

Abstract
Inhalation of trace amounts of ricin toxin, a plant‐derived ribosome‐inactivating protein, results in ablation of alveolar macrophages, widespread epithelial damage, and the onset of acute respiratory distress syndrome (ARDS). While ricin's receptors are ubiquitous, certain cell types are more sensitive to ricin‐induced cell death than others for reasons that remain unclear. For example, we demonstrate in side‐by‐side studies that macrophage‐like differentiated THP‐1 (dTHP‐1) cells are hyper‐sensitive to ricin, while lung epithelium‐derived A549 cells are relatively insensitive, even though both cell types experience similar degrees of translational inhibition and p38 MAPK activation in response to ricin. Using a variety of small molecule inhibitors, we provide evidence that ER stress contributes to ricin‐mediated cytotoxicity of dTHP‐1 cells, but not A549 cells. On the other hand, the insensitivity of A549 cells to ricin was overcome by the addition of (TNF)‐related apoptosis‐inducing ligand (TRAIL; CD253), a known stimulator of extrinsic programmed cell death. These results have implications for understanding the complex pathophysiology of ricin‐induced ARDS in that they demonstrate that intrinsic (e.g., ER stress) and extrinsic (e.g., TRAIL) factors may ultimately determine the fate of specific cell types following ricin intoxication.
Keywords: apoptosis, epithelium, inflammation, lung, macrophage, stress, toxin
Abbreviations
- ARDS
acute respiratory distress syndrome
- ATII
alveolar type II epithelial cells
- AUC
area under the curve (statistical analysis)
- CH
cycloheximide
- dTHP‐1
differentiated THP‐1 cells (as by PMA exposure)
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum‐associated degradation
- ERS
endoplasmic reticulum stress
- ISR
integrated stress response
- MAPK
mitogen‐activated protein kinase
- MR
mannose receptor
- PCD
programmed cell death
- RIDD
regulated IRE1‐dependent decay
- RIP
ribosome‐inactivating protein
- RSR
ribotoxic stress response
- RT
ricin toxin
- RTA
ricin toxin subunit A
- RTB
ricin toxin subunit B
- SAPK
stress‐activated protein kinase
- SRL
sarcin–ricin loop
- Tg
thapsigargin
- TGN
trans‐golgi network
- Tm
tunicamycin
- TRAIL
TNF‐related apoptosis‐inducing ligand, CD253
- UPR
unfolded protein response
1. INTRODUCTION
Acute respiratory distress syndrome (ARDS) is a life‐threatening complication brought about by a variety of insults ranging from physical injury to viral infection to toxin exposure. One such provocateur is ricin, a type II ribosome‐inactivating protein (RIP) or ribotoxin found in castor beans (Ricinus communis). 1 , 2 , 3 Ricin's B subunit (RTB) is a Gal/GalNAc‐specific lectin that adheres to glycoproteins and glycolipids on cell surfaces and promotes the uptake and transport of ricin's A subunit (RTA) to the trans‐Golgi network (TGN) and endoplasmic reticulum (ER). 4 , 5 RTA is liberated from RTB in the ER and translocated into the cell cytoplasm, where it depurinates a conserved residue within the sarcin–ricin loop (SRL) of 28S RNA, thereby arresting ribosome activity. 6 With an estimated K cat ~1500 ribosomes per minute, a single molecule of RTA can wreak havoc on protein synthesis in any given cell type. 7 , 8 Compounding the effects of SRL depurination is the activation of the ribotoxic stress response (RSR) and subsequent stimulation of the MAP kinase pathway, namely p38 and JNK phosphorylation. 9 , 10 , 11 , 12 , 13 , 14 In effect, ribosome damage triggers the onset of pro‐inflammatory and pro‐apoptotic pathways that contributes to ricin‐induced ARDS as evidenced by high levels of pro‐inflammatory cytokines like IL‐1 and IL‐6 in serum and bronchoalveolar lavage (BAL) fluids. 11
While translation inhibition and concomitant activation of RSR are hallmarks of ricin intoxication, evidence indicates that ribosome damage is not the sole determinant of cell fate. In yeast, for example, Li et al identified RTA point mutants that induced ribosome depurination without a cost to cell viability. 15 In the context of the lung, widespread depurination of rRNA in epithelial cells is measurable at time points where the integrity of the epithelium was not yet compromised. 16 Similarly, we reported that the lung‐derived Calu‐3 and A549 epithelial cell lines treated with high doses of ricin remained viable for days, despite significant reductions in protein synthesis. 17 , 18 However, when Calu‐3 and A549 cells were treated concurrently with ricin and (TNF)‐related apoptosis‐inducing ligand (TRAIL; CD253), a known stimulator of extrinsic programmed cell death (PCD), cell viability plummeted. These results suggest that ricin‐induced cell death of A549 cells occurs in response to protein synthesis inhibition plus a secondary insult like TRAIL.
Alveolar‐ and lung tissue‐resident macrophages, in contrast, are hypersensitive to ricin toxin (RT), as evidenced by a virtual ablation of these cell types within hours after ricin exposure by inhalation. 14 , 16 , 18 , 19 , 20 , 21 In the liver, Kupffer cells are similarly sensitive to ricin‐induced killing in vitro and ex vivo. 22 , 23 , 24 The sensitivity of macrophages is borne out in vitro, as ricin has been shown to trigger apoptosis in numerous macrophage and monocytic cell lines of mouse and human origin. 21 , 25 , 26 , 27 , 28 , 29 The sensitivity of macrophages to ricin is due in part to the mannose receptor (MR), which recognizes the mannose side chains or RTA and RTB and facilitates toxin uptake by a mechanism that remains poorly understood. 23 , 30 Nonetheless, other factors likely contribute to the rapid onset of apoptosis observed in macrophages following ricin exposure, especially given the importance of stress and signaling pathways in cell fate determination. Indeed, in this report we implicate ER stress as a driver of macrophage cell death following RT exposure.
2. MATERIALS AND METHODS
2.1. Cell culture conditions
A549 cells (ATCC Cat# CRL‐7909, RRID:CVCL_0023) were grown in Kaighn's Modified Ham's f‐12 media with Pen/Strep and 10% FBS. Cells were passaged at 1:10 after growing to approximately 90% confluence. THP‐1 cells were grown in antibiotic‐free RPMI‐1640 supplemented with 10% HI FBS, 0.2 mM l‐glutamine, and 0.05 mM 2‐mercaptoethanol. The culture was maintained between 2.0 and 8.0 ×105 cells/ml at 37℃ and 5% CO2. Experiments were performed at or below passage 15 in both cell types. THP‐1 cells (ATCC Cat# TIB‐202, RRID:CVCL_0006) were differentiated prior to all experiments using 50 ng/ml PMA. Cells were seeded in complete media plus PMA (Cell Signaling Technology Cat# 4174) and incubated for approximately 65 h. Following PMA‐induced differentiation, cells were washed two times with sterile PBS and returned to the incubator in complete media for 24 h. After the initial recovery period, cells were washed once more with sterile PBS and returned to the incubator in complete media. Experiments with a prolonged pre‐treatment were initiated 6–8 h later, and all other experiments were initiated approximately 24 h later, allowing for a minimum recovery period of 30 h post‐PMA exposure.
2.2. Cytotoxicity and protein synthesis assays
A detailed protocol for the CellTiter‐Glo viability assay, including combinatorial treatment as performed in herein, has been published elsewhere. 31 THP‐1 cells were seeded at 2.5 × 103 cells per well in 96‐well plates and differentiated with PMA in the assay plates as described above. THP‐1 cells were exposed to ricin for 2 h and were allowed 18–22 h of recovery prior to assessing viability. A549 cells were seeded at 10 × 103 cells per well ~24 h prior to the onset of treatment. A549 cells were exposed to ricin for 4 h and were allowed 40–44 h to recover prior to assessing viability. Protein synthesis levels were examined using the O‐propargyl‐puromycin (OPP) integration‐based fluorescent protein synthesis assay kit, following the manufacturer's protocol for the 96‐well plate format (Cayman Biochemical Cat# 601100). A 4‐h ricin exposure period was used for both cell types for the purpose of that assay.
2.3. Cell lysis, protein collection, and western blotting
THP‐1 cells were seeded at ~1.5 × 105 cells/cm2 in 6‐well plates with PMA and allowed to differentiate as described above. A549 cells were seeded at 8.5 × 104 cells/cm2 and allowed to grow until near confluence (~24 h). Cells were treated with ricin (20 ng/ml) and collected by scraping at 3, 6, and 9 h timepoints. Cells were lysed in RIPA buffer containing a protease/phosphatase inhibitor cocktail (Cell Signaling Technology Cat# 5872). To reduce sample viscosity, DNA was sheared by bead beating at 4 m/s for 3 s using 1 mm glass beads. Protein concentration was interpolated by BCA assay (Pierce Cat# 23225). Samples were diluted into reducing sample buffer, boiled, and 20 µg of total protein was separated by electrophoresis on precast 4%–15% polyacrylamide gradient gels (Bio‐Rad Cat# 4561083) in accordance with the manufacturer's protocol.
For western blotting, proteins were transferred to PVDF membranes (Abcam Cat# ab133411) in a high field wet tank transfer. The membranes were retrieved, rinsed briefly in water, and fixed (7% glacial acetic acid 10% MeOH) for 15 min before blocking in TBS plus 0.5% Tween‐20 for 1 h at room temperature. Antibody incubation was performed in TBS plus 0.1% Tween‐20 and 5% BSA, overnight at 4℃. For direct comparison between phospho‐epitope and total protein pool, membranes were first probed with the phospho‐specific antibody (except in the case of IRE1, as signal from the p~antibody was stronger), then stripped, and re‐probed with the associated non‐phospho‐specific antibody to detect the total pool of that protein. Membranes were cut into swatches containing the molecular weight range of target proteins, probed first with α‐p~p38 (1:1000, Cell Signaling Technology Cat# 4511, RRID:AB_2139682), α‐IRE1 (1:1000, Cell Signaling Technology Cat# 3294, RRID:AB_823545), or α‐p~PERK (1:1000, Cell Signaling Technology Cat# 3179, RRID:AB_2095853) followed by and an HRP‐conjugated 2’ antibody (1:5000, Cell Signaling Technology Cat# 7074, RRID:AB_2099233), imaged, then stripped, and re‐probed with α‐p38 (1:1000, Cell Signaling Technology Cat# 8690, RRID:AB_10999090), α‐p~IRE1 (1:1000, Thermo Fisher Scientific Cat# PA1‐16927, RRID:AB_2262241), or α‐PERK (1:1000, Cell Signaling Technology Cat# 3192, RRID:AB_2095847). Membrane swatches probed with α‐DR5 (1:2500, Cell Signaling Technology Cat# 8074, RRID:AB_10950817) were signal bleached with excess ECL, then re‐probed with HRP‐conjugated α‐β tubulin (1:4000, Cell Signaling Technology Cat# 5346, RRID:AB_1950376) to serve as a loading control. Blots were imaged using ECL Plus western blotting substrate (Pierce Cat# 32132) and developed on x‐ray film. Films were digitized on a scanner and ImageJ was used for semi‐quantitative assessment of band density.
Images shown are representative of at least three replicate experiments. The bar graphs depict average results from among these replication sets, which show reproducible trends. However, statistical comparisons were not made due to the inherent loss of quantitative information inherent to film‐based imaging.
2.4. Statistical analysis
All statistical analyses were performed using GraphPad Prism 7.
The EC50 of ricin was calculated from the viability curve by nonlinear regression (variable slope, four parameters) without any imposed constraints. For dTHP‐1 cells, the relative EC50 derived from this nonlinear regression approximates an absolute EC50 as cell viability ranged from <0.1% to approximately 100% over the range of ricin concentrations used. A similar value could not be established for A549 cells, as treatment with ricin alone cannot reduce this cell type to a value approaching 0% viability at any concentration.
Viability and protein synthesis assay data were analyzed using one‐way ANOVA followed by pairwise comparison with correction for multiple comparisons. Brown–Forsythe and Welch ANOVA tests were used for normally distributed data, followed by Dunnett's T3 multiple comparisons test. Data sets with non‐normal distributions were compared using the Kruskal–Wallis one‐way ANOVA followed by Dunn's multiple comparisons test. When assessing the ability of combinatorial treatment to shift the viability of cells across a range of ricin concentrations, multiple unpaired t‐tests with Welch correction were performed, using the two‐stage step‐up method to correct for multiple comparisons via false discovery rate. For ease of visibility in the figures, data were presented with an accompanying area under the curve analysis, giving the total area under each curve plus or minus the 95% confidence interval, rather than indicating the p values at each data point along the curve. AUC values had non‐overlapping 95% confidence intervals in all treatments, which agreed with the significant results obtained from multiple t‐tests between ricin‐only and combinatorial treatment.
Area under the curve was not calculated for the untreated control condition (green reference lines throughout), nor for the groups testing for independent toxicity (orange reference lines) or displaying toxicity of ricin alone (red reference lines). These lines represent the mean group viability with its associated 95% confidence interval and are shown solely for the purpose of comparison with experimental treatments.
3. RESULTS
3.1. Differential sensitivity of A549 and THP‐1 cells to RT
As noted in the introduction, differential sensitivity of various human cell lines to RT has been reported, although it is unclear whether the observed differences are technical (e.g., different toxin potencies) or biological in nature. THP‐1 cells, for example, are reportedly exquisitely sensitive to ricin. The THP‐1 cell line is derived from an acute monocytic leukemia and exhibits multiple monocytic characteristics. 32 When treated with PMA, THP‐1 cells differentiate into macrophage‐like cells. 33 , 34 On the other end of the spectrum, A549 cells are reportedly relatively insensitive to RT, unless sensitized with TNF‐α or TRAIL. 17 , 18 , 31 The A549 cell line is derived from an alveolar cell carcinoma and exhibits characteristics of alveolar type II epithelium (ATII). 35
To compare the sensitivity of the two aforementioned cell types to RT at near identical parameters, PMA‐differentiated THP‐1 (dTHP‐1) cells and A549 cells were each exposed to a range of toxin concentrations (0.5–500 ng/ml) for 4 h, washed, and then examined for cell viability 24 h later. When compared side‐by‐side, the discrepancy in sensitivity of the two cell types to ricin was stark. The viability of dTHP‐1 cells, expressed as area under the curve (AUC) was 1428 ±93, while the viability of the A549 cells was 31,210 ±1100. The absolute half‐maximal inhibitory concentration (IC50; the concentration required to reduce viability by half) of ricin in dTHP‐1 cells was 8.24 ng/ml ±0.54. In contrast, we were unable to establish an IC50 for A549 cells, as no concentration of ricin was reliably capable of reducing viability to <50% (Figure 1A).
FIGURE 1.
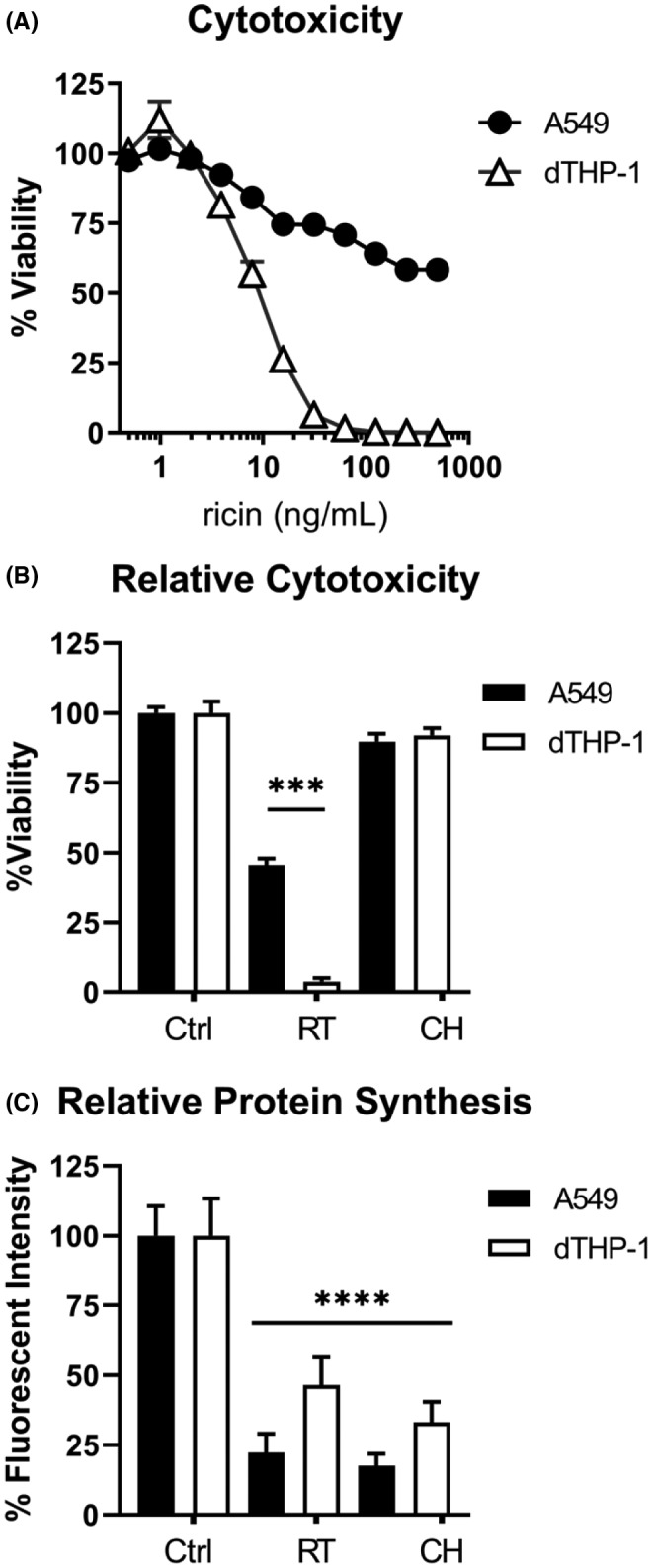
Comparative sensitivity of A549 and dTHP‐1 cells to ricin toxin. (A) Side‐by‐side 96‐well plate cytotoxicity assays were used to compare ricin sensitivity across a range of doses between A549 and dTHP‐1 cells. Cells were exposed to ricin for 4 h, washed, and allowed to recover for 24 h prior to assay development. Data presented are the mean of eight replicate wells with the 95% confidence interval. Viability as expressed by AUC ±95% CI (area under the curve with 95% confidence interval) was significantly different, with a value of 1428 ±93 for THP‐1 cells and 31210 ±1100 for A549 cells. (B) Cytotoxicity and (C) protein synthesis inhibition were compared side‐by‐side in both cell types following 4 h exposure to 20 ng/ml ricin (RT) or 50 µg/ml cycloheximide (CH). Circles represent A549 cells, triangles represent dTHP‐1 cells. In panel (B), viability was significantly reduced in both cell types following ricin treatment (p < 0.0001). When comparing ricin‐treated dTHP‐1 and A549 cells, dTHP‐1 cells exhibited a significantly greater reduction in viability (p < 0.0001). In panel (C), both RT and CH treatment significantly reduced protein synthesis compared to the untreated controls (p < 0.0001). Within each cell type, RT and CH treatment were equally effective at inhibiting protein synthesis. Although ricin treatment had a significantly greater suppressive effect on protein synthesis in A549 cells (p = 0.0065), its lethality was significantly lower in this cell type. (a) N = 7–8, (b) N = 14–16, (c) N = 8–10. The significance of “*** and ****” correspond to <0.001 and <0.0001, respectively
We postulated that the relative insensitivity of A549 cells to ricin as compared to dTHP‐1 cells could simply be due to ineffective toxin uptake and/or inefficient retrograde transport thereby limiting RTA‐mediated ribosome inactivation. 36 To address this possibility, we measured both cell viability and protein synthesis levels side‐by‐side in dTHP‐1 and A549 cells that had been exposed to ricin (20 ng/ml) for 4 h. Under those conditions, A549 cell viability was ~50% and dTHP‐1 cell viability was <5% (Figure 1B). At the same time, protein synthesis, as measured by OPP incorporation, was reduced by 78% ±6.8 and 54% ±10.3, respectively (Figure 1C). This disconnect between ricin‐induced protein synthesis arrest and cell death has been reported by others. 15 , 37 This phenomenon was underscored when dTHP‐1 and A459 cells were treated with cycloheximide (50 µg/ml), a protein synthesis inhibitor that sterically blocks the translocation step of peptide elongation without causing direct ribosome damage. 11 , 38 Protein synthesis in A549 and dTHP‐1 cells was reduced by 82% ±4.4 and 67% ±7.2, respectively, without a significant impact on cell viability (Figure 1B,C). Thus, other factors besides translational inhibition drive sensitivity to ricin‐induced cell death.
We next investigated whether differences in the RSR might account for different sensitivities of A549 and dTHP‐1 cells to toxin‐induced death. Stress‐activated protein kinases activated by the RSR drive ricin‐induced apoptosis, and increased activation in dTHP‐1 cells could explain their hypersensitivity to cell death. To examine this question, we measured the levels of phosphorylated p38 (p~p38) in dTHP‐1 and A549 cells at 3 and 6 h following toxin exposure. Phospho‐p38 is the primary indicator of ricin‐induced RSR. 9 , 20 , 29 In both cell types, the level of p~p38 relative to total p38 levels increased >2‐fold following ricin treatment (Figure 2A–C). Therefore, the relative insensitivity of A549 cells to ricin is not simply due to a failure of toxin uptake or trafficking, or a difference in the efficiency of ribosomal damage within cells.
FIGURE 2.
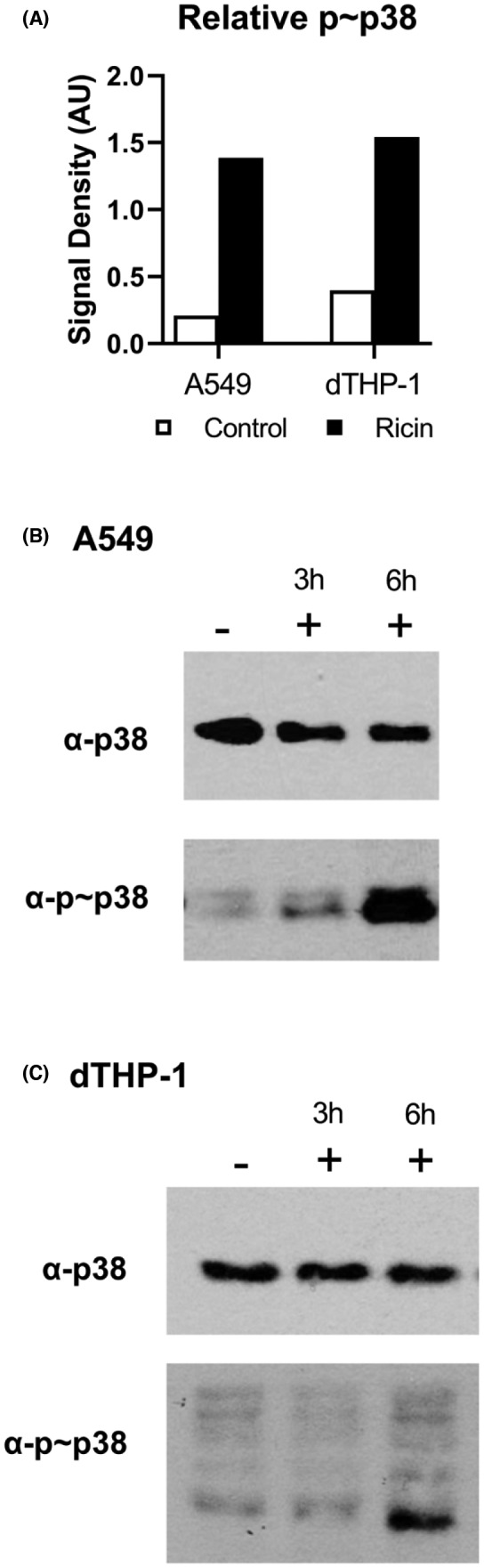
Ribotoxic stress is evident in both A549 and dTHP‐1 cells. (A) The relative abundance of phosphorylated p38 in response to 6 h ricin treatment (black bars, 20 ng/ml) was dramatically increased over the baseline level observed in untreated cells (white bars) in both A549 and dTHP‐1 cells. Bars represent semi‐quantitative densitometric analysis of the phosphorylated form of each protein relative to the total pool of that protein. (C, D) Digitized film images of western blots. Membrane swatches bearing electrophoresed whole cell lysate from A549 cells (left) and dTHP‐1 cells (right) were probed with anti‐phospho‐p38 monoclonal antibody (Cell Signaling Technology Cat# 4511, RRID:AB_2139682) and then stripped and re‐probed with anti‐p38 polyclonal antibody (Cell Signaling Technology Cat# 8690, RRID:AB_10999090). Representative blot results are shown from among three to four replicate experiments. −, indicates the absence of ricin; +, indicates the presence of ricin. Lysates were collected after 3 or 6 h of ricin exposure. Results from 3 h lysates were variable between experiments and were thus excluded from the bar graph. Refer to Figures S1 and S2 to observe the original films from which these images were derived
3.2. Examination of cell type‐specific ricin‐induced ER stress.
We postulated that ER stress might contribute to the differential sensitivity of dTHP‐1 and A459 cells. ER stress can be induced by Ca2+ imbalance, unfolded proteins in the ER lumen, disruption of post‐translational modification processes, and/or disruption of ER‐to‐Golgi trafficking. ER biosynthetic capacity varies by cell type, with highly secretory cells like macrophages operating at or near the ER stress‐activating threshold. 39 , 40 In the case of RT, the unfolding of RTA that occurs in the ER during retro‐translocation may be a stress‐inducing event. 41 , 42 Depurination of ER‐associated ribosomes could also induce stress through disruption of ER proteostasis.
Unfavorable ER conditions are detected by three dedicated sensor proteins: IRE1, PERK, and ATF6. Each sensor is responsible for activating a set of regulatory responses aimed at re‐establishing ER homeostasis, which are collectively referred to as the unfolded protein response (UPR). When UPR reprogramming fails to return the ER to a state of normal function, downstream effectors of both PERK and IRE1 have been shown to trigger apoptosis. 43 , 44 We therefore theorized that if ER stress was contributing to an apoptotic cell fate, these two sensor proteins were most likely to be relevant. As both PERK and IRE1 are subject to activating phosphorylation in response to ER stress, 45 we assessed relative phosphorylation of PERK (p~PERK) and IRE1 (p~IRE1) in A549 and dTHP‐1 cells following exposure to doses of ricin shown to be sufficient to activate p38 mitogen‐activated protein kinase (see above; 20 ng/ml). In dTHP‐1 cells, there were significant increases in the relative abundance of p~IRE1 (Figure 3A,C) and p~PERK (Figure 3B,D), as compared to control (untreated) cells. In A549 cells, however, IRE1 and PERK phosphorylation levels were unchanged following ricin treatment. These results are consistent with ricin triggering an ER stress response in dTHP‐1 cells but not A549 cells.
FIGURE 3.
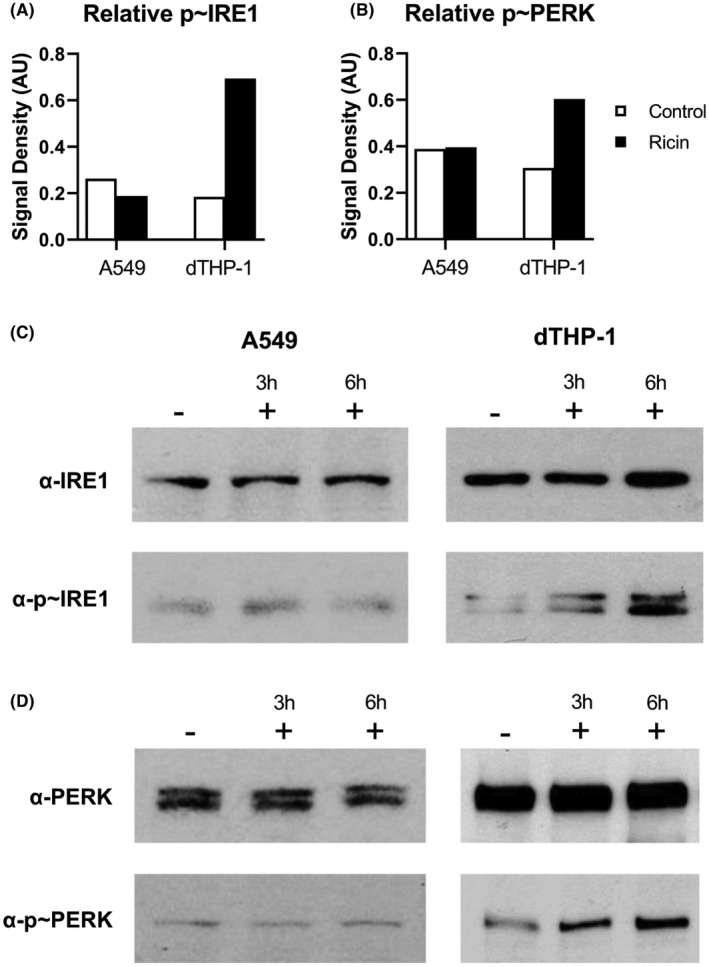
Evidence for ER stress in dTHP‐1 but not A549 cells. The relative abundance of phosphorylated IRE1 (A, C) and phosphorylated PERK (B, D) in response to 6 h ricin treatment (black bars; 20 ng/ml) or controls (white bars) in dTHP‐1 cells. Bars represent the semi‐quantitative signal density of the phosphorylated form of each protein relative to the total pool of that protein probed sequentially on the same membrane swatch. Digitized films of representative western blots selected from among three to four replicate experiments. Membrane swatches were probed with anti‐phospho‐PERK (Cell Signaling Technology Cat# 3179, RRID:AB_2095853) or anti‐IRE1 (Cell Signaling Technology Cat# 3294, RRID:AB_823545), then stripped and re‐probed with anti‐PERK (Cell Signaling Technology Cat# 3192, RRID:AB_2095847) or anti‐phospho‐IRE (Thermo Fisher Scientific Cat# PA1‐16927, RRID:AB_2262241) antibodies. Results at 3 h treatments were variable and thus were not included in the semi‐quantitative graphs in (A) and (B). Refer to Figures S3–S6 to observe the original films from which these images were derived
With increasing severity of ER disruption, UPR processes skew farther toward a pro‐apoptotic cell fate. We therefore expected that if UPR effectors were indeed contributing to ricin‐induced cell death, then chemical induction of severe ER stress would sensitize A549 cells to ricin, and further exacerbate the sensitivity of dTHP‐1 cells. To test this hypothesis, dTHP‐1 and A549 cells were treated with ricin and three commonly utilized endoplasmic reticulum stress (ERS)‐inducing compounds: tunicamycin (Tm), a natural antibiotic that inhibits glycosylation in the Golgi compartment 46 ; dithiothreitol (DTT), a reducing agent that induces protein misfolding throughout the cell and retention of improperly folded proteins in the ER 47 ; and thapsigargin (Tg), a non‐competitive sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor that causes release of Ca2+ from the ER. 48 To control for potential cytotoxic effects caused by aggressive induction of ERS, all combinatorial treatment experiments included a treatment group exposed to the ERS‐inducing compound alone. When the ERS‐only group exhibited reduced viability compared to untreated control cells, the viability of the dual‐treated group was normalized using the independent toxic effect as a baseline to more clearly distinguish synergistic effects of the treatment.
As predicted, we found that each of the three compounds further sensitized dTHP‐1 cells to ricin. Specifically, Tm shifted the viability AUC to 554.5 ±76.5 from 1504 ±103 in ricin‐only treatment, and DTT shifted the viability AUC to 709.1 ±70.1 from 1677 ±73 (Figure 4A,B). Tg significantly enhanced ricin sensitivity at all concentrations exceeding 10 nM without exhibiting independent toxicity (Figure 4C). Tm and Tg co‐treatment were effective with a 2 h pre‐treatment followed by 2 h co‐treatment with ricin, while DTT combinatorial treatment was effective with 2 h pre‐treatment followed by 2 h ricin‐only treatment.
FIGURE 4.
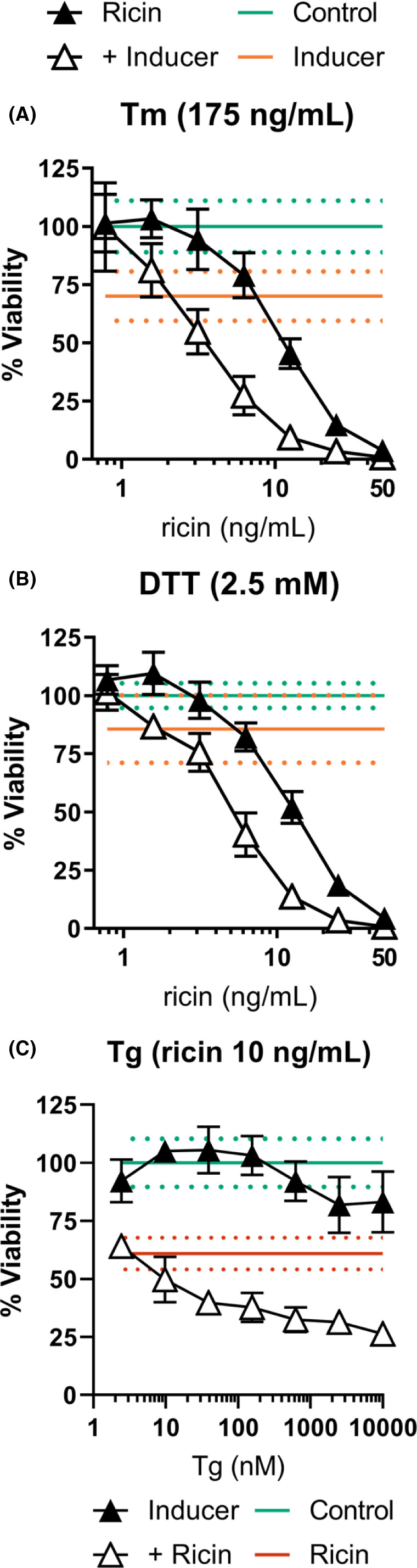
External induction of ER stress further sensitizes dTHP‐1 cells to ricin‐induced death. (A) dTHP‐1 cells treated with Tm (175 ng/ml) for 2 h followed by 2 h Tm +ricin resulted in significantly enhanced ricin‐induced cell death (AUC of 554.5 ±76.5 vs. 1504 ±103 of ricin‐only). (B) Treatment with DTT (2.5 mM) for 2 h followed by a 2 h ricin exposure significantly enhanced ricin‐induced cell death (AUC of 709.1 ±70.1 vs. 1677 ±73 of ricin‐only). (C) Treatment with varying concentrations of Tg for 2 h followed by a 2 h co‐treatment with ricin and Tg. Tg significantly increased ricin‐induced cell death at all concentrations but the two lowest concentrations tested (p ≤ 0.0014) without independent toxicity. Tm and DTT treatment exhibited significant independent toxicity, which was corrected for in the dual‐treatment curves. Closed symbols, ricin‐only; open symbols, dual treatment, error bars represent the 95% confidence interval (CI). Green line and dots represent the mean and 95% CI of control; orange line and dots represent the mean and 95% CI of the indicated ER stress‐inducing compound; red line and dots represent the mean and 95% CI of ricin treatment. Experiments were independently repeated in triplicate with eight replicate wells per treatment condition, with controls for independent effects included in each trial. Representative survival curves are shown. Graphical representation of the unnormalized data and results of pairwise statistical testing for treatment interactions are available in Figure S7
We had previously found that A549 cells, which are less sensitive to ricin‐induced cell death, did not exhibit an ER stress response following ricin exposure. To better understand the relevance of the lack of ER stress associated with ricin exposure in the fate of these cells, we utilized combinatorial treatment with chemical inducers of ER stress in an attempt to increase ricin sensitivity. Treatment with all three compounds significantly sensitized A549 cells to ricin‐induced cell death. Tm co‐treatment shifted the viability AUC to 30,830 ±1609 as compared to 33,962 ±1236 with ricin‐only treatment (Figure 5A). DTT pre‐treatment notably increased the maximum efficiency of ricin‐induced cell death and shifted the viability AUC to 27,082 ±3935 from 41,420 ±2200 in ricin‐only treated cells (Figure 5B). Tg combinatorial treatment shifted the viability AUC to 20,541 ±1344 from 23,326 ±769 (Figure 5C). A549 cells also exhibited lower independent toxicity in response to ER stress‐inducing treatment, tolerating treatments that utilized higher concentrations of the chemical inducers over a longer duration of exposure than dTHP‐1 cells. Tm and Tg combinatorial treatment required 2 h pre‐treatment and 4 h co‐treatment with ricin, while DTT was effective in a 2 h pre‐treatment followed by 4 h ricin‐only treatment. While the increase in ricin‐induced cell death caused by co‐treatment with ERS inducers was statistically significant, it did not reach a degree of killing comparable to that seen in dTHP‐1 cells. This would suggest that while the ability of the ER to cope with ricin‐mediated disruption in A549 cells factors into the determination of cell fate following ricin exposure, it is not the primary determinant of their reduced ricin sensitivity.
FIGURE 5.
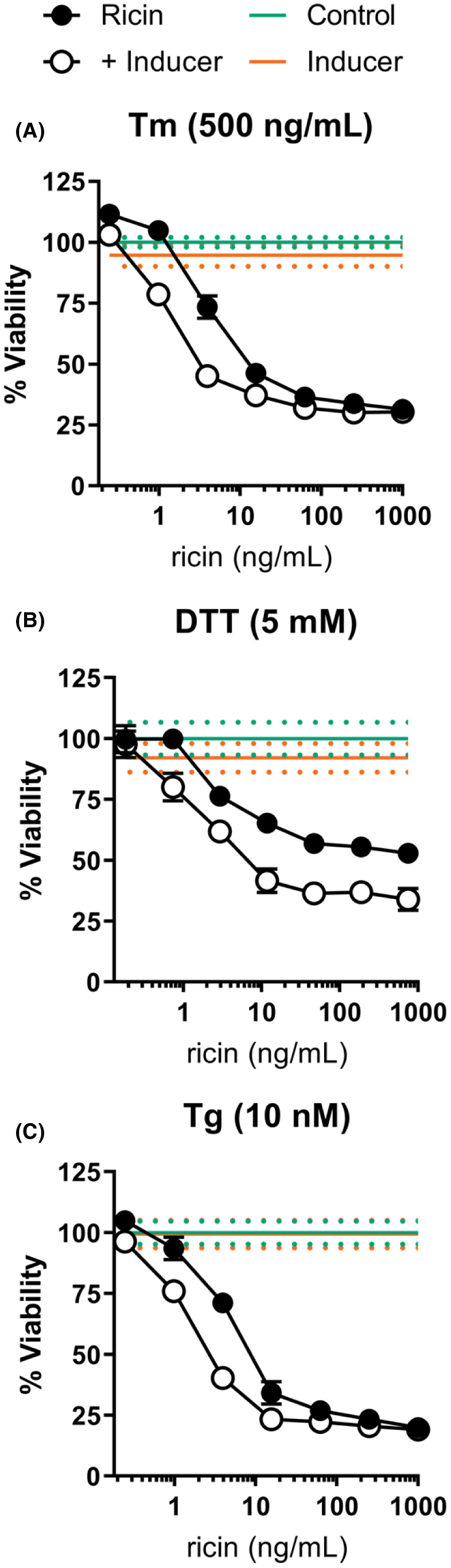
Treatment with ER stress inducers enhanced ricin toxicity in A549 cells. (A) A549 cells treated with Tm (500 ng/ml) for 2 h followed by a 4 h Tm +ricin co‐treatment significantly enhanced ricin‐induced cell death (AUC of 30,830 ±1609 vs. 33,962 ±1236 of ricin‐only). (B) Treatment with DTT (5 mM) for 2 h prior to a 4 h ricin exposure significantly enhanced ricin‐induced cell death (AUC of 27,082 ±3935 vs. 41,420 ±2200 of ricin‐only) (C) Treatment with Tg (10 nM) for 2 h followed by a 4 h Tg +ricin co‐treatment significantly enhanced ricin‐induced cell death (AUC of 20,541 ±1344 vs. 23,326 ±769 of ricin‐only). Closed symbols, ricin‐only; open symbols, dual treatment, error bars represent the 95% CI. The green line and dots represent the mean and 95% CI of control cell viability; orange line and dots represent the mean and 95% CI of cell viability for the indicated ER stress‐inducing compound. Tm and DTT treatment exhibited independent toxicity, which was corrected for in the combinatorial treatment curves. Experiments were independently repeated in triplicate with six to eight replicate wells per treatment condition with controls for independent effects included in each trial. Representative survival curves are shown. Graphical representation of the unnormalized data and results of pairwise statistical testing for treatment interactions are available in Figure S8
3.3. Suppression of ER stress reduced ricin sensitivity of dTHP‐1 cells
Given the apparent interaction between UPR and ricin‐induced cell death, we expected that suppression of ER stress would de‐sensitize dTHP‐1 cells. To test this possibility, we employed the chemical chaperone TUDCA. 49 TUDCA is a bile salt derivative that supplements the folding capacity of the ER, theoretically raising the threshold at which ER dysfunction would elicit a response from the luminal sensor proteins. Indeed, we found that dTHP‐1 cell survival following ricin exposure was substantially enhanced by combinatorial treatment with ≥100 µM TUDCA (Figure 6A). For example, 300 µM TUDCA significantly shifted the viability AUC from 1604 ±174 with ricin alone to 3302 ±207 with combined treatment (Figure 6B). To be effective, TUDCA needed to be provided before (18 h) and during ricin exposure.
FIGURE 6.
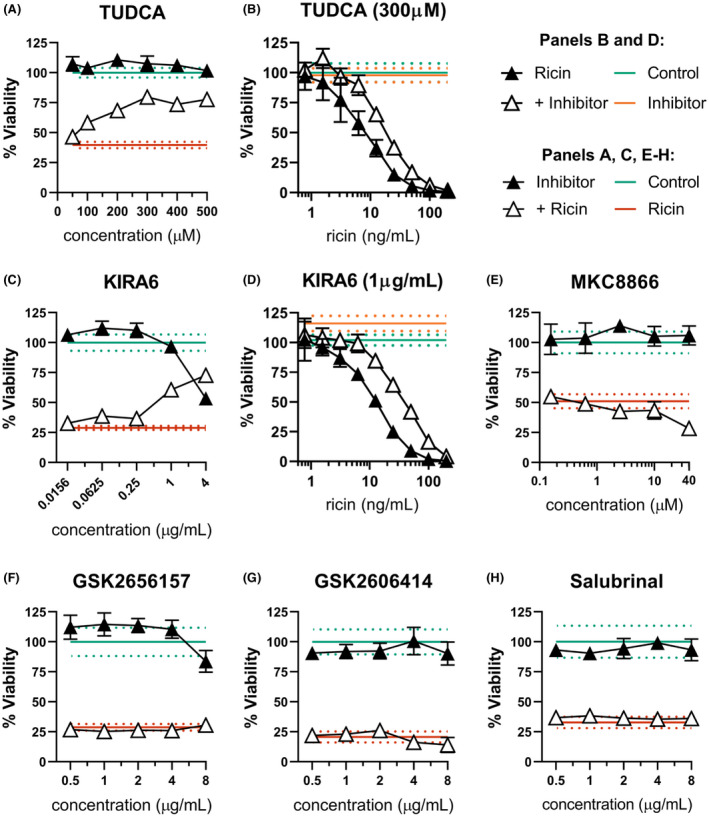
ER stress contributes to dTHP‐1 ricin sensitivity through IRE1 activity. dTHP‐1 cells were pre‐treated with the indicated ERS suppressing compound for 18 h prior to 2 h co‐treatment with ricin (A & C, 10 ng/ml; E–H,12.5 ng/ml). (A) Pre‐treatment with TUDCA was able to partially rescue ricin sensitivity in dTHP‐1 cells. Rescue was statistically significant from 200 µM (p ≤ 0.03). (B) Pre‐treatment with 300 µM TUDCA significantly reduced cytotoxicity of ricin exposure (AUC 3302 ±207 vs. 1604 ±174 of ricin‐only). (C) Specific inhibition of IRE1 with the small molecule KIRA6 partially rescued ricin‐induced dTHP‐1 cell death, with significant independent toxicity at doses exceeding 1 μg/ml. Rescue was statistically significant in all but the lowest doses (p < 0.0001). (D) Pre‐treatment with 1 µg/ml KIRA6 significantly reduced cytotoxicity of ricin exposure (AUC of 5879 ±386 vs. 2165 ±142 of ricin‐only) (E) Pre‐treatment with IRE1 RNase inhibitor MKC8866 enhanced ricin toxicity at 40 µM (p < 0.001) with no effect seen at lower doses. (F and G) Specific inhibition of PERK using the small molecule inhibitors GSK2656157 and GSK2606414 had no effect on ricin‐induced cell death. GSK2656157 exhibited significant independent toxicity above 8 μg/ml, while GSK2606414 was well tolerated. (H) Treatment with Salubrinal had no independent toxicity and no effect on ricin‐induced cell death. Green lines and dots and red lines and dots represent the mean and 95% CI of control‐ and ricin‐treated cells, respectively, while orange lines and dots similarly represent the independent effect of KIRA6 or TUDCA. Closed symbols, ricin‐only; open symbols, dual treatment; error bars represent the 95% CI. Data presented are the mean of at least six replicate wells. For (A), (C), and (E, F), points in the dual‐treatment group have been individually corrected for significant independent effects on viability by the inhibitor treatment as appropriate. Graphical representation of the unnormalized data (panels B, C, and D) and results of pairwise statistical testing for treatment interactions (panels B and D) are available in Figure S9
Though effective in protecting from ricin‐induced cell death, TUDCA’s broadly suppressive activity provides no additional clues as to the relationship between UPR and ricin toxicity. To investigate specific contributions of PERK and IRE1’s respective UPR activities to the observed protection, we co‐treated dTHP‐1 cells with ricin and the IRE1‐specific inhibitor KIRA6 or the PERK‐specific inhibitors GSK2656157 and GSK2606414. We found that KIRA6 significantly reduced ricin‐induced cell death in dTHP‐1 cells (Figure 6C), even after correction for significant independent toxicity at higher concentration. For example, at 1 µg/ml, KIRA6 shifted the viability AUC from 2165 ±142 in ricin‐only treated cells to 5879 ±386 with combinatorial treatment (Figure 6D). Previous reports indicate that ricin is able to prevent IRE1‐mediated alternative splicing of HAC1, yeast homolog of XBP1, the transcription factor that serves as a major downstream effector of IRE1. 50 , 51 This would predict an independence of our observed phenotype from IRE1‐mediated RNase activity, which we tested using MKC8866, which inhibits both alternative splicing and regulated IRE1‐dependent decay (RIDD) of transcripts without hindering the protein's kinase activity. Pre‐treatment with MKC8866 was non‐toxic to cells and enhanced ricin toxicity at high concentration (Figure 6E), suggesting that neither XBP1 activation via alternative splicing nor RIDD are involved in IRE1‐dependent enhancement of ricin‐induced cell death in agreement with the existing literature.
In contrast, co‐treatment with PERK inhibitors GSK2656157 and GSK2606414 did not interact with ricin‐induced cell death, arguing against a role for PERK in the relationship between ricin sensitivity and ERS (Figure 6F,G). Although PERK is activated by ER stress, the regulatory program it enacts is part of the integrated stress response (ISR), and not unique to UPR. Three other ISR kinases can initiate the same shared stress response from other areas of the cell under a wide variety of stress conditions. All four ISR kinases trigger the same regulatory cascade through phosphorylation of their shared substrate, eIF2α. The apparent lack of PERK involvement in ricin‐induced cytotoxicity may therefore be due to redundant activation of PERK’s downstream effector eIF2α by another ISR kinase.
Phospho‐eIF2α has been shown to protect against apoptosis in the context of ER stress, an activity that can be enhanced by the small molecule drug salubrinal which prevents its dephosphorylation. 52 We next treated dTHP‐1 cells with salubrinal to determine if this ISR effector could be leveraged to protect ricin exposed cells. Combinatorial treatment with salubrinal also had no effect on cell viability (Figure 6H), indicating that this pathway may already be saturated, or otherwise dispensable in the determination of cell fate in this context.
3.4. TRAIL sensitizes A549 cells, but not dTHP‐1 cells, to ricin‐induced apoptosis
We previously reported that pre‐treatment of Calu‐3 and A549 cells with TRAIL, a TNF superfamily cytokine active in both homeostasis and pathogenesis in the lung, sensitized them to RT‐induced PCD. Indeed, treatment of A549 cells with TRAlL and ricin shifted the viability AUC to 5278 ±321 from 36,190 ±4027 with maximal killing reaching 95.6% ±0.34 compared with only 69.3% ±4.8 in ricin‐only treated cells (Figure 7A). In dTHP‐1 cells, however, the addition of TRAIL at a range of concentrations had no significant effect on ricin‐induced cell death (Figure 7B,C). The apparent lack of TRAIL sensitization was not due to the absence of TRAIL’s primary death receptor, DR5, as this protein was observed in whole cell lysate of both cell types by western blot (Figure 7D). Unexpectedly, DR5 expression decreased compared to total protein loading over the course of ricin exposure in both cell types, suggesting a preferential degradation of this molecule as a possible means to avoid TNF‐mediated death spiral. This is particularly interesting, as DR5 expression is upregulated downstream of PERK activation in the context of ER stress. 53
FIGURE 7.
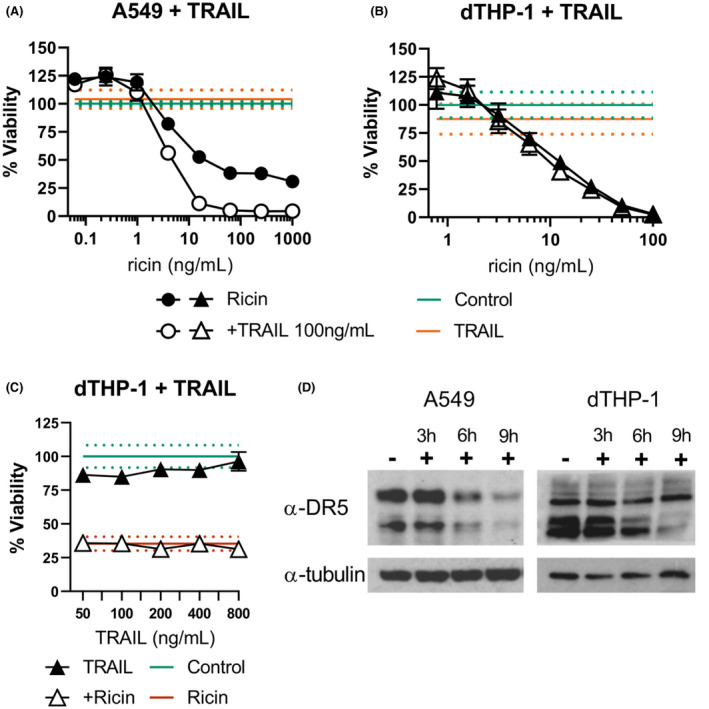
Impact of TRAIL on A549 and dTHP‐1 sensitivities to ricin. (A) Ricin +TRAIL co‐treatment sensitizes A549 cells to ricin‐induced cell death (AUC of 5278 ±321 vs. 36,190 ±4027 for ricin‐only treated), but (B) TRAIL has no effect on ricin cytotoxicity in dTHP‐1 cells (AUC of 1924 ±223 vs. 2161 ±167 for ricin‐only treated). (C) TRAIL co‐treatment failed to influence ricin‐induced dTHP‐1 cell death even at much higher doses (12.5 ng/ml ricin). (D) Western blots analysis of A549 and dTHP‐1 cells demonstrate DR5/TRAILR2 expression in both cell types, the primary death receptor for TRAIL (Cell Signaling Technology Cat# 8074, RRID:AB_10950817). α‐tubulin served as the loading control (Cell Signaling Technology Cat# 5346, RRID:AB_1950376). Closed symbols, ricin‐only; open symbols, dual treatment, error bars represent the 95% CI. Green lines and dots and red lines and dots represent the mean and 95% CI of control‐ and ricin‐treated cells, respectively. Orange lines and dots represent the independent effect of TRAIL. Data presented are the mean of eight replicate wells with the 95% confidence interval. Graphical representation of the unnormalized data and results of pairwise statistical testing for treatment interactions are available in Figure S10. Refer to Figure S11 to observe the original films from which DR5 blot images were derived
4. DISCUSSION
Ricin is internalized by a wide variety of cell types and inactivates ribosomes with high efficiency. 5 However, the cellular and molecular mechanisms ultimately responsible for triggering PCD in response to ricin intoxication remain elusive, but clearly involve more than SRL depurination alone. 15 , 37 In this report, we confirmed that dTHP‐1 and A459 cells are differentially sensitive to ricin‐induced cell death: dTHP‐1 cells resemble alveolar macrophages (and Kupffer cells) in being hypersensitive to ricin intoxication, while A459 cells were confirmed to be relatively resistant to toxin‐induced killing. 18 , 23 Neither protein synthesis inhibition nor RSR activation per se could account for the different cell fates between dTHP‐1 and A549 cells. Rather, we put forth evidence that ER stress contributes of ricin‐induced cell death in dTHP‐1 cells, but not A549 cells. For secretory cells like macrophages, we speculate that perturbation of ER homeostasis, coupled with the ricin‐induced alterations in ribosome function overwhelm pro‐survival signaling and tip the scale toward PCD. 39 , 40 , 54 In the case of A549 cells (and possibly lung epithelial cells in general), we speculate that extracellular ligands such as TRAIL and TNFα synergize with the RSR to trigger ricin‐induced cell killing. 17 , 18 Ultimately, irrespective of cell type, “two‐hits” may be necessary to efficiently trigger ricin‐induced cell death (Figure 8).
FIGURE 8.
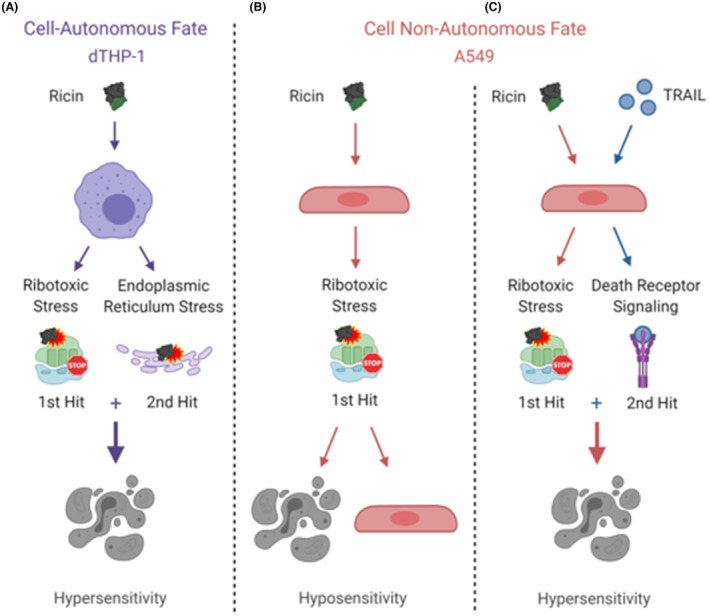
Two two‐hit models to explain cell type‐specific ricin sensitivity. Under normal conditions, dTHP‐1 cells exhibit ricin hypersensitivity (A), while A549 cells exhibit ricin hyposensitivity (B) based on the percent of cultured cells remaining alive after ricin challenge. We have identified ER stress and resulting activation of the unfolded protein response as a contributing factor to the unique sensitivity of dTHP‐1 cells, which are efficiently killed by cell‐autonomous ricin‐induced pro‐apoptotic signaling. Supplementation with TRAIL, a source of extrinsic pro‐apoptotic activation, is sufficient to induce cell non‐autonomous ricin hypersensitivity in A549 cells (C). Addition of TRAIL to dTHP‐1 cells had no effect on their response to ricin, further supporting their fully cell‐autonomous cell fate determination
We propose three (non‐exclusive) mechanisms by which ricin could induce ER stress. First, unfolding of RTA in the ER lumen prior to retro‐translocation may activate the sensor proteins IRE1, PERK, and ATF6 through removal of the chaperone BiP, which is known to bind ricin as a substrate. 55 Second, the subsequent escape of RTA from the ER depends on elements of the ER‐associated degradation (ERAD) pathway responsible for the removal of misfolded proteins from the ER. 41 , 56 , 57 , 58 It is possible that the partial engagement of ERAD by RTA reduces the flux of normal substrates, thereby triggering a stress response. Finally, after RTA had refolded on the cytoplasmic face of the ER membrane, it can, in theory, depurinate both ER‐associated ribosomes and those being recruited to the ER by the signal recognition particle (SRP) system. 37 The inability of stalled, depurinated ribosomes to engage with open translocon complexes could permit Ca2+ leakage and ER disequilibrium. 59 , 60 , 61 Depurination of already engaged ribosomes could also engage IRE1, which appears to surveille nascent peptides during co‐translational translocation and respond directly to proteostatic disturbances sensed in this way. 62 , 63 , 64 Although the mechanism by which ricin induces ER stress remains to be determined, participation of this stress response pathway in ricin‐induced cell death is consistent with the response of THP‐1 cells to Shiga toxin, a ribotoxin identical to ricin in its depurination activity of the SRL. 65
The hypersensitivity of dTHP‐1 cells to ricin was partially dependent on pro‐cell death UPR signaling downstream of IRE1, as evidenced by partial rescue with KIRA6. This effect is not reliant on IRE1’s RNase activity, as demonstrated by use of the inhibitor MKC8866, which suppresses IRE1’s mRNA substrate splicing without impinging on its kinase activity. This is consistent with studies performed in yeast that show ricin inhibits IRE1‐mediated mRNA splicing of its primary transcriptional regulatory effector HAC1, the homolog of mammalian XBP1. 50 , 51 Although activation of both PERK and IRE1 was observed in dTHP‐1 cells in response to ricin, only IRE1‐specific inhibition was sufficient to protect against toxin‐induced cell death. While IRE1 downstream activities are unique to ER stress, PERK acts as the ER stress responsive kinase of the integrated stress response (ISR). The ISR is activated by a wide variety of cellular stress conditions, detected by four specialized sensor kinases (including PERK) that initiate the same signaling cascade through phosphorylation of their common substrate, eIF2α. 11 Ribosome depurination directly activates the ISR kinase PKR, 66 which likely renders the downstream effects of PERK activation redundant in the determination of cell fate following ricin intoxication.
We chose not to examine the phosphorylation of eIF2α or expression of CHOP, which are commonly interpreted as markers of PERK activation in studies of ER stress, as these are general downstream effectors of ISR and interpretation would therefore be confounded by the known participation of PKR in the ribotoxic stress response. Similarly, we did not utilize alternative splicing of XBP1 as a readout of IRE1 activity due to the known inhibition of IRE1‐mediated splicing of the XBP‐1 homolog HAC1 by ricin in yeast. Reliance only upon the direct readout of phosphorylation states of IRE1 and PERK and the effect of specific chemical inhibitors of these proteins does necessarily provide a limited insight into the participation of UPR in the cellular response to ricin. However, this approach avoids known confounding inputs from other cellular processes involved in the response to ricin exposure.
While chemical induction of ERS was able to sensitize A549 cells to ricin, the effect was modest in comparison to the previously described interaction between TRAIL and ricin in A549 cells. This raised the question of whether or not TRAIL sensitization would generalize between cell types in a more substantial way. Participation of the pro‐apoptotic TNF superfamily cytokines TRAIL, TNF‐α, and FASL in the pathogenesis of ricin intoxication has been extensively characterized. Ricin exposure drives the production of TNF‐α in cultured and primary macrophages, 20 , 67 , 68 , 69 lung tissue, 70 BAL, 20 and primary tracheal epithelial cells. 14 In the human airway epithelial cell lines Calu‐3 and A549, TRAIL exposure significantly sensitized cells to ricin‐induced cell death. TNF‐α and FASL had a similarly potent sensitizing effect in A549 cells 17 while in Calu‐3 cells TNF‐α had a more modest effect. 18 Here, we extend these findings to position the activation of extrinsic pro‐apoptotic signaling through TRAIL as a cell type‐specific second hit that substantially determines cell fate in airway epithelial cells. In contrast, TRAIL had no significant effect on ricin‐induced cell death in dTHP‐1 cells, suggesting the role of pro‐apoptotic cytokines in ricin pathogenesis is also cell type‐specific. These findings strongly support previous descriptions of pulmonary exposure, in which macrophages rapidly bind and internalize RT in the lung and initiate pro‐inflammatory and pro‐apoptotic signaling processes, while ATII epithelial cells bind toxin relatively early but do not become apoptotic until many hours later in the pathogenic timeline. 71 We postulate that the lag between toxin reaching these cells and their eventual apoptotic response is due to reliance on pro‐apoptotic cytokine signaling to determine cell fate. The interaction between TRAIL and cell death in A549 cells is of particular biological interest for pulmonary ricin exposure, as soluble TRAIL produced by macrophages is a major driver of epithelial cell death and lung damage in respiratory pathogenesis. 72 , 73 , 74
CONFLICT OF INTEREST
The authors have no financial or other competing interests to declare.
AUTHOR CONTRIBUTIONS
CPR performed experiments, analyzed the results, and prepared the figures. CPR and NJM wrote the manuscript. NJM was responsible for project oversight and obtaining research funding support.
Supporting information
Figures S1‐S11
ACKNOWLEDGMENTS
We thank Dylan Ehrbar for his invaluable feedback on the statistical methods and gratefully acknowledge the Wadsworth Center’s Cell Culture and Media core facility for providing reagents. The graphics in Figure 8 were created with BioRender.com. This work was supported by AI125190 and contract HHSN272201400021C from the National Institutes of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Peterson‐Reynolds C, Mantis NJ. Differential ER stress as a driver of cell fate following ricin toxin exposure. FASEB BioAdvances. 2022;4:60–75. 10.1096/fba.2021-00005
REFERENCES
- 1. Gal Y, Mazor O, Falach R, Sapoznikov A, Kronman C, Sabo T. Treatments for pulmonary ricin intoxication: current aspects and future prospects. Toxins (Basel). 2017:9:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sapoznikov A, Gal Y, Falach R, et al. Early disruption of the alveolar‐capillary barrier in a ricin‐induced ARDS mouse model: neutrophil‐dependent and ‐independent impairment of junction proteins. Am J Physiol Lung Cell Mol Physiol. 2019;316:L255‐L268. [DOI] [PubMed] [Google Scholar]
- 3. Wong J, Magun BE, Wood LJ. Lung inflammation caused by inhaled toxicants: a review. Int J Chron Obstruct Pulmon Dis. 2016;11:1391‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rapak A, Falnes PO, Olsnes S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc Natl Acad Sci USA. 1997;94:3783‐3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sowa‐Rogozińska N, Sominka H, Nowakowska‐Gołacka J, Sandvig K, Słomińska‐Wojewódzka M Intracellular transport and cytotoxicity of the protein toxin ricin. Toxins. 2019;11(6):350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Endo Y, Mitsui K, Motizuki M, Tsurugi K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J Biol Chem. 1987;262:5908‐5912. [PubMed] [Google Scholar]
- 7. Endo Y, Tsurugi K. RNA N‐glycosidase activity of ricin A‐chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J Biol Chem. 1987;262:8128‐8130. [PubMed] [Google Scholar]
- 8. Endo Y, Tsurugi K. The RNA N‐glycosidase activity of ricin A‐chain. The characteristics of the enzymatic activity of ricin A‐chain with ribosomes and with rRNA. J Biol Chem. 1988;263:8735‐8739. [PubMed] [Google Scholar]
- 9. Iordanov MS, Pribnow D, Magun JL, et al. Ribotoxic stress response: activation of the stress‐activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence‐specific RNA damage to the alpha‐sarcin/ricin loop in the 28S rRNA. Mol Cell Biol. 1997;17:3373‐3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Korcheva V, Wong J, Corless C, Iordanov M, Magun B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am J Pathol. 2005;166:323‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vind AC, Genzor AV, Bekker‐Jensen S. Ribosomal stress‐surveillance: three pathways is a magic number. Nucleic Acids Res. 2020;48:10648‐10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vind AC, Snieckute G, Blasius M, et al. ZAKα recognizes stalled ribosomes through partially redundant sensor domains. Mol Cell. 2020;78:700.e707‐713.e707. [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Mader MM, Toth JE, et al. Complete inhibition of anisomycin and UV radiation but not cytokine induced JNK and p38 activation by an aryl‐substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J Biol Chem. 2005;280:19298‐19305. [DOI] [PubMed] [Google Scholar]
- 14. Wong J, Korcheva V, Jacoby DB, Magun B. Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am J Pathol. 2007;170:1497‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li XP, Baricevic M, Saidasan H, Tumer NE. Ribosome depurination is not sufficient for ricin‐mediated cell death in Saccharomyces cerevisiae . Infect Immun. 2007;75:417‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Falach R, Sapoznikov A, Gal Y, et al. Quantitative profiling of the in vivo enzymatic activity of ricin reveals disparate depurination of different pulmonary cell types. Toxicol Lett. 2016;258:11‐19. [DOI] [PubMed] [Google Scholar]
- 17. Hodges AL, Kempen CG, McCaig WD, Parker CA, Mantis NJ, LaRocca TJ. TNF family cytokines induce distinct cell death modalities in the A549 human lung epithelial cell line when administered in combination with ricin toxin. Toxins (Basel). 2019;11:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rong Y, Westfall J, Ehrbar D, LaRocca T, Mantis NJ. TRAIL (CD253) sensitizes human airway epithelial cells to toxin‐induced cell death. mSphere. 2018;3(5):e00399‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown RF, White DE. Ultrastructure of rat lung following inhalation of ricin aerosol. Int J Exp Pathol. 1997;78:267‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korcheva V, Wong J, Lindauer M, Jacoby DB, Iordanov MS, Magun B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol Immunol. 2007;44:2761‐2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lindauer ML, Wong J, Iwakura Y, Magun BE. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL‐1 signaling. J Immunol. 2009;183:1419‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bingen A, Creppy EE, Gut JP, Dirheimer G, Kirn A. The Kupffer cell is the first target in ricin‐induced hepatitis. J Submicrosc Cytol. 1987;19:247‐256. [PubMed] [Google Scholar]
- 23. Mooney B, Torres‐Velez FJ, Doering J, Ehrbar DJ, Mantis NJ. Sensitivity of Kupffer cells and liver sinusoidal endothelial cells to ricin toxin and ricin toxin‐Ab complexes. J Leukoc Biol. 2019;106:1161‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roche JK, Stone MK, Gross LK, et al. Post‐exposure targeting of specific epitopes on ricin toxin abrogates toxin‐induced hypoglycemia, hepatic injury, and lethality in a mouse model. Lab Invest. 2008;88:1178‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong M, Yu H, Wang Y, et al. Critical role of toll‐like receptor 4 (TLR4) in ricin toxin‐induced inflammatory responses in macrophages. Toxicol Lett. 2020;321:54‐60. [DOI] [PubMed] [Google Scholar]
- 26. Gage E, Hernandez MO, O'Hara JM, McCarthy EA, Mantis NJ. Role of the mannose receptor (CD206) in innate immunity to ricin toxin. Toxins (Basel). 2011;3:1131‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiao Z, Ke Y, Li S, et al. Pretreatment with retro‐2 protects cells from death caused by ricin toxin by retaining the capacity of protein synthesis. J Appl Toxicol. 2020;40:1440‐1450. [DOI] [PubMed] [Google Scholar]
- 28. Pratt TS, Pincus SH, Hale ML, Moreira AL, Roy CJ, Tchou‐Wong KM. Oropharyngeal aspiration of ricin as a lung challenge model for evaluation of the therapeutic index of antibodies against ricin A‐chain for post‐exposure treatment. Exp Lung Res. 2007;33:459‐481. [DOI] [PubMed] [Google Scholar]
- 29. Wahome PG, Ahlawat S, Mantis NJ. Identification of small molecules that suppress ricin‐induced stress‐activated signaling pathways. PLoS One. 2012;7:e49075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simmons BM, Stahl PD, Russell JH. Mannose receptor‐mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J Biol Chem. 1986;261:7912‐7920. [PubMed] [Google Scholar]
- 31. Reynolds‐Peterson C, Ehrbar DJ, McHale SM, LaRocca TJ, Mantis NJ. Sensitization of airway epithelial cells to toxin‐induced death by TNF superfamily cytokines. Methods Mol Biol. 2021;2248:19‐42. [DOI] [PubMed] [Google Scholar]
- 32. Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP‐1). Int J Cancer. 1980;26:171‐176. [DOI] [PubMed] [Google Scholar]
- 33. Auwerx J. The human leukemia cell line, THP‐1: a multifacetted model for the study of monocyte‐macrophage differentiation. Experientia. 1991;47:22‐31. [DOI] [PubMed] [Google Scholar]
- 34. Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP‐1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res. 2007;56:45‐50. [DOI] [PubMed] [Google Scholar]
- 35. Lieber M, Smith B, Szakal A, Nelson‐Rees W, Todaro G. A continuous tumor‐cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer. 1976;17:62‐70. [DOI] [PubMed] [Google Scholar]
- 36. Taubenschmid J, Stadlmann J, Jost M, et al. A vital sugar code for ricin toxicity. Cell Res. 2017;27:1351‐1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Szajwaj M, Wawiorka L, Molestak E, et al. The influence of ricin‐mediated rRNA depurination on the translational machinery in vivo – new insight into ricin toxicity. Biochim Biophys Acta Mol Cell Res. 2019;1866:118554. [DOI] [PubMed] [Google Scholar]
- 38. Ennis HL, Lubin M. Cycloheximide: aspects of inhibition of protein synthesis in mammalian cells. Science. 1964;146:1474‐1476. [DOI] [PubMed] [Google Scholar]
- 39. Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU. Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1. Front Mol Biosci. 2019;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Molinari M, Sitia R. The secretory capacity of a cell depends on the efficiency of endoplasmic reticulum‐associated degradation. Curr Top Microbiol Immunol. 2005;300:1‐15. [DOI] [PubMed] [Google Scholar]
- 41. Spooner RA, Watson PD, Marsden CJ, et al. Protein disulphide‐isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem J. 2004;383:285‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Spooner RA, Lord JM. Ricin trafficking in cells. Toxins (Basel). 2015;7:49‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89‐102. [DOI] [PubMed] [Google Scholar]
- 44. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081‐1086. [DOI] [PubMed] [Google Scholar]
- 45. Oslowski CM, Urano F. The binary switch that controls the life and death decisions of ER stressed β cells. Curr Opin Cell Biol. 2011;23:207‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elbein AD. Glycosylation inhibitors for N‐linked glycoproteins. Methods Enzymol. 1987;138:661‐709. [DOI] [PubMed] [Google Scholar]
- 47. Jämsä E, Simonen M, Makarow M. Selective retention of secretory proteins in the yeast endoplasmic reticulum by treatment of cells with a reducing agent. Yeast. 1994;10:355‐370. [DOI] [PubMed] [Google Scholar]
- 48. Price BD, Mannheim‐Rodman LA, Calderwood SK. Brefeldin A, thapsigargin, and AIF4‐ stimulate the accumulation of GRP78 mRNA in a cycloheximide dependent manner, whilst induction by hypoxia is independent of protein synthesis. J Cell Physiol. 1992;152:545‐552. [DOI] [PubMed] [Google Scholar]
- 49. Xie Q, Khaoustov VI, Chung CC, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress‐induced caspase‐12 activation. Hepatology. 2002;36:592‐601. [DOI] [PubMed] [Google Scholar]
- 50. Pierce M, Vengsarkar D, McLaughlin JE, Kahn JN, Tumer NE. Ribosome depurination by ricin leads to inhibition of endoplasmic reticulum stress‐induced HAC1 mRNA splicing on the ribosome. J Biol Chem. 2019;294:17848‐17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parikh BA, Tortora A, Li XP, Tumer NE. Ricin inhibits activation of the unfolded protein response by preventing splicing of the HAC1 mRNA. J Biol Chem. 2008;283:6145‐6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935‐939. [DOI] [PubMed] [Google Scholar]
- 53. Lu M, Lawrence DA, Marsters S, et al. Opposing unfolded‐protein‐response signals converge on death receptor 5 to control apoptosis. Science. 2014;345:98‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Spencer SL, Sorger PK. Measuring and modeling apoptosis in single cells. Cell. 2011;144:926‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gregers TF, Skanland SS, Walchli S, Bakke O, Sandvig K. BiP negatively affects ricin transport. Toxins (Basel). 2013;5:969‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hazes B, Read RJ. Accumulating evidence suggests that several AB‐toxins subvert the endoplasmic reticulum‐associated protein degradation pathway to enter target cells. Biochemistry. 1997;36:11051‐11054. [DOI] [PubMed] [Google Scholar]
- 57. Horrix C, Raviv Z, Flescher E, Voss C, Berger MR. Plant ribosome‐inactivating proteins type II induce the unfolded protein response in human cancer cells. Cell Mol Life Sci. 2011;68:1269‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Slominska‐Wojewodzka M, Gregers TF, Walchli S, Sandvig K. EDEM is involved in retrotranslocation of ricin from the endoplasmic reticulum to the cytosol. Mol Biol Cell. 2006;17:1664‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van Coppenolle F, Vanden Abeele F, Slomianny C, et al. Ribosome‐translocon complex mediates calcium leakage from endoplasmic reticulum stores. J Cell Sci. 2004;117:4135‐4142. [DOI] [PubMed] [Google Scholar]
- 60. Ong HL, Liu X, Sharma A, Hegde RS, Ambudkar IS. Intracellular Ca(2+) release via the ER translocon activates store‐operated calcium entry. Pflugers Arch. 2007;453:797‐808. [DOI] [PubMed] [Google Scholar]
- 61. Johnson CE, Hunt DK, Wiltshire M, et al. Endoplasmic reticulum stress and cell death in mTORC1‐overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol Oncol. 2015;9:675‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Acosta‐Alvear D, Karagöz GE, Fröhlich F, Li H, Walther TC, Walter P. The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. eLife. 2018;7:e43036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Oyadomari S, Yun C, Fisher EA, et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126:727‐739. [DOI] [PubMed] [Google Scholar]
- 64. Plumb R, Zhang ZR, Appathurai S, Mariappan M. A functional link between the co‐translational protein translocation pathway and the UPR. eLife. 2015;4:e07426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee SY, Lee MS, Cherla RP, Tesh VL. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008;10:770‐780. [DOI] [PubMed] [Google Scholar]
- 66. Zhou HR, He K, Landgraf J, Pan X, Pestka JJ. Direct activation of ribosome‐associated double‐stranded RNA‐dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: a new model for ribotoxic stress response induction. Toxins (Basel). 2014;6:3406‐3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Licastro F, Morini MC, Bolognesi A, Stirpe F. Ricin induces the production of tumour necrosis factor‐alpha and interleukin‐1 beta by human peripheral‐blood mononuclear cells. Biochem J. 1993;294(Pt 2):517‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hassoun E, Wang X. Ricin‐induced toxicity in the macrophage J744A.1 cells: the role of TNF‐alpha and the modulation effects of TNF‐alpha polyclonal antibody. J Biochem Mol Toxicol. 2000;14:95‐101. [DOI] [PubMed] [Google Scholar]
- 69. Higuchi S, Tamura T, Oda T. Cross‐talk between the pathways leading to the induction of apoptosis and the secretion of tumor necrosis factor‐alpha in ricin‐treated RAW 264.7 cells. J Biochem (Tokyo). 2003;134:927‐933. [DOI] [PubMed] [Google Scholar]
- 70. David J, Wilkinson LJ, Griffiths GD. Inflammatory gene expression in response to sub‐lethal ricin exposure in Balb/c mice. Toxicology. 2009;264:119‐130. [DOI] [PubMed] [Google Scholar]
- 71. Sapoznikov A, Falach R, Mazor O, et al. Diverse profiles of ricin‐cell interactions in the lung following intranasal exposure to ricin. Toxins (Basel). 2015;7:4817‐4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Braithwaite AT, Marriott HM, Lawrie A. Divergent roles for TRAIL in lung diseases. Front Med (Lausanne). 2018;5:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Herold S, Steinmueller M, von Wulffen W, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage‐expressed TNF‐related apoptosis‐inducing ligand. J Exp Med. 2008;205:3065‐3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Peteranderl C, Morales‐Nebreda L, Selvakumar B, et al. Macrophage‐epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J Clin Invest. 2016;126:1566‐1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S11