

Abstract
The evaluation of bone marrow morphology by experienced hematopathologists is essential in the diagnosis of acute myeloid leukemia (AML); however, it suffers from a lack of standardization and inter-observer variability. Deep learning (DL) can process medical image data and provides data-driven class predictions. Here, we apply a multi-step DL approach to automatically segment cells from bone marrow images, distinguish between AML samples and healthy controls with an area under the receiver operating characteristic (AUROC) of 0.9699, and predict the mutation status of Nucleophosmin 1 (NPM1)—one of the most common mutations in AML—with an AUROC of 0.92 using only image data from bone marrow smears. Utilizing occlusion sensitivity maps, we observed so far unreported morphologic cell features such as a pattern of condensed chromatin and perinuclear lightening zones in myeloblasts of NPM1-mutated AML and prominent nucleoli in wild-type NPM1 AML enabling the DL model to provide accurate class predictions.
Subject terms: Acute myeloid leukaemia, Acute myeloid leukaemia, Translational research
Introduction
A fundamental component in the diagnostic workflow of acute myeloid leukemia (AML) is cytomorphology [1]. The assessment of myeloblast counts and their morphology is essential for correct diagnosis, response assessment, and relapse detection. Cytomorphology may, in some cases, also lead to the suspicion of possible underlying genetics [2], e.g., in acute promyelocytic leukemia with t(15;17) and PML-RARα [3, 4] and AML with t(8;21), inv(16), or t(16;16) [5]. One of the most commonly mutated genes in AML is Nucleophosmin 1 (NPM1). It plays a critical role in disease initiation and is utilized for molecular risk stratification in the recent European Leukemia Net 2017 (ELN2017) recommendations [1]. Mutated NMP1 can be found in a third of all adult AML cases and up to 50–60% in AML with a normal karyotype [6, 7] and is considered a distinct disease entity in the current WHO classification [8]. So far, different morphological subtypes of AML according to the FAB classification [3] have been associated with different frequencies of NPM1 mutations [9]. However, the interpretation of cytomorphologic image data is subjective, time-consuming, and suffers from intra- and inter-observer variability [10, 11]. Artificial neural nets (ANN) have demonstrated excellent capabilities in the processing of large quantities of image data [12]. Deep learning (DL) models are large-scale ANN consisting of a multitude of interconnected parallel processing units called artificial neurons [13, 14]. Especially convolutional neural nets (CNN) achieve outstanding results in image recognition [15]. These capabilities can be used for computer vision purposes in the diagnosis of acute leukemias [16–18].
In our study, we present a CNN-based scalable model that accurately distinguishes between AML cases and healthy subjects from digitalized bone marrow images. Further, our model accurately predicts NPM1 mutation status from bone marrow cytomorphology and unveils distinct morphologic features for the prediction of NPM1 mutation status.
Methods
Data set and molecular analysis
We identified 1251 patients who have been newly diagnosed and treated with AML in the previously reported multicentric trials (AML96 [19]), AML2003 [20], AMLCG1999 [21], AML60+ [22], AMLCG2008 [23], and SORAML [24]) or from the multicentric patient registry of the German Study Alliance Leukemia (SAL, NCT03188874) via retrospective chart review. Eligibility criteria for the AML cohort were newly diagnosed AML according to WHO criteria [8], age ≥18 years, and available biomaterial at initial diagnosis. A control cohort was comprised of 236 bone marrow samples from healthy bone marrow donors who underwent bone marrow donation at our center. Figure 1 shows the set-up of the study cohort and the split of augmented image data between training and test set (4:1). All mentioned studies were previously approved by the Institutional Review Board of the Technical University Dresden. All participants gave their written informed consent according to the Declaration of Helsinki. The preparation of squash slides was performed from anticoagulated bone marrow by experienced laboratory technicians within 2 h after the sample was taken, as recommended by WHO guidelines [25]. Sample staining was performed with the May–Grunwald–Giemsa method [26]. Screening for NPM1 mutations was performed as described previously [27] and a 5% variant allele frequency (VAF) mutation cut-off was used. High-resolution pictures of representative regions of the bone marrow smears (BMS) were taken using the Nikon Eclipse E600 microscope (50-fold magnification) with the Nikon DSFi2 mounted camera and Nikon Imaging Software Elements D4 for image processing. Corresponding regions of interest were manually selected by hematologists and measured 0.1775 × 0.1325 mm. For selection of archived BMS, image acquisition, and upload of images to the database, 10 min of manual labor were needed per case. Per case, a median of 168.5 cells were captured (interquartile range: 124–217). Samples were randomly assigned to either a training or a validation set with a split of 4:1.
Fig. 1. Study cohort.

Patients were identified using retrospective chart review from previous multicenter clinical trials or the German Study Alliance Leukemia (SAL) registry. The control group was comprised of healthy bone marrow donors who underwent unrelated bone marrow donation at our center. Bone marrow smears (BMS) were digitized and sample size was increased by image augmentation (e.g. mirroring or rotating of images). Subsequently, samples were split into a training and test set with a ratio of 4:1.
DL model
A multi-step machine learning workflow with individual DL models for different tasks was set up as shown in Fig. 2. Step 1: BMS were digitalized and uploaded to an online segmentation and labeling platform that we developed for the purpose of this work. A human-in-the-loop cell segmentation approach was performed by hematologists with a Faster Region-based Convolutional Neural Net [28] (FRCNN). First, initial segmentation was done with the VGG Image Annotator [29] tool. Then, the FRCNN was trained with the segmented images and created new segmentation proposals for unsegmented images which were manually corrected by hematologists. The loop was closed by the refinement of segmentation proposals and repeated network training. A quarter of cases was segmented using this human-in-the-loop approach while for the remainder of cases the FRCNN worked autonomously without the need for human intervention for re-segmentation of cells. This way, segmentation quality improved substantially over iterations eliminating the need for manual segmentation in the following unsegmented images. Additionally, hyperparameter optimization was performed automatically using the Optuna [30] framework with a predefined hyperparameter space. Step 2: Feature extraction was performed manually by hematologists. In all, 8500 individual cells were labeled according to lineage, cell type, and characteristics like Auer rods. Features like cell size, eccentricity, and color range were automatically derived by the computer vision algorithms. Step 3: For distinction between AML and healthy control samples based on segmented images, we trained a multitude of DL models for binary predictions of cell types and characteristics that expressed results as ratios (e.g. ratio of myeloblasts among all cells or features such as presence or absence of Auer rods). The aggregated results given by these individual models were used as input for an Ensemble Neural Net (ENN) for final classification decisions. Model architecture for the distinction between AML and healthy control samples was based on the Xception CNN [31] utilizing transfer learning. Xception architecture was modified to receive BMS images (2560 × 1920 pixels) as input at the top level. Fully connected output layers for the 2048-dimensional output vectors of the core Xception architecture were established via hyperparameter optimization. Hyperparameters differed between individual models. Step 4: For NPM1 status prediction, transfer learning with a ResNet50 [32] pretrained on ImageNet was utilized on BMS images. The ResNet50-architecture was modified to use larger input sizes (2000 × 1500 pixels) and the output layer was reshaped to a fully connected layer with two neurons to accommodate the binary classification problem before retraining. Hyperparameter optimization was performed for learning rate, learning rate gamma, momentum, and weight decay. Occlusion sensitivity maps were used to derive information from classification decisions for NPM1 status prediction. DL models were implemented in Python version 3.7.9 with Keras version 2.3.0, TensorFlow version 2.1.2, and PyTorch version 1.5.0. Computations were performed using a high-performance computing system.
Fig. 2. Schematic workflow of the multi-step deep learning platform.

AML patients and healthy controls were identified via retrospective chart review. Representative pictures of bone marrow slides (BMS) were taken and labeled according to diagnosis and NPM1 mutation status. Subsequent image preprocessing, transformation and augmentation led to initial cell border proposals by the Faster Recurrent Convolutional Neural Net (FRCNN) that were manually corrected in order to improve cell borders. The FRCNN was trained iteratively to improve cell border proposals. Automated feature extraction on segmented cells by the image processing module determined characteristics like cell perimeter or nucleus-to-cytoplasm ratio (N:C). Segmented cells were manually labeled according to cell type and characteristics. Manual cell segmentation and labeling was performed with a modified version of the VGG image annotator tool. Thereby, a feature pool of BMS, segmented and labeled cells was generated to iteratively train the image classification module with a split of 2:1 between training and validation set using threefold cross-validation.
Model performance and statistical analysis
For performance analysis of the classification models, we used precision–recall curves and receiver operating characteristics (ROC) with the area under the curve (AUC). Precision is the fraction of true positives among all positive predictions of the DL model while recall is the fraction of all positive predictions of the DL model among all relevant events. The final models were evaluated on the validation set that was withheld from model training. To compare NPM1 VAF the Mann–Whitney U test was used. Computational and statistical analysis was performed in Python (version 3.7.9) and R (version 4.0.3).
Results
DL accurately distinguishes between BMS of AML patients and healthy bone marrow donor samples
We retrospectively identified 1251 AML patients, 386 of which harbored mutated NPM1 according to molecular analyses. Detailed information on patient characteristics and controls is provided in Table 1. A total of 94,162 individual cells were manually segmented to iteratively train the FRCNN. Subsequently, automatic cell segmentation with the FRCNN achieved a mean average precision and a mean average recall of both 0.97 at an intersection over union ratio of 0.5. Inaccuracies were mostly due to overlapping cells. We then applied a CNN-based binary classification model on the previously segmented images to distinguish between cell types and characteristics and aggregated results were used by an ENN to distinguish between AML and healthy donor samples. We found this multistage approach to substantially increase accuracy over simple whole image classification with only one CNN. Single cell-based disease status prediction was tested, but did not yield satisfactory results (AUROC 0.53). To adjust for the moderate sample size and to balance the data set, simple image augmentation techniques like linear transformations or adjustment of color channels and brightness range BMS images were applied. Thereby, we reached an augmented sample size of 5204 AML and 5428 non-AML (healthy donor) BMS images. To prevent overfitting, we used a pooling dropout of 0.25 as suggested by automated hyperparameter optimization. The AML classification model achieved an average AUC for the precision–recall curve of 0.9691 (95% CI: 0.9669–0.9713; Fig. 3A) and an average AUROC of 0.9699 (95% CI: 0.9677–0.9721; Fig. 3B) with a corresponding micro-average accuracy of 0.91. Table 2A shows the distribution of correctly and incorrectly identified AML and control samples in the validation set.
Table 1.
Patient characteristics.
| Parameter | All AML samples | NPM1-mutated AML | NPM1 wild-type AML | Bone marrow donors |
|---|---|---|---|---|
| N | 1251 | 386 | 865 | 236 |
| Age, median (IQR) | 57 (38–67) | 57 (49–66) | 54 (38–64) | 31 (25–39) |
| Sex, n (%) | ||||
| Male | 668 (53.4) | 173 (44.8) | 495 (57.2) | 70 |
| Female | 583 (46.6) | 213 (55.2) | 370 (42.8) | 30 |
| AML type, n (%) | ||||
| De novo | 969 (77.7) | 339 (88.3) | 630 (73) | / |
| sAML | 177 (14.2) | 24 (6.2) | 153 (17.7) | / |
| tAML | 101 (8.1) | 21 (5.5) | 80 (9.3) | / |
| ELN2017 risk, n (%) | ||||
| Favorable | 380 (33) | 277 (78) | 111 (13.8) | / |
| Intermediate | 521 (45.3) | 75 (21.1) | 446 (55.5) | / |
| Adverse | 249 (21.7) | 3 (0.8) | 246 (30.6) | / |
| PB blasts, median (IQR) | 23.5 (4–60) | 38 (11–72.5) | 18 (2–51) | / |
| BM blasts, median (IQR) | 60.5 (40–80) | 73.5 (54–85) | 54 (37–75) | / |
Patient characteristics for the AML and control (bone marrow donors) cohort. The AML cohort is further subdivided by NPM1 mutation status. AML type was defined according to the WHO 2016 classification. sAML secondary AML, tAML therapy-associated AML, PB peripheral blood, BM bone marrow, N/n number, IQR interquartile range.
Fig. 3. Performance measures of the AML classification model and NPM1 prediction.

A Precision–recall curve and B receiver operating characteristic (ROC) for the binary classification of AML vs. healthy bone marrow donor samples from bone marrow slide images obtained by threefold cross-validation (c.v.). Results of the individual c.v. sets are shown in the respective boxes and depicted as light blue, orange, and green graphs. Performance was measured calculating the area under the curve (AUC) for individual validation sets by threefold c.v. and averaging results (Macro avg, blue graph) with one standard deviation (SD) of the mean. C ROC for the binary classification of NPM1 mutation status on bone marrow slide images with fivefold cross-validation. TPR true positive rate, FPR false-positive rate.
Table 2.
Classification accuracy in the validation set.
| (A) AML vs. control | Prediction by deep learning | ||
| Healthy control | AML | ||
| Ground truth | |||
| Healthy control | 43 (89%) | 5 (11%) | |
| AML | 28 (13%) | 223 (87%) | |
| (B) mNPM1 vs. wtNPM1 | Prediction by deep learning | ||
| wtNPM1 | mNPM1 | ||
| Ground truth | |||
| wtNPM1 | 149 (86%) | 24 (14%) | |
| mNPM1 | 11 (14%) | 66 (86%) | |
The model was tested on a validation set of (A) 48 healthy controls and 251 AML patients. Misclassifications were 11% false positives (healthy controls misclassified as AML) and 13% false negatives (AML patients misclassified as healthy). For binary classification of NPM1 mutation status, the validation set was comprised of 77 patients with mutated (m)NPM1 as well as 173 patients with wild-type (wt)NMP1. Misclassifications were 14% false positives (wtNPM1 patients predicted to have mNPM1) as well as 14% false negatives (mNPM1 patients predicted to have wtNPM1). It is to be noted, that dysbalanced patient/control numbers were accounted for by image augmentation to balance the data sets for proper training of the classifiers.
DL accurately predicts NPM1 mutation status and unveils morphologic features
Further, we investigated whether DL could predict the mutational status of NPM1 from bone marrow morphology. BMS image classification at a 50-fold magnification was performed using a ResNet50-architecture using transfer learning. Mirroring and random cropping plus resizing was used to increase sample size and to balance the data. Weight decay of 0.0003 is utilized to prevent overfitting and the data was divided into training and test set with a split of 4:1. The model achieved an accuracy of 0.86 for NPM1 prediction and an AUROC of 0.92 (95% CI: 0.8768–0.9631; Fig. 3C). Table 2B shows the distribution of correctly and incorrectly identified NPM1-mutated and NPM1 wild-type samples in the validation set. Classification on single cells compared to whole image classification did not improve accuracy. To identify key morphological features that led the DL model to the prediction of the respective mutation status we used occlusion sensitivity maps (Fig. 4). This method iteratively blocks pixels of an image from being evaluated by the DL model for classification, which may reduce its predictive capabilities. Thereby, image areas that are essential for high accuracy can be detected as they greatly reduce model performance when being blocked (Fig. 4 ii, iii). By analyzing the heatmaps, we observed that the prediction of mutated NPM1 was associated with a pattern of condensed chromatin and perinuclear lightening zones in myeloblasts (Fig. 4a, orange arrows indicate examples). The prediction of NPM1 wild type was driven by prominent nucleoli (Fig. 4b, yellow arrows indicate examples) which could only very rarely be observed in samples with mutated NPM1 and in that context led to misclassification (false negatives). We further analyzed patient samples from the validation set grouped by NPM1 mutation status and true or false predictions given by the DL model regarding clinical and molecular data. NPM1-mutated AML samples that were correctly identified by the model (true positives) had a significantly higher median VAF than NPM1-mutated AML samples that were misclassified (false negatives) (true positives: 0.41 [95% CI: 0.39–0.42] vs. false negatives: 0.31 [95% CI: 0.1–0.42], p = 0.018, Fig. 5). Further, the rate of patients with therapy-associated AML (tAML) was significantly higher among false negatives compared to true positives (27.3% vs. true positives: 4.1%, p = 0.02) and false negatives had a significantly lower median white blood cell count (WBC) (false negatives 13.16 GPt/l [IQR:2.25–28.57] vs. true positives: 37.48 GPt/l [IQR:17.84–84.95], p = 0.007) and a trend for lower blast counts in peripheral blood (false negatives 25.5% [IQR: 4.75–38.5] vs. true positives: 52.5% [IQR: 16–75.25], p = 0.062). No significant differences for age, sex, ELN2017 risk category, absence or presence of a complex karyotype, bone marrow blast count, Hb, and platelet count were detected. For patients with wild-type NPM1 AML, there was a trend for lower median WBC for true negatives (3.54 GPt/l [IQR: 1.52–19.82] vs. false positives 11.3 GPt/l [IQR:3.95–36.849], p = 0.095), but age, sex, ELN2017 risk category, AML type (de novo, secondary or therapy-associated), absence or presence of a complex karyotype, bone marrow or peripheral blast count, Hb and platelet count showed no differences. As another internal sanity check, we applied the pretrained classifier to the healthy bone marrow donor image data set and found that 214/236 (91%) of cases were correctly identified as NPM1 wild type while only 22/236 (9%) were labeled as NPM1 mutated (false positives). However, we want to point out that the NPM1 classifier has never been trained on healthy controls. Considering its AML-specific training, the very low false-positive rate on newly presented and differently structured image data of healthy controls underlines the distinct morphology picked up by the classifier for correct predictions in NPM1-mutated AML. Interestingly, when we reviewed patient chart data and molecular results in the validation set, we found one sample that was incorrectly labeled as mutated NPM1, but was in fact wild-type NPM1, and the corresponding BMS image was correctly identified as such by the DL model.
Fig. 4. Application of occlusion sensitivity maps to detect features derived by deep learning for the prediction NPM1 mutation status in AML.

Representatives images from bone marrow samples from a a patient with mutated NPM1 AML and b a patient with wild-type NPM1 AML. (i) Bone marrow aspirates were stained with the May–Grunwald–Giemsa method. Pictures were taken at 50-fold magnification using a Nikon ECLIPSE E600 microscope with a mounted Nikon DSFi2 camera. Images were processed using Nikon Imaging Software Elements D4. Scale bar = 10 µm. (ii) Heatmap of occlusion sensitivity maps generated in Python. The scale indicates the importance of certain image areas for correct class prediction: the more intensively green an area is the more important it is for correct class prediction. The heatmap shows a cell-specific class evaluation by the deep learning model. (iii) Masked image overlay of (i) and (ii) generated in Python showing areas the deep learning model focused on. Bright image areas are more important for class predictions. The orange arrows in (a, i) point to examples of cells with condensed chromatin and perinuclear lightening zones the deep learning model picked up for prediction of mutated NPM1. The yellow arrows in (b, i) point to examples of cells with prominent nucleoli the deep learning model picked up for prediction of wild-type NPM1.
Fig. 5. Variant allele frequency of NPM1 true positives and NPM1 false negatives.
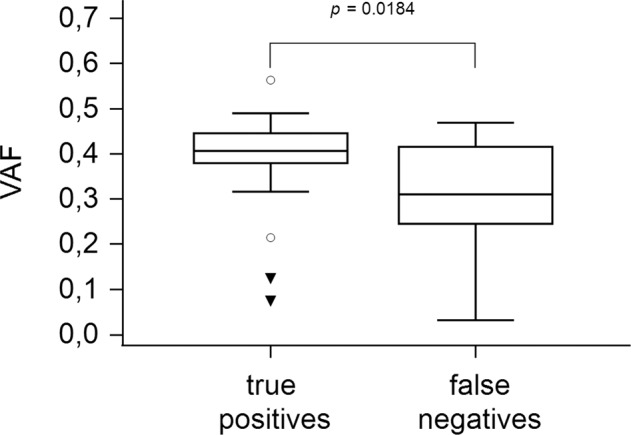
Box-and-whisker plot for variant allele frequency (VAF) of mutated NPM1 AML samples that were correctly identified as such by the deep learning model (true positives) and those that were misclassified (false negatives) compared by the Mann–Whitney U test.
Discussion
We here present a machine learning approach for cell segmentation and image classification which provides a fast, scalable, and highly accurate method to identify AML samples from bone marrow cytomorphology. Our FRCNN achieved a cell segmentation accuracy of 0.97 from BMS. The binary classification model showed an AUC of 0.97 for both the ROC and the precision–recall curve and a micro-average accuracy of 0.91 distinguishing between AML and healthy bone marrow donor samples. Our model can potentially be applied in initial diagnosis when a case of suspected AML is evaluated upon first contact in a hematologic center. It operates autonomously once BMS images are uploaded and detects AML with high accuracy. The model could operate synchronously with lab technicians to flag cases that are highly suspicious of AML for fast evaluation by experts while results from other diagnostic procedures like flow cytometry, cytogenetics, and molecular genetics are still pending. However, a human-in-the-loop approach is still needed as we manually selected representative regions of the BMS for evaluation by the DL model. Also, it is to be noted that bone marrow donors in our cohort were substantially younger than AML patients. Increased age is associated with observable changes in the bone marrow microenvironment such as cellularity, proliferative activity, and apoptosis [33] and such systematic differences could introduce bias to a CNN classifier which needs to be taken seriously not only in our use case but also considering other applications of more subtle changes in bone marrow morphology. Further evaluation of the model using more diverse multicenter data is warranted. Another limitation is the necessity for manual selection of BMS areas representative for disease classification by human judgment. Since this is a potential source of bias, future work will focus on implementing whole slide imaging and an automatization of region-of-interest selection given recent advances such as DL-based automated focusing [34]. Further automatization of BMS development can be achieved using automated BMS staining devices [35].
Furthermore, we used DL to predict the mutation status of NPM1 from cytomorphology alone. For NPM1 prediction our DL model achieved a high accuracy of 0.86 in predicting mutation status. AML with mutated NPM1 has previously been associated with cup-like blast morphology [36, 37]. When analyzing the features that the model used for NPM1 classification with occlusion sensitivity maps, we found so far unreported features like a pattern of condensed chromatin accompanied by perinuclear lightening zones for NPM1-mutated blasts. We observed prominent nucleoli in myeloblasts as a feature the DL model derived to predict wild-type NPM1 AML while these could only rarely be observed in NPM1-mutated AML samples and then led to misclassification by the model. Further, we found a significantly higher VAF in NPM1 true positives while the group of false negatives was comprised of a significantly higher rate of tAML. Wild-type NPM1 serves as a critical structural protein of the nucleolus, but mutations lead to a delocalization to the cytoplasm [38]. This process is partially triggered by insertions causing a frameshift of the C-terminal end of NPM1 and the formation of nuclear export signals [39, 40]. Weakened anchoring and predominant export signals subsequently result in increased nuclear export of NPM1 [38]. Arguably, a cytomorphologic correlate of this process may be the presence of prominent nucleoli in wild-type NPM1 AML and the absence thereof in mutated NPM1 AML—both detected as highly predictive features by our DL model.
Our study shows that DL can derive morphologic features from cytomorphology that predict mutation status. Future work will focus on other clinically important mutations and their morphologic imprint that DL may be able to pick up. In line with our findings, a recent study showed that DL can associate the morphology of myelodysplastic syndromes (MDS) with distinct genetic imprints [41]. However, in order to be integrated into clinical practice, machine-learning models need to be accurate and generalizable. As their development is complex, collaborations between physicians and software engineers is needed in an iterative approach to increase model performance. Since the majority of recently proposed machine-learning models—along with our model—are built on retrospective data, future studies will have to implement such models in a prospective setting to confirm their diagnostic value [42]. Due to the heterogeneity of cell morphology as well as close proximity of cells, disease classification from bone marrow is much more complex than in peripheral blood. Our use case to delineate AML from healthy bone marrow serves as groundwork for more complicated applications of CNNs in bone marrow morphology. AML is defined by bone marrow blast count [1] and CNNs can use a ratio of blasts to accurately detect AML. However, more complex use cases such as reactive bone marrow changes, benign disorders such as vitamin B12 deficiency, or hematologic neoplasms such as MDS are associated with subtle changes of cell morphology [43, 44]. In this scenario, CNNs have to be trained to accurately detect and assign such morphologic changes to the respective disorders. Mori et al. [45] recently used CNN-guided detection of decreased granules—one of the most common dysplastic changes in MDS—and report high accuracy for their classifier based on the ResNet-152. Accordingly, integrated analysis of more complex morphologies can potentially be achieved by feature engineering using a knowledge bank of expert-annotated cells with sufficiently sized training sets per morphologic feature (conceivably in the four- to five-digit number range). Since many hematologic neoplasms are rare disease entities, the development of such a large database requires extensive cooperation and data sharing between institutions and countries to ideally provide an open-source bone marrow database where independent ML models can be trained on analogous to existing cancer data bases such as The Cancer Genome Atlas [46]. Nevertheless, samples need to be properly anonymized to warrant patient data safety. If maintained and funded properly, such a database may vastly accelerate the development of clinically relevant computer vision tools for hematologic diagnostics. Standardization of data acquisition and accessible documentation of methodologies should be implemented to limit bias inherent to local methodologies of digitizing BMS and reporting patient data. Further, an integration of different diagnostic modalities such as cytomorphology both of bone marrow and peripheral blood, flow cytometry as well as genetic and clinical data seems warranted to build ML models that may aid in clinical decision making since evaluating only one modality at a time is insufficient for accurate diagnosis. Ensemble learning could be used to integrate the outputs of different ML models for different diagnostic modalities and provide a comprehensive and interpretable output to the clinician. Future work will focus on the extension of our ML pipeline for other use cases as well as different diagnostic modalities. As our study was limited to our center only, future studies will focus on transferability.
In conclusion, we here present a DL approach for the fast and accurate detection of AML from bone marrow cytomorphology. Our DL model accurately predicts NPM1 mutation status and derived so far unreported morphologic features that indicate absence or presence of NPM1 mutations from myeloblast morphology. This approach can be implemented to aid in clinical decision making, accelerate diagnosis, and may serve as a proof-of-concept for further studies of genetic imprints on disease morphology using DL.
Acknowledgements
We thank all contributing physicians, laboratories, and nurses associated with the German Study Alliance Leukemia and especially participating patients for their valuable contributions. The authors are grateful to the Centre for Information Services and High-Performance Computing TU Dresden for providing its facilities for high throughput calculations.
Author contributions
J.-N.E., J.M.M., K.W., and M.B. designed the study. J.-N.E., J.M.M., A.S.S., K.S., F.K., U.S., J.S., C.R., C.T., and M.B. provided and analyzed patient samples. S.R. T.S., and K.W. developed and implemented the image processing and classification pipeline with neural networks. All authors analyzed and interpreted the data. J.-N.E. wrote the draft. All authors critically revised and edited the manuscript. All authors approved the final version of the manuscript.
Funding information
This study was funded in part by a MeDDrive Grant number 60499 Machine Learning for advanced integrated diagnostics in hematological malignancies to Dr. Middeke from the Technical University Dresden. Open Access funding enabled and organized by Projekt DEAL.
Data availability
De-identified original BMS image data that supported the findings of this study are publicly available under https://www.kaggle.com/sebastianriechert/bone-marrow-slides-for-leukemia-prediction.
Code availability
Python code for the DL models developed and implemented for the purpose of this study is publicly available under https://github.com/TimSchmittmann/DL_detection_of_AML_from_BMS and https://github.com/SebastianRiechert/autofrcnn and https://github.com/SebastianRiechert/npm1-training.
Competing interests
C.T. is CEO and co-owner of AgenDix GmbH, a company performing molecular diagnostics. This study was funded in part by a MeDDrive Grant number 60499 Machine Learning for advanced integrated diagnostics in haematological malignancies from the Technical University Dresden to Dr. Middeke. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jan-Niklas Eckardt, Jan Moritz Middeke.
These authors contributed equally: Karsten Wendt, Martin Bornhäuser.
References
- 1.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bain BJ, Béné MC. Morphological and immunophenotypic clues to the WHO categories of acute myeloid leukaemia. Acta Hematol. 2019;141:232–44. doi: 10.1159/000496097. [DOI] [PubMed] [Google Scholar]
- 3.Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–8. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 4.de Thé H, Chomienne C, Lanotte M, Degos L, Dejean A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature. 1990;347:558–61. doi: 10.1038/347558a0. [DOI] [PubMed] [Google Scholar]
- 5.Nishii K, Usui E, Katayama N, Lorenzo VF, Nakase K, Kobayashi T, et al. Characteristics of t(8;21) acute myeloid leukemia (AML) with additional chromosomal abnormality: concomitant trisomy 4 may constitute a distinctive subtype of t(8;21) AML. Leukemia. 2003;17:731–7. doi: 10.1038/sj.leu.2402871. [DOI] [PubMed] [Google Scholar]
- 6.Falini B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E, et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011;117:1109–20. doi: 10.1182/blood-2010-08-299990. [DOI] [PubMed] [Google Scholar]
- 7.Grimwade D, Ivey A, Huntly BJP. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41. doi: 10.1182/blood-2015-07-604496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 9.Rose D, Haferlach T, Schnittger S, Perglerová K, Kern W, Haferlach C. Specific patterns of molecular mutations determine the morphologic differentiation stages in acute myeloid leukemia (AML) Blood. 2014;124:2388–2388. doi: 10.1182/blood.V124.21.2388.2388. [DOI] [Google Scholar]
- 10.Dasariraju S, Huo M, McCalla S. Detection and classification of immature leukocytes for diagnosis of acute myeloid leukemia using random Forest algorithm. Bioengineering (Basel). 2020;7:120. [DOI] [PMC free article] [PubMed]
- 11.Fuentes-Arderiu X, Dot-Bach D. Measurement uncertainty in manual differential leukocyte counting. Clin Chem Lab Med. 2009;47:112–5. doi: 10.1515/CCLM.2009.014. [DOI] [PubMed] [Google Scholar]
- 12.Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks. Commun ACM. 2017;60:84–90. doi: 10.1145/3065386. [DOI] [Google Scholar]
- 13.Jain AK, Jianchang M, Mohiuddin KM. Artificial neural networks: a tutorial. Computer. 1996;29:31–44. doi: 10.1109/2.485891. [DOI] [Google Scholar]
- 14.Basheer IA, Hajmeer M. Artificial neural networks: fundamentals, computing, design, and application. J Microbiol Methods. 2000;43:3–31. doi: 10.1016/S0167-7012(00)00201-3. [DOI] [PubMed] [Google Scholar]
- 15.Guo Y, Liu Y, Oerlemans A, Lao S, Wu S, Lew MS. Deep learning for visual understanding: a review. Neurocomputing. 2016;187:27–48. doi: 10.1016/j.neucom.2015.09.116. [DOI] [Google Scholar]
- 16.Rodellar J, Alférez S, Acevedo A, Molina A, Merino A. Image processing and machine learning in the morphological analysis of blood cells. Int J Lab Hematol. 2018;40(S1):46–53. doi: 10.1111/ijlh.12818. [DOI] [PubMed] [Google Scholar]
- 17.Matek C, Schwarz S, Spiekermann K, Marr C. Human-level recognition of blast cells in acute myeloid leukaemia with convolutional neural networks. Nat Mach Intell. 2019;1:538–44. doi: 10.1038/s42256-019-0101-9. [DOI] [Google Scholar]
- 18.Ahmed N, Yigit A, Isik Z, Alpkocak A. Identification of leukemia subtypes from microscopic images using convolutional neural network. Diagnostics (Basel). 2019;9. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6787617/. [DOI] [PMC free article] [PubMed]
- 19.Röllig C, Thiede C, Gramatzki M, Aulitzky W, Bodenstein H, Bornhäuser M, et al. A novel prognostic model in elderly patients with acute myeloid leukemia: results of 909 patients entered into the prospective AML96 trial. Blood. 2010;116:971–8. doi: 10.1182/blood-2010-01-267302. [DOI] [PubMed] [Google Scholar]
- 20.Schaich M, Parmentier S, Kramer M, Illmer T, Stölzel F, Röllig C, et al. High-dose cytarabine consolidation with or without additional amsacrine and mitoxantrone in acute myeloid leukemia: results of the prospective randomized AML2003 trial. J Clin Oncol. 2013;31:2094–102. doi: 10.1200/JCO.2012.46.4743. [DOI] [PubMed] [Google Scholar]
- 21.Buchner T, Berdel WE, Haferlach C, Schnittger S, Haferlach T, Serve H, et al. Long-term results in patients with acute myeloid leukemia (AML): the influence of high-dose AraC, G-CSF priming, autologous transplantation, prolonged maintenance, age, history, cytogenetics, and mutation status. Data of the AMLCG 1999 Trial. Blood. 2009;114:485–485. doi: 10.1182/blood.V114.22.485.485. [DOI] [Google Scholar]
- 22.Röllig C, Kramer M, Gabrecht M, Hänel M, Herbst R, Kaiser U, et al. Intermediate-dose cytarabine plus mitoxantrone versus standard-dose cytarabine plus daunorubicin for acute myeloid leukemia in elderly patients. Ann Oncol. 2018;29:973–8. doi: 10.1093/annonc/mdy030. [DOI] [PubMed] [Google Scholar]
- 23.Braess J, Amler S, Kreuzer K-A, Spiekermann K, Lindemann HW, Lengfelder E, et al. Sequential high-dose cytarabine and mitoxantrone (S-HAM) versus standard double induction in acute myeloid leukemia—a phase 3 study. Leukemia. 2018;32:2558–71. doi: 10.1038/s41375-018-0268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–9. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 25.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bain BJ, Clark DM, Wilkins BS. Bone marrow pathology. Wiley; 2019. p. 736.
- 27.Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML) Blood. 2006;107:4011–20. doi: 10.1182/blood-2005-08-3167. [DOI] [PubMed] [Google Scholar]
- 28.Ren S, He K, Girshick R, Sun J. Faster R-CNN: towards real-time object detection with region proposal networks. IEEE Trans Pattern Anal Mach Intell. 2017;39:1137–49. doi: 10.1109/TPAMI.2016.2577031. [DOI] [PubMed] [Google Scholar]
- 29.Dutta A, Zisserman A The VIA annotation software for images, audio and video. In: Proceedings of the 27th ACM International Conference on Multimedia. Association for Computing Machinery (MM’19); 2019. p. 2276–9. Available from: 10.1145/3343031.3350535.
- 30.Akiba T, Sano S, Yanase T, Ohta T, Koyama M. Optuna: a next-generation hyperparameter optimization framework. In: Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. Association for Computing Machinery (KDD’19); 2019. p. 2623–31. Available from: 10.1145/3292500.3330701.
- 31.Chollet F. Xception: Deep learning with depthwise separable convolutions. arXiv:161002357 [cs] [Preprint]. 2017 [cited 2021 Jan 12]. Available from: http://arxiv.org/abs/1610.02357.
- 32.He K, Zhang X, Ren S, Sun J. Deep residual learning for image recognition. arXiv:151203385 [cs] [Preprint]. 2015 [cited 2021 Apr 15]. Available from: http://arxiv.org/abs/1512.03385.
- 33.Ogawa T, Kitagawa M, Hirokawa K. Age-related changes of human bone marrow: a histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech Ageing Dev. 2000;117:57–68. doi: 10.1016/S0047-6374(00)00137-8. [DOI] [PubMed] [Google Scholar]
- 34.Rai Dastidar T, Ethirajan R. Whole slide imaging system using deep learning-based automated focusing. Biomed Opt Express. 2019;11:480–91. doi: 10.1364/BOE.379780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gemen EFA, de Wit NCJ, van Gerven MPB, de Jongh-Leuvenink J. The Sysmex SP1000i for automated bone marrow slide smear staining. Lab Med. 2009;40:719–23. doi: 10.1309/LMK6D4LVJ6NTRCCF. [DOI] [Google Scholar]
- 36.Kroschinsky FP, Schäkel U, Fischer R, Mohr B, Oelschlaegel U, Repp R, et al. Cup-like acute myeloid leukemia: new disease or artificial phenomenon? Haematologica. 2008;93:283–6. doi: 10.3324/haematol.11669. [DOI] [PubMed] [Google Scholar]
- 37.Park BG, Chi H-S, Jang S, Park C-J, Kim D-Y, Lee J-H, et al. Association of cup-like nuclei in blasts with FLT3 and NPM1 mutations in acute myeloid leukemia. Ann Hematol. 2013;92:451–7. doi: 10.1007/s00277-012-1645-5. [DOI] [PubMed] [Google Scholar]
- 38.Falini B, Brunetti L, Sportoletti P, Martelli MP. NPM1-mutated acute myeloid leukemia: from bench to bedside. Blood. 2020;136:1707–21. doi: 10.1182/blood.2019004226. [DOI] [PubMed] [Google Scholar]
- 39.Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A, et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514–23. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 40.Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia. 2009;23:1731–43. doi: 10.1038/leu.2009.124. [DOI] [PubMed] [Google Scholar]
- 41.Nagata Y, Zhao R, Awada H, Kerr CM, Mirzaev I, Kongkiatkamon S, et al. Machine learning demonstrates that somatic mutations imprint invariant morphologic features in myelodysplastic syndromes. Blood. 2020;136:2249–62. doi: 10.1182/blood.2020005488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eckardt J-N, Bornhäuser M, Wendt K, Middeke JM. Application of machine learning in the management of acute myeloid leukemia: current practice and future prospects. Blood Adv. 2020;4:6077–85. doi: 10.1182/bloodadvances.2020002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cazzola M. Myelodysplastic ayndromes. N Engl J Med. 2020;383:1358–74. doi: 10.1056/NEJMra1904794. [DOI] [PubMed] [Google Scholar]
- 44.Bain BJ. Diagnosis from the blood smear. N Engl J Med. 2005;353:498–507. doi: 10.1056/NEJMra043442. [DOI] [PubMed] [Google Scholar]
- 45.Mori J, Kaji S, Kawai H, Kida S, Tsubokura M, Fukatsu M, et al. Assessment of dysplasia in bone marrow smear with convolutional neural network. Sci Rep. 2020;10:14734. doi: 10.1038/s41598-020-71752-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Cancer Genome Atlas Research Network et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
De-identified original BMS image data that supported the findings of this study are publicly available under https://www.kaggle.com/sebastianriechert/bone-marrow-slides-for-leukemia-prediction.
Python code for the DL models developed and implemented for the purpose of this study is publicly available under https://github.com/TimSchmittmann/DL_detection_of_AML_from_BMS and https://github.com/SebastianRiechert/autofrcnn and https://github.com/SebastianRiechert/npm1-training.