

Abstract
Under certain cellular conditions functional proteins undergo misfolding, leading to a transition into oligomers which precede formation of amyloid fibrils. Misfolding proteins are associated with neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. While the importance of lipid membranes in misfolding and disease aetiology is broadly accepted, the influence of lipid membranes during therapeutic design has been largely overlooked. This study utilized a biophysical approach to provide mechanistic insights into the effects of two lipid membrane systems (anionic and zwitterionic) on the inhibition of amyloid-β 40 and α-synuclein amyloid formation at the monomer, oligomer and fibril level. Large unilamellar vesicles (LUVs) were shown to increase fibrillization and largely decrease the effectiveness of two well-known polyphenol fibril inhibitors, (−)-epigallocatechin gallate (EGCG) and resveratrol, however, use of immunoblotting and ion-mobility mass spectrometry revealed this occurs through varying mechanisms. Oligomeric populations in particular were differentially affected by LUVs in the presence of resveratrol, an elongation phase inhibitor, compared to EGCG, a nucleation targeted inhibitor. Ion-mobility mass spectrometry showed EGCG interacts with or induces more compact forms of monomeric protein typical of off-pathway structures, however binding is reduced in the presence of LUVs, likely due to partitioning in the membrane environment. Competing effects of the lipids and inhibitor, along with reduced inhibitor binding in the presence of LUVs, provides a mechanistic understanding for decreased inhibitor efficacy in a lipid environment. Together, this study highlights that amyloid inhibitor design may be misguided if effects of lipid membrane composition and architecture are not considered during development.
Keywords: protein unfolding, amyloid aggregation, amyloid inhibitors, lipid membrane, ion mobility-mass spectrometry
Introduction
Many intrinsically disordered proteins (IDPs) are prone to aggregation in the form of amyloid fibrils, and the accumulation of neuronal plaques containing these aggregates are hallmarks of a variety of neurodegenerative diseases. For example, extracellular plaques composed of amyloid β (Aβ) and intracellular Lewy bodies containing α-synuclein (αS) are associated with Alzheimer’s disease and Parkinson’s disease, respectively. However, despite strong correlative links, the molecular mechanisms by which these IDPs contribute to the disease aetiology is still poorly understood.
In periods of cellular stress, amyloidogenic proteins can enter an aggregation pathway commencing with a nucleation-phase in which misfolding proteins restructure to form aggregation-prone, prefibrillar species. This is followed by an elongation-phase in which monomeric proteins are recruited from solution to form mature fibrils [1,2]. Current research suggests the low molecular weight, soluble, oligomeric species formed early on the aggregation pathway are primarily responsible for neurotoxic effects rather than the mature amyloid fibrils [3–7]. Pre-fibrillar species have been shown to be more toxic than the plaques that they eventually form [6–8], and smaller oligomeric species have been shown to exert enhanced toxicity [9]. While focus has shifted in this regard from fibrils to monomers and oligomers, further research is required to understand the structure of toxic oligomeric species to better guide therapeutic development.
Importantly, many recent studies have highlighted the critical role that membrane composition and curvature plays in amyloid disease propagation through the interactions between misfolded oligomeric species and plasma/organelle membranes. Whether this is due to a unifying mechanism or a variety of molecular processes driving pathogenesis is yet to be fully understood [10–13]. For example, Aβ has been shown to interact with neuronal cell membranes whereby oligomeric forms of the protein cause membrane leakage and transmission of ions, disrupting ionic homeostasis particularly for Ca2+, and resulting in neuronal cell death [11,14–16]. On top of this, studies show that lipid bilayers (and other lipid forms) may increase the aggregation propensity of misfolded proteins as well as alter monomeric and oligomeric structures [17–19]. Also of importance is membrane composition, as differing lipid mixtures induce a variety of different effects including increased rate of αS nucleation in the presence of anionic lipid membranes [20], while shorter chain phospholipids have been shown to halt Aβ fibril formation at low concentrations and accelerate it at higher concentrations [21]. Anionic lipids have been shown to interact with many misfolding proteins such as Aβ, αS and human islet amyloid polypeptide (hIAPP) and increase rates of fibrillar elongation [22,23]. Other components of the cellular membrane such as cholesterol and gangliosides can also influence fibril kinetics and morphology [13,24–26]. The lipidome of neurons is complicated, diverse and is altered with age and disease states. After 20 years of age the total lipid content of the brain decreases, as does the concentration of sphingomyelins and cholesterol, although different neurons are affected more than others, further reinforcing the importance of understanding of amyloid formation in diverse lipid environments [27].
The economic and social burden caused by amyloid diseases makes the hunt for an effective therapeutic incredibly important [28]. Natural polyphenols are of great interest due to their antioxidant effects and have been shown extensively to directly inhibit the aggregation of a wide range of misfolding proteins, particularly Aβ and αS [29,30]. Two compounds that have been widely studied and demonstrated efficacious anti-amyloid activity, (−)-epigallocatechin gallate (EGCG) and resveratrol, are natural products isolated from green tea and red wine, respectively. EGCG has been shown to not only inhibit fibril formation but also remodel Aβ fibrils into smaller, nontoxic, amorphous aggregates [30–33]. Resveratrol, a promising candidate in animal studies [34,35], has been shown to stop Aβ fibril formation through a capping mechanism, preventing large scale fibrilization while having no effect on oligomer formation [36–38]. Despite the promise of these studies, they and many others largely ignore the influence that a lipid environment plays on amyloid formation as well as the effectiveness of inhibitors in this highly dynamic environment [39].
The partitioning preferences of proteins, lipids and small molecules between a phospholipid membrane and the surrounding solvent gives rise to a complex environment with a range of molecular interactions that can naturally influence protein structure, aggregation, fibril formation and inhibition. For example, recent work with hIAPP, a membrane associated amyloidogenic protein, proposed that partitioning of hIAPP and small molecule inhibitors (based on an oligopyridylamide scaffold) between a membrane and solution altered inhibitor efficacy in a manner dependent on the hydrophobicity of the inhibitor [40]. In addition, some inhibitors, including resveratrol, have been shown to influence structural changes in hIAPP that are thought to prevent membrane association of the polypeptide, thereby preventing lipid catalysed aggregation and membrane disruption [41–43]. To better develop effective small molecule inhibitors of misfolding and aggregating proteins we must therefore understand their interactions in the physiological environments in which these inhibitors will be acting.
IDPs are, by definition, highly dynamic and heterogeneous, making them not amenable to study by traditional structural biology techniques such as x-ray crystallography due to the transient nature of the protein structure itself. Other methods like nuclear magnetic resonance spectroscopy, circular dichroism spectroscopy and fluorescence spectroscopy present data on a global average rather than the collection of unique structural states and oligomers that constitute a transient mixture. The combination of nano-electrospray ionisation with native ion-mobility mass spectrometry (IM-MS) allows for gentle ionisation of proteins into the gas-phase, preserving important tertiary and quaternary structures, while separating based on mass to charge (m/z) as well as a rotationally averaged size (or collisional cross section, CCS) [44–46]. It therefore offers a useful alternative analytical method to probe the heterogeneous and dynamic states of IDPs during protein aggregation and investigate the impact of small molecules on this process.
Here we have utilised IM-MS in an integrated biophysical approach to better understand the role of lipid membranes in protein misfolding diseases at distinct stages along the aggregation pathway, from monomeric structure to fibril morphology. We explore differences in the aggregation dynamics of Aβ40 and αS in the presence of two membrane models, namely unilamellar vesicles (LUVs) composed of zwitterionic 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (PC) or a mixture of anionic 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (PG) with PC (herein PCPG). These model systems were chosen as PC is a major phospholipid in human neuronal membranes and PG acts to create a negative bilayer surface charge, which are major components within grey and white matter [27]. The relative efficacy of two small molecule inhibitors, EGCG and resveratrol, was evaluated in these lipid environments and the effects of these variables on protein structure and aggregation propensity were interrogated. This study highlights the interplay between misfolding proteins, phospholipid membranes and fibril inhibitors, and may inform the design of more effective therapies by considering the influence that lipid environments impose on disease development and inhibition.
Results
LUVs increase Aβ40 fibrillization and reduce EGCG and resveratrol inhibitor efficacy
The effects of LUVs consisting of either PC or PCPG on Aβ40 fibril formation were first assessed by performing ThT kinetic assays (Fig. 1A). These aggregation assays were carried out in the presence of LUVs of certain size (i.e. 100 nm polycarbonate membranes were used to create homogenous LUVs) in order to prevent any influences on aggregation kinetics arising from differing vesicle sizes. The size distribution of the LUVs was confirmed by dynamic light scattering (DLS) (Fig. 1B) which displayed a normal and narrow size distribution with diameters of approximately 100–300 nm. ThT experiments in the presence of LUVs alone were also conducted to verify that the addition of lipids did not significantly affect ThT fluorescence properties.
Figure 1. PC and PCPG LUVs alter the aggregation kinetics and fibril formation of Aβ40.
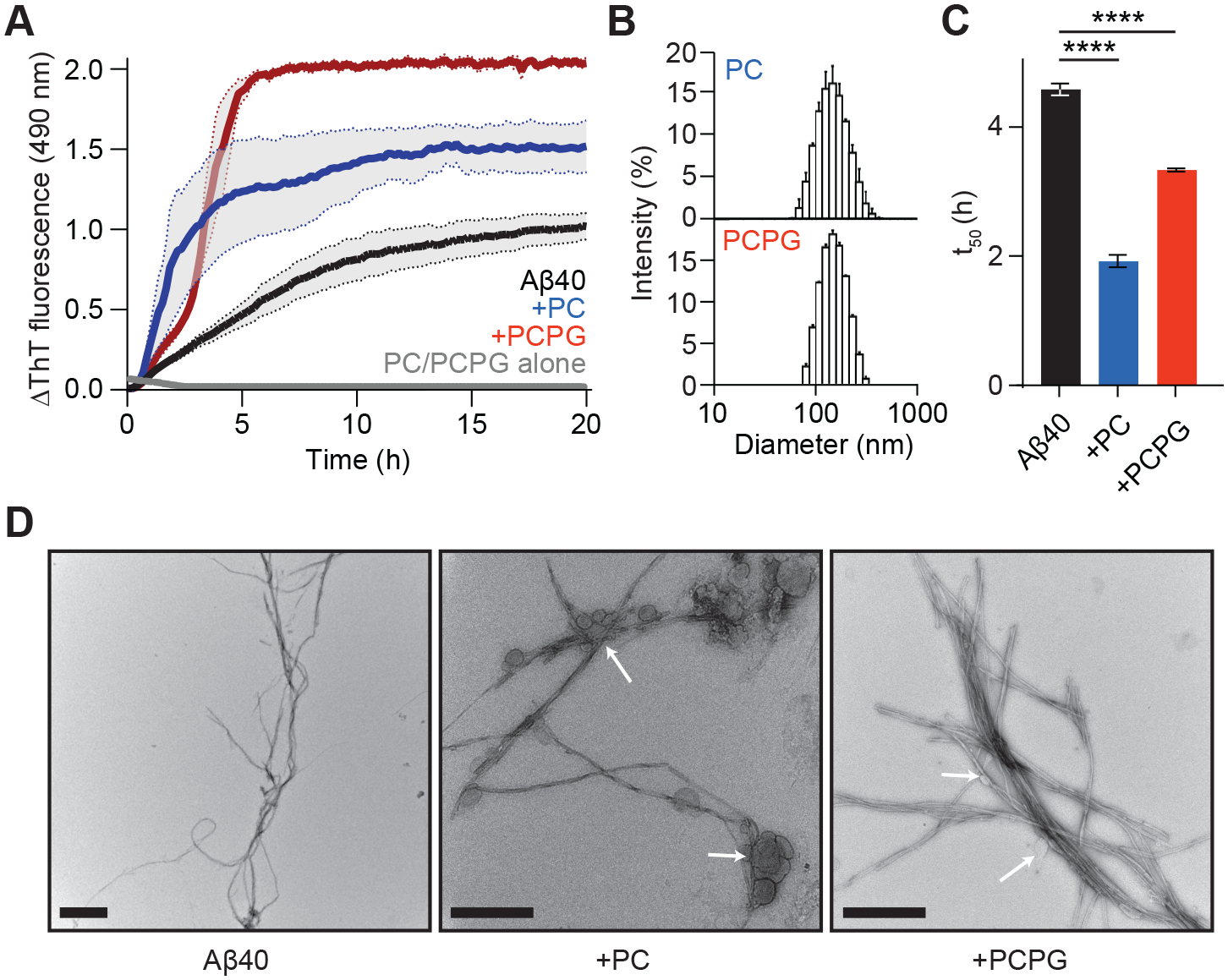
(A) Aβ40 (50 μM) (black) fibril formation was monitored by the change in ThT fluorescence in the presence of either PC (blue) or PCPG (red) LUVs (1:30, Aβ40:LUV). (B) DLS reveals a normal and narrow size distribution for PC (top) and PCPG (bottom) LUVs at a concentration of 1.5 mM, with diameters of approximately 100–300 nm. (C) The time taken to reach half maximum ThT fluorescence (t50) was compared for Aβ40 (50 μM) in the presence of either PC or PCPG LUVs (1:30, Aβ40:LUV). Data are reported as mean ± SEM (n = 3), ****p<0.0001. (D) TEM images show the morphology of Aβ40 alone (left) and in the presence of PC (middle) and PCPG (right) LUVs at the same molar ratios. White arrows highlight points where LUVs are in contact with fibrils. Scale bars represent 500 nm.
Aβ40 showed an overall increase in ThT fluorescence in the plateau phase when incubated with PC and, to a greater extent, PCPG LUVs (1:30 molar ratio, Aβ40:LUV). While this may be due to increased fibril content through a shift in the proportion of recruited monomer, it is not possible to rule out unrelated interactions with the dye [47] or incorporation of lipids into the fibril increasing the aggregate mass [48]. Therefore, we also interrogated the time course of aggregation to gain further insight into the observed differences. The time taken to reach half maximum fluorescence (t50) was calculated for each condition by fitting the data set to a sigmoidal model (Fig. 1C). LUVs significantly reduced the time taken for Aβ40 to reach half maximum fluorescence, with PC having a more pronounced effect. The formation of mature fibrils was confirmed by transmission electron microscopy (TEM) (Fig. 1D). Interestingly, Aβ40 fibrils incubated with LUVs appeared to be vesicle-bound, suggesting LUVs increase the fibrilization propensity of Aβ40 through interactions at the bilayer surface.
Additional ThT assays were conducted to assess the efficacy of small molecule amyloid inhibitors while in the presence of LUVs. Resveratrol and EGCG are known fibril inhibitors broadly effective against a variety of amyloidogenic proteins. However, they have differing modes of inhibition and differing levels of efficacy [49]. EGCG and resveratrol were added at a 1:1 molar ratio which has been used previously to assess inhibitory efficacy of these molecules [33,50]. Upon incubation with LUVs, both EGCG (Fig. 2A) and resveratrol (Fig. 2B) were found to be less effective as fibrilization inhibitors. To quantify the relative efficacy, the fluorescence intensity values were averaged across the final hour of the assay and normalised against the Aβ40 control. A decrease in relative fluorescence was interpreted as an increase in inhibition efficacy (Figs. 2C and 2D).
Figure 2. Inhibition of Aβ40 fibril formation by EGCG and resveratrol is compromised by PC and PCPG LUVs.

The effect of LUVs on Aβ40 (black) fibril inhibition by (A) EGCG (50 μM) in the absence (grey) and presence of PC (blue) and PCPG (green) LUVs, and (B) resveratrol (50 μM) in the absence (brown) and presence of PC (pink) and PCPG (purple) LUVs was assessed through ThT fluorescence assays (1:30, Aβ40:LUV). The extent of fibril inhibition afforded was calculated by comparing the average ThT fluorescence maximum in the final hour of the assay across each treatment for (C) EGCG and (D) resveratrol containing conditions. Fibril formation in the presence of EGCG was confirmed by TEM (E) for Aβ40 with both PC (top) and PCPG (bottom) LUVs (scale bars represent 500 nm). The time taken to reach half ThT maximum fluorescence (t50) was also compared across conditions for (F) EGCG and (G) resveratrol. Data are reported as mean ± SEM (n = 3), *p<0.05, ***p<0.001, ****p<0.0001.
Consistent with previous studies of Aβ40 [33,49], EGCG limits ThT fluorescence at a 1:1 molar ratio, with a 95% decrease in fluorescence at plateau compared to Aβ40 alone. However, when incubated with either LUV at a 1:30 ratio (Aβ40:LUV), the inhibitory efficacy of EGCG was reduced, with a decrease in fluorescence intensity in the plateau phase of 81% and 50% in the presence of PC and PCPG LUVs respectively (Fig. 2B). Interestingly, Aβ40 in the presence of LUVs and EGCG displayed a decrease in ThT fluorescence following approximately 7 and 15 hours of incubation for PCPG and PC respectively, suggestive of decreased mature fibril formation. This observed loss in fluorescence may be attributed to LUV degradation during the assay [51], at which point partitioned EGCG may be released from the bilayer and is then able to remodel mature fibrils into smaller, amorphous aggregates [32,52]. To further complement the ThT assays, TEM data was also collected for Aβ40 aggregated in the presence of EGCG and PC or PCPG LUVs (Fig. 2E). Despite incubation in the presence of the inhibitor, significant fibril formation was observed, again consistent with the notion that the LUVs decrease the inhibitory capacity of the small molecules. In addition to an effect on maximum fluorescence, EGCG also notably shifts the t50 values in the kinetic assay (Fig. 2F), from 5.9 to 7.7 hours for Aβ40 alone and with EGCG respectively. Again, the LUVs attenuate this effect, with t50 decreased compared to the control by 11 % and 127 % for PC and PCPG LUVs, respectively.
The efficacy of resveratrol as an inhibitor of Aβ40 aggregation was also examined in the presence of LUVs. Resveratrol is generally less effective at inhibiting Aβ40 fibril formation than EGCG, with greater ThT fluorescence observed at the same inhibitor concentration. Nevertheless, like EGCG, incubation with LUVs resulted in a further reduction in the inhibitory efficacy of resveratrol (Fig. 2D). However, unlike EGCG, no difference was observed between the zwitterionic (PC) and anionic (PCPG) LUV systems, and no decline in fluorescence was observed over time. This latter point is consistent with the reported mechanism of action of resveratrol; despite being shown to remodel mature fibrils at high concentrations [52], resveratrol principally appears to cap fibril formation but is unable to disassemble fibrils [37]. Interestingly, the t50 for Aβ40 in the presence of resveratrol was reduced (Fig. 2G) reflecting that resveratrol acts in the elongation phase to cap mature fibrils while not interacting with early oligomeric species, therefore not limiting the initial rate of aggregation. Incubation with resveratrol and PC or PCPG LUVs also reduced the t50 compared to Aβ40 alone, with PCPG showing a greater reduction.
Overall, the combined data demonstrate the ability of LUVs to reduce the efficacy of two well-known small molecule inhibitors of fibrilization. PCPG was found to affect Aβ40 inhibitor efficacy more significantly, and anionic LUVs have also been shown in literature to have a greater affinity for Aβ40 [22,53]. For these reasons, we chose to focus only on PCPG LUVs for subsequent experiments in this study.
PCPG LUVs increase αS fibril aggregation and reduce EGCG and resveratrol inhibitor efficacy
To investigate whether the effects of LUVs on fibrillar aggregation and inhibition by EGCG and resveratrol are consistent between protein systems, we performed analogous ThT experiments using αS. Compared to Aβ40, αS aggregates at a much slower rate and allows for in vitro kinetic analysis to be performed in the presence and absence of seed fibrils in a robust manner (Fig. 3A and 3B respectively). This allows us to separately examine the inhibitory action of EGCG and resveratrol during primary nucleation and fibril elongation stages in the presence of LUVs. EGCG and resveratrol were subsequently introduced into these systems to ascertain whether the LUVs had similar effects on inhibitory efficacy against αS aggregation (Fig. 3C and 3D).
Figure 3. PCPG LUVs modulate the inhibitory efficacy of EGCG and resveratrol during seeded and unseeded αS aggregation.

Fibril formation of (A) unseeded αS (150 μM) and (B) seeded αS (50 μM with 2.5% w/w seed fibrils) in the presence of EGCG (50 μM) and resveratrol (50 μM) was monitored through ThT fluorescence assays in the presence and absence of PCPG LUVs (1:30 αS:LUV). The extent of fibril inhibition afforded by EGCG and resveratrol was calculated by comparing the average ThT fluorescence maximum in the final hour of the assay across each treatment for (C) unseeded and (D) seeded assays, and the time taken to reach half maximum ThT fluorescence (t50) was also compared (E and F for seeded and unseeded assays respectively). Data are reported as mean ± SEM (n = 3), *p<0.05.
As observed for Aβ40, in the presence of PCPG LUVs, αS fibrils aggregated faster compared to αS alone during primary nucleation-mediated fibrilization (i.e. unseeded aggregation) (Fig. 3E) demonstrated by a significant reduction in the t50. However, in the presence of seed fibrils (2.5% w/w), the rate of aggregation remained similar to αS alone (Fig. 3F). The data suggests that LUVs primarily modulate the rate of aggregation through alterations in monomeric or early oligomeric structures during the nucleation phase.
In the unseeded assays, the inhibitory activity of resveratrol towards αS aggregation was completely nullified in the presence of PCPG LUVs (Figs. 3A and C), and in fact, αS fibrils with resveratrol and PCPG aggregated faster than αS alone, despite the presence of inhibitor. The fluorescence intensity during the plateau phase measured in the presence of resveratrol was also greatly reduced by PCPG LUVs in the seeded αS system (Fig. 3B and D), however the rate of aggregation remained relatively consistent across conditions for this inhibitor in seeded assays (Fig. 3F). Contrastingly, EGCG was found to be far more effective as an inhibitor against both unseeded and seeded αS aggregation, displaying little ThT fluorescence even in the presence of PCPG LUVs (Fig. 3A and 3B).
LUVs and inhibitors differentially influence the distribution of protein aggregation states
ThT assays are only able to provide insight into the fibril content during the aggregation process, which ignores important details regarding monomeric structure and oligomeric populations. For these reasons, we also carried out immunoblotting experiments to ensure ThT-derived data mirrored relative fibril content across treatments, as well as to gain insight into pre-fibrillar oligomeric populations. Aβ40 fibril-specific antibodies were first used to validate ThT fluorescence data, with the relative intensity (i.e., abundance) of fibrillar species compared to Aβ40 alone as a function of time (Fig. 4A). In general, the relative abundance of fibrils observed over time in the presence of LUVs and/or inhibitors correspond to the ThT kinetic data. Notably, the relative endpoint fibril abundance (Fig. 4B) correlates well with the relative percentage inhibition estimated from ThT fluorescence assays at plateau (c.f. Figs 2A and 2F), lending weight to the ThT assay as a suitable means for estimating fibril inhibitor efficacy, despite the known limitations of this approach.
Figure 4. LUVs alter the abundance of Aβ40 and αS oligomers in the presence of EGCG and resveratrol during fibril formation.
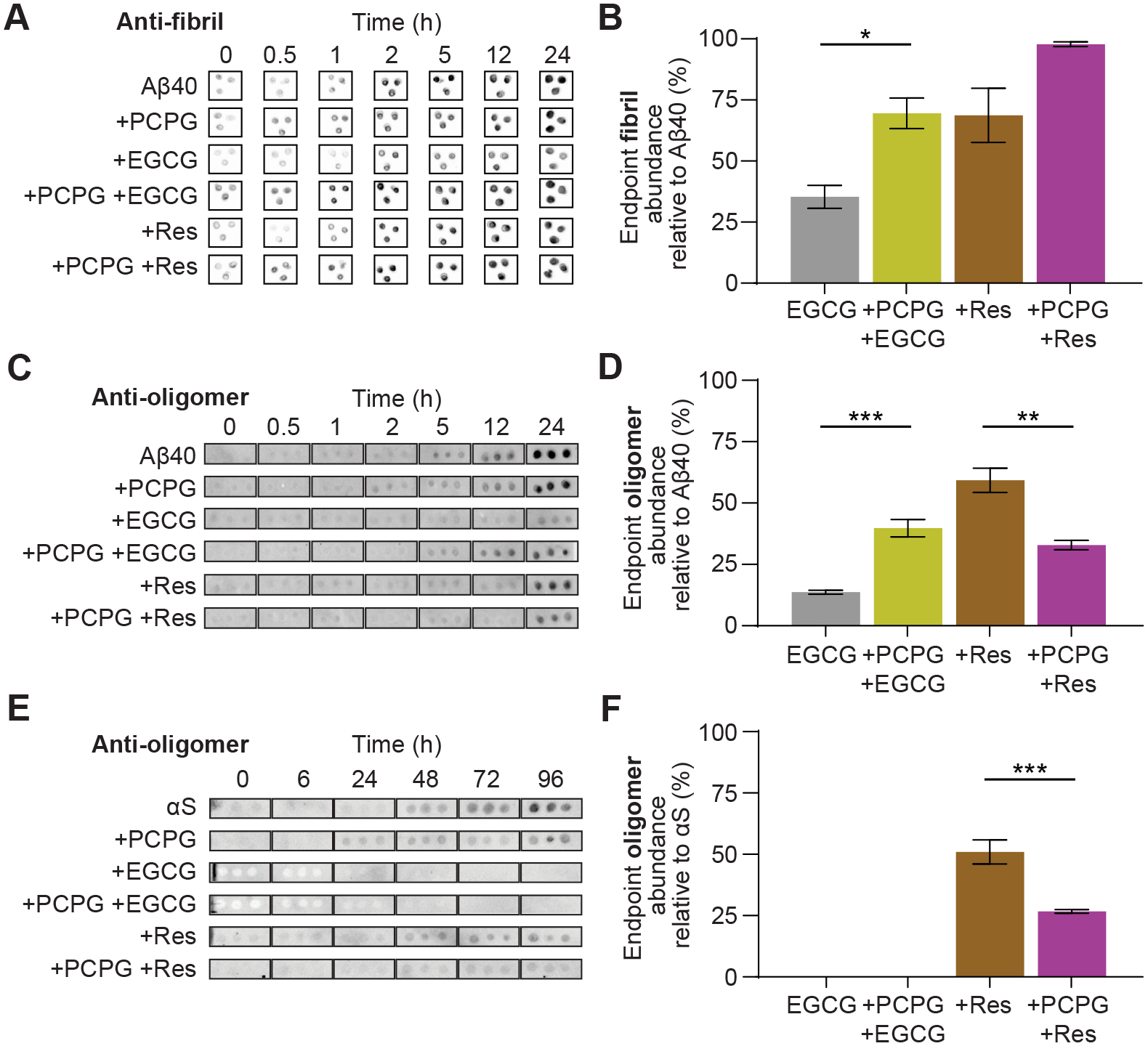
The effect of LUVs on fibril concentrations and fibril inhibition by EGCG (50 μM) and resveratrol (50 μM) was monitored through immuno-dot blot assays. (A) Aβ40 (50 μM) was incubated in the presence and absence of inhibitors and LUVs (1:30 Aβ40:LUV) and the relative abundance of Aβ40 fibrils at the assay endpoint (24 h) was quantified using densitometry (B). Similarly, the effect of LUVs on oligomer formation during aggregation was observed for (C) Aβ40 (50 μM), incubated in the presence and absence of EGCG or resveratrol and LUVs (1:30, Aβ40:LUV), with the abundance of oligomers at 24 h quantified by densitometry (D), and for (E) αS (50 μM) incubated in the presence and absence of EGCG or resveratrol and LUVs (1:30, αS:LUV) and quantified at 96 h by densitometry (D). Data are reported as mean ± SEM (n = 3), *p<0.05, *p<0.01, ***p<0.001.
We also performed immunoblotting to observe the abundance of pre-amyloid oligomers, using an A11 ‘oligomer-specific’ antibody for both Aβ40 and αS in the presence of LUVs and EGCG or resveratrol, in an effort to gain additional mechanistic insight into their effects on early aggregation pathways. While these antibodies are not entirely selective for pre-fibrilar species, having been shown to bind to some extent to fibrils as well as early oligomeric states [54], they can still yield information on early aggregation events that fibril specific assays cannot, particularly for αS which shows an extended lag phase of at least 30 hours by ThT assays.
Oligomer formation is discernibly more evident at earlier timepoints in the presence of PCPG LUVs for both proteins (2 h for Aβ40 and 24 h for αS, which is during the lag-phase observed by ThT assays) (Fig. 4C and 4E). This suggests the enhanced fibril formation observed in ThT assays by PCPG lipids is a result of early formation of oligomeric species which act to nucleate fibril growth. The ability of EGCG and, to a lesser extent resveratrol, to reduce the concentration of oligomeric states compared to the aggregating control was readily observed for both Aβ40 and αS systems (Fig. 4D and 4F respectively), likely contributing (at least in part) to their anti-amyloid activity. Notably however, PCPG LUVs had a significantly different influence on the ability of the inhibitors to reduce oligomer abundance; in the case of resveratrol it served to further decrease oligomer abundance in both systems, while PCPG LUVs diminished the ability of EGCG to reduce Aβ40 oligomer formation with no change for αS. This further reinforces a different mechanism of action of the two inhibitors, and highlights the need to consider more deeply the mode of inhibition of each compound for a given target system.
IM-MS reveals structural changes induced by EGCG complexes with Aβ40 and αS monomers
Thus far, our data suggests that LUVs interact with amyloidogenic proteins early in the aggregation pathway, potentially through changes in the structure of monomeric or low molecular weight oligomeric species. For this reason, we applied native IM-MS to interrogate structural features of soluble protein forms to provide further mechanistic insight into the combined effects of PCPG LUVs on aggregation and inhibition by EGCG and resveratrol. The electrospray ionisation process gives rise to multiply charged protein ions, with the degree of protonation dependent upon solvent-exposed surface area and hence protein foldedness [55]. In native IM-MS experiments, ions are separated not only based on their m/z ratio, but also on rotationally averaged CCS values for a given charge state; the longer that ion takes to traverse the ion mobility sector, the larger its rotationally averaged CCS and vice versa. IM arrival time distributions can therefore be extracted from ion populations of a given m/z to provide structural information based on overall size.
We first analysed any changes in the monomeric structure of Aβ40 due to the addition of EGCG, PCPG LUVs or both (Fig. 5A). EGCG and PCPG were primarily investigated here as they had the greatest effect on both inhibition and aggregation respectively, and resveratrol was not found to form stable complexes with either of the proteins (data not shown). While PCPG showed little effect on the native Aβ40 mass spectrum, the addition of EGCG produced peaks corresponding to Aβ40:EGCG complexes with stoichiometries up to a 1:3 Aβ40:EGCG ratio (Fig. 5A). However, in the presence of both EGCG and PCPG LUVs, only 1:1 Aβ40:EGCG complexes were observed. This reduction in EGCG binding to Aβ40 potentially indicates a mechanism in which PCPG LUVs reduces EGCG inhibitor efficacy by inducing structural changes that decrease EGCG binding affinity, or through a decreased availability due to partitioning of the inhibitor in the lipid environment. No stable complexes were detected between Aβ40 and PC or PG lipids.
Figure 5. Native IM-MS unravels the structural heterogeneity of Aβ40 in the presence of EGCG and PCPG LUVs.
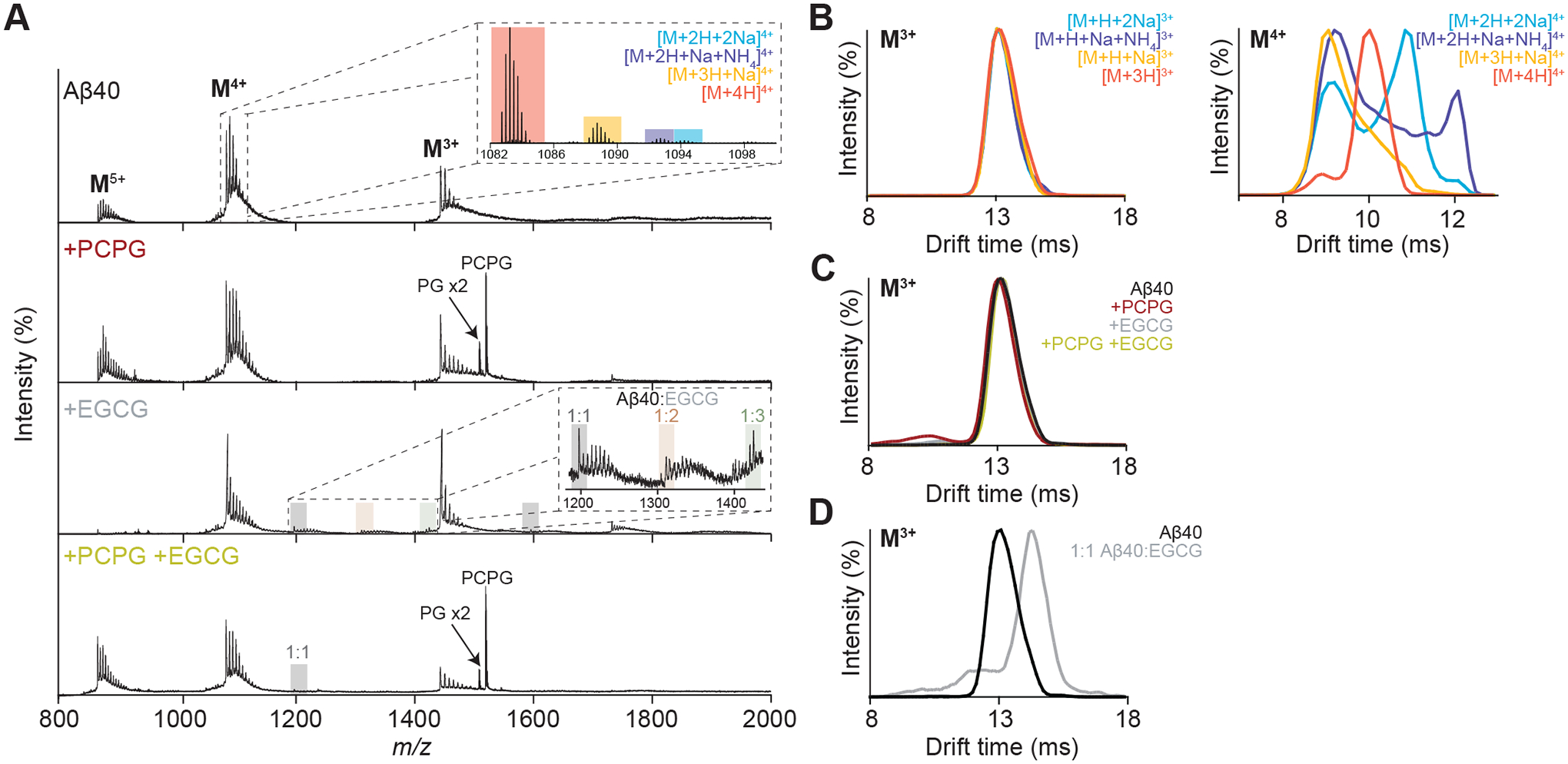
(A) Native MS spectra of Aβ40 (50 μM) in the presence of PCPG (500 μM) and/or EGCG (50 μM). Complexes with EGCG (1:1, 2:1 and 3:1, EGCG:Aβ40) are primarily observed from the M4+ charge state (A, inset). (B) Extracted ATDs for the various Aβ40 adducts giving rise to ions with 3+ (left) and 4+ (right) charge states reveal compaction effects of non-specific metalation specific to the higher charge state. (C) Extracted ATDs for the M3+ ions in the presence of PCPG (500 μM) and/or EGCG (50 μM) demonstrate no conformational change of the monomeric protein in these conditions, while a shift to higher drift time is observed from the ATDs extracted for the M3+ ions of the 1:1 Aβ40:EGCG complex compared to Aβ40 alone (D).
To better understand the overall structural transitions of Aβ40 under these varying conditions, the ion mobility arrival time distributions (ATDs) for the major charge states were extracted and compared. Given the high mass resolution of the spectrometer utilised for this analysis, it was possible to extract ATDs for individual adduct types, and initial comparisons between the most abundant M4+ ions revealed large structural differences depending on the whether ions were adducted with H+, Na+ and NH4+ (Fig. 5B). This observation is consistent with previous studies which demonstrate that nonspecific metal ion adduction to proteins results in ions with more compact conformations than non-metallated forms with the same charge, with the greatest effect observed for intermediate protein charge states [56]. These results suggest that a compaction of the gas-phase ion upon attachment of metal ions may not be a reliable indicator of any conformational changes that occur in solution as a result of changed solution conditions. In contrast, the M3+ ions showed little variation in their ATDs in response to adduct type, thereby indicating a more folded structure less easily influenced by Na+ or NH4+. For this reason, a comparison of the M3+ ATDs was considered more informative to probe for changes in protein structure with varying experimental conditions.
Interestingly, the ATDs of the M3+ monomer ions showed little variation between each condition, suggesting no significant change in the conformation of the soluble monomeric protein in the presence of PCPG vesicles or EGCG (Fig. 5C). Consequently, it is likely the decreased extent of EGCG binding to Aβ40 in the presence of the LUVs as indicated from the native mass spectra is not a result of structural changes but in fact a result of reduced availability of the inhibitor due to partitioning in the membrane. A comparison of the ATDs for the M3+ ions of 1:1 Aβ40:EGCG complexes showed an increase in drift time upon binding of EGCG, however this was proportional to the increase in mass due complexation of EGCG (10% mass increase, 9% drift time increase). This is consistent with previous observations by IM-MS [57,58] which suggest EGCG targets compact protein conformations for binding.
Analogous native IM-MS experiments were performed to evaluate the effects of EGCG and PCPG LUVs on αS. Two charge state distributions were observed for the protein in the native mass spectrum, corresponding to unfolded (higher charged) and partially folded (lower charged) structural populations (Fig. 6A). Much like for Aβ40, αS was shown to form αS:EGCG complexes, with up to 2 EGCG molecules identified bound the protein. These EGCG:protein complexes were more notable at lower charge states (M7+ and M8+), again consistent with a preference for EGCG to bind to more compact protein conformations. A reduction in the abundance of EGCG:protein complexes was once more observed in the presence of PCPG LUVs, which provides further basis for decreased inhibitor efficacy in the presence of LUVs, likely due to partitioning in the membrane environment. As with Aβ40, no stable protein-lipid interactions were detected, however, dimers were more easily observed for αS. A comparison of the relative dimer abundance in each condition revealed that less dimer was present when incubated with EGCG, but incubation with both EGCG and PCPG LUVs restored the relative amount of dimer to levels within error of αS alone (Fig. 6B). This is in slight contrast to immunoblotting assays which suggested PGPG LUVs did not diminish the ability of EGCG to prevent oligomer formation (Fig. 4F), and implies the inhibitory effects of EGCG may arise by action on larger oligomeric states that are not observed in the MS experiment.
Figure 6. Native IM-MS examines the structural heterogeneity of αS in the presence of EGCG and PCPG LUVs.
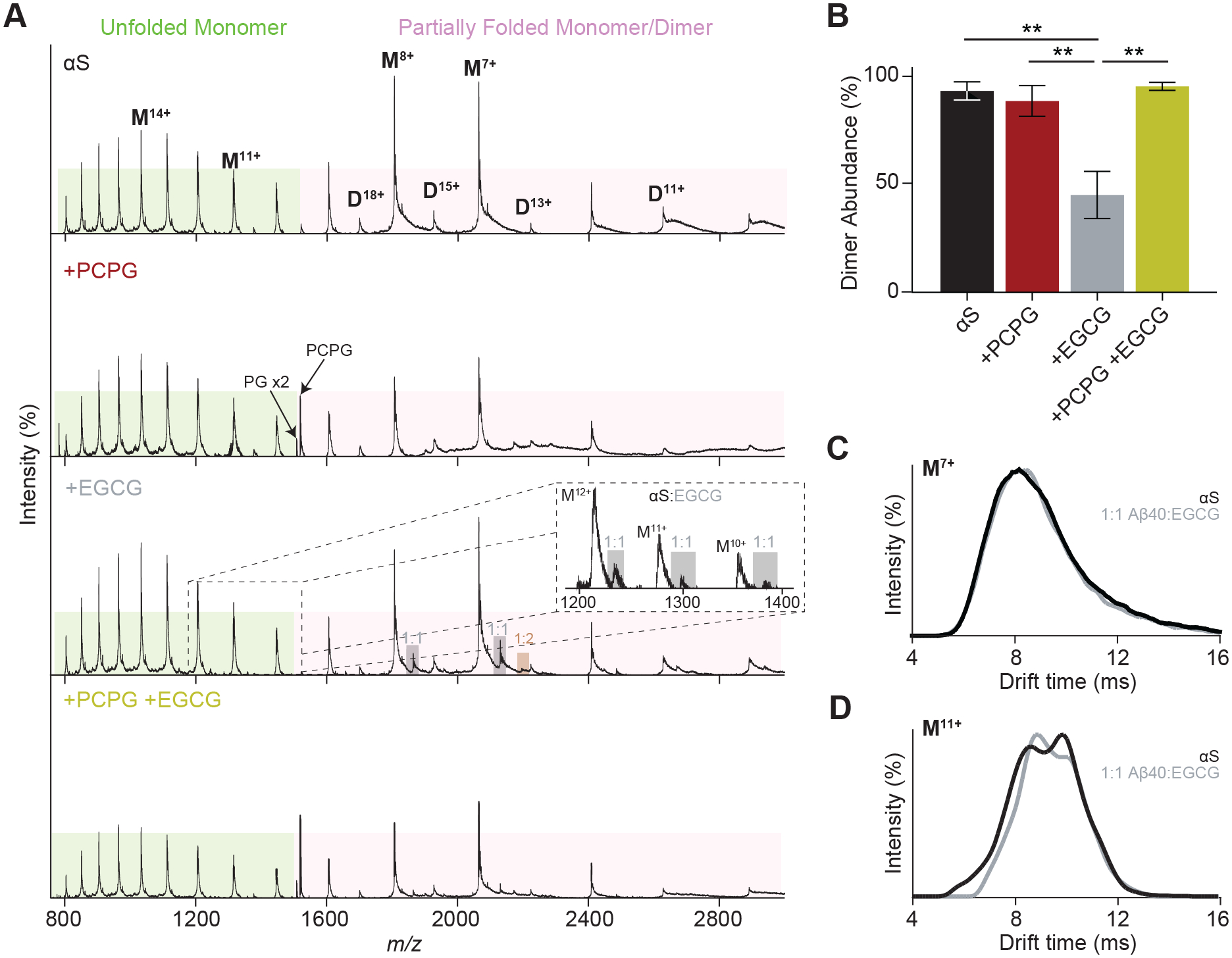
(A) Native MS spectra of αS (50 μM) in the presence of PCPG (500 μM) and/or EGCG (50 μM). Complexes with EGCG are shown (A, inset). (B) Relative dimer intensities were compared between each condition. Data are reported as mean ± SEM (n = 3), **p<0.01. (C) ATD analysis of the αS monomer in the absence of EGCG, in the presence of EGCG (50 μM) and the 1:1 αS:EGCG complex for (C) the more folded M7+ ions and (D) the less folded M11+ ions suggest EGCG preferences formation of more compact structures.
The 7+ and 11+ charge states of αS were analysed by IM-MS (Figs. 6C and 6D) to reflect the partially unfolded αS populations respectively. For the 7+ ions, no variation in ATDs are noted upon EGCG binding, consistent again with a preference for EGCG to bind to already compact structures. In the case of the 11+ ions however a shift in ATD is observed towards lower drift time, suggesting EGCG also has an ability to direct extended conformations to more ordered structures, a phenomenon which has been previously observed and attributed to formation off-pathway species [58]. Since compaction has been noted as a phenomenon occurring before the fibrillar aggregation of amyloid proteins, the high efficacy of EGCG as a fibril inhibitor may be due to targeting this process through complexation [59–61].
Discussion
Effects of the lipid membrane are vitally important considerations in the fibrillar aggregation process which have only gained recent attention and focus. Lipid binding is important for both Aβ40 and αS in their functional states. For example, amyloid precursor protein, the protein from which Aβ40 is derived through proteolysis, is a membrane bound protein [25,62] and αS is thought to play a role as a lipid vesicle tethering protein via an N-terminal double-anchor mechanism [63–65]. Many studies have suggested that lipid membranes initiate aggregation of Aβ40 and αS, acting as a catalytic surfaces for misfolding and fibrilization [20,66,67]. Since potential small molecule inhibitors may be competing against this process, it is important to consider their activity in the lipid environment. On top of this, the aetiology of misfolding diseases is also thought to be due to membrane interactions of misfolding monomer and/or oligomers through disruption, permeation and lysis of neuronal membranes [11,12,68,69]. Therefore, a better understand of this interplay is important to lead to successful therapy design. Given that recent work has shown the effectiveness of EGCG as a fibril inhibitor is environment dependent, including considerations such as oxidation status and pH [70], we investigated how lipid environments may affect the efficacy of two well-known inhibitors of fibrillization, EGCG and resveratrol, towards Aβ40 and αS fibril formation.
We have demonstrated that both PC and PCPG LUVs increase the rate of Aβ40 aggregation. Similarly, PCPG LUVs increased the rate of αS fibrillar aggregation, although only in the absence of preformed seed fibrils. This is consistent with a mechanism in which the lipids primarily influence aggregation in the nucleation phase through a pathway dependent on lipid composition and relative concentration of aggregating protein [20,26,71]. TEM data indicated LUVs were bound to fibrils, potentially due to elongating fibrils propagating from a localised protein concentration on the LUV surface.
Importantly, we have also demonstrated that the lipid environment drastically reduces the ability of EGCG and resveratrol to prevent aggregation in Aβ40, while EGCG only retained inhibitory activity in αS systems. This infers that EGCG is less effective in targeting rapidly aggregating structures, like Aβ40, in contrast to the more slowly aggregating αS. Anionic PCPG LUVs had a more significant effect than zwitterionic PC LUVs in the case of EGCG and Aβ40 aggregation, although both LUVs reduced resveratrol inhibition equally. These differences highlight the different inhibition mechanisms of EGCG and resveratrol, where EGCG inhibits in the nucleation phase and can remodel fibrils, while resveratrol caps fibrils early in the elongation phase [30–33,36–38].
Immunoblotting revealed the appearance of oligomeric Aβ40 and αS occurred earlier in the aggregation time course, and with a much greater abundance preceding the onset of the elongation phase for αS, when in the presence of PCPG LUVs. Recent research has shown the highest energy barrier in fibril elongation is the transition from oligomer to immature fibrils, and the majority of these oligomers do not form fibrils [72]. A higher oligomer concentration would therefore be expected to increase the probability of oligomer to fibril transitions, denoting a mechanistic explanation for the increased aggregation rate. Interestingly, oligomer abundance was higher in the presence of resveratrol alone than both resveratrol and PCPG LUVs, which was contradictory to that observed for EGCG and PCPG. The increased oligomeric concentrations due to PCPG LUVs may outpace the ability for resveratrol to effectively cap fibrils, leading to the greater observed influence of LUVs on resveratrol inhibitor efficacy.
Native MS revealed Aβ40 and αS formed complexes with EGCG while resveratrol did not, consistent with previous work [57,58]. However, the abundance of EGCG-protein complexes was notably reduced in the presence of lipid vesicles. Utilising native IM-MS data we are able to show that PCPG LUVs had little influence on the structure of monomeric protein, and that protein:EGCG complexes increased the extent of structural compaction at less folded, highly charged states but had little effect on the more folded, lower charged states for both Aβ40 and αS. This suggests the ability of LUVs to decrease the effectiveness of the small molecule inhibitors is less related to structural changes and instead presumably a result of decreased availability due to partitioning between the lipid membrane and surrounding solvent. Given both amyloid proteins and small molecule inhibitors will partition between the lipid membrane and surrounding solvent to varying degrees, and since solution aggregation mechanisms are different to those on the bilayer [20,73], this partitioning behaviour is likely to be a significant contributor to variations in inhibitor efficacy for specific protein, inhibitor and lipid composition combinations by altering the extent of protein-inhibitor interactions [41,74].
In summary, we provide further evidence that lipid environments affect the structure and aggregation kinetics of amyloidogenic proteins, Aβ40 and αS, and add insight into inhibition mechanisms of EGCG and resveratrol in this environment. Importantly, LUVs appear to reduce the effects of these small molecule fibril inhibitors by partitioning effects in the membrane environment. This study highlights the importance of the cellular membrane in the fibrillar aggregation of misfolded proteins, together with our relative lack of understanding on the structural effects that this environment induces on amyloid structure and dynamics. A lack of regard for the effects of the lipid environment during therapeutic design ultimately exemplifies how current developments have been ineffective in treating protein misfolding diseases.
Materials and Methods
Human Aβ40 (lyophilised powder) was purchased from Anaspec (Fremont, CA, USA). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol), were purchased from Avanti Polar Lipids (Alabaster, AL, USA). EGCG, resveratrol and all other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified.
Preparation of Aβ40, αS and LUVs
Expression and purification of human αS (UniProt accession number: P37840) was performed as described previously [75]. Pre-formed αS seed fibrils were produced from the monomeric protein preparation as described previously [76]. Aβ40 was pre-treated with NH4OH to ensure monomerisation according to the method described by Ryan, et al [77]. Aliquots were snap frozen and stored at −80 °C and were only defrosted immediately before use. Large unilamellar vesicles (LUVs) were extruded using an Avanti Mini-Extruder with 0.1 μm extruder filters (Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. LUV diameters were confirmed by dynamic light scattering (DLS) and transmission electron microscopy (TEM).
Dynamic light scattering
LUV diameters were measured by dynamic light scattering using a Malvern Zetasizer nano ZS (Malvern Instruments, Malvern, Worcestershire, UK). LUVs were characterised at a concentration of 1.5 mM with a 633 nm laser at a back scattering angle of 173°.
Thioflavin T assays
Fibrillar aggregation was monitored in real-time using a thioflavin T (ThT) fluorescence assay [78]. Primary nucleation-mediated fibrilization of Aβ40 (50 μM) and αS (150 μM) was performed in 100 mM ammonium acetate (pH 7.4) and ThT (40 μM) in the absence and presence of LUVs (1:30 molar ratio; protein:lipid) [24]. Inhibitor concentrations were based on previously established concentrations [33,50]. Fibrilization of αS (50 μM) in the presence of short seed fibrils (2.5 % w/w) was performed to examine the effects on fibrillar aggregation in the absence and presence of LUVs and inhibitors. Samples were plated (50 μL/well) into sealed 384-well microplates (Greiner Bio-One, Kremsmünster, Upper Austria, Austria) and fibril formation was monitored using a FLUOstar Optima microplate reader (BMG Lab Technologies, Melbourne, Victoria, Australia) over a period of 20 h (Aβ40), 25 h (seeded αS) and 120 h (unseeded αS) at 37 °C with shaking (double orbital, 50 rpm) for 30 s prior to fluorescence measurement. ThT fluorescence was measured with an excitation and emission wavelength of 440 and 490 nm, respectively. All data, except for seeded αS assays, were fitted with non-linear sigmoidal dose response to calculate and compare the rate of fibrillar aggregation by finding the time taken to reach half maximum fluorescence (t50). Seeded αS assays were fitted with a one-phase exponential association. The extent of fibril inhibition afforded by each inhibitor with and without LUVs was calculated by comparing the average ThT fluorescence maximum across each treatment in the final hour of the assay. ThT data were analysed and subjected to a Tukey’s multiple comparisons test using Prism 9.0 (GraphPad Software, La Jolla, USA), with all assays performed at least three times and reported as mean ± SEM.
Transmission electron microscopy
Samples were taken immediately at the conclusion of the ThT assay and placed directly (3 μL) onto a carbon-coated 400-mesh nickel TEM grid (ProSciTech, Thuringowa Central, Queensland, Australia) [32]. Grids were washed with 0.22 μm filtered MilliQ water and stained with 2% (w/v) uranyl acetate solution. Samples were viewed using either a CM100 transmission electron microscope (Philips, Eindhoven, North Brabant, Netherlands) or a Tecnai G2 Spirit TEM (FEI, Hillsboro, OR, USA) with a magnification of 45 000×.
Immunoblotting
The relative abundance of various morphological states (i.e. monomer, oligomer or fibril) of Aβ40 and αS was assessed by immuno-dot blotting as described previously [33,79]. Samples were prepared in sealed 384-well microplates (Greiner Bio-One, Kremsmünster, Upper Austria, Austria) and incubated using a FLUOstar Optima microplate reader (BMG Lab Technologies, Melbourne, Victoria, Australia), replicating the conditions of the ThT kinetic assays in the absence of ThT. Briefly, 1.5 μL of sample at various time points during fibril formation were dotted onto nitrocellulose membranes and blocked with 5% skim milk in TBS-T at room temperature for 1 h. Membranes were washed four times prior to incubation with either an A11 anti-oligomer (1:5000 in 2% skim milk TBS-T) (Invitrogen, Carlsbad, CA, USA) or an anti-fibril (ab205339, 1:5000 in 2% skim milk TBS-T)) (Abcam, Cambridge, MA, USA) at 4 °C overnight. An IgG-HRP secondary antibody (403001) and Clarity™ ECL substrate (both Bio-Rad, Hercules, CA, USA) were used for detection. The intensity of the blots was quantified using ImageJ [80], where experiments were performed in triplicate and data reported as mean ± SEM. Data was analysed by one-way ANOVA using Prism 8.0 (GraphPad Software, La Jolla, CA, USA).
Ion-mobility mass spectrometry
Native mass spectra were acquired on a Synapt HDMS G1 Q-TOF instrument, while IM-MS analysis was performed using a Synapt HDMS XS Q-TOF mass spectrometer (Waters Corporation, Manchester, Greater Manchester, UK), both using a nano-electrospray ionisation source. Samples were prepared in 100 mM ammonium acetate (pH 7.0) to a final protein concentration of 50 μM. Aβ40 and αS were incubated in the presence of LUVs (1:10 molar ratio; protein:LUV) and/or inhibitor (1:1 molar ratio; protein:inhibitor) for 10 min at room temperature before analysis. Longer incubation times were avoided to prevent clogging of the nanospray needle due to aggregation, allowing for more consistent data collection. Samples were loaded into platinum-coated borosilicate glass emitters prepared in-house. Gentle ionisation and gas-phase conditions were applied to minimise structural changes prior to detection [81], with instrument parameters as follows: capillary voltage, 1.60 kV; sampling cone, 30 V; extraction cone, 1.5 V; trap/transfer collision energy, 10–15 V; trap gas, 5.5 l/h; backing gas, ~4.5 mbar. The parameters for IM were as follows: Trap DC bias, 20 V; IM cell wave height, 8 V; IM cell wave velocity, 350 m/s; transfer t-wave height, 8 V; transfer t-wave velocity, 250 m/s. Synapt XS settings were consistent with the G1 with the exception of an IM cell wave height of 40 V and IM cell wave velocity of 1100 m/s. Mass spectra and arrival time distributions (ATDs) were viewed using MassLynx (v4.1) and DriftScope (v2.1), respectively (Waters Corporation, Manchester, Greater Manchester, UK). Relative dimeric populations were deconvolved using UniDec [82]. A FWHM of 0.85 m/z, charge range of 1–50 and mass range of 5–500 kDa were input. A curved background subtraction of 100 was also applied.
Acknowledgements
H.M.S. was supported by a Faculty of Sciences Divisional Scholarship from the University of Adelaide. We thank Dr. Mandy Leung for her assistance with DLS data acquisition, and Adelaide Microscopy (University of Adelaide) for technical assistance with TEM. We also thank Flinders Analytical (Flinders University, Australia) for access to IM-MS instrumentation. This research was financially supported in part by an Australian Research Council Discovery Project Grant to T.L.P. (DP170102033) and the United States National Institutes of Health/National Institute of General Medical Sciences (R35 GM128624 to M.T.M.).
Abbreviations:
- αS
α-synuclein
- Aβ
amyloid β
- ATD
arrival time distribution
- CCS
collisional cross section
- DLS
dynamic light scattering
- EGCG
(−)-epigallocatechin gallate
- IDP
intrinsically disordered protein
- IM-MS
ion mobility mass spectrometry
- hIAPP
human islet amyloid polypeptide
- LUVs
large unilamellar vesicles
- PC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol)
- ThT
thioflavin T
- TEM
transmission electron microscopy
Footnotes
Conflicts of Interest: The authors declare that they have no competing interests associated with the contents of this manuscript.
References
- 1.Dobson CM (2004) Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol 15, 3–16. [DOI] [PubMed] [Google Scholar]
- 2.Massi F & Straub JE (2001) Energy landscape theory for Alzheimer’s amyloid β‐peptide fibril elongation. Proteins 42, 217–29. [DOI] [PubMed] [Google Scholar]
- 3.Dodart J-C, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM & Paul SM (2002) Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat Neurosci 5, 452–457. [DOI] [PubMed] [Google Scholar]
- 4.Dahlgren KN, Manelli AM, Blaine Stine W, Baker LK, Krafft GA & Ladu MJ (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
- 5.Lin WH, Ciccotosto GD, Giannakis E, Tew DJ, Perez K, Masters CL, Cappai R, Wade JD & Barnham KJ (2008) Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J Neurosci 28, 11950–11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Günther S, Walsh DM & Wanker EE (2012) Small-molecule conversion of toxic oligomers to nontoxic β-sheet–rich amyloid fibrils. Nat Chem Biol 8, 93–101. [DOI] [PubMed] [Google Scholar]
- 7.Bucciantini M, Giannoni E, Chiti F, Baroni F, Taddei N, Ramponi G, Dobson CM & Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. [DOI] [PubMed] [Google Scholar]
- 8.Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, Klenerman D, Wood NW, Knowles TPJ, Alfonso C, Rivas G, Abramov AY, Valpuesta JM, Dobson CM & Cremades N (2015) Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc Natl Acad Sci U S A 112, E1994–E2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mannini B, Mulvihill E, Sgromo C, Cascella R, Khodarahmi R, Ramazzotti M, Dobson CM, Cecchi C & Chiti F (2014) Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem Biol 9, 2309–2317. [DOI] [PubMed] [Google Scholar]
- 10.Ho CS, Khadka NK, She F, Cai J & Pan J (2016) Polyglutamine aggregates impair lipid membrane integrity and enhance lipid membrane rigidity. Biochim Biophys Acta - Biomembr 1858, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bode DC, Freeley M, Nield J, Palma M & Viles JH (2019) Amyloid-β oligomers have a profound detergent-like effect on lipid membrane bilayers, imaged by atomic force and electron microscopy. J Biol Chem 294, 7566–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairí M, Maglia G & Carulla N (2016) Aβ42 assembles into specific β-barrel pore-forming oligomers in membrane-mimicking environments. Proc Natl Acad Sci 113, 10866–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perissinotto F, Rondelli V, Parisse P, Tormena N, Zunino A, Almásy L, Merkel DG, Bottyán L, Sajti S & Casalis L (2019) GM1 Ganglioside role in the interaction of Alpha-synuclein with lipid membranes: Morphology and structure. Biophys Chem 255, 106272. [DOI] [PubMed] [Google Scholar]
- 14.Kawahara M, Ohtsuka I, Yokoyama S, Kato-Negishi M & Sadakane Y (2011) Membrane incorporation, channel formation, and disruption of calcium homeostasis by Alzheimer’s β-amyloid protein. Int J Alzheimers Dis 2011, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flagmeier P, De S, Michaels TCT, Yang X, Dear AJ, Emanuelsson C, Vendruscolo M, Linse S, Klenerman D, Knowles TPJ & Dobson CM (2020) Direct measurement of lipid membrane disruption connects kinetics and toxicity of Aβ42 aggregation. Nat Struct Mol Biol 27, 886–891. [DOI] [PubMed] [Google Scholar]
- 16.Ugalde CL, Lawson VA, Finkelstein DI & Hill AF (2019) The role of lipids in -synuclein misfolding and neurotoxicity. J Biol Chem 294, 9016–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jayasinghe SA & Langen R (2005) Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 44, 12113–12119. [DOI] [PubMed] [Google Scholar]
- 18.Terzi E, Hölzemann G & Seelig J (1995) Self-association of β-amyloid peptide (1–40) in solution and binding to lipid membranes. J Mol Biol 252, 633–642. [DOI] [PubMed] [Google Scholar]
- 19.Dikiy I & Eliezer D (2012) Folding and misfolding of alpha-synuclein on membranes. Biochim Biophys Acta - Biomembr 1818, 1013–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ & Dobson CM (2015) Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol 11, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee Y-H, La Rosa C, Lim MH & Ramamoorthy A (2017) Reduced lipid bilayer thickness regulates the aggregation and cytotoxicity of amyloid-β. J Biol Chem 292, 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin M-S, Chiu H-M, Fan F-J, Tsai H-T, Wang SSS, Chang Y & Chen W-Y (2007) Kinetics and enthalpy measurements of interaction between β-amyloid and liposomes by surface plasmon resonance and isothermal titration microcalorimetry. Colloids Surf B 58, 231–236. [DOI] [PubMed] [Google Scholar]
- 23.Seeligt J, Lehrmann R & Terzi E (1995) Domain formation induced by lipid-ion and lipid-peptide interactions. Mol Membr Biol 12, 51–57. [DOI] [PubMed] [Google Scholar]
- 24.Sani M-A, Gehman JD & Separovic F (2011) Lipid matrix plays a role in Abeta fibril kinetics and morphology. FEBS Lett 585, 749–754. [DOI] [PubMed] [Google Scholar]
- 25.Williams TL & Serpell LC (2011) Membrane and surface interactions of Alzheimer’s Aβ peptide - insights into the mechanism of cytotoxicity. FEBS J 278, 3905–3917. [DOI] [PubMed] [Google Scholar]
- 26.Habchi J, Chia S, Galvagnion C, Michaels TCT, Bellaiche MMJ, Ruggeri FS, Sanguanini M, Idini I, Kumita JR, Sparr E, Linse S, Dobson CM, Knowles TPJ & Vendruscolo M (2018) Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat Chem 10, 673–683. [DOI] [PubMed] [Google Scholar]
- 27.Naudí A, Cabré R, Jové M, Ayala V, Gonzalo H, Portero-Otín M, Ferrer I & Pamplona R (2015) Lipidomics of human brain aging and Alzheimer’s disease pathology. Int Rev Neurobiol 122, 133–89. [DOI] [PubMed] [Google Scholar]
- 28.2019 Alzheimer’s disease facts and figures (2019) Alzheimer’s Dement 15, 321–387. [Google Scholar]
- 29.Henríquez G, Gomez A, Guerrero E & Narayan M (2020) Potential role of natural polyphenols against protein aggregation toxicity: In vitro, in vivo, and clinical studies. ACS Chem Neurosci 11, 2915–2934. [DOI] [PubMed] [Google Scholar]
- 30.Freyssin A, Page G, Fauconneau B & Bilan A (2018) Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases. Neural Regen Res 13, 955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Q, Zhao Q, Peng J, Yu Y, Wang C, Zou Y, Su Y, Zhu L, Wang C & Yang Y (2019) Peptide-Polyphenol (KLVFF/EGCG) binary modulators for inhibiting aggregation and neurotoxicity of amyloid-β peptide. ACS Omega 4, 4233–4242. [Google Scholar]
- 32.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K & Wanker EE (2010) EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc Natl Acad Sci 107, 7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A & Wanker EE (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol 15, 558–566. [DOI] [PubMed] [Google Scholar]
- 34.Corpas R, Griñán-Ferré C, Rodríguez-Farré E, Pallàs M & Sanfeliu C (2019) Resveratrol induces brain resilience against Alzheimer neurodegeneration through proteostasis enhancement. Mol Neurobiol 56, 1502–1516. [DOI] [PubMed] [Google Scholar]
- 35.Regitz C, Fitzenberger E, Mahn FL, Dußling LM & Wenzel U (2016) Resveratrol reduces amyloid-beta (Aβ1–42)-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur J Nutr 55, 741–747. [DOI] [PubMed] [Google Scholar]
- 36.Feng Y, Wang X ping, Yang S gao, Wang Y jiong, Zhang X, Du X ting, Sun X xia, Zhao M, Huang L & Liu R tian (2009) Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 30, 986–995. [DOI] [PubMed] [Google Scholar]
- 37.Fu Z, Aucoin D, Ahmed M, Ziliox M, Van Nostrand WE & Smith SO (2014) Capping of Aβ42 oligomers by small molecule inhibitors. Biochemistry 53, 7893–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mishra R, Sellin D, Radovan D, Gohlke A & Winter R (2009) Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. ChemBioChem 10, 445–449. [DOI] [PubMed] [Google Scholar]
- 39.Muro E, Atilla-Gokcumen GE & Eggert US (2014) Lipids in cell biology: how can we understand them better? Mol Biol Cell 25, 1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saraogi I, Hebda JA, Becerril J, Estroff LA, Miranker AD & Hamilton AD (2010) Synthetic α-Helix Mimetics as Agonists and Antagonists of Islet Amyloid Polypeptide Aggregation. Angew Chemie Int Ed 49, 736–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maity D, Kumar S, AlHussein R, Gremer L, Howarth M, Karpauskaite L, Hoyer W, Magzoub M & Hamilton AD (2020) Sub-stoichiometric inhibition of IAPP aggregation: a peptidomimetic approach to anti-amyloid agents. RSC Chem Biol 1, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lolicato F, Raudino A, Milardi D & La Rosa C (2015) Resveratrol interferes with the aggregation of membrane-bound human-IAPP: A molecular dynamics study. Eur J Med Chem 92, 876–881. [DOI] [PubMed] [Google Scholar]
- 43.Evers F, Jeworrek C, Tiemeyer S, Weise K, Sellin D, Paulus M, Struth B, Tolan M & Winter R (2009) Elucidating the Mechanism of Lipid Membrane-Induced IAPP Fibrillogenesis and Its Inhibition by the Red Wine Compound Resveratrol: A Synchrotron X-ray Reflectivity Study. J Am Chem Soc 131, 9516–9521. [DOI] [PubMed] [Google Scholar]
- 44.Williams DM & Pukala TL (2013) Novel insights into protein misfolding diseases revealed by ion mobility-mass spectrometry. Mass Spectrom Rev 32, 169–187. [DOI] [PubMed] [Google Scholar]
- 45.Politis A, Stengel F, Hall Z, Hernández H, Leitner A, Walzthoeni T, Robinson CV. & Aebersold R (2014) A mass spectrometry–based hybrid method for structural modeling of protein complexes. Nat Methods 11, 403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharon M & Robinson CV. (2007) The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem 76, 167–193. [DOI] [PubMed] [Google Scholar]
- 47.Hudson SA, Ecroyd H, Kee TW & Carver JA (2009) The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J 276, 5960–5972. [DOI] [PubMed] [Google Scholar]
- 48.Domanov YA & Kinnunen PKJ (2008) Islet Amyloid Polypeptide Forms Rigid Lipid–Protein Amyloid Fibrils on Supported Phospholipid Bilayers. J Mol Biol 376, 42–54. [DOI] [PubMed] [Google Scholar]
- 49.Kantham S, Chan S, McColl G, Miles JA, Veliyath SK, Deora GS, Dighe SN, Khabbazi S, Parat MO & Ross BP (2017) Effect of the biphenyl neolignan honokiol on Aβ42-induced toxicity in Caenorhabditis elegans, Aβ42 fibrillation, cholinesterase activity, DPPH radicals, and iron(II) chelation. ACS Chem Neurosci 8, 1901–1912. [DOI] [PubMed] [Google Scholar]
- 50.Hung VWS, Cheng XR, Li N, Veloso AJ & Kerman K (2013) Electrochemical detection of amyloid-beta aggregation in the presence of resveratrol. J Electrochem Soc 160, G3097–G3101. [Google Scholar]
- 51.Lee JC-M, Bermudez H, Discher BM, Sheehan MA, Won Y-Y, Bates FS & Discher DE (2001) Preparation, stability, and in vitro performance of vesicles made with diblock copolymers. Biotechnol Bioeng 73, 135–145. [DOI] [PubMed] [Google Scholar]
- 52.Chandrashekaran IR, Adda CG, MacRaild CA, Anders RF & Norton RS (2011) EGCG disaggregates amyloid-like fibrils formed by Plasmodium falciparum merozoite surface protein 2. Arch Biochem Biophys 513, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McLaurin JA & Chakrabartty A (1997) Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur J Biochem 245, 355–363. [DOI] [PubMed] [Google Scholar]
- 54.Kumar ST, Jagannath S, Francois C, Vanderstichele H, Stoops E & Lashuel HA (2020) How specific are the conformation-specific α-synuclein antibodies? Characterization and validation of 16 α-synuclein conformation-specific antibodies using well-characterized preparations of α-synuclein monomers, fibrils and oligomers with distinct struct. Neurobiol Dis 146, 105086. [DOI] [PubMed] [Google Scholar]
- 55.Konermann L, Ahadi E, Rodriguez AD & Vahidi S (2013) Unraveling the mechanism of electrospray ionization. Anal Chem 85, 2–9. [DOI] [PubMed] [Google Scholar]
- 56.Flick TG, Merenbloom SI & Williams ER (2013) Effects of Metal Ion Adduction on the Gas-Phase Conformations of Protein Ions. J Am Soc Mass Spectrom 24, 1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hyung SJ, Detoma AS, Brender JR, Lee S, Vivekanandan S, Kochi A, Choi JS, Ramamoorthy A, Ruotolo BT & Lim MH (2013) Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid-β species. Proc Natl Acad Sci U S A 110, 3743–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Konijnenberg A, Ranica S, Narkiewicz J, Legname G, Grandori R, Sobott F & Natalello A (2016) Opposite structural effects of epigallocatechin-3-gallate and dopamine binding to α-synuclein. Anal Chem 88, 8468–8475. [DOI] [PubMed] [Google Scholar]
- 59.Sanders HM, Jovcevski B, Carver JA & Pukala TL (2020) The molecular chaperone β-casein prevents amorphous and fibrillar aggregation of α-lactalbumin by stabilisation of dynamic disorder. Biochem J 477, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das S, Pukala TL & Smid SD (2018) Exploring the structural diversity in inhibitors of α-synuclein amyloidogenic folding, aggregation, and neurotoxicity. Front Chem 6, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pavlova A, Cheng CY, Kinnebrew M, Lew J, Dahlquist FW & Han S (2016) Protein structural and surface water rearrangement constitute major events in the earliest aggregation stages of tau. Proc Natl Acad Sci U S A 113, E127–E136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams TL, Day IJ & Serpell LC (2010) The effect of Alzheimer’s Aβ aggregation state on the permeation of biomimetic lipid vesicles. Langmuir 26, 17260–17268. [DOI] [PubMed] [Google Scholar]
- 63.Georgieva ER, Ramlall TF, Borbat PP, Freed JH & Eliezer D (2010) The lipid-binding domain of wild type and mutant α-synuclein: Compactness and interconversion between the broken and extended helix forms. J Biol Chem 285, 28261–28274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fusco G, Pape T, Stephens AD, Mahou P, Costa AR, Kaminski CF, Kaminski Schierle GS, Vendruscolo M, Veglia G, Dobson CM & De Simone A (2016) Structural basis of synaptic vesicle assembly promoted by α-synuclein. Nat Commun 7, 12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Killinger BA, Melki R, Brundin P & Kordower JH (2019) Endogenous alpha-synuclein monomers, oligomers and resulting pathology: let’s talk about the lipids in the room. npj Park Dis 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shrivastava AN, Aperia A, Melki R & Triller A (2017) Physico-pathologic mechanisms involved in neurodegeneration: Misfolded protein-plasma membrane interactions. Neuron 95, 33–50. [DOI] [PubMed] [Google Scholar]
- 67.Iyer A & Claessens MMAE (2019) Disruptive membrane interactions of alpha-synuclein aggregates. Biochim Biophys Acta - Proteins Proteomics 1867, 468–482. [DOI] [PubMed] [Google Scholar]
- 68.Sciacca MFM, Kotler SA, Brender JR, Chen J, Lee DK & Ramamoorthy A (2012) Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys J 103, 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F, Cremades N, Ying L, Dobson CM & De Simone A (2017) Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443. [DOI] [PubMed] [Google Scholar]
- 70.Sneideris T, Sakalauskas A, Sternke-Hoffmann R, Peduzzo A, Ziaunys M, Buell AK & Smirnovas V (2019) The environment is a key factor in determining the anti-amyloid efficacy of EGCG. Biomolecules 9, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Srivastava AK, Pittman JM, Zerweck J, Venkata BS, Moore PC, Sachleben JR & Meredith SC (2019) β-Amyloid aggregation and heterogeneous nucleation. Protein Sci 28, 1567–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michaels TCT, Šarić A, Curk S, Bernfur K, Arosio P, Meisl G, Dear AJ, Cohen SIA, Dobson CM, Vendruscolo M, Linse S & Knowles TPJ (2020) Dynamics of oligomer populations formed during the aggregation of Alzheimer’s Aβ42 peptide. Nat Chem 12, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Musteikytė G, Jayaram AK, Xu CK, Vendruscolo M, Krainer G & Knowles TPJ (2021) Interactions of α-synuclein oligomers with lipid membranes. Biochim Biophys Acta - Biomembr 1863, 183536. [DOI] [PubMed] [Google Scholar]
- 74.Pithadia A, Brender JR, Fierke CA & Ramamoorthy A (2016) Inhibition of IAPP Aggregation and Toxicity by Natural Products and Derivatives. J Diabetes Res 2016, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Volles MJ & Lansbury PT (2007) Relationships between the Sequence of α-Synuclein and its Membrane Affinity, Fibrillization Propensity, and Yeast Toxicity. J Mol Biol 366, 1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grey M, Linse S, Nilsson H, Brundin P & Sparr E (2011) Membrane interaction of α-synuclein in different aggregation states. J Parkinsons Dis 1, 359–371. [DOI] [PubMed] [Google Scholar]
- 77.Ryan TM, Caine J, Mertens HDT, Kirby N, Nigro J, Breheney K, Waddington LJ, Streltsov VA, Curtain C, Masters CL & Roberts BR (2013) Ammonium hydroxide treatment of Aβ produces an aggregate free solution suitable for biophysical and cell culture characterization. PeerJ 1, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kulig M & Ecroyd H (2012) The small heat-shock protein αB-crystallin uses different mechanisms of chaperone action to prevent the amorphous versus fibrillar aggregation of α-lactalbumin. Biochem J 448, 343–352. [DOI] [PubMed] [Google Scholar]
- 79.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW & Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–9. [DOI] [PubMed] [Google Scholar]
- 80.Schneider CA, Rasband WS & Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu J & Konermann L (2011) Protein–protein binding affinities in solution determined by electrospray mass spectrometry. J Am Soc Mass Spectrom 22, 408–417. [DOI] [PubMed] [Google Scholar]
- 82.Marty MT, Baldwin AJ, Marklund EG, Hochberg GKA, Benesch JLP & Robinson CV. (2015) Bayesian deconvolution of mass and ion mobility spectra: From binary interactions to polydisperse ensembles. Anal Chem 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]