

Abstract
Full activity of soluble methane monooxygenase (sMMO) depends upon formation of 1:1 complex of the regulatory protein MMOB with each alpha subunit of the (αβγ)2 hydroxylase, sMMOH. Previous studies have shown that mutations in the core region of MMOB and in the N- and C-termini cause dramatic changes in the rate constants for steps in the sMMOH reaction cycle. Here, X-ray crystal structures are reported for the sMMOH complex with two double variants within the core region of MMOB, DBL1 (N107G/S110A) and DBL2 (S109A/T111A), as well as two variants in the MMOB N-terminal region H33A and H5A. DBL1 causes a 150-fold decrease in formation rate constant of the reaction cycle intermediate P, whereas DBL2 accelerates the reaction of the dinuclear Fe(IV) intermediate Q with substrates larger than methane by 3–4 fold. H33A also greatly slows P formation, while H5A modestly slows both formation of Q and its reactions with substrates. Complexation with DBL1 or H33A alters the position of sMMOH residue R245, which is part of a conserved hydrogen-bonding network encompassing the active site diiron cluster where P is formed. Accordingly, EPR spectra of sMMOH:DBL1 and sMMOH:H33A complexes differ markedly from that of sMMOH:MMOB, showing an altered electronic environment. In the sMMOH:DBL2 complex, the position of M247 in sMMOH is altered such that it enlarges a molecular tunnel associated with substrate entry into the active site. The H5A variant causes only subtle structure changes despite its kinetic effects, emphasizing the precise alignment of sMMOH and MMOB required for efficient catalysis.
Graphical Abstract
INTRODUCTION
Soluble methane monooxygenase (sMMO) catalyzes the oxygen-dependent hydroxylation of methane to generate methanol in the C1 metabolic cycle of methanotrophs.1, 2 This reaction occurs at a non-heme, oxygen and carboxylate-bridged dinuclear iron cluster contained within the hydroxylase component (sMMOH) of sMMO. The ability of this metallocenter to oxidatively cleave the strong C-H bond of methane, and indeed, the C-D bond of its fully deuterated isotopologue (BDE = 105 and 107 kcal/mole, respectively),3, 4 is conferred by the generation of the reaction cycle intermediate compound Q.5 This exceptionally reactive species contains an oxygen bridged diiron(IV) cluster,6–10 which has no observed precedent in Nature and has only recently been approached in biomimetic synthetic studies.11 It is remarkable that sMMO and Q are able to preferentially react with the hydrocarbon with the strongest aliphatic C-H bond amidst a sea of alternative substrates that are more easily oxidized. This selectivity is combined with insignificant over-oxidation of the methanol product, which is instead selectively oxidized by downstream enzymes to conserve energy and carbon for growth. These characteristics make sMMO an excellent model system to inform the generation of synthetic model complexes for selective strong C-H bond functionalization and Gas-to-Liquid processes.
sMMO is a three protein system: the active site-containing hetero-dimeric (αβγ)2 sMMOH (245 kDa), a cofactorless regulatory protein MMOB (15 kDa) and an electron-transfer FAD and [2Fe2S] containing reductase MMOR (38 kDa).1, 12 MMOR transfers two electrons from NADH to the sMMOH diiron(III) cluster at the beginning of the catalytic cycle, converting it to the diiron(II) state (sMMOHred), an intermediate primed to react with O2 (Scheme 1).5, 13, 14 Formation of a complex with MMOB (sMMOHred:MMOB) allows rapid O2-binding in the active site (intermediate O) and thereafter to the diferrous cluster iron atoms (intermediate, P*).15 P* then converts in a proton-dependent reaction to intermediate P, which is proposed to be a cis-μ−1,2-peroxo-bridged diiron(III) species.5, 14–16 The O-O bond is then broken in another proton-dependent step to form Q.5, 6, 16, 17 Hydrogen atom abstraction from methane by Q would form intermediate R, nominally a Fe2(III,IV)-(μ-O)(OH) species.18, 19 Subsequent radical rebound to the methyl radical would form the product methanol-bound complex termed T.5 Finally, the alcohol product would be released from the active site in the rate-limiting step of the enzyme reaction. All of these intermediates except the short-lived R have been directly observed and the rate constants of their interconversions determined using stopped-flow and rapid freeze-quench techniques.5, 14, 15, 17, 20–22
Scheme 1.
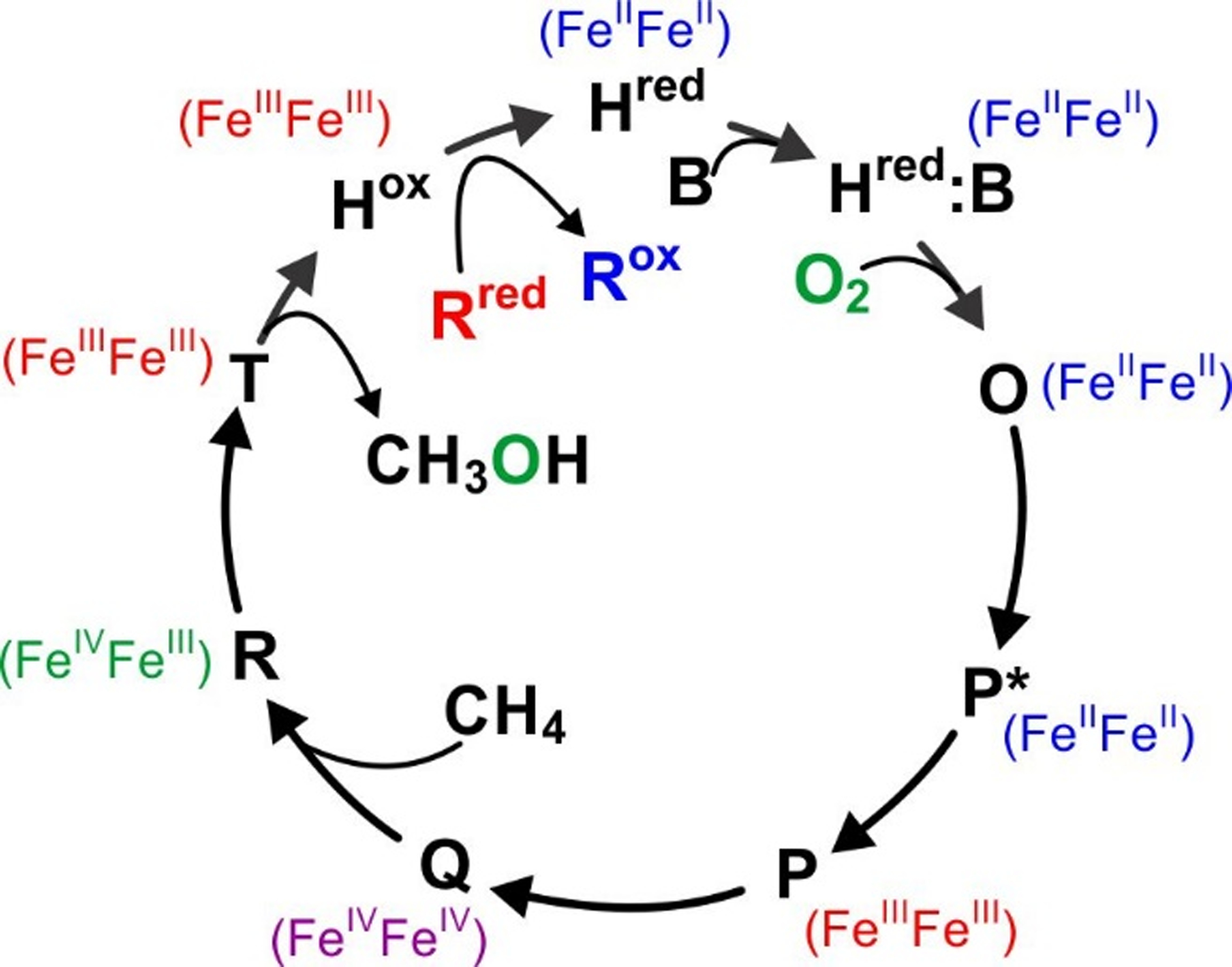
Reaction cycle intermediates of sMMO. The oxidation state of the irons in the diiron cluster are indicated adjacent to the intermediate. H = sMMOH, B = MMOB, R = MMOR
While sMMOH is capable of slowly activating O2 and oxidizing methane in isolation, physiological rates of substrate turnover only occur upon formation of a protein complex with MMOB.13, 23, 24 One of the main effects of this protein-protein interaction upon the sMMOH catalytic cycle is the 1000-fold enhancement of the rate constant for O2 binding and activation at the diiron cluster.5, 22 As a result, the rate-limiting step of catalysis is shifted from O2 binding to product release, leading to a 150-fold increase in turnover number.13 Recent studies have demonstrated that this regulation by MMOB arises from the organization of a small-molecule tunnel for O2 diffusion into the sMMOH active site concurrent with complex formation and diiron cluster reduction.25, 26 The final and perhaps most elegant aspect of MMOB regulation is described by a recently proposed dynamic equilibrium model that explains how sMMO avoids reductive quenching of Q by MMOR.27 MMOB and MMOR were shown to bind with similar affinities to sMMOH and to compete for the same binding site near the buried diiron cluster. The distinct roles of MMOR and MMOB, diiron cluster reduction versus O2 binding, respectively, are manifested only when the correct pair of sMMO components forms a complex. Thus, the diiron cluster is not reduced until MMOB dissociates and is replaced by reduced MMOR, but O2 cannot bind until MMOR dissociates and MMOB binds. In this way the dynamic equilibrium of MMOB and MMOR binding to sMMOH pulls the reaction cycle forward. Transient kinetic studies show that the koff rate constant of the MMOB from sMMOH after oxygen binding is tailored such that the catalytic cycle is completed during the lifetime of the sMMOH:MMOB complex.28, 29 Consequently, reduced MMOR cannot bind sMMOH in the middle of the reaction cycle to aberrantly quench Q.
Our understanding of the regulatory effects of MMOB have been greatly advanced by studies of MMOB variants.15, 21, 27–33 In the absence of a heterologous expression system for soluble sMMOH, MMOB mutants have provided the sole means to perturb sMMO catalysis for protein structure-function studies. Mutagenesis was primarily guided by an NMR study where differential broadening of MMOB resonances in 1H-15N HSQC spectra at different ratios of MMOH and MMOB was used to identify the protein-protein interface.30 MMOB residues at the N-terminal tail (residues 1 – 35), core region (residues 36 – 125) and C-terminal tail (residues 126 – 138) were targeted for site-directed or truncation mutagenesis. In particular, mutation of four residues in the core region to yield what was termed the Quad variant (N107G/S109A/S110A/T111A),28 binary derivatives of the Quad variant termed DBL1 (N107G/S110A) and DBL2 (S109A/T111A),21, 33 two N-terminal region variants, H5A and H33A,28 and the N- and C-terminal truncation variants30, 34 demonstrated dramatic effects of specific mutations on specific steps of the reaction cycle (Figure 1). These effects include: (i) retarding O2-binding (MMOB Δ2–29 and V41R)25, 30, 34, (ii) uncoupling the O2-activation reaction from hydrocarbon oxidation (MMOB Δ126–138)34), (iii) relaxing the size-selective entry of hydrocarbon substrates (Quad, DBL2)21, 28, 33, (iv) disrupting the quantum-tunneling nature of the HAT reaction of Q with methane (Quad)32, and (v) significantly increasing or decreasing the rate constants of reaction cycle steps (H33A, DBL1, Δ126–138, H5A, V41R, V39R)21, 25, 28, 34. This wide-range of impacts was the first indication that significant conformational changes must be imparted within sMMOH by the binding of MMOB.
Figure 1.
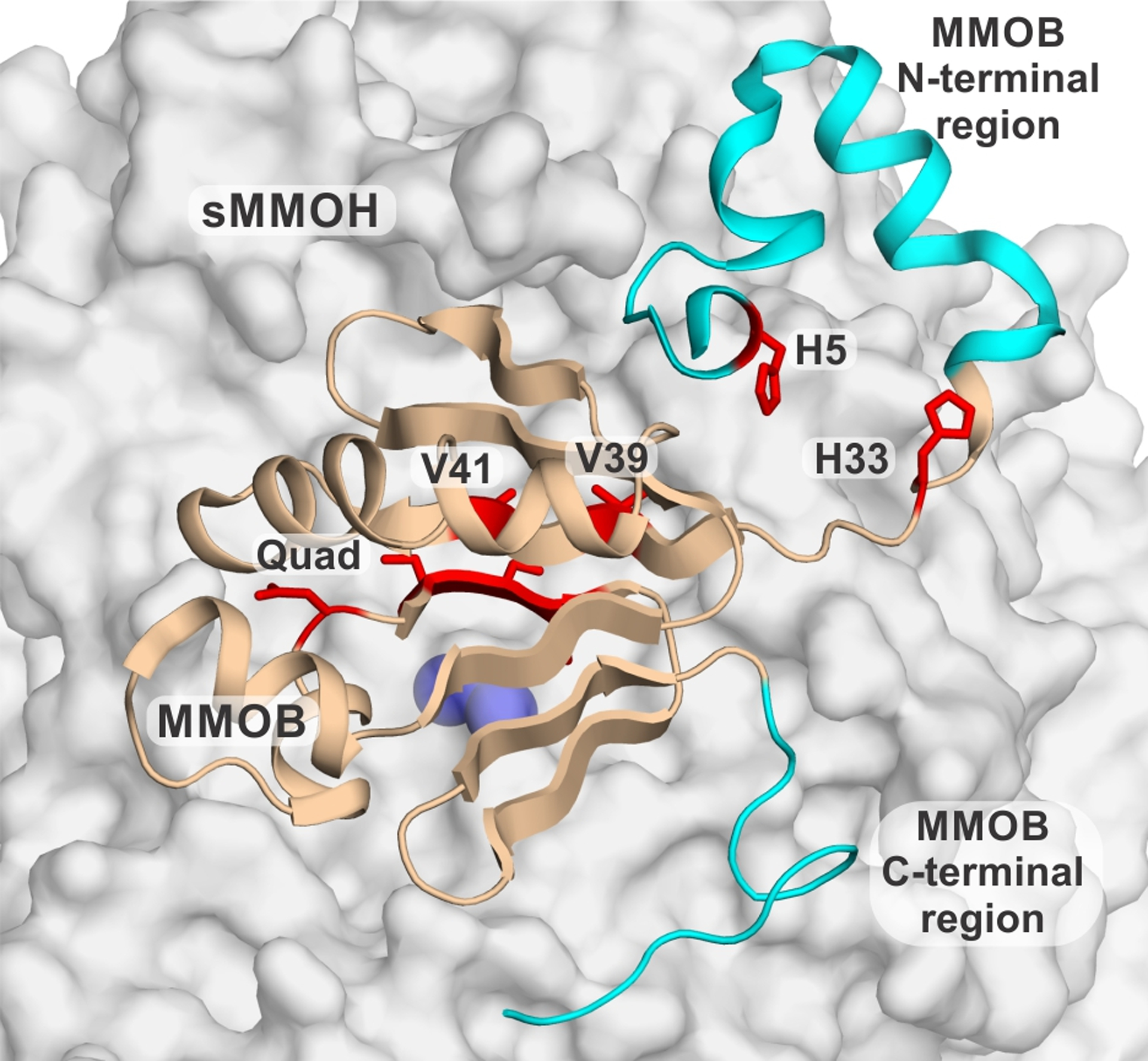
Map of the sites of mutation in MMOB as visualized in the sMMOH:MMOB complex (PDB: 6VK5). MMOB is shown as wheat colored cartoon and sMMOH as a gray surface with the iron atoms as purple spheres. Truncation mutants are represented by cyan colored sections whereas single residue mutants are shown as red sticks.
Recent improvements to the purification of the sMMOH protein that increase specific activity and sample homogeneity8, 15, 35 have allowed the sMMOH:MMOB complex from M. trichosporium OB3b to be crystallized. High-resolution (< 2.0 Å) structures have been obtained from these crystals at both cryogenic temperatures at synchrotrons and at room temperature using Serial Femtosecond Crystallography (SFX) at next-generation X-ray Free-Electron Laser (XFEL) sources.25, 36 These structures have helped delineate the myriad of structural changes occurring within sMMOH and MMOB upon complexation. As expected from early cross-linking studies,24 it was observed that MMOB binds to the α-subunit of sMMOH 12 Å above the active site.36, 37 Two of the four helices providing ligands to the diiron cluster along with two adjacent helices reorganize upon binding MMOB. Some of these conformational changes have been correlated with specific functional effects of MMOB, such as the enhancement of O2-binding through molecular tunnel formation,25 whereas the others remain cryptic.
Here, the regulatory effects of MMOB are further probed by investigating the structures of the complexes of sMMOH with four MMOB variants that affect individual steps of the reaction cycle. An analysis of the protein conformational changes in relation to the wild-type sMMOH:MMOB complex illustrates the relevance of small-molecule tunnels encompassing the sMMOH-MMOB interface in selectively transporting oxygen and methane into the active site. Insight into the role of a conserved hydrogen bond network in modulating the diiron cluster electronics and proton transfer required for reaction cycle intermediate interconversion is also derived from these structures.
Experimental Procedures
Reagents, Protein Expression, and Purification.
Water was purified with a Millipore-Q system. Ultra-high-purity 3-(N-morpholino)-propanesulfonic acid (MOPS) buffer was purchased from Sigma-Millipore. Two different suppliers were used to obtain Na2HPO4 and PEG3350, Hampton Research and Sigma-Millipore. Glass rods and seed bead tubes were supplied by Hampton Research. The sMMOH protein was purified from the native host, Methylosinus trichosporium (Mt) OB3b, as previously reported.35 Plasmids of the MMOB variants; pBWJ400 DBL1, pBWJ400 DBL2, pBWJ400 H5A, and pBWJ400 H33A were transformed into Escherichia coli BL21(DE3) chemically competent cells.21, 28 The proteins were overexpressed and purified using previously described methods.21, 28, 35
Protein Crystallization, Data Collection, Structure Solution, and Refinement.
All protein crystals were grown using the sitting drop vapor diffusion method at room temperature. The sMMOH:DBL2 and sMMOH:H5A protein crystallization conditions consisted of 20% PEG3350 and 0.2 M Na2HPO4 pH 8.8. 500 μl of the crystallization solution was added to each well of a 24-well Cryschem M Plate purchased from Hampton Research. The protein solution consisted of 60 μM sMMOH and 120 μM of either DBL2 or H5A in 100 mM MOPS buffer pH 7.0. 1.5 μl of the crystallization solution was mixed with 1.5 μl protein solution in the sitting well, and then the wells were sealed with Crystal Clear sealing tape purchased from Hampton Research. Bipyramidal crystals formed within 2–3 days. Protein crystals were looped and then transferred to a cryo-solution containing the well solution supplemented with 10% (v/v) ethylene glycol, and then plunged into liquid nitrogen.
The sMMOH:DBL1 and sMMOH:H33A protein crystallization conditions consisted of 21% PEG3350 and 0.2 M Na2HPO4 pH 6.6. 500 μl of the crystallization solution was added to each well of a 24-well Cryschem M Plate. The protein solution consisted of 60 μM sMMOH and 120 μM of H33A or 240 μM DBL1 in 100 mM MOPS buffer pH 7. A seed stock solution was created first by adding 5 μl of the well solution to a drop of wild-type sMMOH:MMOB crystals, crushing the crystals with a glass rod, then transferring the crushed crystals to a Hampton Research seed bead tube containing 30 μl of well solution. The seed bead tube was vortexed for 6 minutes in 30 s intervals. 1.5 μl of the crystallization solution was mixed with 1.5 μl protein solution in the sitting well and then 0.5 μl of the seed stock solution was added. The wells were sealed with Crystal Clear tape and bipyramidal crystals formed in 4–5 days. These crystals were then used to make separate sMMOH:DBL1 and sMMOH:H33A seed stock solutions. The seed stock solutions were then used to set up crystal trays using the same crystallization conditions and protein solutions mentioned above. Bipyramidal crystals formed in 4–5 days and were cryoprotected and frozen as described above.
The crystals were exposed to X-ray radiation, data collected, and the protein structures modeled as described previously.25 PyMOL version 2.3.3 (Schrödinger) was used to visualize, analyze, and design figures of crystal structures. The distribution of the R245 side chain between the on and off conformations (defined below) was assessed through the alternate conformations functionality within COOT followed by occupancy refinement.
Small Molecule Tunnel Analysis.
MOLE 2.538 was used to assess the protein complex structures for identifying small molecule tunnels. The benzoate molecule bound to the diiron cluster was used as the starting point for the calculations. Hetatoms and hydrogen atoms are not taken into account while performing calculations. This precaution ensured that potential tunnels are not obscured by the blocking effect of exogenous molecules that originated from the crystallization conditions or cryoprotectant solution. The probe radius, which controls the level of detail of the calculated surface was maintained at the default value of 3 Å. The cutoff ratio was lowered to 0.5, which meant that tunnels sharing 50 % of the calculated path with one another are considered as one tunnel in order to reduce redundancy. The strategy for using MOLE and PyMOL to identify likely tunnels in sMMOH:MMOB and other proteins is described in Supporting Information.
Electron Paramagnetic Resonance (EPR) Sample Preparation and Measurements.
The mixed-valent state of sMMOH was generated by anaerobically reducing 250 μM sMMOH (500 μM active sites; 1 mM active site iron) in isolation with 250 μM sodium dithionite in the presence of 0.5 mM phenazine methosulfate as mediator for 15 min at room temperature. Aliquots of this reaction were subsequently mixed with stoichiometric equivalents of the various MMOB variants in an anaerobic fashion, resulting in a final working concentration of 200 μM sMMOH (400 μM active sites) and 400 μM MMOB in the EPR samples. The diferrous sMMOH:MMOB complex samples were prepared by reducing preformed complexes of sMMOH (150 μM) and MMOB variants (300 μM) with a catalytic amount of MMOR (10 μM) and NADH (1 mM) under anaerobic conditions at room temperature for 30 min.13 EPR measurements were made using a Bruker Elexsys E-500 spectrometer with an E500-T-DU Digital upgrade and equipped with an Oxford ESR-910 liquid helium cryostat. Data collection conditions are described in the EPR figure legend.
RESULTS
Crystallization and X-ray Crystallography of sMMOH Complexes with MMOB Variants.
MMOB variants were expressed and purified and then co-crystallized with sMMOH at a 1:1 active sites ratio (2 active sites per sMMOH molecule) as described in Experimental Procedures. X-ray crystallography and data refinement yielded structures for sMMOH:DBL2 (Form 1 - PDB:7S6Q, 1.96 Å and Form 2 - PDB:7S7H, 2.40 Å), sMMOH:DBL1 (PDB:7S6S, 1.98 Å), sMMOH:H33A (PDB:7S6T, 1.82 Å) and sMMOH:H5A (PDB:7S6R, 1.89 Å). The refinement statistics are reported in Supporting Information Table S1. Each of the structures except 7S7H contained a benzoate molecule bound to the diiron cluster (see below). Previously published structures of sMMOH:MMOB solved using synchrotron sources25 also had benzoate bound (PDB:6VK4 and 6VK5), facilitating a direct comparison of structures to evaluate changes caused by the MMOB mutations. Attempts to crystallize the Quad variant complex with sMMOH were unsuccessful.
Structure of sMMOH:DBL2.
Two X-ray crystal structures of the diferric sMMOH:DBL2 protein complex were obtained (PDB: 7S6Q and 7S7H). The two structures are nearly identical except for the exogenous ligands bound to the diferric cluster; one of the μ-hydroxo bridging ligands found in the native structure is replaced by benzoate in 7S6Q and ethylene glycol in 7S7H (Figure 2, Omit map shown in Figure S1). The source of the benzoate was determined to be a contaminant found in the PEG3350 used as a precipitant in the crystallization solution. While PEG3350 from an alternative supplier eliminated the benzoate, it was replaced by ethylene glycol from the cryo-protectant solution. A previously published 1.52 Å X-ray crystal structure of Mt sMMOHox (PDB:6VK6) also has an ethylene glycol molecule bound to the diferric cluster.25 However, in that case the ethylene glycol is bound in the gauche conformation, while in the 2.4 Å sMMOH:DBL2 structure, the ethylene glycol ligand is present in the anti conformation. This binding mode causes the ethylene glycol to extend to the outer boundary of the active site cavity, causing a steric clash with the side chain of F188 (Figure S2). This residue shift causes portions of the sMMOH α-subunit to undergo structural rearrangements that have been previously discussed in detail and are not repeated here.25
Figure 2.
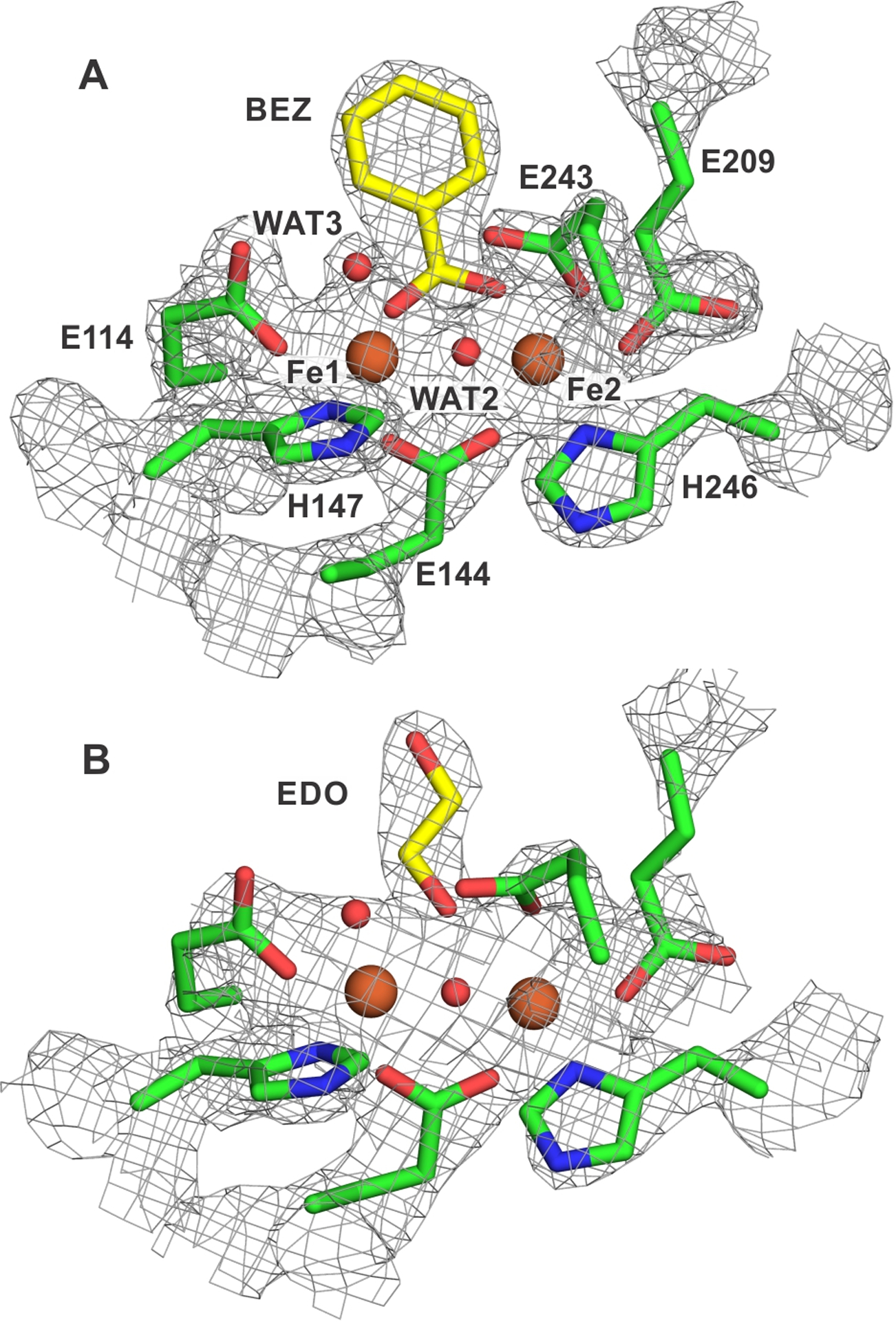
Exogenous ligands bound to the diiron cluster of the sMMOH:DBL2 complex structures. The iron atoms are represented as orange spheres and the first-sphere ligands as sticks, while the 2mFo-DFc electron density map is shown as a gray mesh (σ = 2.0). Shown with carbons in yellow are benzoate (BEZ) bound to the 1.96 Å structure (7S6Q) in panel A and ethylene glycol (EDO) to the 2.4 Å structure (7S7H) in panel B. Omit map in Figure S1.
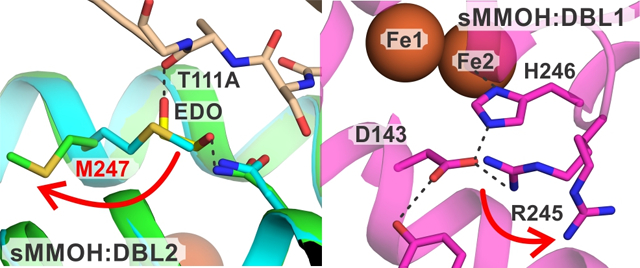
A structural comparison between the sMMOH:DBL2 structure (7S6Q and 7S7H showed the same results) and a previously published sMMOH:MMOB with benzoate bound structure (PDB:6VK5) revealed several structural changes. The β-sheet harboring the T111A/S109A mutation in MMOB shortens in length and becomes twisted when compared to the wild type protein (Figures S3 and S4). This region (MMOB residues 109 to 112) is vital for sMMOH:MMOB interactions as depicted by the reorganization from an unstructured loop in isolated MMOB to a β-sheet in the protein complex.36 The hydrogen bond interactions between MMOB and sMMOH in this region are either abolished (MMOB S109:sMMOH L237) or significantly disrupted (MMOB S110:sMMOH N214) in the sMMOH:DBL2 structure (Figure 3). A small section of helix F of the α-subunit of sMMOH (240 to 249) is reorganized with the largest change undergone by M247 (Figure S4). The sidechain of M247 rotates away from the sMMOH:MMOB interface by 105 degrees (Figure 3) allowing a molecule of ethylene glycol to occupy the vacant position. The ethylene glycol is stabilized by a hydrogen bond with the backbone carbonyl group of MMOB A111. This interfacial region of the protein complex has been termed the hydrophobic dome, which is a microenvironment created upon complex formation thought to aid the binding and transport of non-polar substrates like O2 into the MMOH active site.25 The only other reorganization taking place in sMMOH is restricted to an unstructured loop region (56 to 63) and a portion of helix C (129 to 141) within the α-subunit (Figure S4). The diiron cluster remains unaltered in the sMMOH:DBL2 complex with respect to the bridging ligands, and the first-, second-, and third-sphere residues (Figure S5).
Figure 3.
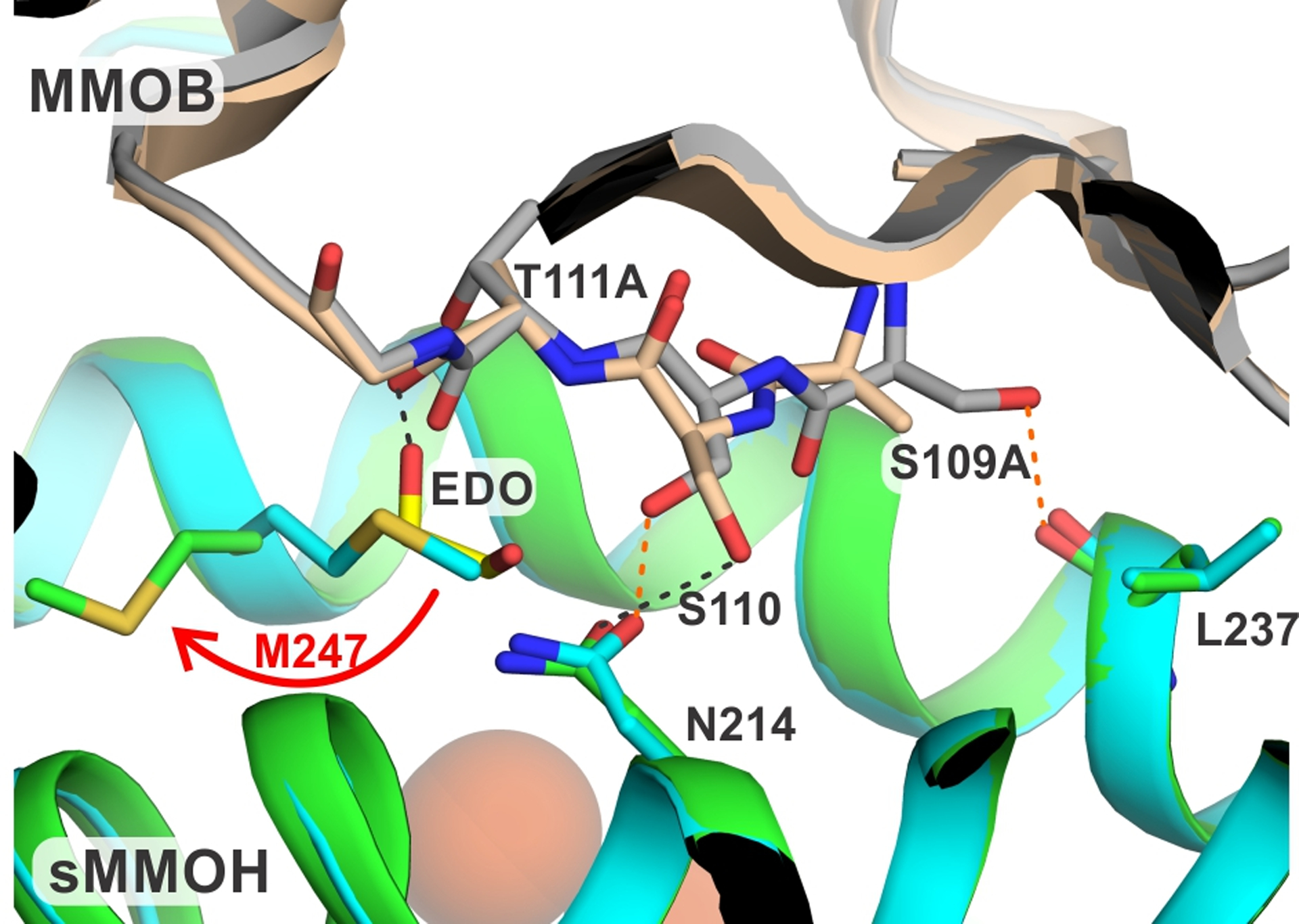
Close up view of the site of DBL2 mutation in the protein complex with sMMOH compared with the wild type complex. The hydrogen bonds in the sMMOH:MMOB complex are shown as orange dashed lines, while they are shown as black dashed lines in the sMMOH:DBL2 complex. Color legend: sMMOH:MMOB complex: sMMOH = cyan, MMOB = gray; sMMOH:DBL2 complex: sMMOH = green, MMOB = wheat.
Structure of sMMOH:DBL1.
Analysis of the structure of sMMOH:DBL1 (PDB:7S6S) revealed loss of several key polar contacts that are present in the structure of sMMOH:MMOB. In particular, the DBL1 mutation (S110A/N107G) in MMOB leads to a loss of the S110 hydrogen bond with N214 of the sMMOH α-subunit (Figure 4). An intra-protein hydrogen bond between the sidechain carbonyl group of N107 and the sidechain hydroxyl group of S45 is also removed by the N107G mutation. While the MMOB β-sheet that contains the important 109 to 111 residues of the Quad region is shortened in the sMMOH:DBL1 complex, the extent of reorganization is not as pronounced as that observed in the sMMOH:DBL2 complex (Figure S4). The short helix preceding the β-sheet has significant backbone changes as well as enhanced dynamics as observed by the nearly 1.5-fold increase in B-factors in this region (Figures S4 and S6). Finally, the 115 to 123 residue region of bound DBL1 undergoes backbone alterations without large sidechain conformational changes (Figure S4).
Figure 4.
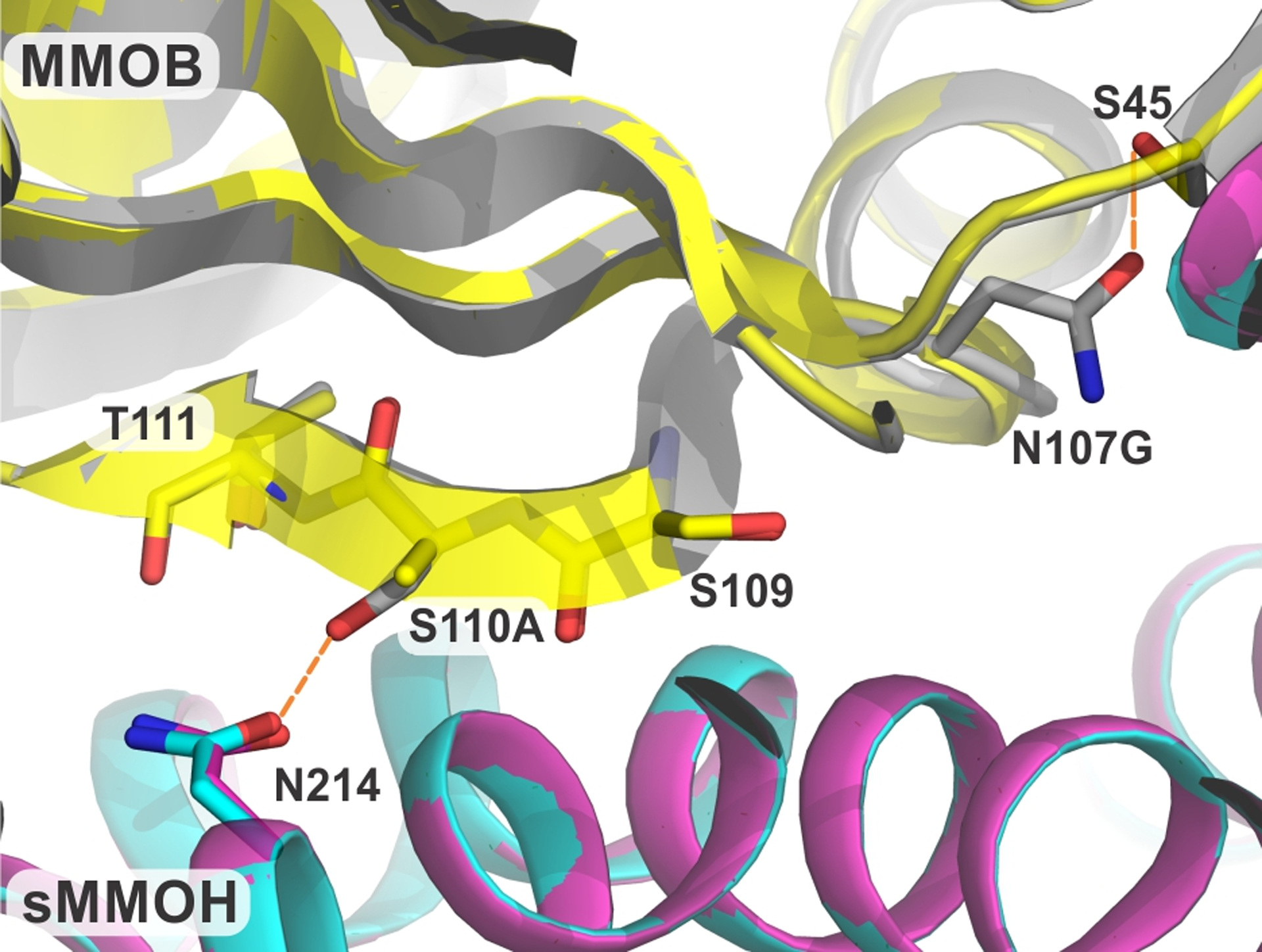
Close up view of the site of DBL1 mutation in the protein complex compared with the wild type protein. The hydrogen bonds in the sMMOH:MMOB complex that are lost in the sMMOH:DBL1 complex are shown as orange dashed lines. Color legend: sMMOH:MMOB complex: sMMOH = cyan, MMOB = gray; sMMOH:DBL1 complex: sMMOH = magenta, MMOB = yellow.
Changes also occur within the structure of sMMOH in the complex. Although the diiron cluster and its first-sphere ligands remain largely unchanged (Figure S5), a variation of note appears in a third-sphere residue that is part of the hydrogen bond network leading to the histidine ligands to the iron atoms. This network is located on the side of the iron cluster away from the substrate binding pocket and is composed of residues that alternate in hydrogen bond donor and acceptor character.36, 39 The Nε position of each histidine ligand is hydrogen bonded to a second-sphere aspartate residue, which in turn has two separate hydrogen bond donors in third-sphere residues. The entire network is strictly conserved in the bacterial multicomponent monooxygenase family. In the protein complex with wild-type MMOB, the sidechain of the third sphere residue R245 is predominantly present in a conformation that stabilizes a salt bridge with D143 (the ‘on’ conformation) (Table S2). However, in the sMMOH:DBL1 structure, the electron density map supports two conformations of the R245 sidechain, the second of which is incapable of bonding to D143. In this rather dynamic ‘off’ conformation, present in approximately 40% occupancy (Table S2), R245 forms a salt bridge with the sidechain of E58 (Figure 5). In the wild-type sMMOH:MMOB complex, the section of helix F (residues 240 to 249) containing R245 is flexible as it transitions from a 310 helix to an α-helix with many of the residues only forming one of two intra-main chain hydrogen bonds.25 This flexibility is compounded in the sMMOH:DBL1 complex, where the loss of additional intra-main chain hydrogen bonds positions the backbone of R245 on a loop section (Figure 5). Along with the sidechain of R245, the backbone carbons of residues 243 to 253 also shift in the mutant MMOB protein complex with respect to the wild-type complex (Figure S4). Finally, an unstructured loop region in the α-subunit of sMMOH (53 to 62) also reorganizes in the sMMOH:DBL1 structure (Figure S4). This loop contains the E58 residue that forms a salt bridge and stabilizes the ‘off’ conformation of R245.
Figure 5.
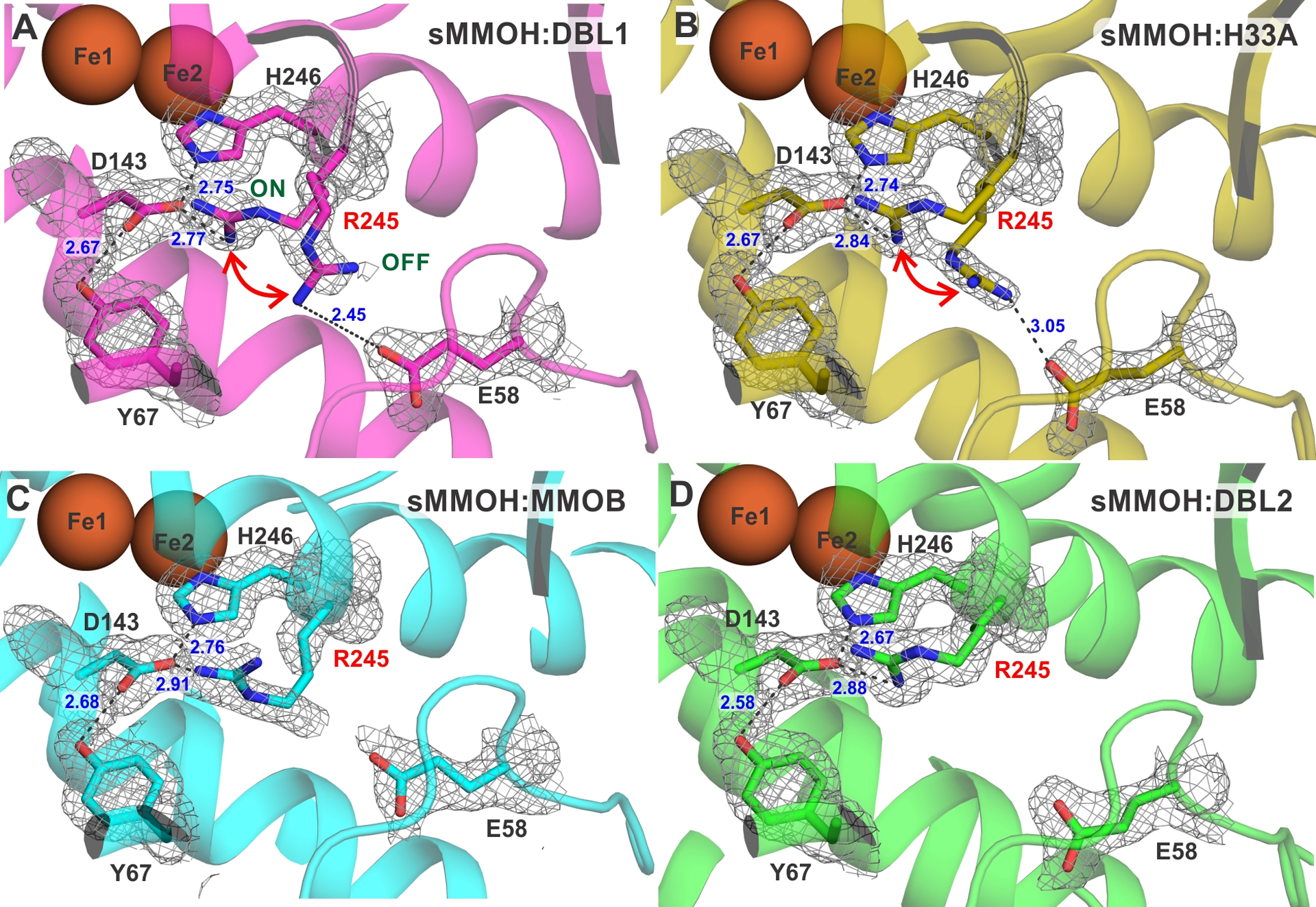
The proximal hydrogen bond network to the H246 ligand of Fe2. The sMMOH:MMOB-variant complex is indicated on the figure (A-D).The 2mFo-DFc electron density map shown as a gray mesh is contoured at 1.1 σ. Salt bridges and hydrogen bonds are represented by black dashed lines. Bonding distances are labeled in blue (Å). An analogous hydrogen bond network is present on the histidine ligand to Fe1 and is not displayed here. The loop backbone of L244 and R245 is colored in gray in panels A and B. The ON and OFF positions for R245 are labeled in panel A.
Structure of sMMOH:H33A.
The X-ray crystal structure of sMMOH:H33A (PDB:7S6T) shows that the loss of the histidine sidechain leads to the loss of an intramolecular hydrogen bonding interaction with the backbone carbonyl of E28 and the sidechain carbonyl of N29 of MMOB (Figure 6). As the complex with sMMOH forms, these interactions and others between the N-terminal MMOB residues, together with those between MMOB and sMMOH residues, cause a loop-to-helix transition in this region.36 The MMOB N-terminus forms a ring-like structure on the sMMOH surface. While the H33A mutation causes an observable main-chain reorganization and increased B-factors within the helical portion of this N-terminus, there are no large conformational changes in this region (Figures S4 and S6).
Figure 6.
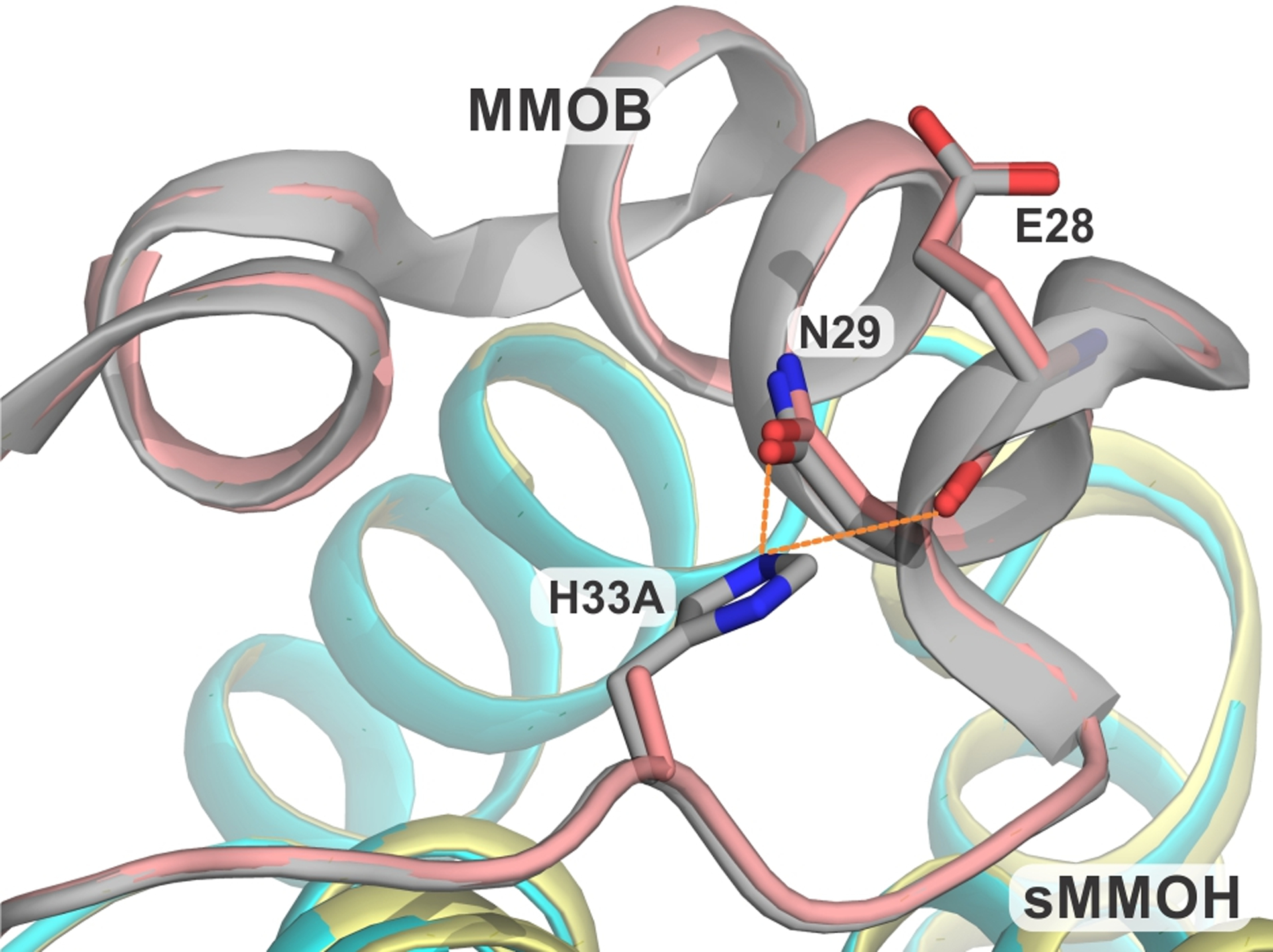
Close up view of the site of H33A mutation in the protein complex compared with the wild type protein. The hydrogen bonds in the sMMOH:MMOB complex are shown as orange dashed lines. Color legend: sMMOH:MMOB complex: sMMOH = cyan, MMOB = gray; sMMOH:H33A complex: sMMOH = sulfur, MMOB = deep-salmon.
Complex formation of H33A with sMMOH causes fewer changes within the sMMOH α-subunit, than described above for DBL1 and DBL2 (Figure S4). The only conformational change of note is the presence of the R245 sidechain in both the ‘on’ and ‘off’ conformations as observed in the sMMOH:DBL1 structure (Figure 5). While significant conformational changes in residues adjoining R245 are not observed as in the sMMOH:DBL1 complex, the R245 backbone is nonetheless present in a loop structure in the middle of helix F due to the loss of intra-main chain hydrogen bonds. The diiron cluster is unaltered as measured by bond lengths and geometrical arrangement of the first-sphere ligands (Figure S5).
Structure of sMMOH:H5A.
The X-ray crystal structure of the sMMOH:H5A complex (PDB: 7S6R) shows the loss of a hydrogen bond with the carboxylate moiety of D81, which is located in the core-region of MMOB (Figure 7). This interaction is replaced by a hydrogen bond between the amino terminus of S2 and D81. Apart from this significant reorganization in the first six residues of MMOB, there are no other large structural changes. The structural changes within the α-subunit of sMMOH caused by binding of H5A are also subtle compared to the effects of the MMOB core mutations described above. There are small changes in the unstructured loop region from residues 55 – 62 and in helix C from residues 129 – 136 (Figure S4). The structure of the sMMOH diiron cluster is largely unaltered in the sMMOH:H5A complex (Figure S5). The observation of modestly altered Q formation and decay kinetics in reactions in the absence of observable structural changes, suggest the occurrence of either very small structural changes or alterations in protein dynamics. They also attest to the precise interaction between sMMOH and MMOB required for efficient and specific catalysis.
Figure 7.
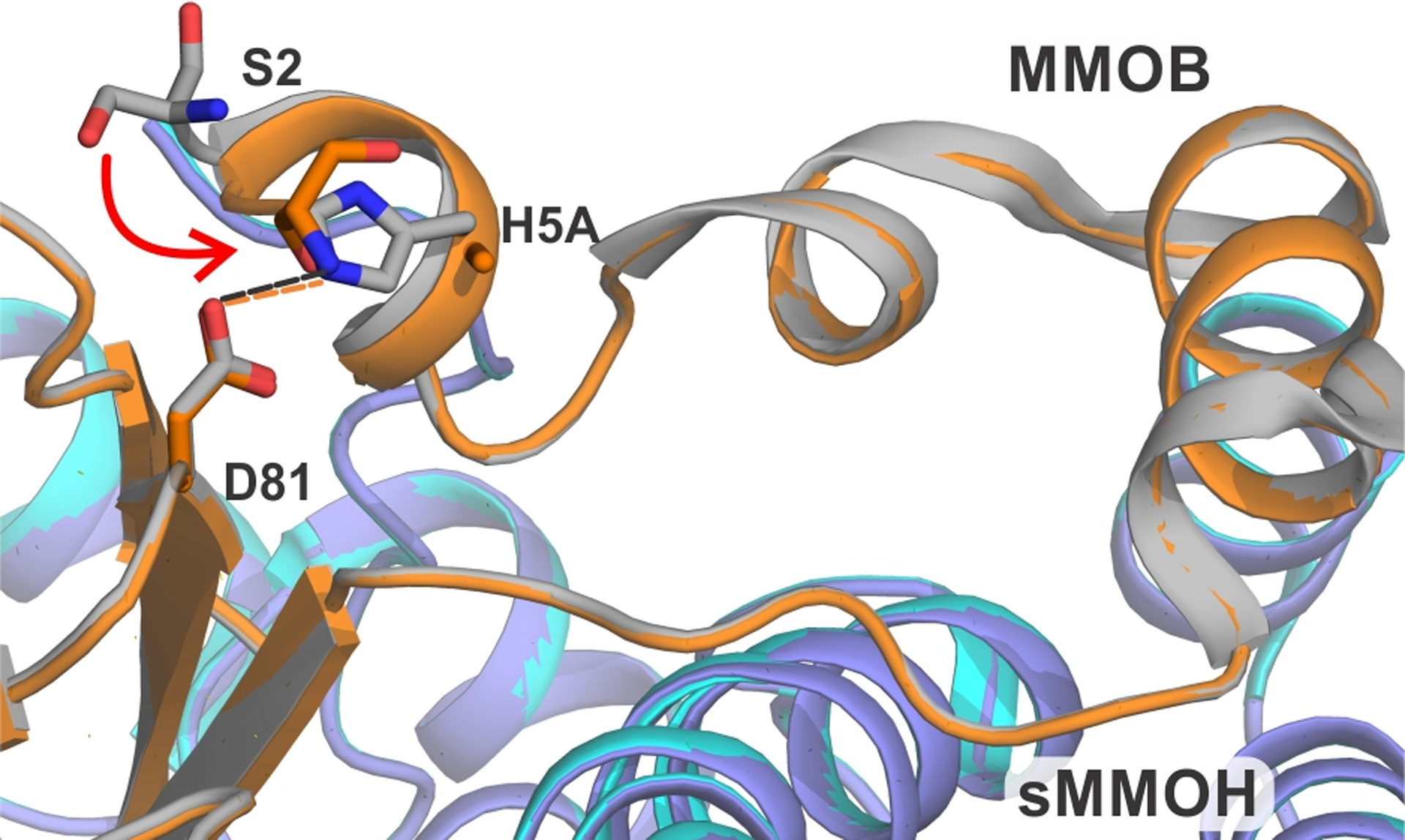
Close up view of the site of H5A mutation in the protein complex compared with the wild type protein. The hydrogen bonds in the sMMOH:MMOB complex are shown as orange dashed lines while they are shown as black dashed lines in the sMMOH:H5A complex. Color legend: sMMOH:MMOB complex: sMMOH = cyan, MMOB = gray; sMMOH:H5A complex: sMMOH = purple-blue, H33A MMOB = orange.
Perturbation of Diiron Cluster Electronics by MMOB Variants.
The mixed-valent state of sMMOH provides a good probe of the electronics of the diiron cluster through an EPR measurement of the zero field splitting D and the J-coupling constant between the iron atoms.24, 40 The observed g-values of the antiferromagnetically coupled diiron system are a function of D/J and are quite sensitive to perturbations in these spin Hamiltonian parameters, especially the more variable parameter J.41 The binding of MMOB to sMMOH triggers a reorganization of the diiron cluster, resulting in a reduction in the J-coupling constant (from ~30 cm−1 to ~5 cm−1) and a resultant shift in the g-values for mixed-valent sMMOH from g = 1.95, 1.86, 1.76 for the isolated component to g = 1.87. 1.77, 1.62 in the sMMOH:MMOB complex (Figure 8A and B).40 The alteration in diiron cluster electronics is recapitulated by the sMMOH:H5A complex (Figure 8F), and to a significant extent in the sMMOH:DBL2 complex (Figure 8E). In stark contrast, the iron atoms within the sMMOH:DBL1 and sMMOH:H33A complexes appear to retain the same coupling constant as sMMOH in isolation (Figure 8C and D). While this could also arise from an inability to form a protein complex, past competition experiments with MMOB and the two MMOB variants have demonstrated that the binding affinity for sMMOH is very high (Kd < 0.1 μM), such that full complex formation is expected at the concentrations used for the EPR experiment.21, 28
Figure 8.
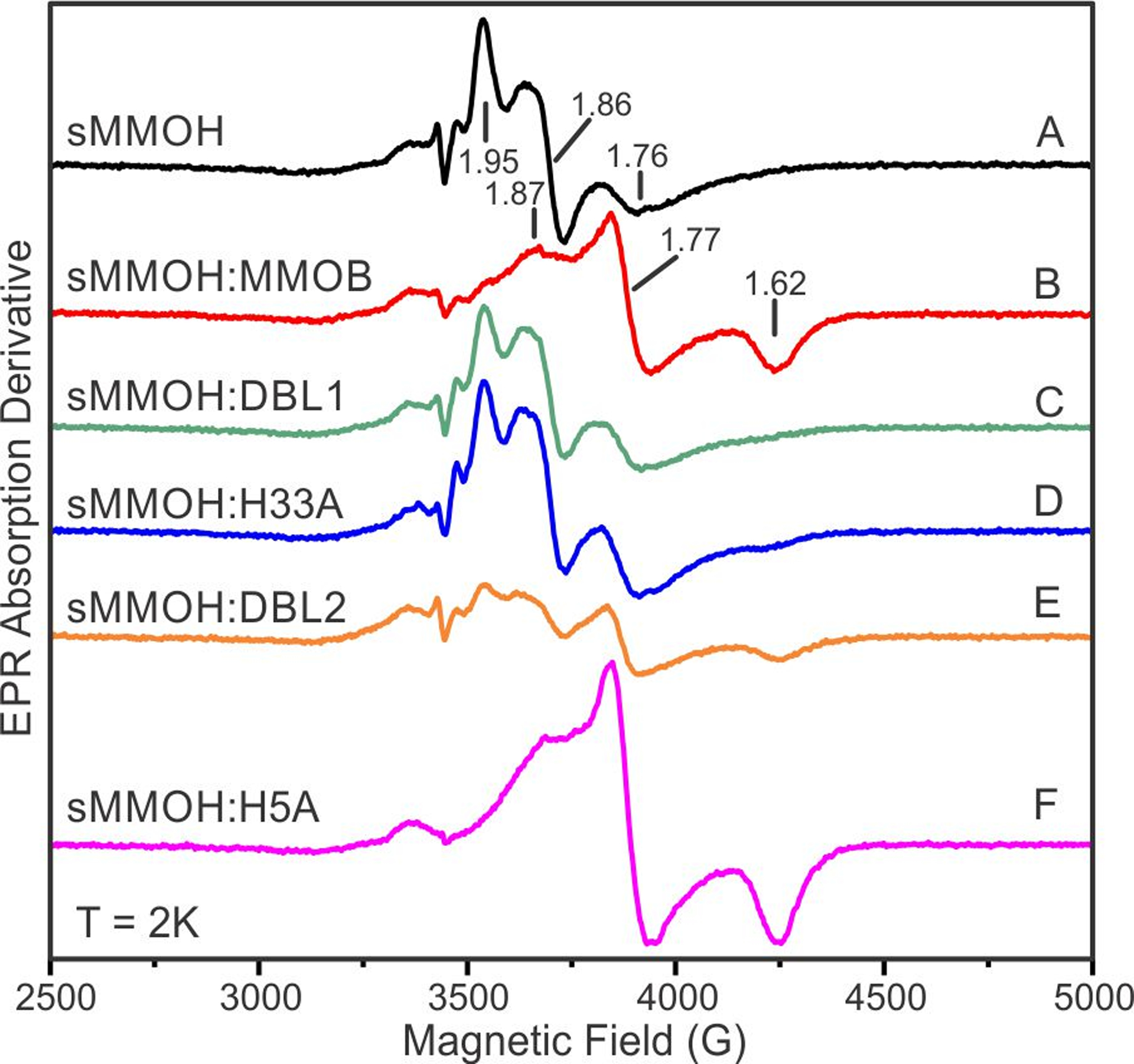
X-band EPR spectra of mixed valent sMMOH in complex with MMOB and MMOB variants. Samples A-F are identified on the figure and were prepared as described in Experimental Procedures. Instrument conditions: Temperature = 2 K, Microwave power = 0.213 mW, Modulation amplitude = 5 G at 100 kHz, Microwave frequency = 9.648 GHz, Sweep time = 90 s.
Upon full reduction of the sMMOH diiron cluster, a resonance near g = 16 is observed due to ferromagnetic coupling of the S = 2 irons.42 This resonance is enhanced ~three-fold when the EPR spectrum is measured with the microwave field aligned parallel with the fixed magnetic field. The g = 16 signal sharpens markedly when diferrous sMMOH is bound to MMOB (Figure S7).24 In accord with the mixed-valent spectra, the spectra of the diferrous sMMOH complexes with MMOB variants that do not allow rapid progress through the P reaction cycle intermediate (DBL1 and H33A) resemble that of diferrous sMMOH alone. Nevertheless, a slight sharpening and increase in intensity is observed in the spectra of these complexes demonstrating that a complex is formed. The spectrum of the diferrous sMMOH:H5A complex that allows rapid progress through intermediate P resembles that of diferrous sMMOH:MMOB.
Effect of the MMOB Variants on Small Molecule Access Tunnels.
Transient kinetic studies of hydrocarbon substrate oxidation have highlighted the specific role of the Quad and DBL2 mutants in enhancing the intramolecular diffusion rate constant of substrates larger than methane.21, 28, 33 The possibility that the mutations create a new or a modified substrate entry route was explored using the sMMOH:DBL2 and sMMOH:MMOB crystal structures and the tunnel finding program MOLE 2.5. An inspection of the sMMOH:DBL2 structure with various combinations of key MOLE 2.5 parameters (interior threshold, bottleneck radius) revealed the presence of three potential small molecule tunnels (Figure 9). The first is a tunnel threading its way through a chain of interior cavities within sMMOH.37 However, this tunnel has been shown to be blocked in the reduced form of sMMOH:MMOB that reacts with O2 at the beginning of the reaction cycle.25 The remaining two tunnels are variations of a single tunnel that begins at the sMMOH active site and emerges at the internal interface with MMOB before reaching the bulk solvent through two divergent paths. The first observed variation of this tunnel was previously named the W308-tunnel after the gating sMMOH W308 residue at the sMMOH:MMOB interface.25 It was shown that this small molecule tunnel (W308-tunnel 1) serves as a route for intramolecular O2 diffusion into the sMMOH active site in the wild-type sMMOH:MMOB complex. Both variations of the W308-tunnel (1 and 2) are lined by strictly conserved and predominantly non-polar residues. The route of W308-tunnel 2 carries it directly past the Quad region of MMOB.
Figure 9.
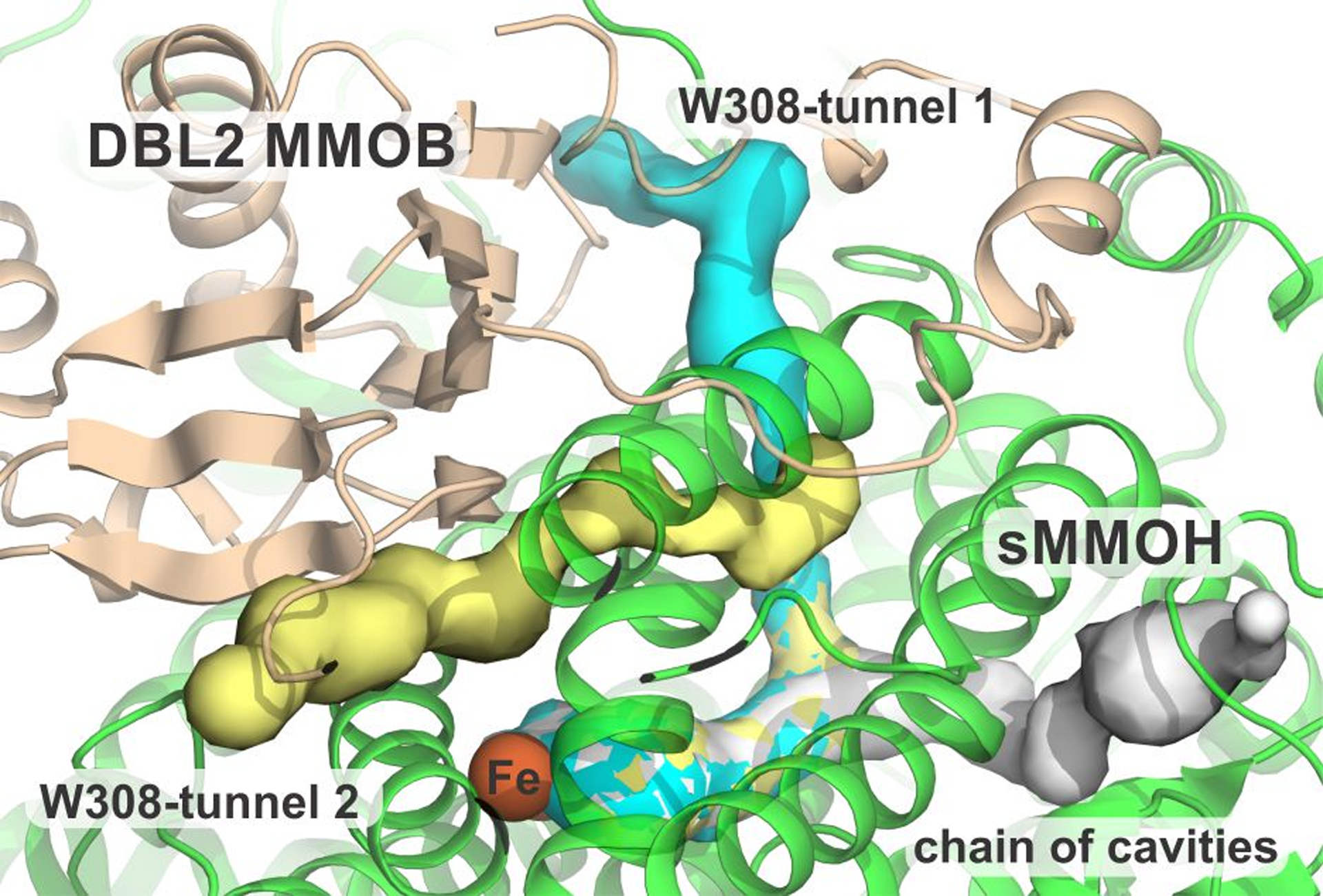
Overview of the three small-molecule tunnels identified in the sMMOH:DBL2 structure. The active site is depicted by the orange spheres of the diiron cluster. Color legend: sMMOH = green, DBL2 MMOB = wheat, chain of cavities tunnel = gray, W308-tunnel 1 = cyan, W308-tunnel 2 = yellow.
The native sMMOH:MMOB crystal structure was probed to assess whether the alternative W308-tunnel 2 is also present in the wild-type protein complex. Indeed, this small molecule tunnel is discovered by MOLE 2.5 if the bottleneck radius is decreased from the value used to discover W308-tunnel 1 (cf. 0.95 Å, 0.85 Å). A comparison of the sMMOH:DBL2 and sMMOH:MMOB structures overlaid with the predicted tunnel shows that it is reorganized by the DBL2 mutation (Figure 10). In the region between the sMMOH active site and the interface with MMOB, the two W308-tunnels in sMMOH:DBL2 are unchanged from the tunnel in the wild-type protein complex. However, after exiting sMMOH into the tightly packed sMMOH-MMOB interface, the W308-tunnel 2 route in the wild-type protein complex is constricted as it passes the Quad region of MMOB, effectively closing this route. The reorganization of this β-sheet in the sMMOH:DBL2 structure, and crucially the conformational change of the M247 sidechain of sMMOH, relieves this bottleneck and opens this alternative route. Importantly, neither the shift in M247 nor the localization of ethylene glycol within the tunnel is observed in the other three mutant sMMOH:MMOB complexes. Past transient kinetic studies have shown that sMMOH:DBL2 (and the complex with the Quad variant) cause a dramatic increase in the rate of reaction of larger adventitious substrates (e.g. ethane, furan or nitrobenzene) with Q.21, 28 In contrast, none of the other MMOB variant complexes exhibit this effect, consistent with the W308-tunnel 2 remaining restricted. The chain of interior cavities in the sMMOH:MMOB complex is unchanged in sMMOH:DBL2 (Figure S8), consistent with the conclusion that it is not directly involved in substrate transit into the active site.
Figure 10.
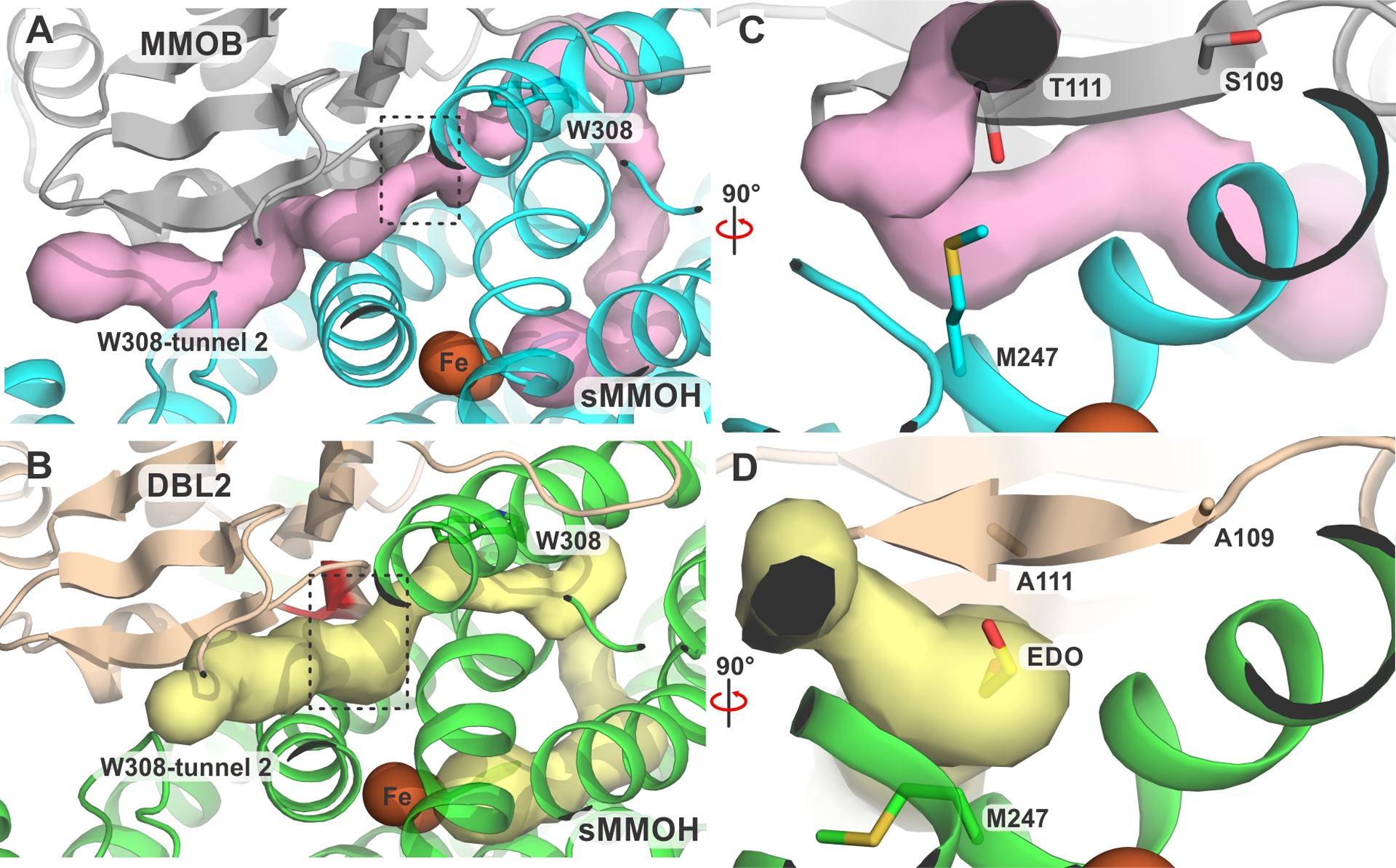
Reorganization of the W308-tunnel 2 by the DBL2 mutation. This branch of the W308-tunnel is observed in the sMMOH:MMOB complex (panel A, pink) with a bottleneck highlighted by the black box, which is relieved in the sMMOH:DBL2 complex (panel B, yellow) by the DBL2 mutations (colored red in panel B). The gating W308 residue in sMMOH is shown as sticks in panel A and B. The regions marked by the black boxes are shown in more detail in panel C (sMMOH:MMOB) and panel D (sMMOH:DBL2). The DBL2 mutant residues and the sMMOH M247 residue regulating the constriction point and the ethylene glycol molecule bound in the tunnel are shown as sticks.
DISCUSSION
The sMMO enzyme serves as one of the best systems in which to investigate O2 activation and strong C-H bond cleavage catalyzed by oxygenases.1 In addition, it has been used to reveal many of the strategies employed by these enzymes to regulate catalysis in order to prevent aberrant oxidative chemistry and preserve the coupling of O2 activation to hydrocarbon oxidation.20, 21, 26–28, 43, 44 Each of these aspects of catalysis is orchestrated by protein-protein interactions in sMMO, especially between the sMMOH and MMOB proteins. Indeed, recent studies suggest that O2 binding, activation, and the reaction of the activated species with hydrocarbons occur in the sMMOH:MMOB complex after dissociation of MMOR.27 The kinetic, regiospecific, and structural changes in sMMOH and sMMOH catalysis caused by MMOB binding have been investigated by several approaches, but none has been more informative than the application of targeted MMOB variants.21, 27–29, 32–34, 37, 44, 45 The crystal structures of the MMOB variants in complex with sMMOH shown in this report reveal discrete and distinct perturbations within sMMOH. Some of these changes can now be correlated with the effects on catalysis and regulation seen during sMMO turnover, and these aspects are discussed here.
Role for the Proximal Hydrogen Bond Network in Modulating Diiron Cluster Electronics.
The perturbations effected by the MMOB variants on the rate constants of intermediate conversion have been tabulated in Table 1 showing the direction and magnitude of the impact. The DBL1 and the H33A mutants affect sMMOH catalysis in a similar manner in that both significantly reduce the rate constant of formation of intermediate P from P*. This effect primarily manifests in the complete absence of observed P accumulation based upon its characteristic 700 nm absorption band. In the sMMOH catalytic cycle regulated by DBL1, the accumulation of Q based upon its characteristic 430 nm absorption band is further reduced by the significantly elevated rate constant of decay in the absence of hydrocarbon substrates. These severe impacts on the rates of intermediate interconversion suggest that, in comparison with the wild-type MMOB complex, significant structural and/or electronic changes must occur at the diiron cluster and associated ligand/residue shells in the presence of H33A and DBL1 MMOB.
Table 1.
Perturbation of the rate constants of the sMMOH catalytic cycle by MMOB variants. The magnitude and direction of the change is relative to the rate constants measured using the wild-type sMMOH:MMOB complex.
| O2 | Substrate | ||||||
|---|---|---|---|---|---|---|---|
| Mechanism: MMOHred:MMOB τ O τ P* τ P τ Q τ T τ MMOHox:MMOB + Product | |||||||
| MMOB Varianta | P Formation | Q Formation | Q Decay No Subb | Q Decay Methane | Q Decay Other Subc | T Decay Other Subd | Ref |
| Quad | UCe | UC | 3x | −7x | 6.8x | 2.5x | 28 |
| DBL2 | −1.2x | UC | 1.3x | −1.5x | 4.3x | 3.1x | 21,31 |
| T111A | 1.3x | UC | UC | −1.3x | 5.7x | 3.1x | 21 |
| DBL1 | −150x | UC | 22x | NOf | NO | −3x | 21 |
| H33A | −30x | −1.6x | UC | −1.3x | −1.3x | −1.5x | 28 |
| H5A | UC | −1.6x | UC | −1.4x | −1.1x | −1.8x | 28 |
| C-termg,h | −30x | UC | −3.3x | −13x | UC | UC | 34 |
Use of the MMOB V41R variant decreases the O formation rate constant 25,000-fold
Sub = substrate
Other sub = nitrobenzene; similar results for furan and ethane
Other sub = nitrobenzene
UC = unchanged
NO = not observed
Deletion of 5 residues from the C-terminal of MMOB
Deletion of 29 residues from the N-terminal of MMOB blocks O formation
A comparison of the metal cluster of sMMOH in complexes with MMOB and the MMOB variants shows very little change in the first and second sphere of the metal coordination (Figure S5). This lack of change may, in part, reflect the stabilizing presence of benzoate bound to the diiron cluster in the active site of the wild-type sMMOH:MMOB complex and in the complexes with the variants due to the crystallization conditions. However, a large change is observed in the third sphere wherein R245 samples at least two orientations in some of the sMMOH:MMOB variant complexes. This shift represents a ~3.7 Å distance change in the guanidino carbon atom positions. The new ‘off’ orientation stabilized by a salt bridge to residue E58 is expected to affect the conserved His:Asp/Glu first and second sphere proximal hydrogen bond network common to ferritin-like diiron metal oxygen-activating enzymes (Figure 5).36 In the bacterial multicomponent monooxygenase (BMM) subfamily comprised of sMMO, propane monooxygenase, toluene monooxygenases, and phenol hydroxylase, this network is extended to contain third-sphere residues such as R245 in sMMO.36, 39 Many of the donor-acceptor distances in the sMMO network are less than 2.8 Å, therefore indicating strong bonds. A role in the structural stabilization and modulation of the electronics of the diiron cluster has been proposed for this extended bonding network in sMMO,36 while a previously predicted role as an electron transfer route has been discounted.36, 39 In sMMOH alone, R245 forms a non-optimal weak salt bridge with D143, more akin to the ‘off’ conformation in the sMMOH:MMOB variant complexes. When the sMMOH:MMOB complex forms, the acidic D143 residue hydrogen bonds or salt bridges to three donor residues: H246 (a ligand to Fe2), R245 and Y67. The D143-R245 salt bridge is observed to strengthen due to a shorter donor-acceptor distance and a more optimal orientation of the acidic and basic headgroups, thereby stabilizing the ‘on’ conformation.36 Close examination of several published sMMOH:MMOB structures shows that R245 ranges from completely bound to as much as 20–40% in the ‘off’ conformation (Table S2) in the diferric complex. The occupancy shifts completely to the ‘on’ conformation in the O2-reactive diferrous structure. Thus, binding of MMOB may simply shift an equilibrium between ‘on’ and ‘off’ conformations. The 40–50% presence of an ‘off’ conformation of R245 in both sMMOH:H33A and sMMOH:DBL1 structures (Table S2) removes the basic R245 donor from the bonding network, thereby affecting the D143:H246 hydrogen bond and indirectly altering the electronics of the diiron cluster. Indeed, the EPR spectrum of the mixed valent states of sMMOH:H33A and sMMOH:DBL1, frozen from solution where no benzoate is bound, show that they fail to alter the environment of the diiron cluster in the manner observed when MMOB is bound (Figure 8A–D). These spectra show that the diiron cluster adopts essentially a single mixed valent electronic state, so the structures seen in the crystal may exaggerate the ‘on’ conformation of R245. It has also been observed that the P* to P transition involves transfer of a proton from a nearby donor in a ‘one hop’ process.15–17, 28 The change in the conformation of R245 may affect the position or environment of the proton donor. The proton-donation process during P* decay in the sMMOH:H33A catalytic cycle does indeed depict a donor moiety with a pKa of ~6.4, lower than that of the donor molecule (pKa = 7.6) in the wild-type complex.15 The same observation of decreased P* to P conversion coupled with a decrease in pKa for the donor has been made for C-terminal truncation variants (Table 1).34 Finally, in a previous study, the N-terminal K15 residue of MMOB was mutated to a cysteine residue, which was then labeled with a 3-bromo-1,1,1-trifluoroacetone (BTFA) label in order to generate a 19F-NMR probe.27 Transient kinetic studies of the sMMOH catalytic cycle with this modified MMOB protein showed that the rate constant of P formation was decreased 2.5 fold while a second proton dependent process, P to Q, was slowed 5 fold. An inspection of the crystal structure of this protein complex (PDB: 7M8R) shows nearly all of the R245 residue to be present in the ‘off’ conformation in both protomers (Table S2).
Elucidation of a Small-Molecule Tunnel for Hydrocarbon Substrate Diffusion.
Transient kinetic studies of the sMMOH catalytic cycle with the DBL2 MMOB variant show that the rate constants of formation of the intermediates, P*, P, and Q are not significantly perturbed. The decay rate constant of Q with methane is marginally reduced, perhaps due to a perturbation in the quantum tunneling contribution to C-H bond breaking as observed for the Quad variant.28 However, when alternative substrates larger than methane are introduced, the decay rate constant of Q is significantly enhanced (Table 1). Product molecules are also released in an accelerated manner from the active site as assessed by measuring turnover numbers and tracking the absorbance change of a chromophoric product.21 The lack of any rate constant perturbation on intermediate interconversion leading to the reactive species of sMMO suggests that DBL2 mimics MMOB in its effects on the electronics of the diiron cluster. This proposal is supported by the similarity of the mixed valent EPR spectra of sMMOH:MMOB and sMMOH:DBL2 and the predominantly ‘on’ conformation of R245 in these complexes. Consequently, the changes in the rate constants for the reaction with substrates larger than methane and dissociation of products suggest a change in the manner by which hydrocarbon substrates are trafficked into, and products out of, the active site of sMMOH at the Q and product release stages of the reaction cycle, respectively.
The nature of hydrocarbon substrate reaction within sMMO is quite unusual. In the reaction with Q, methane oxidation exhibits a D-KIE of ~50, whereas no deuterium isotope effect is observed for other hydrocarbon substrates.3, 20, 46 We have accounted for this and other observations of reaction dynamics and thermodynamics using a “molecular sieve” model in which substrate entry is greatly restricted.1, 26, 28, 33 In the case of methane, this restriction slows the rate constant for entry almost to that for C-H bond breaking, whereas entry of larger substrates with weaker C-H bonds (e.g. ethane) is slowed to the point where it is rate-limiting in the substrate hydroxylation phase of the reaction cycle.26 Indeed, the rate constant of methane binding (~14000 M−1s−1 at 4 °C) is 10 to 100 times slower than typically observed for enzymatic substrate binding reactions.5, 20 Past mutagenesis studies and more recent studies of the sMMOH:MMOB crystal structure show that MMOB plays a key role in regulating substrate entry in a manner compatible with a molecular sieve hypothesis.21, 25, 26, 28 In particular, the Quad variant and its binary subset DBL2 that substitute small for large amino acid side chains cause a large increase in the rate of binding of substrates larger than methane (Table 1). One indication of this effect is the induction of an observable D-KIE for the reaction of ethane with Q when using the Quad or DBL2 variant.32, 33
Recent structural, kinetic and mutagenic studies have shown that O2 enters the sMMOH active site through the W308-tunnel 1 early in the reaction cycle,25 but past kinetic studies showed that methane and other substrates enter only after Q is formed.5, 20 The narrow diameter of W308-tunnel 1 and a surface lined by non-polar residues indicates that it would be suitable for, but also restrict, transit of both dioxygen and methane. However, the current results suggest another possibility. The DBL2 variant does increase the diameter of an access tunnel to the active site of sMMOH, but intriguingly, the relevant tunnel is W308-tunnel 2 rather than 1. Both tunnels share the same last half of the route from W308 into the active site, but only the W308-tunnel 2 travels immediately past the Quad region. This is the region where the key restriction is relieved in the DBL2 variant (Figure 10). The potential role of this tunnel for hydrocarbon substrate entry is supported by several observations. First, the bottleneck residues within the W308-tunnel 2, sMMOH M247 and MMOB T111, are strictly conserved in methane oxidizing hydroxylase and regulatory proteins within the subset of alkane-oxidizing BMM enzymes.36, 47 Second, the major effects of relieving this constriction in W308-tunnel 2 are seen in the kinetics of the hydrocarbon reaction steps of the catalytic cycle. Third, the replacement of only T111 from DBL2 with alanine (T111A) is sufficient to enhance the intra-molecular diffusion rate constant of larger hydrocarbons such as ethane for reaction with Q (Table 1).21 Fourth, the W308-tunnel 2 is lined by all five of the tryptophan residues that are strictly conserved within the methane oxidizers from the BMM enzyme family (Figure S9).25 Fifth, the ethylene glycol molecule bound within W308-tunnel 2 in the DBL2 variant shows that molecules the size of the common adventitious substrates of sMMO can access the interior of the enzyme via this route (Figure 10). In this regard, it is important to note that in the wild-type sMMOH:MMOB complex, the position of M247 would not only restrict access of larger substrates, but it would also exclude polar molecules like ethylene glycol. Lastly, the loss of hydrogen bonding interactions and distortion of the beta sheet in the Quad region that occurs in the sMMOH:DBL2 complex would significantly increase structural flexibility. This change might, in turn, increase the ability of larger substrates to transit the tunnel as observed in single turnover kinetic and steady state experiments.21, 33
Although the characteristics of the W308-tunnel 2 are consistent with it acting as the hypothesized molecular sieve for methane, the current results do not allow a definitive choice between W308-tunnel 1 or 2 for this role. Both tunnels exist in the sMMOH:MMOB complex and both are severely restrictive for molecules even as small as methane. It is not possible to use mutagenesis to test W308-tunnel 1 for hydrocarbon access because mutations effectively block the reaction cycle at the beginning where O2 binds. A previous attempt to obstruct what is now recognized as W308-tunnel 2 was made using the T111Y variant.21 Paradoxically, this variant greatly enhanced the decay rate constant of Q with large substrates, seeming to defeat regulation of substrate access based on size.21 It is likely that this rate enhancement resulted from a breakdown of the tight regulatory control due to severe disruption of the sMMOH:MMOB interface, causing an observed 10-fold weaker affinity for sMMOH.
While an explanation for the mid-cycle entry of methane and other hydrocarbons into the sMMOH active site remains elusive, use of two separately regulated tunnels for O2 and methane would account for the previous observations (Figure 11). Similar proposals for two or more tunnels serving distinct roles have been made for other well characterized enzymes.26, 48–53 However, it is important to note that in the case of sMMOH, the exclusion of methane early in the reaction cycle implies a dynamic aspect to the tunnels. An increase in the cross section of the full length of the methane entry tunnel would be required as the cycle progressed. Such an expansion might occur in response to the dramatic change in oxidation state of the dinuclear cluster irons during the cycle (Scheme 1). The mechanism for tunnel expansion is unknown. However, the release and rotation of sMMOH M247 to expand the tunnel in response to the loss of a hydrogen bonding interaction in the sMMOH:DBL2 complex might serve as a model to show how such an expansion could occur in response to a minor conformational change. There is precedence for such redox-linked opening of the gating residues within the W308-tunnel in sMMOH during the diiron(III) to diiron(II) cluster conversion step.25, 26 Finally, it is important to recognize that even at a single metal center oxidation state, the tunnel diameter will be dynamic due to the flexible nature of proteins. In the current case, the shift of M247 in the MMOH:DBL2 complex increases the diameter of the bottleneck from 1.7 to 2.8 Å, which is formally insufficient to admit CH4 with a quantum diameter of 4 Å.54 However, we propose that the continued restriction is dynamic and is manifested in the very slow rate constant for methane binding, which is decreased by at least one order of magnitude over expected values and is easily monitored by the rate of Q decay on the milliseconds to seconds time scale.
Figure 11.
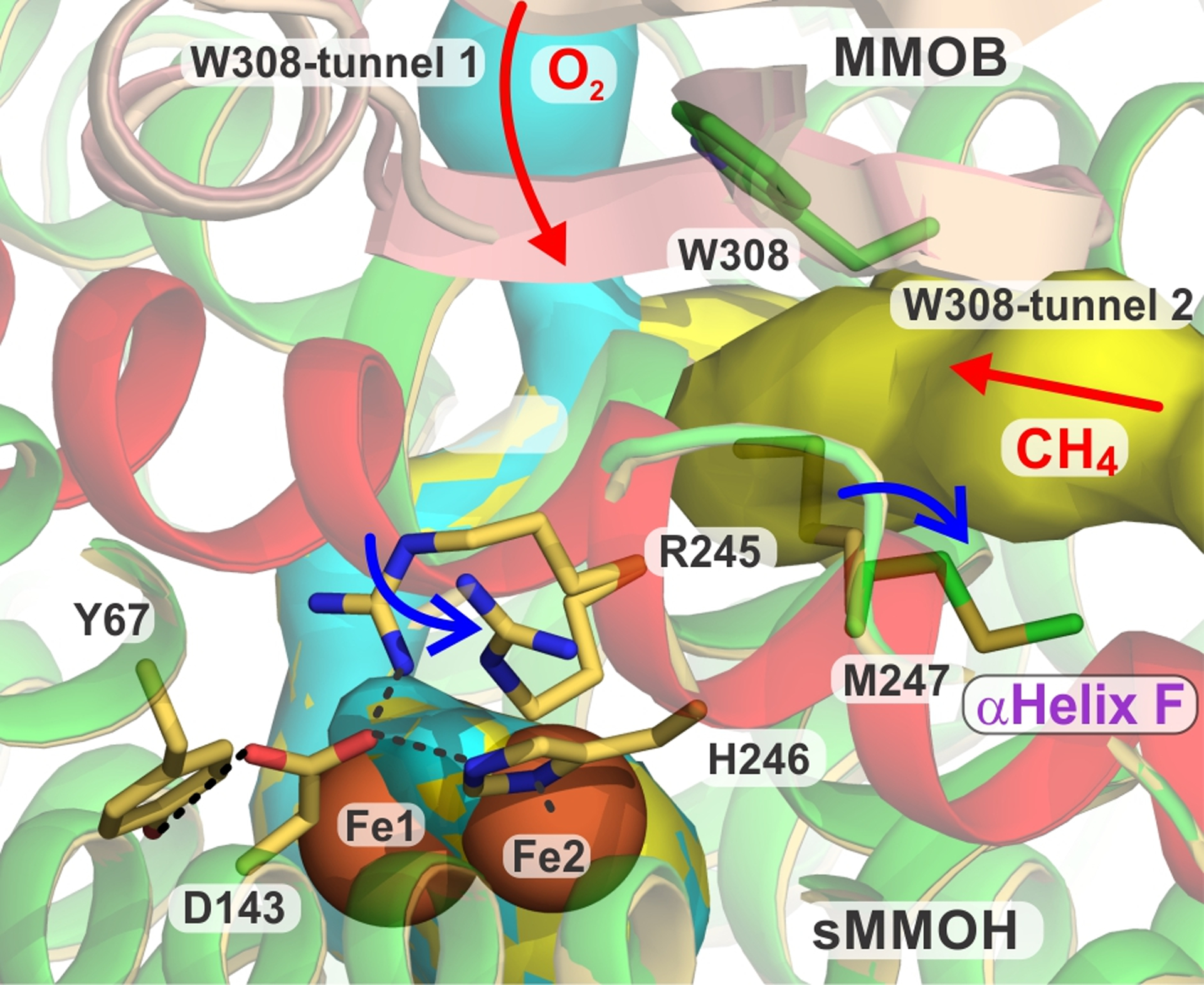
Significance of sMMOH helix F and hypothesis for bifurcated O2 and methane entry into the sMMOH active site. An overlay of 7S6Q and 7S6T is shown. The key F helix is shown in light red with shifting positions of R245 and M247 indicated by blue arrows. Tunnels W308 1 and 2 are shown in cyan and yellow, respectively. The active site is depicted by the orange spheres of the diiron cluster. Color legend: sMMOH = green and sulfur, DBL2 MMOB = wheat, H33A MMOB = deep salmon.
It is significant that the two sMMOH residues that exhibit the most exaggerated change in orientation upon MMOB variant binding, R245 and M247, reside on the same helix F and flank an iron ligand, H246 (Figure 11). Indeed, the majority of the changes observed upon MMOB binding occur in sMMOH helices E, F, and H,36 suggesting that regulation of many aspects of the catalytic cycle depend critically on the structure of this region.
CONCLUSIONS
The X-ray crystal structures presented here show that the mutations in MMOB that cause dramatic changes in the rate constants for intermediate interconversion in the reaction cycle do so by effecting rather subtle reorganization. Most of the changes involve shifts in single amino acid side chain positions or reorganization of short segments of secondary structure. In some cases, increases in B-factors in specific protein regions or a decrease in the number of hydrogen bonds suggest that the mutations also affect the dynamics of protein structure. For an enzyme that must control the access of molecules the size of methane and oxygen, significant rate changes in response to subtle changes in structure and protein flexibility seem quite reasonable. It is interesting to note that regions of MMOB and the alpha subunit of sMMOH, which are far from each other and the active site cavity, function in concert to promote progress through the reaction cycle. It appears that the entire protein has been tuned through evolution to achieve the goal of assuring that the most stable aliphatic hydrocarbon is given selective access to Nature’s most powerful oxidant. The discovery of a second small molecule tunnel passing sMMOH:MMOB interface residues that are known to affect hydrocarbon access presents the intriguing regulatory possibility that O2 and methane use independently regulated access routes. Solution of the structure of Q, the intermediate that binds and oxidizes methane, will be necessary in order to evaluate this possibility.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the financial support of this work from NIH Grants R35-GM118030 (to J.D.L.), R35-GM118047 (to H.A.), and NIH training grant GM08347 (to J.C.J.). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the NIH grant number P30 GM124165. The Pilatus 6M detector on 24-ID-C beamline is funded by a NIH-ORIP HEI grant number S10 RR029205. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the staff at NE-CAT beamlines, Advanced Photon Source, Argonne National Laboratory for assisting data collection.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.1c00673
Table of X-ray crystal structure statistics, nine figures illustrating additional details of the structural perturbations caused by the binding of MMOB variants to sMMOH, table of hydrogen bonding in the conserved structural network supporting the sMMOH diiron cluster.
UniProt and PDB Accession Codes
Methane monooxygenase hydroxylase component (sMMOH, α, β, γ chains) UniProt P27353, A0A1A6FJQ4 and A0A1A6FHH2; Methane monooxygenase regulatory component B (MMOB) UniProt P27356; Methane monooxygenase reductase component (MMOR) UniProt Q53563.The Protein Data Bank accession codes of the coordinates are: 7S6Q (sMMOH:DBL2 form 1), 7S7H (sMMOH:DBL2 form 2), 7S6R (sMMOH:H5A), 7S6S (sMMOH:DBL1), and 7S6T (sMMOH:H33A).
The authors declare no competing financial interest.
REFERENCES
- [1].Banerjee R, Jones JC, and Lipscomb JD (2019) Soluble methane monooxygenase, Ann. Rev. Biochem 88, 409–431. [DOI] [PubMed] [Google Scholar]
- [2].Tinberg CE, and Lippard SJ (2011) Dioxygen activation in soluble methane monooxygenase, Acc. Chem. Res 44, 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nesheim JC, and Lipscomb JD (1996) Large kinetic isotope effects in methane oxidation catalyzed by methane monooxygenase: evidence for C-H bond cleavage in a reaction cycle intermediate, Biochemistry 35, 10240–10247. [DOI] [PubMed] [Google Scholar]
- [4].Ambundo EA, Friesner RA, and Lippard SJ (2002) Reactions of methane monooxygenase intermediate Q with derivitized methanes, J. Am. Chem. Soc 124, 8770–8771. [DOI] [PubMed] [Google Scholar]
- [5].Lee SK, Nesheim JC, and Lipscomb JD (1993) Transient intermediates of the methane monooxygenase catalytic cycle, J. Biol. Chem 268, 21569–21577. [PubMed] [Google Scholar]
- [6].Lee SK, Fox BG, Froland WA, Lipscomb JD, and Münck E (1993) A transient intermediate of the methane monooxygenase catalytic cycle containing a FeIVFeIV cluster, J. Am. Chem. Soc 115, 6450–6451. [Google Scholar]
- [7].Banerjee R, Proshlyakov Y, Lipscomb JD, and Proshlyakov DA (2015) Structure of the key species in the enzymatic oxidation of methane to methanol, Nature 518, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cutsail GE, Banerjee R, Zhou A, Que L, Lipscomb JD, and DeBeer S (2018) High-resolution extended x-ray absorption fine structure analysis provides evidence for a longer Fe···Fe distance in the Q intermediate of methane monooxygenase, J. Am. Chem. Soc 140, 16807–16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Castillo RG, Banerjee R, Allpress CJ, Rohde GT, Bill E, Que L Jr., Lipscomb JD, and DeBeer S (2017) High-energy-resolution fluorescence-detected X-ray absorption of the Q intermediate of soluble methane monooxygenase, J. Am. Chem. Soc 139, 18024–18033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jacobs AB, Banerjee R, Deweese DE, Braun A, Babicz JT, Gee LB, Sutherlin KD, Böttger LH, Yoda Y, Saito M, Kitao S, Kobayashi Y, Seto M, Tamasaku K, Lipscomb JD, Park K, and Solomon EI (2021) Nuclear resonance vibrational spectroscopic definition of the Fe(IV)2 intermediate Q in methane monooxygenase and Its reactivity, J. Am. Chem. Soc 143, 16007–16029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jasniewski AJ, and Que L Jr. (2018) Dioxygen activation by nonheme diiron enzymes: Diverse dioxygen adducts, high-valent intermediates, and related model complexes, Chem. Rev 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Green J, and Dalton H (1986) Steady-state kinetic-analysis of soluble methane mono-oxygenase from Methylococcus-capsulatus (Bath), Biochem. J 236, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fox BG, Froland WA, Dege JE, and Lipscomb JD (1989) Methane monooxygenase from Methylosinus trichosporium OB3b. Purification and properties of a three-component system with high specific activity from a type II methanotroph, J. Biol. Chem 264, 10023–10033. [PubMed] [Google Scholar]
- [14].Liu KE, Valentine AM, Wang DL, Huynh BH, Edmondson DE, Salifoglou A, and Lippard SJ (1995) Kinetic and spectroscopic characterization of intermediates and component interactions in reactions of methane monooxygenase from Methylococcus capsulatus (Bath), J. Am. Chem. Soc 117, 10174–10185. [Google Scholar]
- [15].Banerjee R, Meier KK, Münck E, and Lipscomb JD (2013) Intermediate P* from soluble methane monooxygenase contains a diferrous cluster, Biochemistry 52, 4331–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee SK, and Lipscomb JD (1999) Oxygen activation catalyzed by methane monooxygenase hydroxylase component: Proton delivery during the O-O bond cleavage steps, Biochemistry. 38, 4423–4432. [DOI] [PubMed] [Google Scholar]
- [17].Tinberg CE, and Lippard SJ (2009) Revisiting the mechanism of dioxygen activation in soluble methane monooxygenase from M. capsulatus (Bath): Evidence for a multi-step, proton-dependent reaction pathway, Biochemistry 48, 12145–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Priestley ND, Floss HG, Froland WA, Lipscomb JD, Williams PG, and Morimoto H (1992) Cryptic stereospecificity of methane monooxygenase, J. Am. Chem. Soc 114, 7561–7562. [Google Scholar]
- [19].Gherman BF, Lippard SJ, and Friesner RA (2005) Substrate hydroxylation in methane monooxygenase: quantitative modeling via mixed quantum mechanics/molecular mechanics techniques, J. Am. Chem. Soc 127, 1025–1037. [DOI] [PubMed] [Google Scholar]
- [20].Brazeau BJ, and Lipscomb JD (2000) Kinetics and activation thermodynamics of methane monooxygenase compound Q formation and reaction with substrates, Biochemistry 39, 13503–13515. [DOI] [PubMed] [Google Scholar]
- [21].Brazeau BJ, and Lipscomb JD (2003) Key amino acid residues in the regulation of soluble methane monooxygenase catalysis by component B, Biochemistry 42, 5618–5631. [DOI] [PubMed] [Google Scholar]
- [22].Liu Y, Nesheim JC, Lee S-K, and Lipscomb JD (1995) Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component, J. Biol. Chem 270, 24662–246625. [DOI] [PubMed] [Google Scholar]
- [23].Green J, and Dalton H (1985) Protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath). A novel regulatory protein of enzyme activity, J. Biol. Chem 260, 15795–15801. [PubMed] [Google Scholar]
- [24].Fox BG, Liu Y, Dege JE, and Lipscomb JD (1991) Complex formation between the protein components of methane monooxygenase from Methylosinus trichosporium OB3b. Identification of sites of component interaction, J. Biol. Chem 266, 540–550. [PubMed] [Google Scholar]
- [25].Jones JC, Banerjee R, Shi K, Aihara H, and Lipscomb JD (2020) Structural studies of the Methylosinus trichosporium OB3b soluble methane monooxygenase hydroxylase and regulatory component complex reveal a transient substrate tunnel, Biochemistry 59, 2946–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Banerjee R, and Lipscomb JD (2021) Small-molecule tunnels in metalloenzymes viewed as extensions of the active site, Acc. Chem. Res 54, 2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jones JC, Banerjee R, Shi K, Semonis MM, Aihara H, Pomerantz WCK, and Lipscomb JD (2021) Soluble methane monooxygenase component interactions monitored by 19F NMR, Biochemistry 60, 1995–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wallar BJ, and Lipscomb JD (2001) Methane monooxygenase component B mutants alter the kinetics of steps throughout the catalytic cycle, Biochemistry 40, 2220–2233. [DOI] [PubMed] [Google Scholar]
- [29].Zhang J, Wallar BJ, Popescu CV, Renner DB, Thomas DD, and Lipscomb JD (2006) Methane monooxygenase hydroxylase and B component interactions, Biochemistry 45, 2913–2926. [DOI] [PubMed] [Google Scholar]
- [30].Chang SL, Wallar BJ, Lipscomb JD, and Mayo KH (2001) Residues in Methylosinus trichosporium OB3b methane monooxygenase component B involved in molecular interactions with reduced- and oxidized-hydroxylase component: A role for the N-terminus, Biochemistry 40, 9539–9551. [DOI] [PubMed] [Google Scholar]
- [31].Wallar BJ, and Lipscomb JD (1996) Dioxygen activation by enzymes containing binuclear non-heme iron clusters, Chem. Rev 96, 2625–2657. [DOI] [PubMed] [Google Scholar]
- [32].Brazeau BJ, Wallar BJ, and Lipscomb JD (2001) Unmasking of deuterium kinetic isotope effects on the methane monooxygenase compound Q reaction by site-directed mutagenesis of component B, J. Am. Chem. Soc 123, 10421–10422. [DOI] [PubMed] [Google Scholar]
- [33].Zheng H, and Lipscomb JD (2006) Regulation of methane monooxygenase catalysis based on size exclusion and quantum tunneling, Biochemistry 45, 1685–1692. [DOI] [PubMed] [Google Scholar]
- [34].Zhang J, and Lipscomb JD (2006) Role of the C-terminal region of the B component of Methylosinus trichosporium OB3b methane monooxygenase in the regulation of oxygen activation, Biochemistry 45, 1459–1469. [DOI] [PubMed] [Google Scholar]
- [35].Banerjee R, Komor AJ, and Lipscomb JD (2017) Use of isotopes and isotope effects for investigations of diiron oxygenase mechanisms, Methods Enzymol. 596, 239–290. [DOI] [PubMed] [Google Scholar]
- [36].Srinivas V, Banerjee R, Lebrette H, Jones JC, Aurelius O, Kim I-S, Pham CC, Gul S, Sutherlin KD, Bhowmick A, John J, Bozkurt E, Fransson T, Aller P, Butryn A, Bogacz I, Simon P, Keable S, Britz A, Tono K, Kim KS, Park S-Y, Lee SJ, Park J, Alonso-Mori R, Fuller FD, Batyuk A, Brewster AS, Bergmann U, Sauter NK, Orville AM, Yachandra VK, Yano J, Lipscomb JD, Kern J, and Högbom M (2020) High resolution XFEL structure of the soluble methane monooxygenase hydroxylase complex with its regulatory component at ambient temperature in two oxidation states, J. Am. Chem. Soc 142, 14249–14266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lee SJ, McCormick MS, Lippard SJ, and Cho U-S (2013) Control of substrate access to the active site in methane monooxygenase, Nature 494, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sehnal D, Svobodová Vařeková R, Berka K, Pravda L, Navrátilová V, Banáš P, Ionescu CM, Otyepka M, and Koča J (2013) MOLE 2.0: advanced approach for analysis of biomacromolecular channels, J. Cheminformatics 5, 10.1186/1758-2946-5-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McCormick MS, and Lippard SJ (2011) Analysis of substrate access to active sites in bacterial multicomponent monooxygenase hydroxylases: X-ray crystal structure of xenon-pressurized phenol hydroxylase from Pseudomonas sp. OX1, Biochemistry 50, 11058–11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fox BG, Hendrich MP, Surerus KK, Andersson KK, Froland WA, Lipscomb JD, and Münck E (1993) Mössbauer, EPR, and ENDOR studies of the hydroxylase and reductase components of methane monooxygenase from Methylosinus trichosporium OB3b, J. Am. Chem. Soc 115, 3688–3701. [Google Scholar]
- [41].Sage JT, Xia YM, Debrunner PG, Keough DT, De Jersey J, and Zerner B (1989) Mössbauer analysis of the binuclear iron site in purple acid phosphatase from pig allantoic fluid, J. Am. Chem. Soc 111, 7239–7247. [Google Scholar]
- [42].Hendrich MP, Münck E, Fox BG, and Lipscomb JD (1990) Integer-spin EPR studies of the fully reduced methane monooxygenase hydroxylase component, J. Am. Chem. Soc 112, 5861–5865. [Google Scholar]
- [43].Froland WA, Andersson KK, Lee S-K, Liu Y, and Lipscomb JD (1992) Methane monooxygenase component B and reductase alter the regioselectivity of the hydroxylase component-catalyzed reactions. A novel role for protein-protein interactions in an oxygenase mechanism, J. Biol. Chem 267, 17588–17597. [PubMed] [Google Scholar]
- [44].Wang W, Iacob RE, Luoh RP, Engen JR, and Lippard SJ (2014) Electron transfer control in soluble methane monooxygenase, J. Am. Chem. Soc 136, 9754–9762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang W, and Lippard SJ (2014) Diiron oxidation state control of substrate access to the active site of soluble methane monooxygenase mediated by the regulatory component, J. Am. Chem. Soc 136, 2244–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tinberg CE, and Lippard SJ (2010) Oxidation reactions performed by soluble methane monooxygenase hydroxylase intermediates Hperoxo and Q proceed by distinct mechanisms, Biochemistry 49, 7902–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Osborne CD, and Haritos VS (2019) Beneath the surface: Evolution of methane activity in the bacterial multicomponent monooxygenases, Mol. Phylogen. Evol 139, 106527, 10.1016/j.ympev.2019.106527 [DOI] [PubMed] [Google Scholar]
- [48].Wang PH, Bruschi M, De Gioia L, and Blumberger J (2013) Uncovering a dynamically formed substrate access tunnel in carbon monoxide dehydrogenase/acetyl-CoA synthase, J. Am. Chem. Soc 135, 9493–9502. [DOI] [PubMed] [Google Scholar]
- [49].Doukov TI, Blasiak LC, Seravalli J, Ragsdale SW, and Drennan CL (2008) Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase, Biochemistry 47, 3474–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hino T, Matsumoto Y, Nagano S, Sugimoto H, Fukumori Y, Murata T, Iwata S, and Shiro Y (2010) Structural basis of biological N2O generation by bacterial nitric oxide reductase, Science 330, 1666–1670. [DOI] [PubMed] [Google Scholar]
- [51].Cojocaru V, Winn PJ, and Wade RC (2007) The ins and outs of cytochrome P450s, Biochim. Biophys. Acta Gen. Subj 1770, 390–401. [DOI] [PubMed] [Google Scholar]
- [52].Ebert MCCJC, Dürr SL, Houle AA, Lamoureux G, and Pelletier JN (2016) Evolution of P450 monooxygenases toward formation of transient channels and exclusion of nonproductive gases, ACS Catal. 6, 7426–7437. [Google Scholar]
- [53].Winter MB, Herzik MA, Kuriyana J, and Marletta MA (2011) Tunnels modulate ligand flux in a heme nitric oxide/oxygen binding (H-NOX) domain, Proc. Natl. Acad. Sci. U. S. A. 108, E881–E889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mehio N, Dai S, and Jiang D. e. (2014) Quantum mechanical basis for kinetic diameters of small gaseous molecules, J. Phys. Chem. A 118, 1150–1154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.