


Abstract
APPRIS (https://appris.bioinfo.cnio.es) is a well-established database housing annotations for protein isoforms for a range of species. APPRIS selects principal isoforms based on protein structure and function features and on cross-species conservation. Most coding genes produce a single main protein isoform and the principal isoforms chosen by the APPRIS database best represent this main cellular isoform. Human genetic data, experimental protein evidence and the distribution of clinical variants all support the relevance of APPRIS principal isoforms. APPRIS annotations and principal isoforms have now been expanded to 10 model organisms. In this paper we highlight the most recent updates to the database. APPRIS annotations have been generated for two new species, cow and chicken, the protein structural information has been augmented with reliable models from the EMBL-EBI AlphaFold database, and we have substantially expanded the confirmatory proteomics evidence available for the human genome. The most significant change in APPRIS has been the implementation of TRIFID functional isoform scores. TRIFID functional scores are assigned to all splice isoforms, and APPRIS uses the TRIFID functional scores and proteomics evidence to determine principal isoforms when core methods cannot.
INTRODUCTION
Protein coding genes generate multiple distinct transcript species through alternative splicing (1). The predicted translated gene products—alternative splice isoforms—have unique, but related amino acid sequences. Model genomes are annotated with large numbers of alternative coding transcripts, which can theoretically be translated into isoforms that might associate with different protein partners (2), bind distinct ligands (3), function in specific tissues (4) or even have completely unrelated functions (5,6). The GENCODE v38 human reference set (7), for example, annotates 63,968 sequence distinct protein isoforms for 19 955 coding genes.
One unresolved issue is how many alternative transcripts give rise to functional proteins. Although some transcript variants have ancient origins (8) or are translated in a tissue-specific manner (9), most alternative isoforms have little evidence beyond their expression at the transcript level (10,11) to support a relevant cellular role (12). Most alternative transcripts are primate-derived (8,9) and the vast majority appear not to be under selective pressure (12,13). Reliable proteomics experiments find orders of magnitude less evidence for alternative proteins than would be expected (14).
APPRIS is a database that contains annotations for alternatively spliced proteins for a range of model species (15). APPRIS maps protein structure and function information to splice isoforms and generates cross-species conservation scores. We developed APPRIS within the GENCODE (7) consortium, and GENCODE manual curators use information from the database to update gene models. The functional and structural information provided by APPRIS is limited to the most reliably predicted features, including the presence of Pfam domains (16) and highly conserved functional residues (17).
The primary purpose of APPRIS is the selection of principal isoforms for coding genes (15,18). APPRIS principal isoforms are more than just the representative protein isoform for each gene; we have shown that principal isoforms also best represent the biological reality of the cell (19,20). Selecting a principal isoform for each gene sets APPRIS apart from the other three databases of protein features and annotated alternative splice variants that are currently maintained (21–23).
We have shown that APPRIS principal isoforms coincide with the main variant detected in proteomics experiments (19,20) and with variants selected by Ensembl (24) and RefSeq (25) manual annotators (19). In addition, transcripts that generate APPRIS principal isoforms are under purifying selection while annotated alternative transcript variants, as a whole, are not (12,13). APPRIS principal transcripts even account for all but 0.14% of annotated pathogenic mutations (20).
There are currently no other available methods for determining principal isoforms. Although methods based on RNAseq information have been trialled (26,27), they do not function well (19), and have not been scaled up. Reference variants have been built into the main databases. UniProtKB (28) display isoforms are generally the longest known isoform at the time of their annotation. We have shown that selecting the longest isoform has few advantages over using RNAseq data (19). Ensembl and RefSeq together have generated a set of semi-manually curated main transcripts called MANE Select (20). These transcripts are supported by experimental evidence and are supposed to represent the biology of the coding gene, but are only annotated for the human genome. APPRIS principal isoforms are part of the input to the MANE Select curation process, and MANE Select and principal isoforms selected by APPRIS core methods agree over 97.2% of coding genes.
The presence or absence of evolutionary features is key to determining principal isoforms in APPRIS. The greater the cross species conservation and the more preserved structural and functional features an isoform has, the more likely it is to be selected as the principal isoform (18,29). The structural and functional features and cross species conservation generated by the APPRIS core modules are enough to determine a principal isoform for the vast majority of genes, though on occasions the database has to make use of external information to break a tie.
Here, we detail the updates to the APPRIS database since the last publication. In addition to the many improvements to core methods and outputs in the APPRIS pipeline, we have added a new external machine learning method, TRIFID (30), to help determine Principal Isoforms when the APPRIS core modules require a tie-breaker. As well as its role in determining Principal Isoforms, TRIFID also provides a relative functional score for all splice isoforms. We have also extended APPRIS to the cow and chicken genomes, added extensive proteomics support and incorporated AlphaFold models (31) into the Matador3D module (29).
THE DATABASE
The APPRIS database selects a single reference isoform for protein coding genes for a range of model species. This reference isoform, the principal isoform, is chosen using a rule-based system that makes use of the protein structural and functional features and cross-species alignments provided by the core modules of the APPRIS database (29). The APPRIS principal isoform is almost always the isoform with the most cross-species conservation and the isoform that preserves most of the conserved structural and functional features (Figure 1).
Figure 1.
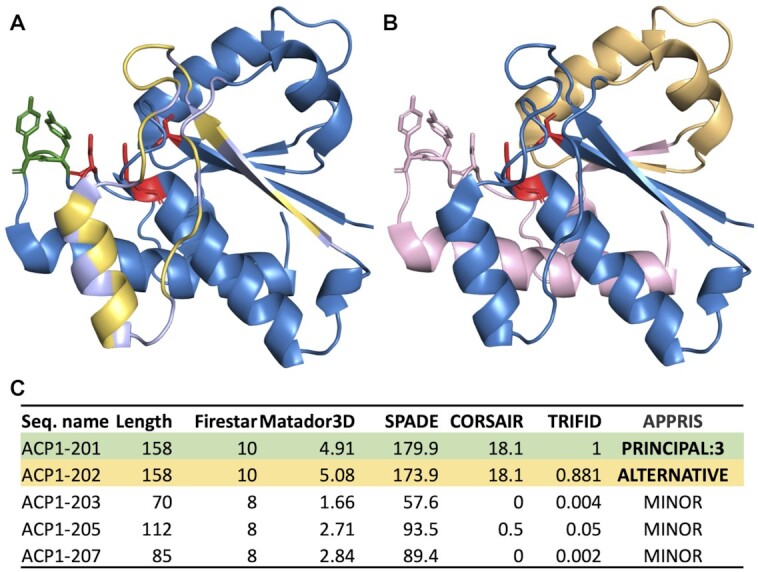
APPRIS annotations for ACP1 from the GENCODE v38 reference set. Panels A and B show PDB structure 4Z99, the resolved structure of isoform A of low molecular weight protein tyrosine phosphatase (ACP1-001 in the GENCODE annotation), onto which has been mapped the effects of alternative splicing for two different annotated isoforms, ACP1-002 and ACP1-005. Known catalytic residues are shown as red sticks, Src-phosphorylated tyrosines are shown in green. Panel A shows variant ACP1-002, which differs from ACP1-001 by a single tandem duplicated homologous exon. Residues identical between ACP1-001 and ACP1-002 in the exon are shown in light blue; those that differ are shown in light yellow. Catalytic and phosphorylated residues are not affected by this exon. Panel B shows variant ACP1-005. In this case the second tandem duplicated exon is read in a different frame leading to a premature stop codon. The unrelated sequence from the frameshifted amino acids is mapped onto the structure in light orange, but these residues are likely to be unfolded. The remaining protein structure of ACP1-001 (shown in light pink) would be lost in this isoform, eliminating one of the catalytic residues and both phosphorylated tyrosines. Isoforms generated from ACP1-003 and ACP1-007 swap even more of the C-terminal region of the principal isoform for unrelated residues and premature stop codons. Both lose the same catalytic and phosphorylated residues as ACP1-005. Panel C shows the scores from the APPRIS modules for the five isoforms. The principal isoform scores are shown with a green background, the ‘alternative’ isoform with an orange background. The scores for variant ACP1-002 are so similar to those of the principal isoform that APPRIS has to determine the principal isoform from external methods. ACP1-001 is chosen as the principal isoform on the strength of proteomics evidence (PRINCIPAL:3). TRIFID predicts that ‘ALTERNATIVE’ variant ACP1-002 is highly likely to be functionally important as a protein. The remaining three isoforms lose protein structure, functional domains and residues and have no detectable cross-species conservation. TRIFID predicts that they will not be functionally relevant at the protein level.
APPRIS consists of four core modules. Matador3D maps PDB protein structures (32) to the annotated splice variants, SPADE maps Pfam functional domains (16), and firestar maps functionally important amino acid residues (17). CORSAIR carries out BLAST (33) searches for orthologues that align without gaps. Two further modules, not used in principal Isoform selection, map trans-membrane helix and signal peptide predictions to the isoforms (29). Principal isoforms are selected in five different steps; those that are selected by the core modules are tagged as ‘PRINCIPAL:1’. In the case of a tie, APPRIS then makes use of external data to select one of the isoforms as principal (29). Isoforms rejected by the core modules are tagged as MINOR, and those that are tied after the core modules, but not later selected as principal, are tagged as ALTERNATIVE (Figure 1).
Refinements to core modules
We have made substantial refinements to the functioning of the APPRIS core modules in order to improve principal isoform selection. For example, SPADE now calculates two scores for Pfam functional domain conservation, a domain integrity score that measures whether or not Pfam domains are intact, and a score based on Pfamscan (16) bitscores. The final APPRIS score is calculated in two phases: first using the bitscore, and using SPADE domain integrity score only if necessary to break ties.
We have made several improvements to the Matador3D module. In particular, we updated the search databases. First we used PDBsum sequences (34) to avoid unreliable unstructured regions, and second we added predicted structures from the EMBL-EBI AlphaFold Protein Structure Database (31). AlphaFold models were first filtered for integrity; trailing unstructured regions at the N- and C-terminals were trimmed and models with more than four consecutive poor scoring amino acid residues inserted into high scoring model regions were rejected (see Supplementary Materials and Figures S1 and S2 for more details). We added AlphaFold models for the six available Bilaterian genomes (human, mouse, rat, zebrafish, Drosophila melanogaster and Caenorhabditis elegans) to the Matador3D search database. The Matador3D module searches against all AlphaFold models as if they were resolved protein structures.
TRIFID functional importance scores
We recently developed TRIFID, a machine learning-based tool to predict the functional importance of splice isoforms. Alternative transcripts predicted by TRIFID to produce functionally important proteins are under purifying selection, while low scoring alternative transcripts generally evolve under neutral selection pressure (30). We have incorporated TRIFID scores into APPRIS and the gene pages show the TRIFID score for principal and alternative isoforms [see supplementary materials]. TRIFID functional importance scores are available for annotated isoforms of all species, but at present are only available for the Ensembl annotations. However, TRIFID scores are available for the RefSeq human reference set, both for version 109 and for version 105 (from build GRCh37).
The use of TRIFID in non-human species is discussed in the supplementary materials.
Additional features for the human reference set
Isoforms from the human reference set are supported by peptide evidence from proteomics experiments. We have substantially expanded the proteomics coverage in the most recent version of the human reference set by including peptides identified in five different proteomics analyses (35–39). In GENCODE v24, we mapped 147 669 peptides to the human reference set; this has grown to 339 442 peptides in GENCODE v38, an increase of almost 130%. This proteomics data is now also used to help determine principal isoforms where the core modules cannot determine a clear principal isoform.
The APPRIS database now also includes a list of known functional alternative isoforms for the human reference set. These small sets of functional isoforms came from large-scale studies that we carried out (8,9) and will be added to as we expand this work. The functional isoforms are tied to a specific version of GENCODE because identifiers sometimes change with the version.
Beyond the core modules
Not all APPRIS principal isoforms are alike. There are five types of principal isoforms, tagged with a score from 1 to 5. The tag depends on the method used to make the final selection. ‘PRINCIPAL:1’ isoforms, as already mentioned, are determined solely using information from the APPRIS core modules, while methods external to the core modules select the remaining principal isoform types.
APPRIS has a new selection process for the PRINCIPAL:2 through to PRINCIPAL:5 isoforms. For all Ensembl annotated species, and for RefSeq versions 109 and 105 of the human reference set, APPRIS uses the TRIFID functional isoform score and the proteomics evidence to break ties. The pipeline that determines the PRINCIPAL:2 through to PRINCIPAL:5 isoforms only activates if the APPRIS core methods have not been able to determine a PRINCIPAL:1 isoform. Although the core methods might not have selected a principal isoform, they will have ranked the isoforms and sorted the isoforms into two groups: those that could be the principal isoform, and those that cannot be the principal isoform. Isoforms rejected by the core methods cannot be the principal isoform. These rejected isoforms are tagged as MINOR and play no part in the PRINCIPAL:2 to PRINCIPAL:5 isoform selection process. The details of the PRINCIPAL:2 to PRINCIPAL:5 selection process can be found in the supplementary materials.
Both the new PRINCIPAL:2 and PRINCIPAL:3 steps are a substantial improvement on the previous selection procedure for human genes, as demonstrated by the 10% improvement in agreement with the MANE Select variant (20). At present, it is only possible to determine PRINCIPAL:3 for the human reference set, but using TRIFID scores to determine PRINCIPAL:2 and PRINCIPAL:4 isoforms is a considerable advance for non-human species where APPRIS was previously forced to use length as the only tie-breaker.
Species annotated in APPRIS
Two new species have been added to the APPRIS database, cow and chicken. This brings the number of Ensembl vertebrate species with annotations in APPRIS to eight (human, chimpanzee, mouse, rat, pig, cow, chicken and zebrafish) to go with the two Ensembl versions of model invertebrate genomes (D. melanogaster (40) and C. elegans (41)). APPRIS also annotates principal isoforms for the RefSeq and UniProtKB annotations of the human, chimpanzee, mouse, rat, pig, and zebrafish gene sets.
Upgrades to the core methods in APPRIS, the addition of the AlphaFold models to the Matador module and the new TRIFID-based selection process have brought about improvements in principal isoform coverage, particularly in species other than human and mouse. The changes in principal isoform coverage for human, mouse, chicken and D. melanogaster can be seen in Figure 2. Using TRIFID to distinguish principal isoforms reclassifies 30% of PRINCIPAL:5 isoforms in cow and 40% of PRINCIPAL:5 isoforms in chicken (Supplementary Figure S3). The changes in all methods can be seen in Supplementary Table S1.
Figure 2.

Improvements in principal isoform coverage in four model species. Bar charts showing the changes in principal isoform coverage with the improvements to the core methods and the change to the new TRIFID-based selection process. The distribution of principal isoforms before the changes is shown in the lighter colour (and labelled ‘Classic’) and the distribution after the changes is in darker bars (‘TRIFID’). Principal isoform types are labelled as P:1 etc. where P:1 is short for PRINCIPAL:1. From top left, (A) human, (B) mouse, (C) chicken, (D) D. melanogaster. There were no P2 or P4 isoforms in chicken or D. melanogaster prior to the improvements because these species are not annotated with CCDS (42) evidence. TRIFID plays no part in the selection of P:1 isoforms, Improvements here are due to the core methods and include the effects of adding AlphaFold models. The TRIFID selection process means that there are now very few P:5 isoforms in any species. Full details can be found in the supplementary materials.
Adding AlphaFold models increased the number of genes with 3D structure coverage by 21% in D. melanogaster, just over 14% in chicken and by 4% in human. AlphaFold models improved PRINCIPAL:1 coverage by just one percentage point in cow and chicken and half a percentage point among human genes, because there are three other core methods besides Matador3D that determine principal isoforms.
The clearest effect of using TRIFID as part of the selection process is that almost all of the genes with less reliably selected PRINCIPAL:5 isoforms now have the more reliable PRINCIPAL:2 or PRINCIPAL:4 isoforms (Figure 2).
DISCUSSION
APPRIS selects a single sequence-unique isoform to be the representative protein for each coding gene. These principal isoforms are chosen based on cross-species conservation and the presence or absence of conserved protein structural and functional features. Until now, large-scale research projects and databases have chosen the longest CDS as the reference variant for coding genes. We have shown that the use of APPRIS principal isoforms substantially improves on this strategy (19,20).
Experimental evidence strongly suggests that most coding genes have a single dominant protein isoform (19). Although there are genes that have clear tissue-specific isoforms (9), most dominant isoforms appear to be dominant regardless of cell type (19). APPRIS is the best predictor of the main protein isoform; APPRIS PRINCIPAL:1 principal isoforms agree with the main experimental proteomics isoform in more than 96% of comparable genes (20), and exons from principal transcripts are under selective pressure, while alternative exons are not (12). What is more, we found that only 48 of the almost 35 000 ClinVar (43) pathogenic or likely pathogenic mutations we analysed (0.14%) affect alternative coding exons, while the rest have their effect on exons that code for APPRIS principal isoforms, or on conserved non-coding regions (20).
Large-scale analyses often require a single representative transcript or isoform for technical reasons. Since results depend on the quality of input data, choosing the APPRIS principal isoform as the main representative should be a critical first step for any genome-wide analysis. APPRIS principal isoforms also capture almost all pathogenic variants, illustrating the importance of using principal isoforms in the clinical interpretation of biomedical data too.
The TRIFID functional relevance scores available via APPRIS are important for researchers working with individual genes: it is not always clear which splice isoform (or isoforms) of a coding gene is functionally important, and this will become more difficult as the number of annotated splice isoforms grow. We have shown that the higher the TRIFID scores for an alternative variant, the more likely it is to be under purifying selection (30). In addition, we have found that exons from the highest scoring TRIFID variants are significantly more likely to have validated pathogenic mutations, while the 85% of alternative exons from transcripts with TRIFID scores of less than 0.2 have practically no pathogenic mutations (20).
APPRIS principal isoforms and the associated annotations are freely accessible through the APPRIS web page and APPRIS WebServices (44), via the GENCODE and Ensembl reference sets, and the UCSC gene browsers (45).
DATA AVAILABILITY
APPRIS principal isoforms, scores and the annotations from the individual methods can be downloaded from the APPRIS web site (https://apprisws.bioinfo.cnio.es/pub/). Ensembl includes principal isoforms for the human, mouse, zebrafish, rat, and pig genomes within its website, BioMart data-mining tool, and API, and human and principal isoforms annotations are available in GENCODE data files.
Principal isoforms for the Ensembl reference set and the predictions from the APPRIS core modules can be visualized in the UCSC Genome Browser with the Principal Splice Isoforms APPRIS Track Hub. Annotations are available for human, chimpanzee, mouse, rat, pig, zebrafish, D. Melanogaster and C. Elegans.
In addition, users can extract APPRIS annotations for specific reference sets (Ensembl, RefSeqGene, UniProtKB) via the APPRIS WebServer and WebServices. The APPRIS WebServices allows users to query further Ensembl vertebrate species (44).
All APPRIS source code is available with distributed version control in a GitHub public-repository (https://github.com/appris/appris/). The APPRIS pipeline is executed on Linux (Ubuntu). However, it can be run on Windows, Mac OS X, or Unix-based systems via its Docker image (appris/core). The APPRIS-Docker image (https://hub.docker.com/r/appris/core) is stored by the software container platform Docker in the public Docker Hub.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Thomas Walsh for all his work on the APPRIS database.
Contributor Information
Jose Manuel Rodriguez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain.
Fernando Pozo, Bioinformatics Institute, Spanish National Cancer Research Centre (CNIO), Madrid, 28029, Spain.
Daniel Cerdán-Vélez, Bioinformatics Institute, Spanish National Cancer Research Centre (CNIO), Madrid, 28029, Spain.
Tomás Di Domenico, Bioinformatics Institute, Spanish National Cancer Research Centre (CNIO), Madrid, 28029, Spain.
Jesús Vázquez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain; CIBER de Enfermedades Cardiovasculares (CIBERCV), 28029 Madrid, Spain.
Michael L Tress, Bioinformatics Institute, Spanish National Cancer Research Centre (CNIO), Madrid, 28029, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Human Genome Research Institute of the National Institutes of Health [2 U41 HG007234]; Spanish Ministry of Science, Innovation and Universities [PGC2018-097019-B-I00]; Carlos III Institute of Health-Fondo de Investigación Sanitaria [PRB3 (IPT17/0019—ISCIII-SGEFI/ERDF, ProteoRed]; ‘la Caixa’ Banking Foundation [HR17-00247]. Funding for open access charge: National Human Genome Research Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1. Smith C.W., Valcárcel J.. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 2000; 25:381–388. [DOI] [PubMed] [Google Scholar]
- 2. Yeh B.K., Igarashi M., Eliseenkova A.V., Plotnikov A.N., Sher I., Ron D., Aaronson S.A., Mohammadi M.. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:2266–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oaxaca-Castillo D., Andreoletti P., Vluggens A., Yu S., van Veldhoven P.P., Reddy J.K., Cherkaoui-Malki M.. Biochemical characterization of two functional human liver acyl-CoA oxidase isoforms 1a and 1b encoded by a single gene. Biochem. Biophys. Res. Commun. 2007; 360:314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Endo M., Druso J.E., Cerione R.A.. The two splice variant forms of Cdc42 exert distinct and essential functions in neurogenesis. J. Biol. Chem. 2020; 295:4498–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hernandez D.A., Bennett C.M., Dunina-Barkovskaya L., Wedig T., Capetanaki Y., Herrmann H., Conover G.M.. Nebulette is a powerful cytolinker organizing desmin and actin in mouse hearts. Mol. Biol. Cell. 2016; 27:3869–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Myers K.R., Yu K., Kremerskothen J., Butt E., Zheng J.Q.. The nebulin family LIM and SH3 proteins regulate postsynaptic development and function. J. Neurosci. 2020; 40:526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Frankish A, Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martinez Gomez L., Pozo F., Walsh T.A., Abascal F., Tress M.L.. The clinical importance of tandem exon duplication-derived substitutions. Nucleic Acids Res. 2021; 49:8232–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodriguez J.M., Pozo F., di Domenico T., Vazquez J., Tress M.L.. An analysis of tissue-specific alternative splicing at the protein level. PLoS Comp. Biol. 2020; 16:e1008287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B.. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reixachs-Solé M., Ruiz-Orera J., Albà M.M., Eyras E.. Ribosome profiling at isoform level reveals evolutionary conserved impacts of differential splicing on the proteome. Nat. Commun. 2020; 11:1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tress M.L., Abascal F., Valencia A.. Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci. 2017; 42:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu T., Lin K.. The distribution pattern of genetic variation in the transcript isoforms of the alternatively spliced protein-coding genes in the human genome. Mol. Biosyst. 2015; 11:1378–1388. [DOI] [PubMed] [Google Scholar]
- 14. Abascal F., Ezkurdia I., Rodriguez-Rivas J., Rodriguez J.M., del Pozo A., Vázquez J., Valencia A., Tress M.L.. Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput Biol. 2015; 11:e1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez J.M., Rodriguez-Rivas J, Di Domenico T., Vázquez J., Valencia A., Tress M.L.. APPRIS 2017: principal isoforms for multiple gene sets. Nucleic Acids Res. 2018; 46:D213–D217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G.A., Sonnhammer E.L.L., Tosatto S.C.E., Paladin L., Raj L.J., Richardson R.D.et al.. Pfam: the protein families database in 2021. Nucleic. Acids. Res. 2020; 49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lopez G., Maietta P., Rodriguez J.M., Valencia A., Tress M.L.. firestar–advances in the prediction of functionally important residues. Nucleic Acids Res. 2011; 39:W235–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tress M.L., Wesselink J-J., Frankish A., López G., Goldman N., Löytynoja A., Massinghamm T., Pardi F., Whelan S., Harrow J., Valencia A.. Determination and validation of principal gene products. Bioinformatics. 2008; 24:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ezkurdia I., Rodriguez J.M., Carrillo-de Santa Pau E., Vázquez J., Valencia A., Tress M.L.. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 2015; 14:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pozo F., Rodriguez J.M., Vazquez J., Tress M.L.. APPRIS principal isoforms and MANE select transcripts in clinical variant interpretation. 2021; bioRxiv doi:20 September 2021, preprint: not peer reviewed 10.1101/2021.09.17.460749. [DOI] [PMC free article] [PubMed]
- 21. Birzele F., Küffner R., Meier F., Oefinger F., Potthast C., Zimmer R.. ProSAS: a database for analyzing alternative splicing in the context of protein structures. Nucleic Acids Res. 2008; 36:D63–D68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shionyu M., Yamaguchi A., Shinoda K., Takahashi K., Go M.. AS-ALPS: a database for analyzing the effects of alternative splicing on protein structure, interaction and network in human and mouse. Nucleic Acids Res. 2009; 37:D305–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martelli P.L., D’Antonio M., Bonizzoni P., Castrignanò T., D’Erchia A.M., D’Onorio De Meo P., Fariselli P., Finelli M., Licciulli F.et al.. ASPicDB: a database of annotated transcript and protein variants generated by alternative splicing. Nucleic Acids Res. 2011; 39:D80–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howe K.L., Achuthan P., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R., Bhai J.et al.. Ensembl 2021. Nucleic Acids Res. 2021; 49:D884–D891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D.et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzàlez-Porta M., Frankish A., Rung J., Harrow J., Brazma A.. Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol. 2013; 14:R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H.D., Menon R., Govindarajoo B., Panwar B., Zhang Y., Omenn G.S., Guan Y.. Functional Networks of Highest-Connected Splice Isoforms: From The Chromosome 17 Human Proteome Project. J. Proteome Res. 2015; 14:3484–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2017; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodriguez J.M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J.J., Lopez G., Valencia A., Tress M.L.. APPRIS: annotation of principal and alternative splice isoforms. Nucleic Acids Res. 2013; 41:D110–D117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pozo F., Martinez-Gomez L., Walsh T.A., Rodriguez J.M., Di Domenico T., Abascal F., Vazquez J., Tress M.L.. Assessing the functional relevance of splice isoforms. NAR Genom. Bioinform. 2021; 3:lqab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burley S.K., Bhikadiya C., Bi C., Bittrich S., Chen L., Crichlow G.V., Christie C.H., Dalenberg K., Di Costanzo L., Duarte J.M.et al.. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021; 49:D437–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J.. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laskowski R.A., Jabłońska J., Pravda L., Vařeková R.S., Thornton J.M.. PDBsum: structural summaries of PDB entries. Protein Sci. 2018; 27:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S.et al.. A draft map of the human proteome. Nature. 2014; 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bekker-Jensen D.B., Kelstrup C.D., Batth T.S., Larsen S.C., Haldrup C., Bramsen J.B., Sørensen K.D., Høyer S., Ørntoft T.F., Andersen C.L.et al.. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 2017; 4:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carlyle B.C., Kitchen R.R., Kanyo J.E., Voss E.Z., Pletikos M., Sousa A.M.M., Lam T.T., Gerstein M.B., Sestan N., Nairn A.C.. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017; 20:1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schiza C., Korbakis D., Jarvi K., Diamandis E.P., Drabovich A.P.. Identification of TEX101-associated proteins through proteomic measurement of human spermatozoa homozygous for the missense variant rs35033974. Mol Cell Proteomics. 2019; 18:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang D., Eraslan B., Wieland T., Hallström B., Hopf T., Zolg D.P., Zecha J., Asplund A., Li L.H., Meng C.et al.. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019; 15:e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thurmond J., Goodman J.L., Strelets V.B., Attrill H., Gramates L.S., Marygold S.J., Matthews B.B., Millburn G., Antonazzo G., Trovisco V.et al.. FlyBase 2.0: the next generation. Nucleic Acids Res. 2019; 47:D759–D765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dubaj Price M., Hurd D.D.. WormBase: a model organism database. Med. Ref. Serv. Q. 2019; 38:70–80. [DOI] [PubMed] [Google Scholar]
- 42. Harte R.A., Farrell C.M., Loveland J.E., Suner M.M., Wilming L., Aken B., Barrell D., Frankish A., Wallin C., Searle S.et al.. Tracking and coordinating an international curation effort for the CCDS Project. Database. 2012; 2012:bas008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Landrum M.J., Chitipiralla S., Brown G.R., Chen C., Gu B., Hart J., Hoffman D., Jang W., Kaur K., Liu C.et al.. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020; 48:D835–D844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodriguez J.M., Carro A., Valencia A., Tress M.L.. APPRIS WebServer and WebServices. Nucleic Acids Res. 2015; 43:W455–W459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M.et al.. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021; 49:D1046–D1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
APPRIS principal isoforms, scores and the annotations from the individual methods can be downloaded from the APPRIS web site (https://apprisws.bioinfo.cnio.es/pub/). Ensembl includes principal isoforms for the human, mouse, zebrafish, rat, and pig genomes within its website, BioMart data-mining tool, and API, and human and principal isoforms annotations are available in GENCODE data files.
Principal isoforms for the Ensembl reference set and the predictions from the APPRIS core modules can be visualized in the UCSC Genome Browser with the Principal Splice Isoforms APPRIS Track Hub. Annotations are available for human, chimpanzee, mouse, rat, pig, zebrafish, D. Melanogaster and C. Elegans.
In addition, users can extract APPRIS annotations for specific reference sets (Ensembl, RefSeqGene, UniProtKB) via the APPRIS WebServer and WebServices. The APPRIS WebServices allows users to query further Ensembl vertebrate species (44).
All APPRIS source code is available with distributed version control in a GitHub public-repository (https://github.com/appris/appris/). The APPRIS pipeline is executed on Linux (Ubuntu). However, it can be run on Windows, Mac OS X, or Unix-based systems via its Docker image (appris/core). The APPRIS-Docker image (https://hub.docker.com/r/appris/core) is stored by the software container platform Docker in the public Docker Hub.