

Abstract
Objectives
Intravenous lidocaine can alleviate painful diabetic peripheral neuropathy (DPN) in some patients. Whether quantitative sensory testing (QST) can identify treatment responders have not been prospectively tested.
Methods
This was a prospective, randomized, double-blind, crossover, placebo-controlled trial comparing intravenous lidocaine to normal saline (placebo) for painful DPN. Thirty-four participants with painful DPN were enrolled and given intravenous lidocaine (5 mg/kg ideal body weight) or placebo as a 40-minute infusion, after a battery of QST parameters were tested on the dorsal foot, with a three-week washout period between infusions.
Results
Thirty-one participants completed both study sessions and were included in the final analysis. Lidocaine resulted in a 51% pain reduction 60-120 minutes after infusion initiation, as assessed on a 0-10 numerical rating scale, while placebo resulted in a 33.5% pain reduction (difference = 17.6%, 95% CI 1.9-33.3%, p = 0.03). Neither mechanical pain threshold, heat pain threshold, nor any of the other measured QST parameters predicted the response to treatment. Lidocaine administration reduced mean Neuropathic Pain Symptom Inventory paresthesia/dysesthesia scores when compared to placebo by 1.29 points (95% CI −2.03 to −0.55, p = 0.001), and paroxysmal pain scores by 0.84 points (95% CI −1.62 to −0.56, p = 0.04), without significant changes in burning, pressing or evoked pain sub-scores.
Discussion
While some participants reported therapeutic benefit from lidocaine administration, QST measures alone were not predictive of response to treatment. Further studies, powered to test more complex phenotypic interactions, may be required to identify reliable predictors of response to pharmacotherapy in patients with DPN.
Keywords: Diabetic Peripheral Neuropathy, DPN, Lidocaine, Chronic Pain, Neuropathic Pain
INTRODUCTION
Diabetic peripheral neuropathy (DPN) is the leading cause of neuropathic pain worldwide and affects more than three million Americans.1 DPN is most often characterized by a mixed small and large fibre neuropathy causing distal symmetric pain, numbness, burning and tingling, accompanied by either sensory loss or tactile/thermal hyperalgesia and allodynia.2 Pain is the most common presenting symptom for patients with diabetic peripheral neuropathy. Painful DPN is difficult to treat, with current pharmacological modalities (e.g. amitriptyline, gabapentin, pregabalin, duloxetine) providing significant relief for only about 20% of afflicted individuals.3, 4
One of the key mechanisms that leads to spontaneous and evoked pain in diabetic peripheral neuropathy is alteration in the expression of voltage-gated sodium channels both peripherally and centrally.5–8 Systemic intravenous administration of lidocaine, a nonselective sodium channel blocker, has shown promise as a useful analgesic adjunct for a multitude of pain conditions, including for painful DPN.9, 10 However, only a minority of patients seem to achieve substantial benefit from intravenous lidocaine infusions, making its indiscriminate use both time and resource intensive.9, 11, 12
The success rate of conventional therapies for the treatment of neuropathy is mixed, which suggests that variability exists among the underlying pain mechanisms on the patient level, and points to individual differences in response to analgesics. For example, in a small study, patients with distal symmetric polyneuropathy (primarily due to DPN) had better response to systemic administration of lidocaine than patients with neuropathic pain due to traumatic nerve injury.13 Recent literature suggests that certain methods of assessing sensory nerve function in neuropathic pain patients may help predict the individual analgesic response.14–17 Despite these promising data, the use of quantitative sensory testing to predict treatment response to lidocaine in a randomized controlled trial of painful DPN has not been published.
MATERIALS AND METHODS
Trial design
This was a prospective, randomized, double-blind, crossover, placebo-controlled trial comparing intravenous lidocaine to normal saline (placebo) for the treatment of diabetic peripheral neuropathy. The study was approved by the Washington University Institutional Review Board and registered with clinicaltrials.gov (NCT02363803) prior to enrollment. Written informed consent was obtained according to the Declaration of Helsinki prior to participation. No important changes were made to the methods outlined below after the onset of enrollment.
Participants
Participants with diabetes mellitus and distal symmetric pain in the lower extremities with a duration of more than three months were recruited for participation in this study. Potential participants were identified from the Washington University Pain Management Center and Diabetes Center, through posters and flyers, and through the Volunteer for Health local registry. Specific inclusion criteria were as follows: 1) Age ≥ 18; 2) Diagnosis of diabetes mellitus (fasting plasma glucose > 126 mg/dL and/or HbA1C >6.5%); 3) Distal symmetric pain in lower extremities with duration of more than three months; 4) Presence of either numbness or at least one sensory disturbance (increased or decreased sensitivity) in the feet; 5) Spontaneous pain with average (past week) intensity of ≥4 on a 0-10 Numerical Rating Scale (NRS).
Specific exclusion criteria were as follows: 1) Not giving consent to participate in the study; 2) Unable to complete a self-reported pain questionnaire; 3) History of moderate to severe renal or liver failure; 4) History of other central or peripheral neurologic disorders; 5) History of cardiac arrhythmias; 6) Contraindication to intravenous lidocaine; 7) Pregnancy or lactation. A comprehensive metabolic panel was drawn in participants who had a history of renal or hepatic disorders and were excluded if a) serum creatinine >1.5 mg/dL (or creatinine clearance <50 mL/min/1.73m2 [by Cockcroft-Gault formula]), b) Alanine aminotransferase (ALT) or Aspartate aminotransferase (AST) were more the twice the upper normal limit, or c) serum albumin below 3.5 g/dL. Participants with a history of cardiac disease had a 12-lead electrocardiogram (EKG) performed to exclude the presence of active arrhythmias.
After obtaining informed consent, demographic data and baseline pain information were recorded. Height and weight were measured. In addition, participants were asked to complete the Brief Pain Inventory (BPI), Hospital Anxiety and Depression Scale (HADS) and Neuropathic Pain Symptom Inventory (NPSI) questionnaires. Eligibility to participate in the trial was confirmed, and only those who met the inclusion/exclusion criteria above were asked to return for subsequent visits. Participants were asked to continue all chronic medications, including chronic pain medications, provided that the dose was stable for at least three weeks. Participants were asked to discontinue PRN analgesics for 24 hours prior to each study visit.
Randomization
Participants were identified and recruited into the study using convenience sampling. Participants who signed informed consent were randomized to the order of interventions, i.e. lidocaine first, or placebo first. Randomization occurred via a computer-generated random sequence in blocks of six, with three patients in each block per arm, prepared by the pharmacist at the institutional investigational drug service.
Blinding
Study drug was prepared to a total volume of 100 mL 0.9% NaCl solution, by an unblinded investigator at the Investigational Drug Service. Each solution was labeled “lidocaine/placebo” and no discernable difference existed between the compounded lidocaine and normal saline solutions. The solutions, after being compounded, were given to blinded investigators to administer to the participants. The investigators, participants, and all ancillary staff present during the study administration were blinded to the treatments provided during the trial. Analysis of data was performed by an unblinded investigator upon completion of the study.
Interventions
Eligible participants attended two study visits during which intravenous lidocaine or placebo infusion were administered, separated by a three-week washout period. Participants were instructed to abstain from food four hours prior to infusion, liquids two hours prior to infusion, and all PRN pain medicines 24 hours prior to infusion. In addition, any changes to the patient’s health status and medication list from their baseline visit were documented at each subsequent visit. Female premenopausal participants submitted a dip-stick pregnancy urinalysis prior to infusion and were withdrawn from the study if pregnant.
Quantitative sensory testing (QST) was performed at each visit prior to infusion onset. To maintain assessment consistency, all QST measurements were made by a single trained coordinator (KF). Quantitative sensory testing was performed as previously described on the dorsal mid-foot.18, 19 If asymmetry in pain intensity existed between extremities, QST was performed on the more painful foot.
Thermal detection thresholds were measured using the Thermal Sensory Analyzer (TSA-II platform – Medoc, Ramat Yishai, Israel). Heat pain threshold (HPT), cold pain threshold (CPT), warm detection threshold (WDT) and cold detection threshold (CDT) were determined using previously published methods.20 A thermode with a contact area of 9.0 cm2 was applied to the tested site, and all thresholds were determined by continuous ramping of temperature from 32°C baseline temperature by 1°C/s until the participant pressed the ‘stop’ button on the device. Predetermined cut-off temperatures were 0°C and 50°C, to minimize thermal damage to the skin. An average threshold was calculated from three consecutive measurements.
Mechanical pain threshold (MPT) was measured using standardized weighted metal probes (Nervetest, MRC systems, Heidelberg, Germany) exerting pressures of 8, 16, 32, 64, 128, 256 and 512 mN. Beginning with an applied force of 8 mN, stimuli were increased in intensity until the sensation induced by increased pressure was described as ‘sharp’. If a participant failed to describe any stimulus, including a 512 mN stimulus, as painful, this was recorded as a value of 1024 mN. The stimulus was then applied in descending stimulus intensities until the participant no longer described the sensation as ‘sharp’. If the participant described an 8 mN pressure intensity as ‘sharp’ during descending stimulus, this was documented as 4 mN. In addition, if the participant could not describe any sensation, painful or otherwise, when using descending stimulus intensities, this was documented as a value of 1024 mN. The process of determining ascending and descending stimulus intensities was repeated for a total of five times, and the final threshold was calculated as the geometric mean of all five ascending and descending stimulus intensities.
Mechanical detection threshold (MDT) was measured with a set of Semmes-Weinstein monofilaments (North Coast Medical, Gilroy, CA, USA). Standardized monofilaments sizes of 1.65 through 6.65, corresponding with target forces of 0.008 to 300 grams, were utilized for assessment. These filaments were applied in a series of three sets of increasing and decreasing stimulus intensities to the target site by asking whether the participant ‘felt’ a specific monofilament. A tactile detection threshold was recorded according to this response. If a participant did not feel a 300 g stimulus on ascending intensity testing, this was documented as a value of 600 grams. If the participant could not feel a descending stimulus of 300 g, this was documented as 300 g. The final threshold was calculated as a geometric mean of all three ascending and descending stimulus intensities.
Wind up ratio (WUR) was measured using standardized weighted metal probes (Nervetest, MRC systems, Heidelberg, Germany). A 256 mN pinprick stimulus was applied to the test site singularly. If a single 256 mN stimulation elicited a pain score of 0 on a 0-10 NRS, a 512 mN probe was used for WUR testing. If a single 256 mN stimulation elicited a pain of intensity ≥ 9 on a 0-10 NRS, a 128 mN probe was used for WUR testing. In all other scenarios, a 256 mN probe was used for WUR testing. Using the correct probe outlined above, a single stimulus was applied with the chosen probe and a verbal pain score was recorded on a 0-10 NRS. After a 10 second wait period, 10 identical pinprick stimuli were applied at a rate of 1/sec to the target site within an area of 1 cm2. The participant was asked for a pain score at the target site, again on a 0-10 NRS. A ratio of pain during repeated stimuli when compared to single stimulus was calculated. This test was repeated two more times, and a geometric mean of the three ratios was calculated to assess WUR.
Vibration was measured with a tuning fork (64 Hz, 8/8 scale) applied at the lateral malleolus area, as previously described.13 A geometric mean of three assessments was calculated to determine vibration detection threshold (VDT).
Conditioned pain modulation (CPM) was assessed using the method of levels via the Q-Sense Thermal Analyzer (Medoc, Ramat Yishai, Israel), equipped with two Peltier thermodes with independent temperature programming. The testing stimulus was determined by the temperature that elicited heat pain of 60 on a 0 to 100 NRS (Pain-60). Cold conditioning stimulus was set at 16°C. At first, the test stimulus was applied at the nondominant forearm twice, 30 seconds apart, and the average pain value on 0 to 100 NRS was obtained. After a 10-minute break, the conditioning stimulus was applied to the dominant forearm for 60 seconds; during the last 30 seconds of conditioning, two Pain-60 heat stimuli were applied to the nondominant forearm. The procedure was repeated twice. Conditioned pain modulation was measured as the difference between NRS of Pain-60 stimuli with and without the contralateral cold conditioning (average score of 2 tests).18
Upon QST testing completion, participants were then asked to rate the spontaneous pain intensity in their lower legs on a scale of 0-10 NRS, with the opportunity to provide non-integer values (e.g. 6.5) if deemed necessary. In addition, participants were asked the following NPSI pain descriptors on a 0-10 scale: burning, squeezing, pins and needles, tingling, stabbing, and electrical shocks. Evoked responses (measured on a 0-10 NRS scale, where 5 is considered normal sensation) to cold (20°C, Rolltemp, Somedic, Norra Mellby, Sweden), warm (40°C, Rolltemp, Somedic, Norra Mellby, Swden), brush (Senselab brush-05, Somedic, Norra Mellby, Sweden) and pinprick (#6.10 Semmes-Weinstein monofilament; North Coast Medical, Gilroy, CA, USA) stimuli were measured on the same dorsal foot utilized for QST testing.13
Intravenous lidocaine (5 mg/kg ideal bodyweight, calculated using the Devine formula21) or normal saline (placebo) was administered via intravenous infusion over 40 minutes. Spontaneous lower extremity pain and evoked responses were measured every 10 minutes after the onset of infusion for the first hour, followed by every 15 minutes for the second hour after infusion initiation. NPSI pain descriptors were also reassessed 60 minutes after initiation of intravenous infusion. All participants were monitored via sphygmomanometer, pulse oximeter and three lead EKG for 120 minutes after infusion initiation. In addition, venous blood samples were collected via an indwelling intravenous catheter on the arm contralateral to the infusion arm, immediately prior to onset of infusion (time point zero), and at 20, 45, 60, 90 and 120 minutes after infusion initiation to assess lidocaine plasma concentrations. A 20% intravenous fat emulsion was readily available for administration in the event of local anesthetic systemic toxicity.
At discharge, participants were given a pain diary and were asked to record spontaneous pain intensity hourly for the first eight hours after infusion initiation, and then daily for three weeks. Participants were contacted by a member of the research team weekly for three weeks after infusion to ascertain pain diary adherence and assess any long-term adverse effects.
After three weeks, participants were asked to return for their second treatment visit, where they received either lidocaine or normal saline, per randomization schedule. Data collection occurred in the same fashion as the participant’s first visit, as described above.
Outcomes
Primary outcome
The primary outcome for this study was the percent change in spontaneous pain scores from time point zero (defined as immediately prior to infusion onset) to 60-120 minutes after infusion onset (assessed every 15 minutes, with final comparison score as an average of these values). The primary outcome was modified after data collection to remove the 40-min timepoint from primary analysis, as that particular timepoint (when the participants are told that the infusion of study drug is completed), was associated with unexpectedly large variability in responses not seen with any other timepoint. No other changes were made to pre-specified primary or secondary end points in the study.
Secondary outcomes
Pre-specified secondary outcomes for the study were the following:
The change in spontaneous pain intensity at 60-120 minutes as a function of baseline heat pain threshold at each visit. The slopes (Pearson coefficients) of the correlation obtained from lidocaine versus normal saline (placebo) were compared.
The change in spontaneous pain intensity at 60-120 minutes as a function of baseline mechanical pain threshold at each visit. The slopes (Pearson coefficients) of the correlation obtained from lidocaine versus normal saline (placebo) were compared.
The change in evoked mechanical and thermal sensation prior to infusion and 60 minutes after infusion initiation, as assessed on a 0-10 numerical rating scale.
The change in Neuropathic Pain Symptom Inventory descriptors of pain prior to infusion and 60 minutes after infusion, as assessed on a 0-10 numerical rating scale.
Adverse effects were assessed by specifically asking for side effects (perioral numbness, dizziness, drowsiness, blurred vision, dry mouth) every 10 minutes for an hour from starting the drug infusion, and by encouraging spontaneous reporting by patients. For events that occurred, symptom duration was documented.
Exploratory analysis centered around assessing whether any other QST measurements taken during testing (CDT, WDT, CPT, WUR, MDT, VDT and CPM) correlated with percent pain reduction at 60-120 minutes after infusion initiation. In addition, participants who achieved >50% pain reduction with lidocaine 60-120 minutes after infusion but not placebo (i.e. “responders”) were compared to those who did not obtain such relief.
All data were initially recorded on paper case report forms, entered to a secure REDCap database, and subsequently verified by an independent investigator.22
Sample size
To identify significant differences in slopes of predictor and response magnitude versus pain reduction in the lidocaine group and placebo group, 34 patients were required for this cross-over trial. The calculation was based on the ability to detect a statistically significant difference between correlation coefficients of the two slopes, with 85% power, and an α = .025, after Bonferroni correction for two comparisons: mechanical and heat pain threshold. The assumption for the correlation coefficients was based on preliminary data obtained in a group of seven patients with DPN receiving treatment with intravenous lidocaine,23 aiming to detect a difference of 14 (SD 18.21) units between correlation coefficients of lidocaine vs placebo curves.
Pharmacokinetic analysis
Blood samples collected during the study were sent for lidocaine level analysis after all participants completed the trial, and the results were unblinded once blood level analysis was performed. Plasma samples were analyzed for lidocaine concentrations using an Agilent 1260 Infinity system equipped with UV diode array detector (Agilent, Santa Clara, CA, USA). A combination of protein precipitation and solid phase extraction (SPE) was used for sample preparation. Plasma samples (100 µL) were mixed with 100 µL of methanol and 10 µL of internal standard (bupivacaine 100 µg/mL in methanol) solution. The samples were vortexed for 20 seconds and centrifuged for 5 minutes at 15700 g. The supernatant was transferred onto Bond Elut Plexa 30 mg (Agilent, Santa Clara, CA, USA) cartridges used for the SPE. The cartridges were preconditioned by eluting 500 µL of methanol followed by 500 µL of water. The samples were loaded on the cartridge, followed by washing with 500 µL of 5% methanol in water. The samples were eluted by applying 400 µL of methanol twice and the eluent was collected and evaporated under gentle stream of N2 gas. Dried samples were reconstituted with 30% methanol in water and 40 µL was injected in the HPLC-UV system. Chromatographic separation was achieved using a Poroshell 120 column (EC-C18, 2.7 µm, 3.0 × 100 mm, Agilent, Santa Clara, CA, USA).24, 25 The mobile consisted of a mixture of 5 mM ammonium acetate (pH 4.0) and acetonitrile at 70:30 ratio. The flow rate was 0.7 mL/min and chromatograms were collected at UV wavelength of 215 nm. The lower limit of quantification for lidocaine was 0.5 µg/mL. The retention time of lidocaine and bupivacaine were 3.4 min and 13.0 min, respectively.
A standard noncompartmental data analysis was performed for each individual lidocaine concentration-time profile. The maximum plasma drug concentration (Cmax) were obtained directly from the experimental data. Terminal half-life, area under the concentration time curve from time zero to infinity (AUC, calculated by linear-up/log-down trapezoidal method), and clearance were calculated using Phoenix WinNonlin version 8.2 (Certara, Princeton, NJ, USA).
Statistical methods
All statistical analyses were performed using SPSS version 26 (IBM Corp., Armonk, N.Y., USA). Continuous variables were assessed for normalcy by graphical inspection, and inferences were made about the underlying distribution. Data assessed as normally distributed were compared using paired t-tests. Data with gross departures from normality were compared using the Wilcoxon signed-rank test. Ordinal data were compared using Wilcoxon signed-rank test, whereas categorical data were compared using McNemar’s test. All QST parameters were normalized and Z-transformed according to methods previously described.26 Z-scores for some parameters were multiplied by −1 to maintain consistency of scores across QST parameters, with positive values indicating “gain of sensation” and negative values indicating “loss of sensation”. Correlations between percent pain reduction and QST parameters were made using bivariate Pearson correlation. Significance testing of correlation coefficients was carried out using Fisher’s Z-transformation. For responder analysis, an arithmetic mean of QST measurements between the two study sessions was calculated obtain a single QST value for each participant. Baseline demographic and QST data were compared using independent samples t-test or Mann-Whitney U test as appropriate.
RESULTS
Recruitment
Recruitment occurred from March 2015 to March 2018. A total of 200 participants were screened for study eligibility (Figure 1). Of these, 55 participants were consented and enrolled into the study. Ten participants voluntarily withdrew from the study prior to randomization and their phase 1 visit. Two participants were excluded for HbA1C values ≤ 6.5. Four participants were excluded for abnormalities on EKG testing, and five participants did not meet pain inclusion criteria prior to phase 1. A total of 34 participants were randomized to either lidocaine or placebo arms in phase 1. Of these, one participant in the lidocaine arm did not return for phase 2 of the study, as he experienced no pain after treatment with the lidocaine infusion over six weeks of follow-up. A total of 33 participants completed both phases of the trial. Two participants (one in each arm) were excluded from final analysis as they exhibited a pain score of zero at the onset of medication infusion during one of the phases of the trial. A total of 31 participants were included in data analysis.
Figure 1:
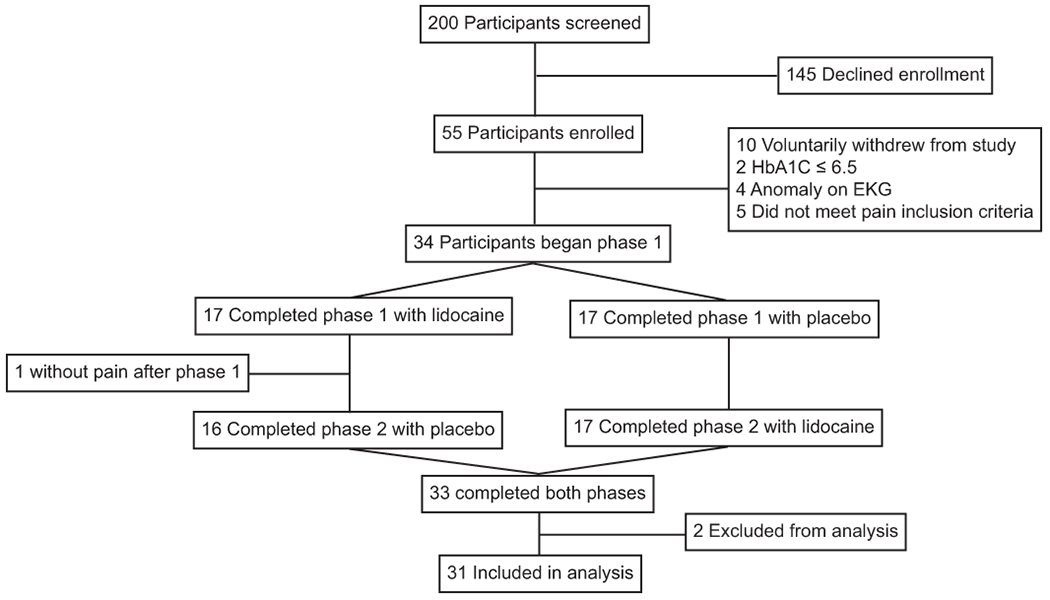
Flowchart
Baseline Data
Participant demographic information is listed in Table 1. Mean participant age was 57 years (SD 11). Approximately half of the participants were female (47%), and predominantly White (71%). The median years with a diagnosis of diabetes in this cohort was 10 (IQR 5-17.5). Hypertension (74%), hypercholesterolemia (53%) and cardiovascular disease (21%) comprised the top three medical comorbidities in this sample. Sixty five percent of participants in this study used analgesic medications for pain control. Overall, 73% of the participants had painful symptoms for greater than two years prior to the study, and 94% described their pain as constant, with a median pain intensity over the previous week as 6.2 on a 0-10 NRS (IQR 5-7.2).
Table 1:
Participant demographic information divided by study medication administration order.
| Placebo during first session (n=17) | Lidocaine during first session (n=17) | Total (n=34) | |
|---|---|---|---|
|
| |||
| Age, mean (SD) | 57.5 (11.9) | 55.6 (9.8) | 56.6 (10.8) |
|
| |||
| Sex, F/M (% female) | 9/8 (53) | 7/10 (41) | 16/18 (47) |
|
| |||
| Race | |||
| Black (%) | 3 (18) | 7 (41) | 10 (29) |
| White (%) | 14 (82) | 10 (59) | 24 (71) |
|
| |||
| Cumulative years with diabetes diagnosis, median (IQR) | 9 (5-17) | 10 (6-18) | 10 (5-17.5) |
|
| |||
| Hemoglobin A1c, mean (SD) | 8.9 (1.9) | 8.0 (2.0) | 8.4 (2.0) |
|
| |||
| Medical comorbidities (%) | |||
| Hypertension | 13 (76) | 12 (71) | 25 (74) |
| Hypercholesterolemia | 10 (59) | 8 (47) | 18 (53) |
| Cardiovascular disease | 6 (35) | 1 (6) | 7 (21) |
| Sleep apnea | 4 (24) | 1 (6) | 5 (15) |
| Asthma | 1 (6) | 2 (12) | 3 (9) |
| Arthritis | 2 (12) | 1 (6) | 3 (9) |
| Anxiety | 2 (12) | 0 (0) | 2 (6) |
| Depressive disorder | 3 (18) | 3 (18) | 6 (18) |
| History of cancer | 3 (18) | 1 (6) | 4 (12) |
|
| |||
| BMI in kg/m2, median (IQR) | 36.9 (29.9-39.2) | 33.0 (26.0-40.8) | 33.4 (28.4-39.7) |
|
| |||
| Baseline analgesic use (%) | |||
| Any analgesic use | 12 (71) | 10 (59) | 22 (65) |
| Strong opioids (e.g. oxycodone, hydrocodone) | 3 (18) | 1 (6) | 4 (12) |
| Weak opioids (e.g. tramadol, codeine) | 0 (0) | 3 (18) | 3 (9) |
| Non-steroidal anti-inflammatory drugs and/or acetaminophen | 8 (47) | 5 (29) | 13 (38) |
| Serotonin-norepinephrine reuptake inhibitors (e.g. duloxetine, venlafaxine) | 5 (29) | 1 (6) | 6 (18) |
| Tricyclic antidepressants (e.g. amitriptyline) | 2 (12) | 0 (0) | 2 (6) |
| Anticonvulsants (e.g. gabapentin) | 9 (53) | 8 (47) | 17 (50) |
| Topical agents (e.g. lidocaine patch, topical steroids) | 1 (6) | 1 (6) | 2 (6) |
| Antispasmodics (cyclobenzaprine) | 0 (0) | 2 (12) | 2 (6) |
|
| |||
| Duration of painful symptoms prior to study (%) | |||
| Six months up to one year | 3 (18) | 2 (12) | 5 (15) |
| More than one year up to two years | 4 (24) | 0 (0) | 4 (12) |
| More than two years up to five years | 5 (29) | 6 (35) | 11 (32) |
| Greater than five years | 5 (29) | 9 (53) | 14 (41) |
|
| |||
| Pain frequency (%) | |||
| Constant | 17 (100) | 15 (88) | 32 (94) |
| Constant with intermittent exacerbations | 0 (0) | 1 (6) | 1 (3) |
| Intermittent | 0 (0) | 1 (6) | 1 (3) |
|
| |||
| Average pain intensity over the previous week 0-10 NRS, median (IQR) | 6 (5-8) | 6.5 (5.5-7) | 6.2 (5-7.2) |
|
| |||
| Neuropathic Pain Symptom Inventory (NPSI) score, mean (SD) | |||
| Burning | 5.3 (2.4) | 5.5 (2.5) | 5.4 (2.4) |
| Pressing | 4.5 (1.9) | 4.3 (2.7) | 4.4 (2.3) |
| Paroxysmal | 5.1 (2.5) | 4.6 (3.1) | 4.9 (2.8) |
| Evoked | 4.7 (2.2) | 4.3 (2.6) | 4.5 (2.4) |
| Paresthesia/dysesthesia | 6.2 (2.8) | 6.1 (3.2) | 6.2 (2.9) |
| Total | 51 (15.6) | 48.4 (19.5) | 49.7 (17.4) |
|
| |||
| Brief Pain Inventory (BPI), mean (SD) | |||
| Pain severity score | 5.7 (1.4) | 5.6 (2.2) | 5.6 (1.8) |
| Pain interference score | 5.3 (1.8) | 5.7 (2.8) | 5.5 (2.3) |
|
| |||
| Hospital Anxiety and Depression Scale (HADS), mean (SD) | |||
| Depression | 5.8 (3.5) | 6.2 (4.6) | 6 (4) |
| Anxiety | 5.8 (3.8) | 7.4 (4) | 6.6 (4) |
Normalized QST data for the participants included in analysis can be found in Figure 2. The population cohort studied predominantly displayed loss of sensation across all tested modalities.
Figure 2:
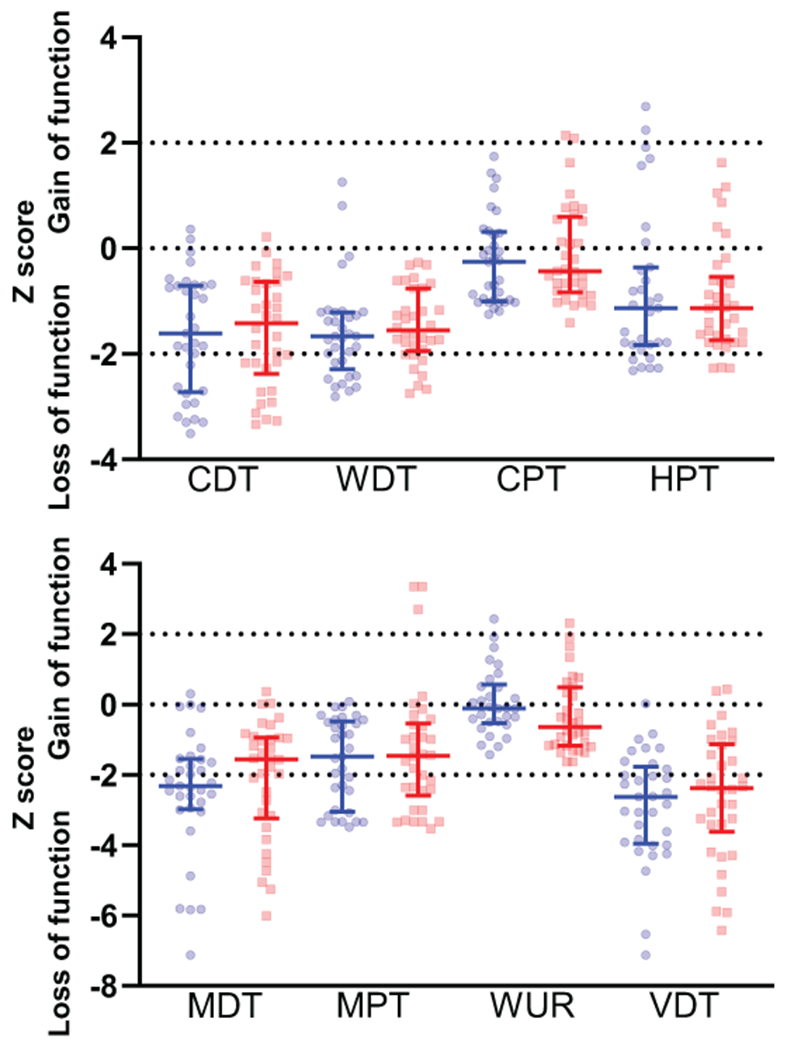
Normalized data for cold detection threshold (CDT), warm detection threshold (WDT), cold pain threshold (CPT), heat pain threshold (HPT), mechanical detection threshold (MDT), mechanical pain threshold (MPT), wind up ratio (WUR), and vibration detection threshold (VDT). N = 34 for all lidocaine sessions and n = 33 for all placebo sessions except for the following measurements: HPT – placebo (n = 31), CPT – lidocaine (n = 33), WUR - placebo (n = 30), WUR – lidocaine (n = 32) as these tests could not be tolerated by some participants. Blue circles denote placebo session; red squares denote lidocaine session. Midpoint line = median, error bars = inter-quartile range
Primary Outcome – Percent pain reduction after 60 to 120 minutes of infusion
Lidocaine infusion resulted in a 51.0% reduction in pain scores while placebo resulted in a 33.5% reduction in pain scores when assessed 60 to 120 minutes after infusion initiation (lidocaine SD 39.9%, placebo SD 45.8%, n = 31, difference 17.6%, p = .03, 95% CI 1.9-33.3%) (Figure 3). A 30% reduction in pain scores was achieved with lidocaine administration in 71.0% of participants during this time frame, compared to 54.8% of participants when placebo was administered (16.2% difference, p = .23, NNT = 6.2). A 50% reduction in pain scores was achieved with lidocaine administration in 54.8% of participants, compared to 38.7% of participants when placebo was administered (16.1% difference, p = .18, NNT = 6.2). Aggregate time course pain intensity data from onset of infusion to 21 days later is depicted in Figure 4.
Figure 3:
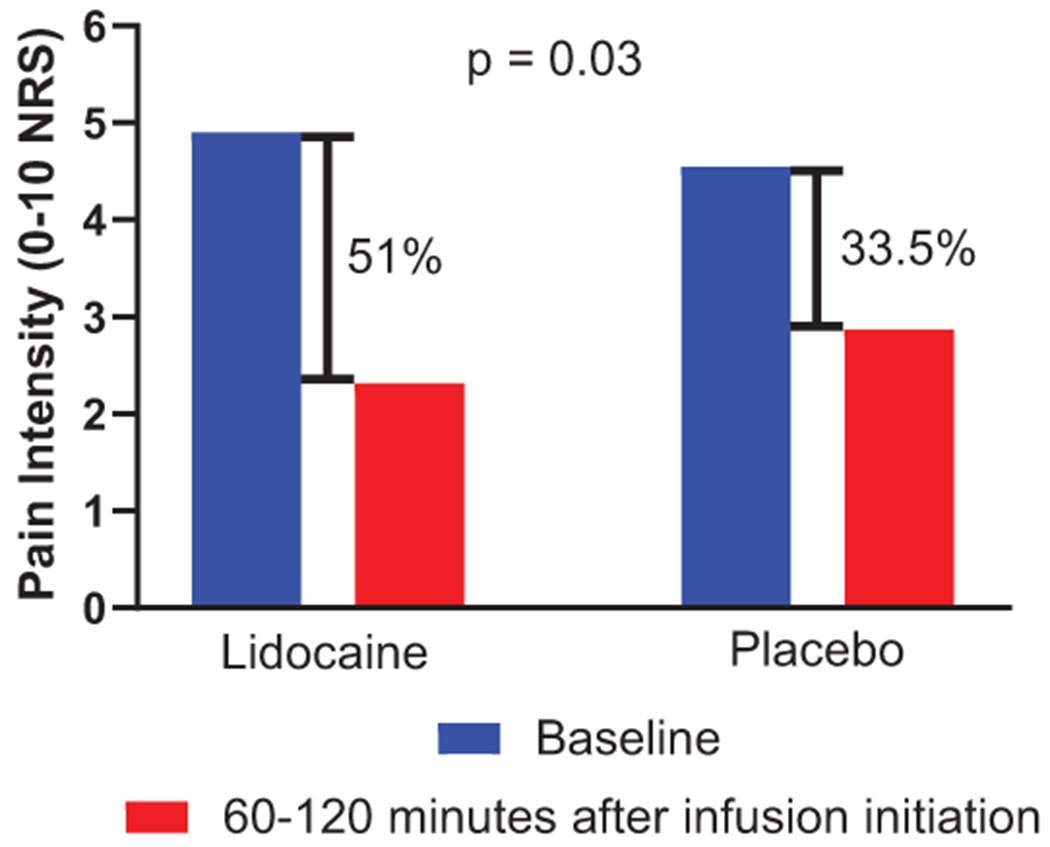
Percent pain reduction in mean spontaneous pain scores from baseline to 60-120 minutes after infusion initiation (n = 31).
Figure 4:
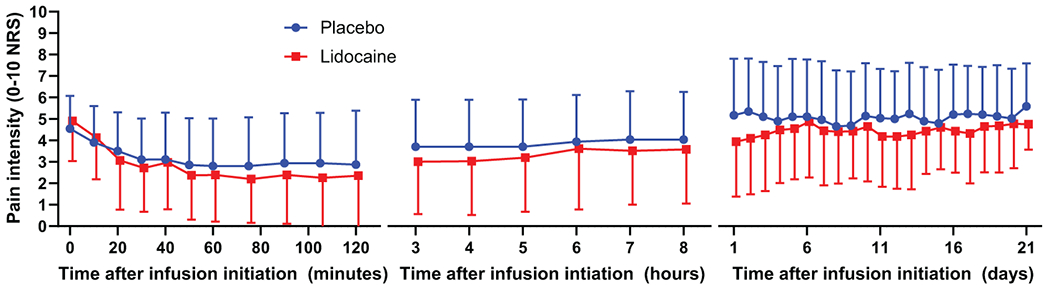
Mean spontaneous pain scores during infusion, 3-8 hours after infusion initiation, and up to 21 days after infusion (n = 31). Error bars = Standard deviation.
Secondary Outcome – Percent pain reduction at 60-120 minutes as a function of mechanical and heat pain thresholds
MPT was not associated with percent pain reduction during either placebo or lidocaine administration (placebo: r = −.276, p = .133; Lidocaine r = −.156, p = .401; difference between correlation coefficients p = .64) (Figure 5). Similarly, HPT was not associated with percent pain reduction during either session (placebo: r = −.062, p = .748; lidocaine: r = .145, p = .454; difference between correlation coefficients p = .45).
Figure 5:
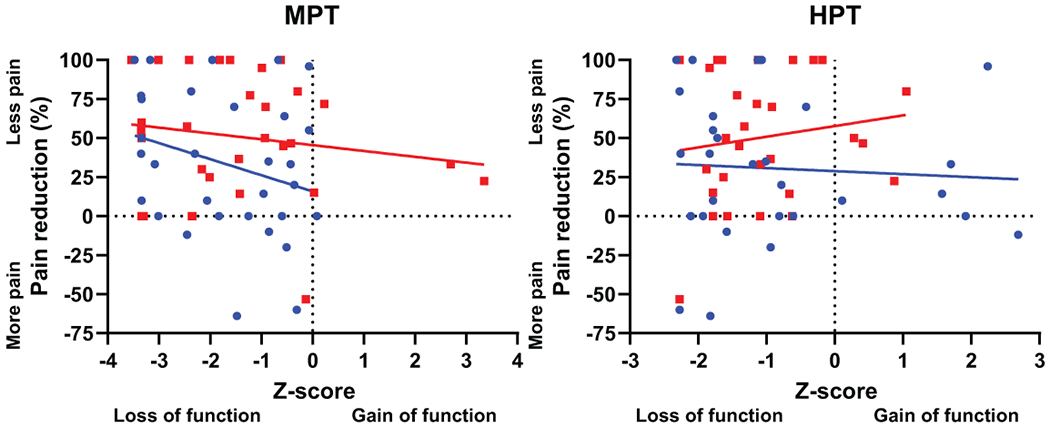
Percent pain reduction 60-120 minutes after infusion as a function of mechanical pain threshold (MPT) and heat pain threshold (HPT), as tested on the most painful foot prior to infusion. Blue circle indicates placebo session; red square indicates lidocaine session. Two participants could not tolerate heat pain threshold probe on skin during testing. MPT regression line information: (n = 31): Placebo (blue): r = −.276, p = .133; Lidocaine (red): r = −.156, p = .401; difference between correlation coefficients p = .64. HPT regression line information (n = 29): Placebo (blue): r = −.062, p = .748; Lidocaine (red): r = .145, p = .454; difference between correlation coefficients p = .45.
Secondary Outcome - Change in evoked mechanical and thermal sensation prior to infusion and 60 minutes after infusion initiation
Lidocaine administration resulted in a 0.84-point reduction in cold intensity on a 0-10 NRS 60 minutes after infusion initiation when compared to placebo (p = 0.04, 95% CI −1.53 to −0.05) (Table 2). In the same timeframe, lidocaine administration did not result in significant reductions in pinprick intensity (difference 0.84 on a 0-10 NRS, p = 0.08, 95% CI −1.78 to 0.10), heat intensity (difference −0.19, p = 0.53, 95% CI −0.82 to 0.43), or brush intensity (difference 0.23, p = 0.59, 95% CI −0.62 to 1.07) when compared to placebo.
Table 2:
Changes in evoked mechanical sensation, evoked thermal sensation, and Neuropathic Pain Symptom Inventory (NPSI) questionnaire descriptors at baseline and 60 minutes after infusion initiation (n = 31). Paired samples t-test utilized to compare groups.
| Placebo | Lidocaine | Lidocaine minus placebo | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pre infusion | 60 minutes | Change (60 min minus pre infusion) | Pre infusion | 60 minutes | Change (60 min minus pre infusion) | Difference | 95% CI | P value | |
| Neuropathic Pain Symptom Inventory subscores | |||||||||
| Burning pain, mean (SD) | 2.90 (2.64) | 1.65 (2.75) | −1.26 (2.44) | 3.00 (2.78) | 1.00 (1.79) | −2.00 (2.25) | −0.74 (3.11) | −1.88 to 0.40 | 0.19 |
| Paroxysmal pain, mean (SD) | 1.15 (1.69) | 0.84 (1.70) | −0.31 (1.15) | 2.00 (2.44) | 0.85 (2.14) | −1.15 (1.92) | −0.84 (2.13) | −1.62 to −0.56 | 0.04 |
| Paresthesia/dysesthesia, mean (SD) | 3.02 (2.66) | 2.13 (2.61) | −0.89 (1.60) | 3.94 (2.76) | 1.76 (2.25) | −2.18 (2.07) | −1.29 (2.01) | −2.03 to −0.55 | 0.001 |
| Evoked mechanical and thermal sensation | |||||||||
| Cold intensity, mean (SD) | 2.74 (2.16) | 3.45 (1.96) | 0.71 (1.59) | 3.00 (2.20) | 2.87 (2.31) | −0.13 (1.26) | −0.84 (2.16) | −1.63 to −0.05 | 0.04 |
| Heat intensity, mean (SD) | 1.87 (1.82) | 1.97 (1.83) | 0.10 (1.37) | 1.87 (1.82) | 1.77 (1.69) | −0.10 (1.19) | −0.19 (1.70) | −0.82 to 0.43 | 0.53 |
| Brush intensity, mean (SD) | 3.83 (1.73) | 3.48 (1.98) | −0.35 (1.97) | 3.55 (1.84) | 3.42 (1.88) | −0.13 (1.45) | 0.23 (2.31) | −0.62 to 1.07 | 0.59 |
| Pinprick intensity, mean (SD) | 3.16 (2.48) | 3.35 (2.51) | 0.19 (2.14) | 3.84 (2.53) | 3.19 (2.68) | −0.65 (1.96) | −0.84 (2.57) | −1.78 to 0.10 | 0.08 |
Secondary Outcome – Change in NPSI Descriptors prior to infusion and 60 minutes after infusion initiation
Lidocaine administration reduced mean NPSI paresthesia/dysesthesia scores when compared to placebo by 1.29 points on a 0-10 NRS (95% CI −2.03 to −0.55, p = 0.001), and paroxysmal pain scores by 0.84 points (95% CI −1.62 to −0.56, p = 0.04), without significantly affecting burning pain score (−0.74, 95% CI −1.88 to 0.40, p = 0.19) (Table 2).
Exploratory Analysis – Percent pain reduction at 60-120 minutes as a function of multiple QST parameters
Supplemental Digital Content Figures 1 and 2 show the QST parameters studied (CDT, WDT, CPT, WUR, MDT, VDT and CPM) as a function of percent pain reduction, with Pearson correlations found on Supplemental Digital Content Table 1. During lidocaine administration CPT (r = .3, p = .11), WUR (.34, p = .09), MDT (r = −.24, p = .19) and CPM (r = .26, p = .17) showed marginal correlations with percent pain reduction, although these correlations were not significantly different from those during placebo administration. NPSI sub-scores were not significantly associated with percent pain reduction with lidocaine vs placebo.
Exploratory Analysis – Responder versus non-responder analysis
Overall, a total of seven participants obtained a >50% reduction in pain 60-120 minutes after infusion with lidocaine but not placebo (i.e. lidocaine responders). Supplemental Digital Content Table 2 displays baseline patient characteristics of these participants compared to those who did not respond to lidocaine infusion. Both groups had similar baseline characteristics and received comparable dosages of lidocaine. Compared to non-responders, lidocaine responders had a general tendency to exhibit less profound thermal sensory loss across QST measurements, with a significant difference in cold detection threshold (Figure 6).
Figure 6:
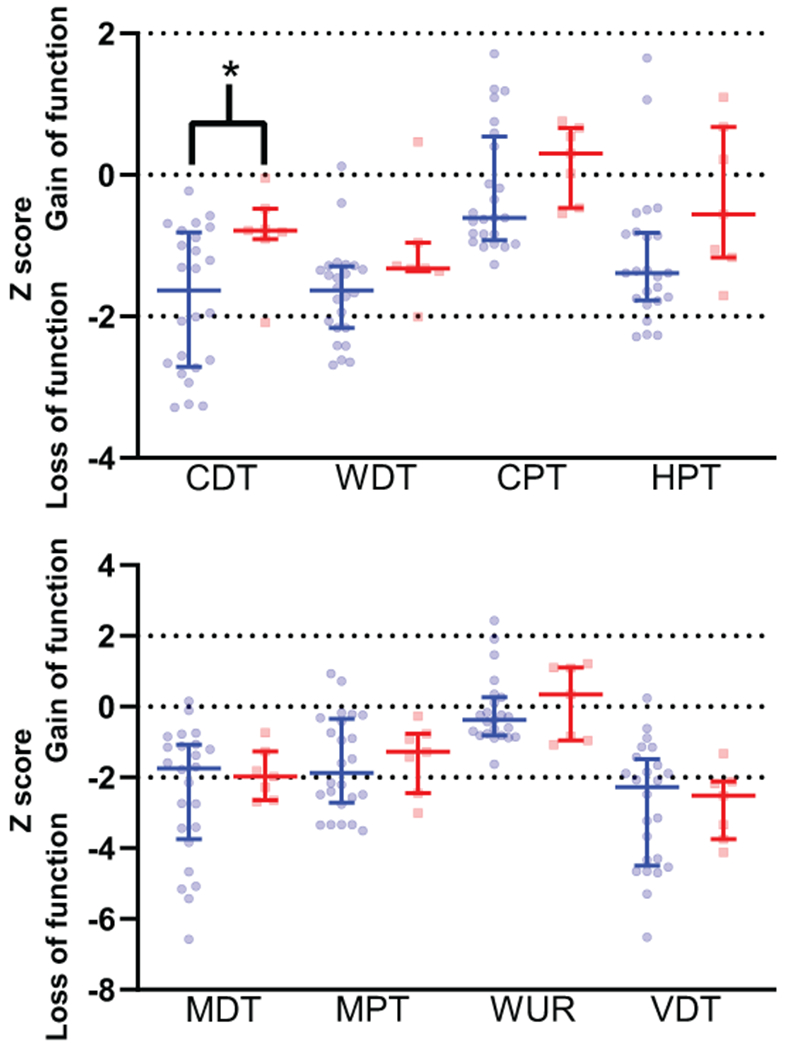
Quantitative sensory testing data, divided by non-responders versus responders (>50% pain reduction at 60-120 minutes with lidocaine but not placebo.) Non-responders denoted with blue, responders denoted with red. * indicates P value < .05. CDT = Cold Detection Threshold (−1.6 [−2.7 to −0.8] versus −0.8 [−0.9 to −0.5]). WDT = Warm Detection Threshold (−1.6 [−2.2 to −1.3] versus −1.3 [−1.4 to −1.0]). CPT = Cold Pain Threshold (−0.6 [−0.9 to 0.5] versus 0.3 [−0.5 to 0.7]). HPT = Heat Pain Threshold (−1.4 [−1.8 to −0.8] versus −0.6 [−1.2 to 0.7]). MDT = Mechanical Detection Threshold (−1.7 [−3.7 to −1.1] versus −2.0 [−2.6 to −1.3]). MPT = Mechanical Pain Threshold (−1.9 [−2.7 to −0.3] versus −1.3 [−2.4 to −0.8]). WUR = Wind Up Ratio (−0.4 [−0.8 to 0.3] versus 0.3 [−1.0 to 1.1]). VDT = Vibration Detection Threshold (−2.3 [−4.5 to −1.50] versus −2.5 [−3.7 to −2.1]).
Adverse Events
Adverse events from this study are summarized in Table 3. All adverse events were mild-to-moderate in nature. The most common adverse event during both lidocaine and placebo administration was dry mouth (13/34 participants during lidocaine administration versus 8/33 during placebo administration). Drowsiness (9/34), perioral numbness (8/34), dysesthesias (7/34) and dizziness (6/34) were more common during lidocaine administration. Only two participants in the study achieved a calculated Cmax lidocaine concentration above 5 μg/mL; both participants described perioral numbness with lidocaine administration. One of these two participants described dysesthesias and slurred speech in addition to perioral numbness. There were no serious adverse events during the study that prompted participant withdrawal; therefore, a number needed to harm could not be determined. In addition, no participants in the study were given 20% intravenous fat emulsion as no serious systemic adverse reactions were observed.
Table 3:
Adverse events
| Lidocaine | Placebo | |||
|---|---|---|---|---|
| Affected/at Risk (%) | Symptom duration in minutes, median (range) | Affected/at Risk (%) | Symptom duration in minutes, median (range) | |
| Dry mouth | 13/34 (40) | 60 (20 to 85) | 8/33 (24) | 50 (39 to 237) |
| Drowsiness | 9/34 (26) | 94 (50 to 529) | 4/33 (12) | 115 (52 to 675) |
| Perioral numbness | 8/34 (24) | 34 (1 to 75) | 2/33 (6) | 30 (18 to 43) |
| Dysesthesia | 7/34 (21) | 16 (2 to 72) | 1/33 (3) | 10 |
| Dizziness | 6/34 (18) | 52 (31 to 475) | 1/33 (3) | 50 |
| Nausea | 2/34 (6) | 46 (22 to 71) | 0/33 (0) | - |
| Slurred speech | 2/34 (6) | 25 (7 to 43) | 0/33 (0) | - |
| Blurred vision | 1/34 (3) | 59 | 0/33 (0) | - |
| Headache | 1/34 (3) | 325 | 1/33 (3) | 486 |
| Arrhythmia | 0/34 (0) | - | 1/33 (3) | 33 |
| Death | 0/34 (0) | - | 0/34 (0) | - |
Lidocaine Pharmacokinetics
Maximum measured lidocaine plasma concentration occurred at 45 minutes after infusion initiation, with a mean concentration of 3.0 μg/mL (SD 1.0) (Figure 7). The results of non-compartment analyses suggest a maximum plasma lidocaine concentration of 3.3 μg/mL (SD 1.1), a half-life of 66.4 (SD 26.2) minutes, a clearance of 1.2 (SD 0.4) L/min, and an area under the concentration-time curve of 327.2 (SD 137.2) min*μg/mL for these participants (n = 32). Lidocaine was not detected in any of the blood samples during placebo administration, verifying appropriate treatment allocation, according to randomization.
Figure 7:
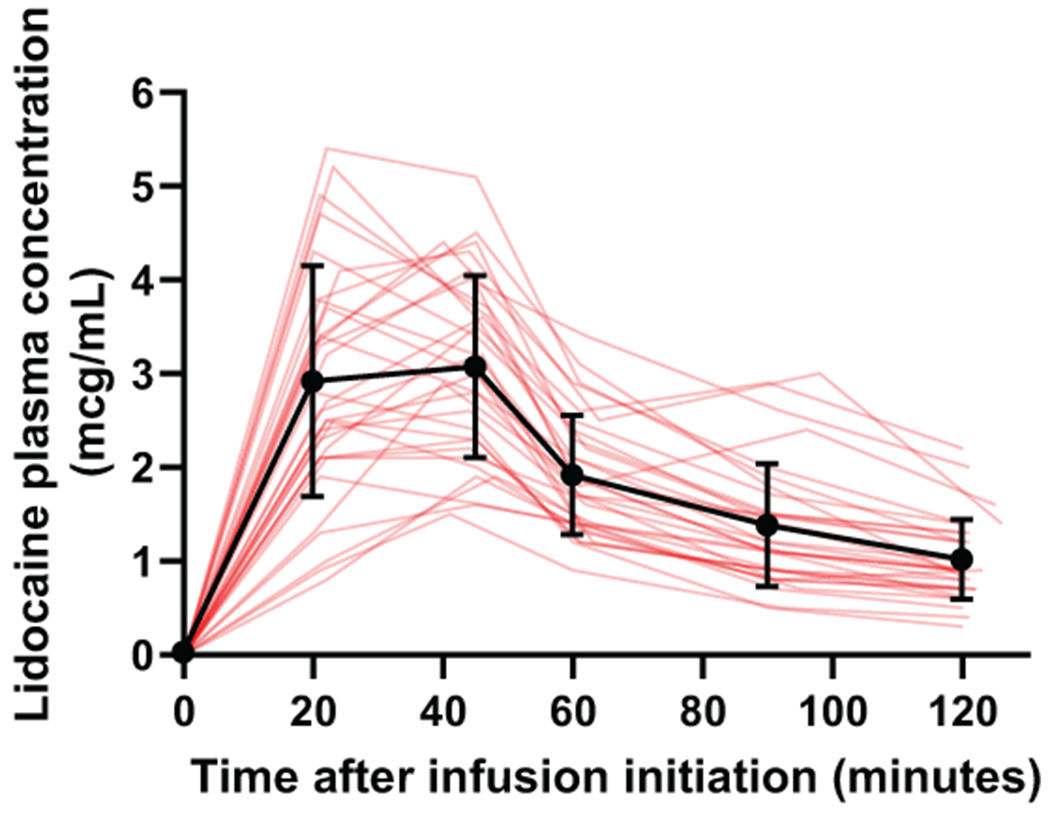
Lidocaine concentrations as a function of time. Thin red lines correspond to individual participants (n = 32). Two participants did not have adequate IV access for repeated blood draws. Black points = mean, error bars = SD
DISCUSSION
In this randomized, double-blind, crossover trial, we compared the analgesic effect of intravenous lidocaine to placebo in patients with painful DPN. Intravenous lidocaine, administered at 5mg/kg IBW dose, resulted in significantly more pain reduction compared to placebo; however, the effect was small and transient. Although the administration of intravenous lidocaine resulted in a >50% decrease in pain severity on average, a high placebo response rate was observed in the study, with a >30% pain reduction from baseline; therefore, the overall pain reduction beyond placebo was modest. The NNT to achieve 30% or 50% short-term relief from lidocaine (not attributable to placebo) was 6.2, which is comparable to other medications used for the management of neuropathic pain.4, 27 The magnitude of relief obtained by lidocaine infusion in the present study was somewhat smaller than that obtained by similar trials in the field; however, the placebo response rate was similar to that found in comparable trials 9, 12. The side effects with lidocaine administration in the current study were mild and not common. Classic side effects such as perioral numbness and drowsiness occurred in 20-30% of participants in this trial, which is lower than that found in comparable trials.12, 13 The plasma concentrations of lidocaine peaked around 3.3 μg/mL, and at the time of primary outcome measure were mostly in the range of 1.5-2 μg/mL. Plasma concentrations are comparable to other similar studies, but may be on the lower end of the therapeutic ranges implicated in analgesic response.13, 28 The IBW dosing approach, utilized to prevent toxicity in obese individuals, may have resulted in a better safety profile but somewhat smaller effect size compared with other studies.
Some studies have reported long-term effects of a single lidocaine infusion in neuropathic pain.29 Except for one participant who reported complete pain relief for at least six weeks after lidocaine infusion (and was subsequently withdrawn from the second treatment as his baseline pain did not resume), we did not observe statistically significant differences in pain reduction between lidocaine and placebo over the three weeks post-intervention period.
QST measurements assessed prior to infusion were consistent across the two study visits. Overall, patients mostly presented with a phenotype of sensory loss across small and large fibers, as seen in lower than average Z-scores for thermal detection/pain thresholds, mechanical detection/pain thresholds, and vibration detection threshold. The phenotypic characterization in the studied sample was similar to that found in the PiNS study of DPN 2. The magnitude of CPM obtained with the dual-thermode method was small, compared with other previously published methods.14, 30
None of the individual QST measures in this study, including the hypothesized mechanical and heat pain thresholds, identified responders to the study intervention in this clinical trial. It is worth noting that several patients presented with severe sensory loss, reaching cut-off values of the thermal and mechanical testing equipment used in this study, which may hinder true interpretation of these participant’s thermal and mechanical threshold limits.
Oxcarbazepine, another sodium channel blocker utilized in the treatment of peripheral neuropathic pain, has shown improved effectiveness in those with an irritable nociceptor phenotype, characterized by hypersensitivity and preserved small nerve fiber function.17 As DPN presentation is characterized by profound hyposensitivity across multiple QST parameters, as previously described and as corroborated within the present trial, a separate analysis of participants with an irritable nociceptor phenotype was not anticipated and likely is not a relevant approach in this patient population.2 However, it is worth noting that the responders to intravenous lidocaine in the current study exhibited less profound sensory loss, which is in line with previous findings that sodium channel blockers may have better utility in neuropathic pain patients with preserved small fiber function.17
While lidocaine suppresses action potential propagation when administered at 10-20 mg/mL concentrations to elicit near complete sodium channel blockade (e.g. perineural administration), it is unlikely that systemic administration can achieve such concentrations without toxicity.28 Lidocaine administration has been shown to reduce ectopic neuronal activity within primary afferent neurons in the dorsal root ganglia (DRG) at dilute concentrations in animal models.31, 32 Selective DRG blockade with dilute 3 mg/mL lidocaine solution has been shown to substantially alleviate phantom limb pain in humans, although the concentrations at the DRG level were likely still higher than those associated with 3 μg/mL peak plasma concentrations with systemic administration.33 Future research measuring local concentration at the DRG with systemic administration may help elucidate whether the main analgesic mechanism of systemic lidocaine results from its suppression of ectopic activity at the DRG level. Central nervous system mechanisms of lidocaine action have also been proposed. Anti-hyperalgesic effects in animal models were observed via the activity of lidocaine’s metabolite, N-ethylglycine, at glycine transporter 1 (GlyT1) to enhance inhibitory transmission in areas of CNS related to pain modulation.34–36 Additional preclinical experiments, including in DPN models, have suggested that lidocaine may alleviate cutaneous hypersensitivity by inhibiting microglial activation in the spinal cord dorsal horn.37, 38 Lidocaine readily crosses the blood brain barrier, and future research should investigate the proposed CNS mechanisms identified in preclinical studies.
We hypothesized that increased hypersensitivity to mechanical and heat pain (i.e. lower MPT and HPT) would be associated with an enhanced analgesic effect of systemic lidocaine in painful DPN. As thermal and mechanical hypersensitivity have been associated with increased expression of voltage-gated sodium channels in sensory neurons, a direct effect of systemic lidocaine on neuronal activity could be associated with a hypersensitivity phenotype. Several factors may have contributed to lack of such association observed in this study. First, while these single phenotypic QST parameters may provide surrogate measures of neuronal hypersensitivity, they are not directly correlated to sensory fiber structure or symptom severity.39 It is also possible that the magnitude of sodium channel blockade required to correlate with reduction of neuronal hypersensitivity is higher than what can be safely achieved with therapeutic doses of intravenous lidocaine. It is also possible that single QST measures are too simplistic to predict the analgesic effect of a drug with multiple possible mechanisms of action.
This study has limitations. This was a single center study with participants recruited through convenience sampling, which may affect generalizability of the trial findings. Although significance was reached on the primary outcome through the use of a cross-over design, the sample size was substantially smaller than what has been recently recommended for pain trials that include phenotypic predictors of response.40 Nevertheless, the correlations obtained between baseline QST measures and pain reduction were weak; these correlations are unlikely to be significant by merely doubling, or even tripling the sample size. While we report on plasma concentration profiles for lidocaine, we did not determine the concentrations of lidocaine metabolite monoethylglycinexylidide (MEGX), which has been reported to modestly correlate with analgesic outcomes in neuropathic pain.41
Notably, there were many strengths to this study. This study was performed using a randomized, double-blinded, placebo-controlled design, which limited treatment bias. All QST procedures were carefully performed by a single, trained member of the research team. Plasma concentrations were analyzed to verify correct treatment allocation, according to randomization. The use of IBW-based dosing resulted in a favorable safety profile, and lidocaine plasma concentrations were comparable to those previously reported in the literature.
In summary, although lidocaine administration resulted in a statistically significant decrease in pain scores when compared to placebo, QST measures were not associated with response to treatment in patients with painful DPN. Considering the potential interactions in various phenotypic and genotypic characteristics that likely determine the response to treatment, it is likely that future studies investigating predictors of response to pharmacological interventions need to be large enough to allow testing for multivariable predictors of response. Such predictors may include not only psychophysical measures, but also demographic, psychosocial, genetic, and other factors.42–44
Supplementary Material
Acknowledgements
We would like to acknowledge Dr. Jacob AuBuchon, Dr. Folasade Oladapo, as well as the outstanding nurses Sandy Finney, Tricia Mischel, Lauren Mistler, Johanna Olusesi, Norma Silk, Teresa Minor, Deborah Chandler, and Amy Fedder for their help with conducting the study.
Funding
The study was supported by a Pilot & Feasibility Award (P30 DK020579) from Washington University Diabetes Research Center. Research reported in this publication was also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1 TR002345. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interest:
Simon Haroutounian has received research support from DISARM therapeutics, and consulting fees from Medoc Ltd and Rafa Laboratories, outside the submitted work. The remaining authors report no conflicts of interest.
Footnotes
Registration and protocol
Registration for this trial along with the study protocol can be found at clinicaltrials.gov (NCT02363803).
Data sharing statement
Data and materials supporting the results or analyses presented in this manuscript will be made available upon reasonable request.
REFERENCES
- 1.Barrett AM, Lucero MA, Le T, Robinson RL, Dworkin RH and Chappell AS. Epidemiology, public health burden, and treatment of diabetic peripheral neuropathic pain: a review. Pain medicine 2007;8 Suppl 2:S50–62. [DOI] [PubMed] [Google Scholar]
- 2.Themistocleous AC, Ramirez JD, Shillo PR, Lees JG, Selvarajah D, Orengo C, Tesfaye S, Rice AS and Bennett DL. The Pain in Neuropathy Study (PiNS): a cross-sectional observational study determining the somatosensory phenotype of painful and painless diabetic neuropathy. Pain 2016;157:1132–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen TS and Finnerup NB. Neuropathic pain treatment: a further step forward. Lancet 2009;374:1218–9. [DOI] [PubMed] [Google Scholar]
- 4.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH and Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet neurology 2015;14:162–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Hehn CA, Baron R and Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012;73:638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black JA, Nikolajsen L, Kroner K, Jensen TS and Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Annals of neurology 2008;64:644–53. [DOI] [PubMed] [Google Scholar]
- 7.Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F and Strichartz GR. The role of sodium channels in chronic inflammatory and neuropathic pain. J Pain 2006;7:S1–29. [DOI] [PubMed] [Google Scholar]
- 8.Meacham K, Shepherd A, Mohapatra DP and Haroutounian S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr Pain Headache Rep 2017;21:28. [DOI] [PubMed] [Google Scholar]
- 9.Kastrup J, Petersen P, Dejgard A, Angelo HR and Hilsted J. Intravenous lidocaine infusion--a new treatment of chronic painful diabetic neuropathy? Pain 1987;28:69–75. [DOI] [PubMed] [Google Scholar]
- 10.Bach FW, Jensen TS, Kastrup J, Stigsby B and Dejgard A. The effect of intravenous lidocaine on nociceptive processing in diabetic neuropathy. Pain 1990;40:29–34. [DOI] [PubMed] [Google Scholar]
- 11.Attal N, Gaude V, Brasseur L, Dupuy M, Guirimand F, Parker F and Bouhassira D. Intravenous lidocaine in central pain: a double-blind, placebo-controlled, psychophysical study. Neurology 2000;54:564–74. [DOI] [PubMed] [Google Scholar]
- 12.Attal N, Rouaud J, Brasseur L, Chauvin M and Bouhassira D. Systemic lidocaine in pain due to peripheral nerve injury and predictors of response. Neurology 2004;62:218–25. [DOI] [PubMed] [Google Scholar]
- 13.Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrom JB and Jensen TS. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 2014;155:1272–9. [DOI] [PubMed] [Google Scholar]
- 14.Yarnitsky D, Granot M, Nahman-Averbuch H, Khamaisi M and Granovsky Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain 2012;153:1193–8. [DOI] [PubMed] [Google Scholar]
- 15.Campbell CM, Kipnes MS, Stouch BC, Brady KL, Kelly M, Schmidt WK, Petersen KL, Rowbotham MC and Campbell JN. Randomized control trial of topical clonidine for treatment of painful diabetic neuropathy. Pain 2012;153:1815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpson DM, Schifitto G, Clifford DB, Murphy TK, Durso-De Cruz E, Glue P, Whalen E, Emir B, Scott GN, Freeman R and Group HIVNS. Pregabalin for painful HIV neuropathy: a randomized, double-blind, placebo-controlled trial. Neurology 2010;74:413–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demant DT, Lund K, Vollert J, Maier C, Segerdahl M, Finnerup NB, Jensen TS and Sindrup SH. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. Pain 2014;155:2263–73. [DOI] [PubMed] [Google Scholar]
- 18.Hincker A, Frey K, Rao L, Wagner-Johnston N, Ben Abdallah A, Tan B, Amin M, Wildes T, Shah R, Karlsson P, Bakos K, Kosicka K, Kagan L and Haroutounian S. Somatosensory predictors of response to pregabalin in painful chemotherapy-induced peripheral neuropathy: a randomized, placebo-controlled, crossover study. Pain 2019;160:1835–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rolke R, Baron R, Maier C, Tolle TR, Treede RD, Beyer A, Binder A, Birbaumer N, Birklein F, Botefur IC, Braune S, Flor H, Huge V, Klug R, Landwehrmeyer GB, Magerl W, Maihofner C, Rolko C, Schaub C, Scherens A, Sprenger T, Valet M and Wasserka B. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): standardized protocol and reference values. Pain 2006;123:231–43. [DOI] [PubMed] [Google Scholar]
- 20.Fruhstorfer H, Lindblom U and Schmidt WC. Method for quantitative estimation of thermal thresholds in patients. Journal of neurology, neurosurgery, and psychiatry 1976;39:1071–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devine BJ. Gentamicin Therapy. Drug Intel Clin Phar 1974;8:650–655. [Google Scholar]
- 22.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N and Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haroutiunian S, Nikolaisen L, Bendtsen TF, Kristensen AD, Finnerup NB and Jensen TS Contribution of peripheral sensory input in maintaining chronic neuropathic pain and predictors of response to intravenous lidocaine. 4th International Congress on Neuropathic Pain. Toronto, ON, Canada, 2013. [Google Scholar]
- 24.Chen L, Liao L, Zuo Z, Yan Y, Yang L, Fu Q, Chen Y and Hou J. Simultaneous determination of nikethamide and lidocaine in human blood and cerebrospinal fluid by high performance liquid chromatography. J Pharm Biomed Anal 2007;43:1757–62. [DOI] [PubMed] [Google Scholar]
- 25.Lotfi H, Debord J, Dreyfuss MF, Marquet P, Ben Rhaiem M, Feiss P and Lachatre G. Simultaneous determination of lidocaine and bupivacaine in human plasma: application to pharmacokinetics. Ther Drug Monit 1997;19:160–4. [DOI] [PubMed] [Google Scholar]
- 26.Magerl W, Krumova EK, Baron R, Tolle T, Treede RD and Maier C. Reference data for quantitative sensory testing (QST): refined stratification for age and a novel method for statistical comparison of group data. Pain 2010;151:598–605. [DOI] [PubMed] [Google Scholar]
- 27.Finnerup NB, Haroutounian S, Baron R, Dworkin RH, Gilron I, Haanpaa M, Jensen TS, Kamerman PR, McNicol E, Moore A, Raja SN, Andersen NT, Sena ES, Smith BH, Rice ASC and Attal N. Neuropathic pain clinical trials: factors associated with decreases in estimated drug efficacy. Pain 2018;159:2339–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao J and Chen LL. Systemic lidocaine for neuropathic pain relief. Pain 2000;87:7–17. [DOI] [PubMed] [Google Scholar]
- 29.Zhu B, Zhou X, Zhou Q, Wang H, Wang S and Luo K. Intra-Venous Lidocaine to Relieve Neuropathic Pain: A Systematic Review and Meta-Analysis. Front Neurol 2019;10:954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nahman-Averbuch H, Yarnitsky D, Granovsky Y, Sprecher E, Steiner M, Tzuk-Shina T and Pud D. Pronociceptive pain modulation in patients with painful chemotherapy-induced polyneuropathy. J Pain Symptom Manage 2011;42:229–38. [DOI] [PubMed] [Google Scholar]
- 31.Koplovitch P and Devor M. Dilute lidocaine suppresses ectopic neuropathic discharge in dorsal root ganglia without blocking axonal propagation: a new approach to selective pain control. Pain 2018;159:1244–1256. [DOI] [PubMed] [Google Scholar]
- 32.Yatziv SL and Devor M. Suppression of neuropathic pain by selective silencing of dorsal root ganglion ectopia using nonblocking concentrations of lidocaine. Pain 2019;160:2105–2114. [DOI] [PubMed] [Google Scholar]
- 33.Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G and Devor M. Peripheral nervous system origin of phantom limb pain. Pain 2014;155:1384–91. [DOI] [PubMed] [Google Scholar]
- 34.Werdehausen R, Mittnacht S, Bee LA, Minett MS, Armbruster A, Bauer I, Wood JN, Hermanns H and Eulenburg V. The lidocaine metabolite N-ethylglycine has antinociceptive effects in experimental inflammatory and neuropathic pain. Pain 2015;156:1647–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saade NE, Al Amin H, Tchachaghian S, Jabbur SJ and Atweh SF. Alteration of GABAergic and glycinergic mechanisms by lidocaine injection in the rostral ventromedial medulla of neuropathic rats. Pain 2010;149:89–99. [DOI] [PubMed] [Google Scholar]
- 36.Muth-Selbach U, Hermanns H, Stegmann JU, Kollosche K, Freynhagen R, Bauer I and Lipfert P. Antinociceptive effects of systemic lidocaine: involvement of the spinal glycinergic system. Eur J Pharmacol 2009;613:68–73. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki N, Hasegawa-Moriyama M, Takahashi Y, Kamikubo Y, Sakurai T and Inada E. Lidocaine attenuates the development of diabetic-induced tactile allodynia by inhibiting microglial activation. Anesth Analg 2011;113:941–6. [DOI] [PubMed] [Google Scholar]
- 38.Cheng KI, Lai CS, Wang FY, Wang HC, Chang LL, Ho ST, Tsai HP and Kwan AL. Intrathecal lidocaine pretreatment attenuates immediate neuropathic pain by modulating Nav1.3 expression and decreasing spinal microglial activation. BMC Neurol 2011;11:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlsson P, Hincker AM, Jensen TS, Freeman R and Haroutounian S. Structural, functional, and symptom relations in painful distal symmetric polyneuropathies: a systematic review. Pain 2019;160:286–297. [DOI] [PubMed] [Google Scholar]
- 40.Gewandter JS, McDermott MP, Mbowe O, Edwards RR, Katz NP, Turk DC and Dworkin RH. Navigating trials of personalized pain treatments: we’re going to need a bigger boat. Pain 2019;160:1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tremont-Lukats IW, Hutson PR and Backonja MM. A randomized, double-masked, placebo-controlled pilot trial of extended IV lidocaine infusion for relief of ongoing neuropathic pain. Clin J Pain 2006;22:266–71. [DOI] [PubMed] [Google Scholar]
- 42.Harris RE, Napadow V, Huggins JP, Pauer L, Kim J, Hampson J, Sundgren PC, Foerster B, Petrou M, Schmidt-Wilcke T and Clauw DJ. Pregabalin rectifies aberrant brain chemistry, connectivity, and functional response in chronic pain patients. Anesthesiology 2013;119:1453–64. [DOI] [PubMed] [Google Scholar]
- 43.Shidahara Y, Natsume T, Awaga Y, Ogawa S, Yamoto K, Okamoto S, Hama A, Hayashi I, Takamatsu H and Magata Y. Distinguishing analgesic drugs from non-analgesic drugs based on brain activation in macaques with oxaliplatin-induced neuropathic pain. Neuropharmacology 2019;149:204–211. [DOI] [PubMed] [Google Scholar]
- 44.Mankovsky T, Lynch M, Clark A, Sawynok J and Sullivan MJ. Pain catastrophizing predicts poor response to topical analgesics in patients with neuropathic pain. Pain Res Manag 2012;17:10–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.