


Abstract
Repeats are prevalent in the genomes of all bacteria, plants and animals, and they cover nearly half of the Human genome, which play indispensable roles in the evolution, inheritance, variation and genomic instability, and serve as substrates for chromosomal rearrangements that include disease-causing deletions, inversions, and translocations. Comprehensive identification, classification and annotation of repeats in genomes can provide accurate and targeted solutions towards understanding and diagnosis of complex diseases, optimization of plant properties and development of new drugs. RepBase and Dfam are two most frequently used repeat databases, but they are not sufficiently complete. Due to the lack of a comprehensive repeat database of multiple species, the current research in this field is far from being satisfactory. LongRepMarker is a new framework developed recently by our group for comprehensive identification of genomic repeats. We here propose msRepDB based on LongRepMarker, which is currently the most comprehensive multi-species repeat database, covering >80 000 species. Comprehensive evaluations show that msRepDB contains more species, and more complete repeats and families than RepBase and Dfam databases. (https://msrepdb.cbrc.kaust.edu.sa/pages/msRepDB/index.html).
INTRODUCTION
Repetitive sequences are abundantly distributed in the genomes of all viruses, bacteria, plants and animals (1). For example, they constitute up to 45% of the genome in Mouse and 50–70% in Human (2). The repetitive sequences of the genome play a central role in the stability of the chromosome, the cell cycle and the regulation of gene expression, and they are also important substrates for genome evolution (3–6). As an example, the number and types of repetitive sequences vary between organisms and may reflect how rapidly an organism evolves to changes in its environment (7,8). Moreover, they are fundamental to the cooperative molecular interactions which form nucleoprotein complexes (9), and have also been implicated in molecular and cellular dysfunction associated with human diseases (10). For instance, the tandem repeat expansion has been associated with >40 monogenic disorders, which has recently been shown to be a major genetic contributor to frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS) and autism spectrum disorder (ASD), the middle of which is the most common form of the motor neuron disease (11,12) and the latter of which is a group of neurodevelopmental disorders characterized by atypical social function, communication deficits, restricted interests and repetitive behaviors (13–15). Besides, the expression of retrotransposition-competent transposable elements can lead to more insertions which can disrupt gene function or alter gene expression, contributing to complex diseases such as lung cancer, pancreatic cancer, ovarian cancer, neurological diseases, blood diseases (16–18), etc. Comprehensive identification, classification and annotation of repeats in genomes can provide accurate and targeted solutions towards understanding and diagnosis of complex diseases, optimization of plant properties and development of new drugs.
To achieve these goals, an accurate and complete repeat database is essential. RepBase (19) and Dfam (20) are two most frequently used repeat databases, but they are not sufficiently complete, because most of the repetitive sequences collected in these two libraries are obtained through some existing detection methods (such as RepeatScout (21) and RepeatMasker (22)). Due to the limitations of sequencing data and the defects in design of the detection principle, existing detection methods cannot accurately and comprehensively obtain the repetitive sequences of various species. For instance, in the Glycine max genome, when the combination of RepBase and Dfam is used as the repetitive sequence database, only 28.47% of bases can be annotated as LTR (Long Terminal Repeat) retrotransposons, whereas the expected proportion should be about 42% (23), which means that about 13.52% of LTR retrotransposons cannot be accurately annotated (Figure 1 and Table 4). Due to the lack of a comprehensive repetitive sequence database of multiple species, the current research in this field is far from being satisfactory.
Figure 1.

The classification and proportion statistics of repetitive sequences in Human, Drosophila and Glycine max genomes annotated by the combination of two databases, Dfam and RepBase, and msRepDB. The Y-axis represents the proportion, the X-axis represents the species. ‘Overall’ represents all types of repetitive sequences, ‘Retroelements’ represents the retroposon elements, ‘DNA transposons’ represents the DNA transposon elements, ‘Unclassified’ represents the repetitive elements that cannot be classified based on the unknown information and ‘Total interspersed repeats’ represents the total interspersed repeats.
Table 4.
Partial comparison of the proportion and detailed classification of detected repeats generated based on two databases of the Glycine max genome
| Combination of RepBase and Dfam [bases masked: 36.11%] | msRepDB [bases masked: 41.54%] | |||||
|---|---|---|---|---|---|---|
| Repeat types | Number of elements | Length occupied | Percentage of sequences | Number of elements | Length occupied | Percentage of sequences |
| Retroelements | 199 220 | 289 032 002 bp | 29.52% | 244 764 | 328 295 871 bp | 33.54% |
| +SINEs | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +Penelope | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +LINEs | 12 626 | 10 304 690 bp | 1.05% | 13 156 | 10 432 965 bp | 1.07% |
| ++CRE/SLACS | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++L2/CR1/Rex | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++R1/LOA/Jockey | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++R2/R4/NeSL | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++RTE/Bov-B | 3 790 | 2 001 199 bp | 0.20% | 3 945 | 2 017 968 bp | 0.21% |
| +++L1/CIN4 | 8 836 | 8 303 491 bp | 0.85% | 9 211 | 8 414 997 bp | 0.86% |
| +LTR elements | 186 594 | 278 727 312 bp | 28.47% | 231 608 | 317 862 906 bp | 32.47% |
| ++BEL/Pao | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| ++Tyl/Copia | 58 199 | 80 563 666 bp | 8.23% | 83 194 | 87 429 549 bp | 8.93% |
| ++Gypsy/DTRS1 | 126 690 | 195 309 037 bp | 19.95% | 140 926 | 225 546 399 bp | 23.04% |
| +++Retroviral | 0 | 0 bp | 0.00% | 340 | 206 126 bp | 0.02% |
| DNA transposons | 58 468 | 41 514 301 bp | 4.24% | 61 139 | 42 789 484 bp | 4.37% |
| +hobo-Activator | 7 612 | 2 233 822 bp | 0.23% | 5 901 | 1 964 869 bp | 0.20% |
| +Tc1-IS630-Pogo | 117 | 56 379 bp | 0.01% | 321 | 75 504 bp | 0.01% |
| +En-Spm | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +MuDR-IS905 | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +PiggyBac | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +Tourist/Harbinger | 923 | 564 171 bp | 0.06% | 1 006 | 582 191 bp | 0.06% |
| +Other | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| Rolling circles | 538 | 252 405 bp | 0.03% | 967 | 740 481 bp | 0.08% |
| Unclassified | 0 | 0 bp | 0.00% | 46 116 | 9 214 511 bp | 0.94% |
| Total interspersed repeats | 330 546 303 bp | 33.77% | 378 050 943 bp | 38.62% | ||
| Small RNA | 2 223 | 902 022 bp | 0.09% | 2 221 | 901 834 bp | 0.09% |
| Satellites | 19 885 | 2 175 759 bp | 0.22% | 9 389 | 6 367 996 bp | 0.65% |
| Simple repeats | 323 670 | 15 236 633 bp | 1.56% | 307 769 | 14 416 738 bp | 1.47% |
| Low complexity | 82 139 | 4 344 053 bp | 0.44% | 75 689 | 3 964 123 bp | 0.40% |
The test results were obtained by using RepeatMasker based on the msRepDB database and the combination of Dfam and RepBase respectively under the default parameter settings.
LongRepMarker (24) is a new framework developed recently by our group for comprehensive identification of genomic repetitive sequences. Comprehensive evaluations carried out in the study of LongRepMarker not only show that LongRepMarker can achieve more satisfactory results than the existing detection methods, but can also discover a large number of new repeat sequences and families. We here propose msRepDB (https://msrepdb.cbrc.kaust.edu.sa/pages/msRepDB/index.html) based on LongRepMarker, which is currently the most comprehensive multi-species repetitive sequence database, covering >80 000 species. msRepDB takes the reference sequence or assembly of species as the input, and generates the masked sequences representing the detected repeats and comprehensive annotation report as the output. When the input data are reference sequences or assemblies, it should be in the FASTA format, and msRepDB matches all subsequences with the database to find out the repeated elements contained in those sequences, as well as their locations and types, and finally masks the repeated elements in the input sequence and generates an annotation report. msRepDB also provides query and download functions. Users can directly retrieve and download the repetitive elements and their classification information from msRepDB according to the taxon name or the family name. In addition, if the user does not have any data, but just a taxon name or a repeat family name, msRepDB will also retrieve all relevant contents from the database and provide download links (Figure 2).
Figure 2.

The function module display of the msRepDB database. The figure mainly shows the four function modules of the msRepDB database, namely ‘Search’, ‘Download’, ‘Online Masking’ and ‘Submit’, and the detailed fields of three detection reports namely ‘Classification Report’, ‘Annotation Report’ and ‘Mapping Report’. For example, the ‘Search’ module provides the following four functions: (1) searching repeats by species name; (2) searching repeats by species name and family name; (3) displaying all annotated repeats of the species; and (4) displaying all annotation repeats of the specific family of the species. As another example, there are six fields in the mapping report: ‘Mapping zero time’, ‘Mapping one time’, ‘Mapping multiple time’, ‘N50’, ‘N75’ and ‘N90’, where ‘Mapping zero time’ indicates the proportion of fragments that cannot be aligned to the reference genome; ‘Mapping one time’ indicates the proportion of fragments that can be aligned to only one location on the reference genome; ‘Mapping multiple time’ indicates the proportion of fragments that can be aligned to many locations on the reference genome; ‘N50’ indicates the length of the longest segment such that all the segments longer than this segment cover at least 50% of the total length of all segments; ‘N75’ indicates the length of the longest segment such that all the segments longer than this segment cover at least 75% of the total length of all segments; and ‘N90’ indicates the length of the longest segment such that all the segments longer than this segment cover at least 90% of the total length of all segments.
MATERIALS AND METHODS
Data collection and identification of repetitive sequences by using LongRepMarker
To obtain a comprehensive repetitive sequence database of multiple species, we must collect the reference genomes or the assemblies of sequencing reads of these species in advance. The NCBI website (https://www.ncbi.nlm.nih.gov/) is an important channel for obtaining these required data. For example, when we enter ‘human’ in the search box on the NCBI homepage and click the search button, the page will turn to the download interface of the Human reference genome ‘GRCH38.P13’. When we continue to follow the prompts to click on the links, we will get a compressed file named ‘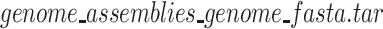 ’. After decompressing this compressed file, we will get a FASTA file named ‘
’. After decompressing this compressed file, we will get a FASTA file named ‘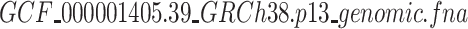 ’, which is the required Human reference genome.
’, which is the required Human reference genome.
As mentioned before, compared with existing detection methods (such as RepeatScout (21), RepeatMasker (22), RepeatModeler2 (25), etc.), LongRepMarker can not only more completely identify repetitive sequences in the genome, but also achieve more prominent performance in discovering new repetitive sequences and families. Therefore, a more comprehensive multi-species repetitive sequence database can be constructed based on the detection results of LongRepMarker (https://github.com/BioinformaticsCSU/LongRepMarker). When the reference genome or the assembly of sequencing reads of species is inputted into the LongRepMarker, it will initiate the following steps to identify and annotate the repetitive sequences contained therein.
Identification of overlap sequences. The repetition relation is a special case of the overlap relation. Thus all possible repetition relationship can be found by searching overlap sequences. Overlap sequences occupy only a small portion of the overall sequences. By finding the overlap sequences between assemblies or chromosomes, the algorithm locates the repetitive sequences faster and more accurately.
Conversion of overlap sequences into unique k-mers . The number and length of sequences will have a great impact on the efficiency of multiple sequence alignment. Generally, the more the number and the longer the length, the greater the computational resource consumption. The unique k-mers (26) (27) are much smaller than overlap sequences both in terms of number and length. Using unique k-mers instead of overlap sequences for mapping can greatly optimize the efficiency of multi-sequence alignment (28).
Generation of the multi-alignment unique k-mers and their coverage regions on overlap sequences. The multi-alignment unique k-mers were first proposed in the paper of LongRepMarker (24), which refers to the unique k-mers that can be aligned to multiple different locations in the overlap sequences. Due to the sequencing bias, the high frequency threshold is often difficult to obtain accurately, which has a great impact on the range of the high frequency k-mers (29–31). However, the multi-alignment unique k-mers are not affected by these factors. By using the multi-alignment unique k-mers to identify repeats in overlap sequences, the algorithm can obtain the repeats in the genomes more comprehensively and stably.
Classification of regions on overlap sequences that can be covered by multi-alignment unique k-mers . Due to the short size of unique k-mers, it is easy to form a coupling alignment (coupling alignment refers to the fusion of unique k-mers that should not be fused together) (32,33). To eliminate the influence of the coupling alignment, the algorithm further classifies the regions on the overlap sequences covered by the multi-alignment unique k-mers into two categories, and filters out the false repetitive sequences, thereby improving the accuracy of the detection results.
Merging fragments with duplication or inclusion. The results of detection methods based on the multiple sequence alignment will inevitably contain redundant elements. In order to make the detection results as pure as possible without any impurities and redundancy, the algorithm merges the detected repetitive fragments with duplication and inclusion relationships (34).
Classification and annotation of the obtained repetitive sequences. When the repetitive sequences are obtained, the algorithm also needs to classify and annotate them, because the repeats without classification and annotation information are meaningless. In this step, a distributed RepeatClassifier (25) developed by our group is used to classify and annotate the obtained repetitive sequences.
Note that LongRepMarker is different from RepeatScout and RepeatModeler2 in detection targets. RepeatScout and RepeatModeler2 both focus on the discovery of repeated families. It is well known that a repeat family is an abstraction of a type of repeat sequence (a one-to-many relationship), and its acquisition must go through the two operations of merging the obtained repeat sequences and taking the consensus sequence. However, the detection goal of LongRepMarker is not to find repeated families, but to comprehensively mine all repeated sequences of the genome (Supplementary Figure S2), and provide a basis for accurately identifying the mutations that exist between different copies. Therefore, in the detection results of LongRepMarker, we merged duplicate copies with high consistency, and saved the duplicate copies with differences as much as possible, and at the same time analyzed the structural variation that occurred in the duplicate copies with differences. Our purpose is to provide a method to study the effect of variations that exist between different duplicate copies on the genetic, evolution and variation of organisms.
Although there will be some redundancy and chimerism in the detection results of LongRepMarker, the repetitive sequences identified by it are still the most complete compared to other existing tools. In order to remove impurities and chimeras in the detection results and output purer repetitive sequences for the database construction, three steps of impurity removal, chimerism removal and consensus sequence construction are carried out after the detection results of LongRepMarker obtained. In this process, the strategies of wicker 80/80/80 rule (as used in RepeatScout), filtering overlaps whose identity is lower than 85% (as used in RepeatScout), and the cutoff score of 225 (as used in RepeatMasker) were used. When the purified repetitive sequences are generated, RepeatClassifier is used to classify and annotate these sequences. After that, the algorithm needs to merge the repeated sequences with its classification and annotation information, and form a file in the FASTA format (35). In this generated file in the FASTA format, the sequence composed of A/T/G/C characters is a repeating sequence, and the sequence starting with an angle bracket above the repeating sequence is the annotation sequence, which contains the corresponding classification and annotation information (36).
Extracting the repetitive sequences and their corresponding families contained in each species from the detection results and storing them in the database
When the purification operation of the previous step is completed, we need to extract the repetitive sequences from the fusion results (files in the FASTA format) and store it in the msRepDB database according to its species name, NCBI accession number, taxid and family information.
DATABASE CONTENT AND USAGE
Home and About
The function of the Home page (Figure 3 A) is to introduce msRepDB, mainly including the application fields of msRepDB, the research and development principles, and the main advantages compared with the existing libraries. The function of the About page is to introduce the main functions and test samples of msRepDB.
Figure 3.

The main functions and usage of msRepDB. Through the ‘Home’ page shown in sub-graph (A), users can understand the research background, research value, and main advantages of msRepDB. Through the ‘Search and Download’ page shown in sub-graph (B), users can retrieve and download the complete repetitive sequences with annotation of the special species by selecting the species taxonomy name and repeat family name. Through the ‘Online Masking’ page shown in sub-graph (C), users can submit the sequence file in the FASTA format to be masked by dragging and dropping or paste the sequence to be masked into the input box on the interface. Through the ‘Submit’ page shown in sub-graph (D), users can update the contents of msRepDB database. Update operations can be divided into the following two types:1) inserting new records and 2) updating existing records in the database. Furthermore, users can also browse the complete functions and detailed instructions of msRepDB through the ‘About’ page, download all tools related to this research through the ‘Tools’ page, and learn about the development team of msRepDB through the ‘Team’ page. Sub-graph (E) shows the screenshot of the complete repetitive sequences with annotation of the special species in the FASTA format. Sub-graph (F) shows the screenshot of the mapping report. Sub-graph (G) shows the screenshot of the classification report. Sub-graph (H) shows the screenshot of the masked sequence file in the FASTA format. Sub-graph (I) shows the screenshot of the extracted masked sequences file in the FASTA format. Sub-graph (J) shows the screenshot of the annotation report.
Search and Download
The functions of the Search and Download page are as follows: (i) by selecting the species taxonomy name, NCBI accession number, taxonomy id and repeat family name in the Search and Download interface, users can retrieve the complete repetitive sequences with classification information of the special species; (ii) by clicking the ‘Download’ button on the interface, users can also download the comprehensive repetitive sequences with classification information of the specific species that they have retrieved to the local disk (Figure 3 B).
Usage example:
Click the ‘select species’ input box to trigger the list of candidate species names;
-
Select or write ‘Drosophila files genus’ in the list box of species taxonomy name, and click the ‘Search’ button;
[Server response]: The server will display all the repetitive sequences and classification information in the genome of Drosophila on the bottom of the interface.
-
Select ‘Drosophila files genus’ in the list box of species taxonomy name, select ‘LTR/Pao’ in the list box of repeat family name, and click the ‘Search’ button;
[Server response]: The server will display all the LTR/Pao-types of repetitive sequences and classification information in the genome of Drosophila on the bottom of the interface.
-
Click the ‘Total families of Drosophila files genus download’ button on the left of the interface;
[Server response]: The server will provide all the repetitive sequences with classification information (LTR/Pao) of the species selected (Drosophila files genus) by the user in the FASTA format (Figure 3 E), and the user can save the downloaded file to the preferred local directory through the ‘Browse’ option.
Online Masking
The functions of the Online Masking are as follows: (i) by dragging and dropping or pasting the sequence to be masked into the input box on the interface, the users can submit the sequence file in the FASTA format to be masked on the ‘Online Masking’ interface; (ii) when the server completes the masking task, it will feed back the masking results to the interface, and the user can obtain detailed masking results and related reports (the annotation mainly includes the classification of the repetitive sequences and their locations in the genome) by browsing and downloading (Figure 3C).
Usage example:
Select ‘Drosophila files genus’ in the list box of species taxonomy name and configure the related parameters;
Download the demo reference genome of Drosophlia (
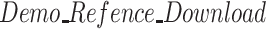 ) to the local disk. The complete Reference download address is https://www.ncbi.nlm.nih.gov/assembly/GCF_000001215.4;
) to the local disk. The complete Reference download address is https://www.ncbi.nlm.nih.gov/assembly/GCF_000001215.4;Upload the file ‘
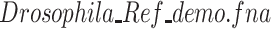 ’ to the server by dragging and dropping from the online masking interface;
’ to the server by dragging and dropping from the online masking interface;-
Click the ‘Submit Masking Job’ button;
[Server response]: When the server receives the submitted file, it will take several to ten minutes to complete masking and generate annotation reports. When the server completes the whole process of online masking, it will prompt that the task has been completed and provide download links for all generated reports.
-
Click the download links (such as ‘Mapping Report’, ‘Classification Report’, ‘N50 & Alignment ratios Report’, ‘Masked sequences by msRepDB’ and ‘Extracted Masked Sequences’) in the online masking interface, and save the masked sequence files and several annotation reports (the annotation mainly includes the classification of the repetitive sequences and their locations in the genome) to the preferred local directory through the ‘Browse’ option;
[Server response]: The server will provide download links for the masked sequence files and all generated reports on the interface (Figure 3F–J).
Submit and tools
The submit function is mainly used to update the contents of the msRepDB database (Figure 3D). Update operations can be divided into the following two types: (i) insert new records into the msRepDB database and (ii) update existing records in the msRepDB database. The data submission operation is completed by the system administrator. Before data submission, the administrator needs to evaluate the submitted data to verify its authenticity and reliability. New data can be entered into the database after passing the assessment. The function of the ‘Tools’ page is to introduce the tools related to our research.
Usage example:
-
Enter the species’ scientific name, taxonomy id, NCBI accession number, and repeat sequence with the family information (Figure 3 D);
[Specific operation]:
Step1: Set the taxonomy id as ‘2517382’;
Step2: Set the species name as ‘Afrixalus weidholzi’;
Step3: Set the repeat sequence with ID and family information as follows
‘>7SLRNA_short_#SINE/Alu
GCCGGGCGCGGTGGCGCGTGCCTGTAGTCCCAGCTA
CTCGGGAGGCTGAGGTGGGAGGATCGCTTGAGTCCA
GGAGTTCTGGGCTGTAGTGCGCTATGCCGATCGGGT
GTCCGCACTAAGTTCGGCATCAATATGGTGACCTCC
CGGGAGCGGGGGACCACCAGGTTGCCTAAGGAGGGG
TGAACCGGCCCAGGTCGGAAACGGAGCAGGTCAAAA
CTCCCGTGCTGATCAGTAGTGGGATCGCGCCTGTGA
ATAGCCACTGCACTCCAGCCTGAGCAACATAGCGAG
ACCCCGTCTCTTAAAAAAAAAAAAAA';
-
Click the ‘Submit’ button on the interface;
[Server response]: When the server receives the submitted information, it will take several seconds to complete the storage and generate feedback information on the interface.
IMPLEMENTATION
The data processing and analysis functions of msRepDB database were implemented using Python v.3.6.9 (www.python.org/getit/) coupled with the SpringBoot integrated framework (https://spring.io/projects/spring-boot). msRepDB runs on a Linux-based Maven server 3.8.1 (Maven is a build automation tool used primarily for java projects, https://maven.apache.org/download.cgi). The database was developed using MySQL 5.7.31 (https://www.mysql.com/), and the web interface was developed using html5 markup language (https://en.wikipedia.org/wiki/HTML5) combined with Bootstrap v.5.0.2 (https://v3.bootcss.com), layUI v.2.6.8 (https://www.layui.com/) and JQuery v.1.11.1 (http://jquery.com) (Supplementary Figures S3– S8). In the process of online masking, two aligners, bwa (37) and minimap2 (38), were used. In this process, the short sequence fragments were aligned using bwa, and the long sequence fragments were aligned using minimap2.
DISCUSSION
Compared with the existing repeat databases, the major improvements of msRepDB are as follows: (i) msRepDB contains more species than RepBase and Dfam databases (i.e. >84 000 in msRepDB versus about 62 000 in the combination of RepBase and Dfam). The comprehensive experiments carried out in the study of LongRepMarker not only show that LongRepMarker can achieve more satisfactory results than the existing detection methods (Supplementary Tables S4–S6, Supplementary Figures S9–S10), but also can discover a large number of new repeat sequences and families. (ii) For a single species, msRepDB contains more complete repeats and families than the existing repeat databases. We have conducted comprehensive experimental evaluations on the coverage and completeness of the msRepDB database. For example, we used the latest version of RepeatMasker (V.4.1.2) to classify and annotate the repeats of the species Human, Mouse, Rice, Glycine max and Drosophila based on the msRepDB database and the combination of the latest RepBase (V.26.06) and Dfam (V.3.3) libraries, respectively. The frequency and length distribution, the multiple alignment ratio, the proportion of coverage over the reference genome and the duplication ratio of the repetitive sequences contained in msRepDB and the combination of Dfam and RepBase databases are shown in Table 1. We can see that the repetitive sequences collected in the msRepDB database have a higher repetition frequency and larger size as a whole. Furthermore, from the perspective of multiple alignment ratio, coverage of the reference genome, and duplication ratio, the repetitive sequences contained in msRepDB are usually more accurate and less redundant than those contained in the combination of Dfam and RepBase databases. Here, the duplication ratio represents the total number of aligned bases in the repetitive sequences divided by the total number of those in the reference. If there are too many repetitive sequences that cover the same regions, the duplication ratio will be greatly increased. This occurs due to multiple reasons, including overestimating repeat multiplicities and overlaps between repetitive sequences.
Table 1.
Partial comparison of the length distribution, multiple alignment ratio, proportion of covering the reference genome and duplication ratio of elements contained in msRepDB database and the combination of Dfam and RepBase
| Length distribution | Mapping | RepeatMasker | Other | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Species | Database | Num | Max | N50 | N75 | N95 | MAR | Non-MAR | Reference | Duplication ratio |
| (bp) | (bp) | (bp) | (bp) | (%) | (%) | (%) | (%) | |||
| H.sapiens(Human) | msRepDB | 1613 | 20 016 | 2858 | 903 | 496 | 88.17% | 11.82% | 47.29% | 0.09% |
| Dfam+RepBase | 1353 | 9043 | 2532 | 786 | 464 | 80.93% | 19.06% | 45.62% | 0.15% | |
| Mouse | msRepDB | 1779 | 15 041 | 3691 | 1061 | 505 | 94.41% | 5.58% | 43.15% | 0.14% |
| Dfam+RepBase | 1407 | 8959 | 2210 | 791 | 437 | 86.28% | 13.71% | 40.58% | 0.21% | |
| Oryza sativa(Rice) | msRepDB | 3556 | 13 922 | 3584 | 1668 | 801 | 98.94% | 1.05% | 50.62% | 3.90% |
| Dfam+RepBase | 3049 | 20 789 | 3879 | 1831 | 892 | 82.81% | 17.18% | 50.50% | 4.14% | |
| D.melanogaster | msRepDB | 477 | 20 014 | 4646 | 2571 | 1153 | 99.65% | 0.34% | 21.86% | 2.40% |
| Dfam+RepBase | 258 | 15 576 | 4802 | 3204 | 1036 | 89.77% | 10.22% | 20.85% | 3.36% | |
| Glycine max | msRepDB | 1226 | 10 856 | 4536 | 3175 | 1130 | 100.00% | 0.00% | 41.31% | 0.44% |
| Dfam+RepBase | 596 | 17 080 | 4688 | 4180 | 3207 | 90.45% | 9.54% | 36.11% | 0.53% | |
‘Num’ represents the number of fragments contained in database. ‘Max(bp)’ represents the length of the longest fragment in database. ‘N50’ represents the length of a fragment, such that all the fragments of at least the same length together cover at least 50% of the total length of all fragments contained in database. ‘N75’ represents the length of a fragment, such that all the fragments of at least the same length together cover at least 75% of the total length of all fragments contained in database. ‘N95’ represents the length of a fragment, such that all the fragments of at least the same length together cover at least 95% of the total length of all fragments contained in database. ‘MAR(%) and Non-MAR(%)’ respectively represent the ratios of multiple alignment and non-multiple alignment. ‘Reference(%)’ represents the proportion of covering the reference genome. ‘Duplication ratio’ represents the total number of aligned bases in the repetitive sequences divided by the total number of those in the reference. If there are too many repetitive sequences that cover the same regions, the duplication ratio will be greatly increased. This occurs due to multiple reasons, including overestimating repeat multiplicities and overlaps between repetitive sequences.
The experimental results in Tables 2, 3 and 4 show that RepeatMasker annotated 3 852 568 Retroelements-type repeats (1 291 793.390 kb in length) on the Human genome based on msRepDB, as compared to 2 800 814 Retroelements-type repeats (1 236 215.277kb in length) for the combination of the state-of-the-art databases (Table 2), annotated 1 828 Statellites-type repeats (1 862.670 kb in length) on the Drosophila genome based on msRepDB, as compared to 1 372 Statellites-type repeats (1 804.199 kb in length) for the combination of the two other databases (Table 3), and annotated 61 139 DNA-transposons-type repeats (42 789.484 kb in length) on the Glycine max genome based on msRepDB, as compared to 58 468 DNA-transposons-type repeats (41 514.301 kb in length) for the combination of the two other databases (Table 4). It can be seen from the experimental results shown in Tables 1– 4, Supplementary Tables S7–S12 and Supplementary Figures S11–S26 that msRepDB is the most complete multi-species repetitive sequence database at present. In order to evaluate the false positive rate of the detection results, we conducted the experiments on the simulated sequencing data for Drosophila, and then we used RepeatMasker to annotate the repeats, and used the annotated set as the ground-truth set to compare with the annotation from RepeatScout and from LongRepMarker (Supplementary Table S13). All the false positives are counted by comparing the ground-truth set of annotations with that of RepeatScout or LongRepMarker.
Table 2.
Partial comparison of the proportion and detailed classification of detected repeats generated based on two databases of the Human genome
| Combination of RepBase and Dfam [bases masked: 45.62%] | msRepDB [bases masked: 47.29%] | |||||
|---|---|---|---|---|---|---|
| Repeat types | Number of elements | Length occupied | Percentage of sequences | Number of elements | Length occupied | Percentage of sequences |
| Retroelements | 2 800 814 | 1 236 215 277 bp | 37.78% | 3 852 568 | 1 291 793 390 bp | 39.48% |
| +SINEs | 1 453 130 | 369 205 643 bp | 11.28% | 1 602 909 | 329 745 622 bp | 10.08% |
| +Penelope | 75 | 14 277 bp | 0.00% | 75 | 14 225 bp | 0.00% |
| +LINEs | 807 771 | 588 058 432 bp | 17.97% | 1 630 986 | 696 100 321 bp | 21.27% |
| ++CRE/SLACS | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++L2/CR1/Rex | 193 908 | 56 822 264 bp | 1.74% | 294 645 | 69 266 031 bp | 2.12% |
| +++R1/LOA/Jockey | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++R2/R4/NeSL | 399 | 95 545 bp | 0.00% | 400 | 95 122 bp | 0.00% |
| +++RTE/Bov-B | 9 890 | 2 788 967 bp | 0.09% | 9 890 | 2 771 539 bp | 0.08% |
| +++L1/CIN4 | 603 337 | 528 287 954 bp | 16.15% | 1 325 814 | 623 904 329 bp | 19.07% |
| +LTR elements | 539 913 | 278 951 202 bp | 8.53% | 618 673 | 265 947 447 bp | 8.13% |
| ++BEL/Pao | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| ++Tyl/Copia | 0 | 0 bp | 0.00% | 12 | 3718 bp | 0.00% |
| ++Gypsy/DTRS1 | 14 309 | 3 767 626 bp | 0.12% | 15 125 | 3 750 523 bp | 0.11% |
| +++Retroviral | 515 395 | 272 547 814 bp | 8.33% | 593 203 | 259 578 662 bp | 7.93% |
| DNA transposons | 425 304 | 102 360 429 bp | 3.13% | 424 193 | 100 612 296 bp | 3.07% |
| +hobo-Activator | 280 952 | 57 692 527 bp | 1.76% | 280 102 | 56 974 131 bp | 1.74% |
| +Tc1-IS630-Pogo | 128 851 | 41 753 772 bp | 1.28% | 128 539 | 40 749 342 bp | 1.25% |
| +En-Spm | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +MuDR-IS905 | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +PiggyBac | 2310 | 554 582 bp | 0.02% | 2285 | 546 552 bp | 0.02% |
| +Tourist/Harbinger | 321 | 59 199 bp | 0.00% | 320 | 59 104 bp | 0.00% |
| +Other | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| Rolling circles | 1614 | 402 976 bp | 0.01% | 3664 | 1 046 162 bp | 0.03% |
| Unclassified | 122 691 | 24 233 010 bp | 0.74% | 225 158 | 30 427 467 bp | 0.93% |
| Total interspersed repeats | 1 362 808 716 bp | 41.65% | 1 422 833 153 bp | 43.48% | ||
| Small RNA | 12 650 | 1 358 026 bp | 0.04% | 10 142 | 979 175 bp | 0.03% |
| Satellites | 15 404 | 82 714 065 bp | 2.53% | 12 135 | 79 167 870 bp | 2.42% |
| Simple repeats | 710 220 | 39 030 544 bp | 1.19% | 663 652 | 37 699 053 bp | 1.15% |
| Low complexity | 102 465 | 6 353 924 bp | 0.19% | 92 549 | 5 565 612 bp | 0.17% |
The test results were obtained by using RepeatMasker based on the msRepDB database and the combination of Dfam and RepBase respectively under the default parameter settings.
Table 3.
Partial comparison of the proportion and detailed classification of detected repeats generated based on two databases of the Drosophila genome
| Combination of RepBase and Dfam [bases masked: 20.85%] | msRepDB [bases masked: 21.86%] | |||||
|---|---|---|---|---|---|---|
| Repeat types | Number of elements | Length occupied | Percentage of sequences | Number of elements | Length occupied | Percentage of sequences |
| Retroelements | 15 330 | 21 048 835 bp | 14.65% | 23 186 | 22 483 014 bp | 15.64% |
| +SINEs | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +Penelope | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +LINEs | 5293 | 5 447 560 bp | 4.49% | 6134 | 6 416 652 bp | 4.46% |
| ++CRE/SLACS | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++L2/CR1/Rex | 811 | 844 019 bp | 0.59% | 870 | 841 783 bp | 0.59% |
| +++R1/LOA/Jockey | 1014 | 1 562 240 bp | 1.09% | 1571 | 1 694 722 bp | 1.18% |
| +++R2/R4/NeSL | 38 | 39 896 bp | 0.03% | 38 | 39 900 bp | 0.03% |
| +++RTE/Bov-B | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +++L1/CIN4 | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +LTR elements | 10 037 | 14 601 275 bp | 10.16% | 16 914 | 16 066 362 bp | 11.18% |
| ++BEL/Pao | 2326 | 3 123 105 bp | 2.17% | 2937 | 3 118 973 bp | 2.17% |
| ++Tyl/Copia | 500 | 740 782 bp | 0.52% | 784 | 733 449 bp | 0.51% |
| ++Gypsy/DTRS1 | 7211 | 10 737 388 bp | 7.47% | 13 243 | 12 190 939 bp | 8.48% |
| +++Retroviral | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| DNA transposons | 4135 | 1 870 086 bp | 1.30% | 4494 | 1 824 527 bp | 1.27% |
| +hobo-Activator | 189 | 75 919 bp | 0.05% | 168 | 76 244 bp | 0.05% |
| +Tc1-IS630-Pogo | 1112 | 609 344 bp | 0.42% | 1108 | 560 858 bp | 0.39% |
| +En-Spm | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +MuDR-IS905 | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +PiggyBac | 23 | 8619 bp | 0.01% | 23 | 8617 bp | 0.01% |
| +Tourist/Harbinger | 0 | 0 bp | 0.00% | 0 | 0 bp | 0.00% |
| +Other | 2243 | 913 674 bp | 0.64% | 2454 | 894 197 bp | 0.62% |
| Rolling circles | 4662 | 999 082 bp | 0.70% | 5232 | 1 028 233 bp | 0.72% |
| Unclassified | 495 | 78 825 bp | 0.05% | 534 | 121 856 bp | 0.08% |
| Total interspersed repeats | 22 997 746 bp | 16.00% | 24 429 397 bp | 17.00% | ||
| Small RNA | 306 | 86 258 bp | 0.06% | 280 | 95 863 bp | 0.07% |
| Satellites | 1372 | 1 804 199 bp | 1.26% | 1828 | 1 862 670 bp | 1.30% |
| Simple repeats | 85 083 | 3 589 418 bp | 2.50% | 83 836 | 3 525 845 bp | 2.45% |
| Low complexity | 10 443 | 488 602 bp | 0.34% | 10 322 | 482 327 bp | 0.34% |
The test results were obtained by using RepeatMasker based on the msRepDB database and the combination of Dfam and RepBase respectively under the default parameter settings.
The latest version of the Dfam database (v3.4) only contains the specific data of 552 species (https://dfam.org/home), which can be further subdivided into unique data and the data fused with RepBase. In addition, the data of other species are directly inherited from RepBase (about 61 518 species). Compared with the latest version of the Dfam database, the msRepDB database currently collects the repetitive sequences of 84 601 species which are obtained based on the corresponding detection results of LongRepMarker after the two processes of removing impurities and chimeras, and constructing the consensus sequences (Supplementary Figure S1, Supplementary Tables S1–S3). From the point of view of data integrity, msRepDB completely covers Dfam and RepBase, while providing data on some previously unlisted species.
The continuous update, as well as the long-term operation and maintenance of the database are fundamental for its utility. Since the establishment of our database, we have collected all available genomes on the websites of NCBI-RefSeq (39) (https://www.ncbi.nlm.nih.gov/refseq/), Ensembl (40) (http://asia.ensembl.org/info/data/ftp/index.html), FungiDB (41) (https://fungidb.org/fungidb/app) etc. based on the species name, NCBI accession number and taxid. The specific update measures are as follows. Firstly, we will further expand the coverage of species, and strive to build the most complete and accurate multi-species repetitive sequence database in field of genomic repetitive sequence research. Secondly, we will continue to improve the performance of the algorithm in the subsequent update process to achieve more accurate repeated sequences detection.
From a functional point of view, msRepDB not only provides a more complete multi-species repeat sequence database for users to view and download, but also provides with online masking and annotation functions, which is a major feature of msRepDB. We have implemented many optimizations on the code of the online masking function, so that it can efficiently process large-scale sequences. With online masking and annotation function, users can directly use msRepDB to accurately and quickly annotate genomes or sequences of interest, and obtain detailed annotation reports without the aid of any other third-party tool. For instance, the online masking will be applied in the following scenarios. Numerous cancers, genetic disorders, neurological disorders, and metabolic disorders, have been associated with the Long Interspersed Element-1 (LINE-1 or L1) retrotransposition (42–44). When RepeatMasker uses msRepDB and the combination of Dfam and RepBase as databases to annotate repetitive sequences in the Human genome, the annotation results based on msRepDB contains 1 325 814 L1/CIN4 retrotransposon elements, with annotated base length of 623 904 329 bp. However, the corresponding annotation results based on the combination of Dfam and RepBase are 603 337 and 528 287 954 bp, respectively (Table 2). The same results can also be obtained through the online masking module of the msRepDB database website. Because the proposed database contains more complete repetitive sequences and efficient use interfaces, we believe that it can provide accurate and targeted solutions towards understanding and diagnosis of complex diseases, optimization of plant properties and development of new drugs, and thus greatly benefit the genome research.
DATA AVAILABILITY
The web interface to the database is available at https://msrepdb.cbrc.kaust.edu.sa/pages/msRepDB/index.html. This website is free, open to all users and no login or password is required.
Supplementary Material
Contributor Information
Xingyu Liao, Computational Bioscience Research Center (CBRC), Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia; Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
Kang Hu, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
Adil Salhi, Computational Bioscience Research Center (CBRC), Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia.
You Zou, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
Jianxin Wang, Hunan Provincial Key Lab on Bioinformatics, School of Computer Science and Engineering, Central South University, Changsha 410083, P.R. China.
Xin Gao, Computational Bioscience Research Center (CBRC), Computer, Electrical and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955, Saudi Arabia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by the National Natural Science Foundation of China [62002388, 61732009, 61772557, U1909208], King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) [FCC/1/1976-18-01, FCC/1/1976-23-01, FCC/1/1976-25-01, FCC/1/1976-26-01, REI/1/0018-01-01, REI/1/4216-01-01, REI/1/4437-01-01, REI/1/4473-01-01, URF/1/4352-01-01, URF/1/4379-01-01, REI/1/4742-01-01, URF/1/4098-01-01], Hunan Provincial Natural Science Foundation of China [2021JJ40787], Hunan Provincial Science and Technology Program [2018wk4001] and 111 Project [B18059]. This work was carried out in part using computing resources at the High Performance Computing Center of Central South University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cox R., Mirkin S.M.. Characteristic enrichment of DNA repeats in different genomes. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:5237–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu J.Y., Shao W., Chang L., Yin Y., Li T., Zhang H., Hong Y., Percharde M., Guo L., Wu Z.et al.. Genomic repeats categorize genes with distinct functions for orchestrated regulation. Cell Rep. 2020; 30:3296–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahmad S.F., Singchat W., Jehangir M., Suntronpong A., Panthum T., Malaivijitnond S., Srikulnath K.. Dark matter of primate genomes: satellite DNA repeats and their evolutionary dynamics. Cells. 2020; 9:2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shapiro J.A., von Sternberg R.. Why repetitive DNA is essential to genome function. Biol. Rev. 2005; 80:227–250. [DOI] [PubMed] [Google Scholar]
- 5. Kaltenegger E., Leng S., Heyl A.. The effects of repeated whole genome duplication events on the evolution of cytokinin signaling pathway. BMC Evol. Biol. 2018; 18:76–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu S., Wang G., Bacolla A., Zhao J., Spitser S., Vasquez K.M.. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 2015; 10:1674–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. George C.M., Alani E.. Multiple cellular mechanisms prevent chromosomal rearrangements involving repetitive DNA. Crit. Rev. Biochem. Mol. Biol. 2012; 47:297–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hall A.C., Ostrowski L.A., Pietrobon V., Mekhail K.. Repetitive DNA loci and their modulation by the non-canonical nucleic acid structures R-loops and G-quadruplexes. Nucleus. 2017; 8:162–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shweta M., Vinod G.. Repetitive sequences in plant nuclear DNA: Types, Distribution, Evolution and Function. Genomics Proteomics Bioinformatics. 2014; 12:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hannan A. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 2018; 19:286–298. [DOI] [PubMed] [Google Scholar]
- 11. DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J.et al.. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron. 2011; 72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alan E.R., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L.et al.. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011; 72:257–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trost B., Engchuan W., Nguyen C.M., Thiruvahindrapuram B., Dolzhenko E., Backstrom l., Mirceta M., Mojarad B.A., Yin Y., Dov A.et al.. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature. 2020; 586:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitra I., Huang B., Mousavi N., Ma M., Lamkin M., Yanicky R., Shleizer-Burko S., Lohmueller K.E., Gymrek M.et al.. Patterns of de novo tandem repeat mutations and their role in autism. Nature. 2021; 589:246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hannan A.J. Repeat DNA expands our understanding of autism spectrum disorder. Nature. 2021; 589:200–202. [DOI] [PubMed] [Google Scholar]
- 16. Beck C.R., Garcia-Perez J.L., Badge R.M., Moran J.V.. LINE-1 elements in structural variation and disease. Annu. Rev. Gen. Hum. Genet. 2011; 12:187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chénais B. Transposable elements and human cancer: a causal relationship?. Biochim. Biophys. Acta. 2013; 1835:28–35. [DOI] [PubMed] [Google Scholar]
- 18. Belancio V.P., Roy-Engel A.M., Deininger P.L.. All y’all need to know ’bout retroelements in cancer. Semin. Cancer Biol. 2010; 20:200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bao W., Kojima K.K., Kohany O.. Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015; 6:11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hubley R., Finn R.D., Clements J., Eddy S.R., Jones T.A., Bao W., Smit A.F., Wheeler T.J.. The Dfam database of repetitive DNA families. Nucleic Acids Res. 2016; 44:D81–D89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Price A.L., Jones N.C., Pevzner P.A.. De novo identification of repeat families in large genomes. Bioinformatics. 2005; 21:i351–i358. [DOI] [PubMed] [Google Scholar]
- 22. Smit A.F.A., Hubley R., Green P.. RepeatMasker Open-4.0. 2015; 1996–2015. [Google Scholar]
- 23. Schmutz J., Cannon S., Schlueter J., Ma J., Mitros T., Nelson W., Hyten D.L., Song Q., Thelen J.J., Cheng J.et al.. Genome sequence of the palaeopolyploid soybean. Nature. 2010; 463:178–183. [DOI] [PubMed] [Google Scholar]
- 24. Liao X., Li M., Hu K., Wu F.-X., Gao X., Wang J.. A sensitive repeat identification framework based on short and long reads. Nucleic Acids Res. 2021; 49:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jullien M.F., Hubley R., Goubert C., Rosen J., Clark A.G., Feschotte C., Smit A.F.. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:9451–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao X., Li M., Luo J., Zou Y., Wu F.-X., Pan Y., Luo F., Wang J.. Improving de novo assembly based on read classification. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2020; 17:177–188. [DOI] [PubMed] [Google Scholar]
- 27. Liao X., Li M., Zou Y., Wu F.-X., Pan Y., Wang J.. An efficient trimming algorithm based on multi-feature fusion scoring model for NGS data. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2020; 17:728–738. [DOI] [PubMed] [Google Scholar]
- 28. Clausen P.T.L.C., Aarestrup F.M., Lund O.. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics. 2018; 19:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koch P., Platzer M., Downie B.R.. RepARK–de novo creation of repeat libraries from whole-genome NGS reads. Nucleic Acids Res. 2014; 42:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chong C., Nielsen R., Wu Y.. REPdenovo: inferring de novo repeat motifs from short sequence reads. PLoS One. 2016; 11:e0150719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liao X., Gao X., Zhang X., Wu F.-X., Wang J.. RepAHR: an improved approach for de novo repeat identification by assembly of the high-frequency reads. BMC Bioinformatics. 2020; 21:463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liao X., Li M., Zou Y., Wu F.-X., Pan Y., Wang J.. Current challenges and solutions of de novo assembly. Quant. Biol. 2019; 7:90–109. [Google Scholar]
- 33. Sohn J.I., Nam J.W.. The present and future of de novo whole-genome assembly. Brief. Bioinformatics. 2018; 19:23–40. [DOI] [PubMed] [Google Scholar]
- 34. Chen Q., Zobel J., Verspoor K.. Duplicates, redundancies and inconsistencies in the primary nucleotide databases: a descriptive study. Database. 2017; 2017:baw163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Page A.J., Taylor B., Delaney A.J., Soares J., Seemann T., Keane J.A., Harris S.R.. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016; 2:e000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bao Z., Eddy S.R.. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002; 12:1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics. 2009; 25:1754-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pruitt K.D., Tatusova T., Maglott D.R.. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007; 35:D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T.et al.. The Ensembl genome database project. Nucleic Acids Res. 2002; 30:38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Basenko E.Y., Pulman J.A., Shanmugasundram A., Harb O.S., Crouch K., Starns D., Warrenfeltz S., Aurrecoechea C., Stoeckert C.J. Jr, Kissinger J.C.et al.. FungiDB: an integrated bioinformatic resource for fungi and oomycetes. J. Fungi. 2018; 4:39–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X., Zhang R., Yu J.. New understanding of the relevant role of LINE-1 retrotransposition in human disease and immune modulation. Front. Cell Dev. Biol. 2020; 8:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solyom S., Ewing A.D., Rahrmann E.P., Doucet T., Nelson H.H., Burns M.B., Harris R.S., Sigmon D.F., Casella A., Erlanger B.et al.. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2012; 22:2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scott E.C., Gardner E.J., Masood A., Chuang N.T., Vertino P.M., Devine S.E.. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 2016; 26:745–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The web interface to the database is available at https://msrepdb.cbrc.kaust.edu.sa/pages/msRepDB/index.html. This website is free, open to all users and no login or password is required.