

Abstract
Background
The search for biomarkers to evaluate ovarian cancer (OC) homologous recombination (HR) function and predict the response to therapy is an urgent clinical need to improve the selection of patients who could benefit from platinum- and olaparib (poly-ADP ribose polymerase inhibitors, PARPi)-based therapies.
Methods
We used a large collection of OC patient-derived xenografts (PDXs) (n = 47) and evaluated their HR status based on BRCA1/2 mutations, BRCA1 promoter methylation and the HRDetect score. RAD51 foci were quantified in formalin-fixed, paraffin-embedded untreated tumour specimens by immunofluorescence and the messenger RNA expression of 21 DNA repair genes by real-time PCR.
Results
Tumour HR deficiency predicted both platinum and olaparib responses. The basal level of RAD51 foci evaluated in geminin-positive/replicating cells strongly inversely correlated with olaparib response (p = 0.011); in particular, the lower the foci score, the greater the sensitivity to olaparib, while low RAD51 foci score seems to associate with platinum activity.
Conclusions
The basal RAD51 foci score is a candidate predictive biomarker of olaparib response in OC patients as it can be easily translatable in a clinical setting. Moreover, the findings corroborate the importance of OC-PDXs as a reliable tool to identify and validate biomarkers of response to therapy.
Subject terms: Ovarian cancer, Tumour biomarkers
Background
Ovarian cancer (OC) is the third most common gynaecological cancer after cervical and uterine cancers [1]. Ninety per cent of OC is epithelial and the high-grade serous and endometrioid histotypes (HGOCs) are the most common [2]. These latter are generally diagnosed at an advanced stage (FIGO stage III/IV), when the tumour has spread through and beyond the peritoneal cavity, negatively affecting prognosis [2, 3]. Cytoreductive surgery followed by adjuvant platinum/paclitaxel chemotherapy has been the standard of care in the past 30 years [2]. In recent years, better knowledge of the biology and molecular characteristics of HGOC has led to the approval of poly-ADP ribose polymerase inhibitors (PARPi) [4, 5]. PARPi inhibit PARP1/2 enzymes, which are involved in the repair of DNA breaks [6]. This inhibition leaves PARP trapped on the DNA with impairment of DNA repair and accumulation of DNA damage, stalling of the replication forks and cell death [7]. Cells with defects in homologous recombination (HR) repair are particularly susceptible to death induced by PARPi [8, 9].
Half of the HGOCs have been reported to have mutation/deletion and lack of expression (due to hypermethylation) of genes involved in the HR repair pathway [10]. HR is a complex, error-free pathway, assigned to repair double-strand DNA breaks [11]. It is a multistep process involving many different proteins (i.e. BRCA1/2, RAD51, Fanconi anaemia genes, etc.) whose mutations or altered expression occur in 40% of HGOCs and result in an HR deficient (HRD) or BRCAness phenotype [10]. PARPi have been shown to be in synthetic lethality with a deficiency in BRCA1/2, meaning that the inhibition of PARP1 enzyme in cancer cells, where also BRCA1/2 function is compromised, causes cell death and this has fostered the clinical development of these compounds in BRCA1/2-mutated tumours, with very interesting results [5, 12]. Two PARPi, olaparib and niraparib, have now been approved for OC, in both relapsed [13, 14] and newly diagnosed tumours [15, 16]. Although they were first indicated in relapsing, platinum-sensitive, BRCA-mutated cancers, their use has expanded, allowing more patients to be treated with these drugs, earlier in their management.
Cumulative clinical and preclinical evidence has, in fact, broadened their efficacy beyond the BRCA (somatic and/or germline) tumour mutational status, including mutations/alterations in other HR genes [17]. Platinum sensitivity and BRCA1/2 mutations are considered surrogates of PARPi sensitivity; however, these correlations are not perfect and, in fact, some PARPi responses have been observed in platinum-resistant tumours, in BRCA wild-type (wt) and HR-proficient (HRP) tumours, while at the same time not all platinum-sensitive or BRCA-mutated tumours benefit [7, 18].
In the past few years, tests have been developed addressing the tumour HRP/HRD status, respectively, but their predictive value is still not sufficient (for recent reviews see [18–20]). These tests rely on the analysis of germline/somatic mutations of genes involved in HR and on genomic profiles secondary to HR defects. Their important limitation is that they capture a snapshot (genomic scar) that measures past events and does not necessarily reproduce the current tumour DNA repair status. These tests fail to take account of a lot of possible mechanisms of resistance (i.e. BRCA reversion mutations, re-expression of protein due to epigenetic events, protection of stalled replication fork) [19, 20].
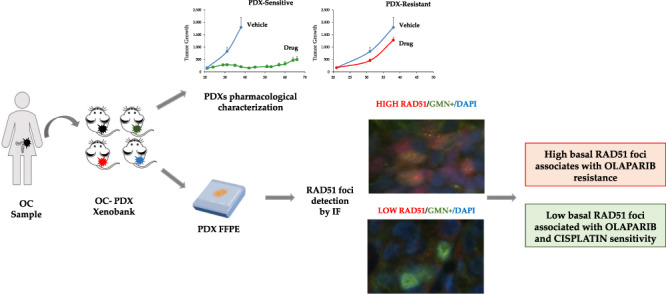
There is an urgent need to set up HRD functional assays to predict PARPi’s treatment benefit. The induction of RAD51 foci formation after DNA damage has been associated with HR repair proficiency, but its clinical translatability suffers—among other things—from the fact that pre- and post-treatment biopsies and ex vivo treatment of primary cell cultures are needed to evaluate the induction of RAD51 foci caused by the treatment [19] and this can be arduous to reconcile with the clinical practice. Recently, the basal percentage of RAD51 foci-positive (RAD51+) tumour cells in the S/G2 phase of the cell cycle (geminin-positive/GMN+ cells) was reported to predict olaparib response in a panel of breast cancer patient-derived xenografts (PDXs) [21, 22]. The basal score of RAD51+ tumour cells in formalin-fixed, paraffin-embedded (FFPE) tumour sections was able to discriminate between olaparib-sensitive and -resistant breast cancer PDXs, i.e. low RAD51 score was associated with olaparib sensitivity.
The present study analysed the ability of the RAD51 foci assay in predicting the sensitivity of patient-derived OC xenograft (OC-PDX) models to cisplatin (DDP) and olaparib, by analysing FFPE OC-PDX samples. Other potential biomarkers (genetic alterations in HR-related genes, BRCA1 promoter hypermethylation and messenger RNA (mRNA) expression of different DNA repair genes) were also investigated. As a whole, BRCA1/2 mutations, BRCA1 promoter hypermethylation and/or an HRDetect score ≥0.7 predicted response to both DDP and olaparib in OC-PDXs; in addition, RAD51 allowed the identification of tumours responding to olaparib with high accuracy, but it did not predict DDP sensitivity. These results support the translation of the RAD51 assay to a clinical setting for PARPi sensitivity, allowing better patient stratification.
Methods
Patient-derived ovarian carcinoma xenobank (OC-xenobank)
In the past decade, a collection of OC-PDXs has been established in our Department of Oncology starting from fresh tumour specimens collected during cytoreductive surgery or paracentesis interventions, as already detailed [23, 24]. Patients provided written informed consent authorising the collection and use of the tissues for research purposes. This xenobank consists of subcutaneously (s.c.) and intraperitoneally (i.p.) transplanted xenografts (Supplementary Table 1). Some of the PDXs derive from relapsing platinum-treated tumours. None of them come from tumours pre-treated with PARPi; however, MNHOC511 was derived from a patient very sensitive (VS) to first-line platinum-based therapy, who relapsed after 30 months and underwent a second-line platinum-based therapy, followed by olaparib given as maintenance. At the time of writing, the patient was still in maintenance therapy with olaparib with no evidence of disease. The xenobank also includes three DDP-resistant models, obtained through multiple in vivo DDP treatment cycles [25]. Forty-seven PDXs have been characterised for DDP response ([23, 24] and present manuscript) and 26 for olaparib response (Bizzaro et al. submitted and present manuscript) (Supplementary Table 1 [26]).
In vivo studies
Procedures involving animals and their care were conducted in conformity with institutional guidelines at the Mario Negri Institute for Pharmacological Research IRCCS (Milan, Italy), which adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D. lg 26/2014; Authorisation no.19/2008-A issued March 6, 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorisation for persons conducting animal experiments (Quality Management System Certificate-UNI EN ISO 9001:2015–Reg, no. 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directive and guidelines (EEC Council Directive 2010/63/UE). All in vivo experiments complied with protocols approved by the Ethics Committee of the Mario Negri Institute for Pharmacological Research IRCCS and the Italian Ministry of Health (approval numbers 510-2016 and 296/2018-PR).
OC-xenobank pharmacological characterisation
DDP (Sigma-Aldrich) was given intravenously at a dose of 5 mg/kg, every 7 days for 3 weeks. Olaparib (Targetmol) was dissolved in 10% v/v dimethylsulfoxide in 10% w/v HP-β-cyclodextrin and diluted in sterile water, given orally at a dose of 100 mg/kg, daily, 5 days a week for 4 weeks. When s.c. engrafted PDXs reached ~100–150 mg tumour weight (TW), mice were randomised (8–10 mice/group) to receive vehicle or specific treatments, i.p. xenografts were treated after 10 days after transplant. For s.c. PDXs, treatment efficacy was expressed as best tumour growth inhibition (T/C), calculated as: T/C% = [(median TW-treated mice/median TW control mice) × 100]. Treatment efficacy in orthotopically transplanted PDXs was expressed as an increase in lifespan (ILS), calculated as: ILS% = [(median survival days of treated mice − median survival days of control mice)/median survival days of treated mice] × 100. As already reported [24, 27, 28], PDXs were classified as VS (T/C ≤ 10% or ILS ≥ 100%), sensitive (S) (10% < T/C < 50% or 40% < ILS < 100%) and resistant (R) (T/C ≥ 50% or ILS ≤ 40%).
BRCA1/BRCA2 mutational status
Genomic DNA was extracted from snap-frozen PDX tumours using the Maxwell® 16 DNA Purification Kit in combination with the Maxwell® 16 Instrument (Promega). In all the samples, the percentage of murine DNA contamination was established by real-time PCR using primers specifically designed to distinguish humans from murine actin-β, and only samples with more than 85% human DNA were processed. BRCA1/BRCA2 mutational status was assessed by next-generation sequencing-targeted resequencing [29] as part of the ongoing molecular characterisation of our OC-PDX repository, and by whole-genome sequencing (WGS) as described in the HRDetect method below [30–32] and validated by Sanger’s method.
HRDetect score
Genomic DNA from ten selected PDX tumours and their respective germline DNA (patients’ blood) were analysed through whole genome sequencing (WSG) at the Wellcome Trust Sanger Institute (Hinxton, UK) as described [30–32]. Briefly, genomic libraries were constructed according to Illumina library protocols and sequencing was done on an Illumina HiSeq X Ten using HiSeq Control Software (HCS). The resulting reads were aligned to the reference germline genome. Mutation calling was done to describe somatic substitutions, indels, structural variants, allele-specific copy number variations and loss of heterozygosity (LOH) across the BRCA1, BRCA2 and PALB2 genes. Mutational signature contributions for substitution signatures 3 and 8, rearrangement signatures 3 and 5, deletions at microhomology and HRD LOH index were calculated for each sample as input into the weighted HRDetect model. The HRDetect algorithm was run as described in [31]. A score ≥0.7 suggests HRD, while <0.7 indicates HR proficiency.
Gene expression analysis
The absolute number of mRNA molecules for the 21 selected genes was quantified by real-time PCR with SYBR green® (Promega) technology, as previously reported [33]. Briefly, RNA was extracted and purified from snap-frozen PDX tumour fragments or ascitic cancer cells with a Maxwell® RSC simplyRNA Tissue Kit (Promega). RNA was retrotranscribed to complementary DNA (cDNA) by using the High Capacity cDNA Archive Kit (Life Technologies). For real-time PCR, optimal primer pairs were chosen (Supplementary Table 2). All the real-time PCRs were run in triplicate, and absolute quantification of the number of mRNA molecules was done on standard curves. Expression data were normalised employing the geometric mean of two housekeeping genes: actin-β (ACTB) and cyclophilin (CYP).
Promoter methylation analysis
One microgram of genomic DNA was treated with sodium bisulfite with an Epitect Plus DNA Bisulfite Kit (Qiagen). To verify successful sodium bisulfite conversion of the samples, a region of calponin promoter was amplified by methylation-specific PCR in all the modified samples, as described [34]. BRCA1 promoter methylation status was confirmed using primers designed ad hoc to distinguish the methylated (M) from the unmethylated (U) region (Supplementary Table 2) [35].
Gene copy number
The CCNE1 gene copy number was calculated in a TaqMan Copy Number Assay (Applied Biosystems) using the PCR thermocycler (ABI-7900, Applied Biosystems). RNAse P copy number was used as a reference gene and DNA extracted from human healthy donors was included as an internal control.
Immunofluorescence (IF) detection of nuclear foci on FFPE OC-PDX samples
To quantify RAD51 and γH2AX nuclear foci, we used an IF-based method [21, 22]. FFPE OC-PDX tissue sections were deparaffinised and antigens were retrieved with DAKO Antigen Retrieval Buffer pH 9.0. The following primary antibodies were used: rabbit anti-RAD51 ab133534 (Abcam), diluted 1:1000; mouse anti-γH2AX monoclonal antibody clone JBW301 (Millipore), diluted 1:200; rabbit anti-GMN 10802-1-AP (Proteintech Group), diluted 1:400; mouse anti-GMN NCL-L (Novo Castra), diluted 1:100. Secondary antibodies were diluted in blocking buffer 1:500, using Alexa Fluor 488 and Alexa Fluor 568 (Life Technologies). Nuclei were stained with 4′,6-diamidino-2-phenylindole (30 ng/mL in phosphate-buffered saline, Sigma-Aldrich). Slides were mounted with Vectashield solution (Vector Labs). Slices were observed using the ECLIPSE Ti2-E (Nikon) fluorescence microscope, with the ×60/1.27 WI Plan APO IR, ∞0.15/0.19 WD 0.18–0.16 objective (Nikon). RAD51 foci were quantified by scoring in blind the percentage of GMN+ tumour cells with five or more foci per nucleus (RAD51+/GMN+). At least 100 GMN+ cells in three different areas of the tissue section were analysed. We used pre-defined thresholds to determine qualitative scores: RAD51+ tumours were >10% RAD51+/GMN+ cells; γH2AX+ tumours had ≥25% γH2AX+/GMN+ cells.
Statistical analyses
Descriptive analyses included a number of observations, mean, standard deviation (SD), median, inter-quartile range, minimum, maximum and number of missing values for continuous variables. Categorical variables were described with frequency, percentage and number of missing values. The agreement between DDP and olaparib responses was assessed by Cohen’s κ index. Response to therapy was analysed as a categorical variable grouped as VS, S or R.
Non-parametric analyses were done when the assumption of normality was not satisfied. The Jonckheere-Terpstra per trend test was used to establish the statistical significance trend between response to therapies (DDP and olaparib) and the percentages of RAD51+/GMN+ cells. Kruskal–Wallis test was used to analyse the associations between continuous percentages of RAD51+/GMN+ cells and responses to DDP and olaparib. Spearman’s correlation coefficients were calculated for the correlation between continuous variables. The positive predictive values (PPVs) and negative predictive values (NPVs) were calculated considering the response to therapies (DDP and olaparib) as dichotomous variables: response VS or S as positive and response R as negative.
A p value < 0.05 was considered significant and correction for multiple comparisons was not applied because of the exploratory nature of the study. Statistical analyses were done with SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
DDP and olaparib responses in the OC-xenobank
The antitumor activity of DDP and olaparib was expressed as T/C value representing the magnitude of tumour growth inhibition by the treatment for s.c. growing tumours (the lower the value, the greater the antitumor effect) and as ILS, ILS over controls for i.p. transplanted tumours (the higher the value, the greater the antitumor effect) [24]. Taking the T/C and ILS together, PDXs could be classified as VS, S and R (Table 1, Bizzato et al. submitted and [23, 24]).
Table 1.
List of the OC-PDXs with the pharmacological and molecular parameters studied.
| #ID PDXs | DDP response | OLA response | BRCA1/2 status | BRCA1 promoter methylation | HRDetect score | % RAD51+/GMN+ cells | % γH2AX+/GMN+ cells | HR status |
|---|---|---|---|---|---|---|---|---|
| MNHOC511 | VS | VS | BRCA1 mut | 0% | 0% | 70% | HRD | |
| MNHOC513 | VS | VS | BRCA1 mut | 0% | 0% | 84% | HRD | |
| MNHOC218 | VS | VS | 0% | 0% | 52% | |||
| MNHOC106 | S | BRCA1 mut | 0% | 0% | 94% | HRD | ||
| MNHOC78 | S | BRCA1 mut | 0% | 0% | 58% | HRD | ||
| MNHOC508 | VS | VS | BRCA2 mut | 0% | 1% | 68% | HRD | |
| MNHOC212 | VS | wt | 100% | 0.999 | 1% | HRD | ||
| MNHOC107 | S | BRCA1 mut | 0% | 2% | 100% | HRD | ||
| MNHOC109 | S | wt | 0% | 2% | 88% | |||
| MNHOC154 | S | BRCA1 mut | 0% | 0.999 | 2% | 60% | HRD | |
| MNHOC76 | R | R | wt | 0% | 2% | 90% | ||
| MNHOC230 | VS | wt | 0% | 0.999 | 3% | HRD | ||
| MNHOC241 | VS | 0% | 3% | 80% | ||||
| MNHOC9 | S | 0% | 3% | |||||
| MNHOC500 | VS | VS | BRCA1 mut | 0% | 4% | 30% | HRD | |
| MNHOC8Y | R | wt | 100% | 4% | 53% | HRD | ||
| MNHOC271 | S | S | 0% | 6% | 66% | |||
| MNHOC266ddpR | R | R | 4% | 7% | 84% | |||
| MNHOC8 | VS | VS | wt | 84% | 20% | 45% | HRD | |
| MNHOC124ddpR | R | R | wt | 0% | 0.119 | 22% | 82% | |
| MNHOC506 | VS | R | wt | 0% | 22% | 66% | ||
| MNHOC143 | S | R | wt | 0% | 26% | 20% | ||
| MNHOC111/2C | R | wt | 0% | 28% | 68% | |||
| MNHOC18 | S | R | wt | 0% | 32% | 76% | ||
| MNHOC84 | R | R | wt | 0% | 32% | 76% | ||
| MNHOC239 | S | R | wt | 0% | 0.075 | 38% | 32% | |
| MNHOC94/2C | R | R | wt | 0% | 38% | 66% | ||
| MNHOC239ddpR | R | R | wt | 0% | 0.156 | 40% | 58% | |
| MNHOC125 | S | BRCA1 mut | 0% | 40% | 98% | HRD | ||
| MNHOC119 | R | wt | 0% | 40% | 78% | |||
| MNHOC22 | VS | R | BRCA1 mut | 0% | 40% | 74% | HRD | |
| MNHOC142 | S | 0% | 40% | 78% | ||||
| MNHOC518 | VS | VS | 93% | 41% | HRD | |||
| MNHOC10 | R | wt | 0% | 42% | ||||
| MNHOC316 | VS | R | 0% | 45% | ||||
| MNHOC315 | R | R | 0% | 45% | ||||
| MNHOC164 | S | 0% | 56% | 9% | ||||
| MNHOC258 | S | 0% | 58% | 90% | ||||
| MNHOC266 | VS | R | BRCA1 mut | 0% | 0.991 | 58% | 96% | HRD |
| MNHOC503 | S | 0% | 62% | 80% | ||||
| MNHOC124 | VS | R | wt | 0% | 0.014 | 66% | 92% | |
| MNHOC182 | R | R | wt | 0% | 70% | 52% | ||
| MNHOC261 | VS | wt | 0% | 0.018 | 78% | |||
| MNHOC94/2TR | R | R | wt | |||||
| MNHOC135 | S | wt | 0% | 0.999 | HRD | |||
| MNHOC520 | S | R | 49% | |||||
| MNHOC79 | R | wt | 0% |
HGOC high-grade ovarian cancer, LG low-grade ovarian cancer, s.c. subcutaneous, i.p. intraperitoneal, DDP cisplatin, OLA olaparib, VS very sensitive, S sensitive, R resistant, mut mutated, wt wild type, HRD homologous recombination deficiency.
The pattern of DDP and olaparib sensitivities is summarised in Fig. 1 for s.c. transplanted PDXs and Fig. 2 for i.p. PDXs. More than two-thirds (70%) of the PDXs were VS/S to DDP, while only one-third of the xenografts (31%) responded to olaparib treatment. In addition, while for DDP-treated OC-PDXs (n = 47), there was a more gradual range of drug responses (16 VS, 17 S, 14 R), olaparib-treated tumours (n = 26) were either clearly VS (n = 7) or not responsive (n = 18) to the drug, with only one showing moderate sensitivity to the drug.
Fig. 1. Responses to cisplatin and olaparib and HR/BRCA profile of the subcutaneously (s.c.) transplanted OC-PDXs.

For tumours grown subcutaneously, the best T/C% for cisplatin (black bars) and for olaparib (grey bars) are shown. Tumours were classified as resistant (R) T/C ≥ 50%; sensitive (S) T/C 10–50% and very sensitive (VS) T/C ≤ 10%. The dashed lines indicate the T/C% thresholds and the three categories of drug response (VS, S and R), as specified in “Methods”. BRCA1/2 mutational status: W wild type, M mutated (grey box); HRDetect score: ≥0.7, HR deficient (grey box); <0.7, HR proficient; BRCA1 promoter methylation status: Met hypermethylated (grey box); U unmethylated, - data not available.
Fig. 2. Responses to cisplatin and olaparib and HR/BRCA profile of the intraperitoneally (i.p.) transplanted OC-PDXs.

Best ILS% for cisplatin (black bars) and olaparib (grey bars) in orthotopic PDXs. Tumours were classified as resistant (R) with ILS% ≤ 40%; sensitive (S) with ILS% 40–100% and very sensitive (VS) with ILS% ≥ 100%. The dashed lines indicate the ILS% thresholds and the three categories of drug response. BRCA1/2 mutational status: W wild type, M mutated (grey box); HRDetect score: ≥0.7, HR deficient (grey box); <0.7, HR proficient; BRCA1 promoter methylation status: Met hypermethylated (grey box); U unmethylated, - data not available.
As a biomarker of olaparib response, response to DDP has been suggested [7]. We tested the predictive role of DDP sensitivity in olaparib’s antitumor effect in our xenobank, but the calculated Cohen κ was low (κ = 0.38), suggesting a lack of concordance. In fact, although all the DDP-resistant tumours (n = 9) failed to respond to olaparib, 9 of the 18 models VS/S to DDP were resistant to the drug. These data confirm their non-overlapping mechanisms of action.
HR status in the OC-xenobank
We analysed BRCA1/2 mutational status, BRCA1 promoter methylation and used a recently developed test, the HRDetect assay, to identify a BRCAness phenotype in PDXs. The mutational status of BRCA1/2 was available for 34 OC-PDXs and 13 of them harboured a BRCA1/2 mutation (Table 1); however, in two of them (MNHOC182 and MNHOC18) there was no LOH and they were considered to have a functional protein (wild-type) (Bizzaro et al. submitted). All 11 BRCA-mutated models were VS/S to DDP (Jonckheere-Terpstra p = 0.019); six of these were also characterised for olaparib response and four were VS to olaparib (Jonckheere-Terpstra p = 0.0114), while the remaining two (the i.p. MNHOC22 and MNHOC266 models) did not respond, despite being very sensitive to DDP (Figs. 1 and 2 and Table 1).
mRNA expression of BRCA1 differed across the PDX tumours (Supplementary Table 3), with a mean of normalised mRNA molecules of 0.044 ± 0.03 and median 0.042 (range 0.0002–0.1448). To understand the large difference in mRNA levels better, we checked for a specific BRCA1 promoter methylation region [35]. Only four models had methylation higher than 80% (MNHOC518, MNHOC8, MNHOC8Y and MNHOC212), while all the others had an unmethylated or scarcely methylated promoter region (Figs. 1 and 2 and Table 1), and their normalised BRCA1 mRNA levels were significantly lower than the average number of mRNA molecules in all the other unmethylated PDXs (p = 0.0013) (Supplementary Table 4). As shown in Table 1, MNHOC518, MNHOC8 and MNHOC212 responded to DDP, while MNHOC8Y was resistant; a response to olaparib was seen in only two of the PDXs (MNHOC518 and MNHOC8).
Ten OC-PDXs were analysed with the HRDetect test. WGS was done and the HRDetect score was calculated (Figs. 1 and 2 and Table 1). Of the five OC-PDXs with a high HRDetect score (≥0.7), two were BRCA1/2 mutated, while the others were not (Table 1). MNHOC212 was BRCA1/2 wild type, but had an HRDetect score higher than 0.7, suggesting an HRD status and, in fact, the BRCA1 mRNA level was very low (Supplementary Table 4). All the PDXs with high HRDetect score were VS/S to DDP, while two out of five PDXs with low scores were resistant to the drug (Table 1).
We classified 17 OC-PDXs as HRD, harbouring mutations in BRCA1/2 and/or BRCA1 promoter hypermethylation and/or an HRDetect score higher than 0.7 [31], and looked for possible association with DDP and olaparib responses. HRD status was associated with both drug responses: 16/17 HRD PDXs responded to DDP (p = 0.001) and 6/8 of olaparib-responsive tumours were HRD (p = 0.0007) (Supplementary Fig. 1).
Association of RAD51 foci with DDP and olaparib antitumor activity
Following a published protocol [21, 22], we analysed and scored the percentages of RAD51+/GMN+ cancer cells in FFPE PDX samples whose sensitivity to DDP (n = 43) (Supplementary Fig. 2) and olaparib (n = 24) (Fig. 3) is known. A representative image of RAD51 foci counted is shown in Fig. 3. The intra-tumour variability was low (data not shown) and RAD51+/GMN+ cells ranged from 0 to 78% (Table 1). We also scored the percentage of cells positive for γH2AX and it was high (mean 68.9, median 70.0) (Table 1), corroborating the reported high genomic instability in ovarian carcinomas [36, 37].
Fig. 3. Percentages of cells positive to RAD51 foci in OC-PDXs and their response to olaparib.

Immunofluorescent staining of RAD51 nuclear foci (red dots) in a geminin-positive (green signal) nucleus. The histogram represents the percentage of RAD51+/GMN+ cells quantified in FFPE OC-PDX samples. For each PDX, the BRCA1/2 mutational status (M mut, W wild type) and sensitivity to olaparib (very sensitive, pale grey; sensitive, grey; resistant, dark grey squares) are reported.
We analysed the percentage of RAD51+/GMN+ cells in tumours classified as HRD and found that they had a significantly lower percentage of RAD51+/GMN+ cells than all the other PDXs (median 2 vs 32%, p = 0.005).
We then looked for associations with drug response and found that the percentage of tumour RAD51+/GMN+ cells was not associated with DDP sensitivity (Kruskal–Wallis test, p = 0.47), even if 15/18 (83%) DDP VS/S PDXs had a low level of RAD51 foci. On the contrary, PDXs VS/S to olaparib had the lowest percentage of RAD51+/GMN+ cells: medians, respectively, 1%, 6% and 38% for VS, S and R (Kruskal–Wallis p = 0.011) (Fig. 4). The PPV and NPV of RAD51 test and olaparib response (VS/S PDXs vs R) were 75% (confidence interval—CI 95%: 43.6–92.1) and 87.5% (CI 95%: 67.5–95.93), respectively, with 83.3% of accuracy value, whereas for RAD51 test and DDP response, the NPV was 36% (CI 95%: 26–47.4), but, more interestingly, the PPV was 83.3% (CI 95%: 63.7–93.4), even if the overall accuracy was 55.8%. Similar results were obtained when considering only the subset of HGOC PDXs (Supplementary Fig. 3).
Fig. 4. RAD51 foci in geminin-positive cells and responses to cisplatin and olaparib in the OC-PDXs.

a, b Percentage of RAD51+/GMN+ cells in the OC-PDXs clustered for their responses to cisplatin (DDP) (a) and olaparib (b) (white bars: very sensitive; pale grey: sensitive; dark grey: resistant). c, d Box plots showing the correlation between RAD51 foci positivity, based on the percentage of RAD51+/GMN+ cells (≤10%, RAD51 negative; >10%, RAD51 positive) and responses to DDP (c) and olaparib (d).
Association of gene expression with DDP and olaparib antitumor activity
In a previous study, we showed that CDK12, XPF and PALB2 mRNA expression levels were associated with DDP sensitivity [33]. Here, we did similar analyses, correlating their mRNA expression with olaparib response, but could not find any correlation. Then, we analysed the expression levels of 21 genes coding for proteins involved in drug efflux, HR and NHEJ pathways and in the mechanism of action of these two drugs (Supplementary Tables 2 and 3), selected based on the available preclinical data on their roles in DDP and olaparib sensitivity/resistance [38].
The genes with mRNA expression significantly associated with DDP antitumor activity were: ARID1A (medians 0.031, 0.024 and 0.014 for VS, S and R, respectively, p = 0.019), ARTEMIS (medians 0.008, 0.006 and 0.004 for VS, S and R, p = 0.02) and USP28 (medians 0.047, 0.026, 0.016 for VS, S and R, p = 0.007) (Supplementary Fig. 4). A trend of association, although not statistically significant, was found for olaparib response and USP28 (median 0.113, 0.027, 0.024 for VS, S and R, p = 0.053). Even though three PDXs showed increased CCNE1 gene copy number (4–5-fold), their CCNE1 mRNA level was not overexpressed; no differences in CCNE1 mRNA expression were observed among the DDP VS, S and R PDXs (Supplementary Table 5).
Discussion
HGOC is very sensitive to platinum-based therapy, as suggested by the fact that more than 75% of the patients respond to first-line chemotherapy [2]. The molecular mechanisms of DDP sensitivity are partly explained by the fact that half of the ovarian carcinomas have defects in HR repair [10]. Both platinum sensitivity and tumour HRD status are clinical predictive biomarkers of PARPi response [5, 39] and HRD status has been approved as a predictive biomarker of PARPi response. Preclinical and clinical evidences have been put forward demonstrating PARPi efficacy beyond BRCA1/2 mutations [17, 19, 39] and some companion tests supported the clinical development of some PARPi [39], although the most appropriate method to evaluate it is still to be defined (for a recent review, see [18, 20]). The identification of other predictive biomarkers of response to both platinum and PARPi is of paramount importance to select patients who will benefit from these therapies.
We have available a platform of more than 60 OC-PDX models, transplanted subcutaneously and orthotopically in immunodeficient mice, that mimic not only the patient’s biological behaviour and pharmacological response to therapy but also the complexity and heterogeneity of human ovarian carcinoma [24]. A substantial number of these PDXs has been tested in vivo for DDP and olaparib responses [24, 33] (Bizzaro et al submitted and present manuscript), making this xenobank a starting point for validating known predictive biomarkers of response but—more interestingly—to identify new ones, as recently reported [23].
Using this xenobank, the present study shows that: (i) the HRD tumour status, based on BRCA1/2 mutational status and/or BRCA1 promoter hypermethylation and/or HRDetect score, predicted both DDP and olaparib responses; (ii) the percentage of RAD51+/GMN+ cells inversely correlated with olaparib sensitivity (the lower the percentage the greater the drug sensitivity), but not with DDP sensitivity; (iii) ARID1A, ARTEMIS and USP28 mRNA expression was associated with DDP antitumor activity; none of the DNA repair gene expression levels investigated was associated with olaparib response, except USP28.
We confirmed that HRD tumour status predicts the response to both DDP and olaparib, as response rates to DDP and olaparib were higher in PDXs with alterations in the HR pathway. These results corroborate the existing clinical data and validate our OC-xenobank as a useful research tool. Even though the sample size of our OC-xenobank is limited, our data suggest basal RAD51 foci levels correlated with olaparib response, although with some exceptions. For example, MNHOC518 and MNHOC8, very sensitive to both DDP and olaparib, had a high percentage of RAD51+/GMN+ cells, while MNHOC266ddpR and MNHOC76, despite the low percentage of RAD51+/GMN+ cells, were resistant to both drugs. The percentage of RAD51+/GMN+ cells seems to predict the response to olaparib better than BRCA1/2 mutations. In fact, only two out of the six BRCA1/2-mutated PDXs resistant to olaparib had also a high percentage of RAD51+/GMN+ cells (MNHOC22 and MNHOC266).
A restoration of HR in an HRD background has been demonstrated by loss of function of proteins (53BP1, CHD4 and REV7) that would restore the DNA end-resection abilities, in the presence of mutated BRCA1, and subsequent recruitment of RAD51 and HR induction [40, 41]. Both these PDXs (MNHOC22 and MNHOC266) have low levels of CHD4 and REV7 mRNA; however, the mutational status of these genes—which would confirm the loss of protein function—is not yet available.
Recent data indicate that PARPi efficacy can rely also on its stimulation on host immune response [42], suggesting that the antitumor activity of PARPi might not solely depend on tumour cell characteristics (i.e. number of RAD51 foci). Although all PDXs grow in nude mice, which lack T cells, PARPi can potentially modulate other murine tumour microenvironment cells (natural killers, macrophages, fibroblasts) [43], resulting in antitumor activity and in part explaining the incomplete correlation.
All (8/8) VS/S PDX models to olaparib were VS/S to DDP. In contrast, 9/18 (50%) PDXs resistant to olaparib were VS/S to DDP. Such discrepancy might be explained by a non-functional nucleotide excision repair (NER) pathway, due to inactivating mutations or epigenetic silencing of NER genes, as recently reported in ovarian [44] and in breast cancers [39]. These non-overlapping, not HR-driven, mechanisms highlight that DDP-based therapy could be active in olaparib-resistant tumours.
A recently set up fluorescent immunohistochemistry method for RAD51 [45] allowed a quantification of nuclear RAD51 protein in two OC patient cohorts (FFPE samples) and found that a high RAD51 score associated with early relapse after platinum treatment, supporting its role as a marker of platinum resistance. In our OC-xenobank, the basal level of RAD51+/GMN+ cells predicted DDP response in 24 out of 43 (56%) PDXs with an overall non-significant correlation, corroborating previous results in a few ovarian and breast cancer PDX models [22]. PDXs with high basal level of RAD51+/GMN+ cells were not enriched in DDP-resistant tumours, in contrast with what was reported by Hoppe et al.; this could be explained by the differences in RAD51 quantification (mean nuclear intensity—nuclear expression score [45] and the number of RAD51 foci quantification—(present manuscript)), and in the outcomes considered (early relapse vs tumour growth inhibition and survival, respectively). We also observed that the PPV of the RAD51 assay, we used was high for DDP response, highlighting the probability of tumours with low RAD51 levels to be VS/S to DDP, even the test has low accuracy. The fact that RAD51 foci levels did not completely predict DDP response suggests that the analysis of foci alone is not sufficient to capture the complexity of DDP-induced DNA damage, whose repair involves not only HR but other pathways, such as NER, base excision repair and translesion synthesis [46–48].
We looked for other possible determinants of DDP and PARPi activity. PARP1 has been shown to be necessary for PARPi to exert their cytotoxic activity and its lack has been described as a mechanism of resistance [49]; however, PARP1 mRNA was clearly detected in all the PDXs. Interestingly, USP28 mRNA levels were inversely associated with DDP activity and a similar pattern was seen for olaparib. USP28 is a de-ubiquitinase involved in many different cancer-related pathways [50] and its lack has been described as a mechanism of PARPi resistance. Studies are ongoing to clarify its role in DDP and olaparib response.
There are several limitations to our study. The sample size of PDXs analysed is limited; however, our study was exploratory in nature and the positive results we obtained confirmed the ones reported in breast cancer PDXs [21, 22] and foster the clinical validation of scoring basal RAD51 foci in OC. The antitumor activity of DDP and olaparib was tested as monotherapy in PDXs not previously treated with platinum-based therapy, to try to have as much as clear data on the simplest experimental setting. Although we realise that this could not completely mirror the clinic, as PARPi are approved as maintenance after first-line platinum-based therapy and in the recurrent setting, the distribution of RAD51 foci number in the few PDXs derived from relapsing, DDP pre-treated tumours is comparable to the chemo-naive-derived PDXs (data not shown), suggesting that DDP treatment seems not to influence the number of RAD51 foci.
In summary, our data underline the importance of OC-PDXs in the identification and validation of biomarkers predictive of response. The molecular characterisation of our xenobank is far from complete; nevertheless, we confirmed the role of HRD in predicting DDP and olaparib sensitivity and the basal level of RAD51+/GMN+ cells as a possible new predictive biomarker of olaparib response. The proposed test is a low-cost, immunofluorescent-based assay. Data on breast cancer clinical specimens ([21, 22] support its clinical applicability, even if it could take some time considering both the need to possibly redefine the threshold to distinguish responsive from not responsive patients and to validate it in both retrospective, where the efficacy of PARPi is known, and prospective trials. Whether these data will be confirmed in ovarian carcinoma specimens with known sensitivity to olaparib, they could point to a breakthrough in the field of predictive biomarkers of PARPi.
Supplementary information
Acknowledgements
We gratefully acknowledged the Italian Association for Cancer Research (AIRC) and 3M Foundation for financial support and to “Pandora”, the ovarian cancer tissue collection (Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Department of Oncology). We also acknowledge Judith Bagott for editing the manuscript.
Author contributions
Conceptualisation and study design: FG, MFA, ER and GD; development of methodology: FG, MC, FR, AL-G, DA and GD; data acquisition: FG, AA, MC, FR, AL-G, DA, VS, SN-Z, RF, ML and MRB; formal and statistical analysis: FG, MFA, GD, ER, DA, SN-Z, AL-G and MRB; manuscript writing, review and revision: FG, GD, AL-G, MFA, ER, VS, DA, MRB and RG; study supervision: GD. All authors read and critically revised the manuscript for intellectual content and approved the final manuscript.
Funding information
The research leading to these results received funding from AIRC (IG ID19797 project—PI Damia Giovanna; IG ID23520—PI Giavazzi Raffaella). AL-G received salary support from the Spanish Association against Cancer (Asociación Española Contra el Cáncer, AECC, INVES20095LLOP) and a grant from “la Caixa” Foundation and European Institute of Innovation and Technology/Horizon 2020 (CaixaImpulse, LCF/TR/CC19/52470003), during the conduct of the study. VS reports personal support from Instituto de Salud Carlos III (ISCIII, CPII19/00033).
Data availability
All data supporting the conclusions of this study have been included within the article and the Supplemental data.
Materials availability
All materials supporting the conclusions of this study have been included within the article and the Supplemental data.
Competing interests
VS reports grants from AstraZeneca, Tesaro and personal fees from Abbvie, outside the submitted work. In addition, AL-G and VS have a patent WO2019122411A1 pending. The other authors declare no competing interests.
Ethics approval and consent to participate
All animal experiments were approved by the Ethical Committee of the Mario Negri Institute for Pharmacological Research IRCCS and the Italian Ministry of Health (approval numbers 510-2016 and 296/2018-PR).
Consent for publication
Not applicable.
Footnotes
The original online version of this article was revised: The original version of this article contained an affiliation error. Affiliation 8 affiliation states: “Unit of Gynecological Oncology Research, European Institute of Oncology, Milan, Italy” and needs to be changed to: “Unit of Gynecological Oncology Research, IEO, European Institute of Oncology IRCCS, Milan, Italy”.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
2/16/2023
A Correction to this paper has been published: 10.1038/s41416-023-02205-1
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01609-1.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. Lancet. 2019;393:1240–53. doi: 10.1016/S0140-6736(18)32552-2. [DOI] [PubMed] [Google Scholar]
- 3.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68:284–96. doi: 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, et al. PARP inhibitors in ovarian cancer. Cancer Treat Rev. 2019;73:1–9. doi: 10.1016/j.ctrv.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Konstantinopoulos PA, Lheureux S, Moore KN. PARP inhibitors for ovarian cancer: current indications, future combinations, and novel assets in development to target DNA damage repair. Am Soc Clin Oncol Educ Book. 2020;40:1–16. doi: 10.1200/EDBK_288015. [DOI] [PubMed] [Google Scholar]
- 6.Bai P. Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol Cell. 2015;58:947–58. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- 7.D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair. 2018;71:172–6. doi: 10.1016/j.dnarep.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 8.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 9.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 10.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–54. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mateo J, Lord CJ, Serra V, Tutt A, Balmana J, Castroviejo-Bermejo M, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30:1437–47. doi: 10.1093/annonc/mdz192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tew WP, Lacchetti C, Ellis A, Maxian K, Banerjee S, Bookman M, et al. PARP inhibitors in the management of ovarian cancer: ASCO Guideline. J Clin Oncol. 2020;JCO2001924. [DOI] [PMC free article] [PubMed]
- 14.Vanacker H, Harter P, Labidi-Galy SI, Banerjee S, Oaknin A, Lorusso D, et al. PARP-inhibitors in epithelial ovarian cancer: actual positioning and future expectations. Cancer Treat Rev. 2021;99:102255. doi: 10.1016/j.ctrv.2021.102255. [DOI] [PubMed] [Google Scholar]
- 15.DiSilvestro P, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Efficacy of maintenance olaparib for patients with newly diagnosed advanced ovarian cancer with a BRCA mutation: subgroup analysis findings from the SOLO1 Trial. J Clin Oncol. 2020;38:3528–37. doi: 10.1200/JCO.20.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez-Martin A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N. Engl J Med. 2019;381:2391–402. doi: 10.1056/NEJMoa1910962. [DOI] [PubMed] [Google Scholar]
- 17.Pilie PG, Gay CM, Byers LA, O’Connor MJ, Yap TA. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25:3759–71. doi: 10.1158/1078-0432.CCR-18-0968. [DOI] [PubMed] [Google Scholar]
- 18.Miller RE, Leary A, Scott CL, Serra V, Lord CJ, Bowtell D, et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann Oncol. 2020;12:1606–22. [DOI] [PubMed]
- 19.Fuh K, Mullen M, Blachut B, Stover E, Konstantinopoulos P, Liu J, et al. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol Oncol. 2020;3:877–86. [DOI] [PubMed]
- 20.Stover EH, Fuh K, Konstantinopoulos PA, Matulonis UA, Liu JF. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol Oncol. 2020;3:887–98. [DOI] [PubMed]
- 21.Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutierrez-Enriquez S, Ducy M, Ibrahim YH, et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med. 2018;10:e9172. [DOI] [PMC free article] [PubMed]
- 22.Cruz C, Castroviejo-Bermejo M, Gutierrez-Enriquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol. 2018;29:1203–10. doi: 10.1093/annonc/mdy099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guffanti F, Alvisi MF, Caiola E, Ricci F, De Maglie M, Soldati S, et al. Impact of ERCC1, XPF and DNA polymerase beta expression on platinum response in patient-derived ovarian cancer xenografts. Cancers. 2020;12:2398–2412. [DOI] [PMC free article] [PubMed]
- 24.Ricci F, Bizzaro F, Cesca M, Guffanti F, Ganzinelli M, Decio A, et al. Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res. 2014;74:6980–90. doi: 10.1158/0008-5472.CAN-14-0274. [DOI] [PubMed] [Google Scholar]
- 25.Ricci F, Brunelli L, Affatato R, Chila R, Verza M, Indraccolo S, et al. Overcoming platinum-acquired resistance in ovarian cancer patient-derived xenografts. Ther Adv Med Oncol. 2019;11:1758835919839543. doi: 10.1177/1758835919839543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bizzaro F et al. VEGF pathway inhibition potentiates PARP inhibitor efficacy in ovarian cancer independent of BRCA status. J Hematol Oncol. 2021 10.1186/s13045-021-01196-x. [DOI] [PMC free article] [PubMed]
- 27.Boven E, Winograd B, Berger DP, Dumont MP, Braakhuis BJ, Fodstad O, et al. Phase II preclinical drug screening in human tumor xenografts: a first European multicenter collaborative study. Cancer Res. 1992;52:5940–7. [PubMed] [Google Scholar]
- 28.Nicoletti MI, Valoti G, Giannakakou P, Zhan Z, Kim JH, Lucchini V, et al. Expression of beta-tubulin isotypes in human ovarian carcinoma xenografts and in a sub-panel of human cancer cell lines from the NCI-Anticancer Drug Screen: correlation with sensitivity to microtubule active agents. Clin Cancer Res. 2001;7:2912–22. [PubMed] [Google Scholar]
- 29.Beltrame L, Di Marino M, Fruscio R, Calura E, Chapman B, Clivio L, et al. Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: a retrospective study. Ann Oncol. 2015;26:1363–71. doi: 10.1093/annonc/mdv164. [DOI] [PubMed] [Google Scholar]
- 30.Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat Commun. 2020;11:2662. doi: 10.1038/s41467-020-16142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517–25. doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016;534:47–54. doi: 10.1038/nature17676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guffanti F, Fratelli M, Ganzinelli M, Bolis M, Ricci F, Bizzaro F, et al. Platinum sensitivity and DNA repair in a recently established panel of patient-derived ovarian carcinoma xenografts. Oncotarget. 2018;9:24707–17. doi: 10.18632/oncotarget.25185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sriraksa R, Chaopatchayakul P, Jearanaikoon P, Leelayuwat C, Limpaiboon T. Verification of complete bisulfite modification using calponin-specific primer sets. Clin Biochem. 2010;43:528–30. doi: 10.1016/j.clinbiochem.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, et al. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J Natl Cancer Inst. 2016;108. [DOI] [PubMed]
- 36.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura N, Shaikh N, Muliaditan D, Soliman TN, McGuinness JR, Maniati E, et al. Specific mechanisms of chromosomal instability indicate therapeutic sensitivities in high-grade serous ovarian carcinoma. Cancer Res. 2020. [DOI] [PubMed]
- 38.Damia G, Broggini M. Platinum resistance in ovarian cancer: role of DNA repair. Cancers. 2019;11:119–33. [DOI] [PMC free article] [PubMed]
- 39.Haunschild CE, Tewari KS. The current landscape of molecular profiling in the treatment of epithelial ovarian cancer. Gynecol Oncol. 2019;11:119–33. [DOI] [PubMed]
- 40.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–4. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee EK, Konstantinopoulos PA. PARP inhibition and immune modulation: scientific rationale and perspectives for the treatment of gynecologic cancers. Ther Adv Med Oncol. 2020;12:1758835920944116. doi: 10.1177/1758835920944116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turinetto M, Scotto G, Tuninetti V, Giannone G, Valabrega G. The role of PARP inhibitors in the ovarian cancer microenvironment: moving forward from synthetic lethality. Front Oncol. 2021;11:689829. doi: 10.3389/fonc.2021.689829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ceccaldi R, O’Connor KW, Mouw KW, Li AY, Matulonis UA, D’Andrea AD, et al. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015;75:628–34. doi: 10.1158/0008-5472.CAN-14-2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoppe MM, Jaynes P, Wardyn JD, Upadhyayula SS, Tan TZ, Lie S, et al. Quantitative imaging of RAD51 expression as a marker of platinum resistance in ovarian cancer. EMBO Mol Med. 2021;13:e13366. doi: 10.15252/emmm.202013366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S, Higgins VJ, Aldrich-Wright JR, Wu MJ. Identification of the molecular mechanisms underlying the cytotoxic action of a potent platinum metallointercalator. J Chem Biol. 2012;5:51–61. doi: 10.1007/s12154-011-0070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slyskova J, Sabatella M, Ribeiro-Silva C, Stok C, Theil AF, Vermeulen W, et al. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018;46:9537–49. doi: 10.1093/nar/gky764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer. 2021;21:37–50. doi: 10.1038/s41568-020-00308-y. [DOI] [PubMed] [Google Scholar]
- 49.Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9:1849. doi: 10.1038/s41467-018-03917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Liu Z, Zhang L, Yang Z, Chen X, Luo J, et al. Targeting deubiquitinase USP28 for cancer therapy. Cell Death Dis. 2018;9:186. doi: 10.1038/s41419-017-0208-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the conclusions of this study have been included within the article and the Supplemental data.
All materials supporting the conclusions of this study have been included within the article and the Supplemental data.