


Abstract
The Human Proteoform Atlas (HPfA) is a web-based repository of experimentally verified human proteoforms on-line at http://human-proteoform-atlas.org and is a direct descendant of the Consortium of Top-Down Proteomics’ (CTDP) Proteoform Atlas. Proteoforms are the specific forms of protein molecules expressed by our cells and include the unique combination of post-translational modifications (PTMs), alternative splicing and other sources of variation deriving from a specific gene. The HPfA uses a FAIR system to assign persistent identifiers to proteoforms which allows for redundancy calling and tracking from prior and future studies in the growing community of proteoform biology and measurement. The HPfA is organized around open ontologies and enables flexible classification of proteoforms. To achieve this, a public registry of experimentally verified proteoforms was also created. Submission of new proteoforms can be processed through email vianrtdphelp@northwestern.edu, and future iterations of these proteoform atlases will help to organize and assign function to proteoforms, their PTMs and their complexes in the years ahead.
INTRODUCTION
Genomic data types probe human biology at the gene and transcript level, yet proteins are the direct source of much activity within our bodies and cells. Current, data on the expression of proteins is not complete as expressed proteins are often highly modified in ways which affect their structure and function. A single human gene usually expresses as a family of related forms of a protein. The term proteoforms has been introduced to refer to the specific molecular species of an expressed and translated gene including the precise combination of sequence variants, alternative splicing events, and post-translational modifications (PTMs) (Figure 1) (1). Proteoforms represent the main determinants of cellular phenotypes in protein-level biology yet remain largely understudied. According to UniProt, the total number of ‘canonical’ (i.e. the representative protein in UniProt for a given gene) human proteins across the entire proteome, is currently 20 386 (2). The number of proteoforms is larger and difficult to estimate because of phenomena such as mRNA splicing, PTMs, coding single-nucleotide polymorphism (cSNPs), and similar events (3).
Figure 1.
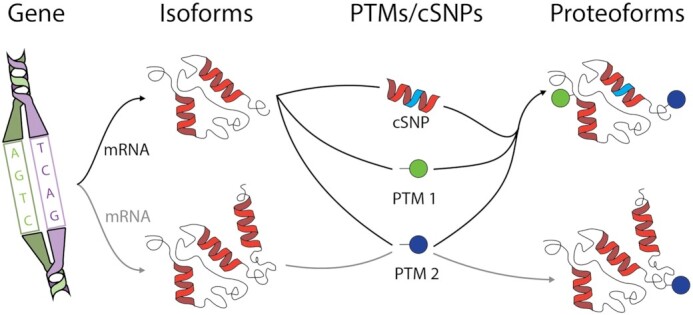
Scheme explaining the concept of a proteoform. A single gene (depicted at left) can be processed to generate different isoforms via alternative splicing of mRNA, which in combination with site-specific post-translational modifications (PTMs) and/or coding single-nucleotide polymorphism (cSNPs) that might be present in the population, generates different proteoforms (at right).
Understanding expressed proteoforms as the molecular phenotype is fundamental. One illustrative example is the case of the KRAS protein, which belongs to the RAS gene family. KRAS is alternatively spliced at the fourth exon, yielding the KRAS4A and KRAS4B isoforms (4). These genes encode for small GTPases which play important roles in cell growth and proliferation via the MAPK and PI3K pathways. The RAS gene family is also among the most frequently mutated in cancer, for example at residues G12, G13 or Q61 in KRAS4. Combined with oncogenic mutations, KRAS undergoes post-translational modification, including farnesylation and carboxymethylation of the C-terminal C185, which is critical for plasma membrane association. Given these sources of variation, there are thousands of possible theoretical proteoforms for the KRAS protein, with <40 mapped thus far in cancer cell lines and tumors (5).
The Protein Ontology (PRO) (6), another major protein knowledgebase, provides proteoform-level permanent identifiers, but only after a proteoform is published in the peer-reviewed literature and has been manually curated. This process leads to high quality annotations but, consequently, lags behind the experimental forefront. Therefore, at present, experimentally verified proteoform related information is mostly locked away from researchers by the lack of a findable, accessible, interoperable and reusable (FAIR) data infrastructure. As highlighted above for the KRAS protein, only those proteoforms representing different protein isoforms coming from alternatively spliced transcripts have their own unique protein identifiers in UniProt. For other types of proteoforms (proteins containing PTMs and protein variants) there is a need to normalize a widely accepted representation and to assign accession numbers, enabling interoperability between protein resources. Naturally, this limitation propagates to other bioinformatics resources that rely on UniProt as the way to represent proteins. Therefore, there is a need to enable a ‘proteoform-centric view’ for proteins, by creating resources and infrastructure for the handling, analysis, and validation of proteoforms. To overcome the shortage of dedicated biological cyberinfrastructure on human proteoforms we have established a Human Proteoform Atlas (HPfA) to serve as a registry for experimentally verified proteoforms and build upon the general Proteoform Atlas hosted by the Consortium of Top-Down Proteomics’ (CTDP) (http://atlas.topdownproteomics.org/).
HUMAN PROTEOFORM ATLAS CONTENTS AND FEATURES
We have established the HPfA, using FAIR principles, to durably store and identify proteoforms as they are experimentally verified from Homo sapiens (UniProt taxon identifier: 9606). The atlas, on-line at http://human-proteoform-atlas.org, is organized around existing open ontologies. It is currently structured to include Anatomy, Blood, Cancer, Cariology, Cell Lines, Cells, Immunology, and Neurologic Atlases, but new atlases can be instantiated as needed.
At present, the Human Proteoform Atlas contains 37 071 unique experimentally verified proteoforms, each of which has been assigned a durable proteoform identifier. These proteoforms come from 30 datasets in 27 peer-reviewed publications (Supplementary Table S1) (7–38), published by 15 unique laboratories over the last seven years (2014 to 2021), and represent 3055 protein isoforms derived from 2465 unique genes (∼13% of the protein-coding human genome). The proteoforms contain 369 separate types of modifications including phosphorylation (∼13% of the total PTMs observed), acetylation (∼43%), disulfides (∼2%), methylation (∼29%), Oxidation (∼2%), Dehydro (∼2%) palmitoylation (<1%), farnesylation (<1%), geranylation (<1%), nitrosylation (<1%), sulfinic acid (<1%) and ADP-ribosylation (<1%).
The Human Proteoform Atlas is built around existing open and community-supported ontologies. Terms from these ontologies are used to classify proteoform entries. Currently, there are 68 ontologic terms used from seven ontologies (Figure 2A), including 11 different tissue types (Figure 2B), and 12 separate ontology terms associated with disease (Figure 2C).
Figure 2.

Proteoform breakdown in the HPfA by ontology in panel (A), tissue in (B) and human disease in (C). Ontology codes are: Uber-anatomy ontology (UBERON), Ontology for MIRNA Target (OMIT), Experimental Factor Ontology (EFO), Human Disease Ontology (DOID), Cell Line Ontology (CLO), Cell Ontology (CL) and BRENDA Tissue Ontology (BTO).
The Human Proteoform Atlas is designed to be open, scalable, and adopted as part of future community-driven activities. It creates the infrastructure needed to maximize dissemination of human proteoforms to the biomedical community while creating a single resource which leverages knowledge of human proteoforms. This repository links to protein-centric databases such as UniProt and has data visualization and integration tools.
User interface
Proteoforms and datasets contained in the Human Proteoform Atlas are broken down into eight current sub-atlases, displaying both dataset and proteoform counts and a description of that Atlas. Atlases are organized around existing open ontologies and can be broken down further by ontology terms or dataset.
A user visiting the site first encounters a view of the eight sub-atlases, with dataset and proteoform counts. If the user is interested in the proteoforms associated with a particular cell type, for example, an eosinophil cell, they may select the Cell atlas which displays a breakdown of all ontological terms from the Cell ontology (CL) in the atlas. Selecting the eosinophil cell ontology term (CL:0000771), proceeds to an overview, including a description of the ontology term, links to EBI (39) and PURL (40), as well as a list of proteoforms associated with eosinophil cells, including protein associations, and modifications. These proteoforms can be further queried using the provided search tool. Proteoforms may be selected to give a detailed description, including biological and chemical proteoform accession numbers, associated proteins, ProForma sequence (41) and post-translational modifications (Figure 3). If the proteoform is from an enhanced dataset (see below), a graphical fragment map is populated and displayed, including various proteoform scores, experimental/observed masses, and dataset information.
Figure 3.

Screenshots of the Human Proteoform Atlas. A proteoform page for PFR00000001011, an unmodified proteoform of the protein NEDD8.
If a user is interested in a specific gene or protein, these may be selected from the gene and protein tabs. Selecting a gene will display a summary page, including the full gene name, its symbol, chromosomal location, and various links to external resources, including but not limited to; HUGO Gene Nomenclature Committee (HGNC) (42), NCBI Reference Sequence Database (REFSEQ) (43), ENSEMBL (44), NCBI, and PubMed (Figure 4A). Selecting a protein displays a protein summary page that includes protein metadata obtained from UniProt (Figure 4B). If available, information about protein functionality, sequence data, and published interactions are also displayed, with the sequence data section displaying different features, including domains and motifs, known mutagenesis information, UniProt PTMs and their locations, and secondary structural information.
Figure 4.

Three screenshots from the Human Proteoform Atlas. (A) A gene entry summary page for NEDD8. (B) The protein summary page for NEDD8 including sequence, function and interaction data from UniProt. (C) A display of seven proteoforms associated with the NEDD8 gene.
A user may also select a specific dataset. Selecting the datasets tab displays a current list of submitted datasets (Figure 5A). When selected, a summary page for a dataset is displayed, featuring gene, protein/proteoform counts, any published manuscripts associated with the dataset (including PubMed links) and whether a dataset is enhanced (Figure 5B).
Figure 5.

Screenshots from the Human Proteoform Atlas. (A) A list of contributed datasets. (B) A dataset summary page.
DEVELOPMENT AND IMPLEMENTATION
We developed two seperate Application Programmable Interfaces (APIs); The Proteoform Repository and the Human Proteoform Atlas (Figure 6). The former, allows the verification and registration of identified proteoforms, via their ProForma (41) or sequence and PTMs, and is administered by the Consortium for Top-Down Proteomics (CTDP). The HPfA API is used to generate the UI for the Human Proteoform Atlas.
Figure 6.

Technical overview of the HPfA. The arrows indicate the flow of data between the Human Proteoform Atlas API and User Interface, the Proteoform Repository API and other external databases.
The Proteoform Repository API
Proteoform (GET) requests are sent to The Proteoform Repository, and a biological and chemical proteoform record number (PFR) returned along with a validated ProForma string. In a collaboration with UniProt, regular reports of added proteoforms are included in the PTM / Processing section of associated protein entries (under Proteomic databases: TopDownProteomics). The Proteoform Repository acts as a standalone API that can be utilized outside of the Human Proteoform Atlas.
The central databases for the Proteoform Repository were created using a MariaDB v10.3 database server hosted at Northwestern University. Web services were created using Microsoft's ASP.NET 5.0 framework using C# v9.0. Documentation of the web API was created using OpenAPI 3.0 and Swagger. All APIs support only ProForma version 2.0 and are not backward compatible with version 1.0. The PFRs generated are guaranteed to be immutable, persistent and unique by consistently processing the ProForma strings received. Each string is validated, translated to use a standard set of modifications, hashed and compared against a database of previously seen ProForma hashes (using a .NET process-level concurrency lock to prevent race conditions).
The Human Proteoform Atlas
The Human Proteoform Atlas (HPfA) consists of a web frontend and database backend established on a cloud platform (Microsoft Azure). The graphical user interface was implemented in Angular 10 using the Angular Material component library. The backend is comprised of a .NET 5 REST API, Entity Framework Core 5 for database access, and a Microsoft SQL Server database. The Human Proteoform Atlas API makes requests to The Proteoform Repository API during datasets submission. Proteoforms are validated and PFRs obtained and incorporated into the SQL database.
External database interactions
The Human Proteoform Atlas UI and backend API both interact with external APIs. The UI utilizes calls to the EBI UniProt API for protein entry information on Protein Overview pages, the HUGO Gene Nomenclature Committee API (42) for gene information, and to the EMBL-EBI Ontology API (39) for ontology data. During dataset submission the Human Proteoform Atlas backend REST API makes calls to the EMBL-EBI Ontology API for ontology term validation.
Data collection and integration
Custom .NET 5 scripts were created to facilitate the internal loading of existing public top-down proteomic data, hand-curated from the literature. In the interest of facilitating data exchange, we have used community-generated data standards including mzIdentML (45) and ProForma for the submission of datasets. Submitted proteoforms do not require full PTM localization (modifications may have a range), corresponding to a level 3 proteoform (or less) as described in Smith et al. (46). Users wanting to submit their own datasets can submit by emailing nrtdp-help@northwestern.edu. Similar to the Proteomics Identification Database (PRIDE) (47), two types of dataset submission are available: standard and enhanced. The standard submission requires at least one target Atlas, a dataset name, any associated ontology terms, at least one associated publication (increasing confidence in the reported proteoforms), and a list of proteoforms as either ProForma or TopPIC strings (48). Optionally, external links to data repositories (such as PRIDE) can also be included, further increasing confidence in reported proteoforms. Enhanced submissions require mzIdentML files (which can be converted from TDReports generated in TDPortal and Thermo Fisher's ProSightPD). Each mzIdentML file may be given its own ontology terms, allowing the grouping of dataset files with different tissues/cell types into one dataset. Enhanced datasets contain spectral information, including fragment scores and coverage, allowing the creation of fragment maps.
FUTURE DIRECTIONS
Given the difficulty of housing all proteoform information from all biological domains in one central location, an open-source framework which creates a freestanding proteoform knowledgebase with the minimum functionality needed to store and display proteoforms would be desirable. This will allow other research communities to establish their own domain-specific proteoform knowledgebases. Associated with the Human Proteoform Project (49–51), proposed by the Consortium for Top-Down Proteomics, we plan to package and adapt an open-source codebase to build robust and scalable proteoform atlases of high-utility.
Currently, dataset submission is coordinated via email and can be cumbersome. Therefore, we plan to work with the community to create a web and/or desktop submission client, allowing ontology validation, client-side proteoform validation and ORCID integration. In addition, the National Resource for Translational and Developmental Proteomics intends to modify their tool, ProSight Lite, to take advantage of the Chemical Proteoform API such that it will be able to easily upload individual user-identified chemical proteoforms.
DATA AVAILABILITY
The Human Proteoform Atlas is a collaborative repository for human proteoforms (http://human-proteoform-atlas.org), with a publicly available API (http://api.human-proteoform-atlas.org). The Proteoform Repository is a publicly available web API that supports ProForma v2.0 submissions (http://www.proteoform.org/api). An open-source codebase is also available for the conversion of tdReports to mzIdentML files (https://github.com/NRTDP/tdReport-to-mzIdentML), and an open-source ProForma parser is available as part of the Consortium for Top-Down Proteomics' open-source software solution (https://github.com/topdownproteomics/sdk).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Paul Danis and the Board of Directors of the Consortium for Top-Down Proteomics for their engagement and support, and Hartmut Schlüter for advancing the Human Proteoform Atlas Initiative and devising the acronym, HPfA.
Notes
Present address: Paul M. Thomas, AbbVie, Inc., 1401 Sheridan Rd, North Chicago,IL 60064, USA.
Contributor Information
Michael A R Hollas, Departments of Molecular Biosciences, Chemistry, and the Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA.
Matthew T Robey, Departments of Molecular Biosciences, Chemistry, and the Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA.
Ryan T Fellers, Departments of Molecular Biosciences, Chemistry, and the Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA.
Richard D LeDuc, Departments of Molecular Biosciences, Chemistry, and the Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA.
Paul M Thomas, Departments of Molecular Biosciences, Chemistry, and the Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA.
Neil L Kelleher, Departments of Molecular Biosciences, Chemistry, and the Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Resource for Translational and Developmental Proteomics [P41 GM108569]; Paul G. Allen Frontiers Program [Award 11715]; National Library of Medicine [R21 LM013097]. Funding for open access charge: National Resource for Translational and Developmental Proteomics [P41 GM108569]; Paul G. Allen Frontiers Program [Award 11715]; National Library of Medicine [R21 LM013097].
Conflict of interest statement. M.T.R., R.T.F. and N.L.K. contribute to the development of commercial proteomics software.
REFERENCES
- 1. Smith L.M., Kelleher N.L., Linial M., Goodlett D., Langridge-Smith P., Ah Goo Y., Safford G., Bonilla L., Kruppa G., Zubarev R.et al.. Proteoform: a single term describing protein complexity. Nat. Methods. 2013; 10:186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Consortium T.U. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2020; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aebersold R., Agar J.N., Amster I.J., Baker M.S., Bertozzi C.R., Boja E.S., Costello C.E., Cravatt B.F., Fenselau C., Garcia B.A.et al.. How many human proteoforms are there. Nat. Chem. Biol. 2018; 14:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ntai I., Fornelli L., DeHart C.J., Hutton J.E., Doubleday P.F., LeDuc R.D., van Nispen A.J., Fellers R.T., Whiteley G., Boja E.S.et al.. Precise characterization of KRAS4b proteoforms in human colorectal cells and tumors reveals mutation/modification cross-talk. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aebersold R., Agar J.N., Amster I.J., Baker M.S., Bertozzi C.R., Boja E.S., Costello C.E., Cravatt B.F., Fenselau C., Garcia B.A.et al.. How many human proteoforms are there. Nat. Chem. Biol. 2018; 14:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Natale D.A., Arighi C.N., Blake J.A., Bona J., Chen C., Chen S.C., Christie K.R., Cowart J., D’Eustachio P., Diehl A.D.et al.. Protein Ontology (PRO): enhancing and scaling up the representation of protein entities. Nucleic Acids Res. 2017; 45:D339–D346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahlf D.R., Compton P.D., Tran J.C., Early B.P., Thomas P.M., Kelleher N.L.. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J. Proteome Res. 2012; 11:4308–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sherma N.D., Borges C.R., Trenchevska O., Jarvis J.W., Rehder D.S., Oran P.E., Nelson R.W., Nedelkov D. Mass spectrometric immunoassay for the qualitative and quantitative analysis of the cytokine Macrophage Migration Inhibitory Factor (MIF). Proteome Sci. 2014; 12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cabras T., Sanna M., Manconi B., Fanni D., Demelia L., Sorbello O., Iavarone F., Castagnola M., Faa G., Messana I.. Proteomic investigation of whole saliva in Wilson's disease. J. Proteomics. 2015; 128:154–163. [DOI] [PubMed] [Google Scholar]
- 10. Chen Y.C., Sumandea M.P., Larsson L., Moss R.L., Ge Y.. Dissecting human skeletal muscle troponin proteoforms by top-down mass spectrometry. J. Muscle Res. Cell Motil. 2015; 36:169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coelho Graça D., Hartmer R., Jabs W., Beris P., Clerici L., Stoermer C., Samii K., Hochstrasser D., Tsybin Y.O., Scherl A.et al.. Identification of hemoglobin variants by top-down mass spectrometry using selected diagnostic product ions. Anal. Bioanal. Chem. 2015; 407:2837–2845. [DOI] [PubMed] [Google Scholar]
- 12. Rehder D.S., Gundberg C.M., Booth S.L., Borges C.R.. Gamma-carboxylation and fragmentation of osteocalcin in human serum defined by mass spectrometry. Mol. Cell. Proteomics. 2015; 14:1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trenchevska O., Sherma N.D., Oran P.E., Reaven P.D., Nelson R.W., Nedelkov D. Quantitative mass spectrometric immunoassay for the chemokine RANTES and its variants. J. Proteomics. 2015; 116:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yassine H.N., Trenchevska O., Ramrakhiani A., Parekh A., Koska J., Walker R.W., Billheimer D., Reaven P.D., Yen F.T., Nelson R.W.et al.. The association of human apolipoprotein C-III sialylation proteoforms with plasma triglycerides. PLoS One. 2015; 10:e0144138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Azizkhanian I., Trenchevska O., Bashawri Y., Hu J., Koska J., Reaven P.D., Nelson R.W., Nedelkov D., Yassine H.N.. Posttranslational modifications of apolipoprotein A-II proteoforms in type 2 diabetes. J. Clin. Lipidol. 2016; 10:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen Y., Hoover M.E., Dang X., Shomo A.A., Guan X., Marshall A.G., Freitas M.A., Young N.L.. Quantitative mass spectrometry reveals that intact histone H1 phosphorylations are variant specific and exhibit single molecule hierarchical dependence. Mol. Cell. Proteomics. 2016; 15:818–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durbin K.R., Fornelli L., Fellers R.T., Doubleday P.F., Narita M., Kelleher N.L.. Quantitation and identification of thousands of human proteoforms below 30 kDa. J. Proteome Res. 2016; 15:976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koska J., Yassine H., Trenchevska O., Sinari S., Schwenke D.C., Yen F.T., Billheimer D., Nelson R.W., Nedelkov D., Reaven P.D.. Disialylated apolipoprotein C-III proteoform is associated with improved lipids in prediabetes and type 2 diabetes. J. Lipid Res. 2016; 57:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ntai I., LeDuc R.D., Fellers R.T., Erdmann-Gilmore P., Davies S.R., Rumsey J., Early B.P., Thomas P.M., Li S., Compton P.D.et al.. Integrated bottom-up and top-down proteomics of patient-derived breast tumor xenografts. Mol. Cell. Proteomics. 2016; 15:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peró-Gascón R., Pont L., Benavente F., Barbosa J., Sanz-Nebot V.. Analysis of serum transthyretin by on-line immunoaffinity solid-phase extraction capillary electrophoresis mass spectrometry using magnetic beads. Electrophoresis. 2016; 37:1220–1231. [DOI] [PubMed] [Google Scholar]
- 21. Pont L., Poturcu K., Benavente F., Barbosa J., Sanz-Nebot V.. Comparison of capillary electrophoresis and capillary liquid chromatography coupled to mass spectrometry for the analysis of transthyretin in human serum. J. Chromatogr. A. 2016; 1444:145–153. [DOI] [PubMed] [Google Scholar]
- 22. Trenchevska O., Yassine H.N., Borges C.R., Nelson R.W., Nedelkov D. Development of quantitative mass spectrometric immunoassay for serum amyloid A. Biomarkers. 2016; 21:743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yassine H.N., Trenchevska O., Dong Z., Bashawri Y., Koska J., Reaven P.D., Nelson R.W., Nedelkov D. The association of plasma cystatin C proteoforms with diabetic chronic kidney disease. Proteome Sci. 2016; 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anderson L.C., DeHart C.J., Kaiser N.K., Fellers R.T., Smith D.F., Greer J.B., LeDuc R.D., Blakney G.T., Thomas P.M., Kelleher N.L.et al.. Identification and characterization of human proteoforms by top-down LC-21 Tesla FT-ICR mass spectrometry. J. Proteome Res. 2017; 16:1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cleland T.P., DeHart C.J., Fellers R.T., VanNispen A.J., Greer J.B., LeDuc R.D., Parker W.R., Thomas P.M., Kelleher N.L., Brodbelt J.S.. High-throughput analysis of intact human proteins using UVPD and HCD on an Orbitrap mass spectrometer. J. Proteome Res. 2017; 16:2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fornelli L., Durbin K.R., Fellers R.T., Early B.P., Greer J.B., LeDuc R.D., Compton P.D., Kelleher N.L.. Advancing top-down analysis of the human proteome using a benchtop quadrupole-Orbitrap mass spectrometer. J. Proteome Res. 2017; 16:609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pont L., Benavente F., Barbosa J., Sanz-Nebot V.. On-line immunoaffinity solid-phase extraction capillary electrophoresis mass spectrometry using Fab′antibody fragments for the analysis of serum transthyretin. Talanta. 2017; 170:224–232. [DOI] [PubMed] [Google Scholar]
- 28. Pont L., Sanz-Nebot V., Vilaseca M., Jaumot J., Tauler R., Benavente F.. A chemometric approach for characterization of serum transthyretin in familial amyloidotic polyneuropathy type I (FAP-I) by electrospray ionization-ion mobility mass spectrometry. Talanta. 2018; 181:87–94. [DOI] [PubMed] [Google Scholar]
- 29. Vialaret J., Schmit P.O., Lehmann S., Gabelle A., Wood J., Bern M., Paape R., Suckau D., Kruppa G., Hirtz C.. Identification of multiple proteoforms biomarkers on clinical samples by routine top-down approaches. Data Brief. 2018; 18:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dai Y., Buxton K.E., Schaffer L.V., Miller R.M., Millikin R.J., Scalf M., Frey B.L., Shortreed M.R., Smith L.M.. Constructing human proteoform families using intact-mass and top-down proteomics with a multi-protease global post-translational modification discovery database. J. Proteome Res. 2019; 18:3671–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu D., Wang Z., Cupp-Sutton K.A., Liu X., Wu S.. Deep intact proteoform characterization in human cell lysate using high-pH and low-pH reversed-phase liquid chromatography. J. Am. Soc. Mass Spectrom. 2019; 30:2502–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Contini C., Firinu D., Serrao S., Manconi B., Olianas A., Cinetto F., Cossu F., Castagnola M., Messana I., Del Giacco S.et al.. RP-HPLC-ESI-IT mass spectrometry reveals significant variations of the human salivary protein profile associated with predominantly antibody deficiencies. J. Clin. Immunol. 2020; 40:329–339. [DOI] [PubMed] [Google Scholar]
- 33. Dubois C., Payen D., Simon S., Junot C., Fenaille F., Morel N., Becher F.. Top-down and bottom-up proteomics of circulating S100A8/S100A9 in plasma of septic shock patients. J. Proteome Res. 2020; 19:914–925. [DOI] [PubMed] [Google Scholar]
- 34. Schmidt A., Farine H., Keller M.P., Sebastian A., Kozera L., Welford R.W.D., Strasser D.S.. Immunoaffinity targeted mass spectrometry analysis of human plasma samples reveals an imbalance of active and inactive CXCL10 in primary Sjögren's syndrome disease patients. J. Proteome Res. 2020; 19:4196–4209. [DOI] [PubMed] [Google Scholar]
- 35. Soler-Ventura A., Gay M., Jodar M., Vilanova M., Castillo J., Arauz-Garofalo G., Villarreal L., Ballescà J.L., Vilaseca M., Oliva R.. Characterization of human sperm protamine proteoforms through a combination of top-down and bottom-up mass spectrometry approaches. J. Proteome Res. 2020; 19:221–237. [DOI] [PubMed] [Google Scholar]
- 36. Yang Z., Shen X., Chen D., Sun L.. Toward a universal sample preparation method for denaturing top-down proteomics of complex proteomes. J. Proteome Res. 2020; 19:3315–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou M., Uwugiaren N., Williams S.M., Moore R.J., Zhao R., Goodlett D., Dapic I., Paša-Tolić L., Zhu Y.. Sensitive top-down proteomics analysis of a low number of mammalian cells using a nanodroplet sample processing platform. Anal. Chem. 2020; 92:7087–7095. [DOI] [PubMed] [Google Scholar]
- 38. Schaffer L.V., Anderson L.C., Butcher D.S., Shortreed M.R., Miller R.M., Pavelec C., Smith L.M.. Construction of human proteoform families from 21 Tesla Fourier transform ion cyclotron resonance mass spectrometry top-down proteomic data. J. Proteome Res. 2021; 20:317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Madeira F., Madhusoodanan N., Lee J., Tivey A.R.N., Lopez R.. Using EMBL-EBI services via web interface and programmatically via web services. Curr. Protoc. Bioinformatics. 2019; 66:e74. [DOI] [PubMed] [Google Scholar]
- 40. Ong E., Xiang Z., Zhao B., Liu Y., Lin Y., Zheng J., Mungall C., Courtot M., Ruttenberg A., He Y.. Ontobee: a linked ontology data server to support ontology term dereferencing, linkage, query and integration. Nucleic Acids Res. 2016; 45:D347–D352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. LeDuc R.D., Schwämmle V., Shortreed M.R., Cesnik A.J., Solntsev S.K., Shaw J.B., Martin M.J., Vizcaino J.A., Alpi E., Danis P.et al.. ProForma: a standard proteoform notation. J. Proteome Res. 2018; 17:1321–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eyre T.A., Ducluzeau F., Sneddon T.P., Povey S., Bruford E.A., Lush M.J.. The HUGO gene nomenclature database, 2006 updates. Nucleic Acids Res. 2006; 34:D319–D321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D.et al.. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R.et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vizcaíno J.A., Mayer G., Perkins S., Barsnes H., Vaudel M., Perez-Riverol Y., Ternent T., Uszkoreit J., Eisenacher M., Fischer L.et al.. The mzIdentML data standard version 1.2, supporting advances in proteome informatics. Mol. Cell. Proteomics. 2017; 16:1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith L.M., Thomas P.M., Shortreed M.R., Schaffer L.V., Fellers R.T., LeDuc R.D., Tucholski T., Ge Y., Agar J.N., Anderson L.C.et al.. A five-level classification system for proteoform identifications. Nat. Methods. 2019; 16:939–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M.et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kou Q., Xun L., Liu X.. TopPIC: a software tool for top-down mass spectrometry-based proteoform identification and characterization. Bioinformatics. 2016; 32:3495–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith L., Agar J., Chamot-Rooke J., Danis P., Ge Y., Loo J., Pasa-Tolic L., Tsybin Y., Kelleher N. The human proteoform project: a plan to define the human proteome. 2020; Preprints doi:19 October 2020, preprint: not peer reviewedhttps://www.preprints.org/manuscript/202010.0368/v1. [DOI] [PMC free article] [PubMed]
- 50. Burnum-Johnson K.E., Conrads T.P., Drake R.R., Herr A.E., Iyengar R., Kelly R.T., Lundberg E., MacCoss M.J., Naba A., Nolan G.P.et al.. New views of old proteins: clarifying the enigmatic proteome. 2021; arXiv doi:17 August 2021, preprint: not peer reviewedhttps://arxiv.org/abs/2108.07660. [DOI] [PMC free article] [PubMed]
- 51. Smith L.M., Agar J.N., Chamot-Rooke J., Danis P.O., Ge Y., Loo J.A., Paša-Tolić L., Tsybin Y.O., Kelleher N.L.The Consortium for Top-Down Proteomics . The human proteoform project: defining the human proteome. Sci. Adv. 2021; 7:eabk0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Human Proteoform Atlas is a collaborative repository for human proteoforms (http://human-proteoform-atlas.org), with a publicly available API (http://api.human-proteoform-atlas.org). The Proteoform Repository is a publicly available web API that supports ProForma v2.0 submissions (http://www.proteoform.org/api). An open-source codebase is also available for the conversion of tdReports to mzIdentML files (https://github.com/NRTDP/tdReport-to-mzIdentML), and an open-source ProForma parser is available as part of the Consortium for Top-Down Proteomics' open-source software solution (https://github.com/topdownproteomics/sdk).