


Abstract
Improved bioassays have significantly increased the rate of identifying new protein-protein interactions (PPIs), and the number of detected human PPIs has greatly exceeded early estimates of human interactome size. These new PPIs provide a more complete view of disease mechanisms but precise understanding of how PPIs affect phenotype remains a challenge. It requires knowledge of PPI context (e.g. tissues, subcellular localizations), and functional roles, especially within pathways and protein complexes. The previous IID release focused on PPI context, providing networks with comprehensive tissue, disease, cellular localization, and druggability annotations. The current update adds developmental stages to the available contexts, and provides a way of assigning context to PPIs that could not be previously annotated due to insufficient data or incompatibility with available context categories (e.g. interactions between membrane and cytoplasmic proteins). This update also annotates PPIs with conservation across species, directionality in pathways, membership in large complexes, interaction stability (i.e. stable or transient), and mutation effects. Enrichment analysis is now available for all annotations, and includes multiple options; for example, context annotations can be analyzed with respect to PPIs or network proteins. In addition to tabular view or download, IID provides online network visualization. This update is available at http://ophid.utoronto.ca/iid.
INTRODUCTION
Protein-protein interaction (PPI) data have numerous applications in molecular biology and biomedicine. Common problems addressed with the help of PPI networks include identifying disease genes (1,2), predicting gene function (3,4), associating genetic variants with traits (5) and identifying drug treatments (6,7). Biomolecular studies increasingly use PPI networks as part of their bioinformatics analysis, alongside tools such as pathway and Gene Ontology (8,9) enrichment. These applications are enabled by a large variety of network visualization and analysis methods, including approaches based on ‘guilt-by-association’ (3,10), network flow (11,12) and more recently, deep learning (13,14).
However, the effectiveness and useability of PPI networks are hampered by several factors including false positive and false negative errors, absence of interaction context and characterization (e.g. stability), and network complexity. False positive rates of early high-throughput PPI studies have been estimated at over 50% (15,16), and also pose a challenge for PPIs supported by a single detection method in a single small-scale study (17). However, recent high-throughput studies (17–20) have much lower false positive rates, comparable to those of PPIs detected by multiple small-scale screens. False negatives are a bigger problem—most species have very few detected PPIs, and the human PPI network, though extensively studied, may still be largely unknown, since the number of detected human PPIs has surpassed estimates of interactome size (16,21) and continues to steadily increase. Importantly, false negatives are not uniformly distributed across proteins (22). The context of interactions, such as their location (e.g. tissue, subcellular localization) and time of occurrence are often not fully known. PPI detection may occur in cell lines and artificial localizations, and does not provide comprehensive annotation across diverse tissues, physiological states and compartments. Without context, interactions may not be relevant for studying a given tissue or disease. Most detection methods also do not provide any characterization of interactions, such as their stability or direction – information that can help analysis methods such as network flow. As the number of detected PPIs has increased, network complexity has made analysis more challenging—network visualizations are often difficult to interpret, network analysis may give overwhelming numbers of hypotheses, and more generally, most proteins appear functionally related since the average network distance between human proteins is about 3.
The previous version of IID (23) focused on reducing false negatives and providing context for PPIs. False negatives were reduced by integrating PPIs from a range of sources: curated databases of experimentally detected PPIs, machine learning studies predicting genome-wide PPIs, and orthology-based PPI predictions. IID provided PPIs in human, 6 model organisms and 11 domesticated species. In most of these organisms, PPIs were annotated with extensive context information including tissues, subcellular localization, diseases, and druggability. IID has been used in several impactful studies, including (24–32).
This update focuses on extending context information, characterizing PPIs with direction, mutation effects, and stability, and improving the interpretability and usability of PPI networks. Context is extended by adding developmental stage annotations, and its use is enhanced by extending how PPIs are annotated with context. PPI databases (33–35), including previous versions of IID, annotate PPIs with a context (e.g. disease, tissue) if both interacting proteins are expressed or otherwise implicated in the context. However, this approach may overlook many relevant PPIs, either because proteins lack context information (e.g. their disease roles are not yet known), or PPIs do not fit into available context categories (e.g. interactions between membrane and cytoplasmic proteins). To address these issues, IID stores two types of associations between a PPI and a context: the usual association where both proteins share the context, and a more flexible association where one or both proteins belong to the context. Users can choose which type of association is used for filtering PPIs. In addition to context, PPIs are now annotated with stability information (stable or transient), direction in pathways, and alterations due to mutations (e.g. increasing strength). Interpretability and usability of networks is improved in four ways: annotation of PPIs with membership in large complexes and conservation across species, extensive network analysis options, and network visualization. Annotation with complexes can help partition large numbers of PPIs into a smaller set of subnetworks with known functions. Conservation information can indicate which organisms are likely to be effective disease models. In addition to topological analysis, IID now provides several types of enrichment analysis for all PPI annotations; enrichment can be calculated among PPIs, network proteins, or partners of query proteins. Network visualization is designed to scale to even very large networks by focusing on the most highly connected network proteins and their connectivity.
MATERIALS AND METHODS
PPI sources
Experimentally detected PPIs are primarily from seven databases: BioGRID (36) 4.3.196, DIP (37), HPRD (38) Release 9, InnateDB (39) 5.4, IntAct (40) 4.2.16, MatrixDB (41) and MINT (42). All databases were downloaded 2021-04-07. Smaller numbers of PPIs are from manual curation of literature and from curated PPIs reported in Lefebvre et al. (43).
Predicted PPIs come from five studies, as previously described (23). Orthologous PPIs are generated by mapping experimentally detected PPIs in each of the eighteen IID species, to orthologous protein pairs in the other 17 species. Mapping is done using 1:1 orthologs downloaded from Ensembl (44) release 103.
Mapping between gene and protein IDs
Mappings between various gene and protein IDs are based on UniProt (45) release 2021_03. For a more complete set of mappings between Ensembl and UniProt IDs, mappings from Ensembl (44) release 103 are also used.
Annotation of PPIs with context: developmental stages, diseases, tissues, subcellular localization
A PPI is annotated with a developmental stage if its two encoding genes are expressed at that stage. A gene is considered expressed at a developmental stage if its mas5-normalized expression is >200, as in Bossi et al. (46). Gene expression levels at developmental stages come from Xie et al. (47), GEO (48) accession GSE18290. Other PPI context annotations, including tissue, subcellular localization and disease, are assigned as described in the previous release (23).
Annotation of PPI conservation, druggability and other properties
Conservation across species
A PPI is considered conserved in another species if both encoding genes have 1:1 orthologs in that species. Orthologs are obtained from Ensembl (44) release 103.
Directionality
Interaction directionality is compiled from the following biological pathway and post-translational modification databases: HumanCyc (49) v20, NetPath (50), PID (51), PhosphositePlus (52) v6.5.9.3 and Reactome (53) v76. Data from these databases are downloaded in BioPAX (54) Level 3 format and converted to extended binary SIF format (SIFnx) using the function ‘toSifnx’ from the package PaxtoolsR (55) v1.14.0 in R (56) v3.6.2. The ‘toSifnx’ function extracts information about interacting gene (protein) pairs within a pathway, including their names and the types of interactions between them. PPIs are annotated with directionality if they have the following types of interactions within pathways: ‘controls-state-change-of’, ‘controls-phosphorylation-of’, ‘controls-transport-of’.
Druggability
PPIs are annotated with drug target class information from UniProt (45) 2021_03, and targeting drugs from DrugBank (57) v5.0, as previously described (23).
Membership in complexes
Complexes comprising ten or more proteins are obtained from CORUM (58) v3.0 and Reactome (53) v76.
Mutation effects
Data describing the effects of mutations on PPIs (59) come from IntAct (40) release 4.2.16. These data are reorganized such that each unique PPI is associated with 12 possible mutation effects (59) (e.g. decreasing strength), accompanied by corresponding mutations. Additionally, each PPI is annotated with a short description of its known mutation effects, using the terms ‘decreasing/disrupting’, ‘increasing’ or ‘no/unknown effect’.
Stability
Stably-interacting PPIs are based on stable complexes identified by Havugimana et al. (60). Transiently interacting PPIs comprise post-translational modification transferases and targets from PhosphositePlus (52) v6.5.9.3.
Enrichment analysis
IID provides frequency and enrichment analysis for all annotations. Enrichment P-values are calculated as hypergeometric probabilities, using custom javascript code (downloadable from the web site). The background for probability calculations is the PPI network supported by the user's selected evidence types. For example, if the user selected ‘experimental’ and ‘predicted’ evidence in their query, then the background will comprise all PPIs that are either experimentally detected or predicted.
Multiple analysis options are available for certain types of annotations. Frequency and enrichment of context annotations (developmental stages, diseases, subcellular localizations, tissues), druggability, and membership in complexes can be analyzed in four ways, as described below.
Frequency and enrichment are calculated among PPIs, and it is assumed that a PPI has an annotation only if both of its proteins have the annotation. The frequency of an annotation (e.g. adipose tissue) is reported as the number and the percentage of retrieved PPIs with the annotation. Enrichment P-values are calculated with the following inputs to a hypergeometric probability function: N = number of PPIs in the background network, M = number of PPIs in the background network with the annotation, n = number of PPIs in the retrieved network, m = number of PPIs in the retrieved network that have the annotation.
Frequency and enrichment are calculated among PPIs, and it is assumed that a PPI has an annotation if either or both proteins have the annotation. Frequency and P-values are calculated as above, but the numbers of annotated PPIs are typically higher.
Frequency and enrichment are calculated among proteins in the retrieved network. The frequency of an annotation is reported as the number and percentage of proteins in the retrieved network with the annotation. Enrichment P-values are calculated with the following inputs to a hypergeometric probability function: N = number of proteins in the background network, M = number of proteins in the background network with the annotation, n = number of proteins in the retrieved network, m = number of proteins in the retrieved network with the annotation.
Frequency and enrichment are calculated among partners of query proteins. Frequency and P-values are calculated as above, but only partners of query proteins are considered.
Frequency and enrichment of four annotation types—conservation across species, directionality, stability, and mutation effects—are calculated only as in option 1.
Topology analysis
Network topology measures are calculated by custom javascript code, downloadable from the web site.
Network visualization
Network visualization is implemented with the vis.js javascript library version 4.21 (visjs.org) and custom javascript code, downloadable from the web site.
RESULTS
IID provides comprehensive, annotated PPI networks in 18 species: human, 6 model organisms and 11 domesticated species. The first aim is to provide interactions for most proteins in these species, so that PPI networks are more widely applicable in biomolecular research. Proteins with few or no detected interactions still represent a large percentage of the human proteome and are especially prevalent in most other species (Figure 1). To increase the coverage of PPI networks (and context as well as interaction replicates), IID includes experimentally detected PPIs from seven curated databases (36–42) and PPIs predicted by machine learning (22,43,61–63) and orthology. Users can select which PPI sources to include, and also a minimum number of publications or bioassays supporting each PPI.
Figure 1.
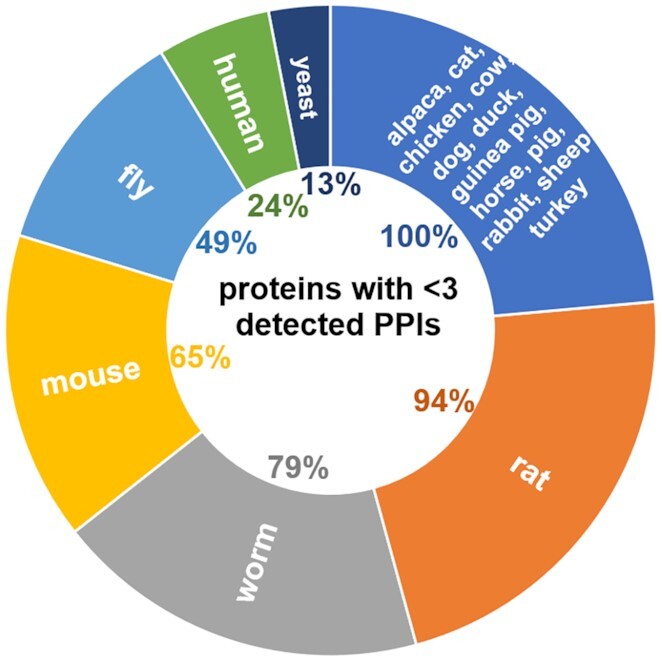
Percentages of low degree proteins in IID species.
Our second aim is to annotate PPIs with comprehensive context and interaction properties, so that users can retrieve subnetworks that are relevant to their research. Ten types of PPI annotations are available including tissues, diseases, and druggability. Annotations are based on a wide range of data sources including gene expression and proteomic studies, and databases of diseases, drugs, pathways, PPIs, proteins and protein complexes. PPI networks filtered by gene expression in relevant tissues have been more effective in bioinformatic workflows such as prioritization of disease genes (64–66). PPIs can be filtered by any number of annotations, and filters can be combined in multiple ways. For example, users can specify that retrieved PPIs should occur in (bone or kidney or liver) and (carcinoma and sarcoma) and (nucleus). In our example (Figure 2), we queried IID with 116 psoriatic arthritis genes from DisGeNET (67) and selected three types of annotations: mutation effects, conservation across species, and druggability. We then calculated degree for the query and interactor proteins in IID and in the retrieved network (Supplementary Table S1). Eleven interacting proteins had a degree higher than 10 and a ratio between the 2 degrees higher than 0.2 (Supplementary Figure S2). Interestingly, the two proteins with the highest ratio were IL-17RD and TRGV9. IL-17RD is a recently discovered receptor for IL-17A (68) that contributes to the proinflammatory pathway controlled by IL-17A, a critical player in the pathogenesis of psoriatic arthritis (69). Notably, TRGV9 is part of the T cell receptor gamma, that forms the complex TCRγδ, known to secrete IL-17 (70) and suggested to have a fundamental role in the development of psoriatic arthritis (71).
Figure 2.

Results of querying IID with 116 psoriatic arthritis genes from DisGeNET (67). This query included three types of annotations: mutation effects, conservation across species, and druggability. (A) Visualization of a subset of retrieved PPIs comprising high degree query proteins (violet) and interaction partners (blue). (B) Mutations with known effects on retrieved PPIs. (C) Conservation of retrieved PPIs across species. (D) Drugs targeting one or both proteins of retrieved PPIs.
Our third objective is to provide online analysis and visualization to help interpret networks, either to refine queries or plan functional studies (Figure 2). Analysis options include calculation of protein degree and clustering coefficients, and enrichment of any PPI annotations. Enrichment can be calculated in several ways, including by considering PPIs or individual proteins. Data can be downloaded in common formats, to include detailed annotation for supplementary material (Supplementary Table S1), or for analysis and visualization with standalone tools, such as NAViGaTOR (72) (Supplementary Figure S1).
Novel features in IID 2021
This update extensively revises and expands the number of PPIs and their annotations, adds network analysis options, and provides network visualization. The number of PPIs increases by almost 50%—from 4,927,742 in the previous release, to 7,369,019.
Context annotations, such as tissues, are now assigned to PPIs in two ways: where both interacting proteins have the annotation (used in IID 2018), and where at least one of the interacting proteins has the annotation. Users can select either approach when filtering PPIs by context, and IID search results indicate whether a PPI’s context annotation is based on both proteins or one, shown as ‘2’ or ‘1’, respectively. The new annotation approach helps address several common problems with the previous approach. One of the main problems is that the context of individual proteins is often unknown. For example, researchers searching for new arthritis proteins may want to know whether their top candidates interact with known arthritis proteins; however, filtering by the usual annotation approach cannot return any results. A second common issue is that relevant PPIs may involve proteins from different contexts; for example, researchers studying membrane proteins may be interested in PPIs where one protein is in the membrane and the other is in the cytoplasm or extracellular space. Similarly, in some scenarios, one protein may not require a context; for example, researchers searching for drug treatments may need PPIs where either protein is a drug target.
This update also adds new types of PPI annotations. Context annotations now include developmental stages, from 1-cell embryo to blastocyst, important for developmental biology, as well as cancer research (73,74). Five other new annotation types do not describe context, but rather the interactions themselves: their conservation across species, direction, membership in large complexes, modification by mutations, and stability. These annotation types, with the exception of complexes, are assigned by only one approach – an annotation can only be present or absent, shown as ‘1’ or ‘0’ in IID results. Conservation annotations indicate whether a PPI may be present in a given species. This information, especially combined with IID’s analysis options, can help identify the best model organisms for functional studies or validation (Figure 2C). Direction annotations and accompanying pathway information are important for network analysis, including flow analysis and signaling studies. Protein complex annotations can make a network more interpretable, since large numbers of PPIs may be grouped into a few functional units. A complex is assigned to both proteins and the pair—a PPI may have one or both proteins in a complex, indicated in IID results as ‘1’ or ‘2’, respectively. Partial membership in a complex can be informative if, for example, many query proteins interact with a complex, but are not within it. Mutation annotations describe the effects of specific mutations on interactions (26,59,75): for example, mutation Trp590Ser in human ABCA1, increases interaction strength between ABCA1 and APOA1. PPIs can be filtered by 12 different mutation effects (Figure 2B), or combinations of these effects. Stability annotations characterize how PPIs carry out their function – as permanent members of larger complexes or as transient interactions. Stability information is also important for detecting PPIs: while stable interactions can be identified by many biological assays, transient PPIs require special techniques.
This update includes several new options for analyzing PPI annotations. Frequency and enrichment of annotations can be analyzed among PPIs, network proteins, or only partners of query proteins. Furthermore, analysis among PPIs has two options: it can assume the previous annotation approach (both proteins having the annotation) or the new approach (one or both proteins having the annotation). Thus, there are a total of four analysis options, referred to as PPIsboth, PPIseither, proteins and partners. These options address different types of questions about a network. The most common questions may deal with network PPIs—for example, in which tissues do the PPIs occur? This is best addressed by the PPIsboth option, which would report numbers of PPIs in each tissue, and their significance. Another PPI-related question might be, which drugs have the biggest impact on the network? Here, the best option may be PPIseither; for each drug, it would report the number and significance of potentially affected PPIs (i.e. PPIs involving at least one of the drug's targets). The proteins option is best for most questions that do not specifically focus on PPIs—for example, which diseases are prevalent in the network? The proteins option would report the frequency and significance of each disease among network proteins. The PPIsboth and PPIseither options may give unintuitive results for such questions. For example, if a disease is annotated to many proteins, but these proteins do not interact with each other, then PPIsboth would report the disease's frequency as zero. Conversely, if a disease is annotated to a single protein, but the protein has a high degree, then, PPIseither would report a high disease frequency. The partners option is helpful when query proteins are a biased sample (e.g. they were selected due to known roles in a disease) and the aim is to understand their partners (e.g. determine prevalent diseases among partners).
Comparison with other PPI resources
IID differs from other PPI resources (such as APID (76), HIPPIE v2.0 (33), HINT (77), iRefWeb (78), MyProteinNet (34), STRING (79) and TissueNet v.2 (35)) by providing more options for reducing false negatives, annotating and filtering PPIs, and analyzing networks. All of these resources integrate PPIs from curated databases, and some provide PPI annotations and network analysis. However, there are differences in terms of available species, options for controlling network false positive and false negative rates, and types of annotations, filtering, and analysis. All resources provide human PPIs, some (HINT (77), iRefWeb (78), MyProteinNet (34)) include model organisms, IID includes model organisms and domesticated species, and two databases (APID (76), STRING (79)) cover over 1000 species. To control false positive rate, some databases assign scores to PPIs, reflecting the quantity and reliability of interaction evidence, while IID enables filtering by types and quantity of evidence. To reduce false negatives, all resources integrate multiple sources of curated PPIs. IID includes physically binding PPIs predicted by machine learning, while STRING (79) and FunCoup (80) include functional interaction predictions. PPI context annotations and filtering are provided by HIPPIE v2.0 (33), MyProteinNet (34), and TissueNet v.2 (35). All three databases include tissue annotations, MyProteinNet (34) includes Gene Ontology (GO) (8,9), and HIPPIE v2.0 (33) includes GO, diseases, directionality and activation/repression annotations. IID provides more extensive annotations and filtering options. Also, IID provides annotations in 18 species, while HIPPIE v2.0 (33) and TissueNet v.2 (35) cover human, and MyProteinNet (34) covers 11 species. Network analysis is supported by HIPPIE v2.0 (33) and STRING (79). HIPPIE v2.0 (33) provides enrichment analysis of diseases and GO in network proteins. STRING (79) provides summary topology statistics for networks, and enrichment analysis of GO, pathways, and protein domains in networks proteins. IID provides both topology and enrichment analysis; it identifies important network nodes, and enables enrichment analysis for all annotations, often in both proteins and PPIs.
DISCUSSION
Although knowledge of PPI networks has been rapidly increasing, applying them in biomolecular research studies can be challenging: proteins of interest may have few available interactions, these interactions may lack context, and the retrieved network may be difficult to interpret. In addition, while false positives continue to be reduced in newer assays, false negatives remain a challenge across most organisms, including human. IID focuses on these issues—it aims to provide comprehensive networks, annotation, and analysis. The overall goal is to help address research problems such as selecting the best model organism, identifying reliable and accurate network-biomarkers, predicting drugs with the strongest impact on a protein network, and finding diseases with a similar mechanism. Future IID updates will focus on extending network analysis options to assist with studies of genetic variants and drug repurposing.
Supplementary Material
Contributor Information
Max Kotlyar, Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute and Data Science Discovery Centre for Chronic Diseases, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada.
Chiara Pastrello, Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute and Data Science Discovery Centre for Chronic Diseases, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada.
Zuhaib Ahmed, Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute and Data Science Discovery Centre for Chronic Diseases, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada.
Justin Chee, Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute and Data Science Discovery Centre for Chronic Diseases, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada.
Zofia Varyova, Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute and Data Science Discovery Centre for Chronic Diseases, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada.
Igor Jurisica, Osteoarthritis Research Program, Division of Orthopedic Surgery, Schroeder Arthritis Institute and Data Science Discovery Centre for Chronic Diseases, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada; Departments of Medical Biophysics and Computer Science, University of Toronto, Toronto, ON M5S 1A4, Canada; Institute of Neuroimmunology, Slovak Academy of Sciences, Bratislava, Slovakia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ontario Research Fund [34876, in part]; Natural Sciences Research Council (NSERC) [203475]; Canada Foundation for Innovation (CFI) [29272, 225404, 33536]; Schroeder Arthritis Institute, Buchan Foundation and Ian Lawson van Toch Fund via the Toronto General and Western Hospital Foundation, University Health Network (in part). The funders had no role in study design, data collection, and analysis, decision to publish or preparation of the manuscript. Funding for open access charge: NSERC [203475].
Conflict of interest statement. None declared.
REFERENCES
- 1. Navlakha S., Kingsford C.. The power of protein interaction networks for associating genes with diseases. Bioinformatics. 2010; 26:1057–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee I., Blom U.M., Wang P.I., Shim J.E., Marcotte E.M.. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011; 21:1109–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tian W., Zhang L. V, Taşan M., Gibbons F.D., King O.D., Park J., Wunderlich Z., Cherry J.M., Roth F.P.. Combining guilt-by-association and guilt-by-profiling to predict Saccharomyces cerevisiae gene function. Genome Biol. 2008; 9(Suppl. 1):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mostafavi S., Morris Q.. Combining many interaction networks to predict gene function and analyze gene lists. Proteomics. 2012; 12:1687–1696. [DOI] [PubMed] [Google Scholar]
- 5. Wu M., Zeng W., Liu W., Lv H., Chen T., Jiang R.. Leveraging multiple gene networks to prioritize GWAS candidate genes via network representation learning. Methods. 2018; 145:41–50. [DOI] [PubMed] [Google Scholar]
- 6. Yeh S.-H., Yeh H.-Y., Soo V.-W.. A network flow approach to predict drug targets from microarray data, disease genes and interactome network - case study on prostate cancer. J. Clin. Bioinforma. 2012; 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Isik Z., Baldow C., Cannistraci C.V., Schroeder M.. Drug target prioritization by perturbed gene expression and network information. Sci. Rep. 2015; 5:17417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al.. Gene ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021; 49:D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cowen L., Ideker T., Raphael B.J., Sharan R.. Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 2017; 18:551–562. [DOI] [PubMed] [Google Scholar]
- 11. Missiuro P.V., Liu K., Zou L., Ross B.C., Zhao G., Liu J.S., Ge H.. Information flow analysis of interactome networks. PLOS Comput. Biol. 2009; 5:e1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jeong H., Qian X., Yoon B.-J.. Effective comparative analysis of protein-protein interaction networks by measuring the steady-state network flow using a Markov model. BMC Bioinforma. 2016; 17:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Camacho D.M., Collins K.M., Powers R.K., Costello J.C., Collins J.J.. Next-generation machine learning for biological networks. Cell. 2018; 173:1581–1592. [DOI] [PubMed] [Google Scholar]
- 14. Gligorijević V., Barot M., Bonneau R.. deepNF: deep network fusion for protein function prediction. Bioinformatics. 2018; 34:3873–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. von Mering C., Krause R., Snel B., Cornell M., Oliver S.G., Fields S., Bork P.. Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 2002; 417:399–403. [DOI] [PubMed] [Google Scholar]
- 16. Hart G.T., Ramani A.K., Marcotte E.M.. How complete are current yeast and human protein-interaction networks. Genome Biol. 2006; 7:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rolland T., Taşan M., Charloteaux B., Pevzner S.J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R.et al.. A proteome-scale map of the human interactome network. Cell. 2014; 159:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huttlin E.L., Ting L., Bruckner R.J., Gebreab F., Gygi M.P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K.et al.. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015; 162:425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huttlin E.L., Bruckner R.J., Paulo J.A., Cannon J.R., Ting L., Baltier K., Colby G., Gebreab F., Gygi M.P., Parzen H.et al.. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017; 545:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luck K., Kim D.-K., Lambourne L., Spirohn K., Begg B.E., Bian W., Brignall R., Cafarelli T., Campos-Laborie F.J., Charloteaux B.et al.. A reference map of the human binary protein interactome. Nature. 2020; 580:402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Venkatesan K., Rual J.-F., Vazquez A., Stelzl U., Lemmens I., Hirozane-Kishikawa T., Hao T., Zenkner M., Xin X., Goh K.-I.et al.. An empirical framework for binary interactome mapping. Nat. Methods. 2009; 6:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kotlyar M., Pastrello C., Pivetta F., Lo Sardo A., Cumbaa C., Li H., Naranian T., Niu Y., Ding Z., Vafaee F.et al.. In silico prediction of physical protein interactions and characterization of interactome orphans. Nat. Methods. 2015; 12:79–84. [DOI] [PubMed] [Google Scholar]
- 23. Kotlyar M., Pastrello C., Malik Z., Jurisica I.. IID 2018 update: context-specific physical protein–protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2019; 47:D581–D589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y., Hagenbuch B.. Protein-protein interactions of drug uptake transporters that are important for liver and kidney. Biochem. Pharmacol. 2019; 168:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yao Z., Aboualizadeh F., Kroll J., Akula I., Snider J., Lyakisheva A., Tang P., Kotlyar M., Jurisica I., Boxem M.et al.. Split Intein-Mediated Protein Ligation for detecting protein-protein interactions and their inhibition. Nat. Commun. 2020; 11:2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Porras P., Barrera E., Bridge A., Del-Toro N., Cesareni G., Duesbury M., Hermjakob H., Iannuccelli M., Jurisica I., Kotlyar M.et al.. Towards a unified open access dataset of molecular interactions. Nat. Commun. 2020; 11:6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yadav A., Vidal M., Luck K.. Precision medicine — networks to the rescue. Curr. Opin. Biotechnol. 2020; 63:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kennedy S.A., Jarboui M.A., Srihari S., Raso C., Bryan K., Dernayka L., Charitou T., Bernal-Llinares M., Herrera-Montavez C., Krstic A.et al.. Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRASG13D. Nat. Commun. 2020; 11:499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shu T., Ning W., Wu D., Xu J., Han Q., Huang M., Zou X., Yang Q., Yuan Y., Bie Y.et al.. Plasma proteomics identify biomarkers and pathogenesis of COVID-19. Immunity. 2020; 53:1108–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silverman E.K., Schmidt H.H.H.W., Anastasiadou E., Altucci L., Angelini M., Badimon L., Balligand J.L., Benincasa G., Capasso G., Conte F.et al.. Molecular networks in network medicine: development and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020; 12:e1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sadegh S., Matschinske J., Blumenthal D.B., Galindez G., Kacprowski T., List M., Nasirigerdeh R., Oubounyt M., Pichlmair A., Rose T.D.et al.. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 2020; 11:3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han Z., Zhang W., Ning W., Wang C., Deng W., Li Z., Shang Z., Shen X., Liu X., Baba O.et al.. Model-based analysis uncovers mutations altering autophagy selectivity in human cancer. Nat. Commun. 2021; 12:3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alanis-Lobato G., Andrade-Navarro M.A., Schaefer M.H.. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res. 2017; 45:D408–D414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Basha O., Flom D., Barshir R., Smoly I., Tirman S., Yeger-Lotem E.. MyProteinNet: build up-to-date protein interaction networks for organisms, tissues and user-defined contexts. Nucleic Acids Res. 2015; 43:W258–W263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Basha O., Barshir R., Sharon M., Lerman E., Kirson B.F., Hekselman I., Yeger-Lotem E.. The TissueNet v.2 database: a quantitative view of protein-protein interactions across human tissues. Nucleic Acids Res. 2017; 45:D427–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oughtred R., Rust J., Chang C., Breitkreutz B.J., Stark C., Willems A., Boucher L., Leung G., Kolas N., Zhang F.et al.. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021; 30:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salwinski L., Miller C.S., Smith A.J., Pettit F.K., Bowie J.U., Eisenberg D.. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004; 32:D449–D451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A.et al.. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009; 37:D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Breuer K., Foroushani A.K., Laird M.R., Chen C., Sribnaia A., Lo R., Winsor G.L., Hancock R.E.W., Brinkman F.S.L., Lynn D.J.. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 2013; 41:D1228–D1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N.et al.. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014; 42:D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Clerc O., Deniaud M., Vallet S.D., Naba A., Rivet A., Perez S., Thierry-Mieg N., Ricard-Blum S.. MatrixDB: integration of new data with a focus on glycosaminoglycan interactions. Nucleic Acids Res. 2019; 47:D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Licata L., Briganti L., Peluso D., Perfetto L., Iannuccelli M., Galeota E., Sacco F., Palma A., Nardozza A.P., Santonico E.et al.. MINT, the molecular interaction database: 2012 Update. Nucleic Acids Res. 2012; 40:D857–D861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lefebvre C., Lim W.K., Basso K., Favera R.D., Califano A.. A Context-Specific Network of Protein-DNA and Protein-Protein Interactions Reveals New Regulatory Motifs in Human B Cells. Systems Biology and Computational Proteomics. 2006; Berlin, Heidelberg: Springer Berlin Heidelberg; 42–56. [Google Scholar]
- 44. Howe K.L., Achuthan P., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R., Bhai J.et al.. Ensembl 2021. Nucleic Acids Res. 2021; 49:D884–D891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. The UniProt Consortium Bateman A., Martin M.-J., Orchard S., Magrane M., Agivetova R., Ahmad S., Alpi E., Bowler-Barnett E.H., Britto R.et al.. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bossi A., Lehner B.. Tissue specificity and the human protein interaction network. Mol Syst Biol. 2009; 5:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie D., Chen C.C., Ptaszek L.M., Xiao S., Cao X., Fang F., Ng H.H., Lewin H.A., Cowan C., Zhong S.. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 2010; 20:804–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.et al.. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Romero P., Wagg J., Green M.L., Kaiser D., Krummenacker M., Karp P.D.. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2005; 6:R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kandasamy K., Sujatha Mohan S., Raju R., Keerthikumar S., Sameer Kumar G.S., Venugopal A.K., Telikicherla D., Navarro D.J., Mathivanan S., Pecquet C.et al.. NetPath: a public resource of curated signal transduction pathways. Genome Biol. 2010; 11:R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schaefer C.F., Anthony K., Krupa S., Buchoff J., Day M., Hannay T., Buetow K.H.. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009; 37:D674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hornbeck P. V, Kornhauser J.M., Latham V., Murray B., Nandhikonda V., Nord A., Skrzypek E., Wheeler T., Zhang B., Gnad F.. 15 years of PhosphoSitePlus®: integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019; 47:D433–D441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R.et al.. The reactome pathway knowledgebase. Nucleic Acids Res. 2020; 48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Demir E., Cary M.P., Paley S., Fukuda K., Lemer C., Vastrik I., Wu G., D’Eustachio P., Schaefer C., Luciano J.et al.. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010; 28:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luna A., Babur Ö., Aksoy B.A., Demir E., Sander C.. PaxtoolsR: pathway analysis in R using pathway commons. Bioinformatics. 2016; 32:1262–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. R Core Team R: A Language and Environment for Statistical Computing. 2020; [Google Scholar]
- 57. Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z.et al.. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018; 46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Giurgiu M., Reinhard J., Brauner B., Dunger-Kaltenbach I., Fobo G., Frishman G., Montrone C., Ruepp A.. CORUM: the comprehensive resource of mammalian protein complexes - 2019. Nucleic Acids Res. 2019; 47:D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. del-Toro N., Duesbury M., Koch M., Perfetto L., Shrivastava A., Ochoa D., Wagih O., Piñero J., Kotlyar M., Pastrello C.et al.. Capturing variation impact on molecular interactions in the IMEx Consortium mutations data set. Nat. Commun. 2019; 10:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Havugimana P.C., Hart G.T., Nepusz T., Yang H., Turinsky A.L., Li Z., Wang P.I., Boutz D.R., Fong V., Phanse S.et al.. A census of human soluble protein complexes. Cell. 2012; 150:1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rhodes D.R., Tomlins S.A., Varambally S., Mahavisno V., Barrette T., Kalyana-Sundaram S., Ghosh D., Pandey A., Chinnaiyan A.M.. Probabilistic model of the human protein-protein interaction network. Nat. Biotechnol. 2005; 23:951–959. [DOI] [PubMed] [Google Scholar]
- 62. Elefsinioti A., Saraç Ö.S., Hegele A., Plake C., Hubner N.C., Poser I., Sarov M., Hyman A., Mann M., Schroeder M.et al.. Large-scale de novo prediction of physical protein-protein association. Mol Cell Proteomics. 2011; 10:M111.010629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang Q.C., Petrey D., Deng L., Qiang L., Shi Y., Thu C.A., Bisikirska B., Lefebvre C., Accili D., Hunter T.et al.. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature. 2012; 490:556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guan Y., Gorenshteyn D., Burmeister M., Wong A.K., Schimenti J.C., Handel M.A., Bult C.J., Hibbs M.A., Troyanskaya O.G.. Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput. Biol. 2012; 8:e1002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Magger O., Waldman Y.Y., Ruppin E., Sharan R.. Enhancing the prioritization of disease-causing genes through tissue specific protein interaction networks. PLoS Comput. Biol. 2012; 8:e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A., Himmelstein D.S., Zhang R., Hartmann B.M., Zaslavsky E., Sealfon S.C.et al.. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015; 47:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Piñero J., Ramírez-Anguita J.M., Saüch-Pitarch J., Ronzano F., Centeno E., Sanz F., Furlong L.I.. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020; 48:D845–D855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Su Y., Huang J., Zhao X., Lu H., Wang W., Yang X.O., Shi Y., Wang X., Lai Y., Dong C.. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci. Immunol. 2019; 4:eaau9657. [DOI] [PubMed] [Google Scholar]
- 69. Wang E.A., Suzuki E., Maverakis E., Adamopoulos I.E.. Targeting IL-17 in psoriatic arthritis. Eur. J. Rheumatol. 2017; 4:272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roller A., Perino A., Dapavo P., Soro E., Okkenhaug K., Hirsch E., Ji H.. Blockade of phosphatidylinositol 3-kinase (PI3K)δ or PI3Kγ reduces IL-17 and ameliorates imiquimod-induced psoriasis-like dermatitis. J. Immunol. 2012; 189:4612–4620. [DOI] [PubMed] [Google Scholar]
- 71. Spadaro A., Scrivo R., Moretti T., Bernardini G., Riccieri V., Taccari E., Strom R., Valesini G.. Natural killer cells and γ/δ T cells in synovial fluid and in peripheral blood of patients with psoriatic arthritis. Clin. Exp. Rheumatol. 2004; 22:389–394. [PubMed] [Google Scholar]
- 72. Brown K.R., Otasek D., Ali M., McGuffin M.J., Xie W., Devani B., Toch I.L., Jurisica I.. NAViGaTOR: Network Analysis, Visualization and Graphing Toronto. Bioinformatics. 2009; 25:3327–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dong J., Kislinger T., Jurisica I., Wigle D.A.. Lung cancer: developmental networks gone awry. Cancer Biol. Ther. 2009; 8:312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Becker-Santos D.D., Thu K.L., English J.C., Pikor L.A., Martinez V.D., Zhang M., Vucic E.A., Luk M.T.Y., Carraro A., Korbelik J.et al.. Developmental transcription factor NFIB is a putative target of oncofetal miRNAs and is associated with tumour aggressiveness in lung adenocarcinoma. J. Pathol. 2016; 240:161–172. [DOI] [PubMed] [Google Scholar]
- 75. Mandilaras V., Garg S., Cabanero M., Tan Q., Pastrello C., Burnier J., Karakasis K., Wang L., Dhani N.C., Butler M.O.et al.. TP53 mutations in high grade serous ovarian cancer and impact on clinical outcomes: a comparison of next generation sequencing and bioinformatics analyses. Int. J. Gynecol. Cancer. 2019; 29:346–352. [DOI] [PubMed] [Google Scholar]
- 76. Alonso-López Di., Campos-Laborie F.J., Gutiérrez M.A., Lambourne L., Calderwood M.A., Vidal M., De Las Rivas J.. APID database: Redefining protein-protein interaction experimental evidences and binary interactomes. Database. 2019; 2019:baz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Das J., Yu H.. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012; 6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Turner B., Razick S., Turinsky A.L., Vlasblom J., Crowdy E.K., Cho E., Morrison K., Donaldson I.M., Wodak S.J.. iRefWeb: interactive analysis of consolidated protein interaction data and their supporting evidence. Database. 2010; 2010:baq023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Szklarczyk D., Gable A.L., Nastou K.C., Lyon D., Kirsch R., Pyysalo S., Doncheva N.T., Legeay M., Fang T., Bork P.et al.. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021; 49:D605–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ogris C., Guala D., Kaduk M., Sonnhammer E.L.L.. FunCoup 4: new species, data, and visualization. Nucleic Acids Res. 2018; 46:D601–D607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.