


Abstract
Small non-coding RNAs (sncRNAs) are pervasive regulators of physiological and pathological processes. We previously developed the human miRNA Tissue Atlas, detailing the expression of miRNAs across organs in the human body. Here, we present an updated resource containing sequencing data of 188 tissue samples comprising 21 organ types retrieved from six humans. Sampling the organs from the same bodies minimizes intra-individual variability and facilitates the making of a precise high-resolution body map of the non-coding transcriptome. The data allow shedding light on the organ- and organ system-specificity of piwi-interacting RNAs (piRNAs), transfer RNAs (tRNAs), microRNAs (miRNAs) and other non-coding RNAs. As use case of our resource, we describe the identification of highly specific ncRNAs in different organs. The update also contains 58 samples from six tissues of the Tabula Muris collection, allowing to check if the tissue specificity is evolutionary conserved between Homo sapiens and Mus musculus. The updated resource of 87 252 non-coding RNAs from nine non-coding RNA classes for all organs and organ systems is available online without any restrictions (https://www.ccb.uni-saarland.de/tissueatlas2).
INTRODUCTION
To advance the understanding of pathological mechanisms on a molecular level, it is essential to first understand the abundance and variability of transcripts (messenger RNAs, mRNAs), proteins, metabolites and other molecules in general. For RNA sequencing data, several popular repositories exist. For example, The Expression Atlas collects gene expression patterns under different biological conditions in 65 organisms (1,2), and The Genotype Tissue Expression (GTEx) project integrates data from 54 types of human organs (3–6). Proteomic resources, such as The Human Protein Atlas, complement these transcriptomic databases (7–9). Moreover, more specialized resources exist, such as those representing dynamic protein patterns related to cell division processes (10).
In contrast to messenger RNAs (mRNAs), a substantial fraction of the genome is transcribed but not translated into proteins. Respective transcripts are commonly referred to as non-coding RNAs (ncRNAs) (11) that were originally considered to result from massive transcription with limited biological meaning. The broad application of high-throughput sequencing (also known as second generation sequencing or massive parallel sequencing) facilitated more accurate profiling of ncRNAs (12). The Encyclopedia of DNA Elements (ENCODE) project provided evidence that 80% of the human genome might be transcribed as ncRNAs (13). Such large-scale data sets facilitated the establishment of many public databases including Rfam (14), NONCODE (15), miRBase (16), GtRNAdb (17) and circBase (18).
In addition, for sncRNAs, cell type specific patterns have been studied with the first landmark publication published by Landgraf in 2007 (19). Likewise, integrated expression studies on miRNAs and their promoters have been generated specifically for humans (20). Motivated by the results of Landgraf et al. we implemented the Human miRNA Tissue Atlas in 2016 (21), which examined dozens of organs and brain regions. More recently, we profiled small non-coding RNAs in 11 mouse tissues (22). Since the publication of our resource in 2016, the Tissue Atlas has been used in hundreds of research studies. Among the most common applications was checking the expression of one or few miRNAs in the different tissues. In part, the tissue atlas was used in review and research articles as reference for the tissue specificity of miRNAs in general (23) or for miRNA expression in specific organs or organ systems, e.g. in the brain (24) or the digestive system (25). In addition, the expression of specific miRNAs such as miR-30 has been checked in the heart (26). In the past 18 months, our web repository was used in studies on COVID-19 patients (27). A more frequent use case was to test whether circulating miRNA biomarkers in human body fluids can be related to specific organs like miR-9–3p, and miR-136–3p plasma levels used for diagnosing mild traumatic brain injury (28). To get a better idea of the application spectrum of our resource and for illustrative reasons, we generated a word cloud from the abstracts of recent studies citing the miRNA tissue atlas (Supplementary Figure S1). Of course, this illustrative representation is dominated by the terms ‘miRNA’ and ‘microRNA’, ‘expression’ and ‘patients’. Further, ‘diseases’ and more specifically ‘cancer’ and ‘SARS-CoV-2’ or ‘COVID-19’ were identified. As specific miRNAs, the word cloud lists ‘miR-221’, ‘miR-222’, ‘miR-21’, ‘miR-200c’, ‘miR-204’ or ‘miR-146’. As solid organ, the liver was among the most prominent ones. While we do not draw scientific conclusion from this illustrative analysis, it nicely demonstrates the very broad application spectrum of our resource.
However, our original analysis was limited in two aspects: (i) technologically, by the use of a microarray platform, thereby limiting detection to known miRNAs while excluding isomiRs, novel miRNAs not known at that time and other small non-coding RNA species, and (ii) biologically, by the use of only two donors. Our former study allows only rather limited conclusions about the tissue specificity of miRNAs and especially of non-miRNA sncRNAs. Moreover, since the original study was performed, it became obvious that isoforms of non-coding RNAs, most dominantly miRNAs, play a dominant role in biological processes like mRNA degradation (29) and that a switch of expression from one miRNA arm to the other might have implications for maladies like Alzheimer’s disease (30). However, as analyses on the specificity of such events were not possible with our original data set, we now introduce an improved sncRNA atlas using deep sequencing tailored for small RNA profiling and present a comprehensive collection of nine classes of non-coding RNA profiles for 21 human tissues from nine organ systems. In brief, we sequenced a total of 188 samples (mean read depth 46.1 M) from six donors, totaling 8.7 Bn reads. The deep coverage allowed us to consider nine different non-coding RNA classes. In detail, we considered 87 252 annotated features (genomic regions encoding sncRNAs according to the respective reference databases). By inclusion of variations of the annotated sncRNAs, most importantly isomiRs, we analyzed altogether 586 581 sncRNAs and variants thereof. To investigate whether the sncRNAs profiles are evolutionary conserved between organisms, as suspected previously (31), we additionally sequenced 58 mouse tissue samples from six organ types from the Tabula Muris collection (32,33).
In the context of our update, it is important to mention the high relevance for the research field which is demonstrated by many other databases with similar or complementary scope. We here provide a selection of databases that might be an alternative or additional source for users of the miRNATissueAtlas2. ExcellmiRDB is a curated online database that provides integrated information about miRNAs levels in biofluids in the context of diseases (34). mESAdb is a comprehensive multi-taxa miRNA expression and sequence analysis database (35). miRmine is a database of human miRNA expression profiles, storing over 300 small RNA sequencing profiles (36). Among the most comprehensive tools, SEAweb represents a compelling and intuitive small RNA expression atlas web application (37). Moreover, databases such as MirGeneDB start to include more and more expression data, becoming universal tools for small RNA researchers (38). Another very broad resource is the RNA atlas, recently published by Mestdagh et al. (39). This atlas stands out by the comprehensiveness, covering small and polyA RNA as well as total RNA from 300 human tissues and different cell lines. The study highlights thousands of previously uncharacterized RNAs, Thereby, an increase of the total number of ncRNAs by approximately 8% is postulated. While the above list is not exhaustive, it demonstrates the importance of respective resources for the field. The key difference to the miRNATissueAtlas2 is our focus on multiple organs from the same donor. We, however, acknowledge that the aforementioned databases excel in other features and are valuable alternative or complementary sources. As reference to the work of others in the field and to support researchers in finding the very best solution for their respective research task we provide a ‘links section’ on the landing page of the miRNATissueAtlas2, pointing to selected important alternatives resources.
MATERIALS AND METHODS
Database implementation
The miRNATissueAtlas2 frontend (webpage) was implemented and deployed via docker, with the Django Web Framework (v2.2) and a PostgreSQL database backend (v11.1). The frontend was built with Webpack (v4.25.1), React JS (v16.13.1), Redux (v4.0.4), Dev Extreme React Grid (v2.6.0), Highcharts (v8.1.0) and material-ui (v6.9.0). The web server utilizes an interactive human anatogram developed by the Gene Expression Team, EMBL-European Bioinformatics Institute (v2.4.0) to enable users to explore various tissues (2). The main visualization panels of the expression features have been realized as Box-Whisker plots. The visualization of dynamic dendrograms has been achieved with Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data (40).
Data analysis
All data included in the miRNATissueAtlas2 were generated using a stand-alone version of our web-based tool miRMaster (41,42). The most recent online version supports the same features as the stand-alone version, most importantly the multi-species support (43). We carried out all analyses using the standard parameter settings as in the online version, allowing one mismatch in the quantification of ncRNAs. The following ncRNA databases are included in our analysis: miRBase version 22.1 (16), Ensembl ncRNA version 100 (44), RNACentral version 15 (45), GtRNAdb version 18.1 (17) and NONCODE version 5 (46). We carried out use cases demonstrating the power of the miRNATissueAtlas2. The downstream processing for the use cases was done using R version 4.0.3 with the packages ggplot2, RColorBrewer, umap, infotheo, viridis, hrbrthemes, biobase, ggridges, forcats, gghalves, ggbeswarm and mapplots. As statistical test, non-parametric Wilcoxon Mann–Whitney test has been performed. If not mentioned explicitly, the P-values were computed in an unpaired and two-tailed manner. To make results comparable between different RNA classes or different organs, z-scores have been computed accordingly. Thereby, data rows were scaled to mean zero and unit variance. z-scores were tail-cut at -2 and 2, which means that all z-scores below -2 and above 2 were set to -2 and 2, respectively. In some cases, dots or bubbles are used to represent the z-scores with the diameter linearly increasing with the z-score. In this case, the values were shifted to non-negative ranges. If mentioned in the manuscript, rows or columns have been re-ordered by hierarchical clustering. In this case, standard complete linkage hierarchical clustering using the Euclidean distance was performed. For statistical evaluations only those organs and organ systems with at least four replicates were considered to limit artificial results. The core measurement for our miRNATissueAtlas is the Tissue Specificity Index (TSI), which is calculated for each single sncRNA either for organs or for organ systems. The TSI for a sncRNA j is computed as follows:
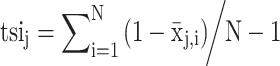 |
Here, N corresponds to the total number of organs or respectively organ systems included in the calculation. The expression intensity of organ or organ system i normalized by the maximal expression of any organ or organ system for this sncRNA j is represented by 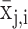 . In case of replicated measurements, the expression of the sncRNA across organs can be estimated by the mean or median across all replicates.
. In case of replicated measurements, the expression of the sncRNA across organs can be estimated by the mean or median across all replicates.
Human sample collection, RNA extraction and sequencing
In this study, the Human miRNATissueAtlas has been updated by inclusion of 188 different tissue samples originating from two female and four male body donors, available from the Gene Expression Omnibus (GSE163534). All bodies were obtained as donations for research and teaching purposes. The local institutional reviewer board (Ethikkommission der Ärztekammer des Saarlandes - Nr 329/20) approved the study. All individuals died from natural causes (cardiac arrest in five of six cases). Concomitant diseases, so far as is known, are listed in Supplementary Table S1 and include cancer in three cases, and sepsis, pulmonary embolism, dementia and heart failure each in one case. Upon arrival at the Department of Anatomy, between 8- and 48-h post-mortem, tissue samples were collected. To increase the resolution for selected organs such as the brain or intestine, further dissection of these organs into several subareas was performed. After collection, samples were immediately stored in RNALater (ThermoFisher) or frozen at -80°C to prevent further degradation. Total RNA including small RNAs was isolated using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s recommendations. In brief, tissues were lysed and homogenized in 700 μl QIAzol Lysis Reagent using 5 mm stainless steel beads and the TissueLyser LT (Qiagen) for 5 min at 50 Hz. After 5 min incubation, samples were mixed with 140 μl chloroform and phase separation was performed at 12 000 × g and 4°C for 15 min. The RNA in the aqueous phase was precipitated by adding 1.5 volumes of 100% ethanol and purified using the RNeasy columns provided with the kit, either manually or semi-automated using the Qiacube instrument with the respective protocols provided for the miRNeasy Mini kit. For quality control, the RNA concentration and purity were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), and the RNA integrity was determined using the Agilent RNA 6000 Nano Kit (Agilent Technologies). The RNA integrity values (RIN) were in an expected range for autopsy samples (RIN: 1 - 8.5). Mouse sample collection has been performed as described in the Tabula Muris Senis original manuscripts (32,47). The RNA extraction has been performed analogous to the human RNA extraction. Human samples were single-end sequenced on the BGISEQ-500RS using the High-throughput Sequencing Set (SE50) (Small RNA) according to the manufacturer’s instructions. The mouse samples have been processed with an advanced pipeline using the higher-throughput SP-960 sample prep system and MGISEQ-400 sequencing system. A detailed description and comparison of both approaches for different RNA sequencing protocols (48,49) yielded slight advantages for the latter sequencing approach in small RNA sequencing without affecting the general expression patterns.
Data availability
All data included in the miRNATissueAtlas2 are freely accessible from the Gene Expression Omnibus (GSE163534).
RESULTS AND DISCUSSION
Extended organ and non-coding RNA scope of the miRNATissueAtlas2
For the update of the miRNATissueAtlas, we aimed to cover the major organ systems (Figure 1A). In total, the online repository contains 188 biological samples from six individuals (two females and four males). The organ represented by the largest number of samples is the brain with 39 biopsies (Figure 1B). These 39 samples comprise 17 distinct brain regions (Figure 1C). Our analyses rely on data of all organs with at least four different biological replicates to achieve a higher statistical reliability. We excluded 22 samples of 14 different organs that were represented by <4 replicates per organ. We also aimed to increase the number of RNA classes in the present update. The database covers 87 252 annotated features (51 Small Cajal body-specific RNA (scaRNAs), 76 ribosomal RNAs (rRNAs), 432 transfer RNA fragments (tRNAs), 1020 small nucleolar RNAs (snoRNAs), 2064 small nuclear RNAs (snRNAs), 2417 miscRNAs, 2656 microRNAs (miRNAs), 28 733 piwi-interacting RNAs (piRNAs) and 49 803 Long intervening noncoding RNAs (lincRNAs) (Figure 1D). Although the last RNA class does not belong to the small non-coding RNAs, a quantification of the lincRNAs was feasible using the deep sequencing data. By including various RNA classes, the title ‘miRNATissueAtlas2’ might seem not be appropriate. However, we decided to keep the original brand since the broadest functionality (e.g. isoform analysis or miRNA family analysis) still is available for miRNAs and for historic reasons. From the data included in the web interface, users can access the expression and tissue specificity of all RNA classes contained in our resource. Notably, for different expression thresholds the number of expressed features varies significantly (Figure 1E) and the ncRNAs split in three classes including (i) those that decrease slowly with increasing thresholds, i.e. tRNA, rRNA and miscRNA, which are highly expressed (80% of features exceeding 2 reads per million mapped reads (RPM)), (ii) those with a moderate loss, i.e. snoRNA, scaRNA, snRNA, lincRNA and miRNA, which are moderately expressed (40–60% of features exceeding 2RPM) and (iii) rather specifically expressed piRNAs that rapidly decrease to below 5% of the expressed features. The observation of specific expression in few samples, namely testis, is in line with their important role in spermatogenesis (50). These and other overview statistics such the average expression of the different RNA species (Figure 1F) can be accessed from the main page of the resource. The similarities and differences of ncRNAs between the organs in general call for a detailed inspection of sncRNA patterns across organs. We thus implemented a new web interface that reflects the new complexity introduced by other RNA classes.
Figure 1.

Broad coverage of different organs and ncRNAs in the miRNATissueAtlas2. (A) In the study, we measured organs from nine organ systems with largest sample numbers for the digestive and the nervous system. (B) Sample counts of the different organs included in the study (bar length and number aside of the bar) together with the number of sub structures (balloon diameter and second number). Notably the numbers do not sum up to 188 samples since organs with <4 replicates were excluded from statistical considerations. (C) For the brain, 17 different sub-structures were measured. Note that the relative locations of hippocampus, substantia nigra and nucleus caudatus are indicated, but these structures are not shown, as they are in a different section. Subfigure was prepared using Motifolio Biology Illustration Toolkit (motifolio.com). (D) Number of representatives from the nine ncRNA classes included in the study. (E) Decrease of expressed representatives (on a percent scale) as a function of an increasing expression threshold (normalized read count). (F) Qualitative consideration of the number of expressed features per ncRNA class and organ. The bubble size scales linear with the number of expressed representatives (as z-scores), and the gray area scales linear with the standard deviation of the number of expressed representatives. Rows and columns are re-ordered with respect to a standard hierarchical clustering.
miRNATissueAtlas2 web interface and functionality
The web interface has six main components that represent the core functionality of miRNATissueAtlas2. These can be selected from the left side of the landing page and contain three views designed mostly for single sncRNAs and three more advanced options for the analysis of sncRNA sets. The first three views include (i) the specificity view, (ii) the patterns view and (iii) the tissue view. From these three views, the users can directly reach the final detailed graphical results for single sncRNAs (Figure 2). The three advanced views include (iv) a network analysis, (v) generation of custom heatmaps and (vi) a downstream in-silico enrichment analysis (Figure 2).
Figure 2.

The web interface and typical results. The upper part of the figure presents the three analyses that are mostly designed for the straightforward analysis of single or few sncRNAs. (1) the ‘specificity view’ where users can filter and select the RNAs of interest, the (2) patterns view and (3) organs view. By clicking on a certain RNA in any of these views, the user is directed to the detailed results page in the middle. From there, the find related functionality allows to identify related miRNAs in Homo Sapiens. On the bottom, the advanced analysis options are shown, including (4) the network view, the (5) custom heat map view and (6) the functional enrichment view.
In the first representation, all features are contained and sorted with respect to their tissue specificity index (TSI). The TSI is an estimate of the tissue specificity, which we already successfully employed in our first tissue atlas (21). The TSI gives each specific ncRNA molecule a numeric value on a scale from 0 to 1, with 1 meaning that expression of the molecule was detected only in one specific tissue, and 0 meaning expression of the molecule was detected in all tissues. Users can select their RNA class of interest and specify a minimal and maximal TSI threshold, depending on whether they are interested in a broadly expressed or a very specific RNA. Further they can select the least number of samples, where the RNA must be expressed in. For the miRNAs, users can also decide whether the microarray or the NGS data set is used. The results table on the bottom is dynamically adjusted as the user changes the input. By selecting an RNA from the table, a detailed representation with expression values across organs is displayed. The data can be displayed in log transformation and in a collapsed and expanded view. On mouseover detailed values are shown to the user. The results table can be downloaded for further downstream processing as we demonstrate in the use cases below. For miRNAs, the isoform view is implemented as an additional feature. For all existing isoforms, the expression can likewise be inspected by selecting the respective isoform. Additionally, correlated miRNAs can be identified using the ‘compare related’ functionality, and the correlation can be displayed as scatter plot. For all RNAs, links to external resources and other information are provided on the top of the page.
The second view is the pattern view. Here, users can select a single RNA or, if groupings are available, also larger entities of RNAs. In the case of miRNAs, for example, complete miRNA families or the two mature miRNAs of one precursor can be viewed. Besides, the same functionality as for the specificity view is available.
The third view is the tissue view. Here, users can select either an organ system or an organ/tissue to see which RNAs are expressed in this tissue. The results table contains all biological replicates, this means if a certain RNA is expressed in one organ in all six bodies, six values are displayed. Thus, by filtering for an RNA of interest, the user immediately gets information on the range of expression and in how many samples the RNA is expressed. Of course, the table can be sorted as all other tables and filters can be specified by users, e.g. only RNAs with an expression over 1000 reads can be displayed. This results table is available for immediate download as in the other views. By selecting a specific RNA in the table users are directed to the RNA specific view where TSI values are provided and the same representation as in the first view is available.
While the first three views are designed mostly for single sncRNAs, in the advanced analysis section, users can compare and display sets of sncRNAs and carry out downstream analyses. The core is the fourth view, a network analysis, where users can provide multiple non-coding RNAs and select multiple organs. The network of these RNAs in the context of the selected tissues is then generated. More precisely, users can select an ‘union’ or ‘intersection’ conjunction. In the first case the subnetwork consists of only RNAs expressed in all selected organs, while in the intersection mode, all RNAs expressed in at least one of the selected organs are used for network generation. However, in the latter case, as the network can become quite large, we additionally implemented an expression filtering, where the user can increase the minimal accepted expression value and only display the connections of all RNAs with expression values exceeding the chosen threshold. Another function to visualize results of multiple sncRNAs is the custom heatmap functionality in the fifth view. Like the network analysis functionality, users can provide a set of small non-coding RNAs and organs for which a clustering is performed with the results displayed as heat maps in an interactive manner. Rows and columns can be sorted according to different criteria and automated clustering is performed. Finally, in case of miRNAs as target molecules, a miEAA enrichment via our existing API can be initiated from either the network or cluster analysis or by manual selection of miRNAs, allowing for even further downstream analyses.
Batch download of data
One widely used functionality in the first version of the Human miRNATissueAtlas was the batch download of data for further processing. We thus extended the download section. While all raw data are accessible from the gene expression omnibus, we provide expression matrices for all RNAs included in the study. These data are identical to those displayed in the web interface. In addition, we provide isoform expression matrices for direct download for the two organism’s human and mouse. Finally, the Tissue Specificity Indices for all RNAs as well as mapping files to unique name identifiers are provided.
Use case 1: ncRNAs from different classes are specific for organs and organ systems
In the previous sections we have introduced the web interface and functionality of miRNATissueAtlas2, demonstrated how analyses on single RNAs can be performed online and described how to access the data for further processing. As a first use case we used the miRNATissueAtlas2 data to make conclusions of specificity of RNAs for organs and organ systems. We first observed that organ systems exhibit specificity depending on the RNA class, with piRNAs, miRNAs, snRNAs and snoRNAs showing the highest specificity (Figure 3A). Our data thus suggest that while respective regulatory ncRNAs are tissue specific, ribosomal and transfer RNAs reveal a significantly lower organ system specificity (P < 10–10). Of all classes, miRNAs show the broadest spectrum of TSI values. Some miRNAs show the overall highest specificity, exceeding piRNAs and lincRNAs but others are more ubiquitously expressed as compared to the tRNAs and rRNAs. Mapping the data to the respective organ systems we identified the nervous and lymphatic systems having highest TSI values and thus largest specificity, while the digestive system, urinary tract, and cardiovascular system are limited with respect to ncRNA specificity (Figure 3B). In the ontology of specificities, we next assessed the specificity for the organs using the different ncRNA classes (Figure 3C). piRNAs peak at a high TSI range, due to the high expression of many piRNAs in the testis, inflating the signals and implying a specificity for the endocrine system. Repeatedly, miRNAs show a more even distribution containing low, medium and highly specific candidates. In contrast to organ systems, tRNAs are more specific for organs, but like the organ systems rRNAs have lowest specificity. The projection on to the organs highlights the testis as the organ with highest specificity (Figure 3D). This is due to the inflation induced by many piRNAs highly expressed only in testis. Importantly, the following organs contain the stomach and bowel from the digestive system. While the digestive system itself was not specific, the individual organs within the digestive systems are. In contrast, the nerves and brain from the nervous system lose in specificity in mapping ncRNAs to the organs. The results for the digestive system and the nervous system in the light of the organ specificity call for a direct comparison of tissue and organs system specificity for all ncRNAs (Figure 3E). We observe the expected correlation of specificity for organs and organ systems with a shift to the organs. At the same time, we identify a substantial fraction of tissue specific ncRNAs that are not specific for the organ systems at all. This finally lead us to the detailed consideration, in which organ which ncRNAs present the highest specificity after scaling the data organ-wise to z-scores (Figure 3F). Basically, all organs have the highest specificity for either miRNAs (e.g. brain, bladder, lung, heart and adipocyte) or lincRNAs (stomach, bowel, skin, spleen and kidney). Other enrichments are the testis, esophagus, and pleurae for piRNAs and the thyroid gland for snoRNAs. The aforementioned observation that sncRNAs can be more specific for organs than organ systems seems to be rather counterintuitive because if a sncRNA is expressed only in one organ it should also be expressed only in one organ system. The reason for this observation lies in the number of replicates in the organ or organ system, the sncRNA is expressed in. As described in the methods section, the expression of a sncRNA in an organ or organ system is estimated by the average expression (mean or median) across replicates. If a sncRNA is expressed only in one organ the average expression of the sncRNA in the organ is high. If the organ system covers many other organs, the average sncRNA in the organ system can become low. As an example, we show the sorted normalized expression of miR-216b-5p in the organs (Figure 3G) and organ systems (Figure 3H). In the first case we find a large difference between the first and second organ making the miRNA rather specific for the pancreas. In contrast, the expression in the endocrine and other organ systems is comparable to each other leading to a low organ system specificity. In this case we used the median expression because the effect becomes more visible here, but the same effect persists with the mean expression. The TSI values for organs and organ systems are provided in Supplementary Table S2.
Figure 3.

Use Case 1: Specificity of ncRNA for organs and organ systems. (A) Ridgeline plots representing the distribution of the organ system specificity for the different ncRNA classes. Tissue specificity (TSI) is calculated between 0 and 1 where 0 represents ncRNAs that are not specific at all and 1 represents perfectly specific sncRNAs. The ncRNA classes are sorted with respect to decreasing average TSI with the most specific class (piRNAs) on top. (B) TSI values for the different organ systems, again sorted with respect to decreasing average TSI and the nervous system as most specific organ system on top. (C) Tissue specificity for the different ncRNA classes but computed on the organ level. While most ncRNA classes form a normal distribution, miRNAs and rRNAs are characterized by an increase towards higher tissue specificity values. The peak for piRNAs is induced by high expression of many piRNAs in testis. (D) Tissue specificity on the organ level. The overall highest specificity is found in testis, again induced by the high expression of piRNAs in this organ. (E) Scatter plot of the specificity on the organ level as compared to the organ system level. The green line represents the angel bisector (i.e. ncRNAs with equal specificity on the organ and organ system level). The orange line represents a smoothing spline with four degrees of freedom. The color of the points represents the difference between organ and organ system specificity. The dark orange dots on top are those ncRNAs with high organ but partially very low organ system specificity. Gray dots in the lower left part are those ncRNAs that reveal a very limited organ and organ system specificity. (F) For each organ and each ncRNA class, the number of specific representatives is shown as bubble using a TSI cutoff of 0.9. The bubble size scales linear with the number of representatives after transforming the numbers per organ to a z-scale (cut at -2 and 2 respectively). (G) For miR-216b-5p, the normalized expression intensity across organs is displayed. From these values, the TSI is computed according to the formula in the Materials and Methods section. In this case, the formula leads to a TSI close to 1. (H) For the same miRNA the normalized expression across organ systems is displayed. Because of an on average low expression in many organ systems the miRNA is not considered as specific for an organ system.
Use case 2: Tissue specificity of sncRNAs is conserved between human and mouse
We also added mouse data to the miRNATissueAtlas2 that are available for download. In detail, we compared brain, bowel, bone, muscle, heart and adipocyte tissue between human (83 of the original samples) and mouse (58 samples from Tabula Muris) (Figure 4A). In general, the tissue specificity between the different ncRNA classes was similar with highest specificity for miRNAs and snoRNAs (Figure 4B). Whereas in humans the largest peak is present for tRNAs at an intermediate tissue specificity, in mouse the piRNAs show an even higher peak at intermediate specificity values. Due to the high degree of conservation for miRNAs (51), we carried out an in-depth comparison. For each miRNA with the identically annotated sequence in human and mouse we computed the tissue specificity and the organ that miRNA is specific for. Next, we iteratively computed the degree of match for increasing values of tissue specificity (Figure 4C). For the lowest possible value of zero, around 60% miRNAs match, i.e. the organ with the highest expression for the miRNA is the same. With increasing tissue specificity this value climbs to 100%, meaning that for the very specific miRNAs the human and mouse results match perfectly. At the threshold of 0.8, the brain has by far the largest number of specific miRNAs matching between mouse and human (Figure 4D).
Figure 4.

Use Case 2: Concordance of the organ specificity between mouse and human. (A) For six tissues, we compared the specificity for 58 mouse samples and 83 samples from the present study. The results for the human specificity of organs and ncRNAs can be different from those presented in the work since we repeated the computations only for the subset of organs present in both organisms to avoid a bias. (B) Back-to-back histogram of the different ncRNA classes in mouse and human. (C) The percentage of organs being most specific for miRNAs in human and mouse as function of the tissue specificity threshold. If no specificity threshold is set, only the most expressed organ is compared, which is identical in around 60% of miRNAs. With increasing lower thresholds (i.e. higher percentage of specific miRNAs) the percentage of most specific organs increases to 100%. The blue line is a smoothed spline with five degrees of freedom. (D) Confusion matrix for an organ specificity threshold of 0.8. Each bubble represents the number of miRNAs for most specific organs in humans and mouse. Actual numbers are provided next to or within the bubbles. Bubbles located on the diagonal are those that match between the organisms.
Limitations of the miRNA TissueAtlats2 and future directions
Our resource shows a few limitations, which we will discuss in the following and that might be important for our users. First, although all individuals included died from a natural cause of death, however, concomitant diseases occurred. This might affect the expression of ncRNAs in different organs but is unlikely to affect the conclusions in general. To account for this, we only included organs in the case studies where we have at least four replicates (166 samples are included), minimizing the importance of outliers. Nevertheless, the online resource contains the full set of organs. Moreover, the expression levels might be affected but not the organ specificities. A further challenge is the observed enrichment for different ncRNA classes by the experimental approach. Even though we aimed to measure small RNAs, we also discuss the occurrence of lincRNAs in the data. These and other longer RNAs might be the direct result of the cell degradome, known mostly for proteins (52,53) but which has been shown to be also existent for RNAs (54,55). Thus, the abundance of the different RNA classes has not necessarily to match the physiological distribution. To reflect this issue, we focus on the comparison of the expression patterns between the organs and scale the results per RNA class. A further challenge is the correct assignment of biopsies to organs and the grouping in the context of this study. For example, the bones also contain bone marrow, a substance that is a reservoir of stem cells with vastly different gene expression programs. Also, adipocytes are measured from marrow adipose tissue (MAT) and other fat tissues. Only for the brain—one of the organs with the most distinguished sub-structures—sufficient samples from different regions were taken to present a high-resolution analysis of the respective sub regions. Moreover, the RNA quality can affect the results of small RNA sequencing studies (56), but a heterogenous quality of RNA samples can hardly be avoided in projects making use of post-mortem whole body samples. Moreover, the tissue specificity is still not an absolute measure, e.g. if another organ is added that had been excluded so far, the tissue specificity values for previous organs could possibly change. Here, the large number of different organs is a certain advantage of our study. Related to this discussion, the organ- and organ system specificity can also be affected by other features, for example how the average expression of a sncRNA across organs or organ systems is computed. To test the influence of the number of included samples, we performed a sub-sampling analysis. We selected 50% of samples randomly and repeated the random selection 100 times. For both, the organ systems and the organs we computed on average moderate coefficient of variation levels. The coefficient of variation for the 100 sub-sampled organ system specificities was 0.16, for the organs 0.09 (Supplemental Figure S2 a,b). The question in how many cases the same organ or organ system has been selected as most specific in the 100 repeated runs is of similar interest (Supplemental Figure S2 c,d). On average, this was the case in 68% for the organ systems and still in 41% for the organs. Considering the large number of organs to select from and the fact that this analysis includes not only highly specific sncRNAs where the selection was consistently close to 100% of cases, the sub-sampling and stability analyses indicate that the number of samples included in the study indeed facilitates a rather stable and reproducible overall tissue specificity analysis.
One final limitation lies in the use of tissue sections from the organs which can contain a broad range of different cell types depending on the heterogeneity of the organ. Countless studies already demonstrated differential gene expression signatures for different cell types within a single tissue using single cell RNA seq. In the same direction, we expect a similar diversity for sncRNA profiles of different cell types within an organ on single cell level. However, compared to the plethora of single cell gene expression studies, analysis of sncRNAs on single cell level is technologically challenging and still in its infancies (57). Nevertheless, a single cell sncRNA atlas would be the logical extension of our current work.
DATA AVAILABILITY
The interactive web app is available at https://www.ccb.uni-saarland.de/tissueatlas2.
All data in this study are freely accessible from the Gene Expression Omnibus (GSE163534).
Supplementary Material
Contributor Information
Andreas Keller, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany; Department of Neurology and Neurobiology, Stanford University, CA 94305, USA.
Laura Gröger, Center for Human and Molecular Biology, Junior Research Group Human Genetics, Saarland University, 66421 Homburg, Germany.
Thomas Tschernig, Institute for Anatomy, Saarland University, 66421 Homburg, Germany.
Jeffrey Solomon, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Omar Laham, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Nicholas Schaum, Department of Neurology and Neurobiology, Stanford University, CA 94305, USA.
Viktoria Wagner, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Fabian Kern, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Georges Pierre Schmartz, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Yongping Li, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Adam Borcherding, Complete Genomics Inc, San Jose, CA 95134, USA.
Carola Meier, Institute for Anatomy, Saarland University, 66421 Homburg, Germany.
Tony Wyss-Coray, Department of Neurology and Neurobiology, Stanford University, CA 94305, USA.
Eckart Meese, Department of Human Genetics, Saarland University, 66421 Homburg, Germany.
Tobias Fehlmann, Clinical Bioinformatics, Center for Bioinformatics, Saarland University, 66123 Saarbrücken, Germany.
Nicole Ludwig, Center for Human and Molecular Biology, Junior Research Group Human Genetics, Saarland University, 66421 Homburg, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The study and open access fees were funded by Saarland University.
Conflict of interest statement. Y.L. and A.B. are employed by MGI and Complete Genomics, developing instruments and chemistry for next generation sequencing. A.K. is member of the scientific advisory board of the company Firalis, developing diagnostic tests based on small RNAs. All other authors declare no competing interests.
REFERENCES
- 1. Petryszak R., Burdett T., Fiorelli B., Fonseca N.A., Gonzalez-Porta M., Hastings E., Huber W., Jupp S., Keays M., Kryvych N.et al.. Expression Atlas update–a database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014; 42:D926–D932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Papatheodorou I., Moreno P., Manning J., Fuentes A.M., George N., Fexova S., Fonseca N.A., Fullgrabe A., Green M., Huang N.et al.. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020; 48:D77–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burgess D.J. Reaching completion for GTEx. Nat. Rev. Genet. 2020; 21:717. [DOI] [PubMed] [Google Scholar]
- 4. Consortium G.T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Consortium G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Consortium G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S.et al.. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010; 28:1248–1250. [DOI] [PubMed] [Google Scholar]
- 8. Ponten F., Jirstrom K., Uhlen M.. The Human Protein Atlas–a tool for pathology. J. Pathol. 2008; 216:387–393. [DOI] [PubMed] [Google Scholar]
- 9. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 10. Cai Y., Hossain M.J., Heriche J.K., Politi A.Z., Walther N., Koch B., Wachsmuth M., Nijmeijer B., Kueblbeck M., Martinic-Kavur M.et al.. Experimental and computational framework for a dynamic protein atlas of human cell division. Nature. 2018; 561:411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eddy S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001; 2:919–929. [DOI] [PubMed] [Google Scholar]
- 12. Metzker M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010; 11:31–46. [DOI] [PubMed] [Google Scholar]
- 13. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R.. Rfam: an RNA family database. Nucleic Acids Res. 2003; 31:439–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu C., Bai B., Skogerbo G., Cai L., Deng W., Zhang Y., Bu D., Zhao Y., Chen R.. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005; 33:D112–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kozomara A., Birgaoanu M., Griffiths-Jones S.. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019; 47:D155–D162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan P.P., Lowe T.M.. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Glazar P., Papavasileiou P., Rajewsky N.. circBase: a database for circular RNAs. RNA. 2014; 20:1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M.et al.. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007; 129:1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Rie D., Abugessaisa I., Alam T., Arner E., Arner P., Ashoor H., Astrom G., Babina M., Bertin N., Burroughs A.M.et al.. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017; 35:872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stahler C., Meese E.et al.. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Isakova A., Fehlmann T., Keller A., Quake S.R.. A mouse tissue atlas of small noncoding RNA. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:25634–25645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bielska A., Niemira M., Kretowski A.. Recent highlights of research on miRNAs as early potential biomarkers for cardiovascular complications of type 2 diabetes mellitus. Int. J. Mol. Sci. 2021; 22:3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen C.Y., Chao Y.M., Lin H.F., Chen C.J., Chen C.S., Yang J.L., Chan J.Y.H., Juo S.H.. miR-195 reduces age-related blood-brain barrier leakage caused by thrombospondin-1-mediated selective autophagy. Aging Cell. 2020; 1:e13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martinez C., Rodino-Janeiro B.K., Lobo B., Stanifer M.L., Klaus B., Granzow M., Gonzalez-Castro A.M., Salvo-Romero E., Alonso-Cotoner C., Pigrau M.et al.. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut. 2017; 66:1537–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bao J., Lu Y., She Q., Dou W., Tang R., Xu X., Zhang M., Zhu L., Zhou Q., Li H.et al.. MicroRNA-30 regulates left ventricular hypertrophy in chronic kidney disease. JCI Insight. 2021; 6:e138027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Centa A., Fonseca A.S., Ferreira S., Azevedo M.L.V., Vaz de Paula C.B., Nagashima S., Machado-Souza C., Miggiolaro A., Baena C.P., de Noronha L.et al.. Deregulated miRNA expression is associated with endothelial dysfunction in post-mortem lung biopsies of COVID-19 patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020; 320:L405–L412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Das Gupta S., Ciszek R., Heiskanen M., Lapinlampi N., Kukkonen J., Leinonen V., Puhakka N., Pitkanen A.. Plasma miR-9-3p and miR-136-3p as Potential Novel Diagnostic Biomarkers for Experimental and Human Mild Traumatic Brain Injury. Int. J. Mol. Sci. 2021; 22:1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sheu-Gruttadauria J., Pawlica P., Klum S.M., Wang S., Yario T.A., Schirle Oakdale N.T., Steitz J.A., MacRae I.J.. Structural basis for target-directed microRNA degradation. Mol. Cell. 2019; 75:1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Backes C., Haas J., Leidinger P., Frese K., Grossmann T., Ruprecht K., Meder B., Meese E., Keller A.. miFRame: analysis and visualization of miRNA sequencing data in neurological disorders. J. Transl. Med. 2015; 13:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fehlmann T., Backes C., Pirritano M., Laufer T., Galata V., Kern F., Kahraman M., Gasparoni G., Ludwig N., Lenhof H.P.et al.. The sncRNA Zoo: a repository for circulating small noncoding RNAs in animals. Nucleic Acids Res. 2019; 47:4431–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schaum N., Lehallier B., Hahn O., Palovics R., Hosseinzadeh S., Lee S.E., Sit R., Lee D.P., Losada P.M., Zardeneta M.E.et al.. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020; 583:596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tabula Muris C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature. 2020; 583:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barupal J.K., Saini A.K., Chand T., Meena A., Beniwal S., Suthar J.R., Meena N., Kachhwaha S., Kothari S.L.. ExcellmiRDB for translational genomics: a curated online resource for extracellular microRNAs. OMICS. 2015; 19:24–30. [DOI] [PubMed] [Google Scholar]
- 35. Kaya K.D., Karakulah G., Yakicier C.M., Acar A.C., Konu O.. mESAdb: microRNA expression and sequence analysis database. Nucleic Acids Res. 2011; 39:D170–D180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Panwar B., Omenn G.S., Guan Y.. miRmine: a database of human miRNA expression profiles. Bioinformatics. 2017; 33:1554–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rahman R.U., Liebhoff A.M., Bansal V., Fiosins M., Rajput A., Sattar A., Magruder D.S., Madan S., Sun T., Gautam A.et al.. SEAweb: the small RNA Expression Atlas web application. Nucleic Acids Res. 2020; 48:D204–D219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fromm B., Domanska D., Hoye E., Ovchinnikov V., Kang W., Aparicio-Puerta E., Johansen M., Flatmark K., Mathelier A., Hovig E.et al.. MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res. 2020; 48:D132–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lorenzi L., Chiu H.-S., Avila Cobos F., Gross S., Volders P.-J., Cannoodt R., Nuytens J., Vanderheyden K., Anckaert J., Lefever S.et al.. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 2021; 10.1038/s41587-021-00936-1. [DOI] [PubMed] [Google Scholar]
- 40. Fernandez N.F., Gundersen G.W., Rahman A., Grimes M.L., Rikova K., Hornbeck P., Ma’ayan A.. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci Data. 2017; 4:170151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fehlmann T., Backes C., Kahraman M., Haas J., Ludwig N., Posch A.E., Wurstle M.L., Hubenthal M., Franke A., Meder B.et al.. Web-based NGS data analysis using miRMaster: a large-scale meta-analysis of human miRNAs. Nucleic Acids Res. 2017; 45:8731–8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fehlmann T., Meese E., Keller A.. Exploring ncRNAs in Alzheimer’s disease by miRMaster. Oncotarget. 2017; 8:3771–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fehlmann T., Kern F., Laham O., Backes C., Solomon J., Hirsch P., Volz C., Muller R., Keller A.. miRMaster 2.0: multi-species non-coding RNA sequencing analyses at scale. Nucleic Acids Res. 2021; 49:W397–W408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P.et al.. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Consortium, R.N. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res. 2020; 49:D212–D220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fang S., Zhang L., Guo J., Niu Y., Wu Y., Li H., Zhao L., Li X., Teng X., Sun X.et al.. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018; 46:D308–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tabula Muris C., Overall c., Logistical c., Organ c.processing, Library, p., sequencing, Computational data, a., Cell type, a. processing, Library, p., sequencing, Computational data, a., Cell type, a. Writing g.et al.. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018; 562:367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y., Fehlmann T., Borcherding A., Drmanac S., Liu S., Groeger L., Xu C., Callow M., Villarosa C., Jorjorian A.et al.. CoolMPS: evaluation of antibody labeling based massively parallel non-coding RNA sequencing. Nucleic Acids Res. 2020; 49:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hahn O., Fehlmann T., Zhang H., Munson C.N., Vest R.T., Borcherding A., Liu S., Villarosa C., Drmanac S., Drmanac R.et al.. CoolMPS for robust sequencing of single-nuclear RNAs captured by droplet-based method. Nucleic Acids Res. 2020; 49:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grivna S.T., Pyhtila B., Lin H.. MIWI associates with translational machinery and PIWI-interacting RNAs (piRNAs) in regulating spermatogenesis. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:13415–13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. He L., Hannon G.J.. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004; 5:522–531. [DOI] [PubMed] [Google Scholar]
- 52. Lopez-Otin C., Overall C.M.. Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002; 3:509–519. [DOI] [PubMed] [Google Scholar]
- 53. Wolf-Levy H., Javitt A., Eisenberg-Lerner A., Kacen A., Ulman A., Sheban D., Dassa B., Fishbain-Yoskovitz V., Carmona-Rivera C., Kramer M.P.et al.. Revealing the cellular degradome by mass spectrometry analysis of proteasome-cleaved peptides. Nat. Biotechnol. 2018; 36:1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ma X., Yin X., Tang Z., Ito H., Shao C., Meng Y., Xie T.. The RNA degradome: a precious resource for deciphering RNA processing and regulation codes in plants. RNA Biol. 2020; 17:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jackowiak P., Nowacka M., Strozycki P.M., Figlerowicz M.. RNA degradome–its biogenesis and functions. Nucleic Acids Res. 2011; 39:7361–7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ludwig N., Becker M., Schumann T., Speer T., Fehlmann T., Keller A., Meese E.. Bias in recent miRBase annotations potentially associated with RNA quality issues. Sci. Rep. 2017; 7:5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hucker S.M., Fehlmann T., Werno C., Weidele K., Luke F., Schlenska-Lange A., Klein C.A., Keller A., Kirsch S.. Single-cell microRNA sequencing method comparison and application to cell lines and circulating lung tumor cells. Nat. Commun. 2021; 12:4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data included in the miRNATissueAtlas2 are freely accessible from the Gene Expression Omnibus (GSE163534).
The interactive web app is available at https://www.ccb.uni-saarland.de/tissueatlas2.
All data in this study are freely accessible from the Gene Expression Omnibus (GSE163534).