

Abstract
Runt-related transcription factor 1 (RUNX1), which is also known as acute myeloid leukemia 1 (AML1), has been frequently found with genomic aberrations in human leukemia. RUNX1 encodes a transcription factor that can regulate the expression of hematopoietic genes. In addition, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) performs an important function for malignant tumors in immune surveillance. However, the regulatory mechanism of TRAIL expression remain to be fully elucidated. In the present study, tetradecanoylphorbol 13-acetate-treated megakaryocytic differentiated K562 cells was used to examine the effect of RUNX1 on TRAIL expression. Luciferase assay series of TRAIL promoters for the cells co-transfected with RUNX1 and core-binding factor β (CBFβ) expression vectors were performed to evaluate the nature of TRAIL transcriptional regulation. Electrophoresis mobility shift assay of the RUNX1 consensus sequence of the TRAIL promoter with recombinant RUNX1 and CBFβ proteins was also performed. BloodSpot database analysis for TRAIL expression in patients with acute myeloid leukemia were performed. The expression of TRAIL, its receptor Death receptor 4 and 5 and RUNX1 in K562 cells transfected with the RUNX1 expression vector and RUNX1 siRNA were evaluated by reverse transcription-quantitative PCR (RT-qPCR). TRAIL and RUNX1-ETO expression was also measured in Kasumi-1 cells transfected with RUNX1-ETO siRNA and in KG-1 cells transfected with RUNX1-ETO expression plasmid, both by RT-qPCR. Cell counting, lactate dehydrogenase assay and cell cycle analysis by flow cytometry were performed on Kasumi-1, KG-1, SKNO-1 and K562 cells treated with TRAIL and HDAC inhibitors sodium butyrate or valproic acid. The present study demonstrated that RUNX1 is a transcriptional regulator of TRAIL. It was initially found that the induction of TRAIL expression following the megakaryocytic differentiation of human leukemia cells was RUNX1-dependent. Subsequently, overexpression of RUNX1 was found to increase TRAIL mRNA expression by activating its promoter activity. Additional analyses revealed that RUNX1 regulated the expression of TRAIL in an indirect manner, because RUNX1 retained its ability to activate this promoter following the mutation of all possible RUNX1 consensus sites. Furthermore, TRAIL expression was reduced in leukemia cells carrying the t(8;21) translocation, where the RUNX1-ETO chimeric protein interfere with normal RUNX1 function. Exogenous treatment of recombinant TRAIL proteins was found to induce leukemia cell death. To conclude, the present study provided a novel mechanism, whereby TRAIL is a target gene of RUNX1 and TRAIL expression was inhibited by RUNX1-ETO. These results suggest that TRAIL is a promising agent for the clinical treatment of t(8;21) AML.
Keywords: runt-related transcription factor 1, tumor-necrosis factor-related apoptosis inducing ligand, transcription, acute myeloid leukemia 1, apoptosis
Introduction
Numerous transcription factors have been found to mediate key roles in hematopoiesis (1). Runt-related transcription factor 1 (RUNX1), which is also known as acute myeloid leukemia (AML)1, is one of the key transcription factors for hematopoiesis. RUNX1 was first cloned from the breakpoint of the t(8;21) chromosome translocation in AML cases of the AML-M2 subtype of the French-American-British (FAB) classification (2). Chromosome aberrations and mutations in RUNX1 has been frequently reported to associate with leukemia pathogenesis (3,4). The t(8;12) translocation rearranges the RUNX1 locus to generate a fusion protein RUNX1-ETO (5), which trans-dominantly interferes with normal RUNX1 function to facilitate leukemogenesis (6,7). Point mutations in this locus that result in loss-of-function or dominant-negative mutations in this gene were frequently found in cases of sporadic AML, myelodysplastic syndrome/myeloproliferative neoplasms or pedigrees of familial platelet disorder with predisposition to AML development (8-12).
Conventional disruption experiments previously revealed that RUNX1 is required for the initial mid-gestation development of hematopoietic stem cells from the hemogenic endothelium (13-15). In addition, conditional knock-out and knock-in techniques applied in adult mouse models demonstrated the possible function of RUNX1 in regulating the size of circulating and bone marrow myeloid precursor cell populations, platelet production and T-/B-lymphoid development and function (16-19). RUNX1 encodes a DNA-binding subunit of the hetero-dimeric transcriptional factor complex known as core-binding factor (CBF) (20,21). The N-terminus of RUNX1, the Runt domain, is responsible for both association with the β subunit CBFβ and binding to DNA at the consensus sequence TGT/cGGT. (22). By contrast, domains at the C-terminus serve as the recruiting sites for regulatory co-factors (23,24).
RUNX1 performs its biological functions through the transcriptional regulation of its target genes, including colony-stimulating factor 1 (CSF1/M-CSF) receptor, hematopoietic transcription factors, such as PU.1, CCAAT/enhancer binding protein α, cell cycle-related genes, such as p14ARF and p21WAF1, microRNA (miR)-24, miR-223, IL-3 and granzyme B (25-28). RUNX1 expression was found to be upregulated during the differentiation of K562 leukemia cells following treatment with tetradecanoylphorbol 13-acetate (TPA) (29). TPA activates protein kinase C and promotes cancer, such as papilloma and squamous cell carcinoma, alongside the tumor initiator 7,12-dimetylbenzanthracene (DMBA) (30).
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a secretory and/or membrane-bound protein that has been extensively studied (31). TRAIL induces apoptosis by binding to its specific receptors, death receptor 5 (DR5) and DR4, on the target cell surface, where the resultant signal mediates TRAIL-induced apoptosis by interacting with Fas associated via death domain and activating caspases (32). TRAIL is a member of the TNF family of cytokines that induces apoptosis (32). Previously, TNF-α, a TNF family member, was reported to alter the lactate dehydrogenase (LDH) isotype in non-Hodgkin's lymphomas and cell surface CD marker profiles upstream of apoptosis induction in K562 cells (33,34). A number of previous studies also support the notion that TRAIL has the capability to preferentially kill tumor cells instead of non-cancerous normal cells, such that recombinant TRAIL is now under exploration in a number of on-going clinical trials for the treatment of malignant neoplasms (35,36). TRAIL efficacy was found to be increased by co-treatment with other therapeutic agents, such as histone deacetylase inhibitors (HDACis) in Jurkat T-cell leukemia cells (37). Histone deacetylases (HDACs) reduce histone acetylation and serve as transcriptional repressors (38). Oncoproteins, such as EWS-Fli1, aberrantly disrupt physiological transcriptional conditions by cooperating with HDACs (38). Therefore, HDACis can inhibit the function and expression of oncogenes and have potential as anti-tumor agents. To date, five HDACis, namely valproic acid (VPA), vorinostat, panobinostat, belinostat, and romidepsin, have been approved by the Food and Drug Administration in the United States for the treatment of cutaneous T-cell lymphomas, multiple myeloma or peripheral T-cell lymphomas (39). TRAIL is endogenously expressed by numerous cell types in the immune system, including natural killer (NK) cells, T lymphocytes, natural killer T cells, dendritic cells, and macrophages (40). This protein participates in the immune surveillance system against malignant tumors (40), because TRAIL- or TRAIL-R-deficient mice frequently develop lymphomas, exhibit xenografted carcinogen-induced tumors that grow faster and show increased lymph node metastases in xenograft models (41-43). Cloning of the promoter region of the human TRAIL gene revealed its structure, which contains putative binding sites for a number of transcription factors, including nuclear factor of activated T-cells, activator protein 1 (AP-1) and specificity protein 1 (Sp1) (44). Specificity protein 1, NF-κB, FOXO3a, c-Myc and p53 have all been reported to regulate the transcription of the TRAIL gene (45-47). However, details of the regulatory mechanisms underlying TRAIL expression remain to be fully elucidated. In the RUNX1-ETO-inducible U937 pro-monocytic leukemia cell line, RUNX1-ETO induction was reported to upregulate TRAIL expression (48). However, the effects of wild-type RUNX1 on TRAIL regulation under physiological conditions remain unclear.
In the present study, the transcriptional relationship between RUNX1 and TRAIL was investigated using RT-qPCR and luciferase reporter assays in a panel of leukemia cell lines. In addition, the biological function of TRAIL on leukemia cell lines was assessed.
Materials and methods
DNA and plasmid constructs
Hemagglutinin (HA)-tagged mouse RUNX1b encoded in the pRc/CMV plasmid (Invitrogen; Thermo Fisher Scientific, Inc.) was previously described (49). HA-tagged mouse RUNX1b was first digested by HindIII and EcoRI before being filled-in, where the complementary nucleotides were added to the 5′-overhang of the digest-end using DNA polymerase I (Klenow; New England Biolabs). The filled-in DNA fragment was then subjected to blunt-end ligation to the NotI-digested/filled-in site of the pRc/CMV plasmid. The sequence of the plasmid construct was analyzed and confirmed by Sanger sequencing using the 3500 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) and BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). FLAG-tagged mouse CBFβ (CBFβ-FLAG) encoded in pRc/CMV was also produced as previously described (10,50). The mouse CBFβ-FLAG was amplified by PCR using AmpliTaq Gold™ DNA polymerase (Thermo Fisher Scientific, Inc.) before being ligated into the TA-cloning site of the pCR2.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.). The CBFβ-FLAG DNA was then excised from the plasmid by XbaI and SpeI and subjected to the cohesive-end ligation at the XbaI site of the pRc/CMV plasmid. The sequence of the construct was analyzed and confirmed by Sanger sequencing using the 3500 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) and BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). Runx1 and CBFβ are highly homologous between human and mouse (51). The amino acid sequence of human RUNX1 holds 96.4% sequence identity to that of murine RUNX1 (accession no. NP_001001890.1 for human RUNX1; accession no. NP_001104493.1 for murine RUNX1). Similarly, the amino acid sequence of human CBFβ has 98.3% sequence identity to mouse CBFβ (accession no. NP_001746.1 for human CBFβ; accession no. NP_001154930.1 for mouse CBFβ). Mouse RUNX1 and CBFβ can both regulate human transcription of RUNX1 target genes, since ectopic human RUNX1 expression has been previously demonstrated to rescue the hematopoietic function lost in RUNX1-deficient mouse cells (23,52,53). Therefore, RUNX1 and CBFβ are considered to be functionally interchangeable between humans and mice, at least under these previous experimental conditions. RUNX1-ETO cDNA kindly donated by Dr. Misao Ohki (Chromatin Function in Leukemogenesis Project and Cancer Genomics Division, National Cancer Center Research Institute, Tokyo, Japan) was amplified with KpnI site-linked oligonucleotides (forward, 5′-GGG GTA CCG TGA TGC CGT ATC CCC GTA GAT GCC-3′ and reverse, 5′-GGG GTA CCC TAG CGA GGG GTT GTC TCT ATG GTG-3′) using PrimeSTAR GXL DNA polymerase (cat. no. R050A; Takara Bio Inc.) by PCR (98°C for 10 sec, followed by 30 cycles of 98°C for 10 sec, 60°C for 15 sec, 68°C for 60 sec, followed by 68°C 5 min). It was then digested by KpnI and subcloned into the KpnI site of the pcDNA3.1 plasmid (Invitrogen; Thermo Fisher Scientific Inc.). The sequence of construct was confirmed by Sanger sequencing using 3500 the Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) and BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). pcDNA3.1 and pRc/CMV are both mammalian expression vectors that express exogenous genes by using the same CMV promoter and enhancer sequences. Therefore, expression levels of the exogenous genes following transfection with these two vectors are considered to be comparable. However, the pcDNA3.1 plasmid contains more restriction enzyme sites in its multiple cloning site compared with pRc/CMV. To generate the pTRAIL PF reporter construct, the human wild-type TRAIL promoter obtained from the human genome library, a λPS library from human peripheral blood leukocytes (MoBiTec GmbH) using lambda phage λPS vector containing human genome DNA was inserted into the reporter plasmid pGV-B2, which contains a firefly luciferase gene (Toyo B-Net Co., Ltd.; https://www.toyo-b-net.co.jp/bio/product-pgv2.html). The human TRAIL promoter 5′-deletion mutants pTRAIL/-1523, pTRAIL/-819 and pTRAIL/-165 (40) were kindly donated by Dr B Mark Evers (Department of Surgery, University of Texas Medical Branch, Galveston, USA), which were inserted into the reporter plasmid pGL2m, derived from pGL2-basic by removal of the cryptic AP-1 site. Site-directed mutagenesis of the plasmids was performed using PCR-based methods. Mt1, where the wild-type sequence of ACCACA was changed to ATAACG and Mt2, where the wild-type sequences TGTGGT was changed to CGTTAT, were generated by site-directed mutagenesis (54) as aforementioned. The plasmid was amplified by inverse-PCR with oligonucleotide primers (Mt1 forward, 5′-GTA GCA TGA GAA AAA taA CgT ATG GAA GTT TCA G-3′ and reverse, 5′-CTG AAA CTT CCA TAc GTt aTT TTT CTC ATG CTA C-3′ and Mt2 forward, 5′-GAG GCT TAG AGC TCc GTt aTA GAA TGA GGA TAT G-3′ and reverse, 5′-CAT ATC CTC ATT CTA taA CgG AGC TCT AAG CCT C-3′) containing the planned mutation shown in small letters using PrimeStar GXL DNA polymerase (cat. no. R050A; Takara Bio Inc.), before the template plasmid was destroyed by digestion with DpnI. These amplified PCR products underwent spontaneous circularisation due to the 'sticky' 5′ ends in the primer sequences. These circularised DNA were used to transform competent DH5α E.coli cells to produce intact recombinant plasmids. The constructs were confirmed by Sanger sequencing using the 3500 Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.) and BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Cell Culture
HeLa cells (Riken BioResource Center) were cultured in DMEM (FUJIFILM Wako Pure Chemical Corporation) with 10% FBS (Sigma-Aldrich; Merck KGaA) and 2 mmol/l glutamine (Gibco; Thermo Fisher Scientific, Inc.). The leukemia cell line K562 was cultured in DMEM/F-12 (FUJIFILM Wako Pure Chemical Corporation) with 10% FBS and 2 mmol/l glutamine. The Kasumi-1 leukemia cell line (55) carrying t(8;21), which expresses the RUNX1-ETO fusion protein (kindly provided by Dr. Toshiya Inaba, Department of Molecular Oncology and Leukemia Program Project, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan) and KG-1, an AML cell line without RUNX1-ETO (Riken BioResource Center), were cultured in RPMI-1640 with 10% FBS. SKNO-1 leukemia cells carrying the t(8;21) translocation and express the RUNX1-ETO fusion protein, were obtained from the Japanese Collection of Research Bioresources Cell Bank and cultured in RPMI-1640 supplemented with 10% FBS and 10 ng/ml granulocyte macrophage (GM)-CSF (cat. no. 300-03; PeproTech, Inc.). All cells were maintained at 37°C in humidified air with 5% CO2.
Differentiation of K562 cells
K562 cells were treated with 25 nM TPA (cat. no. P8139; Sigma-Aldrich; Merck KGaA) for 48 h at 37°C (29), subjected to the centrifugation by cytospin-preparation at 78 × g for 6 min at room temperature (Shandon Cytospin 3 Centrifuge; Thermo Fisher Scientific, Inc.) and then processed with May-Gruenwald-Giemsa staining (Giemsa for 30 min at room temperature) followed by light microscopic examination with an Eclipse E800 optical microscope (Nikon Corporation). The magnification was ×400. The morphological changes of differentiation, including increased cell size and cytoplasm-to-nucleus ratio, formation of numerous vesicles and lobulated nuclei, was observed. TPA was dissolved in DMSO, which was added into control samples to a final concentration of 0.1% volume of medium.
Luciferase assay
Expression plasmids [RUNX1 in pRc/CMV (0.75 µg) and CBFβ in pRc/CMV (0.25 µg)] and reporter plasmids (1.25 µg; pTRAIL PF, pTRAIL/-1523, pTRAIL/-819, pTRAIL/-165, Mt1, Mt2 and pGL2m) were transfected into 5×104 HeLa and K562 cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). phRL-TK plasmid (0.0625 µg; cat. no. E6241; Promega Corporation) was co-transfected as an internal control. After incubation at 37°C for 48 h, the cells were lysed using passive lysis buffer contained in the Dual-Luciferase® Reporter Assay System (cat. no. E1960; Promega Corporation). Luciferase activities were measured using the Dual-Luciferase® Reporter Assay System with a luminometer (Lumat LB 9507; Titertek-Berthold). The cell lysate (20 µl) were mixed with luciferase assay reagent II (50 µl) and Photinus pyralis luciferase activity was measured with a luminometer. Stop & Glo reagent (50 µl) was then added to the mixture and Renilla reniformis luciferase activity driven by the TK promoter was measured with a luminometer. The luciferase activities were normalized by the activity of TK.
Transfections
For K562 cells (1×106), the expression plasmids for RUNX1 in pRc/CMV (3 µg) and for CBFβ in pRc/CMV (2 µg) or RUNX1 small interfering RNA (siRNA; 300 pmol) was transfected by electroporation (program T-016 of Nucleofector™ 2b device; Lonza Group, Ltd.) in 100 µl buffer contained within the AMAXA Cell Line Nucleofector Kit V (cat. no. VCA-1003; Lonza Group, Ltd.) in a cuvette equipped with aluminium electrodes of the kit at room temperature. Nucleofector™ 2b device and AMAXA Cell Line Nucleofector Kit V were used for the transfection of K562, Kasumi-1 and KG-1 cells. For Kasumi-1 cells (1×106), RUNX1-ETO siRNA (300 pmol) was transfected using program L-014 of Nucleofector™ 2b in 100 µl RPMI-1640. For KG-1 cells (1×106), RUNX1-ETO in pcDNA3.1 plasmid (2 µg) was transfected using program V-001 of Nucleofector™ 2b (Lonza) in 100 µl RPMI-1640. After electroporation, all cells were immediately collected with 500 µl RPMI1640 containing 10% FBS as recovery medium at room temperature. K562 cells were analyzed 48 h after RUNX1 and CBFβ transfection or 72 h after RUNX1 RNA siRNA transfection after electroporation. Kasumi-1 and KG-1 cells were analyzed 24 h after electroporation.
RUNX1 siRNA (assay ID, HSS141472; sense, 5′-UCU GGU CAC UGU GAU GGC UGG CAA U-3′ and antisense, 5′-AUU GCC AGC CAU CAC AGU GAC CAG A-3′) was purchased from Invitrogen; Thermo Fisher Scientific, Inc. Stealth RNAi™ siRNA Negative Control Hi GC duplex was used as negative control (cat. no. 12935400; Invitrogen, Thermo Fisher Scientific, Inc.). Whenever RUNX1 cDNA and CBFβ cDNA were transfected, the empty pRc/CMV plasmid was used as a negative control because RUNX1 cDNA and CBFβ cDNA were inserted into this vector.
For luciferase reporter assays, RUNX1 cDNA and CBFβ cDNA were expressed as effectors. Empty pRc/CMV was used as the corresponding negative control. RUNX1-ETO siRNA was synthesized by Eurofins Scientific (Sense, 5′-CCU CGA AAU CGU ACU GAG AAG-3′ and antisense, 5′-UCU CAG UAC GAU UUC GAG GUU-3′) and transfected. As a negative control, Stealth RNAi™ siRNA Negative Control Hi GC duplex was also used. RUNX1-ETO cDNA, which was inserted into the pcDNA3.1 plasmids, had empty pcDNA3.1 as the control vector.
Measurement of mRNA expression
Expression plasmids were transfected into K562 cells using Nucleofector™ 2b. After 48 h, total RNA was isolated from cells using Isogen (cat. no. 311-02501) or Isogen II (cat. no. 311-07361; both Nippon Gene Co., Ltd.). cDNA was generated using SuperScript III (cat. no. 18080044) or IV (cat. no. 18090010) reverse transcriptases (Thermo Fisher Scientific, Inc.), oligo (dT)15 primer (cat. no. 3805; Takara Bio, Inc.) and dNTP mixture (cat. no. 4030; Takara Bio, Inc.) at 55°C for 10 min followed by 80°C for 10 min. cDNA was quantified using Thunderbird™ SYBR™ qPCR mix (cat. no. QPS-201; Toyobo Life Science), specific primers synthesized by Eurofins and StepOnePlus (Thermo Fisher Scientific, Inc.). The thermocycling conditions were 95°C for 10 sec, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min, then 95°C for 15 sec, 60°C for 1 min and 95°C for 15 sec. Primer sequences are listed as follows: TRAIL forward, 5′-GAG AGT AGC AGC TCA CAT AAC T-3′ and reverse, 5′-TCA TGG ATG ACC AGT TCA CCA TT-3′; RUNX1 forward, 5′-CTG AGC CCA GGC AAG ATG AG-3′ and reverse, 5′-GTC TTG TTG CAG CGC CAG TG-3′; CBFβ forward, 5′-TCG AGA ACG AGG AGT TCT TCA GGA-3′ and reverse, 5′-AGG CGT TCT GGA AGC GTG TCT-3′; RUNX1/ETO forward, 5′-CAC TGT CTT CAC AAA CCC AC-3′ and reverse, 5′-ATG AAC TGG TTC TTG GAG CTC CTT-3′; DR4 forward, 5′-ACC TTC AAG TTT GTC GTC GTC-3′ and reverse, 5′-CCA AAG GGC TAT GTT CCC ATT-3′; DR5 forward, 5′-GCC CCA CAA CAA AAG AGG TC-3′ and reverse, 5′-AGG TCA TTC CAG TGA GTG CTA-3′; β-actin forward, 5′-GCT GTG CTA CGT CGC CCT G-3′ and reverse, 5′-GGA GGA GCT GGA AGC AGC C-3′ and GAPDH forward, 5′-TGC ACC ACC AAC TGC TTA GC-3′ and reverse, 5′-GGC ATG GAC TGT GGT CAT GAG-3′. β-actin and GAPDH were used as the reference genes. The quantification method used was relative standard curve (56).
Cell viability assay
The recombinant human soluble TRAIL protein was a lyophilized powder purchased from PeproTech, Inc. The recombinant TRAIL was dissolved in water, diluted in PBS and used for experimentation. Equivalent volumes of PBS was added in the control groups. Cells were treated with recombinant human soluble TRAIL for 3 days (at 10, 50, or 100 ng/ml) at 37°C, before cell numbers were counted using a Bürker-Turk counting chamber (Erma, Inc.) under an Olympus CK40 inverted light microscope (Olympus Corporation), capable of phase contrast observation, using ×100 magnification. Lactate dehydrogenase (LDH) assay was performed using the Cytotoxicity LDH Assay Kit-WST (cat. no. CK12; Dojindo Molecular Technologies, Inc.). Collected cells (2×105 cells) were suspended in PBS with 0.1% Triton X-100 and 10 µg/ml of propidium iodide (PI) at room temperature for 5 min, before DNA content was analyzed by BD FACS Canto II and BD FACSDiva software v6.1.3 (BD Biosciences) (57). TRAIL was co-treated with 1 mM sodium butyrate (NaB; Sigma-Aldrich; Merck KGaA), 2 mM VPA (cat. no. 197-09722; FUJIFILM Wako Pure Chemical Corporation) and 20 µM caspase inhibitor z-VAD-fmk (cat. no. 3175-v; Peptide Institute, Inc.) for 72 h at 37°C (37). NaB and VPA were dissolved in distilled water, whilst z-VAD-fmk was dissolved in DMSO. When zVAD-fmk was used, solvent DMSO was added in control samples and the final concentration of DMSO was 0.1% volume of medium.
Preparation of recombinant proteins
To produce hexa-histidine (His6)-tagged RUNX1 proteins, partial cDNA encoding the Runt domain (amino acids 24-189) of mouse RUNX1b originally in the pRc/CMV plasmid was amplified by PCR using PrimeSTAR GXL DNA polymerase (cat. no. R050A; Takara Bio Inc.), digested by KpnI and inserted into the pQE30 plasmid (Qiagen, Inc.). To generate His6-tagged CBFβ, full length mouse CBFβ originally in the pRc/CMV plasmid was amplified by PCR using PrimeSTAR GXL DNA polymerase (cat. no. R050A; Takara Bio Inc.), digested by KpnI and inserted into the pET3a vector (Qiagen, Inc.). His6-tagged proteins of RUNX1 containing the Runt domain (amino acids 24-189) and CBFβ were expressed in the XL-1 Blue strain of E.coli and resuspended in a buffer (4 ml) containing 0.1 M sodium phosphate, 10 mM Tris, 6 M guanidine-HCl, 0.1% NP-40, 1 mM PMSF and 10 mM 2-mercaptoethanol and adjusted to Ph 8.0 with 6 N NaOH. The total volume of the lysate was mixed with 300 µl Ni-NTA agarose beads (>15 µmol Ni2+/ml gel; 50% slurry; cat. no. 145-09681; FUJIFILM Wako Pure Chemical Corporation) for 1 h at 4°C and purified using a 100-ml syringe (cat. no. 08-649; Nipro Medical Corporation) with 0.34 mm chromatography paper (cat. no. 3030917; Cytiva) as a column. The column was washed with buffer A plus 0.8 mM imidazole and His-tagged proteins were eluted at 4°C in buffer B consisting of buffer A with 250 mM imidazole and 20% glycerol. These procedures were performed as described previously (58). The eluted proteins (10 µl) were resolved on 12.5% SDS-PAGE and stained with Coomassie brilliant blue staining. The His-tagged protein (His-tagged RUNX1, 18 kDa; His-tagged CBFβ, 21 kDa) amounts were estimated by comparing with the BSA (66 kDa; Sigma-Aldrich; Merck KGaA) band signal intensity visually on the SDS-PAGE gel.
Electrophoresis mobility shift assay (EMSA)
EMSA was performed using the LightShift Chemiluminescent EMSA kit (cat. no. 20148 and 89880; Thermo Fisher Scientific, Inc.). DNA probes (Eurofins Scientific) were prepared by annealing the following oligonucleotides: Control (CT), biotin-5′-GTC TGT GGT TTC TGT GGT TTC TGT GGT TT-3′ and 5′-AAA CCA CAG AAA CCA CAG AAA CCA CAG AC-3′; Wild-type (WT)1, biotin-5′-GAA AAA CCA CAT ATG G-3′ and 5′-CCA TAT GTG GTT TTT C-3′; mutant (MT)1, biotin-5′-GAA AAA TAA CGT ATG G-3′ and 5′-CCA TAC GTT ATT TTT C-3′; WT2, biotin-5′-AGC TCT GTG GTA GAA T-3′ and 5′-ATT CTA CCA CAG AGC T-3′ and MT2, biotin-5′-AGC TCC GTT ATA GAA T-3′ and 5′-ATTCTATAACGGAGCT-3′. The DNA probes (0.8 pmol) and recombinant His6-tagged RUNX1 (15 ng) and His6-tagged CBFβ (60 ng) were reacted in 1X binding buffer (cat. no. 20148A) and 50 ng/ml poly (dI-dC; cat. no. 20148E) supplied by the manufacturer in a 10-µl reaction volume at 15°C for 30 min. The reaction mixture was resolved on a 10% polyacrylamide gel in 0.5X TBE buffer at 4°C and transferred onto a nylon membrane (cat. no. 10416196; Cytiva), fixed using a UV cross-linker (UV Stratalinker 2400 (Stratagene; Agilent Technologies, Inc.) under the auto cross-link condition of 1.2 mJ (×100), 25-50 sec at room temperature and then reacted with streptavidin-HRP (cat. no. 89880) for 15 min at room temperature. The signal was detected using the Chemiluminescent substrate solution (cat. no. 89880) using an ImageQuant LAS 500 (Cytiva).
Bioinformatics analysis
TRAIL expression in bone marrow samples from patients with AML was analyzed by surveying the BloodPool: AML samples with normal cells data set of the BloodSpot (https://servers.binf.ku.dk/bloodspot/) database (59) using data of GSE42519 (60), GSE13159 (61,62), GSE15434 (63), GSE61804 (64), GS E14468 (65-67) and The Cancer Genome Atlas (TCGA) database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga).
Statistical analysis
Experiments were repeated three times and statistically analyzed using a two-tailed, unpaired Student's t-test by Microsoft Excel 2016 or 2019 (Microsoft Corporation) or one-way ANOVA followed by Tukey's post hoc test by RStudio Desktop 2021.09.1+372 (RStudio; https://www.rstudio.com). Samples were adjudged to be significantly different at P<0.05. The results were presented as the mean ± standard deviation.
Results
TRAIL expression is increased after K562 differentiation induced by TPA stimulation in a RUNX1-dependent manner
The human leukemia cell line K562 can be induced to differentiate into cells of a megakaryocytic lineage, where RUNX1 expression was found to be upregulated during this process (29). To investigate if TRAIL expression was also affected by this differentiation process, K562 cells was stimulated with 25 nM tetradecanoylphorbol 13-acetate (TPA) for 48 h, following which morphological changes, including increased cell size and cytoplasm-to-nucleus ratio, many vesicles and lobulated nuclei, were observed (Fig. 1A). TRAIL expression was next measured in these cells. TRAIL expression was significantly increased after TPA stimulation, which appeared to be at least partially dependent on RUNX1 (Fig. 1B), since RUNX1 knockdown significantly reversed the TPA-induced increase in TRAIL. In the presence of TPA, RUNX1 expression was also significantly reduced by transfection with siRNA targeting RUNX1 compared with that in cells transfected with negative siRNA (Fig. 1C). The effects of RUNX1 siRNA transfection on K562 cell viability and cell cycle progression without TPA treatment was subsequently examined. RUNX1 siRNA did not alter cell viability or cell cycle progression compared with those in cells after control siRNA transfection in this experimental condition (Fig. S1A and B). In addition, TRAIL transcriptional induction in K562 cells after differentiation may be dependent on increased RUNX1 expression. However, the effects of RUNX1 siRNA were negligible without TPA treatment, which could be explained by the low basal endogenous RUNX1 expression levels in K562 cells.
Figure 1.

TRAIL expression is increased after K562 differentiation model in a RUNX1-dependent manner. (A) May-Gruenwald-Giemsa staining of the TPA-treated K562 cells. The cells were treated with TPA, attached to glass slides using Cytospin 3 and examined using May-Gruenwald-Giemsa staining. Scale bars, 100 µm. (B and C) RUNX1 siRNA was transfected into K562 cells by electroporation. After 24 h, the cells were treated with TPA for 48 h for differentiation after which the relative expression of (B) TRAIL and (C) RUNX1 was measured by RT-qPCR using actin for normalization. (D and E) RUNX1 and CBFβ expression plasmids were co-transfected into K562 cells by electroporation. After 48 h, total RNA was prepared and the relative expression of (D) TRAIL and (E) RUNX1 was measured by RT-qPCR using actin for normalization. **P<0.01. TPA, tetradecanoylphorbol 13-acetate; siRNA, small interfering RNA; RUNX1, Runt-related transcription factor 1; CBFβ, core-binding factor β; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; RT-qPCR, reverse transcription-quantitative PCR.
To verify the dependency of this observed TRAIL transcriptional induction on RUNX1 expression, mouse RUNX1 was exogenously expressed in K562 cells. The transfection efficiency of the individual RUNX1 and CBFβ plasmids into K562 cells was first confirmed by the observed increased RUNX1 and CBFβ expression compared with that in their corresponding control plasmid-transfected cells (Fig. S1C). The transfection efficiency in K562 cells was ~80%. As shown in Fig. 1D and E, ectopic expression of mouse RUNX1 along-side its dimerization partner, CBFβ, into K562 cells resulted in a significant increase in endogenous TRAIL and RUNX1 mRNA expression. However, the expression of the TRAIL receptors DR4 and DR5 was not affected by ectopic RUNX1 and CBFβ expression (Fig. S1D). Therefore, these data suggest that TRAIL expression induction is dependent on RUNX1 expression.
RUNX1 activates the TRAIL promoter
To determine if RUNX1 can activate TRAIL transcription through the TRAIL promoter sequences, promoter constructs of human TRAIL was subjected to a series of luciferase reporter assays. Human TRAIL promoter sequences containing 1523 bases upstream of the transcriptional start site of the TRAIL gene (Fig. 2A) were cloned as previously described (44), where it was established that this fragment can function as a regulatory cis-element (44). Although the transcription of a number of TRAIL variants was reported to start ~30 bp downstream of the start site, the previous report (44) stated the transcription start site to be at the +1 position of the TRAIL promoter. Luciferase reporter assay using this DNA fragment of pTRAIL PF revealed that co-expression with RUNX1 and its co-factor CBFβ significantly activated the human TRAIL promoter (Fig. 2B). The transfection efficiency in HeLa cells was ~30%. Since the effector plasmids and reporter plasmids were co-transfected into the cells, sufficient signal values were detected even though the transfection efficiency was low. This activation occurred in a CBFβ-dependent manner, because ectopic transfection with either CBFβ or RUNX1 alone only weakly and insignificantly activated TRAIL transcription, unlike when both were transfected together (Fig. 2C). Therefore, the RUNX1/CBFβ combination may function as a transcription factor complex of the human TRAIL gene. This is consistent with a previous report, which stated that RUNX1 encodes a DNA-binding subunit of the CBF complex (20,21), where the CBF complex requires both RUNX1 and CBFβ to fully function as a transcription factor.
Figure 2.

RUNX1 upregulates TRAIL transcription independent of the consensus RUNX1 sequences on the TRAIL promoter. (A) Nucleotide sequence of the human TRAIL promoter element are depicted. RUNX1-binding consensus sequences, ACCACA and TGTGGT, are shown in red boxes (Sites 1 and 2). The transcription start site is indicated by the red arrow. (B and C) RUNX1 increased TRAIL promoter activity. (B) RUNX1 and CBFβ expression plasmids were transfected alongside the TRAIL promoter-luciferase reporter plasmid (pTRAIL) into HeLa cells. At 48 h post-transfection, cell lysates were prepared and luciferase assays were performed. (C) The RUNX1 and/or CBFβ plasmids were transfected alongside the pTRAIL-promoter construct in HeLa cells before luciferase activities were measured. Results were normalized to Renilla luciferase driven by the TK promoter. (D) Site-directed mutagenesis of sites 1 and 2 was used to generate mutant 1 and mutant 2, respectively. (E) pTRAIL/-819 luciferase constructs encoding the TRAIL promoter with the Mt1 or Mt2 mutations were transfected into HeLa cells alongside the RUNX1 and CBFβ expression plasmids, before their activities were measured and compared with those of the pTRAIL construct pTRAIL/-819. pGL2m, an empty reporter plasmid, was used as the control. (F) Electrophoresis mobility shift assay was performed to detect the formation RUNX1 protein-DNA complexes. Recombinant His-tagged RUNX1 and His-tagged CBFβ produced in E. coli were incubated with probes containing sequences shown in (D). CT is a control probe with the RUNX1 consensus sequence. The positions for of the protein-DNA complexes (shift bands) and free probes are indicated on the right side of gel image. *P<0.05 and **P<0.01. RUNX1, Runt-related transcription factor 1; CBFβ, core-binding factor β; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; WT, wild-type; Mt, mutant.
Consensus RUNX1 sequences on the TRAIL promoter do not affect RUNX1-mediated TRAIL transcription
RUNX1 functions as a transcription factor by specifically binding to DNA at the TGT/cGGT consensus sequences (22). As shown in Fig. 2A, two consensus sites were found in the complete human TRAIL promoter sequence (Fig. 2A). To analyze if these sites are important for the regulatory effects of RUNX1 on TRAIL expression, mutations were introduced into these sites by PCR-based site-directed mutagenesis (Fig. 2D), with the aim of interfering with RUNX1 binding to these mutated sequences. Luciferase reporter assays using these mutant constructs of the 819-bp TRAIL promoter sequences showed comparable transcriptional activation by RUNX1 between wild type pTRAIL/-819 and mutant types Mt1 and Mt2, indicating that neither site were important for TRAIL transcriptional regulation by RUNX1 (Fig. 2E).
Subsequently, EMSA was performed to directly assess if RUNX1 can bind to the promoter sequences used for Fig. 2E. RUNX1 was unable to bind to the mutant sequences, including MT1, MT2 and WT2 sequences (Fig. 2F). By contrast, RUNX1 protein was found to bind the WT1 sequence (Fig. 2F). However, this binding was not responsible for the transactivation of TRAIL promoter by RUNX1, since the luciferase activities of the MT1 construct, which is the corresponding mutant for WT1, was not affected (Fig. 2E).
To assess if these sequences are dispensable for the transcription control of TRAIL, deletion mutants of the TRAIL promoter sequences were subjected to luciferase reporter assay following the ectopic expression of RUNX1 and CBFβ. The transfection efficiency was ~30% in HeLa cells and ~10% in K562 cells. Luciferase in the deletion construct up to the -819 position was activated by the presence of the RUNX1/CBFβ transcription factor complex (Fig. 3A and B). Luciferase in the longer construct containing sequences up to the -1523 position was also activated (Fig. 3A and B). However, transcription of the shorter deletion mutant up to the -165 position was also activated by RUNX1 and CBFβ ectopic expression. These results were reproducible in both HeLa and K562 cells (Fig. 3A and B). Therefore, these consensus sites were concluded to be dispensable for the transcriptional activation of the human TRAIL gene by RUNX1.
Figure 3.
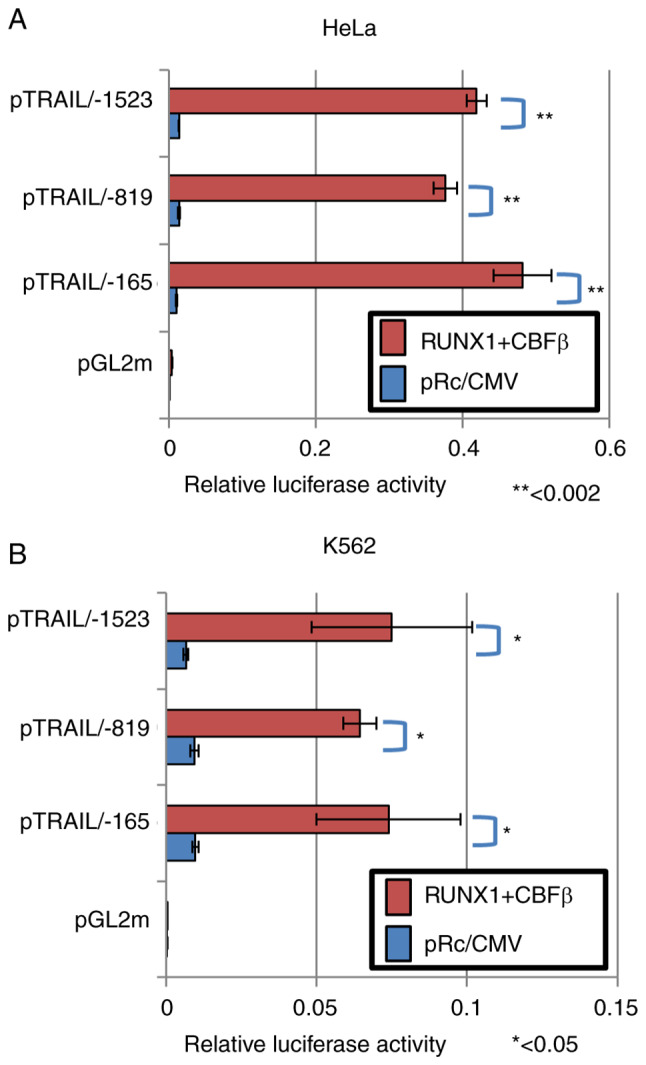
RUNX1 upregulates TRAIL transcription through the region -165 downstream juxtaposed to the transcription start site. RUNX1 and CBFβ expression plasmids were transfected alongside the luciferase reporter plasmids encoding the TRAIL promoter deletion mutants into (A) HeLa cells and (B) K562 cells. At 48 h post-transfection, cell lysates were prepared and luciferase assays were performed. Results were normalized to Renilla luciferase driven by the TK promoter. Empty vectors pRc/CMV and pGL2m were used as controls. RUNX1, Runt-related transcription factor 1; CBFβ, core-binding factor β; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand. *P<0.05 and **P<0.005.
TRAIL expression is reduced in leukemia cells with the RUNX1-ETO protein
The aforementioned findings gave rise to the investigation into how TRAIL expression is regulated in cells where RUNX1 function is impaired. TRAIL expression in patients with t(8;21)AML was next measured. A public database of gene expression profiles of healthy individuals and patients with malignant hematopoiesis was analyzed through BloodSpot database (59) using data of GSE42519 (60), GSE13159 (61,62), GSE15434 (63), GSE61804 (64), GSE14468 (65-67) and TCGA database. TRAIL expression was found to be significantly decreased in AML samples with t(8;21) compared with that in other-type AML samples or normal cells (Fig. 4A), which were obtained from the bone marrow.
Figure 4.

TRAIL expression is reduced in t(8;21)AML patients and cell lines. (A) BloodSpot revealed that TRAIL expression is lower in the t(8;21) subtype of AML among other types of leukemia samples analyzed. The expression of TRAIL was analyzed by the Microarray Innovations in Leukemia (MILE) study GSE13159. (B) RT-qPCR results of RUNX1-ETO and TRAIL expression in Kasumi-1 and SKNO-1 cells compared with KG-1 cells. Data were standardized by GAPDH expression. (C) RUNX1-ETO expression was knocked down by RUNX1-ETO siRNA in Kasumi-1 cells. In total, 24 h after siRNA transfection, RUNX1-ETO and TRAIL mRNA levels were analyzed by RT-qPCR. Data were standardized by GAPDH expression. (D) After 24 h of RUNX1-ETO overexpression in KG-1 cells, RUNX1-ETO and TRAIL mRNA levels were analyzed by RT-qPCR and normalized to that of GAPDH expression. **P<0.01 and ***P<0.001. AML, acute myeloid leukemia; Normal, AML with a normal karyotype; Complex, AML with a complex karyotype; RT-qPCR, reverse transcriptionquantitative PCR; si, small interfering; RUNX1, Runt-related transcription factor 1; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Next, TRAIL expression in the Kasumi-1 and SKNO-1 t(8;21) AML cell lines, which express the RUNX1-ETO chimeric protein and trans-dominantly interfere with normal RUNX1 function (6). TRAIL mRNA expression was markedly significantly reduced in Kasumi-1 and SKNO-1 cells compared with that in the RUNX1-ETO-negative AML cell line KG-1 (Fig. 4B). RUNX1-ETO knockdown in Kasumi-1 cells significantly increased TRAIL expression compared with that in cells transfected with the control siRNA (Fig. 4C). The transfection efficiency was ~65%. In addition, RUNX1-ETO overexpression in KG-1 cells, which do not contain the t(8;21) translocation, significantly reduced TRAIL expression compared with that in cells transfected with the empty plasmid (Fig. 4D). The transfection efficiency was ~60%. This suggest that RUNX1-ETO negatively regulates TRAIL expression in AML cells with t(8;21).
Recombinant TRAIL exerts cytocidal effects on t(8;21) AML cells
It was subsequently found that treatment with the exogenous recombinant TRAIL protein suppressed the proliferation of Kasumi-1 cells. Treatment with the recombinant TRAIL protein for 3 days reduced the number of viable Kasumi-1 cells whilst increasing LDH release in a TRAIL concentration-dependent manner (Fig. 5A and B), suggesting that cell death was induced. Cell cycle profiling revealed that the sub-G1 phase fraction of Kasumi-1 cells was increased whereas the G1 phase cell fraction was decreased in a TRAIL concentration-dependent manner (Fig. 5C). These inhibitory effects mediated by TRAIL treatment were significantly reversed by caspase inhibitor zVAD-fmk treatment, suggesting that these effects were caused by increasing apoptosis. By contrast, these inhibitory, possibly apoptotic effects of TRAIL were significantly and weakly observed in K562 cells, which have no RUNX1 mutations (Fig. S2), but was not observed in KG-1 cells, which are negative for RUNX1-ETO expression (Fig. S3). Furthermore, another AML cell line, SKNO-1, which also expresses RUNX1-ETO, was found to have its viability reduced and sub-G1 population increased by exogenous TRAIL treatment, which was reversed by zVAD-fmk treatment (Fig. S4).
Figure 5.

Recombinant TRAIL treatment inhibits the proliferation of the RUNX1-ETO-expressing Kasumi-1 leukemia cells. (A-C) Kasumi-1 cells (1×105) were treated with recombinant TRAIL with or without zVAD-fmk for 3 days. (A) Cell count using the Bürker-Turk counting chamber and (B) Lactate dehydrogenase assay. (C) Cell cycle profiling by flow cytometry analysis revealed that the proportion of cells in the sub-G1 fraction was increased by recombinant TRAIL. zVAD-fmk was used to block apoptosis. Representative histograms (upper panels) and a bar graph (a lower panel) are presented. **P<0.01 and *P<0.05. TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Combination of TRAIL and HDACi efficiently kills Kasumi-1 cells
Inhibition of cell proliferation and increased sub-G1 Kasumi-1 cell populations were potentiated further after HDACis NaB or VPA, were combined with TRAIL (Fig. 6A and B). Since HDACis, including NaB and VPA, can upregulate DR5 expression and sensitize malignant human tumor cells such as Jurkat T-cell leukemia, to TRAIL-induced apoptosis (37), TRAIL signaling appeared to activate Kasumi-1 cell death. The combination of TRAIL and HDACis was found to exert superior effects on cell viability reduction and cell death induction compared with those mediated by either alone in a dose-dependent manner in Kasumi-1 cells (Fig. S5).
Figure 6.

Combination of TRAIL and HDAC inhibitors efficiently kills Kasumi-1 cells. (A and B) Kasumi-1 cells were treated with 100 ng/ml of TRAIL and/or HDAC inhibitors (1 mM NaB or 2 mM VPA) for 3 days. (A) Cell numbers were counted using the Bürker-Turk counting chamber. (B) Cell cycle profiling was analyzed by flow cytometry. **P<0.01. TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; HDAC, histone deacetylase; VPA, valproic acid.
Therefore, the effects of TRAIL on cell survival likely depend on the presence of the RUNX1-fusion protein.
Discussion
The present study demonstrated that TRAIL is a novel target gene of RUNX1, as supported by the observation that TRAIL expression was increased following TPA-induced K562 megakaryocytic differentiation, which is dependent on RUNX1. Previous reports stated that TRAIL is expressed in megakaryocytes and platelets, where it regulates megakaryocytic development (68-70). However, the upstream regulation mechanism of TRAIL expression remains unclear. The present study revealed that RUNX1 serves as a transcriptional regulator of TRAIL during leukemia cell differentiation. Increasing RUNX1-ETO expression was previously found to increased TRAIL expression in the RUNX1-ETO-inducible U937 cell line (48). However, the present study demonstrated that wild-type RUNX1 physiologically functioned as a positive regulator of TRAIL expression whereas RUNX1-ETO suppressed TRAIL expression in AML cells harboring t(8;21). This discrepancy in results may be caused by differences in the cellular background or experimental system. RUNX1 may indirectly upregulate TRAIL transcription without dependence on consensus RUNX1 binding sites in its promoter region. RUNX1 was previously found to regulate high mobility group AT-hook 2 promoter activity without the need for consensus sequences, likely by interacting with other intermediary factors (71), which supports the findings from the present study. The RNA sequencing approach should be applied in future studies to clarify this hypothesis, which can be used to visualize the network of transcription factors and signal transduction components within complex cellular system. This may shed light on the underlying mechanism of the RUNX1-TRAIL axis.
A number of transcription factors that can regulate TRAIL expression have been identified (45-47). The majority of these transcription factors regulate TRAIL expression by binding to their DNA sequence in the promoter region (45-47). By contrast, the present study suggests that RUNX1 can be categorized as a transcription factor that regulates TRAIL expression in an indirect manner. It will therefore be important to examine if this indirect mechanism also exists in the expression of other genes associated with hematopoietic malignancy.
RUNX1 was initially recognized as a transcription factor that mediates myeloid functions such as myeloperoxidase-induction and macrophage-colony stimulating factor 1-receptor upregulation (5,6). Further analyzes suggested that RUNX1 is also a regulator of T-cell activation (16,17). Previous studies using RUNX1 gene-modified mice demonstrated that RUNX1 serves an essential role in lymphoid function (16,17). One such finding was that granzyme B (28), which is a secreted protease that functions as an important effector molecule of activated cytotoxic T-cells, was identified as a RUNX1 target gene. In the present study, TRAIL was identified to be another RUNX1 target that functions as an important effector molecule for lymphoid function. The molecular mechanism of the immune system related to TRAIL should be investigated further with focus on the involvement of other members of the RUNX family.
Elucidation of the molecular mechanisms through which RUNX1 expression is induced by TPA stimulation in K562 cells remains a question that needs to be addressed. It has been extensively reported that RUNX1 has two promoters, proximal and distal, where this two-promoter organization is strongly conserved into target genes of the RUNX2 and RUNX3 families through evolution (50,72). Findings from the present study therefore serves as an initial starting point for further investigation into the mechanisms involved.
Since TRAIL preferentially kills cancer cells such as colon, lung, breast, central nervous system, skin and kidney cancers, recombinant TRAIL has been considered to be a promising anti-cancer agent (73). Although several types of malignancies remain resistant to TRAIL, induction of its receptor DR5, may also be a viable option for sensitizing these tumor cells to TRAIL (37,74,75). The present study in conjunction with BloodSpot data revealed that TRAIL expression is reduced in t(8;21) AML samples, suggesting that RUNX1 mutations caused the TRAIL downregulation. Furthermore, compensation of TRAIL expression reduced cell viability in Kasumi-1 and SKNO-1 cells, both of which carry the t(8;21) translocation. By contrast, the WT RUNX1-expressing AML cell line KG-1 and the chronic myelogenous leukemia cell line K562 were resistant to TRAIL treatment. K562 was previously reported to be resistant to TNF-α whereas NK cells caused K562 cell death (76). This suggest that K562 cells possess a common mechanism of death ligand resistance. Death ligands, such as TNF-α, can not only induce apoptosis, but can also induce cell proliferation depending on the receptor profile (77). Although TRAIL may also confer such a property, in the present study TRAIL only reduced cell viability and cell cycle progression. The possible proliferative effect of TRAIL warrants further investigation, especially in a clinical setting. In the present study, sub-G1 analysis was used to evaluate cell death, which was also previously used to analyze TRAIL function (37,55,64,65). Annexin V/PI staining is an alternative method that can be used to assess cell death, which distinguishes between apoptosis and necrosis. To confirm apoptosis induction in detail, Annexin V/PI assay accompanied by sub-G1 analysis should be performed and is a limitation of the present study.
The sensitivity of Kasumi-1 to TRAIL was found to be increased by the combined treatment with HDACi. Cell viability assay was performed to assess the effects of TRAIL alongside different concentrations of HDACis, which found that cell viability was reduced by combined treatment. HDACis was previously reported to upregulate TRAIL receptor expression and sensitize target cells to TRAIL-induced apoptosis in Jurkat cells. In addition, HDACis may regulate the oncogenic pathway to induce AML cell death because RUNX1-ETO can bind to HDACs to abrogate the normal transcriptional function of RUNX1 (38). It is therefore important to analyze TRAIL function in normal myeloid/lymphoid development and to evaluate the possibility of TRAIL as a novel treatment method for this type of leukemia. However, the lack of in vivo validation is a limitation of the present study. To elucidate the mechanism of malignant transformation and therapy of AML, further in vivo analysis is required. In addition, results from clinical trials testing the effects of TRAIL on AML are expected to yield promising results.
Taken together, the present study demonstrated TRAIL to be a transcriptional target of RUNX1, where the effects are likely to be indirect. Additionally, TRAIL treatment may be useful for inducing apoptosis on leukemia cells that expresses the RUNX1-ETO mutant. The present study therefore provides novel insights into the regulation of TRAIL expression and roles of the transcription factor RUNX1.
Supplementary Data
Acknowledgments
The authors would like to thank Dr Toshiya Inaba, Department of Molecular Oncology and Leukemia Program Project, Research Institute for Radiation Biology and Medicine, Hiroshima University, Hiroshima, Japan for valuable advice on Kasumi-1 and SKNO-1 culture conditions and distribution of Kasumi-1 cells. The authors are grateful to Dr Misao Ohki, Chromatin Function in Leukemogenesis Project and Cancer Genomics Division, National Cancer Center Research Institute, Tokyo, Japan, for supplying RUNX1-ETO cDNA. The authors would like to thank Dr B Mark Evers, Department of Surgery, University of Texas Medical Branch, Galveston, USA, for the supply of human TRAIL promoter 5′-deletion mutants pTRAIL/-1523, pTRAIL/-819 and pTRAIL/-165.
Abbreviations
- RUNX1
Runt-related transcription factor 1
- AML1
acute myeloid leukemia 1
- TRAIL
tumor-necrosis factor-related apoptosis inducing ligand
Funding Statement
The present study was supported by JSPS KAKENHI (grant nos. JP15K09487, JP16K09857, JP16K10038, JP17K09936 and JP19K08846). It was also supported in part by Shimizu Foundation for Immunology and Neuroscience Grant for 2016, Children's Cancer Association of Japan Financial Support for 2016 and Research Institute for Production Development Grant for 2017.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
TY designed the study and performed the biological analyzes. KY performed the luciferase assays. and KT, YK, AES, NK, AM and TS analyzed experimental data. TO designed the study. TY and TO can confirm the authenticity of all the raw data. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Orkin SH, Zon LI. Hematopoiesis: An evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci USA. 1991;88:10431–10434. doi: 10.1073/pnas.88.23.10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059–1064. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyoshi H, Kozu T, Shimizu K, Enomoto K, Maseki N, Kaneko Y, Kamada N, Ohki M. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993;12:2715–2721. doi: 10.1002/j.1460-2075.1993.tb05933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank R, Zhang J, Uchida H, Meyers S, Hiebert SW, Nimer SD. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene. 1995;11:2667–2674. [PubMed] [Google Scholar]
- 7.Meyers S, Lenny N, Hiebert SW. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol Cell Biol. 1995;15:1974–1982. doi: 10.1128/MCB.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osato M, Asou N, Abdalla E, Hoshino K, Yamasaki H, Okubo T, Suzushima H, Takatsuki K, Kanno T, Shigesada K, Ito Y. Biallelic and heterozygous point mutations in the runt domain of the AML1/PEBP2alphaB gene associated with myeloblastic leukemias. Blood. 1999;93:1817–1824. doi: 10.1182/blood.V93.6.1817.406k36_1817_1824. [DOI] [PubMed] [Google Scholar]
- 9.Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 10.Mizutani S, Yoshida T, Zhao X, Nimer SD, Taniwaki M, Okuda T. Loss of RUNX1/AML1 arginine-methylation impairs peripheral T cell homeostasis. Br J Haematol. 2015;170:859–873. doi: 10.1111/bjh.13499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsumura T, Nakamura-Ishizu A, Muddineni SSNA, Tan DQ, Wang CQ, Tokunaga K, Tirado-Magallanes R, Sian S, Benoukraf T, Okuda T, et al. Hematopoietic stem cells acquire survival advantage by loss of RUNX1 methylation identified in familial leukemia. Blood. 2020;136:1919–1932. doi: 10.1182/blood.2019004292. [DOI] [PubMed] [Google Scholar]
- 12.DiFilippo EC, Coltro G, Carr RM, Mangaonkar AA, Binder M, Khan SP, Rodriguez V, Gangat N, Wolanskyj A, Pruthi RK, et al. Spectrum of abnormalities and clonal transformation in germline RUNX1 familial platelet disorder and a genomic comparative analysis with somatic RUNX1 mutations in MDS/MPN overlap neoplasms. Leukemia. 2020;34:2519–2524. doi: 10.1038/s41375-020-0752-x. [DOI] [PubMed] [Google Scholar]
- 13.Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84:321–330. doi: 10.1016/S0092-8674(00)80986-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang Q, Stacy T, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA. 1996;93:3444–3449. doi: 10.1073/pnas.93.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marín-Padilla M, Speck NA. Cbfa2 is required for the formation of intra-aortic hematopoietic clusters. Development. 1999;126:2563–2575. doi: 10.1242/dev.126.11.2563. [DOI] [PubMed] [Google Scholar]
- 16.Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, Ito Y, Littman DR. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/S0092-8674(02)01111-X. [DOI] [PubMed] [Google Scholar]
- 17.Ichikawa M, Asai T, Saito T, Seo S, Yamazaki I, Yamagata T, Mitani K, Chiba S, Ogawa S, Kurokawa M, Hirai H. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med. 2004;10:299–304. doi: 10.1038/nm997. [DOI] [PubMed] [Google Scholar]
- 18.Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106:494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bruijn M, Dzierzak E. Runx transcription factors in the development and function of the definitive hematopoietic system. Blood. 2017;129:2061–2069. doi: 10.1182/blood-2016-12-689109. [DOI] [PubMed] [Google Scholar]
- 20.Wang S, Wang Q, Crute BE, Melnikova IN, Keller SR, Speck NA. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol Cell Biol. 1993;13:3324–3339. doi: 10.1128/mcb.13.6.3324-3339.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci USA. 1993;90:6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyers S, Downing JR, Hiebert SW. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: The runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanno T, Kanno Y, Chen LF, Ogawa E, Kim WY, Ito Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol Cell Biol. 1998;18:2444–2454. doi: 10.1128/MCB.18.5.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitabayashi I, Yokoyama A, Shimizu K, Ohki M. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. EMBO J. 1998;17:2994–3004. doi: 10.1093/emboj/17.11.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otto F, Lübbert M, Stock M. Upstream and downstream targets of RUNX proteins. J Cell Biochem. 2003;89:9–18. doi: 10.1002/jcb.10491. [DOI] [PubMed] [Google Scholar]
- 26.Rossetti S, Sacchi N. RUNX1: A microRNA hub in normal and malignant hematopoiesis. Int J Mol Sci. 2013;14:1566–1588. doi: 10.3390/ijms14011566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imperato MR, Cauchy P, Obier N, Bonifer C. The RUNX1-PU.1 axis in the control of hematopoiesis. Int J Hematol. 2015;101:319–329. doi: 10.1007/s12185-015-1762-8. [DOI] [PubMed] [Google Scholar]
- 28.Wargnier A, Legros-Maida S, Bosselut R, Bourge JF, Lafaurie C, Ghysdael CJ, Sasportes M, Paul P. Identification of human granzyme B promoter regulatory elements interacting with activated T-cell-specific proteins: implication of Ikaros and CBF binding sites in promoter activation. Proc Natl Acad Sci USA. 1995;92:6930–6934. doi: 10.1073/pnas.92.15.6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elagib KE, Racke FK, Mogass M, Khetawat R, Delehanty LL, Goldfarb AN. RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood. 2003;101:4333–4341. doi: 10.1182/blood-2002-09-2708. [DOI] [PubMed] [Google Scholar]
- 30.Verma AK, Wheeler DL, Aziz MH, Manoharan H. Protein kinase cepsilon and development of squamous cell carcinoma, the nonmelanoma human skin cancer. Mol Carcinog. 2006;45:381–388. doi: 10.1002/mc.20230. [DOI] [PubMed] [Google Scholar]
- 31.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–682. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 32.LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 33.Jurisic V, Bumbasirevic V, Konjevic G, Djuricic B, Spuzic I. TNF-alpha induces changes in LDH isotype profile following triggering of apoptosis in PBL of non-Hodgkin's lymphomas. Ann Hematol. 2004;83:84–91. doi: 10.1007/s00277-003-0731-0. [DOI] [PubMed] [Google Scholar]
- 34.Jurisic V, Srdic-Rajic T, Konjevic G, Bogdanovic G, Colic M. TNF-α induced apoptosis is accompanied with rapid CD30 and slower CD45 shedding from K-562 cells. J Membr Biol. 2011;239:115–122. doi: 10.1007/s00232-010-9309-7. [DOI] [PubMed] [Google Scholar]
- 35.Stuckey DW, Shah K. TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol Med. 2013;19:685–694. doi: 10.1016/j.molmed.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Twomey JD, Kim SR, Zhao L, Bozza WP, Zhang B. Spatial dynamics of TRAIL death receptors in cancer cells. Drug Resist Updat. 2015;19:13–21. doi: 10.1016/j.drup.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–6271. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 38.Farooqi AA, Naqvi SK, Perk AA, Yanar O, Tabassum S, Ahmad MS, Mansoor Q, Ashry MS, Ismail M, Naoum GE, Arafat WO. Natural agents-mediated targeting of histone deacetylases. Arch Immunol Ther Exp (Warsz) 2018;66:31–44. doi: 10.1007/s00005-017-0488-0. [DOI] [PubMed] [Google Scholar]
- 39.Zhou M, Yuan M, Zhang M, Lei C, Aras O, Zhang X, An F. Combining histone deacetylase inhibitors (HDACis) with other therapies for cancer therapy. Eur J Med Chem. 2021;226:113825. doi: 10.1016/j.ejmech.2021.113825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falschlehner C, Schaefer U, Walczak H. Following TRAIL's path in the immune system. Immunology. 2009;127:145–154. doi: 10.1111/j.1365-2567.2009.03058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zerafa N, Westwood JA, Cretney E, Mitchell S, Waring P, Iezzi M, Smyth MJ. TRAIL deficiency accelerates hematological malignancies. J Immunol. 2005;175:5586–5590. doi: 10.4049/jimmunol.175.9.5586. [DOI] [PubMed] [Google Scholar]
- 42.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol. 2002;168:1356–1361. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 43.Finnberg N, Klein-Szanto AJ, El-Deiry WS. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest. 2008;118:111–123. doi: 10.1172/JCI29900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q, Ji Y, Wang X, Evers BM. Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem Biophys Res Commun. 2000;276:466–471. doi: 10.1006/bbrc.2000.3512. [DOI] [PubMed] [Google Scholar]
- 45.Allen JE, El-Deiry WS. Regulation of the human TRAIL gene. Cancer Biol Ther. 2012;13:1143–1151. doi: 10.4161/cbt.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Azahri NS, Kavurma MM. Transcriptional regulation of tumour necrosis factor-related apoptosis-inducing ligand. Cell Mol Life Sci. 2013;70:3617–3629. doi: 10.1007/s00018-013-1264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nebbioso A, Carafa V, Conte M, Tambaro FP, Abbondanza C, Martens J, Nees M, Benedetti R, Pallavicini I, Minucci S, et al. c-Myc modulation and acetylation is a key HDAC inhibitor target in cancer. Clin Cancer Res. 2017;23:2542–2555. doi: 10.1158/1078-0432.CCR-15-2388. [DOI] [PubMed] [Google Scholar]
- 48.Barbetti V, Tusa I, Cipolleschi MG, Rovida E, Sbarba PD. AML1/ETO sensitizes via TRAIL acute myeloid leukemia cells to the pro-apoptotic effects of hypoxia. Cell Death Dis. 2013;4:e536. doi: 10.1038/cddis.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okuda T, Takeda K, Fujita Y, Nishimura M, Yagyu S, Yoshida M, Akira S, Downing JR, Abe T. Biological characteristics of the leukemia-associated transcriptional factor AML1 disclosed by hematopoietic rescue of AML1-deficient embryonic stem cells by using a knock-in strategy. Mol Cell Biol. 2000;20:319–328. doi: 10.1128/MCB.20.1.319-328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukushima-Nakase Y, Naoe Y, Taniuchi I, Hosoi H, Sugimoto T, Okuda T. Shared and distinct roles mediated through C-terminal subdomains of acute myeloid leukemia/Runt-related transcription factor molecules in murine development. Blood. 2005;105:4298–4307. doi: 10.1182/blood-2004-08-3372. [DOI] [PubMed] [Google Scholar]
- 51.Ito Y. Oncogenic potential of the RUNX gene family: 'Overview'. Oncogene. 2004;23:4198–4208. doi: 10.1038/sj.onc.1207755. [DOI] [PubMed] [Google Scholar]
- 52.Kanno Y, Kanno T, Sakakura C, Bae SC, Ito Y. Cytoplasmic sequestration of the polyomavirus enhancer binding protein 2 (PEBP2)/core binding factor alpha (CBFalpha) subunit by the leukemia-related PEBP2/CBFbeta-SMMHC fusion protein inhibits PEBP2/CBF-mediated transactivation. Mol Cell Biol. 1998;18:4252–4261. doi: 10.1128/MCB.18.7.4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goyama S, Yamaguchi Y, Imai Y, Kawazu M, Nakagawa M, Asai T, Kumano K, Mitani K, Ogawa S, Chiba S, et al. The transcriptionally active form of AML1 is required for hematopoietic rescue of the AML1-deficient embryonic para-aortic splanchno-pleural (P-Sp) region. Blood. 2004;104:3558–3564. doi: 10.1182/blood-2004-04-1535. [DOI] [PubMed] [Google Scholar]
- 54.Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991;77:2031–2036. doi: 10.1182/blood.V77.9.2031.2031. [DOI] [PubMed] [Google Scholar]
- 56.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 57.Horinaka M, Yoshida T, Tomosugi M, Yasuda S, Sowa Y, Sakai T. Myeloid zinc finger 1 mediates sulindac sulfide-induced upregulation of death receptor 5 of human colon cancer cells. Sci Rep. 2014;4:6000. doi: 10.1038/srep06000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kagoshima H, Akamatsu Y, Ito Y, Shigesada K. Functional dissection of the alpha and beta subunits of transcription factor PEBP2 and the redox susceptibility of its DNA binding activity. J Biol Chem. 1996;271:33074–33082. doi: 10.1074/jbc.271.51.33074. [DOI] [PubMed] [Google Scholar]
- 59.Bagger FO, Sasivarevic D, Sohi SH, Laursen LG, Pundhir S, Sønderby CK, Winther O, Rapin N, Porse BT. BloodSpot: A database of gene expression profiles and transcriptional programs for healthy and malignant haematopoiesis. Nucleic Acids Res. 2016;44(D1):D917–D924. doi: 10.1093/nar/gkv1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rapin N, Bagger FO, Jendholm J, Mora-Jensen H, Krogh A, Kohlmann A, Thiede C, Borregaard N, Bullinger L, Winther O, et al. Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood. 2014;123:894–904. doi: 10.1182/blood-2013-02-485771. [DOI] [PubMed] [Google Scholar]
- 61.Kohlmann A, Kipps TJ, Rassenti LZ, Downing JR, Shurtleff SA, Mills KI, Gilkes AF, Hofmann WK, Basso G, Dell'orto MC, et al. An international standardization programme towards the application of gene expression profiling in routine leukaemia diagnostics: The microarray innovations in LEukemia study prephase. Br J Haematol. 2008;142:802–807. doi: 10.1111/j.1365-2141.2008.07261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné MC, De Vos J, Hernández JM, Hofmann WK, Mills KI, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the international microarray innovations in leukemia study group. J Clin Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klein HU, Ruckert C, Kohlmann A, Bullinger L, Thiede C, Haferlach T, Dugas M. Quantitative comparison of micro-array experiments with published leukemia related gene expression signatures. BMC Bioinformatics. 2009;10:422. doi: 10.1186/1471-2105-10-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warnat-Herresthal S, Perrakis K, Taschler B, Becker M, Baßler K, Beyer M, Günther P, Schulte-Schrepping J, Seep L, Klee K, et al. scalable prediction of acute myeloid leukemia using high-dimensional machine learning and blood transcriptomics. iScience. 2020;23:100780. doi: 10.1016/j.isci.2019.100780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taskesen E, Bullinger L, Corbacioglu A, Sanders MA, Erpelinck CA, Wouters BJ, van der Poel-van de Luytgaarde SC, Damm F, Krauter J, Ganser A, et al. Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: Further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2011;117:2469–2475. doi: 10.1182/blood-2010-09-307280. [DOI] [PubMed] [Google Scholar]
- 67.Taskesen E, Babaei S, Reinders MM, de Ridder J. Integration of gene expression and DNA-methylation profiles improves molecular subtype classification in acute myeloid leukemia. BMC Bioinformatics. 2015;16(Suppl 4):S5. doi: 10.1186/1471-2105-16-S4-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang L, Wang L, Zhao CH, Zhu XJ, Hou Y, Jun P, Hou M. Contributions of TRAIL-mediated megakaryocyte apoptosis to impaired megakaryocyte and platelet production in immune thrombocytopenia. Blood. 2010;116:4307–4316. doi: 10.1182/blood-2010-02-267435. [DOI] [PubMed] [Google Scholar]
- 69.Melloni E, Secchiero P, Celeghini C, Campioni D, Grill V, Guidotti L, Zauli G. Functional expression of TRAIL and TRAIL-R2 during human megakaryocytic development. J Cell Physiol. 2005;204:975–982. doi: 10.1002/jcp.20358. [DOI] [PubMed] [Google Scholar]
- 70.Crist SA, Elzey BD, Ludwig AT, Griffith TS, Staack JB, Lentz SR, Ratliff TL. Expression of TNF-related apoptosis-inducing ligand (TRAIL) in megakaryocytes and platelets. Exp Hematol. 2004;32:1073–1081. doi: 10.1016/j.exphem.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 71.Lam K, Muselman A, Du R, Harada Y, Scholl AG, Yan M, Matsuura S, Weng S, Harada H, Zhang DE. Hmga2 is a direct target gene of RUNX1 and regulates expansion of myeloid progenitors in mice. Blood. 2014;124:2203–2212. doi: 10.1182/blood-2014-02-554543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rini D, Calabi F. Identification and comparative analysis of a second runx3 promoter. Gene. 2001;273:13–22. doi: 10.1016/S0378-1119(01)00579-0. [DOI] [PubMed] [Google Scholar]
- 73.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yoshida T, Shiraishi T, Nakata S, Horinaka M, Wakada M, Mizutani Y, Miki T, Sakai T. Proteasome inhibitor MG132 induces death receptor 5 through CCAAT/enhancer-binding protein homologous protein. Cancer Res. 2005;65:5662–5667. doi: 10.1158/0008-5472.CAN-05-0693. [DOI] [PubMed] [Google Scholar]
- 75.Yoshida T, Maoka T, Das SK, Kanazawa K, Horinaka M, Wakada M, Satomi Y, Nishino H, Sakai T. Halocynthiaxanthin and peridinin sensitize colon cancer cell lines to tumor necrosis factor-related apoptosis-inducing ligand. Mol Cancer Res. 2007;5:615–625. doi: 10.1158/1541-7786.MCR-06-0045. [DOI] [PubMed] [Google Scholar]
- 76.Jurisić V, Spuzić I, Konjević G. A comparison of the NK cell cytotoxicity with effects of TNF-alpha against K-562 cells, determined by LDH release assay. Cancer Lett. 1999;138:67–72. doi: 10.1016/S0304-3835(99)00011-7. [DOI] [PubMed] [Google Scholar]
- 77.Jurisic V, Bogdanovic G, Kojic V, Jakimov D, Srdic T. Effect of TNF-alpha on Raji cells at different cellular levels estimated by various methods. Ann Hematol. 2006;85:86–94. doi: 10.1007/s00277-005-0010-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.