

ABSTRACT
Macroautophagy/autophagy is triggered by various starvation and stress conditions. The phospholipid phosphatidylinositol-3-phosphate (PtdIns3P) is essential for the formation of the autophagosome both in yeast and mammals. The class III phosphatidylinositol 3-kinase, PIK3C3C in humans or Vps34 in yeast, produces PtdIns3P by phosphorylating the 3ʹ-OH position of phosphatidylinositol (PtdIns). In order to synthesize PtdIns3P for the initiation of autophagy, PIK3C3/Vps34 has a heterotetrameric core, the PIK3C3 complex I (hereafter complex I) composed of PIK3C3/Vps34, PIK3R4/Vps15, BECN1/Vps30, and ATG14/Atg14. A fifth component of complex I, NRBF2 in mammals and Atg38 in yeast, was found and has been characterized in the past decade. The field has been expanding from cell and structural biology to mouse model and cohort studies. Here I will summarize the structures and models of complex I binding NRBF2/Atg38, its intracellular roles, and its involvement in health and disease. Along with this expansion of the field, different conclusions have been drawn in several topics. I will clarify what has and has not been agreed, and what is to be clarified in the future.
KEYWORDS: atg38, membranes, nrbf2, pik3c3, ptdins3p, vps34
Introduction
NRBF2 (nuclear receptor binding factor 2) was originally found as an interacting protein of PPAR (peroxisome proliferator activated receptor) [1,2]. Therefore, it was also named comodulator of PPAR and RXRA/RXRα (COPR) 1 and 2 [2]. Later it started drawing attention because of its involvement in autophagy [3]. In 2013, the Ohsumi laboratory isolated the yeast Atg38 as a class III phosphatidylinositol 3-kinase (PtdIns3K, or Vps34) complex I-specific binding protein [4]. Vps34 is a lipid kinase, phosphorylating the 3-OH position of phosphatidylinositol (PtdIns) to generate PtdIns3P [5]. The Vps34 complex I (hereafter complex I) is essential for the initiation of autophagy, and composed of Vps34 (PIK3C3/VPS34 in human), Vps15 (PIK3R4/VPS15/p150 in human), Vps30/Atg6 (BECN1/Beclin1 in human), and Atg14 (ATG14/BARKOR in human), whereas complex II replaces Atg14 with Vps38 (UVRAG in human), having a role on endocytic trafficking (Figure 1). The authors postulated that NRBF2 would be the mammalian Atg38 ortholog. Immediately after this, three groups reported the identification of NRBF2 as a mammalian complex I-binding protein [6–8]. It has been ten years since the NRBF2 involvement in autophagy was found, and the research fields have been expanding from cell biology to structural biology, pathology, and neurobiology.
Figure 1.
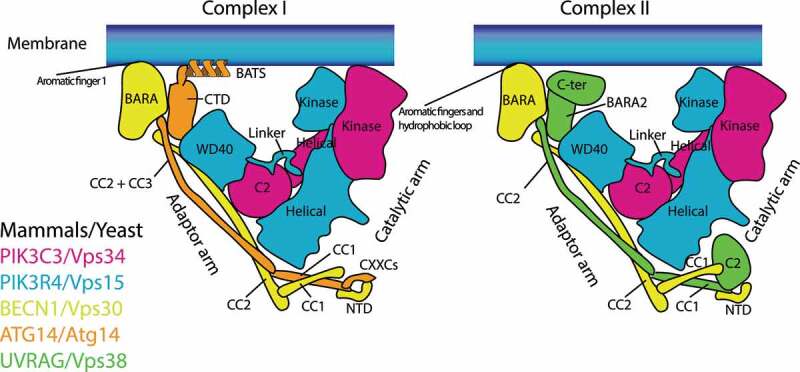
Schematic structural models of mutually exclusive complex I (left) and complex II (right). PIK3C3/Vps34, PIK3R4/Vps15, and BECN1/Vps30 are found in both complexes, whereas ATG14/Atg14 is complex I-specific, and UVRAG/Vps38 is complex II-specific. Complex I associates with membranes via the aromatic finger 1 in the BECN1 BARA domain and the BATS domain in ATG14 (which does not exist in Atg14). Complex II associates with membranes via two aromatic finger motifs (aromatic fingers 1 and 2) and the hydrophobic loop, all of which are in the BECN1 BARA domain. C2: C2 domain; CC: Coiled-coil; BARA: BARA domain; BATS: BATS domain; NTD: N terminal domain; CTD: C terminal domain; CXXCs: CXXC motifs; WD40: WD40 domain
In this review I will summarize the basic architecture of NRBF2/Atg38, its intracellular roles, and its involvement in health and disease. Naturally, different conclusions among studies also have been seen. In particular, the exact subunit(s) in complex I interacting with NRBF2/Atg38, activation/inhibition of complex I by NRBF2, and the binding stoichiometry between complex I and NRBF2 are topics that have not been agreed upon, and I will discuss what should be clarified.
Structures of NRBF2/Atg38
NRBF2/COPR2 [2], is composed of a microtubule-interacting and targeting (MIT) domain at its N terminus, and a coiled-coil homodimerization domain at the C terminus. These two domains are flanked by an intrinsically disordered region (IDR, Figure 2 left). There are three NRBF2 isoforms: Isoform 2, also known as COPR1 [2], lacks the IDR, whereas isoform 3 lacks the first two helices of the MIT domain; instead 28 unknown amino acid residues are appended in front of the third helix of the MIT domain (see below). So far there are no reports regarding the physiological roles of these isoforms.
Figure 2.
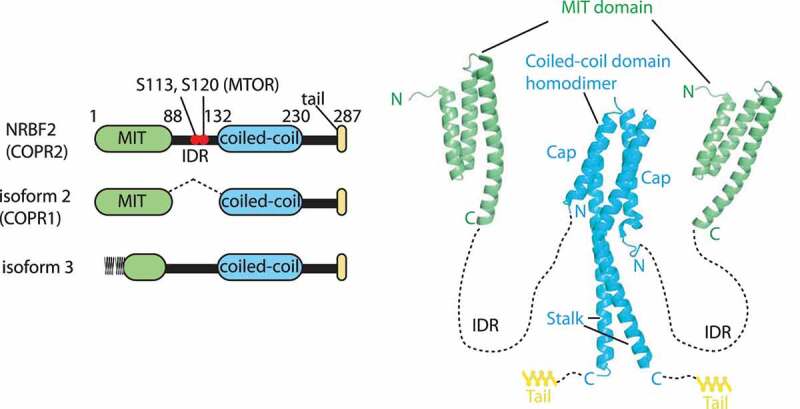
Structures of NRBF2/Atg38. left: schematic representations of NRBF2 and its isoforms (in human residue numbers). The S113 and S120 residues in the intrinsically disordered region (IDR) are phosphorylated by MTOR. Right: structures of the NRBF2 MIT domain (PDB: 4ZEY) and Atg38 coiled-coil domain (PDB: 5KC1). NRBF2/Atg38 makes a stable homodimer via the two segments, the cap and stalk in the coiled-coil domain
There is good agreement that the MIT domain is responsible for binding to complex I [4,7–10]. The MIT domain is composed of three helices (Figure 2 right). As for isoform 3, although the appended amino acid sequence is not similar to the sequence in the corresponding region of NRBF2, a secondary structure prediction site (HHpred: https://toolkit.tuebingen.mpg.de/tools/hhpred) predicted helices 1 and 2 of the NRBF2/Atg38 MIIT domains to be most similar to the appended sequence, indicating that the N-terminal region of isoform-3 may also have an MIT domain assembly. It would be interesting to see if isoform-3 still binds to complex I, or some other protein(s). Both NRBF2 and Atg38 form strong homodimers via the C-terminal coiled-coil domain [4,8–10] (Figure 2 right). Homodimerization is essential for efficient binding to, and for the integrity of, the complex in yeast and mammals [4,7,10]. The structure of this region is mushroom-like shaped, with a four-helix bundle segment (cap) at its N terminus, and a coiled-coil segment at its C terminus (stalk). Following the stalk, there is a helical extension (tail) both in yeast and mammals. Homodimerization and localization to autophagic puncta of Atg38 were affected when more than two segments of the cap, stalk, and tail were deleted simultaneously [10].
The S113 and S120 residues in the IDR of human NRBF2 are phosphorylated by MTOR (mechanistic target of rapamycin kinase) [11] (Figure 2 left). For the role of these phosphorylations, see the below sections. Although the exact amino acid residue is unknown, NRBF2 is also phosphorylated by ULK1 [11].
NRBF2/Atg38-binding subunit (s) in complex I – from a methodological point of view
As summarized in Table 1, all of the complex I subunits have been confusingly proposed as NRBF2/Atg38-binding partners. This might be due to the methods being employed, which are mostly immunoprecipitation (IP), hydrogen deuterium exchange mass spectrometry (HDX-MS), or cryo-electron microscopy (cryo-EM). For all methods it is important to bear in mind that except for PIK3C3 in humans [12] and Vps30 in yeast [13], each of the complex I and II subunits is not stable by itself, therefore singly expressed/purified proteins might not be correctly folded, resulting in soluble aggregation. Also, domain deletion can cause conformational changes of the altered protein, as well as the whole complex assembly. IP is the most popular method in cell biology to detect protein-protein interactions. However, because complex I is a stable complex, cellular IPs might not be able to differentiate direct interactions from indirect interactions among the complex I subunits. HDX-MS is a mass-spectrometry based, label-free method, and purified intact proteins can be used to map the interaction sites at 5–10 amino acids resolution [14]. Of note, results should be carefully analyzed to differentiate a direct interaction from indirect conformational changes caused by the direct interaction. Also, for performing HDX-MS and analyzing the data, it is recommended to follow the community’s guidelines [14]. With the resolution revolution in Cryo-EM one is able to routinely visualize macromolecules at 3–5 Å resolution [15]. The resolution depends on various factors including the stability, flexibility, and size of the sample. Since complex I is flexible, and undergoes various conformational changes, the resolution has not been dramatically improved until recently [9,16–19]. Furthermore, NRBF2 and Atg38 are small molecules (32 kDa and 26 kDa, respectively), and it has been difficult to definitively assign their locations to the cryo-EM density [17]. In spite of these technical difficulties, ATG14/Atg14 was found as an NRBF2/Atg38-binding subunit in all studies except for Cao et al. [6] (Table 1). This is reasonable, given that ATG14/Atg14 is the complex I-specific subunit. The complex I-Atg38 binding is independent of nutrient condition in yeast [4]. Also, mammalian NRBF2 was identified in non-starvation conditions [6–8,11], indicating mammalian complex I-NRBF2 binding also does not depend on nutrient condition. Ma et al. found that residues S113 and S120 in the IDR of NRBF2 are phosphorylated by MTOR in nutrient-rich conditions. The phosphorylated NRBF2 binds to PIK3C3-PIK3R4, making complex I inactive, whereas starvation dephosphorylates these residues, allowing NRBF2 to bind to BECN1-ATG14, leading to an active form of the complex [11]. How the phosphorylated NRBF2 and PIK3C3-PIK3R4 are assembled in rich medium condition remains to be elucidated. Interestingly, previous in vitro studies used bacterially purified NRBF2/Atg38, which are post-translational modification (PTM)-free, and identified Vps30-Atg14 or BECN1-ATG14 as NRBF2/Atg38-interacting subunits [9,10]. These results are consistent with the dephosphorylated NRBF2 interaction with BECN1-ATG14 during starvation by Ma et al. [11]. This binding subunit question will ultimately be answered by cryo-EM or crystallography at higher resolution, while various factors such as PTMs and the binding stoichiometry between complex I and NRBF2 (see below) need to be taken into account.
Table 1.
Summary of NRBF2/Atg38-binding candidates
| Reference | Binding subunit (s) | Method |
|---|---|---|
| Cao et al. [6] | PIK3R4 (WD40 domain), but not PIK3C3, BECN1, nor ATG14 | IP in HEK293T cells and in vitro translation |
| Araki et al. [4] | Vps34 and Atg14 | Yeast two-hybrid and in vitro affinity isolation |
| Lu et al. [7] | ATG14, UVRAG | in vitro GST affinity isolation (ATG14), HA-NRBF2 affinity isolation from TREx-293 cells followed by peptide identification with mass-spectrometry (UVRAG) |
| Zhong et al. [8] | BECN1, ATG14 | IP in HepG2 or HeLa cells |
| Young et al. [9] | BECN1 (BH3 in NTD), ATG14 (CC1), PIK3R4 (Helical) | HDX-MS, EM (negative stain) |
| Ohashi et al. [10] | Vps30 (CC1), Atg14 (CC1) | HDX-MS |
| Ma et al. [11] | PIK3C3-PIK3R4 (when NRBF2 S113 and S120 are phosphorylated), BECN1-ATG14 (when dephosphorylated) | IP with WT, non-phosphorylatable or phosphomimetic NRBF2 and each of singly expressed complex I subunits in HEK293 T cells |
| Young et al. [17] | BECN1 (BH3 in NTD), ATG14 (CC1), PIK3R4 (Helical) | HDX-MS, cryo-EM with complex I (MIT fused to BECN1, MBP-MIT+MIT fused to BECN1, or complex I + FL NRBF2) |
Outside of complex I, UVRAG was also found to be an NRBF2-binding protein, and coexpression of PIK3C3-PIK3R4-UVRAG-NRBF2 (without BECN1) increased PtdIns3K activity [7] (Table 1). How NRBF2 modulates the assembly and activity of complex II remains to be seen.
Binding stoichiometry between complex I and NRBF2/Atg38
Both yeast and human complex I can be stably purified without Atg38 and NRBF2 [9,10,19,20], indicating Atg38 and NRBF2 are not essential for the assembly of complex I in vitro. On the other hand, both in yeast and mammalian cells, the deletion or knockdown of ATG38/NRBF2 causes partial dissociation of complex I [4,7,8]. This indicates that there might be a specific step where complex I requires Atg38/NRBF2 for its efficient assembly. The yeast complex I binds to Atg38 with a 1:2 stoichiometry (one copy of complex I bound to one homodimer of Atg38) in vivo [4]. Also, for both yeast complex I-Atg38 and human complex I-NRBF2, a study on in vitro reconstitution coupled with size exclusion chromatography – multiangle light scattering (SEC-MALS) has been reported [10]. SEC-MALS is a method for determining the absolute molar mass of molecules in solution. This revealed that yeast complex I binds to Atg38 with a 1:2 stoichiometry independent of reconstitution condition (Figure 3). Whereas in the case of the human proteins, the binding stoichiometry depends on the relative abundance of NRBF2 to complex I. When complex I is more abundant than NRBF2, complex I is homodimerized by a NRBF2 homodimer (2:2), whereas the stoichiometry becomes 1:2 when NRBF2 is more abundant than complex I [10] (Figure 3). Structurally, Young et al. proposed the 2:2 stoichiometry model and hypothesized that this stoichiometry has important consequences for complex I to interact with membranes [9], whereas the same group also proposed the 1:2 stoichiometry model based on the observation that 2 copies of the MIT domain are required to fully activate complex I [17]. This in vitro stoichiometry change remains to be seen in vivo. One possibility is that in mammalian cells the binding stoichiometry may be regulated in a spatio-temporal manner depending on the local concentration of complex I and NRBF2.
Figure 3.
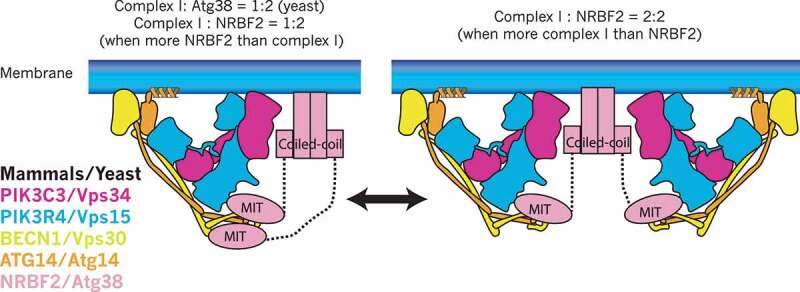
Binding stoichiometry between complex I and NRBF2/Atg38. In mammals, the stoichiometry between complex I and NRBF2 is 1:2 (one copy of complex I and one homodimer of NRBF2, left) when NRBF2 is more abundant than complex I, whereas it can be 2:2 (two copies of complex I and one homodimer of NRBF2, right) when complex I is more abundant than NRBF2. In yeast the stoichiometry is always 1:2 regardless of the comparative abundance between complex I and Atg38. It must be noted that the complex I subunit (S) binding to the MIT domain is still speculative
Membrane binding role of NRBF2/Atg38
Both yeast and human complexes I and II are Y-shaped, bearing PIK3C3/Vps34 and PIK3R4/Vps15 as the catalytic arm, and BECN1/Vps30 and ATG14/Atg14 (complex I) or UVRAG/Vps38 (complex II) as the adaptor arm [13,16,20] (Figure 1). The BATS domain of ATG14 (which does not exist in yeast Atg14) and the aromatic finger motifs in the BARA domain of BECN1, both of which are in the adaptor arm, are essential for the activity and membrane binding of complexes I and II, respectively [12,13,18,21,22] (Figures 1 and 3). The C-terminal coiled-coil domain of Atg38 is essential for the localization of Atg38 to autophagic puncta and to the pellet fraction in subcellular fractions [10], indicating that this region might be responsible for membrane binding (Figure 3). Since yeast Atg14 lacks the BATS domain, the activity of yeast complex I is much weaker than that of yeast complex II on flat membranes [13], which stands in contrast to human complexes I and II [12]. Therefore, membrane binding may be a more important role for Atg38 in yeast than it is for NRBF2 in mammals. However, the atg38∆ mutant only reduced, not eliminated, the colocalization of the complex I subunits and Atg18, an effector of complex I to the PAS [4], indicating that there might be unknown factors that help complex I localize to the PAS in addition to Atg38. It is possible that Atg38 might become essential at a specific step during autophagy. It is not clear how NRBF2 contributes to membrane binding in mammals. In rich media, 81% of NRBF2 in HeLa cells is cytosolic (http://mapofthecell.biochem.mpg.de/about.html), suggesting that NRBF2 may become membrane-localized upon starvation.
NRBF2/Atg38 contribution to PtdIns3P synthesis
It is not clear how Atg38 contributes to PtdIns3P synthesis in yeast. In yeast, PtdIns3P synthesis is solely dependent on Vps34 and Vps15, and deletions of VPS30, ATG14, and VPS38 show negligible effects [23]. Also, the autophagy-defective phenotype of atg38∆ is milder than that of atg14∆ [4]. These indicate that Atg38 might not be essential for the production of PtdIns3P, although it is still possible that Atg38 could affect the kinase activity of complex I. On the other hand, mammalian PtdIns3P synthesis is dependent not only on PIK3C3 complexes, but also partially on the class II PI3Ks [24]. Zhong et al. measured intracellular PtdIns3P levels, and found that in contrast to yeast, ATG14 siRNA significantly reduced PtdIns3P levels, whereas NRBF2 siRNA significantly increased PtdIns3P levels during serum starvation, hypothesizing that NRBF2 is a catalytic suppressor of complex I [8]. The opposite has also been reported by several groups. Lu et al. reported that NRBF2 facilitates complex I activity in cells [7]. In vitro studies from the Hurley group showed that adding excess amounts of NRBF2 to complex I increased the kinase activity [9,17], although the fold increase in the activation significantly differs between the two papers from this group [9,17] (Table 2). Interestingly, as described above, Ma et al. reported that the NRBF2 contribution to the activity depends on the phosphorylation status of NRBF2 by MTOR [11] (Table 2). In contrast to this study, which showed that phosphorylated NRBF2 inactivates complex I by binding to PIK3R4, Young et al. reported that bacterially purified, PTM-free NRBF2 activates complex I by binding to the helical solenoid of PIK3R4 [17] (Tables 1 and 2). While the NRBF2 binding to PIK3R4 is a common observation between Ma et al. and Young et al., how these opposite conclusions were drawn between the cellular (Ma et al.) and in vitro (Young et al.) studies needs to be clarified in the future.
Table 2.
Summary of NRBF2 contribution to PIK3C3 kinase activity
| Reference | activate/inhibit | Method | substrate |
|---|---|---|---|
| Lu et al. [7] | activate | IP from mice ±NRBF2, or IP with coexpressed PIK3C3-PIK3R4-ATG14 or PIK3C3-PIK3R4-UVRAG ± NRBF2 in HEK293T cells, then detect with TLC | sonicated PI |
| Zhong et al. [8] | inhibit | PtdIns3P detection with ELISA (Echelon) using cell extract from RPE-1 cells (± NRBF2 siRNA) | none |
| Young et al. [9] | activate (10-fold increase for both FL and MIT) | ADP-Glo assay kit (Promega) with purified complex I and NRBF2 (FL and MIT domain) at the ratio of 1:25 | sonicated liposomes (∼50–100 nm) containing 22 μM PI |
| Young et al. [17] | activate (2-fold increase for both FL and MIT) | ADP-Glo assay kit (Promega) with purified complex I and NRBF2 (FL and MIT domain) at the ratio of 1:63 | sonicated liposomes with PI + PS at 1:6 |
| Ma et al. [11] | activate (with non-phosphorelatable), inhibit (with phosphomimetic) | IP with flag-tagged NRBF2 constructs, stably expressed in MEFs, then detect with TLC | sonicated PI (Avanti 840,042 C) |
| Cai et al. [27] | activate | IP with CCZ1 in mouse brains and N2a cells ± NRBF2, then detect with a PtdIns3P ELISA kit (unknown) | Not described |
Eukaryotic cells are compartmentalized by biological membranes, mainly composed of glycerophospholipids, phosphatidylcholine (PC), phophatidylethanolamine (PE), phosphatidylserine (PS), and phosphatidylinositol (PtdIns) [25,26]. We recently found that lipid composition and size (membrane curvature) greatly affect the activity of human complexes I and II [12]. None of the above studies has used a lipid substrate with a physiologically relevant lipid composition for the lipid kinase assay (Table 2). How NRBF2 affects complex I activity with a physiologically relevant substrate needs to be seen in the future.
NRBF2/Atg38 involvement in autophagy
Of the several types of autophagy, here I will discuss macroautophagy (hereafter referred to as autophagy). Autophagy is triggered by starvation, and its process is composed of sequential stages: Initiation, nucleation, expansion, closure, maturation, and degradation (Figure 4, colored in red). The initiation of autophagosome biogenesis is negatively regulated by MTOR complex 1 (MTORC1) that phosphorylates the ULK1 complex [27–30] and complex I [11,31] (also see NRBF2/Atg38-binding subunit(s) in complex I and NRBF2/Atg38 contribution to PtdIns3P synthesis). Upon starvation MTORC1 is inactivated, which leads to activation of the ULK1 complex. ULK1 complex activation recruits complex I, which produces PtdIns3P at highly curved ER structures called omegasomes (1. Initiation in Figure 4)[32]. The locally enriched PtdIns3P leads to nucleation of phagophores by recruiting downstream effectors such as WIPIs and DFCP (2. Nucleation in Figure 4), which is followed by membrane expansion, closure (3. Expansion and 4. Closure in Figure 4), and autophagosome fusion with lysosomes to become autolysosomes (5. Maturation in Figure 4). Finally, the cargos are degraded (6. Degradation in Figure 4) [33,34]. Complex I and PtdIns3P are essential for the initiation of autophagy both in yeast and mammals [23,35–38]. During starvation or starvation-mimicking conditions, NRBF2/Atg38 colocalizes with autophagosome markers [4,6–8]. NRBF2 knockdown significantly reduced the localization of autophagosome markers and increased the SQSTM1/p62 levels [6,7,11,39]. Also, atg38∆ reduced the autophagic activity, although it is milder than atg14∆ [4]. For the above reasons, and assuming NRBF2/Atg38 is a binding protein of complex I that is essentially involved in the initiation of autophagy, it is conceivable that NRBF2/Atg38 is a positive regulator for the initiation of autophagy. On the other hand, Zhong et al. reported that NRBF2 is a negative regulator, based on the reduced SQSTM1 levels and increased intracellular PtdIns3P levels in NRBF2 knockdown cells during starvation [8] (see above and Table 2). Interestingly, the same study also reported that NRBF2 puncta formation can be independent of the ULK1 complex [8], which is essential for the localization of complex I to the autophagosome [40]. This indicates NRBF2 could be involved in the initiation of autophagy either with complex I or in an ULK1 and complex I–independent manner. In Schizosaccharomyces pombe, Atg38 is known to interact with Atg8 via an Atg8-family-interacting motif (AIM) which is not conserved in other species. This interaction is important for autophagosome expansion and autophagy flux [41].
Figure 4.
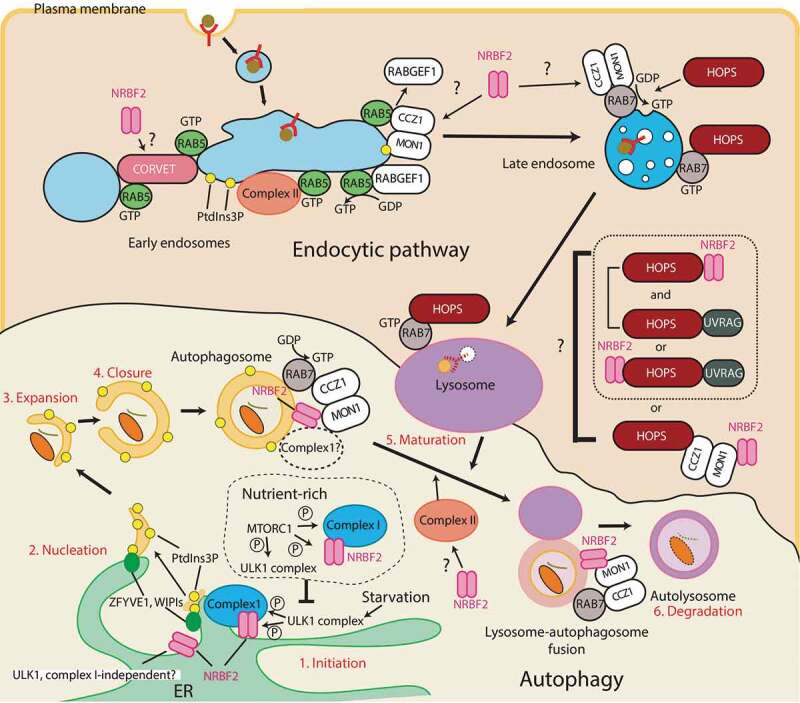
Autophagy and endocytic pathways that involve NRBF2. Autophagy: In nutrient-rich conditions, MTORC1 inhibits autophagy by phosphorylating the ULK1 complex and complex I-NRBF2 complex. Upon starvation, MTORC1 is inactivated, leading to the ULK1 complex activation that recruits complex I-NRBF2, which in turn produces PtdIns3P (1. Inititation). PtdIns3P recruits its effectors such as DFCPs and WIPIs at omegasomes that causes nucleation of phagophores (2. Nucleation). The phagophore expands (3. Expansion), then closes (4. Closure) to become an autophagosome. The autophagosome fuses with lysosomes to become an autolysosome (5. Maturation). The cargos are eventually degraded in the autolysosome (6. Degradation). As well as the initiation step, NRBF2 is also involved in the autophagy maturation step by associating with the MON1-CCZ1 complex. The NRBF2-MON1-CCZ1 facilitates the GEF activity of RAB7, which is involved in the maturation step. Circled P: phosphorylation; ?: the direct involvement of NRBF2 has not been confirmed. Endocytic pathway: During endocytosis, lipids and surface proteins including ligand-receptor complexes are internalized. The endocytosed vesicle first fuses with the early endosome marked by RAB5. The early endosome matures into the late endosome marked by RAB7, then the late endosome eventually fuses with lysosomes to degrade the endocytosed cargos. The early to late endosome transition is mediated by the MON1-CCZ1 complex. The CORVET and HOPS complexes facilitate the tethering of early endosomes and late endosomes, respectively. NRBF2 was found to interact with VPP33A, the common subunit between the CORVET and HOPS complexes. Also, NRBF2 is known to interact with the MON1-CCZ1 complex during autophagy and phagocytosis, but it remains to be seen whether this interaction also occurs in the endocytic pathway or not. The complex II-specific UVRAG subunit is also known to bind to the HOPS complex independently of complex II. It has been unclear whether the NRBF2 and UVRAG interactions with the HOPS complex are mutually exclusive or not
Knockout or knockdown of NRBF2 also impairs the generation of autolysosomes, indicating that NRBF2 can also be involved in the autophagosome maturation step [7,42]. NRBF2 deficiency prohibits RAB7 recruitment to the autophagosome that is thought to be important for the maturation step during autophagy [43]. NRBF2 binds to the MON1-CCZ1 complex, a guanine nucleotide exchange factor (GEF) that activates RAB7 by allowing RAB7 to bind GTP (see the below chapters for more details) [42,44]. The NRBF2-associated MON1-CCZ1 GEF activity is increased in a starvation-induced autophagy condition, and decreased with the MON1-CCZ1 complex purified from nrbf2−/- mouse brain [42]. The NRBF2-MON1-CCZ1 interacts with complex I subunits in mouse brains under non-starvation conditions [42]. It remains to be seen at which organelle this interaction occurs, and whether complex I can be also involved in the autophagosome maturation step with NRBF2 during starvation (Figure 4). In contrast to this finding, Kuchitsu et al. showed that RAB7 is dispensable for the autophagosome maturation step during starvation [45]. In this case, NRBF2 should have an additional role in the maturation step to show the impairment of the autolysosome generation in the absence of NRBF2.
NRBF2 involvement in the endocytic pathway
The endocytic pathway is essential for keeping cellular homeostasis. This pathway is mainly used for internalizing lipids and surface proteins including ligand-receptor complexes (endocytosis). During endocytosis the internalized vesicle is first fused with the early endosome marked by RAB5, then the early endosome matures into the late endosome marked by RAB7. The late endosome eventually fuses with lysosomes to degrade the content [46] (Figure 4). The transition from early to late endosomes marked by RAB5 and RAB7 is mediated by the SAND-1/MON1-CCZ1 complex (SAND-1 for C. elegans, and MON1 for mammals), which is an effector of RAB5 that displaces the RAB5 GEF, RABGEF1/Rabex5, from membranes. At the same time the SAND-1/MON1-CCZ1 complex is a GEF for RAB7 (Figure 4) [47–50]. Membrane fusion on early endosomes and late endosomes/lysosomes are facilitated by the tethering complexes class C core vacuole/endosome tethering (CORVET) and homotypic fusion and vacuole protein sorting (HOPS) complex, respectively [51,52]. These complexes share common subunits (Vps11, Vps16, Vps18, and Vps33), and have unique subunits Vps8 and Vps3 for the CORVET complex, and Vps41 and Vps39 for the HOPS complex [51,52]. On early endosomes, complex II is recruited by RAB5 to synthesize PtdIns3P [53,54]. Also, Vps21 (RAB5 ortholog in yeast) recruits the CORVET complex via Vps8 and Vps3 [55–57]. Whereas the HOPS complex is recruited to late endosomes/lysosomes by RAB7 (Figure 4) [51,52,58]. The HOPS complex also interacts with the SAND-1/MON1-CCZ1 complex [50,51]. SAND1 can bind to PtdIns3P, and the intracellular SAND1 localization is dependent of PtdIns3P [50]. An endocytic pathway BioID study identified the PIK3C3 complex subunits including NRBF2, but not ATG14 as interacting proteins of VPS33A, the core subunit of the human CORVET/HOPS complexes [59]. Because ATG14 was not found in this study, it is likely that this interaction could be independent of complex I. As described above and below, during autophagy and phagocytosis NRBF2 is reported to bind to the MON1-CCZ1 complex, which binds to the HOPS complex. It remains to be elucidated whether BioID detected the direct interaction between NRBF2 and the CORVET/HOPS complex, or the indirect interaction mediated by the MON1-CCZ1 complex (Figure 4). The complex II-specific UVRAG subunit is also known to bind to the HOPS complex independently of complex II [60]. This interaction was found in the maturation steps of autophagy and endocytosis, although its direct involvement in the autophagosome maturation step was not confirmed by Jiang et al. [61]. Whether the HOPS complex containing NRBF2 and UVRAG are mutually exclusive or not also remains to be seen (Figure 4).
NRBF2 in health and disease
According to the Human Protein Atlas (https://www.proteinatlas.org/) and Yasumo et al. [1], NRBF2 mRNA is expressed ubiquitously in human organs, particularly at high levels in the blood, liver, and muscle tissues. The NRBF2 protein can also be detected ubiquitously in all organs (Human Protein Atlas). The Proteomics DB (https://www.proteomicsdb.org/proteomicsdb/#overview) shows that all three NRBF2 isoforms are highly expressed in the placenta, spleen, B-lymphocyte, and natural killer cells. nrbf2−/- null mutant mice are viable, and no enhanced mortality, whereas these mice showed focal liver necrosis [7]. Although the phenotype of atg14 null mutant mice has not been reported, this nrbf2 KO mice phenotype is very different from the embryonic lethal phenotypes of pik3c3, pik3r4, and becn1 null mutant mice, and the neonatal lethal phenotypes of null mutant mice lacking essential ATG genes [62,63]. This indicates that the NRBF2 contribution to autophagic activity during embryogenesis and neonatal stages might be minor compared to other major autophagy players.
NRBF2 involvement in inflammatory bowel diseases (IBD) and phagocytosis
Maintaining the homeostasis of intestinal epithelial cells is essential for all metazoans, and its failure is one of the causes of IBD represented by Crohn disease and ulcerative colitis [64,65]. Maintenance of the endocytic pathway by PIK3C3 or PtdIns3P is essential for epithelial cell polarity in Drosophila and Caco-2 organoids [66], and for polarized distribution of cell-junction proteins in intestinal epithelial cells, because the deficiency of pik3c3 causes IBD-like features in zebrafish [67]. Inflammation is prevented by removing pathogens, immune complexes, and an excessive number of apoptotic (dying) cells via phagocytosis. Phagocytosis occurs in a series of several steps. First, the particle needs to be recognized by the host cell via specific receptors. Then, the particle is surrounded by the plasma membrane of the host cell, ingested and detached from the plasma membrane. Next, the phagosome fuses with early endosomes followed by late endosomes, transforming from the early phagosome marked by RAB5, to the late phagosome marked by RAB7. The late phagosome eventually fuses with lysosomes, generating a phagolysosome to degrade the particle (Maturation in Phagocytosis, Figure 5) [68,69]. PIK3C3 involvement in the maturation step from early phagosome formation to phagolysosome formation has been shown in RAW macrophages and C. elegans [70,71]. Similar to the endocytic pathway, PIK3C3 is recruited to early phagosomes by RAB5. Also, RAB5 recruits the MON1-CCZ1 complex that in turn activates RAB7 (Figure 5; see also NRBF2 involvement in endocytic pathway) [47,49,72]. Moreover, the Green laboratory found LC3-associated phagocytosis (LAP), a noncanonical autophagy that can be triggered by various extracellular stimuli such as pathogens, apoptotic cells, immune complex, and viruses [73–75]. LAP requires LC3 and its conjugation (lipidation) system (such as ATG12–ATG5, and ATG16L1, Figure 5), as well as reactive oxygen species (ROS) synthesized by the NOX2 complex (CYBB/NOX2, CYBA/p22phox, NCF1/p47phox, NCF4/p40phox, NCF2/p67phox, and RAC1), but the ULK1 complex and ATG9 are dispensable [74]. Also, complex II (but not complex I) along with its specific binding protein RUBCN/Rubicon are essential for the PtdIns3P synthesis on this phagosome (LAPosome) [74]. The recruitment hierarchy of the above protein complexes during LAP is summarized in Figure 5.
Figure 5.
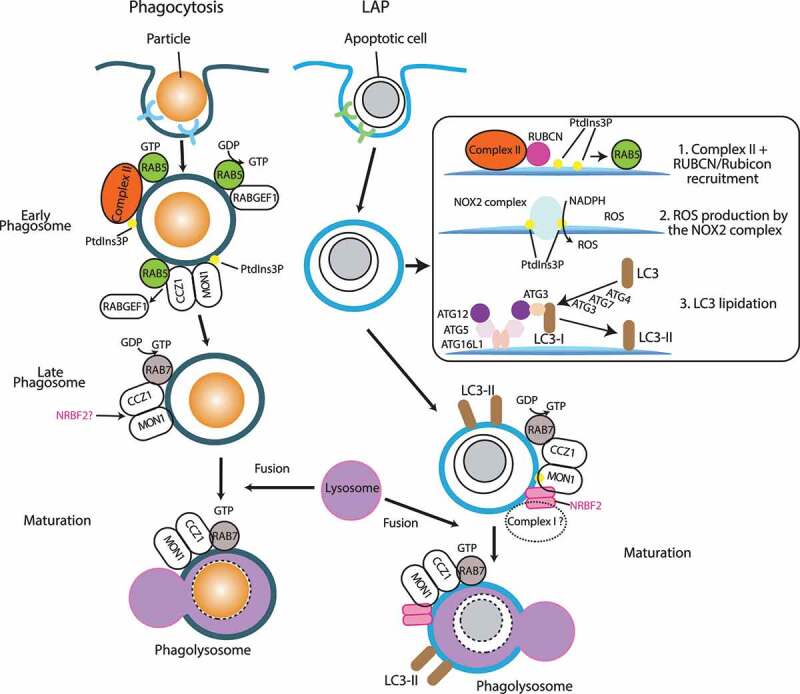
Phagocytosis and LAP pathways where PIK3C3 complexes and NRBF2 are involved. In phagocytosis, the closed phagosome first fuses with early endosomes to become the early phagosome marked by RAB5. The early phagosome matures into the late phagosome, and it eventually fuses with lysosomes to become the phagolysosome (maturation), then the particle is degraded. Similar to the endocytic pathway, RAB5 recruits complex II to the early phagosome, and the RAB5-RAB7 transition is mediated by the MON1-CCZ1 complex. In LAP, complex II and RUBCN/Rubicon are recruited to the closed phagosome (LAPosome, step1), where RUBCN is essential for the recruitment of complex II. In contrast to phagocytosis, the complex II- RUBCN complex is required for the recruitment of RAB5 [74]. The PtdIns3P synthesized by the complex II- RUBCN complex recruits the NOX2 complex, which synthesizes ROS from NADPH on the LAPosome (step2). This is followed by LC3 lipidation (step 3). Along with LC3-II [74], RAB7 activated by the MON1-CCZ1 complex facilitates LAPosome fusion with lysosomes (maturation) to degrade the particle. In BMDM from mice, NRBF2 is required for the maturation step of LAP by facilitating the GEF activity of the MON1-CCZ1 complex to activate RAB7
Wu et al. found that when nrbf2−/- mice were fed with 2% dextran sulfate sodium (DSS), a chemical colitis inducer where oral administration has been used a model for acute and chronic colitis [76], the mice exhibited an increased inflammatory response, and also a significant accumulation of apoptotic cells in the colon [44]. The phagosomes in bone marrow-derived macrophages (BMDM) from the nrbf2−/- mice exhibited increased RAB5A recruitment and delayed RAB7 recruitment, indicating an impairment of the phagosome maturation step. This was due to the reduction in NRBF2 binding to the MON1-CCZ1 complex. The NRBF2-MON1-CCZ1 interaction facilitates the GEF activity of RAB7 (see NRBF2/Atg38 involvement in autophagy and NRBF2 involvement in endocytic pathway). Also, the authors showed an interaction between the MON1-CCZ1 complex and PIK3C3 [44], although it remains to be seen whether this interaction includes the whole of complex I or PIK3C3 on its own, since PIK3C3 by itself is relatively stable. These phagosomes were stained by LC3, and LC3 staining was increased on the phagosomes from the nrbf2−/- mice, indicating NRBF2 regulates the maturation step of LAP [44]. It remains to be seen how LAP, which is driven by complex II + RUBCN, is (potentially) matured by the complex I-NRBF2 complex. Figure 5 summarizes the phagocytosis and LAP pathways where PIK3C3 complexes and NRBF2 are involved.
NRBF2 in cancer
Autophagy can be either tumor-suppressive or -promoting depending on the stage of tumorigenesis. During the early stage of tumorigenesis, autophagy suppresses tumor-inducing factors such as inflammation, DNA damage, and ROS [77]. The tumor suppressor concept of autophagy was originally derived from the finding that BECN1 was deleted at high rates in sporadic human breast cancers and ovarian cancers [78]. However, Laddha et al. reported that the deletions contain either only BRCA1, the well-known tumor suppressor, or both BRCA1 and BECN1, but not BECN1 alone [79]. This indicates that BRCA1 deletions are the main drivers of tumorigenesis, and so BECN1’s role as a tumor supressor remains unclear [80]. Indeed, the COSMIC (https://cancer.sanger.ac.uk/cosmic) and ICGC data portal (https://dcc.icgc.org/) databases do not show a significant number of somatic mutations in the BECN1 locus compared to the loci encoding the other PIK3C3 complex subunits such as PIK3C3, PIK3R4, and UVRAG. Somatic cancer mutations in the coding regions of PIK3C3 complex subunits, and their potential effects on the stability and activity of the complexes are described elsewhere [81]. During the late stage of tumorigenesis, cancer cells use autophagy to promote their growth and maligancy [77]. Therefore, autophagy has been targeted for cancer therapy.
A survey on the ICGC data portal revealed that NRBF2 only has five high impact somatic mutations (mutations that change amino acid sequences), which is the lowest frequency among the genes encoding complex I and II subunits, and none of the five mutations are categorized as clinically significant. Mutations in NRBF2 (in fact all of the PIK3C3 complex subunit-encoding genes) also cannot be found in the Cancer Gene Census (https://cancer.sanger.ac.uk/census) in the COSMIC database. Therefore, NRBF2 is unlikely a cancer driver. Interestingly, the D125N mutation, located at the IDR, has been found from 4 independent samples in COSMIC database [82–84]. The reason why this mutation is enriched remains to be seen. Also, the disease-protective haplotype in one of the correlated, highly trait-associated variants at the 10q21.2 breast cancer risk locus is associated with reduction of the NRBF2 promoter activity, whereas the other genes encoding PIK3C3 complex subunits were not found in this study [85].
NRBF2 in neurobiology
Although no somatic mutations associated with neurodegenerative diseases in the coding regions of the PIK3C3 complex subunits have been reported, several studies have implied that the reduced activity and stability of complex I could be associated with neurodegenerative diseases [86–89]. NRBF2 involvement was not mentioned in these studies. The expression levels of NRBF2, along with BECN1, PIK3C3 and PIK3R4, are reduced in Alzheimer disease (AD) post mortem brains [90]. A bioinformatics study suggested that gene expression of RBM8A, one of the exon junction complex (EJC) subunits, is reduced in AD, which is associated with downregulation of NRBF2, PIK3R4, and BECN1 [91]. For more than two decades, amyloid plaque, composed of amyloid beta (Aβ), has served as a hallmark of AD. NRBF2 is involved in the degradation of Aβ and amyloid beta precursor protein C-terminal fragments (APP-CTFs) in cell models and mice models for AD [39,42,90]. Although this is beyond the scope of this review, the majority of clinical trials targeting Aβ as an AD treatment have failed even when amyloid plaques are eliminated by drug treatment. This indicates that Aβ could be a clinical feature, rather than a cause of the disease [92,93]. In sharp contrast, Aβ has also been shown to have antimicrobial properties [94–96]. Loss of NRBF2 also causes memory and long-term potentiation deficits in mice [90,97]. It is not clear whether these learning and memory impairments also depend on complex I or not.
NRBF2 in health
As mentioned above, NRBF2 mRNA expression is high in the blood and muscles. NRBF2 mRNA expression level is increased after cycling exercise [98]. A significant number of single nucleotide polymorphisms (SNPs) were found in the NRBF2 gene of Tibetan pigs, which are adapted to high altitude, compared to Wuzhishan pigs, a low altitude control breed [99]. It has also been reported that hypoxia induces SUMOylation of PIK3C3, which enhances the assembly of complex I [100]. It will be interesting to see whether NRBF2 is actively involved in this assembly or not.
The SNPs in NRBF2 were also found to be associated with plasma fatty acid metabolism, especially γ-linoleic acid, one of the Omega6 (n6) polyunsaturated fatty acids (PUFAs) [101,102].
Concluding remarks
In spite of its stable interaction with complex I, nrbf2 KO phenotypes in cultured cells and mice are less severe than the strong KO phenotypes of the other complex I subunits. This indicates that NRBF2/Atg38 may become important at specific steps in autophagy, or in specific organs in mammals – as indicated by its potential involvement in IBD. Alternatively, NRBF2/Atg38 may become essential in the absence of some other components. As mentioned above, several discrepancies such as the identity of the NRBF2/Atg38-binding subunit(s) in complex I, activation/inhibition of complex I by NRBF2, and the binding stoichiometry are likely to be derived from the methods and experimental conditions that the researchers employed. Also, the NRBF2 effect on the GEF activity of the CCZ1-MON1 complex has been examined only by using immunoprecipitated MON1-CCZ1 complex [42,44]. The GEF assays will be ideally performed with purified recombinant CCZ1-MON1 complex to exclude the possibility of the contribution of other endogenous proteins coimmunoprecipitated with the MON1-CCZ1 complex to the GEF activity. I hope this review will help raise awareness of these issues for researchers so that they might be solved by future research. Additionally, there have been no hereditary diseases reported for which NRBF2 could be responsible. The recent and rapid development of information technology and the development of human disease databases may be able to reveal these.
Acknowledgments
I would like to thank Shirley Tremel, Anna Howes, and Roger L. Williams for critically reading the manuscript.
Funding Statement
This work was supported by the Medical Research Council [MC_U105184308].
Disclosure statement
The authors report no conflict of interest.
Abbreviations
| AD | Alzheimer disease |
| ATG | autophagy related |
| ATG14 | autophagy related 14 |
| Atg38 | autophagy related 38 |
| BECN1 | beclin 1 |
| BMDM | bone marrow-derived macrophages |
| CORVET | class C core vacuole/endosome tethering |
| Cryo-EM | cryo-electron microscopy |
| HDX-MS | hydrogen-deuterium exchange mass-spectrometry |
| HOPS | homotypic fusion and vacuolar protein sorting complex |
| IBD | inflammatory bowel disease |
| IP | immunoprecipitation |
| LAP | LC3-associated phagocytosis |
| LAPosome | LAP-engaged phagosome |
| MIT | microtubule-interacting and targeting |
| MTORC1 | mechanistic target of rapamycin kinase complex 1 |
| NRBF2 | nuclear receptor binding factor 2 |
| PAS | phagophore assembly site |
| PIK3C3 | class III phosphatidylinositol 3-kinase |
| PIK3R4 | phosphoinositide-3-kinase regulatory subunit 4 |
| PtdIns3K | class III phosphatidylinositol 3-kinase |
| PTM | post-translational modification |
| ROS | reactive oxygen species |
| TLC | thin layer chromatography |
| ULK1 | unc-51 like autophagy activating kinase |
| UVRAG | UV radiation resistance associated |
| VPS | vacuolar protein sorting |
References
- [1].Yasumo H, Masuda N, Furusawa T, et al. Nuclear receptor binding factor-2 (NRBF-2), a possible gene activator protein interacting with nuclear hormone receptors. Biochim Biophys Acta. 2000;1490(1–2):189–197. [DOI] [PubMed] [Google Scholar]
- [2].Flores AM, Li L, Aneskievich BJ.. Isolation and functional analysis of a keratinocyte-derived, ligand-regulated nuclear receptor comodulator. J Invest Dermatol. 2004;123(6):1092–1101. [DOI] [PubMed] [Google Scholar]
- [3].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Araki Y, Ku WC, Akioka M, et al. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol. 2013;203(2):299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schu PV, Takegawa K, Fry MJ, et al. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260(5104):88–91. [DOI] [PubMed] [Google Scholar]
- [6].Cao Y, Wang Y, Abi Saab WF, et al. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem J. 2014;461(2):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lu J, He L, Behrends C, et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun. 2014;5(1):3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhong Y, Morris DH, Jin L, et al. Nrbf2 protein suppresses autophagy by modulating Atg14L protein-containing Beclin 1-Vps34 complex architecture and reducing intracellular phosphatidylinositol-3 phosphate levels. J Biol Chem. 2014;289(38):26021–26037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Young LN, Cho K, Lawrence R, et al. Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc Natl Acad Sci U S A. 2016;113(29):8224–8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ohashi Y, Soler N, Garcia Ortegon M, et al. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy. 2016;12(11):2129–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ma X, Zhang S, He L, et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy. 2017;13(3):592–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ohashi Y, Tremel S, Masson GR, et al. Membrane characteristics tune activities of endosomal and autophagic human VPS34 complexes. Elife. 2020;9. 10.7554/eLife.58281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rostislavleva K, Soler N, Ohashi Y, et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science. 2015;350(6257):aac7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Masson GR, Burke JE, Ahn NG, et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat Methods. 2019;16(7):595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Henderson R. From electron crystallography to single particle CryoEM (nobel lecture). Angew Chem Int Ed Engl. 2018;57(34):10804–10825. [DOI] [PubMed] [Google Scholar]
- [16].Ma M, Liu JJ, Li Y, et al. Cryo-EM structure and biochemical analysis reveal the basis of the functional difference between human PI3KC3-C1 and -C2. Cell Res. 2017;27(8):989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Young LN, Goerdeler F, Hurley JH. Structural pathway for allosteric activation of the autophagic PI 3-kinase complex I. Proc Natl Acad Sci U S A. 2019;116(43):21508–21513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chang C, Young LN, Morris KL, et al. Bidirectional control of autophagy by BECN1 BARA domain dynamics. Mol Cell. 2019;73(2):339–53 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stjepanovic G, Baskaran S, Lin MG, et al. Vps34 kinase domain dynamics regulate the autophagic PI 3-kinase complex. Mol Cell. 2017;67(3):528–34 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baskaran S, Carlson LA, Stjepanovic G, et al. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. Elife. 2014;3. 10.7554/eLife.05115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang W, Choi W, Hu W, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22(3):473–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fan W, Nassiri A, Zhong Q. Autophagosome targeting and memxbrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci U S A. 2011;108(19):7769–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kihara A, Noda T, Ishihara N, et al. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Devereaux K, Dall’Armi C, Alcazar-Roman A, et al. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PLoS One. 2013;8(10):e76405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].van Meer G, de Kroon AI. Lipid map of the mammalian cell. J Cell Sci. 2011;124(1):5–8. [DOI] [PubMed] [Google Scholar]
- [26].van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cai CZ, Yang C, Zhuang XX, et al. NRBF2 is a RAB7 effector required for autophagosome maturation and mediates the association of APP-CTFs with active form of RAB7 for degradation. Autophagy. 2020;1–19. 10.1080/15548627.2020.1760623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ganley IG, Lam Du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy. 2013;9(12):1983–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. [DOI] [PubMed] [Google Scholar]
- [35].Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Matsunaga K, Morita E, Saitoh T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190(4):511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Noda T, Matsunaga K, Yoshimori T. Atg14L recruits PtdIns 3-kinase to the ER for autophagosome formation. Autophagy. 2011;7(4):438–439. [DOI] [PubMed] [Google Scholar]
- [38].Itakura E, Kishi C, Inoue K, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun Q, Fan W, Chen K, et al. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105(49):19211–19216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang C, Cai CZ, Song JX, et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy. 2017;13(12):2028–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yu ZQ, Sun LL, Jiang ZD, et al. Atg38-Atg8 interaction in fission yeast establishes a positive feedback loop to promote autophagy. Autophagy. 2020;16(11):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jager S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(20):4837–4848. [DOI] [PubMed] [Google Scholar]
- [44].Wu MY, Liu L, Wang EJ, et al. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy. 2020;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kuchitsu Y, Homma Y, Fujita N, et al. Rab7 knockout unveils regulated autolysosome maturation induced by glutamine starvation. J Cell Sci. 201131(7):1–10. [DOI] [PubMed] [Google Scholar]
- [46].Elkin SR, Lakoduk AM, Schmid SL. Endocytic pathways and endosomal trafficking: a primer. Wien Med Wochenschr. 2016;166(7–8):196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010;464(7289):778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gerondopoulos A, Langemeyer L, Liang JR, et al. BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol. 2012;22(22):2135–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Nordmann M, Cabrera M, Perz A, et al. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol. 2010;20(18):1654–1659. [DOI] [PubMed] [Google Scholar]
- [50].Poteryaev D, Datta S, Ackema K, et al. Identification of the switch in early-to-late endosome transition. Cell. 2010;141(3):497–508. [DOI] [PubMed] [Google Scholar]
- [51].Solinger JA, Spang A. Tethering complexes in the endocytic pathway: CORVET and HOPS. Febs J. 2013;280(12):2743–2757. [DOI] [PubMed] [Google Scholar]
- [52].Balderhaar HJ, Ungermann C. CORVET and HOPS tethering complexes - coordinators of endosome and lysosome fusion. J Cell Sci. 2013;126(6):1307–1316. [DOI] [PubMed] [Google Scholar]
- [53].Christoforidis S, Miaczynska M, Ashman K, et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol. 1999;1(4):249–252. [DOI] [PubMed] [Google Scholar]
- [54].Buckles TC, Ohashi Y, Tremel S, et al. Regulation of a class iii pi3k lipid kinase (VPS34 Complex II) by the small G protein Rab5A: single molecule analysis reveals a dual regulatory mechanism. Biophys J. 2020;(11). doi: 10.1016/j.bpj.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Peplowska K, Markgraf DF, Ostrowicz CW, et al. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev Cell. 2007;12(5):739–750. [DOI] [PubMed] [Google Scholar]
- [56].Pawelec A, Arsic J, Kolling R. Mapping of Vps21 and HOPS binding sites in Vps8 and effect of binding site mutants on endocytic trafficking. Eukaryot Cell. 2010;9(4):602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Markgraf DF, Ahnert F, Arlt H, et al. The CORVET subunit Vps8 cooperates with the Rab5 homolog Vps21 to induce clustering of late endosomal compartments. Mol Biol Cell. 2009;20(24):5276–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Brocker C, Kuhlee A, Gatsogiannis C, et al. Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci U S A. 2012;109(6):1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hunter MR, Hesketh GG, Benedyk TH, et al. Proteomic and biochemical comparison of the cellular interaction partners of human VPS33A and VPS33B. J Mol Biol. 2018;430(14):2153–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liang C, Lee JS, Inn KS, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25(8):1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kuma A, Komatsu M, Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017;13(10):1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nemazanyy I, Blaauw B, Paolini C, et al. Defects of Vps15 in skeletal muscles lead to autophagic vacuolar myopathy and lysosomal disease. EMBO Mol Med. 2013;5(6):870–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380(9853):1590–1605. [DOI] [PubMed] [Google Scholar]
- [65].Larabi A, Barnich N, Nguyen HTT. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. 2020;16(1):38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].O’Farrell F, Lobert VH, Sneeggen M, et al. Class III phosphatidylinositol-3-OH kinase controls epithelial integrity through endosomal LKB1 regulation. Nat Cell Biol. 2017;19(12):1412–1423. [DOI] [PubMed] [Google Scholar]
- [67].Zhao S, Xia J, Wu X, et al. Deficiency in class III PI3-kinase confers postnatal lethality with IBD-like features in zebrafish. Nat Commun. 2018;9(1):2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Botelho RJ, Grinstein S. Phagocytosis. Curr Biol. 2011;21(14):R533–8. [DOI] [PubMed] [Google Scholar]
- [69].Uribe-Querol E, Rosales C. Control of phagocytosis by microbial pathogens. Front Immunol. 2017;8:1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vieira OV, Botelho RJ, Rameh L, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155(1):19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kinchen JM, Doukoumetzidis K, Almendinger J, et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol. 2008;10(5):556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Langemeyer L, Borchers AC, Herrmann E, et al. A conserved and regulated mechanism drives endosomal Rab transition. Elife. 2020;9. 10.7554/eLife.56090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. [DOI] [PubMed] [Google Scholar]
- [74].Martinez J, Malireddi RK, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [75].Martinez J. LAP it up, fuzz ball: a short history of LC3-associated phagocytosis. Curr Opin Immunol. 2018;55:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mizoguchi E, Low D, Ezaki Y, et al. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intest Res. 2020;18(2):151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Poillet-Perez L, White E. Role of tumor and host autophagy in cancer metabolism. Genes Dev. 2019;33(11–12):610–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. [DOI] [PubMed] [Google Scholar]
- [79].Laddha SV, Ganesan S, Chan CS, et al. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res. 2014;12(4):485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ohashi Y, Tremel S, Williams RL. VPS34 complexes from a structural perspective. J Lipid Res. 2019;60(2):229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Bi WL, Greenwald NF, Abedalthagafi M, et al. Genomic landscape of high-grade meningiomas. NPJ Genom Med. 2017;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6(1):6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485(7399):502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Darabi H, McCue K, Beesley J, et al. Polymorphisms in a putative enhancer at the 10q21.2 breast cancer risk locus regulate NRBF2 expression. Am J Hum Genet. 2015;97(1):22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Zhang T, Dong K, Liang W, et al. G-protein-coupled receptors regulate autophagy by ZBTB16-mediated ubiquitination and proteasomal degradation of Atg14L. Elife. 2015;4:e06734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wold MS, Lim J, Lachance V, et al. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington’s disease models. Mol Neurodegener. 2016;11(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Sanchez-Martin P, Lahuerta M, Viana R, et al. Regulation of the autophagic PI3KC3 complex by laforin/malin E3-ubiquitin ligase, two proteins involved in Lafora disease. Biochim Biophys Acta Mol Cell Res. 2020;1867(2):118613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lachance V, Wang Q, Sweet E, et al. Autophagy protein NRBF2 has reduced expression in Alzheimer’s brains and modulates memory and amyloid-beta homeostasis in mice. Mol Neurodegener. 2019;14(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zou D, Li R, Huang X, et al. Identification of molecular correlations of RBM8A with autophagy in Alzheimer’s disease. Aging (Albany NY). 2019;11(23):11673–11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Liu PP, Xie Y, Meng XY, et al. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther. 2019;4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Modrego P, Lobo A. A good marker does not mean a good target for clinical trials in Alzheimer’s disease: the amyloid hypothesis questioned. Neurodegener Dis Manag. 2019;9(3):119–121. [DOI] [PubMed] [Google Scholar]
- [94].Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kumar DK, Choi SH, Washicosky KJ, et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid-beta peptide hint at new ways to treat Alzheimer’s disease. Front Aging Neurosci. 2018;10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ouyang X, Ahmad I, Johnson MS, et al. Nuclear receptor binding factor 2 (NRBF2) is required for learning and memory. Lab Invest. 2020;100(9):1238–1251. [DOI] [PubMed] [Google Scholar]
- [98].Mahoney DJ, Parise G, Melov S, et al. Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. Faseb J. 2005;19(11):1498–1500. [DOI] [PubMed] [Google Scholar]
- [99].Dong K, Yao N, Pu Y, et al. Genomic scan reveals loci under altitude adaptation in Tibetan and Dahe pigs. PLoS One. 2014;9(10):e110520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Yao Y, Li H, Da X, et al. SUMOylation of Vps34 by SUMO1 promotes phenotypic switching of vascular smooth muscle cells by activating autophagy in pulmonary arterial hypertension. Pulm Pharmacol Ther. 2019;55:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Guan W, Steffen BT, Lemaitre RN, et al. Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ Cardiovasc Genet. 2014;7(3):321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hu Y, Li H, Lu L, et al. Genome-wide meta-analyses identify novel loci associated with n-3 and n-6 polyunsaturated fatty acid levels in Chinese and European-ancestry populations. Hum Mol Genet. 2016;25(6):1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]