

ABSTRACT
ALS (amyotrophic lateral sclerosis), the most common motor neuron disease, causes muscle denervation and rapidly fatal paralysis. While motor neurons are the most affected cells in ALS, studies on the pathophysiology of the disease have highlighted the importance of non-cell autonomous mechanisms, which implicate astrocytes and other glial cells. In ALS, subsets of reactive astrocytes lose their physiological functions and become toxic for motor neurons, thereby contributing to disease pathogenesis. Evidence of astrocyte contribution to disease pathogenesis are well established in cellular and animal models of familial ALS linked to mutant SOD1, where astrocytes promote motor neuron cell death. The mechanism underlying astrocytes reactivity in conditions of CNS injury have been shown to involve the MTOR pathway. However, the role of this conserved metabolic signaling pathway, and the potential therapeutic effects of its modulation, have not been investigated in ALS astrocytes. Here, we show elevated activation of the MTOR pathway in human-derived astrocytes harboring mutant SOD1, which results in inhibition of macroautophagy/autophagy, increased cell proliferation, and enhanced astrocyte reactivity. We demonstrate that MTOR pathway activation in mutant SOD1 astrocytes is due to post-transcriptional upregulation of the IGF1R (insulin like growth factor 1 receptor), an upstream positive modulator of the MTOR pathway. Importantly, inhibition of the IGF1R-MTOR pathway decreases cell proliferation and reactivity of mutant SOD1 astrocytes, and attenuates their toxicity to motor neurons. These results suggest that modulation of astrocytic IGF1R-MTOR pathway could be a viable therapeutic strategy in SOD1 ALS and potentially other neurological diseases.
Abbreviations: ACM: astrocyte conditioned medium; AKT: AKT serine/threonine kinase; ALS: amyotrophic lateral sclerosis; BrdU: thymidine analog 5-bromo-2’-deoxyuridine; CNS: central nervous system; EIF4EBP1/4EBP1: eukaryotic translation initiation factor 4E binding protein 1; GFAP: glial fibrillary acidic protein; IGF1R: insulin like growth factor 1 receptor; INSR: insulin receptor; iPSA: iPSC-derived astrocytes; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta;MTOR: mechanistic target of rapamycin kinase; NES: nestin; PPK1: 3-phosphoinositide dependent protein kinase 1; PI: propidium iodide; PPP: picropodophyllotoxin; PTEN: phosphatase and tensin homolog; S100B/S100β: S100 calcium binding protein B; SLC1A3/ EAAT1: solute carrier family 1 member 3; SMI-32: antibody to nonphosphorylated NEFH; SOD1: superoxide dismutase 1; TUBB3: tubulin beta 3 class III; ULK1: unc-51 like autophagy activating kinase 1.
KEYWORDS: Astrocytes, autophagy, EIF4EBP1, IGF1R, motor neurons, mtor, PPP, SOD1G93A, Torin1, ULK1
Introduction
MTOR (mechanistic target of rapamycin kinase) is an evolutionarily conserved serine-threonine kinase that regulates numerous intracellular and extracellular signals involved in cell metabolism, growth, proliferation, survival [1], and macroautophagy/autophagy [2,3]. MTOR nucleates into two different multi-protein complexes, MTOR complex 1 (MTORC1) that is nutrient-sensitive [4], and MTOR complex 2 (MTORC2), which is growth factor-sensitive, but nutrient-insensitive [5]. Due to its pleiotropic effects on cellular homeostasis, it is not surprising that alterations of MTOR signaling have been associated with a number of pathologies from cancer to neurodegeneration [6].
MTOR dysregulation has been proposed to play a pathogenic role in ALS (amyotrophic lateral sclerosis), an untreatable disease that causes motor neurons degeneration, paralysis, and death. While motor neurons are the most affected cells in ALS, studies on the pathophysiology of the disease highlight the importance of non-cell autonomous mechanisms implicating astrocytes [7]. Under conditions of CNS (central nervous system) stress or injury, astrocytes change their morphology and properties in order to increase neuroprotection and preserve neuronal activity [8]. However, when the stress is prolonged and extensive, such as in the case of ALS, astrocytes lose their protective functions and become reactive, acquiring a distinct morphology, increased proliferation, pro-inflammatory and cytotoxic properties, and thereby participating in the demise of motor neurons [9,10].
Studies suggest the existence of a causal relationship between MTOR signaling and astrocyte reactivity in neurodegeneration and CNS injury. For instance, in tuberous sclerosis it was reported that MTOR activation correlates with reactive astrocytosis and gliosis, contributing to tuber formation [11]. Further, activation of MTOR signaling is detected in reactive astrocytes, in both in vitro and in vivo models of spinal cord injury [12]. Moreover, in a model of hypoxic-ischemic brain damage, it is reported that MTOR participates in inducing proliferation of reactive astrocytes [13]. Based on this evidence, we hypothesized that MTOR activation could also play an important role in astrocyte reactivity and toxicity to motor neurons in ALS. We started to investigate this hypothesis, in a cell culture system, using human iPSA (iPSC-derived astrocytes) harboring G93A mutant SOD1 (superoxide dismutase 1) associated with an autosomal dominant familial form of ALS. We found that in mutant astrocytes elevated levels of IGF1R (insulin like growth factor 1 receptor) stimulated MTOR activity through AKT-mediated phosphorylation. Importantly, we showed that pharmacological or genetic inhibition of the IGF1R-MTOR signaling pathway in SOD1G93A iPSA resulted in attenuation of astrocyte toxicity to motor neurons, suggesting that this pathway could be targeted therapeutically in ALS astrocytes.
Results
The MTOR pathway is activated in SOD1G93A iPSA
To investigate the MTOR pathway in ALS astrocytes, we used human iPSA cells generated by targeted mutagenesis of the SOD1 gene in an iPSC line from a healthy control individual, resulting in a G93A mutant SOD1, which we had previously described [14]. By immunofluorescence, both control and G93A iPSA lines expressed astrocyte markers, including the SLC1A3/EAAT1 (solute carrier family 1 member 3), the most prevalent glutamate transporter expressed in astrocytes, NES (nestin), a type VI intermediate filament protein expressed in astroglial stem cells, and S100B/S100β (S100 calcium binding protein B), a glial-specific marker primarily expressed in astrocytes (Fig S1A).
Comparing control and SOD1G93A iPSA, we found that mutant cells have higher MTOR phosphorylation at serine 2448 (Ser2448) relative to total MTOR (Figure 1A), which is indicative of MTOR pathway activation [15]. This difference was not associated with an increase in MTOR mRNA level (Fig S1B), indicating that MTOR activation in SOD1G93A iPSA was due to post-transcriptional modifications. Next, we investigated the phosphorylation of the MTOR substrate ULK1 (unc-51 like autophagy activating kinase 1), which is specifically phosphorylated at Ser757 by MTOR [3]. SOD1G93A iPSA had significantly higher levels of phosphorylated ULK1 at Ser757 than control, relative to total ULK1, confirming that MTOR was activated in mutant cells (Figure 1B). Consistent with increased ULK1 phosphorylation, in SOD1G93A iPSA we found decreased levels of the lipidated form of MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta; LC3-II) (Figure 1C), which is associated with mature autophagosomes [15]. In addition, control and SOD1G93A iPSA were fixed and immunostained with MAP1LC3B antibody. When autophagy is active, LC3-II redistributes to puncta associated with autophagosomes, in contrast to LC3-I, which has a prevalently cytosolic localization [16]. The average number of puncta per cell was decreased in SOD1G93A iPSA, confirming that autophagosome-associated LC3-II was significantly lower than in control (Figure 1D).
Figure 1.
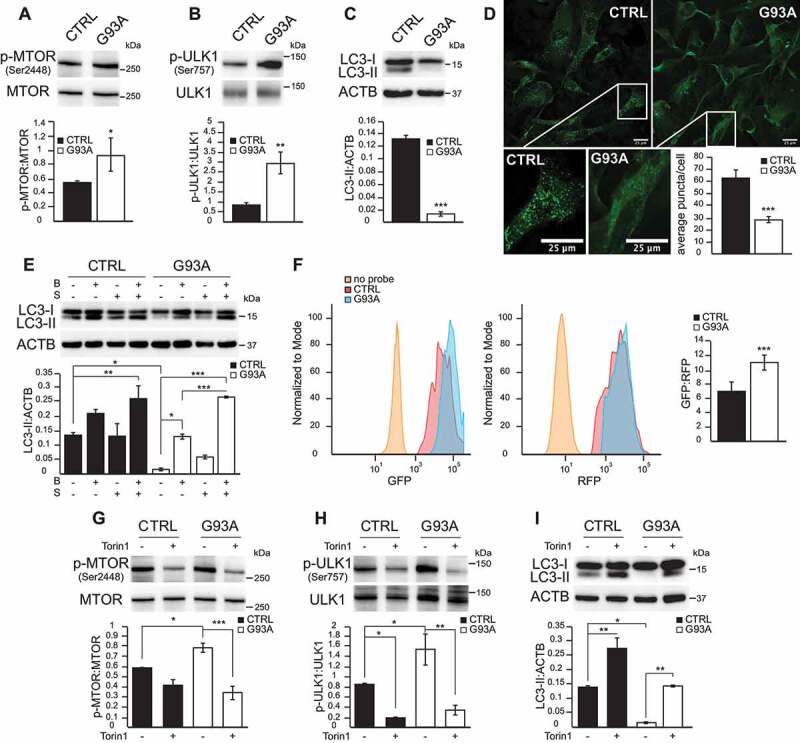
The MTOR pathway is activated in SOD1G93A iPSA. (A) Representative western blot and quantification of control (CTRL) and SOD1G93A (G93A) iPSA of phosphorylated (P)-MTOR (Ser2448) protein levels normalized by total MTOR (n = 4), (B) p-ULK1 (Ser757) protein levels normalized by total ULK1 (n = 5), (C) LC3-II protein levels normalized to ACTB/β-actin (n = 6). (D) Representative images of CTRL and G93A iPSA immunostained with antibody against MAP1LC3B. Scale bars: 25 μm. Quantification of average LC3 puncta/cell (n = 108, 115 cells). (E) CTRL and G93A iPSA treated with 100 nM bafilomycin A1 for 2 h (B) and/or nutrient deprivation for 4 h (S). Representative western blot and quantification of LC3-II protein levels normalized to ACTB (n = 3). (F) CTRL and G93A iPSA were transduced with a retroviral construct carrying the GFP-LC3-RFP-LC3ΔG probe. Representative histograms of GFP and RFP expression and quantification of GFP:RFP median fluorescence intensity (MFI) ratio (n = 6). CTRL and G93A iPSA were treated with 250 nM Torin1 (+) or vehicle (-) for 24 h. (G) Representative western blot and quantification of p-MTOR (Ser2448) protein levels normalized to total MTOR (n = 3), (H) p-ULK1 (Ser757) protein levels normalized to total ULK1 (n = 3), (I) LC3-II protein levels normalized to ACTB (n = 3). P values <0.05 by Mann–Whitney test (A, B, D) or by unpaired Student’s t test (C) or by one-way ANOVA test with Sidak’s correction between indicated groups (E, G, H, I) are shown
To assess if the lower amount of autophagosomes was due to impairment or to inhibition of the autophagy process, we evaluated autophagic fluxes by blocking lysosome acidification with bafilomycin A1 in basal conditions or during nutrient deprivation. In SOD1G93A iPSA, bafilomycin A1 alone induced a significant increase in the levels of LC3-II, which were further elevated when bafilomycin A1 was combined with nutrient deprivation, reaching levels comparable to control cells (Figure 1E). To further demonstrate that autophagy flux was impaired in SOD1G93A iPSA, we employed a ratiometric fluorescent probe, GFP-LC3-RFP-LC3ΔG, delivered by retroviral transduction and evaluated the flux by flow cytometry. This probe is cleaved by endogenous ATG4 proteases into equimolar amounts of GFP-LC3 and RFP-LC3ΔG. GFP-LC3 is then degraded by autophagy, while RFP-LC3ΔG remains in the cytosol, serving as an internal control. Thus, autophagic flux can be estimated by calculating the GFP:RFP signal ratio, which is inversely correlated with autophagic activity [17]. This approach confirmed that the autophagic flux was significantly decreased in SOD1G93A iPSA compared to control cells, as the GFP:RFP ratio was increased by 60% in mutant cells (Figure 1F).
These results suggested that mutant astrocytes are autophagy competent, but their autophagy initiation was downregulated, presumably by MTOR activation. To test this hypothesis, we inhibited MTOR pharmacologically with the selective catalytic ATP-competitive MTOR inhibitor Torin1 [18]. Treatment with Torin1 (250 nM for 24 h) effectively inactivated MTOR in both control and SOD1G93A iPSA, as indicated by decreased phosphorylation of Ser2448 MTOR (Figure 1G) and Ser757 ULK1 (Figure 1H), and resulted in significantly increased LC3-II levels (Figure 1I). Autophagy stimulation by Torin1 for 24 h increased LC3-II levels in SOD1G93A iPSA compared to control cells (800 fold vs. 8 fold increase), although Torin1-stimulation in mutant cells did not produce the same absolute levels of LC3-II observed in stimulated control cells.
MTOR activation results in the phosphorylation of EIF4EBP1/4EBP1 at Thr37/46 [19], a key regulator of protein translation and cell growth [20,21]. EIF4EBP1 phosphorylation was significantly higher in SOD1G93A iPSA relative to control cells (Figure 2A). Increased MTOR-dependent EIF4EBP1 phosphorylation was associated with a remarkable increase in the proliferation of mutant astrocytes, measured by the BrdU (thymidine analog 5-bromo-2ʹ-deoxyuridine) incorporation into newly synthesized DNA (Figure 2B). Next, we inhibited MTOR to further investigate the role of MTOR-dependent EIF4EBP1 phosphorylation on iPSA proliferation. Torin1 decreased Thr37/46 EIF4EBP1 phosphorylation in both control and mutant iPSA (Figure 2C) and significantly lowered cell proliferation of SOD1G93A iPSA (Figure 2D).
Figure 2.
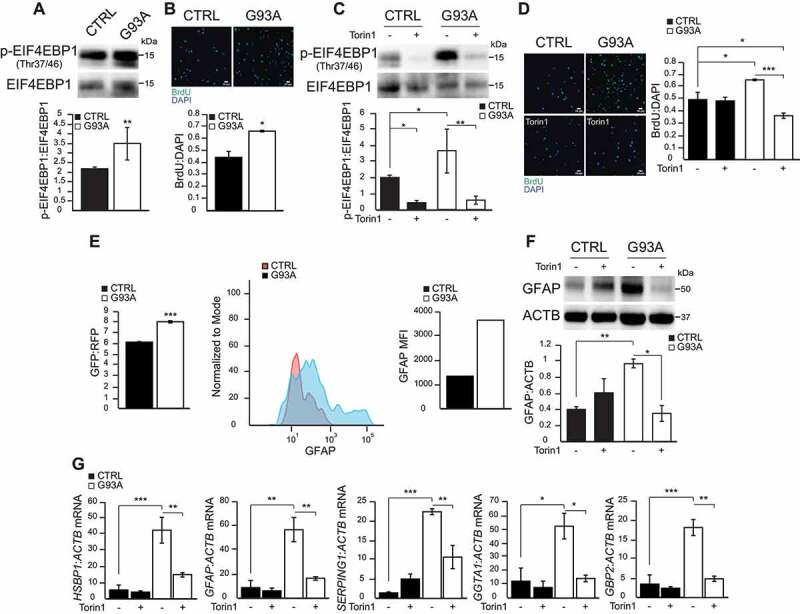
EIF4EBP1 phosphorylation, cell proliferation, and GFAP expression are elevated in SOD1G93A iPSA. (A) Representative western blot and quantification of CTRL and G93A iPSA of p-EIF4EBP1 (Thr37/46) protein levels normalized to total EIF4EBP1 (n = 6). (B) Representative images of CTRL and G93A iPSA stained with BrdU (green) and DAPI (blue), and quantification of cell proliferation expressed as BrdU:DAPI ratio (n = 9). CTRL and G93A iPSA were treated with 250 nM Torin1 (+) or vehicle (-) for 24 h. (C) Representative western blot and quantification of p-EIF4EBP1 (Thr37/46) protein levels normalized to total EIF4EBP1 (n = 3). (D) Representative images of CTRL and G93A iPSA stained with BrdU (green) and DAPI (blue), and quantification of cell proliferation (n = 9). (E) Quantification of GFP:RFP fluorescence ratio of GFP-LC3-RFP-LC3ΔG transduced cells FACS sorted based on GFP and RFP double positivity (n = 3). Representative histograms and quantification of GFAP MFI of FACS-sorted cells pooled from three transduction experiments. (F) Representative western blot and quantification of GFAP protein levels normalized to ACTB (n = 3). (G) HSBP1, GFAP, SERPING1, GGTA1, GBP2 mRNA levels normalized to ACTB (n = 3), to assess astrocytes reactivity. P values <0.05 by Mann–Whitney test (A) or by unpaired Student’s t test (B) or by one-way ANOVA test with Sidak’s correction between indicated groups (C, D, F, G) are shown
We had previously shown that SOD1G93A iPSA increase expression of GFAP (glial fibrillary acidic protein) [14], an intermediate astrocytic filament, which is upregulated in reactive astrocytes [8,10]. Here, we investigated GFAP levels in iPSA expressing GFP-LC3-RFP-LC3ΔG probe, isolated by FACS (Fluorescence-activated cell sorting) using RFP as positive marker to identify transduced cells. The double positive GFP-RFP population was sorted and immediately permeabilized to allow the intracellular staining with anti-GFAP antibody. Sorted SOD1G93A iPSA cells showed higher GFP:RFP ratio and also higher expression of GFAP compared to control cells (Figure 2E), indicating that lower autophagy flux corresponded to higher levels of GFAP. Importantly, Torin1 decreased GFAP protein expression in mutant iPSA (Figure 2F), suggesting a link between MTOR activation, cell proliferation, and GFAP upregulation in SOD1G93A iPSA. To confirm the connection between MTOR and astrocyte reactivity, we measured transcripts of selected markers of reactive astrocytes panel before and after Torin1 treatment. Specifically we quantified expression of HSBP1 and GFAP, markers of pan-reactivity and SERPING1, GGTA1 and GBP2, markers of A1 reactive astrocytes, which are particularly harmful for neurons and synapses [22]. All astrocyte reactivity markers were remarkably increased in SOD1G93A compared to control iPSA, and significantly reduced by Torin1 (Figure 2G). Taken together, these data revealed that the MTOR pathway is activated in SOD1G93A iPSA, resulting in inhibition of autophagy, increased cell proliferation, and astrocyte reactivity. Moreover, they indicated that pharmacological inhibition of MTOR reverts these phenotypes.
Next, to assess if MTOR activation in SOD1G93A iPSA is linked to the burden of mutant SOD1, we silenced total SOD1 expression (i.e., both mutant and wild type alleles) for 48 h by shSOD1 lentivirus transduction. Upon silencing, the total SOD1 mRNA (Figure 3A) and protein levels (Figure 3B) were decreased by approximately 60% in both control and SOD1G93A iPSA. However, SOD1 silencing resulted in decreased Ser2448 phosphorylation of MTOR (Figure 3C), Thr37/46 phosphorylation of EIF4EBP1 (Figure 3D), cell proliferation (Figure 3E), and astrocyte reactivity marker expression (Figure 3F) in SOD1G93A iPSA, but not in control cells. Together, these results suggested that MTOR activation and its downstream consequences are dependent on mutant SOD1 burden.
Figure 3.
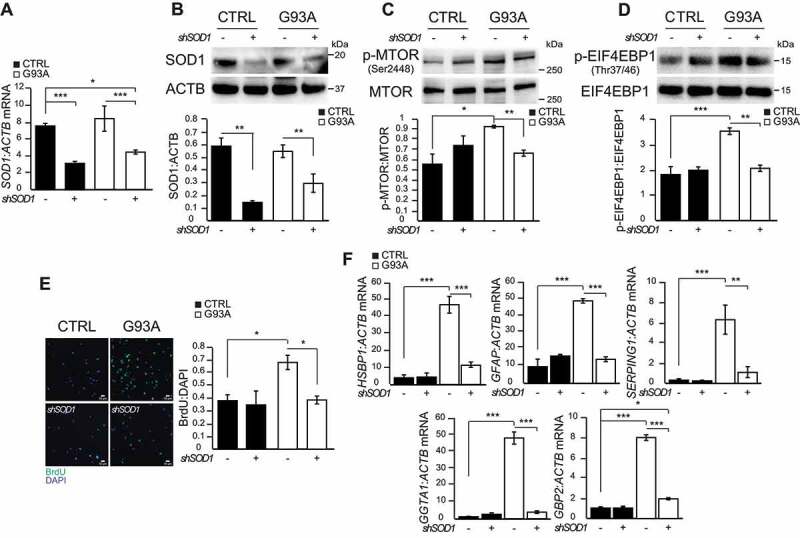
SOD1 silencing in SOD1G93A iPSA decreases MTOR activity. CTRL and G93A iPSA were transduced with shSOD1 lentiviral particles (+) or negative control (-) for 48 h prior to the assays. (A) SOD1 mRNA levels normalized to ACTB (n = 3). (B) Representative western blot and quantification of SOD1 protein levels normalized to ACTB (n = 3), (C) p-MTOR (Ser2448) protein level normalized to total MTOR (n = 3), (D) p-EIF4EBP1 (Thr37/46) protein level normalized to total EIF4EBP1 (n = 3). (E) Representative images of CTRL and G93A iPSA stained with BrdU (green) and DAPI (blue), and quantification of cell proliferation (n = 7). (F) HSBP1, GFAP, SERPING1, GGTA1, GBP2 mRNA levels normalized to ACTB (n = 3). P values <0.05 by one-way ANOVA test with Sidak’s correction between indicated groups are shown
IGF1R is increased in ALS iPSA
AKT, a pleiotropic serine/threonine kinase, activates MTOR through Ser2448 phosphorylation [23,24]. Since we observed elevated Ser2448 MTOR phosphorylation in SOD1G93A iPSA (Figure 1A), and because MTOR activation by AKT is reported in a model of spinal cord injury [12], we investigated AKT activation, which can be stimulated via phosphorylation at Thr308 by PDPK1 (3-phosphoinositide dependent protein kinase 1) [25]. In SOD1G93A iPSA, we detected an increase in PDPK1-dependent AKT activation, as indicated by elevated levels of Thr308 phosphorylation (Figure 4A).
Figure 4.
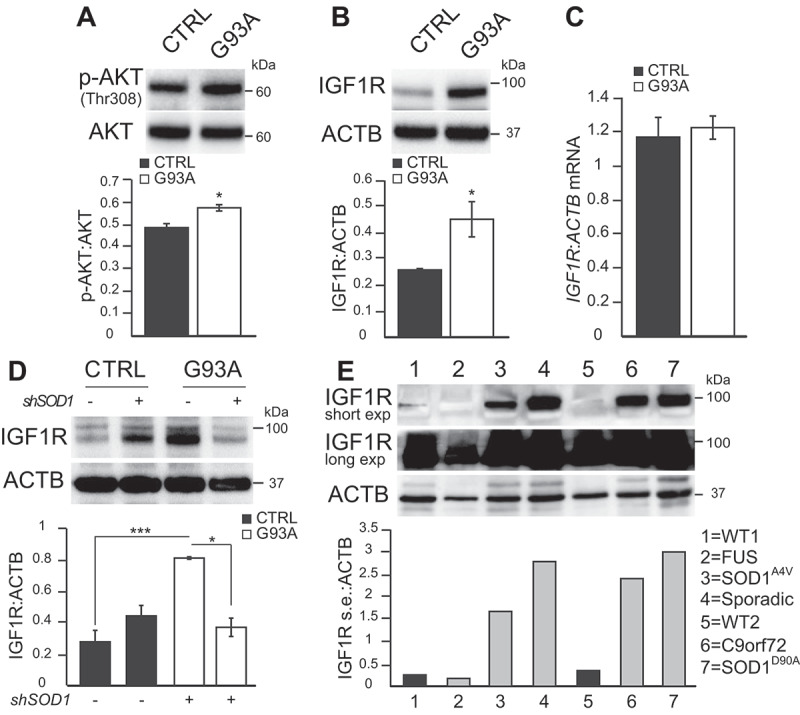
IGF1R levels are increased in SOD1G93A iPSA. (A) Representative western blot and quantification of CTRL and G93A iPSA of p-AKT (Thr308) protein levels normalized to total AKT (n = 3), (B) IGF1R protein levels normalized to ACTB (n = 6). (C) IGF1R mRNA levels normalized to ACTB (n = 3). (D) CTRL and G93A iPSA were transduced with shSOD1 lentiviral particles (+) or negative control (-) for 48 h prior to the assays. Representative western blot and quantification of IGF1R protein levels normalized to ACTB (n = 4). P values <0.05 by unpaired Student’s t test (A-B) or by one-way ANOVA test with Sidak’s correction between the indicated groups (D) are shown. (E) Representative western blot of WT lines from healthy subjects (lane 1–5) and ALS iPSA (FUS, SOD1AV4, Sporadic, C9orf72, SOD1D90A in lane 2, 3, 4, 6, 7, respectively) and quantification of IGF1R protein levels normalized to ACTB
Elevated AKT Thr308 phosphorylation could be the result of loss of the tumor suppressor PTEN (phosphatase and tensin homolog) [26]. However, we did not find significant differences in PTEN protein levels between mutant and control iPSA (Fig S2A). Therefore, we investigated regulatory elements upstream of PTEN and PDPK1 pathway. One of the best known upstream regulators of this pathway is the IGF1R. IGF1R bound to its ligands activates a signaling pathway involving a series of kinases that ultimately results in MTOR phosphorylation and activation [27,28]. Strikingly, we observed a significant increase in the levels of IGF1R in SOD1G93A iPSA compared to control cells (Figure 4B). This protein increase did not correspond to increased mRNA levels (Figure 4C), suggesting post-transcriptional mechanisms of IGF1R upregulation. Furthermore, SOD1 silencing in SOD1G93A iPSA significantly decreased IGF1R (Figure 4D). On the contrary, silencing SOD1 in control iPSA resulted in a slight, not statistically significant, increase in IGF1R levels, indicating that IGF1R upregulation was dependent on the burden of mutant SOD1. To assess the specificity of this observation, we also measured the levels of INSR (insulin receptor), which is structurally related to IGF1R [29]. We did not detect any significant difference in the levels of INSR between mutant and control iPSA (Fig S2B), suggesting a selective involvement of IGF1R in mutant cells. Furthermore, treatment with Torin1 did not affect IGF1R levels in mutant iPSA (Fig S2C), indicating that alterations of IGF1R levels do not depend on MTOR.
Interestingly, IGF1R upregulation was not only associated with SOD1G93A mutation, because we found a remarkable increase of IGF1R levels also in human patient-derived iPSA, harboring other SOD1 mutations (SOD1A4V and SOD1D90A), and in iPSA derived from a sporadic ALS patient and a patient harboring C9orf72 hexanucleotide expansion, as compared to iPSA derived from healthy subjects (Figure 4E). This observation, however, did not apply to all ALS cases, since we did not detect increased levels of IGF1R in FUS (fused in sarcoma) mutant iPSA. We also noted that IGF1R levels were variable among cell lines, and in the case of one healthy control (lane 5) and FUS (lane 2) cells, the protein could only be detected after a very long exposure of the western blot. This evidence indicated that dysregulation of IGF1R levels may be a common event in a subset of iPSC-derived human astrocytes with diverse forms of ALS, which could be involved in activating MTOR signaling.
Downregulation of IGF1R reverses the abnormal phenotypes of SOD1G93A iPSA
To further investigate the involvement of IGF1R in MTOR activation, we silenced IGF1R in SOD1G93A iPSA (Figure 5A), which resulted in a decrease of both MTOR (Figure 5B) and ULK1 phosphorylation (Fig S3A). In parallel with the decrease in MTOR activation, IGF1R silencing in SOD1G93A iPSA caused a significant decrease in cell proliferation (Figure 5C) and in GFAP levels (Figure 5D), and the expression of selected markers of reactive astrocytes panel (Figure 5E). Moreover, we tested the effects of a known small molecule IGF1R inhibitor, PPP (picropodophyllotoxin). PPP inhibits the phosphorylation of Tyr1136 of IGF1R [30], which is crucial for receptor activation. It was recently suggested that PPP increases the rate of degradation of IGF1R [31]. In line with this interpretation, we found that PPP (15 μM for 24 h) induced a significant decrease in IGF1R protein levels in SOD1G93A iPSA (Figure 5F). As a consequence of PPP treatment, MTOR phosphorylation was decreased in both control and SOD1G93A iPSA (Figure 5G). Moreover, PPP treatment decreased ULK1 phosphorylation in SOD1G93A iPSA (Fig S3B). IGF1R inhibition also resulted in a reduction in cell proliferation of SOD1G93A iPSA (Figure 5H) and in a remarkable decline in GFAP expression, in both control and mutant iPSA (Figure 5I). Together, these results indicated that IGF1R plays a key role in MTOR activation and its downstream consequences in mutant iPSA.
Figure 5.
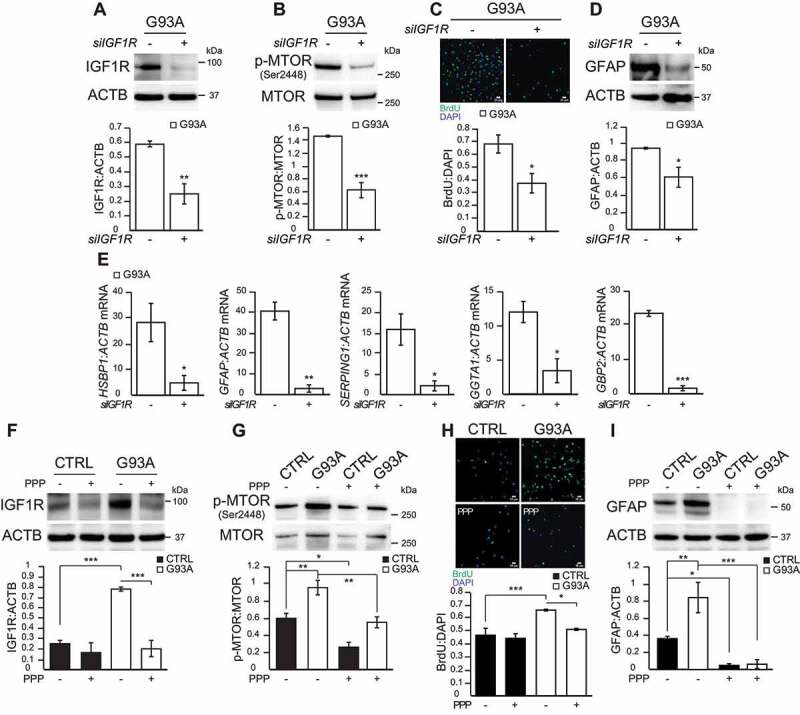
Genetic and pharmacological manipulation of IGF1R in SOD1G93A iPSA reverses proliferation and astrocytic reactivity. G93A iPSA were transfected with siRNA for IGF1R (+) or for Renilla luciferase (-) for 48 h prior to the assays. (A) Representative western blot and quantification of IGF1R protein levels normalized to ACTB (n = 3), (B) p-MTOR (Ser2448) protein level normalized to total MTOR (n = 3). (C) Representative images of CTRL and G93A iPSA stained with BrdU (green) and DAPI (blue), and quantification of cell proliferation (n = 6). (D) Representative western blot and quantification of GFAP protein levels normalized to ACTB (n = 3). (E) HSBP1, GFAP, SERPING1, GGTA1, GBP2 mRNA levels normalized to ACTB (n = 3). CTRL and G93A iPSA were treated with 15 μM PPP (+) or vehicle (-) for 24 h. (F) Representative western blot and quantification of IGF1R protein levels normalized to ACTB (n = 3), (G) p-MTOR (Ser2448) protein level normalized to total MTOR (n = 4). (H) Representative images of CTRL and G93A iPSA stained with BrdU (green) and DAPI (blue), and quantification of cell proliferation (n = 9). (I) Representative western blot and quantification of GFAP protein levels normalized to ACTB (n = 4). P values <0.05 by unpaired Student’s t test (A-E) or by one-way ANOVA test with Sidak’s correction between indicated groups (F-I) are shown
Inhibition of IGF1R-MTOR pathway protects motor neurons from SOD1G93A iPSA toxicity
We found that downregulation of the MTOR pathway through direct inhibition of MTOR or IGF1R prevents key phenotypes in SOD1G93A iPSA that recapitulate reactive astrocytosis, such as increased expression of GFAP and other reactive astrocyte markers and cell proliferation. These findings suggested that manipulation of the IGF1R-MTOR pathway could be an approach to counteract the non-cell autonomous toxicity of human mutant astrocytes to motor neurons. To test this hypothesis, we assessed the effects of IGF1R-MTOR pathway inhibition in mutant astrocytes on their toxicity to motor neurons. We recently reported that ACM (astrocyte conditioned medium) from SOD1G93A iPSA reduces the viability of motor neurons derived from human healthy iPSC [14]. Therefore, here we employed a similar approach, where SOD1G93A iPSA were grown in the presence of Torin1, PPP, or IGF1R silencing; then, ACM was collected and used to challenge motor neurons. After 6 days of exposure to ACM, motor neurons were fixed and immunostained for TUBB3 (tubulin beta 3 class III) and the non-phosphorylated NEFH (neurofilament heavy) detected with antibody SMI-32. We observed that, compared to control ACM, motor neurons exposed to SOD1G93A iPSA ACM had a number of TUBB3 positive neurites that showed a patchy or absent SMI-32 staining (Figure 6A,B), indicating ongoing degeneration of the processes [32]. Next, we assessed cell viability by loading motor neurons with calcein to stain live cells and PI (propidium iodide) to stain the nucleus of dead cells. Cell viability was evaluated measuring the percentage of live cells (calcein positive and PI negative) over the total number of cells. As expected, SOD1G93A iPSA ACM decreased significantly the viability of motor neurons relative to control ACM (Figure 7A,B,J). On the other hand, ACM from SOD1G93A iPSA treated with Torin1 prior to ACM collection was significantly less toxic to motor neurons, as indicated by the absence of SMI-32 alterations (Figure 6C) and loss of cell viability (Figure 7C,J). The protective effects of Torin1 were comparable to those obtained in a parallel experiment in which SOD1 was silenced in SOD1G93A iPSA and compared to cells transfected with siRNA against Renilla luciferase, as control (Figure 6E, F; Figure 7G,H,J). Mutant iPSA ACM toxicity also was markedly decreased by both PPP treatment and IGF1R silencing (Figure 6D,G; Figure 7D,I,J). Notably, both Torin1 and PPP protected motor neuron viability only when added to astrocytes, and not when added to the ACM after it was harvested (Figure 7E,F,J), indicating that these drugs exert their neuroprotective effect through astrocytes and not directly on motor neurons. Strikingly, motor neurons were more effectively protected with ACM obtained from mutant iPSA under IGF1R downregulation conditions than with ACM obtained from control iPSA (Figure 7J). Taken together, these results suggested that there could be a therapeutic value in modulating the IGF1R-MTOR pathway to attenuate SOD1G93A astrocyte toxicity.
Figure 6.
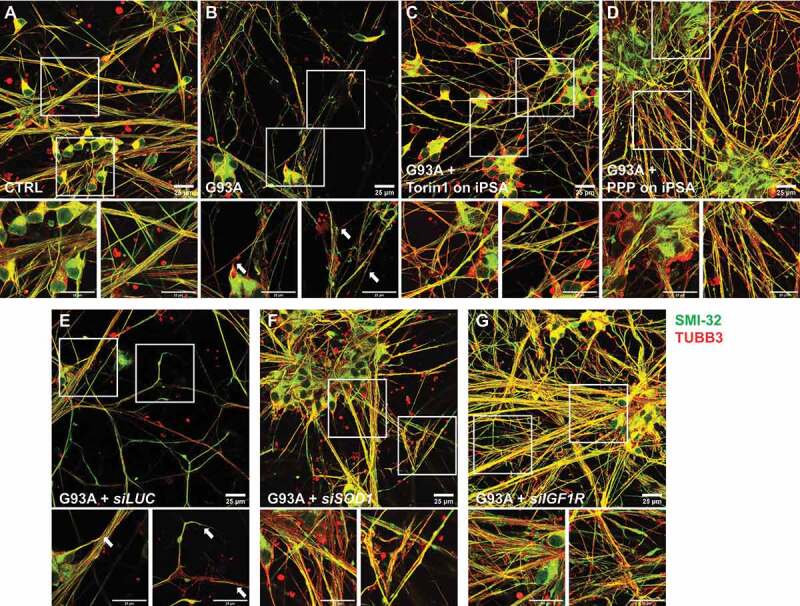
IGF1R-MTOR pathway inhibition protects motor neurons from processes degeneration. CTRL and G93A iPSA were treated with 250 nM Torin1 or 15 μM PPP. G93A iPSA were silenced for SOD1 or IGF1R. iPS-derived motor neurons were exposed to two doses of 25% ACM for 6 days, then immunostained with antibodies against TUBB3 (red) and SMI-32 (green). Representative fields of merged channel acquisitions of motor neurons cultured in ACM from untreated CTRL (A), untreated G93A (B), Torin1 treated G93A (C), PPP treated G93A (D), luciferase silenced G93A (E), SOD1 silenced G93A (F), and IGF1R silenced G93A (G). Insets are higher magnification images of neuronal soma (left panels) and processes (right panels). Scale bars: 25 μm. Arrows indicate regions of SMI-32 loss
Figure 7.
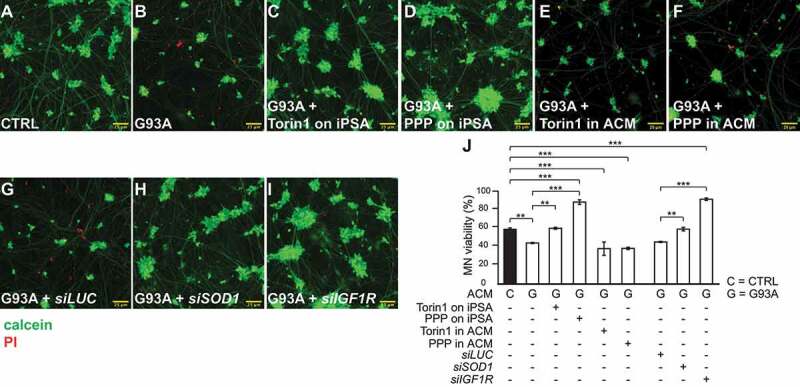
IGF1R-MTOR pathway inhibition protects motor neurons from SOD1G93A iPSA toxicity. CTRL and G93A iPSA were treated with 250 nM Torin1 or 15 μM PPP. G93A iPSA were silenced for SOD1 or IGF1R. iPS-derived motor neurons were exposed to two doses of 25% ACM for 6 days, then loaded with calcein (green) and PI (red) for 10 min, and imaged. Representative images of merged channel acquisitions of calcein and PI (see Fig S4 for single channel images) of motor neurons cultured in ACM from untreated CTRL (A), untreated G93A (B), Torin1 treated G93A (C), PPP treated G93A (D), Torin1 added to G93A ACM (E), PPP added to G93A ACM (F), luciferase silenced G93A (G), SOD1 silenced G93A (H), and IGF1R silenced G93A (I). Scale bars: 25 μm. (J) Quantification of motor neuron (MN) viability, expressed as percentage of live neurons over the total number of neurons (n = 9 individual wells per condition, 25 imaged fields per well). P values <0.05 by one-way ANOVA test with Sidak’s correction between indicated groups are shown
Discussion
MTOR is a key regulator of metabolism, autophagy, cell growth, and aging [33], and alterations of MTOR activity have been associated with a number of diverse neurodegenerative diseases [34,35], including ALS. However, CNS cell-type specific MTOR involvement in ALS has not been elucidated. Functionally, therapeutic modulation of MTOR activity has been attempted in ALS mouse models, with discrepant outcomes. On one hand, MTOR inhibition in mutant SOD1 transgenic mice hastens disease progression and increases motor neuron degeneration [36,37]. On the other hand, MTOR inhibition is protective in a transgenic mouse model of neuron-specific TDP-43 overexpression [38]. These results highlight the complexity of the global effects of MTOR manipulation, and understanding the biological implications of these approaches likely requires a more in-depth analysis of the effects at the cell-type level. Despite the evidence that MTOR is implicated in astrocyte reactivity in various neuropathological conditions [11–13] and that reactive astrocytes play a crucial role in ALS pathogenesis, no studies so far have addressed the effects of modulating MTOR signaling in ALS astrocytes. Addressing this gap is essential because MTOR modulation in astrocytes may have different effects on disease than ubiquitous treatment, and its consequences may differ among cell types. For example, the process of MTOR-dependent autophagy is differentially regulated in CNS cell types, as indicated by studies describing the regulation of metabolic stress-induced autophagy in neurons and astrocytes [39,40]. Moreover, neuronal autophagy appears to be largely unaffected by MTOR inhibition, while autophagy is strongly activated in primary astrocytes after MTOR inhibition [41].
In this study, we investigated astrocytic MTOR pathway involvement in non-cell autonomous toxicity, using a human astrocyte model, harboring G93A mutant SOD1 causative of familial ALS. Our results indicated that in mutant astrocytes there was constitutive activation of MTOR, which induced the stimulation of two key downstream MTOR effectors, ULK1 and EIF4EBP1. The increase in p-MTOR in SOD1G93A iPSA relative to control cells was moderate, but it resulted in a strong activation of ULK1 and EIF4EBP1, confirming that even relatively small increases in MTOR activation can trigger a strong response in its downstream signaling pathways, as reported in the autophagy guidelines [15]. In agreement with the known anabolic effects of MTOR pathway activation [1], mutant astrocytes showed a remarkable decrease in autophagy and increase in cell proliferation. In addition, mutant astrocytes displayed a strong increase in the expression of markers of reactive astrocytosis, including GFAP, which is shown to be elevated in astrocytes in parallel to MTOR activation in neuropathological conditions [11–13].
We found that SOD1G93A astrocyte treatment with the selective MTOR inhibitor Torin1 restored autophagy, normalized cell proliferation, and attenuated reactive astrocytosis, as shown by a drastic decrease in marker expression. In addition, silencing SOD1 in mutant astrocytes showed inhibition of MTOR signaling and a comparable decrease in the expression of astrocytosis markers, highlighting an unprecedented link between MTOR activation, astrocytic reactivity, and mutant SOD1 burden in SOD1G93A astrocytes.
To test the hypothesis that MTOR activation is responsible for mutant SOD1 astrocyte toxicity to motor neurons, we used an established motor neuron toxicity assay, in which healthy human iPSC-derived motor neurons were exposed to ACM. As previously shown [14], ACM from SOD1G93A iPSA decreased motor neuron viability, but treatment of mutant astrocytes with Torin1 significantly decreased this toxicity, suggesting that MTOR inhibition in astrocytes could be a therapeutic target in SOD1 ALS.
In order to understand the mechanism of MTOR activation in mutant SOD1 astrocytes, we investigated pathways upstream of MTOR. Increased phosphorylation of AKT at Thr308 suggested an involvement of IGF1R, a transmembrane tyrosine kinase receptor, since IGF1R binding to its ligands activates a cascade of kinases that result in AKT and MTOR phosphorylation and activation [27,28]. IGF1R controls a wide variety of functions related to CNS development and maturation, synaptic plasticity, and neuroprotection after injuries [42,43]. However, studies on ALS focus on IGF1, the most selective and potent IGF1R ligand, rather than on the receptor. This is because a decrease in IGF1 is observed in different ALS models, and thus studies propose IGF1 delivery as therapeutic approach. Positive effects are obtained by retrograde viral delivery of IGF1 to motor neurons [44] or by muscle-specific expression of IGF1 [45]. Based on this evidence, recombinant human IGF1 is tested in ALS clinical trials, which unfortunately fails to provide therapeutic benefits [46]. Later, it is found that IGF1 levels in skeletal muscles of sporadic ALS patients is severely decreased, whereas IGF1R expression is significantly increased [47]. These findings could be explained by the observation that, when IGF1R is increased, free measurable IGF1 decreases [48]. Interestingly, it is shown that in spinal cord of SOD1G93A mice IGF1R highly co-localizes with reactive astrocytes [49], suggesting that IGF1R levels could be more directly linked with astrocyte reactivity and ALS pathophysiology than the ligand. Indeed, we found a remarkable increase in IGF1R levels in mutant iPSA and demonstrated that genetic or pharmacological inhibition of IGF1R downregulated MTOR signaling recapitulating the neuroprotective effects of MTOR inhibition. Notably, we found a similar increase in IGF1R levels in other patient-derived ALS iPSA lines, harboring two different SOD1 mutations, as well as in iPSA with C9orf72 expansion and sporadic ALS iPSA. Future work on additional astrocytes derived from diverse forms of familial and sporadic ALS will be needed to further extend these results. Nevertheless, it is possible that increased IGF1R levels are a common molecular feature, and possibly a common pathogenic mechanism, of various forms of ALS. It needs to be noted that we did not see increased IGF1R levels in FUS mutant iPSA. FUS belongs to a category of familial ALS mutants that affect RNA binding proteins [50], and it is possible that the underlying mechanisms of this group of diseases do not involve IGF1R. Additional ALS astrocyte lines with different mutations in RNA-binding proteins need to be studied to address this possibility.
In mutant SOD1G93A iPSA, we observed a significant increase of IGF1R protein, but not in its mRNA expression. Hence, changes in IGF1R levels were likely the result of altered protein turnover. It is shown that MTOR is involved in plasma membrane recycling pathways [51], but alterations of receptor recycling in mutant iPSA appear to be specific to IGF1R, since the levels of INSR, which shares structural similarities with IGF1R [29], were not significantly different in mutant and control cells. The specific downregulation of IGF1R turnover could be ascribed to the selective role of SNX-BAR5/6 (sorting nexins with a Bin, Amphiphysin, and Rvs [BAR] domain) protein, which have been shown to mediate non-core retromer dependent recycling of IGF1R, but not INSR [52]. Therefore, mutant SOD1 may interfere with the degradation of IGF1R, either by preventing its delivery to the lysosomes via SNX-BAR5/6 mediated trafficking or by altering MTOR activity directly. However, MTOR inhibition did not significantly alter IGF1R levels in SOD1G93A iPSA, suggesting that MTOR activation was a downstream effect of increased IGF1R. Based on these considerations, we proposed that trafficking alterations were the leading mechanism of IGF1R dysregulation. The involvement of SNX-BAR5/6 mediated trafficking of IGF1R in ALS astrocytes is a promising putative mechanism that remains to be further elucidated.
Both genetic and pharmacological inhibition of IGF1R decreased MTOR activity in mutant iPSA, and resulted in decreased cell proliferation and astrocytic activation markers, thereby attenuating reactive phenotypes of SOD1G93A astrocytes. Based on these findings, we studied the non-cell autonomous toxicity of mutant iPSA to motor neurons in response to IGF1R inhibition in mutant iPSA, which resulted in prevention of mutant ACM toxicity, whereas treatment of the motor neurons with MTOR or IGF1R inhibitors did not confer protection. These results indicated that modulation of the IGF1R-MTOR pathways was protective on motor neurons through its effects on astrocytes. Since astrocyte toxicity was assessed using ACM, we could speculate that modulation of IGF1R was able to favorably modify mutant astrocyte cell secretion, by reducing the emission of toxic factor, enhancing the release of neurotrophic factors, or both. Future work will be needed to determine the mechanisms underlying modifications in secretory activity and the nature of the secreted factors. Furthermore, experimentation using in vivo models of ALS will be necessary to demonstrate preclinical efficacy of this strategy to attenuate non-cell autonomous toxicity.
In summary, here we demonstrated that human astrocytes harboring SOD1G93A linked to familial ALS showed activation of the MTOR pathway due to increased levels of IGF1R. MTOR activation resulted in decreased autophagy and increased astrocytic reactivity, cell proliferation, and motor neurons toxicity, which can be attenuated by inhibiting IGF1R or MTOR. Therefore, this work unveiled a new potential role of astrocytic IGF1R-MTOR pathway in ALS pathophysiology. These findings highlighted the therapeutic potential of targeting two check points in this pathway, IGF1R and MTOR, to modulate ALS astrocytic toxicity in SOD1 mutants and potentially other forms of ALS.
Materials and methods
Cell culture
The SOD1G93A iPSA line and its isogenic control line were from the CDI MyCell Disease product (CDI, 01434) and previously characterized by us [14]. CDI iPSA lines were maintained in DMEM (high glucose, pyruvate; ThermoFisher Scientific, 11995073) supplemented with 10% FBS (VWR International, 16777–014), N2 supplement (ThermoFisher Scientific, 17502001), Glutamax (ThermoFisher Scientific, 17502001), and penicillin-streptomycin (ThermoFisher Scientific, 15140122), and plated on Matrigel (Corning, 354277)-coated plates. The other iPSA used in this study were de-identified lines gifted to us by Dr. Nicholas Maragakis (Johns Hopkins University) and grown as previously described [53].
SOD1G93A iPSA line and isogenic control cells were split with TrypLE (ThermoFisher Scientific, 354277), plated for experiments and assays were performed 5–7 days after plating. iPSA were used within 25–40 days in culture.
Wild-type iPSC-derived motor neurons (CDI, 01279) were grown according to the manufacturer’s protocol. Briefly, they were thawed and plated on poly-D-lysine (Millipore Sigma, P0899) and Matrigel-coated plates, in motor neuron medium (CDI, 01279) composed of iCell Neural base medium, iCell Neurons medium supplement, and iCell Nervous System supplement.
Cell treatments, transfections, and gene silencing
iPSA were treated with 15 μM PPP (picropodophyllotoxin; Santa Cruz Biotechnology, sc-204008A) dissolved in DMSO (Millipore Sigma, D8418) for 24 h, or 250 nM Torin1 (Selleckchem, S2827) dissolved in DMSO for 24 h.
For gene silencing, we used endoribonuclease-prepared siRNA pools (MISSION esiRNA), composed of a heterogeneous mixture of siRNAs all targeting the same mRNA sequence. Cells were transfected with MISSION siRNA transfection reagent (Millipore Sigma, S1452), according to the manufacturer’s protocol, for 48 h prior to the assays. To silence human SOD1 we used Millipore Sigma, EHU050511. Alternatively, we used human shSOD1 lentiviral particles (Millipore Sigma, SOD1 SHCLNV-NM_000454, clone TRCN0000018344) and pLKO.1-puro Empty Vector Control Transduction Particles (Millipore Sigma, SHC001V) as negative control, added to culture medium for 48 h. To silence human IGF1R we used siRNA pools (Millipore Sigma, EHU028861). siRNA targeting Renilla luciferase (Millipore Sigma, EHURLUC) was used as negative control for the siRNA experiments.
Western blots
Cells were lysed in RIPA buffer containing 25 mM Tris, pH 7.4, 125 mM NaCl, 1 mM EGTA, 1% Triton X-100 (Millipore Sigma, 93443), 0.5% sodium deoxycholate (Millipore Sigma, D6750), 0.1% sodium dodecyl sulfate (Millipore Sigma, L3771), phosphatase inhibitor and protease inhibitor cocktail (ThermoFisher Scientific, 78441) for 40 min on ice, and centrifuged at 16,000 g for 20 min at 4°C. The supernatant was collected and protein concentration determined by standard BCA protein assay.
Western blots were performed on the ThermoFisher Scientific western flow system. Thirty μg of proteins (heated at 95°C for 10 min) were loaded on NuPAGE 4–12% Bis-Tris Protein Gels. Proteins were transferred onto PVDF membranes, blocked in 5% BSA (Millipore Sigma, A9647) in TBS-T (0.5 M tris base, 1.5 M NaCl, 1% TWEEN 20), immunoblotted with appropriate primary and horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch; goat anti-rabbit, 111–035-144; goat anti-mouse, 115–035-146; 1:5000), and visualized with chemiluminescent substrates on a Bio-Rad Chemidoc Touch. Membranes were stripped using BlotFresh western Blot Stripping reagent (SignaGen Laboratories, SL100324). The following primary antibodies were used: ACTB/β-actin (Millipore Sigma, A5316), LC3B (Millipore Sigma, L7543), p-MTOR (Ser2448; Cell Signaling Technology, 2971), MTOR (Cell Signaling Technology, 2983), p-ULK1 (Ser757; Cell Signaling Technology, 6888), ULK1 (Cell Signaling Technology, 8054), p-EIF4EBP1 (Thr37/46; Cell Signaling Technology, 2855), EIF4EBP1 (Cell Signaling Technology, 9452), p-AKT (Thr308; Cell Signaling Technology, 9275), p-AKT (Ser473; Cell Signaling Technology, 4060), AKT (Cell Signaling Technology, 4691), PTEN (Cell Signaling Technology, 9559), IGF1R (Cell Signaling Technology, 3018), INSR (Cell Signaling Technology, 3025), GFAP (Dako/Agilent, Z0334). All primary antibodies were used at 1:1000. Quantification of band intensity was performed with ImageJ software.
Autophagy flux measurements by flow cytometry using ratiometric LC3 fluorescent tandem probe
iPSA were retrovirally transfected with GFP-LC3-RFP-LC3ΔG [17]. After 48 h of transduction, cells were harvested and transferred to siliconized 1.5 ml tubes (131–615 CH WATSON). The cells were centrifuged at 2,300 g for 2 min, suspended in ice-cold PBS (ThermoFisher Scientific, 10010049), and analyzed on a BD FACS LSR or Fortessa instrument. At least 10,000 events for each sample were acquired using DIVA software (version 1.0). Data were processed with FlowJo software (version 10.6.2). Cells were collected, washed with PBS and sorted on a BD FACS Aria7 Instrument. After sorting, the cells were permeabilized using 0.5% BSA and 0.05% Triton X-100 in PBS and stained with anti-GFAP AlexaFluor 647 antibody (1:100, ThermoFisher Scientific, 51–9792-82) for 30 min at 4°C, then washed and analyzed on a BD FACS LSR or Fortessa instrument.
RNA extraction, reverse transcription, and quantitative real-time PCR
Total RNA was extracted from iPSA using the SV Total RNA Isolation Kit (Promega, Z3105), following the manufacturer instructions. RNA was quantified with an Eppendorf Bio Plus photometer. Using an equal amount of RNA from each sample, complementary DNA was generated with ImProm-II Reverse Transcription System (Promega, A3800) and analyzed by real-time qPCR using SYBR green chemistry (ThermoFisher Scientific, 4309155). Real-time PCR standard curves were constructed with serial dilutions of cDNA from analyzed samples using at least five dilution points. The efficiency of all primer sets was between 95 and 105%. ACTB was used as an internal control for cDNA quantification and normalization of the amplified products. The primers were designed and analyzed with Primer3. Primer sequences were as follows: HSBP1-Fw: TAACGCTTGGTCCTCACACC; HSBP1-Rv: CCCACAGAGGCTGATGTTGT; GFAP-Fw: CCCTCACCAAATTCCACCCG; GFAP-Rv: TTGAGTTGCAGGTAGCAGGG; SERPING1-Fw: TGGTGACCAGAAGTTTGGAGTC; SERPING1-Rv: TGTTGTTGCGACCTTCCCTT; GGTA1-Fw: GGATCGCCCTAAACTCTCGG; GGTA1-Rv: TTCATTATTTTCTCAGTGAAGTCCC; GBP2-Fw: CATCACTCCTGCCAAGTGGT; GBP2-Rv: ACAGATCATGCAGCCTCCAC; SOD1-Fw: AGATGACTTGGGCAAAGGTG; SOD1-Rv: TACACCACAAGCCAAACGAC; MTOR-Fw: AGTGGACCAGTGGAAACAGG; MTOR-Rv: TTCAGCGATGTCTTGTGAGG; ACTB-Fw: ATAGCACAGCCTGGATAGCAACGTAC; ACTB-Rv: CACCTTCTACAATGAGCTGCGTGTG.
Immunocytochemistry
For MAP1LC3B immunocytochemistry, cells were fixed and permeabilized with cold MeOH (methanol):acetone (1:1) solution for 15 min. Upon 3 washes in PBS, cells were blocked with 1% BSA and 0.3% Triton X-100 in PBS for 1 h, then incubated with MAP1LC3B primary antibody (1:100; Cell Signaling Technology, 2775) in blocking solution overnight at 4°C. Upon 3 PBS washes, cells were incubated in fluorophore-conjugated secondary antibody (Jackson ImmunoResearch, Cy3 donkey anti-rabbit, 711–165-152, 1:500) for 1 h at room temperature. Cells were washed 3 times in PBS and mounted on slides for imaging with Fluoromount-G (Southern Biotech, 0100–01). Z-stack of the whole cell was acquired, with a step size of 0.21 μm on a Leica TCS SP5 confocal microscope equipped with 63x objective (N/A 1.40). Image analysis of the number of the puncta per cell was performed with the “3D Object counter” function of ImageJ software. The threshold was set manually and objects with a volume below 500 or above 5 voxels were counted as puncta. Data are presented as average of puncta per cell.
For all other immunocytochemistry experiments, cells were fixed with 4% PFA (paraformaldehyde) for 10 min. Upon washes in PBS, cells were permeabilized with 0.3% Triton X-100 in PBS for 10 min, then blocked with 1% BSA and 10% NGS in PBS for 1 h and incubated in appropriate primary antibodies in blocking solution overnight at 4°C. Upon 3 PBS washes, cells were incubated in fluorophore-conjugated secondary antibodies (1:500) for 1 h at room temperature. Cells were washed in PBS and mounted on slides for imaging with Fluoromount-G. Z-stack of the whole cell was acquired, with a step size of 0.5 μm on a Leica TCS SP5 confocal microscope equipped with 63x objective (N/A 1.40). Representative Z-projection images are shown in figures. Primary antibodies used were SLC1A3 (Abcam, ab416), NES (Abcam, ab22035), S100B (Abcam, ab4066), TUBB3 (Cell Signaling Technology, 5666), SMI-32 (BioLegend, 801701). All primary antibodies were used 1:100.
Cell proliferation
For cell proliferation assays, 5 × 103 iPSA/well were plated in 96 well plates in iPSA medium for 24 h, then cultured in medium containing 250 nM Torin1 or 15 μM PPP (or vehicle), together with 10 μM BrdU (5-bromo-2´-deoxyuridine; ThermoFisher Scientific, B23151) labeling solution for 24 h. For silencing conditions, 5 × 103 iPSA/well were plated in 96 well plates in iPSA medium for 2 h, then transfected with siRNA for IGF1R (or siRNA for Renilla luciferase as negative control) or transduced with shSOD1 lentiviral particles (or pLKO.1-puro Empty Vector as negative control) for 24 h, then 10 μM BrdU was added for 24 h. Then, iPSA were fixed with 4% PFA for 15 min, washed in PBS, permeabilized with 0.1% Triton X-100 in PBS for 20 min, incubated with 1 N HCl and then 2 N HCl, adjusted pH with phosphate-citric acid buffer pH 7.4 and incubated with anti-BrdU primary antibody-FITC conjugated overnight (ThermoFisher Scientific, B35130). Upon washes in PBS, followed by Hoechst incubation (DAPI, 1:1000; ThermoFisher Scientific, R37165) for 10 min, cells were imaged on the ImageXpress Pico Automated Imaging System equipped with a 4x objective (NA 0.13) (Molecular Devices, San Jose, CA), BrdU- and DAPI-positive nuclei were counted using the dedicated ImageXpress Pico application. Cell proliferation is expressed as the ratio between BrdU positive nuclei over DAPI positive nuclei.
Motor neuron survival in ACM (astrocyte conditioned medium)
1.5 x 104 iPSA/well were plated in 24-well plates in motor neuron medium for 4 days, then iPSA were treated with 250 nM Torin1 or 15 μM PPP, or vehicle for 24 h. For gene silencing, 1.5 × 104 iPSA/well were plated in 24 well plates in motor neuron medium for 3 days, then transfected with siRNA for IGF1R or SOD1, or siRNA for Renilla luciferase as negative control, for 48 h. ACM from each condition was collected and centrifuged at 1200 g for 5 min to remove cell debris and frozen at −20°C.
3.8 x 104 iCell motor neurons/well were plated in 96 well plates in motor neuron medium. 2 days post-plating, 75% of medium was replaced with fresh motor neuron medium containing 25% ACM. At day 5, 75% of medium was replaced for the second time, with fresh motor neuron medium containing 25% of the same ACM. At day 8, motor neurons were incubated with calcein (ThermoFisher Scientific, C3099), PI (propidium iodide; ThermoFisher Scientific, P3566) and Hoechst (DAPI) diluted 1:1000 in culture medium for 10 min at room temperature. Fluorescence (FITC, TRITC, DAPI) and phase contrast images of motor neurons were acquired with the ImageXpress Pico Automated Imaging System equipped with a 20x objective (NA 0.40), in 5% CO2 and 37°C controlled environment. Images were processed using Fiji distribution of ImageJ. Quantification of live and dead cells was performed as described previously [14]. Data are expressed as percentage of live motor neurons (PI negative) over the total number of Hoechst positive motor neurons.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM). Normality of the data was investigated by Shapiro-Wilk test and statistical significance was calculated accordingly, using GraphPad. Statistical analysis information is indicated in the figure legends and p values <0.05 was considered significant and shown in the figures as * p < 0.05, ** p < 0.001, *** p < 0.0001.
Supplementary Material
Acknowledgments
We thank Dr. Nicholas Maragakis (Johns Hopkins University) for some of the iPSC patient-derived astrocytes used in this manuscript. We thank Dr. Cècile Vindis (INSERM, Institute for Cardiovascular and Metabolic Diseases, Toulouse, France) for critically reading the manuscript.
Funding Statement
This work was supported by the National Institute of Neurological Disorders and Stroke [R01NS093872]; National Institute of Neurological Disorders and Stroke [R01NS062055].
Disclosure statement
The authors declare no conflicts of interest.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Laplante M, Sabatini DM.. MTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–73. [DOI] [PubMed] [Google Scholar]
- [3].Kim J, Kundu M, Viollet B, et al. AMPK and MTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kim DH, Sarbassov DD, Ali SM, et al. MTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–75. [DOI] [PubMed] [Google Scholar]
- [5].Zinzalla V, Stracka D, Oppliger W, et al. Activation of MTORC2 by association with the ribosome. Cell. 2011;144(5):757–68. [DOI] [PubMed] [Google Scholar]
- [6].Laplante M, Sabatini DM. MTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–967. [DOI] [PubMed] [Google Scholar]
- [10].Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics. 2010;7(4):471–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sosunov AA, Wu X, Weiner HL, et al. Tuberous sclerosis: a primary pathology of astrocytes? Epilepsia. 2008;49(Suppl 2):53–62. [DOI] [PubMed] [Google Scholar]
- [12].Codeluppi S, Svensson CI, Hefferan MP, et al. The Rheb-MTOR pathway is upregulated in reactive astrocytes of the injured spinal cord. J Neurosci. 2009;29(4):1093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].He M, Shi X, Yang M, et al. Mesenchymal stem cells-derived IL-6 activates AMPK/MTOR signaling to inhibit the proliferation of reactive astrocytes induced by hypoxic-ischemic brain damage. Exp Neurol. 2019;311:15–32. [DOI] [PubMed] [Google Scholar]
- [14].Granatiero V, Konrad C, Bredvik K, et al. Nrf2 signaling links ER oxidative protein folding and calcium homeostasis in health and disease. Life Sci Alliance. 2019;2(5):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. [DOI] [PubMed] [Google Scholar]
- [18].Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of MTORC1. J Biol Chem. 2009;284(12):8023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yip CK, Murata K, Walz T, et al. Structure of the human MTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38(5):768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roux PP, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspect Biol. 2012;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Morita M, Gravel S-P, Hulea L, et al. MTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle. 2015;14(4):473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Navé BT, Ouwens DM, Withers DJ, et al. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–31. [PMC free article] [PubMed] [Google Scholar]
- [24].Sekulic A, Hudson CC, Homme JL, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60(13):3504–3513. [PubMed] [Google Scholar]
- [25].Sarbassov DD, et al. Phosphorylation and regulation of Akt/PKB by the rictor-MTOR complex. Science. 2005;307(5712):1098–1101. [DOI] [PubMed] [Google Scholar]
- [26].Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Menon S, Dibble C, Talbott G, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of MTORC1 at the lysosome. Cell. 2014;156(4):771–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Manning BD, Tee AR, Logsdon MN, et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–62. [DOI] [PubMed] [Google Scholar]
- [29].Menting JG, Lawrence C, Kong G-W, et al. Structural congruency of ligand binding to the insulin and insulin/type 1 insulin-like growth factor hybrid receptors. Structure. 2015;23(7):1271–1282. [DOI] [PubMed] [Google Scholar]
- [30].Vasilcanu D, Girnita A, Girnita L, et al. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene. 2004;23(47):7854–62. [DOI] [PubMed] [Google Scholar]
- [31].Fellows AD, Rhymes ER, Gibbs KL, et al. IGF 1R regulates retrograde axonal transport of signalling endosomes in motor neurons. EMBO Rep. 2020;21(3):e49129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Blizzard CA, Lee KM, Dickson TC. Inducing chronic excitotoxicity in the mouse spinal cord to investigate lower motor neuron degeneration. Front Neurosci. 2016;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sabatini DM. Twenty-five years of MTOR: uncovering the link from nutrients to growth. Proc Natl Acad Sci U S A. 2017;114(45):11818–11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Swiech L, Perycz M, Malik A, et al. Role of MTOR in physiology and pathology of the nervous system. Biochim Biophys Acta. 2008;1784(1):116–132. [DOI] [PubMed] [Google Scholar]
- [35].Switon K, et al. Molecular neurobiology of MTOR. Neuroscience. 2017;341:112–153. [DOI] [PubMed] [Google Scholar]
- [36].Zhang X, Li L, Chen S, et al. Rapamycin treatment augments motor neuron degeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7(4):412–25. [DOI] [PubMed] [Google Scholar]
- [37].Saxena S, Roselli F, Singh K, et al. Neuroprotection through excitability and MTOR required in ALS motoneurons to delay disease and extend survival. Neuron. 2013;80(1):80–96. [DOI] [PubMed] [Google Scholar]
- [38].Wang IF, Tsai KJ, Shen CK. Autophagy activation ameliorates neuronal pathogenesis of FTLD-U mice: a new light for treatment of TARDBP/TDP-43 proteinopathies. Autophagy. 2013;9(2):239–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pamenter ME, Perkins GA, McGinness AK, et al. Autophagy and apoptosis are differentially induced in neurons and astrocytes treated with an in vitro mimic of the ischemic penumbra. PLoS One. 2012;7(12):e51469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moruno-Manchon JF, Uzor N-E, Ambati CR, et al. Sphingosine kinase 1-associated autophagy differs between neurons and astrocytes. Cell Death Dis. 2018;9(5):521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kulkarni A, et al. Differential regulation of autophagy during metabolic stress in astrocytes and neurons. Autophagy. 2019;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nieto Guil AF, Oksdath M, Weiss LA, et al. IGF-1 receptor regulates dynamic changes in neuronal polarity during cerebral cortical migration. Sci Rep. 2017;7(1):7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sosa L, Dupraz S, Laurino L, et al. IGF-1 receptor is essential for the establishment of hippocampal neuronal polarity. Nat Neurosci. 2006;9(8):993–5. [DOI] [PubMed] [Google Scholar]
- [44].Kaspar BK, et al. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839–42. [DOI] [PubMed] [Google Scholar]
- [45].Dobrowolny G, Giacinti C, Pelosi L, et al. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J Cell Biol. 2005;168(2):193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sorenson EJ, Windbank AJ, Mandrekar JN, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008;71(22):1770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lunetta C, Serafini M, Prelle A, et al. Impaired expression of insulin-like growth factor-1 system in skeletal muscle of amyotrophic lateral sclerosis patients. Muscle Nerve. 2012;45(2):200–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wilczak N, De Vos RA, De Keyser J. Free insulin-like growth factor (IGF)-I and IGF binding proteins 2, 5, and 6 in spinal motor neurons in amyotrophic lateral sclerosis. Lancet. 2003;361(9362):1007–11. [DOI] [PubMed] [Google Scholar]
- [49].Chung YH, Joo KM, Shin CM, et al. Immunohistochemical study on the distribution of insulin-like growth factor I (IGF-I) receptor in the central nervous system of SOD1(G93A) mutant transgenic mice. Brain Res. 2003;994(2):253–9. [DOI] [PubMed] [Google Scholar]
- [50].Mathis S, Goizet C, Soulages A, et al. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci. 2019;399(p):217–226. [DOI] [PubMed] [Google Scholar]
- [51].Dauner K, Eid W, Raghupathy R, et al. MTOR complex 1 activity is required to maintain the canonical endocytic recycling pathway against lysosomal delivery. J Biol Chem. 2017;292(14):5737–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kvainickas A, et al. Cargo-selective SNX-BAR proteins mediate retromer trimer independent retrograde transport. J Cell Biol. 2017;216(11):3677–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Haidet-Phillips AM, et al. Gene profiling of human induced pluripotent stem cell-derived astrocyte progenitors following spinal cord engraftment. Stem Cells Transl Med. 2014;3(5):575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.