

ABSTRACT
Targeted protein degradation (TPD) provides unprecedented drug discovery strategies, but it is incapable of degrading non-protein pathogenic biomolecules. We have previously developed the concept of autophagosome-targeting compounds (ATTEC), which can target pathogenic proteins to autophagic degradation. Since macroautophagy (autophagy hereafter) is capable of degrading a wide spectrum of substrates including non-protein biomolecules, ATTEC should also be capable of targeting those non-protein biomolecules for autophagic degradation. Here in our most recent study, we have demonstrated this possibility using lipid droplets (LDs) as an exemplar target. LDs are intracellular structures storing neutral lipids, which can be degraded by autophagy. Based on the concept of ATTEC, compounds binding with both the LDs and the key phagophore and autophagosome protein LC3 may target LDs to autophagic degradation. We designed and synthesized such compounds by connecting the identified LC3-binding molecules to known LD-binding probes via a chemical linker. At micromolar concentrations, these compounds drastically reduced LDs via autophagy through the predicted mechanism, and rescued LD-related phenotypes in cells and in two independent mouse models with hepatic lipidosis. Our proof-of-concept study demonstrates the possibility of harnessing autophagy to degrade non-protein biomolecules by ATTEC.
KEYWORDS: ATTEC, autophagy, degrader, lipid droplets, NASH, obesity
Targeted protein degradation (TPD) moves drug discovery into a new era by its capability of targeting previously “undruggable” targets. Meanwhile, there has been a lack of technologies focusing on degradation to target non-protein biomolecules, which are also the cause of many diseases. For example, abnormal accumulation of neutral lipids is involved in many diseases such as obesity, cardiovascular disease, fatty liver disease, and neurodegenerative disorders. Thus, strategies to degrade these molecules (or relevant organelles) may greatly expand the target spectrum for drug discovery.
We have previously established the concept of autophagosome-tethering compounds (ATTEC) as a potential new technology for TPD. Autophagy is capable of degrading non-protein substrates, including lipid droplets (LDs), which are ubiquitous lipid-storing cellular structures with a neutral lipid core. Thus, we investigated whether we can apply the ATTEC concept to degrade LDs, as a proof-of-concept study to test the potential of ATTEC in degrading non-protein biomolecules.
In our recently published study [1], we designed LD-targeting ATTEC compounds (LD∙ATTECs) by linking the previously discovered LC3-binding molecules with known LD detection probes, which specifically recognize intracellular LDs. We designed four different LD∙ATTECs and then investigated the compounds’ effects in a number of different cellular and animal models.
We observed near-complete clearance of oleic-acid (OA)-induced intracellular LDs by LD∙ATTECs at ~5 to 15 μM concentrations in mouse embryonic fibroblasts and neuroblastoma cells (SH-SY5Y). In addition, LD∙ATTECs are also capable of degrading endogenous large LDs in 3T3-L1 differentiated adipocytes. The LD clearance by LD∙ATTECs is mediated through autophagy, because autophagy inhibition by NH4Cl or chloroquine treatment or by ATG5 knockdown almost completely abolishes the LD-lowering effects.
We further tested whether the mechanism of action (Figure 1) of LD∙ATTECs is mediated through tethering of LDs to LC3 and to the phagophore. We confirmed the target engagement of LC3 (more specifically LC3B because our LC3-binding compounds bind to LC3B) by a number of different approaches. First, we confirmed that the compounds lacking the LC3-binding moiety lack the LD-clearance capability in all the different cells and animals tested, suggesting that the binding with LC3 is required for LD-clearance. Second, the knockout of Lc3b largely abolishes the LD-lowering effects of LD∙ATTECs, confirming that the target-engagement of LC3 is required. Third, in the in vitro experiments using recombinant purified proteins, we confirmed that these compounds directly interact with LC3 and enable the interaction between LC3 and triacylglycerol (TAG), which is the major lipid component of LDs. Fourth, we observed a significant increase of colocalization between LDs and autophagosomes following LD-ATTECs treatment in the cells.
Figure 1.
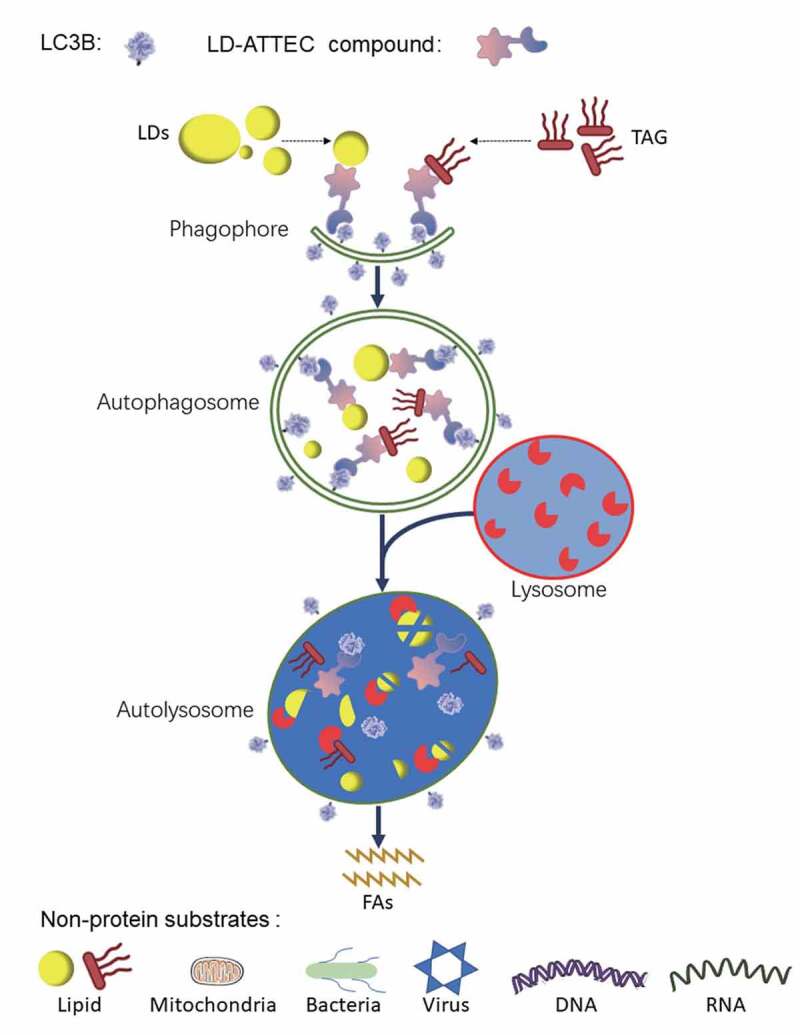
A schematic illustration of the mechanism of action of LD·ATTEC-mediated lipid degradation. LD·ATTECs bind to LDs or neutral lipids (using TAG as a typical example) via hydrophobic interactions and the phagophore protein LC3 simultaneously, leading to formation of the LD/TAG–LD·ATTEC–LC3 ternary complex, engulfment of the complex within autophagosomes, and subsequent autophagic degradation of LDs/neutral lipids after autophagosome-lysosome fusion, providing an energy source for the cells. Conceptually, ATTEC could be applied to non-protein substrates such as lipids, organelles, pathogens, and nucleic acids. FAs, fatty acids
We then investigated the specificity of LD∙ATTECs and revealed that they do not change polar lipids in cells and in animal tissues, and thus do not impair cytoplasmic or intracellular membranes. This is consistent with our design using the neutral-lipid specific-binding moiety for target engagement of LDs. In addition, LD∙ATTECs did not influence global autophagy activity, as indicated by direct measurement of autophagy flux or by proteomic analysis. The proteomic analysis also revealed significant lowering of PLIN2, which is a marker protein for LDs, consistent with the LD lowering.
Encouraged by the cellular data, we investigated the potential effects of LD∙ATTECs in animal models with aberrant accumulation of LDs. We injected these compounds versus controls intraperitoneally (ip) into two different mouse models of metabolic disorders. One is a genetic model (db/db mice, C57BL/6J-Leprdb/Leprdb) with obesity, and the other is nonalcoholic steatohepatitis (NASH) mouse model generated by a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD, 60 kcal % fat) feeding for 10 weeks. For both models, the injection of LD∙ATTECs for two weeks significantly reduces the whole-body weight, body fat:lean ratio, liver weight, liver LDs, and serum TAG and cholesterol levels. In addition, the liver fibrosis developed in the NASH mice is significantly rescued as well, confirming potential therapeutic benefits of LD∙ATTECs.
Taken together, our study expanded the degradation technologies for LDs based on the concept of ATTEC. Autophagy is a powerful cellular degradation machinery capable of degrading many different types of non-protein substrates such as lipids, organelles, pathogens, and nucleic acids. Conceptually, ATTEC could be applied to many different categories of substrates, expanding the target spectrum of degradation technologies.
Funding Statement
This work was supported by the National Natural Science Foundation of China [31970748]; National Natural Science Foundation of China [81925012].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Fu Y, Chen N, Wang Z, et al. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 2021. DOI: 10.1038/s41422-021-00532-7 [DOI] [PMC free article] [PubMed] [Google Scholar]