


Abstract
Rapid advances in high-throughput sequencing technologies have led to the discovery of thousands of extrachromosomal circular DNAs (eccDNAs) in the human genome. Loss-of-function experiments are difficult to conduct on circular and linear chromosomes, as they usually overlap. Hence, it is challenging to interpret the molecular functions of eccDNAs. Here, we present CircleBase (http://circlebase.maolab.org), an integrated resource and analysis platform used to curate and interpret eccDNAs in multiple cell types. CircleBase identifies putative functional eccDNAs by incorporating sequencing datasets, computational predictions, and manual annotations. It classifies them into six sections including targeting genes, epigenetic regulations, regulatory elements, chromatin accessibility, chromatin interactions, and genetic variants. The eccDNA targeting and regulatory networks are displayed by informative visualization tools and then prioritized. Functional enrichment analyses revealed that the top-ranked cancer cell eccDNAs were enriched in oncogenic pathways such as the Ras and PI3K-Akt signaling pathways. In contrast, eccDNAs from healthy individuals were not significantly enriched. CircleBase provides a user-friendly interface for searching, browsing, and analyzing eccDNAs in various cell/tissue types. Thus, it is useful to screen for potential functional eccDNAs and interpret their molecular mechanisms in human cancers and other diseases.
INTRODUCTION
Extrachromosomal circular DNA (eccDNA) originates from chromosomal DNA but is independent of it. EccDNA is ubiquitous in various eukaryotes and plays multiple biological roles in different cell types (1). Researchers have known for decades that eccDNA occurs in tumor cells. However, comprehensive studies on their structure and function have returned relatively little useful information because of the limitations of available detection and analysis technology (2,3). Other researchers have exploited recently developed high-throughput sequencing technology and eccDNA identification methods (4), revealing strong associations between eccDNA and cancers: (i) the eccDNA mediates oncogene over-expression by increasing gene copy numbers and transcript levels which lead to tumor deterioration (5–9). Tumor cells contain abundant oncogene-harboring eccDNA that is often amplified by co-amplification with adjacent enhancers. This mechanism leads to oncogene over-expression (6–8). Oncogene-harboring eccDNAs are mobile enhancers that globally amplify chromosomal transcription (5). Moreover, they have loose, open chromatin and a circular topology. Thus, multiple regulatory elements may remotely regulate transcription and enable the oncogenes on the eccDNAs to be transcribed efficiently (9); (ii) the eccDNA mediates drug resistance in cancer cells either by amplifying the drug resistance gene on eccDNA, or by using the reversible loss of the gain-of-function mutation of the drug resistance gene carried by eccDNA (10,11); (iii) the eccDNA can drive tumor gene heterogeneity and accelerate tumor genome evolution (12). Oncogene-derived eccDNAs, which lack centromeres, may be asymmetrically delivered to progeny cells via amitosis. Cells with relatively more oncogene-derived eccDNAs have a growth advantage and are enriched in rapidly proliferating tumor cells. Computational simulation has confirmed this phenomenon (13). Neuroblastoma eccDNAs accelerate somatic genome rearrangement and oncogene remodeling via chimeric circularization and reintegration into linear chromosomes (14).
The foregoing studies indicated that eccDNAs are promising biomarkers for cancer diagnosis and prognosis and are, therefore, research hotspots (15). Meanwhile, the number of human eccDNAs generated by the computational analysis of sequencing data has increased. Therefore, it has been challenging for researchers to compile available data to investigate their functions. In fact, the biological functions of most eccDNAs remain unknown and loss-of-function experiments are difficult to perform, as circular and linear chromosomes overlap in most cases. Moreover, the formation and regulation of most eccDNAs remain unclear and their targeting genes, genetic variants and chromatin characteristics in various cell types have seldom been explored. Therefore, an integrated database and analysis platform is required for human eccDNAs.
In the present study, we developed the novel platform CircleBase (http://circlebase.maolab.org) that compiles and interprets human eccDNAs from available public resources and predicts the regulatory networks between eccDNAs and genetic/epigenomic factors by integrating relevant databases (Figure 1). CircleBase is a powerful, convenient tool that explains the generation mechanisms underlying various eccDNA functions and facilitates the exploration of cancer cell heterogeneity and genome diversity. We are dedicated to maintaining CircleBase and extending the range of organisms and cell types to keep the resource updated.
Figure 1.

Workflow of CircleBase construction.
MATERIALS AND METHODS
eccDNA collection
CircleBase comprises 601 036 eccDNAs (candidates larger than 50M were removed) gleaned from 13 published papers on PubMed (https://pubmed.ncbi.nlm.nih.gov/) and includes the following information: (i) chromosomal localizations of eccDNAs based on reference genomes hg19 and hg38; (ii) conditions or treatments; (iii) sample types; (iv) sequencing library types and (v) validation strategies. The eccDNA localizations were collected from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) (16,17) and Genome Sequence Archive (GSA, https://ngdc.cncb.ac.cn/gsa/) (18).
Gene annotation
AnnotatePeaks.pl in the HOMER package (v. 4.11.1, http://homer.salk.edu) (19) was used with its default parameters for eccDNA gene annotation based on the genomic position. The annotation results included element annotation, distance to TSS, nearest promoter ID, NCBI gene ID, nearest Refseq ID, nearest Ensembl ID, gene name, gene alias, gene description and gene type.
Targeting genes
3 049 674 enhancer-target pairs from human variation annotation database (VARAdb) (20) were collected to determine the target genes of the eccDNAs located in the enhancers and super-enhancers. There were 9 879 737 enhancer-target networks in 935 samples determined using the JEME algorithm (https://github.com/yiplabcuhk/JEME) (21) and 284,834 enhancer-gene links identified using the GeneHancer database (22).
Regulatory elements
Regulatory elements are anchored by chromatin looping to gene promoter regions to regulate gene transcription. Enhancers and super-enhancers are positive regulatory elements that maintain cell type-specific gene expression and control cell fate during development (23). There were 65 950 super-enhancers from dbSUPER (24), 2 839 656 enhancers from EnhancerAtlas (25), 22 60 114 enhancers/super-enhancers from SEA (26) and 331 146 super-enhancers from SEdb (27). They were collected in CircleBase, and their target genes were annotated according to their source databases. Additionally, 98 274 452 chromatin states were predicted using the core 25-state ChromHMM model (28). ChromHMM was trained using the data imputed for 12 epigenetic marks across all 127 reference epigenomes in the ENCODE project (29).
Chromatin interactions
Chromatin loops mediated by the CTCF/cohesin complex connect regulatory elements to their target genes. In CircleBase, 28 442 796 chromatin interaction records were collected from three databases: (i) 25 222 085 pairs of chromatin interactions were identified using the EpiTensor algorithm in OncoBase (30). These included 2 847 794, 5 691 699 and 16 682 592 promoter–promoter, enhancer–promoter and enhancer–enhancer interactions, respectively. (ii) 3 095 881 pairs of chromatin interactions were collected from the 4DGenome database (31). They were experimentally determined via Hi-C, ChIA-PET, IM-PET, 3C, 4C and 5C. (iii) 124 830 ChIA-PET narrow peaks were detected from ENCODE (29) in MCF-7, HeLa-S3, K562, NB4 and HCT116 cancer cells.
Logistic function for score normalization
The scores were differently defined in the various annotation databases. Therefore, they were transformed to harmonize their ranges within [0,1]. The Z-score for each record in each dataset was calculated as follows:
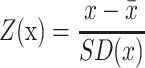 |
(1) |
where 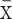 and SD(x) are the mean and standard deviation for all samples, respectively. A logistic function was then used to convert the Z-score as follows:
and SD(x) are the mean and standard deviation for all samples, respectively. A logistic function was then used to convert the Z-score as follows:
 |
(2) |
where Z is the Z-score of each sample, e is the base of natural logarithm.
Diffusion system for prioritizing target genes
The network-based diffusion algorithm PageRank was used to prioritize the target genes for each eccDNA (30). An improved version of PageRank was applied as follows:
 |
(3) |
where Weightv→u is used to measure the weight of the edge. Weight = 1 for the edge derived from the enhancer/super-enhancer interactions determined by the regulatory elements section. Weight = 0.5 for the edge derived from the chromatin connections determined by the chromatin interactions section. In the targeting genes section, the original enhancer-target scores derived from VARAdb and JEME were used as the weight values, and the enhancer-target scores from GeneHancer were normalized to [0, 1] via the foregoing logistic function. The mean weight scores were used as ultimate edge scores when a single eccDNA was implicated in >1 connection record.
Regulatory networks
Regulatory networks were constructed by combining the enhancer-gene pairs from the targeting genes, regulatory elements and chromatin interactions sections. Regulatory element targets overlapping with eccDNA were defined as eccDNA targets. Redundant eccDNA–gene pairs were removed. Genes interacting with eccDNA were ranked using Google PageRank and circle sizes were positively correlated with PageRank scores (32). An interactive view was prepared for the gene prioritization related to certain eccDNAs in a two-layer network ranked by PageRank score in the regulatory network section (30). PageRank ranking was applied to all genes regulated by certain eccDNA and all other eccDNAs regulating them. The network view showed the two-layer networks. The eccDNAs or genes related to the sub-networks were listed in the table following the interactive view.
Functional enrichment
A functional enrichment analysis was performed using the R package clusterProfiler version 4.0.2 (33). A gene ontology analysis was performed with the clusterProfiler enrichGO function (34). Kyoto Encyclopedia of Genes and Genomes pathways were analyzed with the clusterProfiler enrichKEGG function (35). Terms were considered significantly enriched when their Benjamini–Hochberg-adjusted P-values were <0.05 (36).
Chromatin accessibility
Chromatin accessibility reflected both aggregate TF binding and the regulatory potential of a genetic locus (37). In CircleBase, (i) 52 078 883 accessible regions were detected by ATAC-seq in over 1400 samples from ATACdb (38); (ii) 3 181 274 accessible regions were detected by ATAC-seq in Cistrome (39); (iii) 1 051 532 accessible regions were detected by ATAC-seq in 23 cancer types from The Cancer Genome Atlas (TCGA) (40) and (iv) 62 154 007 accessible regions were detected by DNase-seq in 243 samples from ENCODE (29).
Epigenetic regulations
There were 156 379 641 peaks collected from 7734 ChIP-seq samples of 952 TFs in ChIP-Atlas (41), Cistrome (39), ENCODE (29), GTRD (42) and ReMap (43). Moreover, 5 911 338 and 64 669 729 histone modification peaks were obtained from 185 and 979 ChIP-seq samples from ENCODE (29) and Roadmap (44), respectively. The targets of ChIP-seq experiments from ENCODE (29) included H2A.Z, H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me2, H3K4me3, H3K79me2, H3K9ac, H3K9me1, H3K9me3 and H4K20me1. The targets of ChIP-seq experiments from Roadmap included H2A, H2AK5ac, H2AK9ac, H2BK120ac, H2BK12ac, H2BK15ac, H2BK20ac, H2BK5ac, H3K14ac, H3K18ac, H3K23ac, H3K23me2, H3K27ac, H3K27me3, H3K36me3, H3K4ac, H3K4me1, H3K4me2, H3K4me3, H3K56ac, H3K79me1, H3K79me2, H3K9ac, H3K9me1, H3K9me3, H3T11ph, H4K12ac, H4K20me1, H4K5ac, H4K8ac and H4K91ac.
Genetic variants
Genetic variants were collected from 1 245 959 177 human single-nucleotide variants (SNVs) in dbSNP release 155 (45), 8 775 948 common single-nucleotide polymorphisms (SNPs) from the Genome Aggregation Database (gnomAD) Project v. 2.1 (46), 272 607 risk SNPs from the genome-wide association study (GWAS) catalog (47) and 314 238 risk SNPs from GWASdb v. 2.0 (48). Disease-related genetic variants including 81 666 495 somatic mutations of human cancers from OncoBase (30) and 670 082 de novo mutations from the Gene4Denovo database (49), were also collected.
Expression quantitative trait locus (eQTL)
The eQTL are genomic loci that regulate gene expression levels and play vital roles in deciphering gene regulation and spatiotemporal specificity (50). Correlation between genotype and tissue-specific gene expression level may be used to interpret the effect of gene variant on gene expression in human tissues or cancers. Here, 71 478 479 significant SNP–gene pairs (false discovery rate (FDR) < 0.05) in 49 human tissues were compiled from the GTEx Project v. 8.0 (51). Additionally, 1 412 029 significant cis-eQTL-gene and trans-eQTL-gene pairs in 33 cancer types were collected from the PancanQTL database (52).
Interactive circular visualization
High-resolution chromatin interactions, TF binding clusters, somatic mutations, enhancers, super-enhancers, and their predicted targets were illustrated in a circular ideogram layout plotted with BioCircos (http://bioinfo.ibp.ac.cn/biocircos/index.php) (53). The ideogram was then implemented for circular visualization of various biological data such as genomic features, genetic variants, gene expression, and biomolecular interactions.
eccDNA prioritization scoring system
The number of annotated hits (records) assigning a score to each corresponding eccDNA was used for each regulatory category. Considering that eccDNA has k hits per regulatory category (F), and μ and σ are the fitted parameters of the corresponding Gaussian model, the eccDNA score in this category was calculated as follows:
 |
(4) |
The score of each category was normalized to [0, 1] using the foregoing logistic function. The final score S of each eccDNA would be the average of normalized scores for all six regulatory categories (Equation 5) since normalized scores between some categories are not independent (Supplemental Figure S1). The calculation of the scoring system was implemented using the SciPy package v1.6.3 in Python v3.7.6 (54).
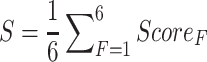 |
(5) |
System design and database construction
CircleBase (http://circlebase.maolab.org) was developed by combining jQuery with the PHP-based web framework CodeIgniter (https://codeigniter.com). It was based on previously reported databases and platforms (30,50,55,56). All datasets in CircleBase were stored either in MySQL database or as flat files. Academic users may freely access related data and analytical results through the web interface. The liftOver module in the UCSC toolkit (https://genome.ucsc.edu/cgi-bin/hgLiftOver) (57) was used to convert genomic coordinates among various versions of the human genome. In details, eccDNA locations from the paper PMID 34193237 were lifted over from hg38 to hg19 while the rest were lifted over from hg19 to hg38. In addition, super-enhancer locations from the SEA database were lifted over from hg38 to hg19.
Data and code availability
The accession, DOI, version and permalink information of the foregoing databases was listed in Supplementary Table S1. To make the codes open-access, we deposited the source codes of CircleBase in GitHub (https://github.com/leishenggit/CircleBase). The full tables of human eccDNAs are available in the Download module of CircleBase (http://circlebase.maolab.org/welcome/download).
DATABASE FEATURES AND APPLICATIONS
Statistics and features
CircleBase compiled available eccDNAs (n = 601 036) from public resources and comprehensively interpreted those in various cancer cell lines (n = 423 018), cancer tissues (n = 6498), and healthy individuals (n = 171 596) (Figure 2A). These eccDNAs were detected by whole genome sequencing (WGS, n = 427 064), Circle-Seq (n = 145 588), cf-eccDNA-seq (n = 25 995), ATAC-seq (n = 1783), whole exome sequencing (WES, n = 467) and others (Figure 2B). We collected 114 611 eccDNAs from ES2, 84 629 from LnCap, 69 420 from U937, 57 780 from OVCAR8, 41 221 from C4-2 and 38 876 from HeLaS3 cell lines (Figure 2C). We collected 136 619 eccDNAs from the muscle, 34 977 from the plasma, 939 from the connective tissue, 754 from the brain, 712 from the breast, 648 from the esophagus, 599 from the lung, 435 from the skin, 427 from the stomach, 362 from the bladder, 301 from the head and neck, 340 from the ovaries, 250 from the uterine corpus endometrial, 153 from the liver, 157 from the pancreas, 149 from the prostate, and others (Figure 2D).
Figure 2.

Percentage of eccDNAs in each category and distribution of ranking scores. (A) Percentage of eccDNAs in three cell groups including cancer cell line, cancer tissue, and healthy individual. (B) Percentage of eccDNAs in different library types such as WGS, Circle-Seq, ATAC-seq, WES and ChIA-PET. (C) Percentage of eccDNAs in different cell lines such as ES2, LnCap, U937, OVCAR8, C4-2, HeLaS3, PC-3 and C4-2B. (D) Percentage of eccDNAs in different human tissues such as muscle, plasma, connective tissue, brain, breast, esophagus and lung. (E) Density distribution of eccDNAs in different ranking scores.
Unlike our previous study (55), the hits numbers of eccDNAs per chromosome were not suitable for the Poisson distribution (Supplemental Figures S2-S7). The parameter lambda of the Poisson distribution is approximated by the average value of the observed data, and then it is substituted into the R function rpois to generate the theoretical values. But the hits numbers per chromosome could be fitted to a Gaussian distribution by using Box–Cox transformation (Supplemental Figures S8-S13). The parameters of the normal distribution, that is, the mean and variance are approximated by the mean and variance of the observed data, and then they are substituted into the R function rnorm to generate the theoretical values. Importantly, the mean values of six scores after logistic transformation conform to a normal distribution with range of [0, 1] (Figure 2E). Therefore, CircleBase helps prioritize putative functional eccDNAs by incorporating high-throughput experimental datasets from ENCODE (29), computational predictions, and manual annotations. It classifies these genetic and epigenetic annotations into targeting genes, epigenetic regulations, regulatory elements, chromatin accessibility, chromatin interactions and genetic variants (Figure 1). It predicts regulatory networks between eccDNAs and genetic/epigenomic factors by integrating relevant databases. These data sources are combined to score eccDNAs and help interpret the potential functions of the eccDNAs in the human genome.
Website interface
CircleBase comprises the Home, Search, Stats, Manual, Download, Links and Contact modules. A brief introduction of CircleBase and the workflow are shown in the Home module. Data are integrated in the Search module of CircleBase and can be searched by gene symbol, genomic region, eccDNA ID, NCBI gene ID and Ensembl gene ID. Moreover, examples are provided for new users. When users search their target eccDNAs, the target genes, regulatory elements, chromatin interactions, regulatory networks, functional enrichments, chromatin access, epigenetic regulations, genetic variants and eQTL, and the summary are displayed at the click of a mouse (Figure 3). In the Stats module, eccDNA distributions under various criteria including cell line and cell group, tissue, and library type are exhibited in a pie chart. In the Manual module, steps of how to use CircleBase are shown. In the Download module, all eccDNA data used are available for user download. In the Links Module, related databases such as TCGA (40), International Cancer Genome Consortium (ICGC) (58), and others are provided. In the Contact module, the e-mail addresses and research fields of related authors are described in detail, enabling users to communicate with our team.
Figure 3.

Web interface of the Search module in CircleBase.
Significance and applications
The top 1000 eccDNAs scored by the ranking system were used to perform functional enrichment analyses (59) including 791 and 209 eccDNAs from cancer cells and normal tissues, respectively. The top-ranked cancer cell eccDNAs were enriched in oncogenic pathways (Figure 4A) including Rap1 signaling (hsa04015), Ras signaling (hsa04014), Melanoma (hsa05218), MAPK signaling (hsa04010), PI3K-Akt signaling (hsa04151), Regulation of actin cytoskeleton (hsa04810), Phospholipase D signaling (hsa04072), Gastric cancer (hsa05226), Pathways in cancer (hsa05200) and Circadian entrainment (hsa04713). By contrast, eccDNAs from normal tissues had no significant enrichment (Figure 4B). The foregoing results validated the ranking system used in CircleBase.
Figure 4.

Functional enrichment analysis of the top 1000 eccDNAs. (A) Functional enrichment analysis of 791 eccDNAs from cancer cells. (B) Functional enrichment analysis of 209 eccDNAs from normal tissues.
Here, we considered eccDNA ecc595499 (chr10:81044184–81134986) in gastric cancer and eccDNA ecc597923 (chr2:176959007–177342179) in ovarian cancer as examples (60). CircleBase showed the targeting gengs, chromatin interactions in their loci and flanking regions, and regulatory network (Figures 5A–C; Figures 6A–C). Our platform predicted several targeting genes of ecc595499 enriched in multiple molecular functions including carbohydrate binding, S100 protein binding, MHC class II protein complex binding, translation elongation factor activity, cyclosporin A binding, MHC protein complex binding, peptidyl-prolyl cis-trans isomerase activity, cis-trans isomerase activity, calcium-dependent phospholipid binding, nuclear receptor coactivator activity, ribosome binding, and drug binding (Figure 5D). Our platform disclosed dozens of ecc597923 targeting genes enriched in multiple biological processes related to embryonic organ development such as appendage morphogenesis, limb morphogenesis, appendage development, limb development, embryonic limb morphogenesis, and embryonic appendage morphogenesis (Figure 6D). Experimental validation of eccDNA function is currently in progress and we believe that by using the regulatory networks and functional predictions from CircleBase, researchers will be able to elucidate the mechanisms underlying the functions of eccDNAs in human cancers and other diseases in the near future.
Figure 5.

Illustration of the eccDNA ecc595499 in gastric cancer. (A). BioCircos view of targeting genes. (B) BioCircos view of chromatin interactions. (C) Regulatory network. The interacting genes with the eccDNA are ranked by Google PageRank and the size of the circle is positively correlated with the PageRank score. (D) Molecular function enrichment of eccDNA ecc595499 targeting genes.
Figure 6.

Illustration of the eccDNA ecc597923 in ovarian cancer. (A) BioCircos view of targeting genes. (B) Regulatory network. The interacting genes with the eccDNA are ranked by Google PageRank and the size of the circle is positively correlated with the PageRank score. (C) BioCircos view of chromatin interactions. (D) Biological process enrichment of eccDNA targeting genes.
DISCUSSION AND PERSPECTIVES
Cancer cells can rapidly adapt to changes in the tumor micro-environment by amplifying the oncogenes on their eccDNA (61). Numerous experimental methods have been developed to detect eccDNA including Circle-Seq based on next-generation sequencing (62) and SMOOTH-seq based on third-generation sequencing platforms (63). Several computational methods have been developed to analyze eccDNAs including AmpliconArchitect (64), ECdetect (6) to identify eccDNAs from whole-genome sequencing data, and Circle-Map (65) and Circle_finder (66) to detect eccDNAs in Circle-seq and ATAC-seq data, respectively. Integrated databases and annotation platforms are required for human eccDNAs. However, none of these have been developed or published to date. As far as we know, CircleBase is the first database for eccDNA and has several advantages. It (i) is fitted with a highly interactive visualization function for eccDNAs and their related annotations; (ii) has a ranking system based on a Gaussian distribution model for better decision-making and (iii) provides comprehensive eccDNA annotations as follows:
It furnishes annotations for eccDNA targeting genes based on bioinformatics predictions by JEME (21) and EpiTensor in the OncoBase platform (30).
It incorporates epigenome information from ENCODE (29) and the Roadmap (44) epigenomics project with the chromatin status of the eccDNA locus in the linear genome.
It fine-maps the genetic basis of the eccDNA using disease-related and common SNPs from TCGA (40), gnomAD (46), dbSNP (45), GWASdb (48) and others.
It annotates eccDNAs to regulatory elements, such as promoters, enhancers, and super enhancers via ChromHMM (28), EnhancerAtlas (25), dbSUPER (24) and others.
It measures the chromatin accessibility of the eccDNA locus with ATACdb (38), Cistrome (39) and others.
It establishes chromatin interaction networks for the eccDNA locus using Hi-C processed data from 4DGenome (31), OncoBase (30) and others.
In conclusion, CircleBase curates eccDNAs in the human genome from various cell types and provides a scoring system to prioritize the eccDNAs based on comprehensive annotations. Our aims are to extend the ranges of annotations and cell types and continue updating CircleBase.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Ms Tao Wang from School of Life Sciences of Central South University for her help in this project. We thank Mr Mingcong You from Baiyining Medicine in Beijing for his valuable discussions regarding this work.
Contributor Information
Xiaolu Zhao, Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing, China; National Clinical Research Center for Obstetrics and Gynecology, Peking University Third Hospital, Beijing, China; Key Laboratory of Assisted Reproduction (Peking University), Ministry of Education, Beijing, China; Beijing Key Laboratory of Reproductive Endocrinology and Assisted Reproductive Technology (Peking University Third Hospital), Beijing, China.
Leisheng Shi, Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, and China National Center for Bioinformation, Beijing, China; University of Chinese Academy of Sciences, Beijing, China.
Shasha Ruan, Department of Clinical Oncology, Renmin Hospital of Wuhan University, Wuhan, Hubei, China; The First Clinical College of Wuhan University, Wuhan, Hubei, China.
Wenjian Bi, Department of Medical Genetics, School of Basic Medical Sciences, Peking University, Beijing, China.
Yifan Chen, Institute of Medical Innovation and Research, Peking University Third Hospital, Beijing, China; Biobank, Peking University Third Hospital, Beijing, China.
Lin Chen, State Key Laboratory of Natural and Biomimetic Drugs, Department of Chemical Biology, School of Pharmaceutical Sciences, Peking University, Beijing, China.
Yifan Liu, Department of Biochemistry & Molecular Medicine, University of Southern California Keck School of Medicine, Los Angeles, CA, USA.
Mingkun Li, Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, and China National Center for Bioinformation, Beijing, China; University of Chinese Academy of Sciences, Beijing, China; Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, China.
Jie Qiao, Center for Reproductive Medicine, Department of Obstetrics and Gynecology, Peking University Third Hospital, Beijing, China; National Clinical Research Center for Obstetrics and Gynecology, Peking University Third Hospital, Beijing, China; Key Laboratory of Assisted Reproduction (Peking University), Ministry of Education, Beijing, China; Beijing Key Laboratory of Reproductive Endocrinology and Assisted Reproductive Technology (Peking University Third Hospital), Beijing, China; Beijing Advanced Innovation Center for Genomics, Beijing, China.
Fengbiao Mao, Institute of Medical Innovation and Research, Peking University Third Hospital, Beijing, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDINGS
Clinical Medicine Plus X - Young Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities [PKU2021LCXQ015]; research start-up funding, Peking University Third Hospital [BYSYYZD2021001]; National Natural Science Foundation of China [32170493, 32170656, H16-82003137]. Funding for open access charge: Clinical Medicine Plus X - Young Scholars Project, Peking University, the Fundamental Research Funds for the Central Universities [PKU2021LCXQ015]; research start-up funding, Peking University Third Hospital [BYSYYZD2021001]; National Natural Science Foundation of China [32170493, 32170656, H16-82003137].
Conflict of interest statement. None declared.
REFERENCES
- 1. Cohen S., Yacobi K., Segal D.. Extrachromosomal circular DNA of tandemly repeated genomic sequences in Drosophila. Genome Res. 2003; 13:1133–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hotta Y., Bassel A.. Molecular size and circularity of DNA in cells of mammals and higher plants. Proc. Natl. Acad. Sci. U.S.A. 1965; 53:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cox D., Yuncken C., Spriggs A.I.. Minute chromatin bodies in malignant tumours of childhood. Lancet. 1965; 1:55–58. [DOI] [PubMed] [Google Scholar]
- 4. Wu S., Bafna V., Mischel P.S.. Extrachromosomal DNA (ecDNA) in cancer pathogenesis. Curr. Opin. Genet. Dev. 2021; 66:78–82. [DOI] [PubMed] [Google Scholar]
- 5. Zhu Y., Gujar A.D., Wong C.-H., Tjong H., Ngan C.Y., Gong L., Chen Y.-A., Kim H., Liu J., Li M.. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell. 2021; 39:694–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Turner K.M., Deshpande V., Beyter D., Koga T., Rusert J., Lee C., Li B., Arden K., Ren B., Nathanson D.A.. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature. 2017; 543:122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morton A.R., Dogan-Artun N., Faber Z.J., MacLeod G., Bartels C.F., Piazza M.S., Allan K.C., Mack S.C., Wang X., Gimple R.C.. Functional enhancers shape extrachromosomal oncogene amplifications. Cell. 2019; 179:1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schneider S., Hiemstra J., Zehnbauer B., Taillon-Miller P., Le Paslier D., Vogelstein B., Brodeur G.. Isolation and structural analysis of a 1.2-megabase N-myc amplicon from a human neuroblastoma. Mol. Cell Biol. 1992; 12:5563–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu S., Turner K.M., Nguyen N., Raviram R., Erb M., Santini J., Luebeck J., Rajkumar U., Diao Y., Li B.et al.. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature. 2019; 575:699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ståhl F., Wettergren Y., Levan G.. Amplicon structure in multidrug-resistant murine cells: a nonrearranged region of genomic DNA corresponding to large circular DNA. Mole. Cell Biol. 1992; 12:1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nathanson D.A., Gini B., Mottahedeh J., Visnyei K., Koga T., Gomez G., Eskin A., Hwang K., Wang J., Masui K.. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014; 343:72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verhaak R.G., Bafna V., Mischel P.S.. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat. Rev. Can. 2019; 19:283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith G., Taylor-Kashton C., Dushnicky L., Symons S., Wright J., Mai S.. c-Myc-induced extrachromosomal elements carry active chromatin. Neoplasia. 2003; 5:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koche R.P., Rodriguez-Fos E., Helmsauer K., Burkert M., MacArthur I.C., Maag J., Chamorro R., Munoz-Perez N., Puiggròs M., Garcia H.D.. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 2020; 52:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhu J., Zhang F., Du M., Zhang P., Fu S., Wang L.. Molecular characterization of cell-free eccDNAs in human plasma. Scient. Rep. 2017; 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edgar R., Domrachev M., Lash A.E.. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.et al.. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen T., Chen X., Zhang S., Zhu J., Tang B., Wang A., Dong L., Zhang Z., Yu C., Sun Y.et al.. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteomics Bioinformatics. 2021; 10.1016/j.gpb.2021.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duttke S.H., Chang M.W., Heinz S., Benner C.. Identification and dynamic quantification of regulatory elements using total RNA. Genome Res. 2019; 29:1836–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan Q., Liu Y.J., Bai X.F., Han X.L., Jiang Y., Ai B., Shi S.S., Wang F., Xu M.C., Wang Y.Z.et al.. VARAdb: a comprehensive variation annotation database for human. Nucleic Acids Res. 2021; 49:D1431–D1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao Q., Anyansi C., Hu X., Xu L., Xiong L., Tang W., Mok M.T.S., Cheng C., Fan X., Gerstein M.et al.. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017; 49:1428–1436. [DOI] [PubMed] [Google Scholar]
- 22. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M.et al.. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017; 2017:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun Y., Zhou B., Mao F., Xu J., Miao H., Zou Z., Phuc Khoa L.T., Jang Y., Cai S., Witkin M.et al.. HOXA9 reprograms the enhancer landscape to promote leukemogenesis. Cancer Cell. 2018; 34:643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khan A., Zhang X.. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016; 44:D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao T., Qian J.. EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2020; 48:D58–D64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen C.G., Zhou D.S., Gu Y., Wang C., Zhang M.Y., Lin X.Y., Xing J., Wang H.L., Zhang Y.. SEA version 3.0: a comprehensive extension and update of the Super-Enhancer archive. Nucleic Acids Res. 2020; 48:D198–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang Y., Qian F.C., Bai X.F., Liu Y.J., Wang Q.Y., Ai B., Han X.L., Shi S.S., Zhang J., Li X.C.et al.. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 2019; 47:D235–D243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ernst J., Kellis M.. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017; 12:2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R.et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li X., Shi L., Wang Y., Zhong J., Zhao X., Teng H., Shi X., Yang H., Ruan S., Li M.. OncoBase: a platform for decoding regulatory somatic mutations in human cancers. Nucleic Acids Res. 2019; 47:D1044–D1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Teng L., He B., Wang J., Tan K.. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics. 2016; 32:2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ghoshal G., Barabasi A.L.. Ranking stability and super-stable nodes in complex networks. Nat. Commun. 2011; 2:394. [DOI] [PubMed] [Google Scholar]
- 33. Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L.. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. The Innovation. 2021; 2:100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al.. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanehisa M., Goto S., Furumichi M., Tanabe M., Hirakawa M.. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010; 38:D355–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benjamini Y., Hochberg Y.. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B. 1995; 57:289–300. [Google Scholar]
- 37. Klemm S.L., Shipony Z., Greenleaf W.J.. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019; 20:207–220. [DOI] [PubMed] [Google Scholar]
- 38. Wang F., Bai X.F., Wang Y.Z., Jiang Y., Ai B., Zhang Y., Liu Y.J., Xu M.C., Wang Q.Y., Han X.L.et al.. ATACdb: a comprehensive human chromatin accessibility database. Nucleic Acids Res. 2021; 49:D55–D64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zheng R., Wan C., Mei S., Qin Q., Wu Q., Sun H., Chen C.H., Brown M., Zhang X., Meyer C.A.et al.. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019; 47:D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W.et al.. The chromatin accessibility landscape of primary human cancers. Science. 2018; 362:eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C.. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018; 19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kolmykov S., Yevshin I., Kulyashov M., Sharipov R., Kondrakhin Y., Makeev V.J., Kulakovskiy I.V., Kel A., Kolpakov F.. GTRD: an integrated view of transcription regulation. Nucleic Acids Res. 2021; 49:D104–D111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheneby J., Menetrier Z., Mestdagh M., Rosnet T., Douida A., Rhalloussi W., Bergon A., Lopez F., Ballester B.. ReMap 2020: a database of regulatory regions from an integrative analysis of Human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res. 2020; 48:D180–D188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R.et al.. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010; 28:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alfoldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P.et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E.et al.. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019; 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li M.J., Liu Z., Wang P., Wong M.P., Nelson M.R., Kocher J.P., Yeager M., Sham P.C., Chanock S.J., Xia Z.et al.. GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2016; 44:D869–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao G., Li K., Li B., Wang Z., Fang Z., Wang X., Zhang Y., Luo T., Zhou Q., Wang L.et al.. Gene4Denovo: an integrated database and analytic platform for de novo mutations in humans. Nucleic Acids Res. 2020; 48:D913–D926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mao F., Xiao L., Li X., Liang J., Teng H., Cai W., Sun Z.S.. RBP-Var: a database of functional variants involved in regulation mediated by RNA-binding proteins. Nucleic Acids Res. 2016; 44:D154–D163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. GTEx Consortium Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348:648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gong J., Mei S., Liu C., Xiang Y., Ye Y., Zhang Z., Feng J., Liu R., Diao L., Guo A.Y.et al.. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2018; 46:D971–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cui Y., Chen X.W., Luo H.X., Fan Z., Luo J.J., He S.M., Yue H.Y., Zhang P., Chen R.S.. BioCircos.js: an interactive Circos JavaScript library for biological data visualization on web applications. Bioinformatics. 2016; 32:1740–1742. [DOI] [PubMed] [Google Scholar]
- 54. Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J.et al.. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020; 17:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mao F., Liu Q., Zhao X., Yang H., Guo S., Xiao L., Li X., Teng H., Sun Z., Dou Y.. EpiDenovo: a platform for linking regulatory de novo mutations to developmental epigenetics and diseases. Nucleic Acids Res. 2018; 46:D92–D99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang T., Ruan S.S., Zhao X.L., Shi X.H., Teng H.J., Zhong J.N., You M.C., Xia K., Sun Z.S., Mao F.B.. OncoVar: an integrated database and analysis platform for oncogenic driver variants in cancers. Nucleic Acids Res. 2021; 49:D1289–D1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M.et al.. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021; 49:D1046–D1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020; 578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liao Y.X., Wang J., Jaehnig E.J., Shi Z.A., Zhang B.. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Rese. 2019; 47:W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim H., Nguyen N.P., Turner K., Wu S.H., Gujar A.D., Luebeck J., Liu J.H., Deshpande V., Rajkumar U., Namburi S.et al.. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020; 52:891-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bailey C., Shoura M., Mischel P., Swanton C.. Extrachromosomal DNA—relieving heredity constraints, accelerating tumour evolution. Ann. Oncol. 2020; 31:884–893. [DOI] [PubMed] [Google Scholar]
- 62. Moller H.D. Circle-Seq: isolation and sequencing of chromosome-derived circular DNA elements in cells. Methods Mol. Biol. 2020; 2119:165–181. [DOI] [PubMed] [Google Scholar]
- 63. Fan X., Yang C., Li W., Bai X., Zhou X., Xie H., Wen L., Tang F.. SMOOTH-seq: single-cell genome sequencing of human cells on a third-generation sequencing platform. Genome Biol. 2021; 22:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deshpande V., Luebeck J., Nguyen N.-P.D., Bakhtiari M., Turner K.M., Schwab R., Carter H., Mischel P.S., Bafna V.. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat. Commun. 2019; 10:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Prada-Luengo I., Krogh A., Maretty L., Regenberg B.. Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads. BMC Bioinformatics. 2019; 20:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kumar P., Kiran S., Saha S., Su Z., Paulsen T., Chatrath A., Shibata Y., Shibata E., Dutta A.. ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Sci. Adv. 2020; 6:eaba2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession, DOI, version and permalink information of the foregoing databases was listed in Supplementary Table S1. To make the codes open-access, we deposited the source codes of CircleBase in GitHub (https://github.com/leishenggit/CircleBase). The full tables of human eccDNAs are available in the Download module of CircleBase (http://circlebase.maolab.org/welcome/download).