


Abstract
De novo mutations, a consequence of errors in DNA repair or replication, have been reported to accumulate with age in normal tissues of humans and model organisms. This accumulation during development and aging has been implicated as a causal factor in aging and age-related pathology, including but not limited to cancer. Due to their generally very low abundance mutations have been difficult to detect in normal tissues. Only with recent advances in DNA sequencing of single-cells, clonal lineages or ultra-high-depth sequencing of small tissue biopsies, somatic mutation frequencies and spectra have been unveiled in several tissue types. The rapid accumulation of such data prompted us to develop a platform called SomaMutDB (https://vijglab.einsteinmed.org/SomaMutDB) to catalog the 2.42 million single nucleotide variations (SNVs) and 0.12 million small insertions and deletions (INDELs) thus far identified using these advanced methods in nineteen human tissues or cell types as a function of age or environmental stress conditions. SomaMutDB employs a user-friendly interface to display and query somatic mutations with their functional annotations. Moreover, the database provides six powerful tools for analyzing mutational signatures associated with the data. We believe such an integrated resource will prove valuable for understanding somatic mutations and their possible role in human aging and age-related diseases.
INTRODUCTION
Previous studies of somatic mutations in human tissues have been mostly focused on tumors (1). As clonal outgrowths, tumors reflect somatic mutations in the original normal cell and those added during its clonal expansion after neoplastic transformation. Patterns of mutation frequencies, spectra and distribution across the genome vary dramatically between different cancers and even between different tumours of the same cancer (2). Thousands of cancers have been sequenced, with databases built for facilitating analysis of mutational patterns, sources, and clinical outcomes, e.g. TCGA (The Cancer Genome Atlas), ICGC (International Cancer Genome Consortium), COSMIC (Catalogue Of Somatic Mutations In Cancer), and OncoKB (Precision Oncology Knowledge Base) (3–6). These databases have been valuable resources for understanding somatic mutations in tumours and promoting further basic and clinical research (7).
In contrast to cancers, studying somatic mutations in normal tissues remains a challenge because each cell harbors its own unique mutation spectrum. Somatic mutation analysis requires either single-cell sequencing or surrogate approaches, such as sequencing clonal outgrowths or taking advantage of mutation expansion, either through genetic drift or as a consequence of a growth advantage (8,9). Using these methods, studies have shown somatic mutations accumulate in normal somatic cells during human aging, and the speeds of accumulation are accelerated in smoking, UV exposure, and under ulcerative colitis, inflammatory bowel, or cirrhosis diseases (10–23).
Thus far only one database has been developed for somatic mutations in normal cells and tissues, DSMNC (a Database of Somatic Mutations in Normal Cells) (24). This database has been updated for the last time on August 2018, contains somatic mutations in six human tissue types and does not provide additional, but important analytical tools for mutational signature analysis. Clearly, there is a significant need for an up-to-date database that catalogues all publicly available small variants, including single nucleotide variations (SNVs) and small insertions and deletions (INDELs), in normal somatic cells of humans, with available tools for analysis of mutational patterns and signatures.
Here, we present SomaMutDB, a database of somatic mutations in normal human tissues. SomaMutDB catalogs 2.42 million SNVs and 0.12 million INDELs identified in nineteen normal tissues and cell types reported, using 2838 single cells, clones or biopsies from 374 human subjects. The database contains (a) an interactive genome browser to browse mutations across the genome, (b) multiple user-friendly ways to search mutations of interest and (c) a computing infrastructure providing six mutational signature analysis tools, which help users to extract signatures from their data and comparing them with known reference signatures from cancers (3,25). We believe that SomaMutDB provides a convenient platform to search, browse and analyze somatic mutations in human normal samples.
MATERIALS AND METHODS
System design and implementation
The webserver of SomaMutDB is built on Nginx (version r24-2) and Gunicorn (version 19.10.0; a Python WSGI HTTP Server for UNIX) with Flask (version 1.1.2; a WSGI web application framework) (Figure 1A). Mutations and their annotations are stored in MySQL (version 8.0.25) and accessed by Flask. The ‘Analysis’ function utilizes SGE (Sun Grid Engine; version 8.1.9) to manage users’ jobs and is controlled also by Flask. Jbrowse2 (version 1.0.3) (26) is integrated to browse somatic mutations and related annotation along the genome. An interactive graphic user interface was designed with jQuery (version 3.1.0). For access, SomaMutDB supports many commonly used web browsers, e.g. Google Chrome, Safari or Firefox.
Figure 1.
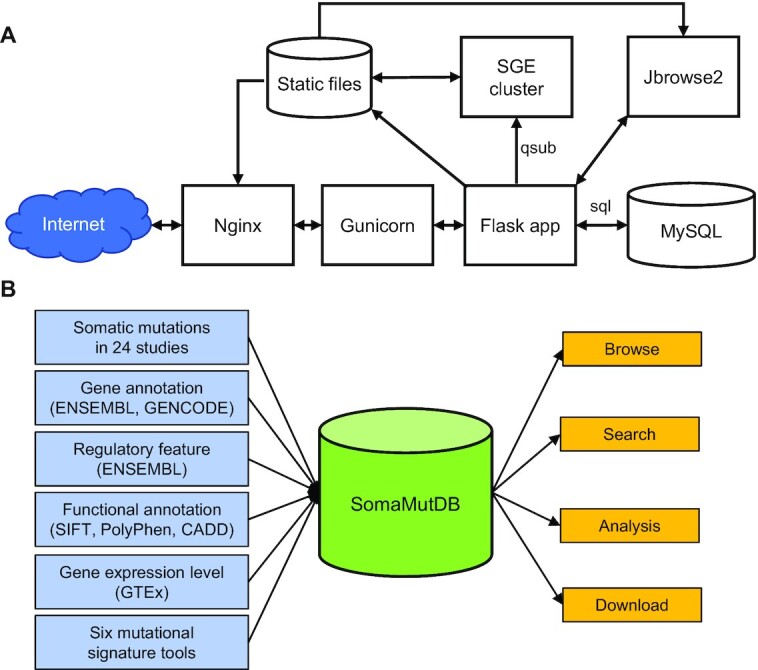
Database construction and content. (A) The structure of SomaMutDB. (B) Illustration of collected data and features.
Data sources
We collected somatic mutations from 24 published studies that made their somatic mutation data publicly available (10–23,27–36) (Table 1, Figure 1B). For now, data are limited to somatic mutations reported for normal tissues and cells, not for tumors or other abnormalities. This is because mutation frequencies and spectra of abnormal samples, e.g., genetic deficiency, can be significantly different from those in normal cells. For example, we excluded somatic mutation data on cells or biopsies of carcinoma in situ from a bladder cancer study (15), inflamed colon epithelium of colon diseases (16,17), liver diseases (19), smokers or ex-smokers from the bronchial epithelium studies (21), embryo samples with trisomy 21 (32), and placenta samples with abnormal parameters (34). From all selected samples we collected whole-genome and -exome data and re-analyzed sequencing data generated from genome amplifications with the same variant calling pipeline (see Data processing). Besides somatic mutations, we also integrated the annotations of gene and regulatory elements from ENSEMBL (37) and GENCODE (38), as well as median expression levels of genes for each normal tissue and cell type reported by GTEx (39).
Table 1.
Summary of somatic mutations (as of 12 April 2021)
| Tissue/cell type | # Individual | # Sample | # SNV | # INDEL |
|---|---|---|---|---|
| Adipocytes | 4 | 20 | 22 556 | 1379 |
| Bladder | 100 | 680 | 142 537 | 4324 |
| Blood | 19 | 157 | 102 693 | 1490 |
| Bone marrow | 2 | 109 | 113 877 | 142 |
| Brain | 21 | 180 | 59 014 | 1250 |
| Colon | 85 | 521 | 1 184 598 | 52 885 |
| Embryonic stem cell | 3 | 39 | 7768 | 665 |
| Endometrium | 28 | 257 | NA | 23 585 |
| Fibroblast | 22 | 44 | 8987 | 1436 |
| iPSC | 3 | 5 | 3652 | 38 |
| Kidney | 6 | 25 | 37 078 | 4157 |
| Liver | 23 | 243 | 271 647 | 7165 |
| Lung | 7 | 256 | 311 208 | 12 719 |
| Placenta | 5 | 106 | 17 000 | 1786 |
| Skeletal muscle | 8 | 33 | 25 653 | 1807 |
| Skin | 16 | 78 | 59 293 | 1669 |
| Small Intestine | 13 | 31 | 25 821 | 19 |
| Testicle | 1 | 19 | 194 | NA |
| Ureter | 28 | 35 | 23 942 | 75 |
Data processing
Sequencing data generated from genome amplifications were re-analyzed with SCcaller (version 2.0.0) (35). Briefly, adapter and low-quality reads were trimmed by Trim Galore (version 0.6.4). The trimmed reads were aligned to the human reference genome (GRCh37 with decoy or GRCh38) by BWA-MEM (version 0.7.17) (40). Duplications were removed using Samtools (version 1.9) (41). The known INDELs (1000 Genomes Project, phase 1) and SNPs (dbSNP) were downloaded from public GATK Google bucket (https://console.cloud.google.com/storage/browser/gcp-public-data–broad-references). The reads around known INDELs were local-realigned, and base quality scores were recalibrated based on known SNVs and INDELs, both via GATK (version 3.5.0) (42). Germline heterozygous SNPs and INDELs were identified in their corresponding bulk whole-genome sequences using Haplotypecaller (GATK quality score ≥30, ≥20× depth and with dbSNP annotation). Somatic mutations were then identified using SCcaller based on the amplification bias estimated from the germline heterozygous SNPs after filtering out all germline SNPs. We kept somatic SNVs at ≥20× sequencing depth, and INDELs with ≥30× sequencing depth and variant quality score ≥25. Those SNVs or INDELs reported by dbSNP were filtered out using snpEff (version 5.0c) (43).
For all collected somatic mutations, we adopted the mutations reported in the original literature except for the following three studies (Table 1). First, in a study on colon (17) we manually corrected sample ID mismatches. Further, the publicly available mutations of this study (stored in Mendeley Data) were not filtered and we processed the data according to the filtering thresholds of its original publication (read depth ≥ 5× and allele supporting reads ≥ 2). Second, in two studies by Franco I, et al. (13,14) somatic mutations were reported at group level, e.g., a young versus an old group, without sample ID or individual ID. For mutations reported in these studies, we present data as they were published in supplementary tables because no further individual-level information was available.
All reported somatic mutations that had been called based on the human reference genome GRCh37 were converted to the GRCh38 version using CrossMap (version 0.5.2) (44). Mutations found in small chromosome contigs were excluded and those in chromosomes 1–22, X and Y in GRCh38 were kept.
Functional annotation
We performed functional annotations of somatic mutations using multiple tools. We first annotated the somatic mutations using the Ensembl Variant Effect Predictor (VEP, version 102) (45). Among the multiple effects for each mutation, we selected the one that was most impactful by setting ‘pick_order’ as ‘rank,biotype,mane,tsl,appris,length’. The SIFT and PolyPhen scores for functional effects of coding mutations were predicted and collected from the annotations. The co-location to regulatory elements was identified also using VEP. The deleteriousness of somatic mutations was calculated using CADD (version 1.6) (46).
Mutational signature analysis tools
Mutational signatures are combinations of mutation subtypes arising from specific mutagenesis processes (3). SNVs can be classified into six major types: C > A, C > G, C > T, T > A, T > C and T > G. Considering the neighbouring bases flanking the substitutions, SNVs can be further divided into 96 subtypes (47). Similar as for SNVs, INDELs can be divided into 83 subtypes by considering length of INDELs, affected nucleotides (C or T) and the number of repetitive elements of the repetitive or microhomology region when occurring in such a region (48).
SomaMutDB provides six signature analysis tools for different usages (Supplementary Table S1). Using MutationalPatterns (version 3.0.1) (49), SomaticSignatures (version 2.26.0) (50), hdp (0.1.5) (51), signature_tools_lib (version 0.0.0.9000) (25), and SigProfiler (version 1.1.1) (52) mutational signatures can be extracted based on one or more of the following algorithms: non-negative matrix factorization (NMF), principal component analysis, hierarchical Bayesian Dirichlet process. Extracted or previously known signatures can be fitted into a collection of somatic mutations by MutationalPatterns, signature_tools_lib, SigProfiler, and mmsig (customized on version 0.0.0.9000) (53) using non-negative least squares, Kullback-Leibler divergence, simulated annealing, and expectation maximization. Finally, to compare extracted and reference signatures, cosine similarity can be calculated using MutationalPatterns (49). The reference signatures from COSMIC and Signal databases were collected in SomaMutDB and are downloadable, including mutational signatures from cancers or those related to environmental mutagenesis (3,25).
RESULTS
Database content and usage
SomaMutDB is a database of somatic mutations dedicated to storing, browsing, searching and analyzing SNVs and INDELs obtained by different approaches for detecting somatic mutations in normal tissues and cells. In its current version (as of April 2021; Table 1), SomaMutDB incorporates 2 417 518 SNVs and 116 591 INDELs from 19 normal tissues and cell types in 374 individuals. The age of individuals ranges from 0 to 106 years, with 98 embryo samples (Supplementary Figure S1). Most individuals are between 30 and 79 years old (76.8%). Of all the samples, 47.1% and 50.2% were male and female, respectively. Among the 2838 samples, 390 were whole-genome amplified single cells, 748 were single-cell clones, and 1700 were collected from natural clones using laser capture microdissection. All the above information can be downloaded from the database.
In SomaMutDB, somatic mutations can be interactively browsed and searched in various ways (Figure 2). To browse somatic mutations, SomaMutDB has an interactive and user-friendly browser—Jbrowse2 (Figure 2A). Users can zoom in/out and scroll to any interested regions along the genome to view mutations in the context of many other genomic features. To simplify the search, we provided somatic mutations in different categories, viz., by different sample types (e.g. T or B lymphocytes from blood) or by location (e.g. sigmoid or transverse colons) in each tissue and cell type. Detailed descriptions and functional annotations of mutations can be found in the right panel named ‘ATTRIBUTES’ after clicking a mutation. For each mutation, users can query its corresponding sample by checking the ‘MF’ column in ‘SAMPLES’ panel. We also provided genomic feature annotations in the same browser, including gene features, regulatory features and median gene expression level of specific tissues.
Figure 2.

Interactive browsing and searching of SomaMutDB. (A) Overview of the interactive browser using an example of a SNV in the NOTCH1 gene in human transverse colon. Regulatory features, gene features, gene expression level and detailed information of the SNV are shown in the same webpage. (B) Screenshots of search functions and results in SomaMutDB. The search results present detailed information of somatic mutations in the TP53 gene.
In addition to the browser, users can search mutations of interest in the ‘Search’ webpage (Figure 2B). Users can search somatic mutations according to a gene name, a list of gene names, chromosome regions and specific types of tissues. We prepared multiple lists of genes with known functions or annotations, e.g., aging, cell senescence, DNA repair, transcription factors, cancer driver genes (3,54–56), to help users select mutations in a particular list of genes. The search results include information on the mutations, such as chromosome, genomic coordinate, genotype, tissue type, sex and age of donor, gene identity (symbol) and molecular consequence(s). Additional functional annotations of somatic mutations can also be selected through a button using gear icon (Figures 2 and Supplementary Figure S2). All search results can be downloaded for further analysis. Furthermore, users can download the literature sources of each somatic mutation listed in SomaMutDB.
To help users extract and analyze mutational signatures, SomaMutDB contains six signature analysis tools (Figure 3 and Supplementary Table S1). For the ‘Extract’ and ‘Fit’ functions, users can select one signature analysis tool and upload their customized set of somatic mutations to our database for signature analysis. Default parameters for analysis are pre-set according to the manuals of each tool, which can be customized by users. Analysis jobs will be submitted to our computing cluster. Users will be noticed by email when their job is completed and will be provided with a link to access the results in the same email. The results are provided as figures with the corresponding data files (illustrated in Supplementary Figure S3 and Supplementary Table S2), parameters used for analysis and all intermediate files by analysis tool. Users can also view results using the ‘View result’ function (Figure 3) with job ID in the email.
Figure 3.

Screenshots of web interfaces of the ‘Analysis’ function. Extract, similarity and fit functions can be selected in ‘Analysis’ webpage. Users’ data can be uploaded, and default setting of tools can be directly changed on the website. The figures of results can be viewed in the database with job ID through ‘View result’.
Case study on signature analysis
To illustrate usage of the ‘Analysis’ function, we present an example using data in publication (23). First, we uploaded the raw SNV data in vcf format and corresponding sample information, and chose NMF method in MutationalPatterns tool (set ‘Range of signature number to estimate’ to ‘2:2’). The results computed by SomaMutDB showed that the relative contribution of C > A mutations is higher in liver cells from young subjects or in liver stem cells (LSC) than in the same cell type from aged subjects. Also, the aged group has more T > C mutations (Figure 4A). Signature analysis indicates that signature S1 dominates the aged group and signature S2 is the major pattern for mutations in the other two groups (Figure 4B and C). To compare these two signatures to known cancer signatures, they were uploaded together with the COSMIC cancer signatures (version 3, without artifact signatures) using the ‘Similarity’ function. The results showed that signature S1 is highly correlated with SBS5 caused by aging and signature S2 is associated with the oxidative stress-related signatures SBS18 and SBS36 (Figure 4D). Finally, we uploaded the SNV matrix data (obtained from the results after the ‘Extract’ step) and the COSMIC cancer signatures in the ‘Fit’ function and chose the strict NMF method in MutationalPatterns tool with default setting. The results showed that SBS5 contributes more to somatic mutations in the aged group, whereas SBS18 and SBS36 are more similar to the young and LSC groups (Figure 4E). Taken together, SomaMutDB provided the exact same results as those obtained independently in our previous publication (23).
Figure 4.

Re-analysis of public liver data using SomaMutDB. (A) Distributions of 6 major types of SNVs in liver samples from three groups, young and aged differentiated hepatocytes and young liver stem cells (LSC). (B) Contributions of de novo extracted mutational signatures in the three liver groups identified by MutationalPatterns using the NMF method. (C) Basepair contributions of two mutational signatures extracted. Signature S1 enriched with T > C transitions, and Signature S2 enriched with C > A transversions at non-CpG sites. (D) Heatmap of cosine similarities between de novo extracted signatures and COSMIC signatures (version 3.0). Signature S1 is associated with SBS5, which is an aging signature found in tumors, while signature S2 correlated with signatures SBS18 and SBS36, which are related to oxidative stress. In users’ application, signatures in row and column can be plotted in the same order as uploaded files provided by the users. (E) An example of ‘Fit’ function: contribution of COSMIC signatures to the liver SNVs estimated by MutationalPatterns using the strict NMF method.
CONCLUSIONS AND FUTURE DEVELOPMENTS
Here, we present SomaMutDB, a somatic mutation database for normal tissues. It features (a) storage of ∼2.53 million SNVs and INDELs in normal cells of 19 human tissues and cell types obtained from whole-genome or -exome sequencing of single cells, clonal outgrowths or naturally expanded mutations analysed in biopsies; (b) allowing online browsing for mutations across the genome and searching for somatic mutations based on functional annotations in genes and regulatory elements; and (c) deciphering mutational signatures using six powerful signature analysis tools. SomaMutDB provides a comprehensive data resource and a set of interactive analysis tools to facilitate genomic research of somatic mutations in normal human tissues and cell types, which would benefit researchers in studying the mechanisms of somatic mosaicism during aging or other conditions. In the future, we will keep adding more somatic mutations in normal tissues not only based on current single cell-based approaches but also using other technologies, such as Nanorate sequencing (57). We believe that SomaMutDB will be of broad interest to researchers working on somatic mutations of normal tissues.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Shailesh M. Shenoy, Donnie Frid and Eliezer Sylvestre of the IT Department at Albert Einstein College of Medicine for assistance with database implementation.
Contributor Information
Shixiang Sun, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, USA.
Yujue Wang, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, USA.
Alexander Y Maslov, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, USA; Laboratory of Applied Genomic Technologies, Voronezh State University of Engineering Technology, Voronezh, Russia.
Xiao Dong, Institute on the Biology of Aging and Metabolism, and Department of Genetics, Cell Biology and Development, University of Minnesota, Minneapolis, MN, USA.
Jan Vijg, Department of Genetics, Albert Einstein College of Medicine, Bronx, NY, USA; Center for Single-Cell Omics, School of Public Health, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health [P01 AG017242, P01 AG047200, P30 AG038072, U01 ES029519, U19 AG056278 to J.V., R00 AG056656 to X.D.]; Glenn Foundation for Medical Research (to J.V.). Funding for open access charge: NIH [5P01AG017242-26].
Conflict of interest statement. A.Y.M., X.D. and J.V. are co-founders and shareholders of SingulOmics Corp.
REFERENCES
- 1. Vijg J., Dong X.. Pathogenic mechanisms of somatic mutation and genome mosaicism in aging. Cell. 2020; 182:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N.et al.. The repertoire of mutational signatures in human cancer. Nature. 2020; 578:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E.et al.. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chakravarty D., Gao J., Phillips S.M., Kundra R., Zhang H., Wang J., Rudolph J.E., Yaeger R., Soumerai T., Nissan M.H.et al.. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 2017; 2017: 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tomczak K., Czerwinska P., Wiznerowicz M.. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn.). 2015; 19:A68–A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang J., Baran J., Cros A., Guberman J.M., Haider S., Hsu J., Liang Y., Rivkin E., Wang J., Whitty B.et al.. International Cancer Genome Consortium Data Portal–a one-stop shop for cancer genomics data. Database (Oxford). 2011; 2011:bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dagogo-Jack I., Shaw A.T.. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018; 15:81–94. [DOI] [PubMed] [Google Scholar]
- 8. Vijg J. From DNA damage to mutations: all roads lead to aging. Ageing Res. Rev. 2021; 68:101316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ellis P., Moore L., Sanders M.A., Butler T.M., Brunner S.F., Lee-Six H., Osborne R., Farr B., Coorens T.H.H., Lawson A.R.J.et al.. Reliable detection of somatic mutations in solid tissues by laser-capture microdissection and low-input DNA sequencing. Nat. Protoc. 2021; 16:841–871. [DOI] [PubMed] [Google Scholar]
- 10. Blokzijl F., de Ligt J., Jager M., Sasselli V., Roerink S., Sasaki N., Huch M., Boymans S., Kuijk E., Prins P.et al.. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016; 538:260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J., Yandava C.N.et al.. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018; 359:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saini N., Giacobone C.K., Klimczak L.J., Papas B.N., Burkholder A.B., Li J.L., Fargo D.C., Bai R., Gerrish K., Innes C.L.et al.. UV-exposure, endogenous DNA damage, and DNA replication errors shape the spectra of genome changes in human skin. PLos Genet. 2021; 17:e1009302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Franco I., Johansson A., Olsson K., Vrtacnik P., Lundin P., Helgadottir H.T., Larsson M., Revechon G., Bosia C., Pagnani A.et al.. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat. Commun. 2018; 9:800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Franco I., Helgadottir H.T., Moggio A., Larsson M., Vrtacnik P., Johansson A., Norgren N., Lundin P., Mas-Ponte D., Nordstrom J.et al.. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol. 2019; 20:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lawson A.R.J., Abascal F., Coorens T.H.H., Hooks Y., O’Neill L., Latimer C., Raine K., Sanders M.A., Warren A.Y., Mahbubani K.T.A.et al.. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science. 2020; 370:75–82. [DOI] [PubMed] [Google Scholar]
- 16. Nanki K., Fujii M., Shimokawa M., Matano M., Nishikori S., Date S., Takano A., Toshimitsu K., Ohta Y., Takahashi S.et al.. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature. 2020; 577:254–259. [DOI] [PubMed] [Google Scholar]
- 17. Olafsson S., McIntyre R.E., Coorens T., Butler T., Jung H., Robinson P.S., Lee-Six H., Sanders M.A., Arestang K., Dawson C.et al.. Somatic evolution in non-neoplastic IBD-affected colon. Cell. 2020; 182:672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moore L., Leongamornlert D., Coorens T.H.H., Sanders M.A., Ellis P., Dentro S.C., Dawson K.J., Butler T., Rahbari R., Mitchell T.J.et al.. The mutational landscape of normal human endometrial epithelium. Nature. 2020; 580:640–646. [DOI] [PubMed] [Google Scholar]
- 19. Brunner S.F., Roberts N.D., Wylie L.A., Moore L., Aitken S.J., Davies S.E., Sanders M.A., Ellis P., Alder C., Hooks Y.et al.. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. 2019; 574:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tang J., Fewings E., Chang D., Zeng H., Liu S., Jorapur A., Belote R.L., McNeal A.S., Tan T.M., Yeh I.et al.. The genomic landscapes of individual melanocytes from human skin. Nature. 2020; 586:600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshida K., Gowers K.H.C., Lee-Six H., Chandrasekharan D.P., Coorens T., Maughan E.F., Beal K., Menzies A., Millar F.R., Anderson E.et al.. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature. 2020; 578:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang L., Dong X., Lee M., Maslov A.Y., Wang T., Vijg J.. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:9014–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brazhnik K., Sun S., Alani O., Kinkhabwala M., Wolkoff A.W., Maslov A.Y., Dong X., Vijg J.. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci. Adv. 2020; 6:eaax2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miao X., Li X., Wang L., Zheng C., Cai J.. DSMNC: a database of somatic mutations in normal cells. Nucleic Acids Res. 2019; 47:D971–D975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Degasperi A., Amarante T.D., Czarnecki J., Shooter S., Zou X., Glodzik D., Morganella S., Nanda A.S., Badja C., Koh G.et al.. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat Cancer. 2020; 1:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buels R., Yao E., Diesh C.M., Hayes R.D., Munoz-Torres M., Helt G., Goodstein D.M., Elsik C.G., Lewis S.E., Stein L.et al.. JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 2016; 17:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee-Six H., Obro N.F., Shepherd M.S., Grossmann S., Dawson K., Belmonte M., Osborne R.J., Huntly B.J.P., Martincorena I., Anderson E.et al.. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018; 561:473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rouhani F.J., Nik-Zainal S., Wuster A., Li Y., Conte N., Koike-Yusa H., Kumasaka N., Vallier L., Yusa K., Bradley A.. Mutational history of a human cell lineage from somatic to induced pluripotent stem cells. PLos Genet. 2016; 12:e1005932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kwon E.M., Connelly J.P., Hansen N.F., Donovan F.X., Winkler T., Davis B.W., Alkadi H., Chandrasekharappa S.C., Dunbar C.E., Mullikin J.C.et al.. iPSCs and fibroblast subclones from the same fibroblast population contain comparable levels of sequence variations. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:1964–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li R., Du Y., Chen Z., Xu D., Lin T., Jin S., Wang G., Liu Z., Lu M., Chen X.et al.. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science. 2020; 370:82–89. [DOI] [PubMed] [Google Scholar]
- 31. Xing D., Tan L., Chang C.H., Li H., Xie X.S.. Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2013106118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hasaart K.A.L., Manders F., van der Hoorn M.L., Verheul M., Poplonski T., Kuijk E., de Sousa Lopes S.M.C., van Boxtel R.. Mutation accumulation and developmental lineages in normal and Down syndrome human fetal haematopoiesis. Sci. Rep. 2020; 10:12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thompson O., von Meyenn F., Hewitt Z., Alexander J., Wood A., Weightman R., Gregory S., Krueger F., Andrews S., Barbaric I.et al.. Low rates of mutation in clinical grade human pluripotent stem cells under different culture conditions. Nat. Commun. 2020; 11:1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coorens T.H.H., Oliver T.R.W., Sanghvi R., Sovio U., Cook E., Vento-Tormo R., Haniffa M., Young M.D., Rahbari R., Sebire N.et al.. Inherent mosaicism and extensive mutation of human placentas. Nature. 2021; 592:80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dong X., Zhang L., Milholland B., Lee M., Maslov A.Y., Wang T., Vijg J.. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods. 2017; 14:491–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bae T., Tomasini L., Mariani J., Zhou B., Roychowdhury T., Franjic D., Pletikos M., Pattni R., Chen B.J., Venturini E.et al.. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science. 2018; 359:550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howe K.L., Achuthan P., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R., Bhai J.et al.. Ensembl 2021. Nucleic Acids Res. 2021; 49:D884–D891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J.et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Consortium G.T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020; 369:1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M.et al.. Twelve years of SAMtools and BCFtools. Gigascience. 2021; 10:giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.et al.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M.. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012; 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao H., Sun Z., Wang J., Huang H., Kocher J.P., Wang L.. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics. 2014; 30:1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F.. The ensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rentzsch P., Schubach M., Shendure J., Kircher M.. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021; 13:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A.et al.. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Petljak M., Alexandrov L.B., Brammeld J.S., Price S., Wedge D.C., Grossmann S., Dawson K.J., Ju Y.S., Iorio F., Tubio J.M.C.et al.. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019; 176:1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blokzijl F., Janssen R., van Boxtel R., Cuppen E.. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018; 10:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gehring J.S., Fischer B., Lawrence M., Huber W.. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015; 31:3673–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Y., Roberts N.D., Wala J.A., Shapira O., Schumacher S.E., Kumar K., Khurana E., Waszak S., Korbel J.O., Haber J.E.et al.. Patterns of somatic structural variation in human cancer genomes. Nature. 2020; 578:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Islam S.M.A., Wu Y., Díaz-Gay M., Bergstrom E.N., He Y., Barnes M., Vella M., Wang J., Teague J.W., Clapham P.et al.. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. 2021; bioRxiv doi:13 December 2020, preprint: not peer reviewed 10.1101/2020.12.13.422570. [DOI] [PMC free article] [PubMed]
- 53. Rustad E.H., Nadeu F., Angelopoulos N., Ziccheddu B., Bolli N., Puente X.S., Campo E., Landgren O., Maura F.. mmsig: a fitting approach to accurately identify somatic mutational signatures in hematological malignancies. Commun Biol. 2021; 4:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tacutu R., Thornton D., Johnson E., Budovsky A., Barardo D., Craig T., Diana E., Lehmann G., Toren D., Wang J.et al.. Human ageing genomic resources: new and updated databases. Nucleic Acids Res. 2018; 46:D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Knijnenburg T.A., Wang L., Zimmermann M.T., Chambwe N., Gao G.F., Cherniack A.D., Fan H., Shen H., Way G.P., Greene C.S.et al.. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018; 23:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 175:598–599. [DOI] [PubMed] [Google Scholar]
- 57. Abascal F., Harvey L.M.R., Mitchell E., Lawson A.R.J., Lensing S.V., Ellis P., Russell A.J.C., Alcantara R.E., Baez-Ortega A., Wang Y.et al.. Somatic mutation landscapes at single-molecule resolution. Nature. 2021; 593:405–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.