

Summary
Various genetic and environmental risk factors have been implicated in the pathogenesis of amyotrophic lateral sclerosis (ALS). Despite this, the cause of most ALS cases remains obscure. In this review, we describe the current evidence implicating genetic and environmental factors in motor neuron degeneration. While the risk exerted by many environmental factors may appear small, their effect could be magnified by the presence of a genetic predisposition. We postulate that gene-environment interactions account for at least a portion of the unknown etiology in ALS. Climate underlies multiple environmental factors, some of which have been implied in ALS etiology, and the impact of global temperature increase on the gene-environment interactions should be carefully monitored. We describe the main concepts underlying such interactions. Although a lack of large cohorts with detailed genetic and environmental information hampers the search for gene-environment interactions, newer algorithms and machine learning approaches offer an opportunity to break this stalemate. Understanding how genetic and environmental factors interact to cause ALS may ultimately pave the way towards precision medicine becoming an integral part of ALS care.
Keywords: Amyotrophic lateral sclerosis, Epidemiology, Interactions, Cause, Genetics, Environment, Climate
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurological disease that primarily affects motor neurons leading to paralysis and death from respiratory failure. With a prevalence of 4·9 –12 cases per 100,000 population1 and an incidence of 3·1 cases per 100,000 person-years,2 ALS is a relatively rare disease. It is also a disease of aging, with symptoms typically commencing around age 65.3 These characteristics (a fatal, rare disease of the aged) hint at the underlying etiology and suggest a roadmap to search for ALS risk factors. Today, we know the genetic causes of approximately 20% of cases. The remaining cases are likely caused by a mixture of multiple genetic and environmental factors4 (Figure. 1).
Figure 1.
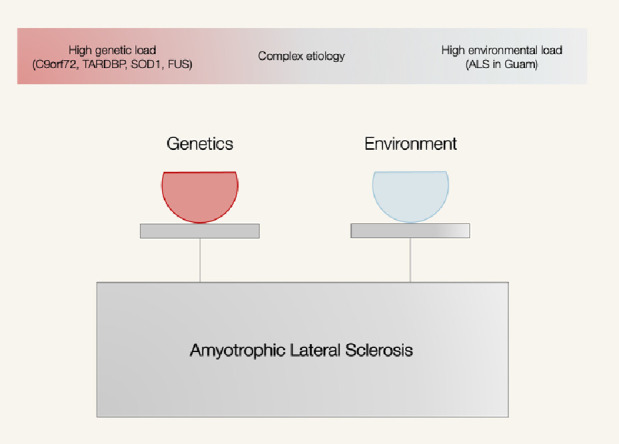
Amyotrophic lateral sclerosis (ALS) is likely to be a complex disease arising from variable interactions between genetic factors and environmental factors. ALS in Guam refers to the geographical cluster of ALS cases on the island of Guam.118
Here, we aim to summarise what is known about the environmental and genetic ALS risk factors. We also explore how these factors potentially interact to trigger motor neuron death.
ALS phenotypes
ALS clinical presentation is highly variable across patients. For example, several aspects of the ALS phenotype vary widely, such as the onset age, the extent of damage to the upper or the lower motor neuron, the most affected body region, progression rate, and the occurrence of non-motor symptoms, the most frequent being cognitive impairment.5, 6, 7 Despite this, most studies examining the cause of ALS treat patients as a monolith by assuming that the same risk factors are operating in each case. The more significant numbers analyzed by this approach increase the statistical power. However, the assumption is not necessarily valid and different environmental and genetic causes may operate within different ALS patient subgroups. If this is the case, treating them as a single group risks diluting the very risks one is trying to detect.
The different distribution of these phenotypes across diverse cohorts probably contributes to the discordant results observed across studies (see below). Even though ALS risk factors are undoubtedly granular and may operate at a local community level, there is no easy solution to this problem. Clustering based on clinical characteristics may not be the most suitable criterion when looking for etiology.6
Mendelian randomization studies
Most studies looking for ALS etiology are observational in nature. As such, many issues potentially limit the validity of their results, such as selection bias, reverse causality, and confounders. These issues are not unique to ALS and are present in the epidemiological study of any rare disease.
More recently, Mendelian randomization has emerged as a valuable tool to overcome such hidden confounders.8 Mendelian randomization studies rely on the random assortment of genes from parents to offspring during conception, as described by Mendel's second law. Since the inheritance of one trait will be independent of the inheritance of other traits, genetic variants related to one exposure will not be linked to those related to potential confounders and can be used as a proxy.9,10 Single nucleotide polymorphisms (SNPs) related to a particular exposure are retrieved from the summary statistics of previous GWA studies and then used as a separate outcome GWAS to assess the relationship between the exposure and the outcome.11
However, Mendelian randomization studies are not free from limitations, and three major assumptions must hold for the results to be valid: (i) genetic variants should be associated with the exposure (relevance assumption); (ii) they must not be associated with confounders (independence assumption); and (iii) they must not influence the outcome through other pathways than the exposure considered (exclusion restriction assumption).8,9 The first assumption is usually quantified using the F-statistics, while the remaining two are addressed through sensitivity measures such as the MR-PRESSO and Cochran's Q test.11 A practical consideration is that Mendelian randomization requires large datasets to detect meaningful effects, and, ideally, a positive outcome should be replicated in an independent dataset.
Furthermore, the SNPs used as instrumental variables of the exposure are identified based on a specific linkage disequilibrium structure. Hence their validity may not be the same across multiple populations. Ideally, the two GWASes should be selected from the same population to avoid this pitfall. Finally, Mendelian Randomization studies could be limited when considering time-varying exposures, such as behaviours. In these cases, GWAS would be performed at a specific time point while the behaviour could change before and after that point in response to many internal and external stimuli.11,12 The confounding effect of time (or aging) is a recurring theme when tackling gene-environment-climate relationships.
Genetic factors
Heritability estimates provide a measure of how much phenotypic or disease risk variance can be explained by genetics. Estimates from twin studies13,14 and pedigree studies15,16 give higher heritability estimates compared to those from population-level genome-wide SNP data.17 The difference reflects the contribution of rare variants captured in family studies and perhaps the relatively small numbers of cases involved in twin and pedigree studies yielding wide confidence intervals. With only 30 genes reliably identified to cause or modify ALS risk,18 a significant proportion of heritability in ALS is still missing.
Despite this knowledge gap, the known genes are already converging on multiple functional pathways. These pathways include global protein and RNA homeostasis dysfunction, oxidative stress and inflammation, mitochondrial dysfunction, impaired cytoskeletal integrity, and altered axonal transport dynamics (18,19; summarized in Table 1). Interestingly, many of these pathways are reactive and responsive to exogenous influences. For example, exposure to pesticides, cigarette smoke, traumatic brain injury, and physical activity can induce oxidative stress and inflammation,20 which could have a knock-on effect on cellular pathways with pre-existing lower tolerance to dysregulation due to predisposing genetic factors. In a recent Mendelian randomization study, physical activity was correlated with the altered expression of many genes, including C9orf72 and was inversely proportional to the onset age among patients with C9orf72 expansion.21 Exposure to cigarette smoking has been shown to cause heritable epigenetic changes resulting in altered gene expression and triggering abnormal inflammatory response.22,23
Table 1.
Crosstalk of genes in functional pathways impacted in ALS.
| Pathways | Genes | Molecular impact on disease |
|---|---|---|
| Protein homoeostasis | C9orf72, SQSTM1, TBK1, CCNF, FUS, TARDBP, OPTN, UBQLN2, VCP, CHMP2B, SOD1, VAPB, SIGMAR1 | Protein aggregation and accumulation due to mutation-induced protein misfolding and/or aberrant out-of-frame protein production hampered by impaired removal via autophagy or ubiquitin-proteosomal pathway. |
| RNA homeostasis | C9orf72, SQSTM1, TBK1, CCNF, FUS, TARDBP, ATXN2, SETX, ANG, hnRNPA1, MATR3 | Altered RNA splicing producing cyptic or aberrantly spliced transcripts and dysregulated transcriptome causing differrential and pathogenic expression of genes. Aggregation of abberant transcripts into RNA foci could act as a sink resulting in depletion of other transcripts resulting in further dysregulation of the transcriptome and proteome. These could have knock-on effects on other cellular processes. |
| Oxidative stress & inflammation | SQSTM1, TBK1, FUS, TARDBP, UBQLN2, VCP, SOD1 | Free radicals could induce abnormal modification of proteins causing it to aggregate and/or altering protein function, cause DNA and organelle damage. Inflammation induced by microglia could aggravate oxidative stress response. Persistent and uncontrolled oxidative stress response drives the formation of irreversible stress granules that can sequester RNA-binding proteins and accelerate pathogenic aggregation of proteins. |
| Cellular energetics | CHCHD10, TBK1, OPTN, VCP, SOD1 | Structural and functional mitochondrial abnormalities cause defective energy production, abnormal calcium handling, dynamics, and apoptotic signaling. Mitochondria can be damaged by reactive oxygen species, mitochondrial DNA damage, and impaired fission and fusion. |
| Cytoskeletal integrity & intracellular transport | C9orf72, SQSTM1, CCNF, FUS, TARDBP, UBQLN2, TUBA4A, SOD1, ALS2, SPG11, VAPB, FIG 4, SIGMAR1, PFN1, ANXA11, KIF5A | Intracellular trafficking of organelles within the soma and dendrites are important in maintaining a functional intracellular signaling and cross-talk between neurons at the synaptic junctions. Destabilized cytoskeletal scaffold, impaired machineries for axonal transport and endo-trafficking can affect homeostasis of other cellular processes potentially amplifying pathogenic processes already underway. |
Taken together, there is a theory of evidence that suggests that the risks conferred purely by genetics, especially by variants with minor effects operating in sporadic ALS, are not sufficient to cause disease. Instead, they require an external trigger to push the system into an irreversible cascade of cell death and loss. This theory could usefully explain some of the variability in penetrance observed from genetic studies.
Environmental factors
Given that the environmental exposure of an individual (the “exposome”24) is essentially infinite, it is perhaps not surprising that a plethora of environmental factors has been implicated in ALS pathogenesis.4,25,26 These hypotheses predominantly arose from anecdotal cases, patients' medical history, risk factors identified in other diseases, spatial epidemiology studies,27 and biological plausibility based on our incomplete knowledge of the molecular mechanisms involved in neurodegeneration.4 A common criticism of these suspects is that they arise from a “streetlight's effect,” reminiscent of Captain Louis Renault “rounding up the usual suspects” in Casablanca, rather than being data driven. Clearly, many environmental factors remain to be explored, and there is an unmet need to develop methods to investigate the exposome systematically.28
In contrast to genetic studies, evidence nominating an environmental factor is seldom successfully replicated. These discordant findings could be due to the patients’ different ancestral origins leading to differential susceptibility to that environmental factor. It could also be driven by environmental exposures being local, circumscribed events within a geographical area. More often, however, methodological differences account for most of this discordance. Selection bias could yield a false relationship or hide a true one. The use of prevalent cases in these studies is concerning, as detected relationships may be driven by survival rather than by risk. Confounders could differentially influence the results across the studies depending on their adjustment (known) and distribution (unknown). Reduced sample size could lead to a type II error, hiding genuine differences and exacerbating false-positive associations. Finally, measures chosen to assess an exposure could be differently susceptible to information bias (recall bias being the most common).
Compounding these study design effects, most analyses assume a linear model, but this may not be the case for all environmental factors. Looking at other diseases, the relation between alcohol consumption and acute coronary syndrome is J-shaped, for example.29 Additional complexity arises from the timing and duration of the exposure, circumstances that are rarely taken into account but could strongly influence the risk and vary across studies.4 An exposure that happened decades before disease onset (e.g., sports-related head injury as a teenager) will be more challenging to study than one that occurs immediately before onset (e.g., bone fracture). Finally, as mentioned above, differing phenotypes could underlie different etiologies.6,7,30 In conclusion, each epidemiological study collecting environmental study is likely closer to being an entirely new experiment rather than a replication of a previous observation.
It should also be considered that collecting and analyzing information across multiple environmental factors could provide insights into disease pathogenesis that would have been missed by considering single exposures. This reinforces the need to focus on the individual exposome, despite some issues that are difficult to overcome. Firstly, many tools are needed to assess the exposures, both through internal (-omics characterization) and external measurements. Furthermore, the collection of many variables will need analysis to employ appropriate dimensionality-reduction approaches. Finally, to characterize an individual's lifetime exposome would require either multiple measures overtime or a smaller number of measures known to be stable over extended periods.31,32 Registries could offer a convenient solution to the issue of collecting numerous, accurate, and sequential data on exposures.
Despite these caveats, several environmental factors have been robustly implicated in the pathogenesis of ALS, and we review the evidence for each here.
Cigarette smoking
At this stage, there is sufficient epidemiological and genetic data indicating that cigarette smoking is a bona fide risk factor for ALS.12,33,34 Studies showed an increased risk ranging from 1·4334 to two-fold for individuals who smoked compared to never-smokers.35 Compellingly, the risk was further increased when comparing current smokers to individuals who never smoked.35,36 Furthermore, most studies showed a pack-years dose-response relationship,34 although the relationship may rely more on the exposure duration than intensity.33,36 Some studies also showed that lowering the age of smoking initiation could further increase the risk.34 Finally, even the number of years since smoking cessation was associated with a decreased risk.33,36
This is not to say that every study has agreed with smoking as a risk factor. Negative and weak data for smoking as an ALS risk factor have also been published.37, 38, 39 A systematic review based on eighteen papers did not support an overall association between smoking and ALS, though it did show a significantly higher risk among women (RR 1·66).40
While the ease in assessing smoking exposure has aided in studying this risk factor in ALS, factors associated with a benign cardiovascular profile (physical activity) or occupation may have acted as confounders. However, a recent study showed a shared genetic risk between smoking and ALS,41 while a Mendelian randomization study suggested that confounders do not influence this relationship.42 These genetic data underscored the epidemiological data implicating cigarette smoking in ALS pathogenesis and provided an early example of the power of genomics to resolve such lifestyle factors.
How smoking increases the ALS risk remains unclear, though theories abound. A single cigarette smoke puff contains considerable quantities of oxidative molecules.43 Accordingly, one hypothesis is that smoking could overwhelm the antioxidant capacity of cells. Furthermore, some chemicals present in cigarette smoke (lead and formaldehyde) have a neurotoxic effect. Finally, smoking inhibits paraoxonase (PON), an enzyme able to reduce the damage from oxidative stress.40,44 This would explain the increased risk for women for whom oxidation mechanisms seem to be upregulated, leading to an increased level of intermediate oxidant metabolites.40 The change in the smoking habits among women during the last decades could have contributed to the increase of the ALS incidence reported among this subgroup.45
Physical activity
There are multiple lines of evidence pointing to excessive physical activity increasing ALS risk.4 For example, patients often highlighted their high fitness levels before disease onset, and there are anecdotal reports of prominent athletes, including the eponymous Lou Gehrig after whom the disease is named, developing ALS.4 Furthermore, patients usually show a favorable cardiovascular profile,46 which could be interpreted as an effect of engaging in physical activity. The relationship has been further fueled by the evidence of a six-fold higher ALS risk among former Italian professional soccer players47 and a four-fold higher mortality rate for ALS among what Americans refer to as Football compared to the general population.48 Finally, the hypothesis could be biologically plausible as strenuous exercise could place increased strain on the motor system necessary to undertake these activities, possibly through an oxidative stress mechanism.49,50
Similar to smoking, the evidence implicating physical activity in the etiology of ALS has been contradictory to date. Four systematic reviews did not find a relationship51,52 or reported inconclusive data.50,53 Even when considering physical activity types separately (occupational, leisure-time, varsity athleticism, professional sport), the reports remained conflicting.53 Moreover, it is unclear which level of exercise is required to drive ALS risk.54 A questionnaire-led, population-based, case-control study including 636 patients and 2166 controls showed no association for vigorous, occupational or cumulative physical activity. In contrast, leisure-time physical activity was significantly higher among ALS patients (OR 1·08, 95% CI 1·02 - 1·14, p = 0·008).55 The peculiar organization of the motor cortex could favour both the athletic prowess and the risk of developing ALS.4 Thus, performing physical activity (leisure-time) would be associated with ALS while occupational may not. A recent PET study may help unravel this conundrum, as it showed distinct changes in the brain metabolism of ALS patients who practiced sport compared to those who did not. Despite having the same level of disability, the former group showed a more diffuse metabolic change in the cerebellar, frontotemporal, and corticospinal tracts, suggesting that these patients could be coping better with the neurodegenerative process.56
Methodological issues may have contributed to the inconsistency of these results. The studies varied widely in sample size, and most of them were retrospective and prone to recall bias and confounders. Other confounders, such as smoking, cardiovascular comorbidities, body mass index (BMI) and hypercholesterolemia,57 were inconsistently accounted for across studies. There is even no consensus on how to quantify the intensity of physical activity, with various methods being deployed, including questionnaires, information on previous occupations, Metabolic Equivalents, Compendium of Physical Activities, or frequency of sweating.53 Also, it is unclear why other types of professional athletes apart from soccer and football players are not at risk.58 Even among soccer players, only some positions, such as midfielder, showed a higher risk for ALS.58 Though initially greeted with skepticism,59 these data have been borne out.
It has been suggested that the increased risk among soccer and football players could be mediated by exposure to pesticides from the field, head traumas, or the use of performance-enhancing drugs.60 However, genomics seems to suggest that physical activity is indeed involved. A previous study using a linkage disequilibrium score regression showed that strenuous physical activity might contribute to ALS risk. Notably, and in keeping with some aspects of the available epidemiological data, light physical activity was protective against the risk of developing ALS.41 Moreover, a recent Mendelian randomization study found that physical activity is directly linked to the ALS risk, suggesting a causal relationship.21
Dyslipidemia
Genetic data have been quite conclusive in asserting that hyperlipidemia is an ALS risk factor. A recent Mendelian randomization study ruled out the role of confounders and reverse causality by showing that increased plasma LDL cholesterol is directly correlated to the risk of developing ALS.41 The finding was confirmed separately in European and Asian populations, adding further weight to this risk factor.61 As a consequence, it has been postulated that lowering blood cholesterol levels could reduce the ALS risk.
In contrast to the genetic data, previous epidemiological data reported conflicting results. While some studies showed higher levels of total cholesterol, LDL and LDL/HDL ratio62, 63, 64 and HDL,63 others found no differences.65,66 Some studies even reported an opposite ratio.67,68 A recent meta-analysis involving 3291 ALS patients and 3367 controls showed no pooled differences in any of the lipid markers considered except for lower levels of total cholesterol among Asian patients.69 Similar results came from a meta-analysis involving 1930 cases and 3635 controls which did not find significant differences for lipid markers, even when considering Caucasian and Asian ethnicities separately.66 Against this, the most extensive study to date analyzing nearly 700 drugs based on Medicare data that included 10,450 ALS cases and 104,500 controls showed that a broad range of antihyperlipidemic drugs with different mechanisms of action were associated with a lower ALS risk. These data strongly point to hyperlipidemia influencing ALS risk, rather than a secondary effect from any particular drug class.70
However, there are several reasons to trust genetic over retrospective epidemiological studies in this case. First, the lipid profile is part of a more global cardiovascular profile, including BMI,71,72 glycaemia,73 cardiovascular comorbidities, and cigarette smoking. Each of these factors could have acted as a confounder in the retrospective epidemiological studies. Second, ALS is characterized by a hypermetabolic state,74 and abnormal lipid profile observed after patients develop symptoms could merely reflect secondary changes arising from the degenerative process. Such reverse causality could affect epidemiological studies but not properly designed genetic studies. Finally, the epidemiological studies typically employed sample sizes that were several folds smaller than those utilized in genome-wide studies and Mendelian randomization analyses.
Other environmental factors
Many other environmental factors have been proposed as influencing the risk of developing ALS, including heavy metals,75 pesticides, electromagnetic fields,76,77 occupations,78 drugs,79 and cyanotoxins.80 Evidence for these factors has been weaker; however, they could be involved in less common pathogenetic mechanisms (see below) and should not be overlooked.
Climate
The effect of climate change on the environment must be carefully considered. Earth is racing towards at least a 1·5 °C temperature increase over the following decades, resulting in myriad ecological and geopolitical changes. The resultant environmental upheavals will have lasting effects on public health concerning food security and water access, the geographical distribution of vectors responsible for zoonotic infections, exposures to industrial pollutants, and even the pattern of renal, cardiovascular and pulmonary conditions.81 For example, the environmental conditions conducive for of malaria transmission will extend over many areas of the United States, South America, Europe, and Australia by the end of the century.82
Neurological disorders should also be included in that rubric, given the relative importance of the local environment in their pathogenesis. Much importance has been placed on the butterfly effect, whereby minor, locally defined events have outsized effects on distant environs. However, we must not forget that the opposite is also true: planetary climate change due to an excessive release of greenhouse gases into the atmosphere will alter local air pollution patterns, a risk factor for many neurodegenerative diseases, including ALS. Shifting industrial bases due to food scarcity and economic forces arising from climate changes could compound these environmental disruptions. Humidity will increase because of the higher rate of water evaporation from the earth's surface. A previous study showed that the birth seasonality among four ALS cohorts across the two hemispheres correlated with humidity rates during the year. Presciently, the authors argued in 2012 that this aspect could be influenced by an imminent climate change.83 Finally, a climate temperature increase could directly or indirectly lead to increased oxidative stress, changes in cerebrovascular hemodynamics, excitotoxicity, and microglial activation, all implicated in neurological conditions, such as ALS.84
The effect of climate changes may not just be isolated to the environmental aspect of gene-environment interactions. Population displacement due to climate change could cause the exposure of immigrants with certain genetic factors to new external agents to which they had not been previously exposed. Such hazardous materials, neurotoxins and infections could adversely affect their nervous system.85,86 While the local population has evolved a tolerance level to their local environment, migrants to the area may lack the same protection, making them more susceptible.
Such considerations are speculative and unexplored at present. However, the 2021 United Nations Change Conference (COP26) highlighted the potentially devasting effects of climate change on the environment. Consequently, it is worth keeping this hypothetical scenario in mind as we could be facing it soon.
Gene-environment interactions
A fundamental conundrum in studying environmental and lifestyle exposures is that they tend to be common in the community (e.g., smoking, exercise, and hyperlipidemia), and yet ALS is ostensibly rare. Gene-environment interactions offer a possible explanation for this and may be particularly relevant when considering rare, sporadic diseases that occur in later life. Under this model, not every person exposed to an environmental factor would develop the disease. Instead, only those individuals with an underlying genetic predisposition would be at risk. Environmental factors alone or genetic factors alone would be insufficient to lead to motor neuron degeneration. Instead, the interactions between these environmental and genetic factors drive the issue.87 While such interactions could be rare in the general population, they could explain many cases in the ALS subpopulation. In the absence of a deeper understanding of this mechanism, the sporadic occurrence of the disease in the community would look stochastic. In reality, it hints at a much more complex pathogenic system.
Various methodologies have been developed to model gene-environment interactions and are described elsewhere.88, 89, 90, 91, 92, 93, 94, 95 The most straightforward approach would be examining factors already known to be associated with ALS. For example, the risk exerted by an environmental exposure associated with ALS could vary according to the presence or not of one of the genetic variants listed in Table 1.89,90 Epidemiological studies could explore this hypothesis using stratified analysis in which the risk of developing the disease is calculated across the possible combinations of the two factors. If the risk among individuals doubly exposed is greater than the product of the risk given by the two factors individually, then a multiplicative interaction can be inferred.89, 90, 91,95 For example, both asbestos and cigarette smoking increase the risk of developing lung cancer, but those exposed to both have a risk that far exceeds either alone.96 In another example, estrogen use was globally protective against the risk of Alzheimer's Disease, but the risk was only truly decreased among ε4-negative women (HR = 0·59; 95% CI, 0·36–0·99).97
The gene-environment interactions may also exist on an additive scale.95 In this paradigm, the joint exposure risk would differ from the sum of the single exposures risks.89, 90, 91,95 It is still debated whether the multiplicative or additive measure more truly reflects the underlying biological interactions, and each may be valid depending on circumstances.95 For example, if the etiology involves multiple steps (as is postulated for ALS98,99), two factors acting at the same stage will fit an additive model. In contrast, factors acting at different stages will fit a multiplicative hypothesis.89 If both exposures are needed for the disease to develop, interaction on an additive scale should be searched.95,100 However, interactions on a multiplicative scale have been more widely used, perhaps for the practical reason that they are prominent in magnitude and easier to detect.95
Our discussion so far has been predicated on the notion that each interacting factor is associated with ALS on its own. However, there are many complex interactions in nature where this is not the case. For example, individuals with glucose-6-phosphate dehydrogenase deficiency are usually asymptomatic as the genetic predisposition exerts no effect on its own. However, these patients develop fever, dark urine, and abdominal pain when they consume fava beans.90 The LIPC gene was associated with high cholesterol levels only after adding dietary fat to the model.101 A polymorphism of the serotonin transporter gene (SLC6A4) promoter moderates the influence of stressful life events on depression while not exerting an effect in the absence of stressors.102 Many other examples come from studies assessing the risk of asthma, lung cancer, type 2 diabetes, and heart diseases.88 These types of interaction are much more difficult to detect, particularly in a rare disease such as ALS.
Genetics could also predispose to an environmental exposure which in turn causes the disease. For example, an individual could be predisposed to commencing cigarette smoking, which will increase the ALS risk. This is not a gene-environment interaction in the proper sense but rather a covariance (or gene-environment correlation). This covariance could also act the other way around, such as the case of older paternal age increasing the risk of mutagenesis103 or somatic mutations accumulating within the brain as the person ages.104 This latter mechanism may be an elegant explanation for the apparent discrepancy between the low heritability estimates and our failure to date to identify significant environmental factors underlying ALS.
Nevertheless, such possibilities should be carefully considered as they could reflect the underlying biology or act as a noise obscuring other findings.105 Gene-environment interactions could also explain the phenotypic variability among siblings carrying the same genetic mendelian mutation. Additional genetic variants or environmental exposures could determine a different age of onset106 or a different disease,107 or even prevent the disease from happening (reduced penetrance) .90
To date, there have been a limited number of studies examining gene-environment interactions in ALS. A study involving 143 ALS patients and an equal number of controls found that genetic variants in the PON1 gene increased the risk of ALS among individuals exposed to pesticides.108 A second study explored the interaction of the same gene using population density as a proxy of environmental exposure to pesticides. The authors found a significant interaction between density population and a marker rs854560 (L55M) at the genotypic level.109 However, it is not easy to draw meaningful conclusions from these publications. Instead, these studies highlight the difficulties in designing studies to examine gene-environment interactions studies in ALS.
First, there is a paucity of accurately measured data for both genetic and environmental factors. While genome sequencing has become progressively more affordable,110 the same cannot be said for the exposome, where only limited data are available.24 More recently, spatial data analysis has emerged to infer an environmental effect.6,27,111 Furthermore, metabolomics and epigenetics are increasingly used as proxies of environmental exposures. Particularly in the case of metabolomics, it can be challenging to distinguish an environmental effect precipitating the disease from a secondary effect of the disease itself.28 On the other hand, epigenetics could offer an extra layer to consider when seeking the effect of environmental factors on genetics. Such information would complement the genomic sequence information; environmental factors can alter gene expression through epigenetic modifications (such as DNA methylation, histone modifications, chromatin remodeling) and each can be quantified with appropriate methods.112 Nevertheless, the nature of quantifiable measures continues to improve and may offer an opportunity to understand gene-environment interactions in ALS more completely.
As a second major issue, many samples are required to study ALS. This is due to its rarity, the high number of exposure variables that need to be considered, the low frequency of genetic variants involved, and the minimal effect that the various factors are expected to have. A priori knowledge could partially reduce the sample size needed; data collection could be limited to environmental data known to act on pathways involved in the disease pathogenesis. However, this hypothesis-based approach limits the discovery of new variants and environmental exposures.88 As an alternative strategy, a polygenic risk score could be used to measure genetic predisposition. A recent study investigated the interaction between genetic susceptibility, as assessed by the weighted sum of previously associated loci, and the exposure to household coal for the risk of developing lung cancer among Asian females. Using two different cohorts, the authors found that the risk given by the genetic predisposition was weaker among coal ever-users than never-users, suggesting a multiplicative interaction among these two factors.113
However, detecting an effect becomes more complicated if the timing of the exposure is relevant. Under that model, environmental pathogens would influence the ALS risk only during sensitive periods or if the exposure acts for a sufficient length of time.4,28,114 This possibility could explain why ALS risk increases with age reaching its peak around the seventh decade of life. A cumulative effect of environmental exposures could be necessary for the disease to develop, along with structural and functional changes linked to the aging of motor neurons.115 Being the third variable in the ALS equation, time likely interacts with the genetic predisposition, influencing the age of onset.4
Finally, here we focused on gene-environment interactions, but interactions could also occur between two (or more) environmental exposures or genetic variants (epistasis).88 Recent and more sophisticated methods to explore large-scale gene-environment interactions have been developed and could help solve the riddle of dealing with many variables.116,117
Conclusions
Gene and environmental factors could be variably involved in ALS pathogenesis. Exploring their interactions will likely give some critical insights. So far, the field has dealt chiefly with simplistic etiological models of ALS in which nature and nurture were considered as a dichotomy.28 Furthermore, environmental factors are typically studied at diagnosis, whereas their window of action could precede the onset by many years. While simple is not necessarily wrong, studying one factor at a time has potentially hidden effects that will become apparent when multiple factors are considered simultaneously. Knowledge of gene-environment interactions would allow the identification of susceptible individuals who could then be targeted for specific interventions. For example, they could be advised to avoid the culprit exposure during their lives. This precision medicine approach for ALS could lessen the risk of developing the disease or delay onset by decades.28
Outstanding questions
Many questions must be addressed as we travel along this Road to Utopia. Which exposures and genetic variants make the most sense to examine early on? How should we make our selection? While such a candidate-based approach would dramatically lower the need for data, it would hamper discovering new genetic and environmental factors hidden due to our limited knowledge of biological processes. The next question follows from this observation: is it feasible to study gene-environment interactions over many exposures in a rare disease such as ALS? The era of big data, along with the easy-to-access data repositories, offers us perhaps the first real glimmer of hope to undertake such a study. Recently published algorithms can search for interactions on a genome-wide and exposome basis even in the setting of limited sample sizes and power.116,117 Machine Learning techniques may reveal unexpected data-driven correlations and prioritize a smaller number of factors for study.119, 120, 121 However, in the end, the data lake will likely need to expand in size dramatically. A concerted effort is required to make environmental and genome-wide data from large population-based settings publicly available.28
Search strategy and selection criteria
Data for the review were identified through searches from PubMed with the following search terms: “Amyotrophic Lateral Sclerosis”, “ALS”, “environment*”, “genetic*”, “interaction*”. Only articles in English with published up to August 2021 were included; references of the collected published studies were also considered. Selection of the most appropriate references was made by the authors.
Declaration of Competing Interest
Rosario Vasta and Ruth Chia declare no conflicts of interests.
Bryan J. Traynor holds the US, Canadian and European patents on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion in C9orf72. He is an associate editor of Brain and sits on the editorial boards of Neurobiology of Aging, the Journal of Neurology, Neurosurgery, and Psychiatry, and EClinicalMedicine.
Adriano Chiò serves on the editorial advisory board of Amyotrophic Lateral Sclerosis and Neurological Sciences and has received research support from the Italian Ministry of Health (Ricerca Finalizzata), Regione Piemonte (Ricerca Finalizzata), University of Turin and the European Commission (Health Seventh Framework Programme) and serves on scientific advisory boards for Mitsubishi Tanabe, Roche, Denali Pharma, Cytokinetics, Biogen, and Amylyx and has a research contract with Biogen.
Acknowledgments
Contributors
RV and RC wrote the manuscript and designed and produced the figure and the table. BJT and AC outlined, supervised, and edited the draft and approved of the final version to be submitted. All authors approved the final version of the manuscript.
Acknowledgments
This work was supported by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016–02362405); the Progetti di Rilevante Interesse Nazionale program of the Ministry of Education, University and Research (grant 2017SNW5MB); the European Commission's Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867); and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy. The Susa and Val di Susa Rotary club also contributed. This work was also supported by the Intramural Research Program of the NIH, National Institute on Aging (Z01-AG000949–02), and by the National Institute of Neurologic Disorders and Stroke (1ZIANS003154). The funders had no role in data collection or analysis and did not participate in writing or approving the manuscript.
References
- 1.Vasta R., Moglia C., Manera U., Canosa A., Grassano M., Palumbo F., et al. What is amyotrophic lateral sclerosis prevalence? Amyotroph Lateral Scler Frontotemporal Degener. 2021:1–6. doi: 10.1080/21678421.2021.1936557. [DOI] [PubMed] [Google Scholar]
- 2.Chiò A., Logroscino G., Traynor B.J., Collins J., Simeone J.C., Goldstein L.A., et al. Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology. 2013;41:118–130. doi: 10.1159/000351153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardiman O., Al-Chalabi A., Chio A., Corr E.M., Logroscino G., Robberecht W., et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 4.Al-Chalabi A., Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 5.Chiò A., Calvo A., Moglia C., Mazzini L., Mora G., PARALS study group Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatr. 2011;82:740–746. doi: 10.1136/jnnp.2010.235952. [DOI] [PubMed] [Google Scholar]
- 6.Vasta R., Canosa A., Manera U., Di Pede F., Cabras S., De Marchi F., et al. Do ecological factors influence the clinical presentation of amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2021 doi: 10.1136/jnnp-2020-325625. [DOI] [PubMed] [Google Scholar]
- 7.Chiò A., Moglia C., Canosa A., Manera U., D'Ovidio F., Vasta R., et al. ALS phenotype is influenced by age, sex, and genetics: a population-based study. Neurology. 2020;94:e802–e810. doi: 10.1212/WNL.0000000000008869. [DOI] [PubMed] [Google Scholar]
- 8.Davies N.M., Holmes M.V., Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi: 10.1136/bmj.k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bandres-Ciga S., Noyce A.J., Traynor B.J. Mendelian randomization-A journey from obscurity to center stage with a few potholes along the way. JAMA Neurol. 2019 doi: 10.1001/jamaneurol.2019.3419. [DOI] [PubMed] [Google Scholar]
- 10.Smith G.D., Ebrahim S. “Mendelian randomization”: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 11.Julian T.H., Boddy S., Islam M., Kurz J., Whittaker K.J., Moll T., et al. A review of Mendelian randomization in amyotrophic lateral sclerosis. Brain. 2021 doi: 10.1093/brain/awab420. awab420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armon C. Smoking is a cause of ALS. High LDL-cholesterol levels? Unsure. Ann Neurol. 2019 doi: 10.1002/ana.25469. [DOI] [PubMed] [Google Scholar]
- 13.Graham A.J., Macdonald A.M., Hawkes C.H. British motor neuron disease twin study. J Neurol Neurosurg Psychiatry. 1997;62:562–569. doi: 10.1136/jnnp.62.6.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Chalabi A., Fang F., Hanby M.F., Leigh P.N., Shaw C.E., Ye W., et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry. 2010;81:1324–1326. doi: 10.1136/jnnp.2010.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wingo T.S., Cutler D.J., Yarab N., Kelly C.M., Glass J.D. The heritability of amyotrophic lateral sclerosis in a clinically ascertained United States research registry. PLoS ONE. 2011;6:e27985. doi: 10.1371/journal.pone.0027985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan M., Heverin M., McLaughlin R.L., Hardiman O. Lifetime risk and heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2019;76:1367. doi: 10.1001/jamaneurol.2019.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller M.F., Ferrucci L., Singleton A.B., Tienari P.J., Laaksovirta H., Restagno G., et al. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol. 2014;71:1123. doi: 10.1001/jamaneurol.2014.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ranganathan R., Haque S., Coley K., Shepheard S., Cooper-Knock J., Kirby J. Multifaceted genes in amyotrophic lateral sclerosis-frontotemporal dementia. Front Neurosci. 2020;14:684. doi: 10.3389/fnins.2020.00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chia R., Chiò A., Traynor B.J. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17:94–102. doi: 10.1016/S1474-4422(17)30401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachmann M.C., Bellalta S., Basoalto R., Gómez-Valenzuela F., Jalil Y., Lépez M., et al. The challenge by multiple environmental and biological factors induce inflammation in aging: their role in the promotion of chronic disease. Front Immunol. 2020;11 doi: 10.3389/fimmu.2020.570083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Julian T.H., Glascow N., Barry A.D.F., Moll T., Harvey C., Klimentidis Y.C., et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine. 2021;68 doi: 10.1016/j.ebiom.2021.103397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corley J., Cox S.R., Harris S.E., Hernandez M.V., Maniega S.M., Bastin M.E., et al. Epigenetic signatures of smoking associate with cognitive function, brain structure, and mental and physical health outcomes in the Lothian Birth Cohort 1936. Transl Psychiatry. 2019;9:248. doi: 10.1038/s41398-019-0576-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zong D., Liu X., Li J., Ouyang R., Chen P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenetics Chromatin. 2019;12:65. doi: 10.1186/s13072-019-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wild C.P. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14:1847–1850. doi: 10.1158/1055-9965.EPI-05-0456. [DOI] [PubMed] [Google Scholar]
- 25.Ingre C., Roos P.M., Piehl F., Kamel F., Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol. 2015;7:181–193. doi: 10.2147/CLEP.S37505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oskarsson B., Horton D.K., Mitsumoto H. Potential environmental factors in amyotrophic lateral sclerosis. Neurol Clin. 2015;33:877–888. doi: 10.1016/j.ncl.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spencer P.S., Lagrange E., Camu W. ALS and environment: clues from spatial clustering? Rev Neurol (Paris) 2019;175:652–663. doi: 10.1016/j.neurol.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Traynor B.J., Singleton A.B. Nature versus nurture: death of a dogma, and the road ahead. Neuron. 2010;68:196–200. doi: 10.1016/j.neuron.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pitsavos C., Makrilakis K., Panagiotakos D.B., Chrysohoou C., Ioannidis I., Dimosthenopoulos C., et al. The J-shape effect of alcohol intake on the risk of developing acute coronary syndromes in diabetic subjects: the CARDIO2000 II study. Diabet Med. 2005;22:243–248. doi: 10.1111/j.1464-5491.2004.01384.x. [DOI] [PubMed] [Google Scholar]
- 30.Rosenfeld J., Swash M. What's in a name? Lumping or splitting ALS, PLS, PMA, and the other motor neuron diseases. Neurology. 2006;66:624–625. doi: 10.1212/01.wnl.0000205597.62054.db. [DOI] [PubMed] [Google Scholar]
- 31.Niedzwiecki M.M., Walker D.I., Vermeulen R., Chadeau-Hyam M., Jones D.P., Miller G.W. The exposome: molecules to populations. Annu Rev Pharmacol Toxicol. 2019;59:107–127. doi: 10.1146/annurev-pharmtox-010818-021315. [DOI] [PubMed] [Google Scholar]
- 32.Wild C.P. The exposome: from concept to utility. Int J Epidemiol. 2012;41:24–32. doi: 10.1093/ije/dyr236. [DOI] [PubMed] [Google Scholar]
- 33.Peters S., Visser A.E., D'Ovidio F., Vlaanderen J., Portengen L., Beghi E., et al. Effect modification of the association between total cigarette smoking and ALS risk by intensity, duration and time-since-quitting: Euro-MOTOR. J Neurol Neurosurg Psychiatry. 2019 doi: 10.1136/jnnp-2019-320986. [DOI] [PubMed] [Google Scholar]
- 34.Wang H., O'Reilly É.J., Weisskopf M.G., Logroscino G., McCullough M.L., Thun M.J., et al. Smoking and risk of amyotrophic lateral sclerosis: a pooled analysis of 5 prospective cohorts. Arch Neurol. 2011;68:207–213. doi: 10.1001/archneurol.2010.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson L.M., McGuire V., Longstreth W.T., Matkin C. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. I. Cigarette smoking and alcohol consumption. Am J Epidemiol. 2000;151:156–163. doi: 10.1093/oxfordjournals.aje.a010183. [DOI] [PubMed] [Google Scholar]
- 36.Gallo V., Bueno-De-Mesquita H.B., Vermeulen R., Andersen P.M., Kyrozis A., Linseisen J., et al. Smoking and risk for amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann Neurol. 2009;65:378–385. doi: 10.1002/ana.21653. [DOI] [PubMed] [Google Scholar]
- 37.Weisskopf M.G., McCullough M.L., Calle E.E., Thun M.J., Cudkowicz M., Ascherio A. Prospective study of cigarette smoking and amyotrophic lateral sclerosis. Am J Epidemiol. 2004;160:26–33. doi: 10.1093/aje/kwh179. [DOI] [PubMed] [Google Scholar]
- 38.Fang F., Bellocco R., Hernán M.A., Ye W. Smoking, snuff dipping and the risk of amyotrophic lateral sclerosis–a prospective cohort study. Neuroepidemiology. 2006;27:217–221. doi: 10.1159/000096956. [DOI] [PubMed] [Google Scholar]
- 39.Opie-Martin S., Jones A., Iacoangeli A., Al-Khleifat A., Oumar M., Shaw P.J., et al. UK case control study of smoking and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21:222–227. doi: 10.1080/21678421.2019.1706580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alonso A., Logroscino G., Hernán M.A. Smoking and the risk of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2010;81:1249–1252. doi: 10.1136/jnnp.2009.180232. [DOI] [PubMed] [Google Scholar]
- 41.Bandres-Ciga S., Noyce A.J., Hemani G., Nicolas A., Calvo A., Mora G., et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann Neurol. 2019;85:470–481. doi: 10.1002/ana.25431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhan Y., Fang F. Smoking and amyotrophic lateral sclerosis: a Mendelian randomization study. Ann Neurol. 2019;85:482–484. doi: 10.1002/ana.25443. [DOI] [PubMed] [Google Scholar]
- 43.Yanbaeva D.G., Dentener M.A., Creutzberg E.C., Wesseling G., Wouters E.F.M. Systemic effects of smoking. Chest. 2007;131:1557–1566. doi: 10.1378/chest.06-2179. [DOI] [PubMed] [Google Scholar]
- 44.Weisskopf M.G., Ascherio A. Cigarettes and amyotrophic lateral sclerosis: only smoke or also fire? Ann Neurol. 2009;65:361–362. doi: 10.1002/ana.21700. [DOI] [PubMed] [Google Scholar]
- 45.Chiò A., Mora G., Moglia C., Manera U., Canosa A., Cammarosano S., et al. Secular trends of amyotrophic lateral sclerosis: the Piemonte and Valle d'Aosta register. JAMA Neurol. 2017 doi: 10.1001/jamaneurol.2017.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turner M.R., Wotton C., Talbot K., Goldacre M.J. Cardiovascular fitness as a risk factor for amyotrophic lateral sclerosis: indirect evidence from record linkage study. J Neurol Neurosurg Psychiatry. 2012;83:395–398. doi: 10.1136/jnnp-2011-301161. [DOI] [PubMed] [Google Scholar]
- 47.Chiò A., Benzi G., Dossena M., Mutani R., Mora G. Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain. 2005;128:472–476. doi: 10.1093/brain/awh373. [DOI] [PubMed] [Google Scholar]
- 48.Lehman E.J., Hein M.J., Baron S.L., Gersic C.M. Neurodegenerative causes of death among retired National Football League players. Neurology. 2012;79:1970–1974. doi: 10.1212/WNL.0b013e31826daf50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bastos A.F., Orsini M., Machado D., Mello M.P., Nader S., Silva J.G., et al. Amyotrophic lateral sclerosis: one or multiple causes? Neurol Int. 2011;3:e4. doi: 10.4081/ni.2011.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harwood C.A., McDermott C.J., Shaw P.J. Physical activity as an exogenous risk factor in motor neuron disease (MND): a review of the evidence. Amyotroph Lateral Scler. 2009;10:191–204. doi: 10.1080/17482960802549739. [DOI] [PubMed] [Google Scholar]
- 51.Hamidou B., Couratier P., Besançon C., Nicol M., Preux P.M., Marin B. Epidemiological evidence that physical activity is not a risk factor for ALS. Eur J Epidemiol. 2014;29:459–475. doi: 10.1007/s10654-014-9923-2. [DOI] [PubMed] [Google Scholar]
- 52.Armon C. Sports and trauma in amyotrophic lateral sclerosis revisited. J Neurol Sci. 2007;262:45–53. doi: 10.1016/j.jns.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 53.Lacorte E., Ferrigno L., Leoncini E., Corbo M., Boccia S., Vanacore N. Physical activity, and physical activity related to sports, leisure and occupational activity as risk factors for ALS: a systematic review. Neurosci Biobehav Rev. 2016;66:61–79. doi: 10.1016/j.neubiorev.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 54.Wang M.D., Little J., Gomes J., Cashman N.R., Krewski D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology. 2016 doi: 10.1016/j.neuro.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 55.Huisman M.H.B., Seelen M., de Jong S.W., Dorresteijn K.R.I.S., van Doormaal P.T.C., van der Kooi A.J., et al. Lifetime physical activity and the risk of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2013;84:976–981. doi: 10.1136/jnnp-2012-304724. [DOI] [PubMed] [Google Scholar]
- 56.Canosa A., D'Ovidio F., Calvo A., Moglia C., Manera U., Torrieri M.C., et al. Lifetime sport practice and brain metabolism in Amyotrophic Lateral Sclerosis. Neuroimage Clin. 2020;27 doi: 10.1016/j.nicl.2020.102312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turner M.R. Increased premorbid physical activity and amyotrophic lateral sclerosis: born to run rather than run to death, or a seductive myth? J Neurol Neurosurg Psychiatry. 2013;84:947. doi: 10.1136/jnnp-2013-304935. [DOI] [PubMed] [Google Scholar]
- 58.Chio A., Calvo A., Dossena M., Ghiglione P., Mutani R., Mora G. ALS in Italian professional soccer players: the risk is still present and could be soccer-specific. Amyotroph Lateral Scler. 2009;10:205–209. doi: 10.1080/17482960902721634. [DOI] [PubMed] [Google Scholar]
- 59.Al-Chalabi A., Leigh P.N. Trouble on the pitch: are professional football players at increased risk of developing amyotrophic lateral sclerosis? Brain. 2005;128:451–453. doi: 10.1093/brain/awh426. [DOI] [PubMed] [Google Scholar]
- 60.Vanacore N. Neurodegenerative causes of death among retired National Football League players. Neurology. 2013;80:1266–1267. doi: 10.1212/01.wnl.0000428873.10254.b7. [DOI] [PubMed] [Google Scholar]
- 61.Zeng P., Zhou X. Causal effects of blood lipids on amyotrophic lateral sclerosis: a Mendelian randomization study. Hum Mol Genet. 2019;28:688–697. doi: 10.1093/hmg/ddy384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dupuis L., Corcia P., Fergani A., Gonzalez De Aguilar J.-.L., Bonnefont-Rousselot D., Bittar R., et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70:1004–1009. doi: 10.1212/01.wnl.0000285080.70324.27. [DOI] [PubMed] [Google Scholar]
- 63.Delaye J.B., Patin F., Piver E., Bruno C., Vasse M., Vourc'h P., et al. Low IDL-B and high LDL-1 subfraction levels in serum of ALS patients. J Neurol Sci. 2017;380:124–127. doi: 10.1016/j.jns.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 64.Chełstowska B., Barańczyk-Kuźma A., Kuźma-Kozakiewicz M. Dyslipidemia in patients with amyotrophic lateral sclerosis - a case control retrospective study. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:195–205. doi: 10.1080/21678421.2020.1832119. [DOI] [PubMed] [Google Scholar]
- 65.Chiò A., Calvo A., Ilardi A., Cavallo E., Moglia C., Mutani R., et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology. 2009;73:1681–1685. doi: 10.1212/WNL.0b013e3181c1df1e. [DOI] [PubMed] [Google Scholar]
- 66.Huang R., Guo X., Chen X., Zheng Z., Wei Q., Cao B., et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: a study from south-west China and a meta-analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:359–365. doi: 10.3109/21678421.2015.1047454. [DOI] [PubMed] [Google Scholar]
- 67.Chen X., Wei Q.Q., Chen Y., Cao B., Ou R., Hou Y., et al. Clinical disease stage related changes of serological factors in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:53–60. doi: 10.1080/21678421.2018.1550516. [DOI] [PubMed] [Google Scholar]
- 68.Sutedja N.A., van der Schouw Y.T., Fischer K., Sizoo E.M., Huisman M.H.B., Veldink J.H., et al. Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82:638–642. doi: 10.1136/jnnp.2010.236752. [DOI] [PubMed] [Google Scholar]
- 69.Liu J., Luo X., Chen X., Shang H. Lipid profile in patients with amyotrophic lateral sclerosis: a systematic review and meta-analysis. Front Neurol. 2020;11 doi: 10.3389/fneur.2020.567753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pfeiffer R.M., Mayer B., Kuncl R.W., Check D.P., Cahoon E.K., Rivera D.R., et al. Identifying potential targets for prevention and treatment of amyotrophic lateral sclerosis based on a screen of medicare prescription drugs. Amyotroph Lateral Scler Frontotemporal Degener. 2019:1–11. doi: 10.1080/21678421.2019.1682613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moglia C., Calvo A., Grassano M., Canosa A., Manera U., D'Ovidio F., et al. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry. 2019;90:666–673. doi: 10.1136/jnnp-2018-319611. [DOI] [PubMed] [Google Scholar]
- 72.Paganoni S., Deng J., Jaffa M., Cudkowicz M.E., Wills A.-.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44:20–24. doi: 10.1002/mus.22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vasta R., D'Ovidio F., Logroscino G., Chiò A. The links between diabetes mellitus and amyotrophic lateral sclerosis. Neurol Sci. 2021;42:1377–1387. doi: 10.1007/s10072-021-05099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dupuis L., Pradat P.-.F., Ludolph A.C., Loeffler J.-.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011;10:75–82. doi: 10.1016/S1474-4422(10)70224-6. [DOI] [PubMed] [Google Scholar]
- 75.Cicero C.E., Mostile G., Vasta R., Rapisarda V., Signorelli S.S., Ferrante M., et al. Metals and neurodegenerative diseases. A systematic review. Environ Res. 2017;159:82–94. doi: 10.1016/j.envres.2017.07.048. [DOI] [PubMed] [Google Scholar]
- 76.Filippini T., Hatch E.E., Vinceti M. Residential exposure to electromagnetic fields and risk of amyotrophic lateral sclerosis: a dose-response meta-analysis. Sci Rep. 2021;11:11939. doi: 10.1038/s41598-021-91349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luna J., Leleu J.-.P., Preux P.-.M., Corcia P., Couratier P., Marin B., et al. Residential exposure to ultra high frequency electromagnetic fields emitted by Global System for Mobile (GSM) antennas and amyotrophic lateral sclerosis incidence: a geo-epidemiological population-based study. Environ Res. 2019;176 doi: 10.1016/j.envres.2019.108525. [DOI] [PubMed] [Google Scholar]
- 78.D'Ovidio F., d'Errico A., Calvo A., Costa G., Chiò A. Occupations and amyotrophic lateral sclerosis: are jobs exposed to the general public at higher risk? Eur J Public Health. 2017;27:643–647. doi: 10.1093/eurpub/ckx006. [DOI] [PubMed] [Google Scholar]
- 79.D'Ovidio F., d'Errico A., Farina E., Calvo A., Costa G., Chiò A. Amyotrophic lateral sclerosis incidence and previous prescriptions of drugs for the nervous system. Neuroepidemiology. 2016;47:59–66. doi: 10.1159/000448618. [DOI] [PubMed] [Google Scholar]
- 80.Bradley W.G., Mash D.C. Beyond Guam: the cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph Lateral Scler. 2009;10(Suppl 2):7–20. doi: 10.3109/17482960903286009. [DOI] [PubMed] [Google Scholar]
- 81.Atwoli L., Baqui A.H., Benfield T., Bosurgi R., Godlee F., Hancocks S., et al. Call for emergency action to limit global temperature increases, restore biodiversity, and protect health: wealthy nations must do much more, much faster. Neurology. 2021 doi: 10.1212/WNL.0000000000012691. [DOI] [PubMed] [Google Scholar]
- 82.Colón-González F.J., Sewe M.O., Tompkins A.M., Sjödin H., Casallas A., Rocklöv J., et al. Projecting the risk of mosquito-borne diseases in a warmer and more populated world: a multi-model, multi-scenario intercomparison modelling study. Lancet Planet Health. 2021;5:e404–e414. doi: 10.1016/S2542-5196(21)00132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pamphlett R., Fang F. Season and weather patterns at time of birth in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(5):459–464. doi: 10.3109/17482968.2012.700938. [DOI] [PubMed] [Google Scholar]
- 84.Zammit C., Torzhenskaya N., Ozarkar P.D., Calleja Agius J. Neurological disorders vis-à-vis climate change. Early Hum Dev. 2021;155 doi: 10.1016/j.earlhumdev.2020.105217. [DOI] [PubMed] [Google Scholar]
- 85.Merino J.G. Climate change. Neurology. 2021;97:657. doi: 10.1212/WNL.0000000000012692. –657. [DOI] [PubMed] [Google Scholar]
- 86.McMichael C. Human mobility, climate change, and health: unpacking the connections. Lancet Planet Health. 2020;4:e217–e218. doi: 10.1016/S2542-5196(20)30125-X. [DOI] [PubMed] [Google Scholar]
- 87.Rothman K.J. Causes. Am J Epidemiol. 1976;104:587–592. doi: 10.1093/oxfordjournals.aje.a112335. [DOI] [PubMed] [Google Scholar]
- 88.Moffitt T.E., Caspi A., Rutter M. Measured gene-environment interactions in psychopathology: concepts, research strategies, and implications for research, intervention, and public understanding of genetics. Perspect Psychol Sci. 2006;1:5–27. doi: 10.1111/j.1745-6916.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- 89.Ottman R. Gene-environment interaction: definitions and study designs. Prev Med. 1996;25:764–770. doi: 10.1006/pmed.1996.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ottman R. An epidemiologic approach to gene-environment interaction. Genet Epidemiol. 1990;7:177–185. doi: 10.1002/gepi.1370070302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rothman K.J., Greenland S., Walker A.M. Concepts of interaction. Am J Epidemiol. 1980;112:467–470. doi: 10.1093/oxfordjournals.aje.a113015. [DOI] [PubMed] [Google Scholar]
- 92.Ahlbom A., Alfredsson L. Interaction: a word with two meanings creates confusion. Eur J Epidemiol. 2005;20:563–564. doi: 10.1007/s10654-005-4410-4. [DOI] [PubMed] [Google Scholar]
- 93.Siemiatycki J., Thomas D.C. Biological models and statistical interactions: an example from multistage carcinogenesis. Int J Epidemiol. 1981;10:383–387. doi: 10.1093/ije/10.4.383. [DOI] [PubMed] [Google Scholar]
- 94.Greenland S. Interactions in epidemiology: relevance, identification, and estimation. Epidemiology. 2009;20:14–17. doi: 10.1097/EDE.0b013e318193e7b5. [DOI] [PubMed] [Google Scholar]
- 95.Rothman K.J., Lash T.L., Haneuse S., VanderWeele T.J. 4th ed. LWW; 2021. Modern Epidemiology. [Google Scholar]
- 96.Saracci R. Asbestos and lung cancer: an analysis of the epidemiological evidence on the asbestos-smoking interaction. Int J Cancer. 1977;20:323–331. doi: 10.1002/ijc.2910200302. [DOI] [PubMed] [Google Scholar]
- 97.Yaffe K., Haan M., Byers A., Tangen C., Kuller L. Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology. 2000;54:1949–1954. doi: 10.1212/wnl.54.10.1949. [DOI] [PubMed] [Google Scholar]
- 98.Al-Chalabi A., Calvo A., Chio A., Colville S., Ellis C.M., Hardiman O., et al. Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 2014;13:1108–1113. doi: 10.1016/S1474-4422(14)70219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiò A., Mazzini L., D'Alfonso S., Corrado L., Canosa A., Moglia C., et al. The multistep hypothesis of ALS revisited: the role of genetic mutations. Neurology. 2018;91:e635–e642. doi: 10.1212/WNL.0000000000005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knol M.J., van der Tweel I., Grobbee D.E., Numans M.E., Geerlings M.I. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol. 2007;36:1111–1118. doi: 10.1093/ije/dym157. [DOI] [PubMed] [Google Scholar]
- 101.Ordovas J.M., Corella D., Demissie S., Cupples L.A., Couture P., Coltell O., et al. Dietary fat intake determines the effect of a common polymorphism in the hepatic lipase gene promoter on high-density lipoprotein metabolism: evidence of a strong dose effect in this gene-nutrient interaction in the Framingham study. Circulation. 2002;106:2315–2321. doi: 10.1161/01.cir.0000036597.52291.c9. [DOI] [PubMed] [Google Scholar]
- 102.Caspi A., Sugden K., Moffitt T.E., Taylor A., Craig I.W., Harrington H., et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 103.van Os J., Rutten B.P., Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–1082. doi: 10.1093/schbul/sbn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lodato M.A., Woodworth M.B., Lee S., Evrony G.D., Mehta B.K., Karger A., et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karg K., Sen S. Gene × environment interaction models in psychiatric genetics. Curr Top Behav Neurosci. 2012;12:441–462. doi: 10.1007/7854_2011_184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cooper-Knock J., Hewitt C., Highley J.R., Brockington A., Milano A., Man S., et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135:751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cooper-Knock J., Kirby J., Highley R., Shaw P.J. The spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:326–339. doi: 10.1007/s13311-015-0342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Morahan J.M., Yu B., Trent R.J., Pamphlett R. A gene-environment study of the paraoxonase 1 gene and pesticides in amyotrophic lateral sclerosis. Neurotoxicology. 2007;28:532–540. doi: 10.1016/j.neuro.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 109.Diekstra F.P., Beleza-Meireles A., Leigh N.P., Shaw C.E., Al-Chalabi A. Interaction between PON1 and population density in amyotrophic lateral sclerosis. Neuroreport. 2009;20:186–190. doi: 10.1097/WNR.0b013e32831af220. [DOI] [PubMed] [Google Scholar]
- 110.Carr G. Special report: the human genome: biology 2.0. Economist. 2010;395:50. [Google Scholar]
- 111.Vasta R., Calvo A., Moglia C., Cammarosano S., Manera U., Canosa A., et al. Spatial epidemiology of amyotrophic lateral sclerosis in Piedmont and Aosta Valley, Italy: a population-based cluster analysis. Eur J Neurol. 2018 doi: 10.1111/ene.13586. [DOI] [PubMed] [Google Scholar]
- 112.Tammen S.A., Friso S., Choi S.W. Epigenetics: the link between nature and nurture. Mol Aspects Med. 2013;34:753–764. doi: 10.1016/j.mam.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Blechter B., Wong J.Y.Y., Agnes Hsiung C., Hosgood H.D., Yin Z., Shu X.O., et al. Sub-multiplicative interaction between polygenic risk score and household coal use in relation to lung adenocarcinoma among never-smoking women in Asia. Environ Int. 2021;147 doi: 10.1016/j.envint.2020.105975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bradley W.G., Andrew A.S., Traynor B.J., Chiò A., Butt T.H., Stommel E.W. Gene-environment-time interactions in neurodegenerative diseases: hypotheses and research approaches. Ann Neurosci. 2018;25:261–267. doi: 10.1159/000495321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pandya V.A., Patani R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: a cellular perspective. Brain. 2020;143:1057–1072. doi: 10.1093/brain/awz360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thomas D. Gene–environment-wide association studies: emerging approaches. Nat Rev Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Westerman KE, Pham DT, Hong L, Chen Y, Sevilla-González M, Sung YJ, et al. GEM: scalable and flexible gene-environment interaction analysis in millions of samples. 2020. 10.1101/2020.05.13.090803. [DOI] [PMC free article] [PubMed]
- 118.Steele J.C., McGeer P.L. The ALS/PDC syndrome of Guam and the cycad hypothesis. Neurology. 2008;70:1984–1990. doi: 10.1212/01.wnl.0000312571.81091.26. [DOI] [PubMed] [Google Scholar]
- 119.Upstill-Goddard R., Eccles D., Fliege J., Collins A. Machine learning approaches for the discovery of gene-gene interactions in disease data. Brief Bioinform. 2013;14:251–260. doi: 10.1093/bib/bbs024. [DOI] [PubMed] [Google Scholar]
- 120.Koo C.L., Liew M.J., Mohamad M.S., Salleh A.H.M. A review for detecting gene-gene interactions using machine learning methods in genetic epidemiology. Biomed Res Int. 2013;2013 doi: 10.1155/2013/432375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao Q., Li D., Huang X., Ren B., Yue L., Du B., et al. Identifying gene-environment interactions on the efficacy of folic acid therapy for hyperhomocysteinemia based on prediction model. Nutr Res. 2020;77:54–61. doi: 10.1016/j.nutres.2020.03.001. [DOI] [PubMed] [Google Scholar]