (Figures 2E and 2F). The interaction between WLS and UPR sensor proteins, ERN1, EIF2AK3 and ATF6, was further confirmed by immunofluorescence confocal imaging of BMDCs from wild-type and wls-null mice (Figure 2G). However, no such molecular complexes were detected in BMDCs lacking WLS (Figures 2E and 2F). Also, addition of WNT1 and WNT3A in the culture did not impact the assembly of WLS-associated complex (Figures 2H and 2I).
It was also noted that CANX and CALR were detected in the WLS-associated complex (Figure 2D) of wild-type BMDCs, but the expression of both proteins was either low or barely detectable in wls-deficient BMDCs (Figure 2A), suggesting the likely involvement of WLS in regulating the CANX-CALR cycle. The CANX-CALR cycle is known to be involved in one of the rate-limited steps in protein glycosylation and plays a central role in folding and quality control of glycosylated cargo. The dysregulation of this cycle may cause ER stress via dysregulated protein glycosylation and ER quality control. The loss of CANX-CALR cycle in wls-null BMDCs may, therefore, suggest that the primary cause of ER stress noted in wls-null BMDCs may be the aberrant protein glycosylation.
In support of the importance of protein glycosylation in WLS-associated complex formation, treatment of the cells with an ER glycosylation inhibitor, tunicamycin (TM; inhibiting N-linked glycosylation), resulted in disassembly of the WLS-associated complex, while the ER UPR inducers, brefeldin A (BFA) and thapsigargin (TG), had no effect on the WLS-UPR-CANX-CALR complex assembly (Figure 3A-C), except that the interaction of WLS and ATF6 was lost in the presence of either TM or BFA. Further, while TM treatment resulted in lower levels of calcium mobilization from the ER in WT BMDCs, wls-deficient BMDCs treated with TM showed further decrement in calcium mobilization after CCL2 stimulation, but not BFA and DTT (dithiothreitol) treatment, as compared to wild-type BMDCs and those without TM treatment (Figure 3D-F). Also, while the level of UPR sensors at the resting state did not differ between wls-deficient and wild-type BMDCs (Figure 3G-I), lower levels of total Xbp1 (Xbp1t), spliced Xbp1 (Xbp1s), and Hspa5 were found in wls-deficient BMDCs following treatment with TM, and as in wild-type BMDCs, DDIT3/CHOP signaling was highly activated in TM-treated wls-null BMDCs (Figure 3J). Further, wls-deficient BMDCs treated with TM showed a significantly greater number of autophagic cells than those treated with other ER stress inducers, even though treatment with all ER UPR inducers resulted in similar numbers of apoptotic cells in wls-deficient and wild-type BMDCs (Figures 3K and 3L). These results suggested the potential importance of proper glycosylation in WLS-associated complex formation.
Figure 3.
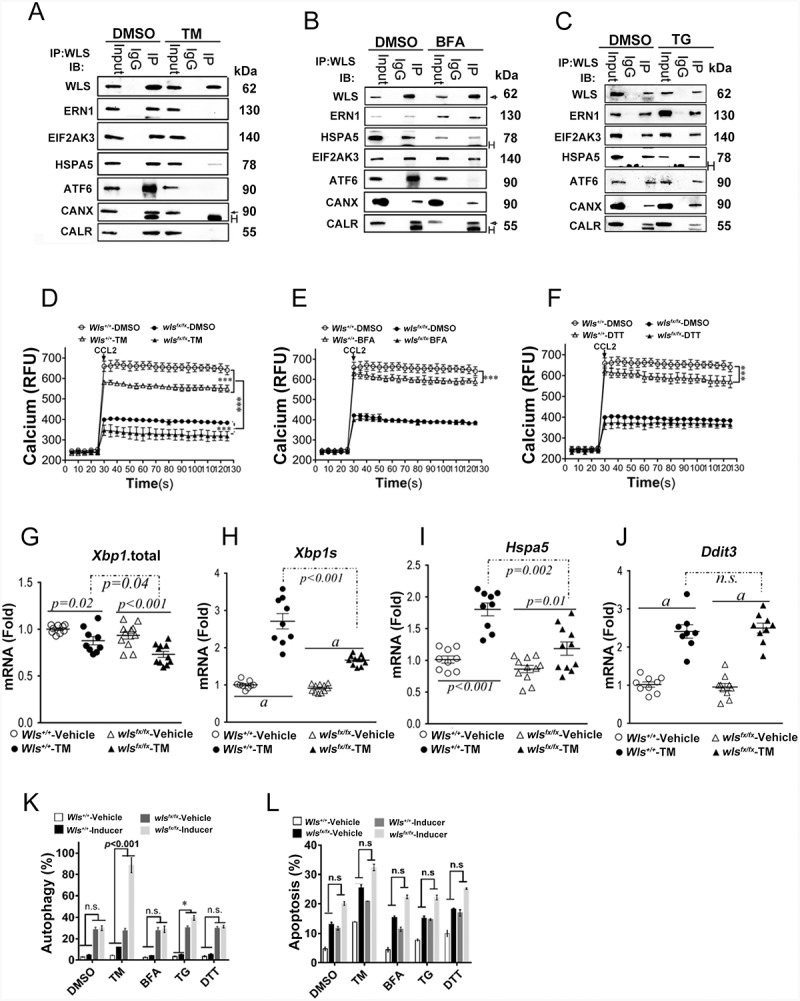
WLS coordinates a multi-protein complex and regulates ER stress response in BMDCs
(A-C) Western blot analysis of WLS, ERN1, EIF2AK3, ATF6, HSPA5, CANX, and CALR in anti-WLS immunoprecipitates and BMDC cells by TM, BFA, and TG treatment, respectively. (D-F) Level of intracellular calcium in wild-type or wls-null BMDCs following treatment with TM, BFA and DTT, respectively. (G-J) Relative levels of mRNAs for total Xbp1.total (Xbp1t), spliced Xbp1 (Xbp1s), Hspa5, and Ddit3 in wild-type and wls-null BMDCs treated with vehicle control or TM (15 μM) (p < 0.001; n.s., not significant; N ≥ 10 in each panel). (K-L), percentage of autophagic and apoptotic BMDCs with wild-type and wls-null genotypes after TM, BFA, TG, or DTT treatment, respectively.
WLS-mediated molecular chaperone complex modulates protein glycosylation and cytokine secretion in BMDCs
Next, to investigate the effect of the WLS-associated complex on protein glycosylation and glycoprotein quality control, the level and the pattern of glycosylation were analyzed. Results showed that significantly lower levels of total glycosylation were noted in wls-null BMDCs than those in wild-type BMDCs (Figure 4A). On comparing different glycan modification of total proteins in wild-type BMDCs, wls-deficient BMDCs showed a significantly lower level of N- and O-linked, α-1,2-linked fucopyranosyl and α-1,6-linked monnopyranosyl glycans, while minor modifications were noted on α-2-3,6,8-linked N-acetylneuraminic glycans (Figure 4B). As compared with wls-deficient BMDCs, wild-type BMDCs treated with TM showed significantly greater inhibition of total glycosylation level, whereas no inhibitory effect of total glycosylation level was observed in both wls-deficient and wild-type BMDCs treated with two other ER stress inducers, BFA and DTT (Figure 4A). Further, using a global glycosylation chip assay of secretory and membrane proteins, the glycosylation level of 507 proteins was compared between wls-deficient and wild-type BMDCs (N = 3 mice in each group; Figure 4C). Of these proteins, 223 exhibited a significantly lower glycosylation level than did the wild-type counterpart, while 15 major signaling proteins were shown to be hypo-glycosylated in wls-deficient BMDCs (Figures 4D and 4E), including proteins in the WNTs signaling pathway. As shown in Figures 4F and 4G, the glycosylation level of WNT pathway members was significantly lower in wls-deficient BMDCs, with concomitant lower concentrations of WNT in the culture medium (Figure 4H). The glycosylation of proteins involved in glucose metabolism was also significantly altered in wls-deficient BMDCs, while the levels of cellular ATP, glucagon, and glucose were significantly lower in wls-deficient than in wild-type BMDCs (Figure 4I-K). The glycosylation level of DC-associated cytokines and receptors was also significantly lower in wls-deficient than in wild-type BMDCs (Figures 4L and 4M). Moreover, membrane potential in DCs was detected by cationic JC-1 probe as a functional marker. The results showed that the level of mitochondria membrane potential in wls-null BMDCs was lower than that of WT BMDCs (Fig. S1N).
Figure 4.
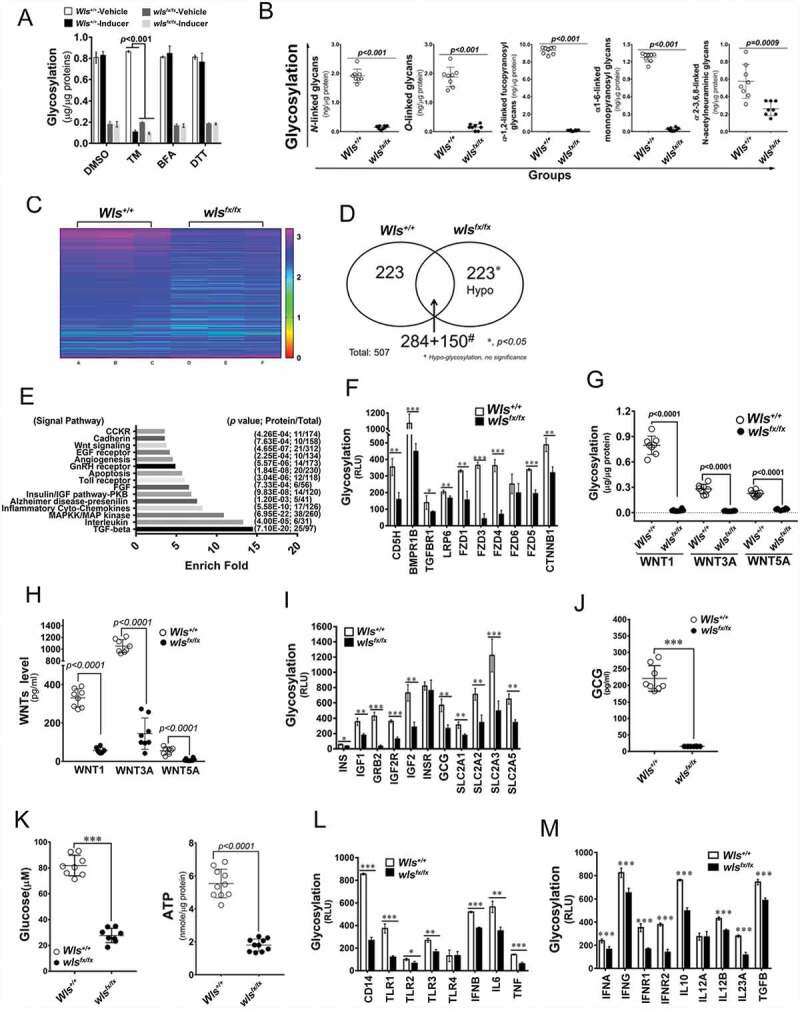
WLS-mediated molecular chaperone supercomplex modulates protein glycosylation, cytokine secretion, and cell fate in BMDCS. (A) Total glycosylation levels as measured by ELISA in BMDCs with wild-type and wls-null genotypes after TM, BFA, or DTT treatment. (B) Glycosylation levels of different kinds of glycan modification of total proteins determined by phenol-sulfuric acid method. (C) Heatmap of 507 biomarkers in wild-type and wls-null (wlsfx/fx) BMDCs; (D) Comparison of the number of hypoglycosylated proteins between wild-type and wls-null cells. #, hypo-glycosylation, no significance (*, p< 0.05); (E) GO biological process over-representation analysis with 15 signal pathways between wild-type and wls-null cells; (F) Glycosylation levels of CDH5/VE-Cadherin, BMPR1B, TGFBR1, LRP6, FZD1/Frizzled-1, FZD3, FZD4, FZD5, FZD6, and CTNNB1/β-catenin in wild-type and wls-null cells. (G) Glycosylation levels as measured by ELISA in anti-WNT1, WNT3A, and WNT5A immunoprecipitates of wild-type and wls-null BMDC. (H) Levels of WNT1, WNT3A, and WNT5A as measured by ELISA in wild-type and wls-null BMDCs. (I) Glycosylation levels of INS (insulin), IGF1, GRB2, IGF2R, INSR, GCG (glucagon), SLC2A1/GLUT1, SLC2A2, SLC2A3, and SLC2A5 in wild-type and wls-null cells. (J-K) Levels of GCG, glucose, and ATP as measured by ELISA in wild-type and wls-null BMDCs. (L) Glycosylation levels of CD14, TLR1, TLR2, TLR3, TLR4, IFNB, IL6, and TNF in wild-type and wls-null cells. (M) Glycosylation levels of IFNA, IFNG, IFNAR1, IFNGR2, IL10, IL12A, IL23A and TGFA in wild-type and wls-null cells
Mapping the binding domain of UPR sensors and glycosylation regulators on WLS
To provide the structural features of WLS-associated protein complex and to map the potential interaction domains, wild-type and a series of WLS mutants with a tagged sequence encoding GFP (Figure 5A) were generated and analyzed in transfected cells with the respective expression constructs. In this set of experiments, HEK 293 cells were tested as a model, as BMDCs were not amenable to transfection. After transfection, WLS complexes were immunoprecipitated using anti-GFP mAbs. As shown in Figure 5B, differential loss of WLS binding to a panel of the UPR sensor proteins was observed, suggesting the presence of a multi-protein complex associated with WLS. Based on the binding pattern, WLS was found to interact with ERN1, HSPA5, ATF6, and EIF2AK3 via its signal domain (SD), the predicted lipocalin fold (PLF), and the C-terminal domain (CT), respectively (Figure 5B). WLS was also found to interact with the glycosylation regulators CANX and CALR via its C-terminal domain (Figure 5B).
Figure 5.
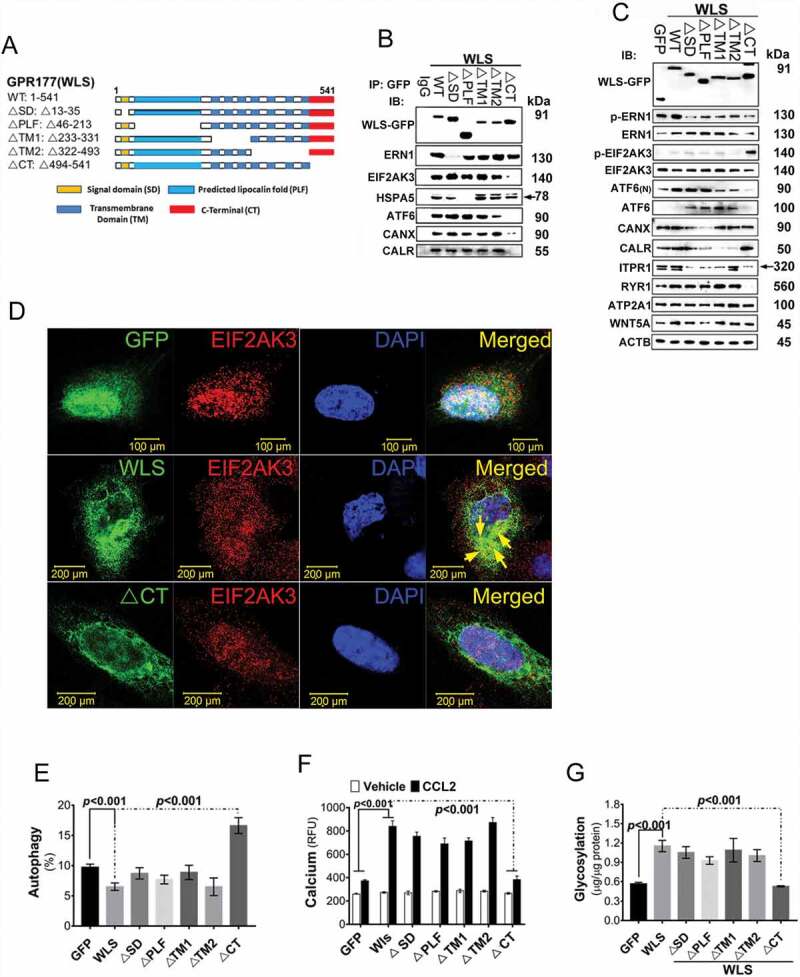
WLS coordinates a multi-protein complex and regulates ER stress response in A549 cells. (A) Wild-type and various deletion mutants of WLS as indicated. (B) Western blot analysis of UPR sensors and CANX-CALR complex expression in anti-GFP immunoprecipitates from wild-type and mutant WLS-transfected A549 cells. Arrow, HSPA5. (C) Western blot analysis of UPR sensors and calcium regulators (ITPR1, RYR1, and ATP2A1) in A549 cells transfected with wild-type or mutant WLS (Arrow, ITPR1). (D) Confocal microscopy imaging of the interaction between WLS and EIF2AK3 in A549 cells transfected with wild-type or WLSΔCT mutants. WLS, green; EIF2AK3, red; and nuclei, blue. (E) Percentage of autophagic A549 cells transfected with wild-type or WLS mutants. (F) Intracellular calcium levels in A549 cells transfected with wild-type or WLS mutants. (G) glycosylation levels as measured by ELISA in A549 cells transfected with wild-type or WLS mutants. Data were generated from three independent experiments for each panel (***, p< 0.001)
Further, forced WLS expression was found to activate the ERN1 and HSPA5 signaling pathways, but these activities were abolished in cells transfected with WLS mutants that had no or limited binding to ERN1 and HSPA5, respectively (Figure 5B, 5C and S2A-C). Total and spliced XBP1 and the downstream signaling molecules, ERN1 and HSPA5, were found to be downregulated in cells with WLSΔSD and ΔPLF mutant’s expression (Fig. S2A-C). HSPA5 expression was also repressed in cells with WLSΔPLF mutant expression, and no HSPA5 was detected in immune precipitates of the WLS complex (Figure 5B and S2C). In contrast, cells with WLSΔCT expression increased EIF2AK3-EIF2A signals and decreased the expression of ERN1, ATF6, ITPR1, and RYR1 (Figure 5C, S2D, and S2E), while DDIT3 and PPP1R15A/GADD34, which are downstream effectors of EIF2AK3 activation, were upregulated in cells with the WLSΔCT mutant expression relative to those with wild-type WLS (Figure 5C, S2D and S2E).
Interactions between WLS and the UPR sensors, ERN1 and EIF2AK3, were further investigated using immunofluorescent confocal imaging, in which the wild-type WLS, but not the WLSΔSD or CT, showed perinuclear co-localization with ERN1 or EIF2AK3, respectively (Figure 5D and S2F). Wild-type WLS transfectants showed a lower percentage of autophagic cells and correspondingly lower levels of the autophagy markers LC3B, ATG5, ATG12, and ATG16L1 (Figure 5E and S2G). In contrast, WLSΔCT mutants showed increased levels of autophagy and autophagy marker expression. Further, upon CCL2 stimulation, cells with wild-type WLS showed increased intracellular calcium levels, whereas WLSΔCT mutants showed no detectable change in intracellular calcium levels under the same conditions (Figure 5F). Similarly, cells transfected with wild-type WLS showed increased total glycosylation levels, whereas WLSΔCT mutants showed no detectable change in the total glycosylation levels under the same conditions (Figure 5G). These results suggested, therefore, that WLS may serve as an essential chaperon integrating a multi-protein complex in a domain-selective manner.
Wls-deficient DCs fail to prime Th1, Th2 and Th17 responses in vivo
After noting the substantial loss of ER glycoprotein quality control mechanism in wls-null DCs in vitro, we investigated the in vivo correlates in two common immunological disease models. Firstly, the impact of DC-specific wls deficiency on a model of myelin oligodendrocyte glycoprotein 35–55 (MOG35–55)-induced experimental autoimmune encephalomyelitis (EAE) was examined (Fig. S3A for schematic experimental protocol). Results showed that mice with DC-specific wls deficiency had lower levels of spinal injuries and EAE scores than did their wild-type counterparts (Figure 6A). Analysis of spinal cord–infiltrating cells showed that mice with DC-specific wls deficiency had significantly lower total numbers of DCs, plasmacytoid dendritic cells (pDCs), and conventional dendritic cells (cDCs) (Figure 6B-D) but similar numbers of macrophages and neutrophils as compared to wild-type mice (Fig. S3B and S3C). Further, the number of infiltrating Th1, Th17, and Th1/Th17 (CD4+ IFNG+ IL17A+) cells was significantly lower in DC-specific wls-deficient mice than in wild-type mice (Figure 6E). Histological analyses also demonstrated a lower degree of spinal cord inflammation and parenchymal vacuolar degeneration in the white matter of MOG-immunized DC-specific wls-null mice than in wild-type mice (Figure 6F). Collectively, these results suggested that DC-specific wls deficiency may exert a significant effect on the development of pathogenic Th1, Th17, and Th1/Th17 cells.
Figure 6.
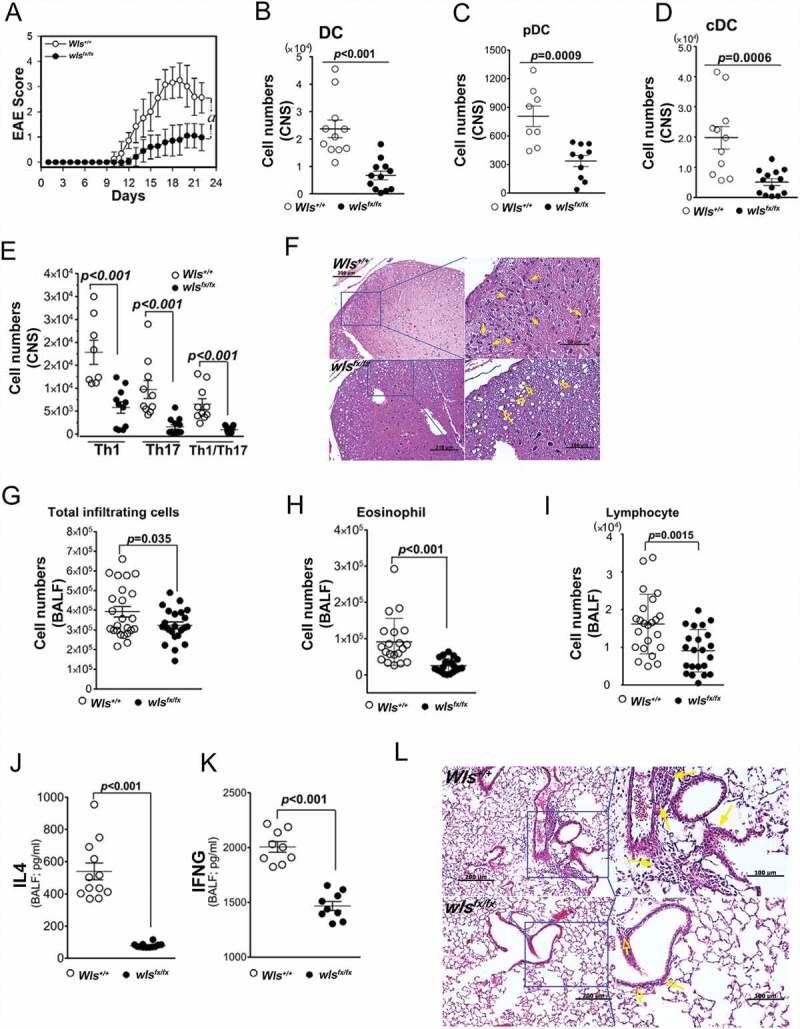
In vivo mouse EAE model. (A) Clinical score (means ± SEM) of MOG35–55-induced EAE in wild-type or DC-specific, wls-null (wlsfx/fx) mice (N = 8 mice/group; ***p < 0.0001). (B-E) Number of DCs, pDCs, cDCs, and CNS-infiltrating Th1, Th17, or Th1/Th17 (CD4+ IFNG+ IL17A+) CD4+ T cells from MOG35–55-induced wild-type and DC-specific wls-null mice on day 21 (N = 8 mice/group; p < 0.001 in two-tailed unpaired t-test; n.s., not significant n ≥ 12). (F) Histological analysis of spinal cords of wild-type and wls-null mice with EAE using hematoxylin & eosin (H&E) staining. (G-K) Total number of inflammatory cells and cell differentials in BALFs after OVA challenge (p < 0.001; n.s., not significant). (L) H&E staining of lung tissues in wild-type and wls-null mice (N = 12)
Secondly, a mouse model of ovalbumin (OVA)-induced pulmonary Th2-associated allergic inflammation was used to further investigate the effect of wls deficiency in vivo (Fig. S3D for schematic experimental protocol). Analysis of the cell populations in the bronchoalveolar lavage fluids (BALFs) of OVA-challenged, DC-specific wls-null mice showed significantly lower levels of infiltrating eosinophils and lymphocytes (Figure 6G-I), but no apparent change was noted in the number of macrophages or neutrophils (Fig. S3E and S3F). In addition, the levels of IL4 and IFNG in the BALF of OVA-challenged wls-null mice were significantly lower than those of wild-type mice (Figures 6J and 6K). After OVA challenge, histological analysis of lung tissues showed a much lower degree of inflammatory infiltrate in DC-specific wls-null mice than in wild-type mice (Figure 6L). These results suggested, therefore, that DCs with wls deficiency were unable to mount both Th1- and Th2-associated responses in vivo.
Discussion
Collectively, these results demonstrated, for the first time, that independent of WNT signaling, WLS is essential in the regulation of ER quality control pathway in dendritic cells via its chaperon-like activity, integrating ER stress sensing (ERN1, HSPA5, EIF2AK3, and ATF6), CANX-CALR cycle and calcium regulation to ensure ER homeostasis (see Figure 7 for schematic illustration). The profound impact of wls deficiency on DC’s maturation and immunity was shown both in vitro and in vivo. Our results also showed that the WLS-associated complex in the ER served as a critical platform via a glycan-dependent and domain-selective manner, wherein WLS, via its C-terminal domain, interacts with CANX, CALR, ATF6, EIF2AK3 and two calcium regulators, ITPR1 and RYR1, while ERN1 interacts with the SD portion and HSPA5 with the PLF domain of WLS.
Figure 7.
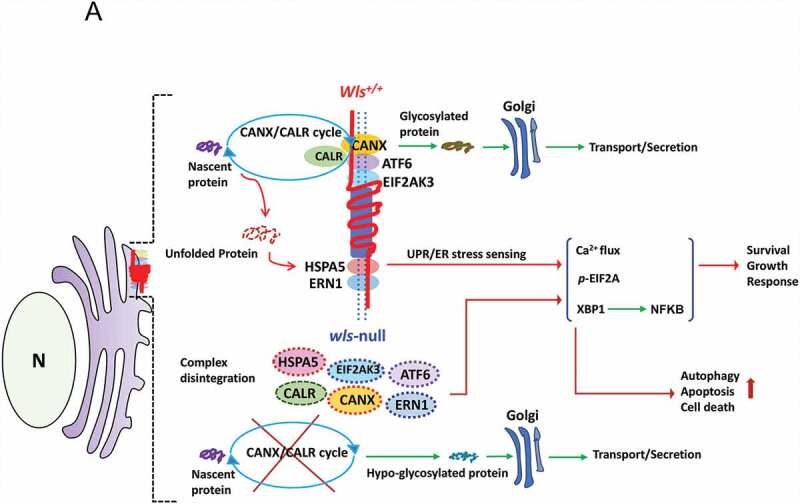
A schematic diagram illustrates a hypothetical model of WLS as an essential chaperone docking ER stress sensors and CANX-CALR complex and controlling ER homeostasis. According to our results, WLS integrates the CANX-CALR cycle, UPR sensors (ERN1, EIF2AK3, ATF6, HSPA5) and calcium regulators (not depicted in the scheme) via different interacting domains. Functionally, the WLS-associated complex controls glycoprotein quality, ER homeostasis and, at least in part, cellular survival, growth and response, whereas WLS deficiency releases UPR sensors and dissociates the CANX-CALR complex, resulting in global hypo-glycosylation and activation of UPR signaling, leading to autophagy, apoptosis and cell death
Interestingly, WLS-associated complex was shown to be particularly vulnerable under glycosylation-dependent ER stress, as demonstrated in cells treated with TM, but not BFA. TM is a naturally occurring antibiotic and known to induce ER stress by inhibiting the first step in the biosynthesis of N-linked glycans in the proteins, resulting in many misfolded proteins [19], but the mechanism through which aberrant glycoproteins activate the UPR and affect ER homeostasis is unclear. Another ER stress inducer, BFA, on the other hand, is an ER-Golgi transport inhibitor and has been shown to cause accumulation of proteins in the ER, ER stress, and ultimately apoptosis. Our results suggested that disruption of WLS-associated complex under TM-induced ER stress, particularly the dissociation of WLS and CANX-CALR cycle, led to the loss of glycoprotein quality control and initiated the UPR. This was further supported by the finding that WLS deficiency resulted in the loss of CANX-CALR cycle and activation of UPR. Also, the fact that TM treatment disrupted WLS-associated complex suggests that the complex formation may be itself via, in part, glycan-dependent interaction. Functionally, wls deficiency in DCs led to global hypo-glycosylation and increased levels of autophagy, loss of DC’s immune priming effects, thereby disrupting the immune response in models of autoimmune and allergic diseases. Based on these findings, a series of events can be envisaged, wherein upon TM-mediated ER stress, the loss of WLS-associated complex, including HSPA5, releases UPR sensors and the CANX-CALR complex to initiate downstream UPR signaling and block calcium flux, leading to alteration of cellular maturation, growth and functional deficiency. Accordingly, loss of WLS expression or interaction between WLS and HSPA5 may favor cell death over the restoration of homeostasis under conditions of TM-mediated ER stress.
Many important processes guiding protein glycosylation and folding occur in the ER, including glucose and mannose trimming from N-glycan, disulfide bond formation, and peptidyl-propyl bond formation [22]. Deficiencies in protein glycosylation can lead to the accumulation of unfolded (or misfolded) protein, induction of UPR, and ER stress [23]. In this regard, CANX-CALR cycle is involved in one of the rate-limited steps in protein glycosylation and plays a central role in folding and quality control of glycosylated cargo [24,25]. Although the detailed regulatory mechanism underlying CANX function remains to be fully defined, CANX deficiency has been reported to reduce the survival within 48 h of birth, and those survived suffered from multiple motor disorders [26]. Also, CANX is known to be involved in calcium homeostasis by interacting with the calcium regulatory channel in mitochondria-associated membrane, which modulates energetic respiration demand and inter-organelle communication [27,28]. Our results from analysis of WLS’ function in DCs revealed an intimate relationship between WLS, CANX-CALR cycle and two calcium regulators, ITPR1 and RYR1 in the ER, highlighted the importance of WLS in the process of glycoprotein quality control and its fundamental role in controlling cellular homeostasis.
UPR, the major ER stress response signaling pathway, is induced by dysfunctional ER homeostasis and determines cell fate [20], while HSPA5 facilitates UPR signaling and maintains the permeability barrier of the ER during protein translocation, targeting misfolded proteins for retrograde translocation. HSPA5 is shown to interact with ERN1, thereby preventing ERN1 dimerization and repressing ERN1-mediated downstream XBP1 function and calcium homeostasis [29,30], ultimately repressing UPR activity [31]. Also, the loss of HSPA5 increases ER stress and promotes cell death [32]. This collective evidence underscores the importance of HSPA5 in the WLS-mediated chaperone complex, serving as the major repressor of ERN1 and acting as a glycan-mediated initiator to coordinate the consequent change in protein structure, leading to complex dissociation. In fact, UPR has recently been recognized for its role in immune cell differentiation and function, including those associated with infections, tumors, and autoimmune responses [14,33]. For example, the UPR sensor XBP1 is abundantly and constitutively spliced in DCs. Also, the loss of XBP1 function leads to DC apoptosis [34], and functional loss of ERN1 endonucleases has been shown to cause the loss of type I conventional DCs in the intestine [35]. While the role of UPR-associated stress response in DCs remains to be completely defined, our results are consistent with these established mechanisms of action and provide a novel mechanistic insight on the critical role of WLS in UPR signaling and its subsequent responses.
EIF2AK3, another essential type I transmembrane protein, is activated by auto-phosphorylation during terminal UPR [14,18]. As with ERN1, EIF2AK3 activity is repressed by its interaction with HSPA5. EIF2AK3 remains dissociated from HSPA5 until ER stress, which induces EIF2AK3 oligomerization and autophosphorylation of the cytosolic kinase domain [20]. EIF2AK3-DDIT3-ATF4 signaling is tightly controlled, and constitutively activated EIF2AK3 signaling inevitably favors ER-stress-mediated calcium imbalance and autophagy by inducing the expression of several autophagy genes in vitro and in vivo [36,37]. Our results showed that constitutive EIF2AK3 activation occurred in DCs with WLS deficiency, while ERN1 and ATF6 were comparatively inhibited, resulting in EIF2AK3 auto-phosphorylation and subsequent hypo-glycosylation and DDIT3–EIF2A signaling. These findings were recapitulated by the analysis of cells with the WLSΔCT mutant expression, wherein the loss of ERN1 and ATF6 was noted in wls-deficient cells.
Consistent with our finding that DDIT3-EIF2AK3 signaling was activated in wls-null DCs, another study reports that EIF2AK3-DDIT3-induced EIF2AK4/GCN2 (eukaryotic translation initiation factor 2 alpha kinase 4; a serine/threonine-protein kinase) and phosphorylation of EIF2A result in autophagy, inhibition of protein synthesis, and inflammasome activation in DCs [38]. Therefore, these findings suggest that WLS may provide a barrier against EIF2AK3 activation during early ER stress, which is maintained until WLS-mediated deconstruction is induced by uncontrolled ER damage. Finally, in addition to UPR stress response, we observed that abnormal protein glycosylation ablated several important cellular signaling molecules in DCs, including Toll-like receptors, chemokines, and cytokines involved in priming and inflammation, resulting in the loss of Th1, Th2, and Th17 immune responses in different disease models. Collectively, the current results suggest a novel role of WLS as a potentially critical chaperone that controls protein glycosylation, ER homeostasis, and immunity.
Materials and Methods
Plasmids, cell lines, and other materials
WLS full-length cDNA (accession number AK 159,207) tagged with EGFP was subcloned into the pEGFP/c1 vector (Clontech, 6084–1) [39]. The method of production and infection with lentiviruses was performed according to the National RNAi Core Facility Platform (NRCFP)’s protocol. For lentivirus infection, 2–5 × 105 cells were infected with lentiviruses at the indicated multiplicity of infection (MOI) of 0.2–4. HEK293 and A549 cell lines were subcultured and maintained according to ATCC (CRL-1573™ and CCL-185™) protocols. Transfection was performed using a Lipofectamine transfection kit (GIBCO/BRL, 11,668–500).
Generation of wlsfx/fx mice
In brief, the Wlsfx allele was created by inserting loxP sites flanking exon 3 of Wls. Mice homozygous for Wlsfx were generated by National Taiwan University (Taipei, TW). Wlsfx/fx mice were interbred with stud males (Wlsfx; Itgax [Integrin Subunit Alpha X]-Cre) to generate the desired genotype. Itgax-Cre mice (B6.Cg-Tg(Itgax-cre)1–1Reiz/J) homozygous for Cre recombinase linked to the Itgax promoter was commercially available from Jackson Laboratory [40]. Knockout mice used for the experiments were confirmed to have the desired genotype via standard genotyping techniques.
Generation of bone marrow-derived dendritic cells (BMDCs)
Bone marrow cells from femurs and tibiae of Itgax-Cre-Wls+/+ and wlsfx/fx were isolated and cultured in DC media (RPMI 1640 [Thermo Fisher Scientific, 2,187,034] supplemented with 10% FBS [Thermo Fisher Scientific, 16,000,044], 100 U/ml penicillin, 100 mg/ml streptomycin [Thermo Fisher Scientific, 15,140,122], and 20 ng/ml recombinant mouse CSF2/GM-CSF [R&D, 415-ML-020]) and plated at 5 × 105 cells/ml. On day 7, the cells were treated with or without 100 ng/ml LPS (Sigma-Aldrich, L2654) for the indicated times or for 24 h to generate mature BMDCs.
Extraction of different glycan modification
The 500 µg of total proteins extracted from wild-type or wls-null (wlsfx/fx) BMDC were added to a 30 k centrifugal filter unit (Merck Millipore, MRCF0R030). Then, urea solution (8 M; Sigma-Aldrich, U5378) was added so as to bring the total volume to 400 µl. Samples were centrifuged at 16,000 x g for 15 min to remove cytosolic contaminants, and 200 µl of urea solution was added to wash it twice. Next, after adding 200 µl of ammonium bicarbonate (50 mM; Sigma-Aldrich, 09830), the tubes were centrifuged at 16,000 x g for 15 min to exchange buffer. The different glycan modifications were released from the samples by the addition of 6 UN of PNGase F (P0704S), O-glycosidase (P0733S), α1-6 mannosidase (P0727S), α2-3,6,8 neuraminidase (P0720S), or α1-2 fucosidase (P0724S), all from New England Biolabs, in 200 μl of NH4HCO3 solution (25 mM) at 37°C; they were digested overnight (16 h) under shaking conditions and finally centrifuged at 16,000 x g for 15 min to get the released glycans in a new tube.
T-cell proliferation study
For DC-T co-culture, syngeneic CD4+ T cells were isolated from the spleens, and co-cultured with BMDCs at 1:1, 5:1 and 10:1 ratios to obtain a range of stimulation, and incubated in a humidified 37°C, 5% CO2 incubator for 24 h or as required. Cell proliferation analysis was performed using the fluorescein isothiocyanate (FITC) bromodeoxyuridine (BrdU) Flow Kit (BD Biosciences, 559,619), according to the manufacturer’s protocol. After treatment and BrdU labeling, cells were harvested and washed twice with PBS. To fix the cells, 0.7 ml of cold ethanol (70%) was added dropwise to a tube containing 0.3 ml of cell suspension in PBS and the cell suspension incubated on ice for 1 h. Subsequently, the cells were washed twice with PBS and incubated with 100 μl of staining buffer contained 0.1 mg/ml propidium iodide (BD Biosciences, 559,619), 2 mg/ml RNaseA (Thermo Fisher Scientific, EN0531) and BrdU-FITC for 1 h at 37°C in the dark. The cells were then analyzed by flow cytometry (BD Biosciences, 559,619).
Cytokine analysis
This was conducted using the mouse IL6, TNF, WNT1, WNT3A, IL4, IL12A, TGFB, IL10, IL23A and IFNG ELISA kit from R&D Systems and Cusabio (R&D Systems, M6000B, MTA00B, [CUSABIO, CSB-EL026128MO], [CUSABIO, CSB-EL026136MO], M4000B, [CUSABIO, CSB-E07360m], MB100B, M1000B, M2300, MIF00, respectively), according to the manufacturer’s instructions. The supernatants were then subjected to ELISA in triplicates. IL6, TNF, WNT1, WNT3A, IL4, IL12A, TGFB, IL10, IL23A and IFNG/INFγ concentrations were computed with reference to standard curves derived from purified IL6, TNF, WNT1, WNT3A, IL4, IL12A, TGFB, IL10, IL23A and IFNG supplied in the ELISA Kit.
Detection of autophagy
Cyto ID® staining tests assay was performed to detect the autophagy of mouse dendritic cell by using Cyto ID® Autophagy detection kit (Enzo Life Sciences, ENZ-KIT175-0200) following the manufacturer’s instructions. The cells were seeded into 100 mm Petri dish at a density of 5 × 105 cells per dish. Cells were harvested, washed with PBS, resuspended in 1× assay buffer and stained with Cyto-ID® green dye for 30 min at room temperature in the dark. Then, the cells were centrifuged at 300 x g for 5 min, and then supernatant was discarded. The cells were washed with 1× assay buffer, and centrifuged at 300 x g for 5 min. The cells were resuspended in 1× assay buffer and analyzed by Flow Cytometry within 30 min of staining.
Transmission electron microscopy (TEM)
BMDCs were placed into 2.5% glutaraldehyde (Sigma-Aldrich, G6257) for at least 2 h in 4°C. The cells were post-fixed in 1% osmium tetroxide (Sigma-Aldrich, 201,030), dehydrated in acetone (Sigma-Aldrich, 650,501) and embedded in epoxy resin (Sigma-Aldrich, O5500). Resin-embedded blocks were cut into 50–60 nm ultrathin sections. The ultrathin sections were stained with both uranyl acetate and lead citrate. The changes in the endoplasmic reticulum were examined with a transmission electron microscope.
RNA extraction and semi-quantitative real-time PCR
Total RNA was isolated from the cells with Trizol reagent (Thermo Fisher Scientific, 15,596,026), as previously described [41,42]. The WLS, XBP1, total, XBP1s, HSPA5, HSP90B1, DDIT3, DNAJB9 and PPP1R15A mRNA expressions from HEK293 cells were detected with SYBR Green Quantitative RT-PCR kit (Applied Biosystems, 4,309,155). All reactions and data analyses were performed according to the manufacturer’s instructions.
Western blotting and immunohistochemical analysis
Western blotting and immunohistochemical (fluorescence) staining were performed as described previously [8,43]. The primary antibodies used in this study were actin monoclonal antibodies (1:5000 dilution; Sigma–Aldrich, A1978), FITC-conjugated anti-rabbit IgG, and rhodamine-conjugated anti-mouse IgG, alkaline phosphatase-conjugated anti-rabbit IgG antibody (1:500 dilution; Jackson ImmunoResearch Laboratories, 111–095-003, 115–025-146, 111–055-003), AHR (aryl hydrocarbon receptor) goat polyclonal antibody (1:200 dilution; Santa Cruz Biotechnology, sc-8088), ERN1, EIF2AK3, ATF6, EIF2A, p-EIF2A, ATG4, ATG16L1, ATG12, PIK3C3, LC3B and p-EIF2AK3 rabbit polyclonal antibody (1:500 dilution; Cell Signaling Technology, 3294, 5683, 65,880, 5324, 5199, 7613, 8089, 4180, 4263, 12,741, 3191, respectively). WLS and HSPA5 rabbit polyclonal antibody (1:500 dilution; Abcam, ab72385 and ab21685). All of the experiments were repeated at least three times.
Preparation of cell populations of spleen, spinal cord and CNS
Twenty mice (Itgax-Cre-Wls+/+ and wlsfx/fx each), 10 weeks, were induced by i.p. injection with LPS derived from E. coli (Sigma-Aldrich, L2880). LPS was dissolved in phosphate-buffered saline (PBS) (Sigma-Aldrich, 806,552) and administered by i.p. with a dose of 5 μg/g body weight in 100 μL in in vivo experiments. Control mice were injected PBS. Sixteen hours after LPS injection, mice were sacrificed and spleens were aseptically removed. Spleen cells were used for flow cytometry analysis after red blood cell lysis. Cell staining for flow cytometry was performed at 4°C after Fc-block step for 15 min with the following monoclonal antibodies: FITC-conjugated anti- ITGAX/CD11c, BV421-conjugated anti- ADGRE1/F4/80, PE-conjugated anti-ITGAM/CD11b, PerCP-Cy5.5-conjugated anti-PTPRCR/B220, PE-Cy7-conjugated anti-PTPRC/CD45, APC-conjugated anti-MHCII, APC-Cy7-conjugated anti-Ly-6 G/Gr1 (Biolegend, 117,305, 123,131, 101,207, 103,235, 103,115, 107,613, 108,423, respectively) for 20 min (Fig. S3B). All data were analyzed with FlowJo.
EAE induction and evaluation of EAE score
Male Itgax-Cre-Wls+/+ and wlsloxp/loxp mouse, aged 10–12 weeks and weighing 22–30 g, were purchased from the National Lab. Experimental Animal Center (NLAC, Taipei, Taiwan) and housed in the pathogen-free laboratory of Kaohsiung Medical University. The mice were maintained under a 12-h light-dark cycle with free access to food and water. All experimental protocols were performed in accordance with the Kaohsiung Medical University guidelines for the care and use of laboratory animals and the National Institutes of Health policy on laboratory animal welfare. EAE was induced as previously described [44] and all efforts were made to minimize animal suffering. Briefly, under anesthesia, the mice were immunized subcutaneously in both hind footpads and in the back with a mixture of MOG35-55 (Sigma-Aldrich, M4449) (amino acid sequence: Met-Glu-Val-Gly-Trp-Tyr-Arg-Ser-Pro-Phe-SerArg-Val-Val-His-Leu-Tyr-Arg-Asn-Gly-Lys) (Experimental Animal Center of Kaohsiung Medical University) spinal cord homogenate (0.1 ml) and an equal volume of Freund’s incomplete adjuvant (Sigma-Aldrich, AR002) containing Mycobacterium tuberculosis H37Ra (400 μg; BD Difco, 231,141). The day of immunization was designated Day 0. The animals were weighed twice a day, and neurological deficits were assessed every day according to a five point scoring system [45]. by two independent blinded observers. The following scores were assigned: 0 = no signs, 1 = flaccid tail, 2 = impairment of fighting reflex and/or loss of muscle tone in hind limbs, 3 = complete paralysis of hind limbs, 4 = paraplegia, and 5 = moribund state/death.
OVA-induced asthma model
OVA-induced asthma was performed as described in our previous report with a minor modification [46]. Briefly, the mice were sensitized with an i.p. injection of 50 μg/mouse of OVA (grade V; Sigma-Aldrich, 41,235) and 5 mg of aluminum hydroxide (Sigma-Aldrich, 239,186) in 100 μl of pyrogen-free phosphate-buffered saline (PBS, pH 7.3) or equal amounts of PBS on days 1 and 14. Then, the mice were challenged with 5% aerosolized OVA or PBS through an air-compressing nebulizer in a plexiglass chamber (Yuyue, Danyang, Jiangsu, 403A) for 30 min for three successive days (days 21–23). Mice were divided into two groups: (A) PBS/PBS mice that were sensitized and challenged with PBS; (B) OVA/OVA mice that were sensitized and challenged with OVA. Mice were sacrificed on day 24 for the analysis of inflammatory infiltrates, cytokine production and WLS expression.
Cytokines measurements
Bronchoalveolar lavage fluid (BALF) and serum were harvested as described in our previous report [46]. The IL4, IL5, IL6, IL10, IL22 and IFNG levels in the BALFs were measured using sandwich enzyme-linked immunosorbent assays (ELISA) following the manufacturer’s instructions (R&D Systems, M4000B, M5000, M6000B, M1000B, M2200, MIF00, respectively).
Statistical analysis
Quantitative variables are presented as mean ± SD. Significance of differences was determined using a two-sample t-test. Statistical analysis of categorical variables was performed using Chi-squared analysis, one-way analysis of variance, and Fisher’s exact analysis. Differences with a p value < 0.05 were considered significant.
Supplementary Material
Acknowledgments
We are grateful for the support from the Bio-Bank, Medical Research Department, E-DA Hospital, Taiwan. This work was supported in part by research grants KMUH 106-6R33, KMUH107-7R46 and KMUH107-7R34 from the Kaohsiung Medical University Hospital, Kaohsiung Medical University Research Center Grant (KMU-TC108A04-5) and MOST-105-2314-B-037-70-MY2, MOST-106-2314-B-037- 090-MY2, MOST-107-2314-B-037-063-MY3, MOST-107-2314-B-037-028-MY3, MOST108-2314-B-037 −065 -MY3, 109-2326-B-003 −001 -MY3 from the Ministry of Science and Technology, Taiwan, Kaohsiung Medical University grant (KMU-DK108012), National Health Research Institutes, Taiwan (EOPP10-014), Kaohsiung Medical University “The Talent Plan” (106KMUOR04), Taiwan, and Shenzhen Science and Technology Peacock Team Project (KQTD20170331145453160).
Funding Statement
This work was supported by the Ministry of Science and Technology, Taiwan [MOST-109-2326-B-003 −001 -MY3]; Ministry of Science and Technology, Taiwan [MOST-107-2314-B-037-028-MY3].
Supplementary material
Supplemental data for this article can be accessed here.
Conflict of interest
All authors declare no conflict of interest
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Banziger C, Soldini D, Schutt C, et al. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006;125(3):509–522. . [DOI] [PubMed] [Google Scholar]
- [2].Wu BT, Wen SH, Hwang SP, et al. Control of Wnt5b secretion by Wntless modulates chondrogenic cell proliferation through fine-tuning fgf3 expression. J Cell Sci. 2015;128(12):2328–2339. [DOI] [PubMed] [Google Scholar]
- [3].Staal FJ, Luis TC, Tiemessen MM.. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol. 2008;8(8):581–593. [DOI] [PubMed] [Google Scholar]
- [4].Steinman RM, Granelli-Piperno A, Pope M, et al. The interaction of immunodeficiency viruses with dendritic cells. Curr Top Microbiol Immunol. 2003;276:1–30. [DOI] [PubMed] [Google Scholar]
- [5].Banchereau J, Steinman RM.. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. [DOI] [PubMed] [Google Scholar]
- [6].Zhang P, Zhou L, Pei C, et al. Dysfunction of Wntless triggers the retrograde Golgi-to-ER transport of Wingless and induces ER stress. Sci Rep. 2016;6(1):19418. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Augustin I, Goidts V, Bongers A, et al. The Wnt secretion protein Evi/Gpr177 promotes glioma tumourigenesis. EMBO Mol Med. 2012;4(1):38–51. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chiou SS, Wang LT, Huang SB, et al. Wntless (GPR177) expression correlates with poor prognosis in B-cell precursor acute lymphoblastic leukemia via Wnt signaling. Carcinogenesis. 2014;35(10):2357–2364. . [DOI] [PubMed] [Google Scholar]
- [9].Zhou C, Sun Y, Guo S, et al. Wls Expression Correlates with Tumor Differentiation and TNM Stage in Hepatocellular Carcinoma. Dig Dis Sci. 2018;63(1):166–172. . [DOI] [PubMed] [Google Scholar]
- [10].Fu J, Jiang M, Mirando AJ, et al. Reciprocal regulation of Wnt and Gpr177/mouse Wntless is required for embryonic axis formation. Proc Natl Acad Sci U S A. 2009;106(44):18598–18603. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cao J, Zhang L, Wan Y, et al. Ablation of Wntless in endosteal niches impairs lymphopoiesis rather than HSCs maintenance. Eur J Immunol. 2015;45(9):2650–2660. . [DOI] [PubMed] [Google Scholar]
- [12].Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528–531. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhong Z, Zylstra-Diegel CR, Schumacher CA, et al. Wntless functions in mature osteoblasts to regulate bone mass. Proc Natl Acad Sci U S A. 2012;109(33):E2197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018;69(2):169–181. [DOI] [PubMed] [Google Scholar]
- [15].Lindholm D, Korhonen L, Eriksson O, et al. Recent Insights into the Role of Unfolded Protein Response in ER Stress in Health and Disease. Front Cell Dev Biol. 2017;5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313(5783):104–107. [DOI] [PubMed] [Google Scholar]
- [17].Osorio F, Tavernier SJ, Hoffmann E, et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat Immunol. 2014;15(3):248–257. . [DOI] [PubMed] [Google Scholar]
- [18].Grootjans J, Kaser A, Kaufman RJ, et al. The unfolded protein response in immunity and inflammation. Nat Rev Immunol. 2016;16(8):469–484. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Banerjee A, Lang JY, Hung MC, et al. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J Biol Chem. 2011;286(33):29127–29138. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bertolotti A, Zhang Y, Hendershot LM, et al. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2(6):326–332. . [DOI] [PubMed] [Google Scholar]
- [21].Song S, Tan J, Miao Y, et al. Crosstalk of ER stress-mediated autophagy and ER-phagy: involvement of UPR and the core autophagy machinery. J Cell Physiol. 2018;233(5):3867–3874. . [DOI] [PubMed] [Google Scholar]
- [22].Lowe JB. Glycosylation, immunity, and autoimmunity. Cell. 2001;104(6):809–812. [DOI] [PubMed] [Google Scholar]
- [23].Reily C, Stewart TJ, Renfrow MB, et al. Glycosylation in health and disease. Nat Rev Nephrol. 2019;15:346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci U S A. 1994;91(3):913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81(3):425–433. [DOI] [PubMed] [Google Scholar]
- [26].Denzel A, Molinari M, Trigueros C, et al. Early postnatal death and motor disorders in mice congenitally deficient in calnexin expression. Mol Cell Biol. 2002;22(21):7398–7404. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lynes EM, Raturi A, Shenkman M, et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J Cell Sci. 2013;126(17):3893–3903. . [DOI] [PubMed] [Google Scholar]
- [28].Roderick HL, Lechleiter JD, Camacho P. Cytosolic phosphorylation of calnexin controls intracellular Ca(2+) oscillations via an interaction with SERCA2b. J Cell Biol. 2000;149(6):1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kimata Y, Oikawa D, Shimizu Y, et al. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J Cell Biol. 2004;167(3):445–456. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Oikawa D, Kimata Y, Kohno K. Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J Cell Sci. 2007;120(9):1681–1688. [DOI] [PubMed] [Google Scholar]
- [31].Amin-Wetzel N, Saunders RA, Kamphuis MJ, et al. A J-Protein Co-chaperone Recruits BiP to Monomerize IRE1 and Repress the Unfolded Protein Response. Cell. 2017;171(7):1625–37 e13. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hussien Y, Podojil JR, Robinson AP, et al. ER Chaperone BiP/GRP78 Is Required for Myelinating Cell Survival and Provides Protection during Experimental Autoimmune Encephalomyelitis. J Neurosci. 2015;35(48):15921–15933. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Han J, Kaufman RJ. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017;31(14):1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. 2007;204(10):2267–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tavernier SJ, Osorio F, Vandersarren L, et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat Cell Biol. 2017;19(6):698–710. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].B’Chir W, Maurin AC, Carraro V, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–7699. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Marciniak SJ, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Woo CW, Kutzler L, Kimball SR, et al. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat Cell Biol. 2012;14(2):192–200. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wang LT, Wang SJ, Hsu SH. Functional characterization of mammalian Wntless homolog in mammalian system. Kaohsiung J Med Sci. 2012;28(7):355–361. [DOI] [PubMed] [Google Scholar]
- [40].Esterhazy D, Loschko J, London M, et al. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T(reg) cells and tolerance. Nat Immunol. 2016;17(5):545–555. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang LT, Chiou SS, Chai CY, et al. Intestine-Specific Homeobox Gene ISX Integrates IL6 Signaling, Tryptophan Catabolism, and Immune Suppression. Cancer Res. 2017;77(15):4065–4077. . [DOI] [PubMed] [Google Scholar]
- [42].Wang LT, Chiou SS, Chai CY, et al. Transcription factor SPZ1 promotes TWIST-mediated epithelial-mesenchymal transition and oncogenesis in human liver cancer. Oncogene. 2017;36(31):4405–4414. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hsu SH, Wang LT, Lee KT, et al. Proinflammatory Homeobox Gene, ISX, Regulates Tumor Growth and Survival in Hepatocellular Carcinoma. Cancer Res. 2013;73(2):508–518. . [DOI] [PubMed] [Google Scholar]
- [44].Bailey SL, Schreiner B, McMahon EJ, et al. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol. 2007;8(2):172–180. . [DOI] [PubMed] [Google Scholar]
- [45].Lin GJ, Huang SH, Chen YW, et al. Melatonin prolongs islet graft survival in diabetic NOD mice. J Pineal Res. 2009;47(3):284–292. . [DOI] [PubMed] [Google Scholar]
- [46].Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199–221. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.