


Abstract
As an increasing number of noncoding RNAs (ncRNAs) have been suggested to encode short bioactive peptides in cancer, the exploration of ncRNA-encoded small peptides (ncPEPs) is emerging as a fascinating field in cancer research. To assist in studies on the regulatory mechanisms of ncPEPs, we describe here a database called SPENCER (http://spencer.renlab.org). Currently, SPENCER has collected a total of 2806 mass spectrometry (MS) data points from 55 studies, covering 1007 tumor samples and 719 normal samples. Using an MS-based proteomics analysis pipeline, SPENCER identified 29 526 ncPEPs across 15 different cancer types. Specifically, 22 060 of these ncPEPs were experimentally validated in other studies. By comparing tumor and normal samples, the identified ncPEPs were divided into four expression groups: tumor-specific, upregulated in cancer, downregulated in cancer, and others. Additionally, since ncPEPs are potential targets for neoantigen-based cancer immunotherapy, SPENCER also predicted the immunogenicity of all the identified ncPEPs by assessing their MHC-I binding affinity, stability, and TCR recognition probability. As a result, 4497 ncPEPs curated in SPENCER were predicted to be immunogenic. Overall, SPENCER will be a useful resource for investigating cancer-associated ncPEPs and may boost further research in cancer.
Graphical Abstract
Graphical Abstract.
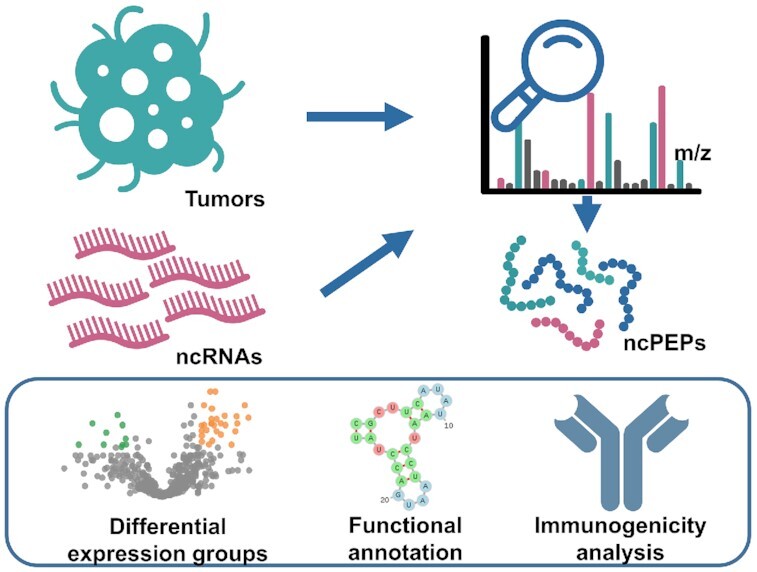
SPENCER: a comprehensive database for small peptides encoded by non-coding RNAs in cancer patients.
INTRODUCTION
As reported in the ENCODE project, up to 80% of the human genome has the capacity to be transcribed into ncRNA (1). ncRNAs are a class of RNA molecules that are widely involved in many fundamental biological processes such as genomic modulation (2), environmental responses (3), and body development (4). A growing body of evidence has shown that dysfunction of ncRNAs may lead to various human diseases including cancer (5,6). Hence, tremendous efforts have been made to explore the relationship between ncRNAs and cancer. Since ncRNAs are generally considered to lack the capability to encode functional proteins, most relevant studies have focused on only the functional role of ncRNAs as transcripts themselves in different cancer types (6). However, with the development of proteomics and translatomics technologies, researchers in many studies have highlighted the coding potential of ncRNAs (7), and identified a series of small biologically active peptides translated from short open reading frames (sORFs) in ncRNAs. Moreover, accumulating evidence has revealed the importance of several ncPEPs in the pathogenesis and progression of cancer (8). For example, the lncRNA HOXB-AS3 can encode a 53-amino acid (aa) peptide, and is reported to reduce the growth of colon cancer (9). In addition, CASIMO1, a microprotein of 10 kDa, encoded by another lncRNA, plays a critical role in breast cancer cell proliferation and is implicated in cellular lipid homeostasis (10). In addition to lncRNAs, some circRNAs can also be translated into functional peptides implicated in cancer development. For example, circPPP1R12A can be translated into a 73-aa protein that promotes the metastasis of colon cancer (11). Therefore, annotating the expression status and functions of ncPEPs in tumor samples may be a worthwhile strategy to decipher the pathogenesis of cancer and provide valuable information for cancer studies.
Recently, utilizing cancer immunotherapy to eradicate cancer cells has become the most investigated subject in cancer research (12). Cancer immunotherapy aims to engage the immune system against targets that are expressed in only tumor cells. One important class of such targets is neoantigens, which can be processed and presented by the major histocompatibility complex (MHC) on the cell surface and recognized by T cell receptors (TCRs) to stimulate a highly specific antitumor immune response (13). Cancer immunotherapy based on neoantigens has become a new popular research focus, and mutation-derived neoantigens likely play a principal role in this concept (14). However, the therapeutic effects of mutation-derived neoantigens are limited to patients with tumors with a high-mutation burden (15). Therefore, there is a great demand for a comprehensive analysis of cancer antigens and the identification of new classes of neoantigens, especially for patients with a low tumor mutational burden. Since some ncPEPs are expressed in only tumor tissues (16), they have great potential as novel neoantigens in the cancer immunotherapy. Recently, the lncRNA meloe was demonstrated to produce three polypeptides in melanoma, MELOE-1, MELOE-2 and MELOE-3 (17). Moreover, MELOE-1 and MELOE-2 were experimentally verified to have prominent immunogenicity, and potentially ideal tumor neoantigens for immunotherapy (18,19). Furthermore, a series of tumor vaccines targeting validated ncPEPs was recently developed, and immunogenicity and efficacy were evaluated in mouse models of cancer (20). Accordingly, identifying ncPEPs from cancer tissues and evaluating their potential immunogenicity is a promising direction of cancer research, and the results may provide new perspectives for cancer immunotherapy.
With increasing attention on ncPEPs, a large number of novel ncRNA translation products have been identified and validated. However, the relevant information is scattered among innumerable published articles, which is inconvenient for researchers exploring the functional roles of ncPEPs. Currently, several databases, such as ncEP (21), FuncPEP (22), ARA-PEPs (23) and cncRNAdb (24), have been developed to collect and curate experimentally verified ncPEPs from published articles. In addition, some other data resources, including sORFs.org (25), SmProt (26), MetamORF (27) and PsORF (28), have collected potential ncRNA-encoded sORFs or peptides from high-throughput experiments such as ribosome profiling and mass spectra. Although valuable information is provided by the above data resources, they were constructed mainly for general purposes, and more in-depth investigations on the functional roles of ncPEPs in cancer development have not been performed. At present, a specific resource for the systematic study of cancer-related ncPEPs remains absent. Therefore, the development of an integrated database dedicated to cancer-associated ncPEPs is urgent needed.
In this study, we present SPENCER (http://spencer.renlab.org), a novel database that allows the exploration and visualization of ncPEPs in cancer patients (Figure 1). An intact set of cancer-related MS data was collected from multiple proteomic databases. By integrating all the validated human protein sequences from UniProt and all the possible sORF sequences translated from known ncRNAs, a comprehensive searchable database was constructed. Using this database, MaxQuant (29) was applied to search for ncPEPs from the collected MS data. To further investigate the underlying relationship between ncPEPs and cancer, quantitative analysis of each collected study was performed to identify ncPEPs that were differentially expressed between tumors and paracancerous/normal tissues. Finally, to explore the potential utility of ncPEPs as targets in neoantigen-based cancer immunotherapy, we further evaluated their immunogenicity using a robust pipeline and included the prediction results in SPENCER.
Figure 1.

Overall design and construction of SPENCER. From published literature and available databases, we collected ∼450 000 ncRNA sequences and ∼60 MS datasets in SPENCER (upper left). MaxQuant was applied to search for ncPEPs in the collected MS data. Meanwhile, differential expression analysis of the identified small peptides between tumors and normal/paracancerous tissues was performed. To investigate the potential roles of small peptides in cancer, SPENCER integrates detailed annotation of the associated ncRNAs with information from external resources, including function, category and structure (upper right). In addition, using a robust pipeline, the immunogenicity of all the identified ncPEPs was predicted based on three feature scores. Finally, we integrated and visualized the data obtained above to build the SPENCER database (lower).
MATERIALS AND METHODS
Identification of ncPEPs
Mass spectrometry datasets were collected from PRIDE (30), MassIVE.quant (31), JPOST (32), Panorama (33), PeptideAtlas (34) and iProX (35). Only cancer-related datasets with tumors and normal/paracancerous tissues were included in our study. Raw data and associated sample information were downloaded for subsequent analysis (Supplementary Table S1).
To identify ncPEPs from the collected MS datasets, we first constructed a human proteomic sequence database by combining known protein sequences and predicted translation products from ncRNAs. In detail, we collected an intact set of ncRNA transcript sequences from RNAcentral (36) and identified 6 088 980 possible sORFs in the assembled transcripts using a stand-alone version of NCBI’s ORFfinder (37). Based on the identified sORFs, amino acid sequences were translated using the standard codon table of amino acids. Subsequently, sequences of these ncRNA-encoded candidates were integrated with all the validated human protein sequences retrieved from UniProt (38) to construct a complete human proteomic database. Notably, to remove redundancy from the proteomic database, we conducted sequence alignments between every ncRNA-encoded candidate and coding protein and filtered out small peptides matching a validated protein sequence. Then, MaxQuant (29) was applied to search for ncPEPs in each collected MS data using the proteomic database. The parameters, including digestion, label type, and modification type, were set according to the detailed experimental information of the MS data being analyzed. The false discovery rate (FDR) of the peptide-spectrum match (PSM) was set to 1% to reduce false identifications. Using the default razor protein FDR, a peptide that could belong to different proteins was assigned to the protein with the highest likelihood. If a detected mass spectrum was assigned to an ncRNA-encoded candidate, the corresponding peptide was recognized as an ncPEP.
Expression quantification and differential expression analysis
For each identified ncPEP, the peptide intensity calculated by MaxQuant was regarded as its expression level. To reduce the bias introduced by different processing methods among samples, the peptide intensities were first normalized. For label-free MS data, we used the delayed normalization algorithm (39) to normalize peptide intensity. For MS data acquired with labeling technologies such as tandem mass tag (TMT), isobaric tag for relative and absolute quantitation (iTRAQ), stable isotope labeling by amino acids in cell culture (SILAC), and demethylation labeling, the peptide intensities were normalized by comparison with the intensity of the reference channel. Based on the normalized intensities, we then performed differential expression analysis for each identified ncPEP by comparing the normalized intensity between tumorous and normal/paracancerous tissues. To determine the statistical significance of results, a two-sample t-test was performed for each peptide, and the Benjamini-Hochberg procedure was used to calculate the adjusted P value. In addition, the fold change (FC) in peptide expression was calculated as the ratio of the average intensity between the tumor and control groups. Peptides with an adjusted P value <0.05 and log2FC >1 were determined to be upregulated in cancer, while those with an adjusted P value <0.05 and log2FC <−1 were considered downregulated in cancer. In particular, peptides exclusively expressed in the tumor samples were considered tumor-specific. In addition to the above three conditions, the remaining ncPEPs were labeled as others. Finally, using the above results, we categorized the identified ncPEPs into the four groups.
Functional annotation of ncPEPs
To comprehensively study the potential role of ncPEPs in cancer regulatory mechanisms, we annotated the peptides with functional and structural information. We first matched all the ncPEP transcripts with known ncRNAs downloaded from 12 data resources (Supplementary Table S2) to obtain basic information, including RNA type, gene name, and genome location. Moreover, CPAT (40) was applied to predict the coding potential of each ncRNA-origin sORF. Meanwhile, we used the RNAfold tool in the ViennaRNA suite (41) to predict the secondary structure of each ncRNA transcript. To validate the reliability of our identified ncPEPs, a sequence similarity search strategy was constructed to check whether the identified peptides had been reported in other studies. A series of reported ncPEPs were manually collected from other studies, covering translation products identified by ribosome profiling (Ribo-seq), mass spectrometry, western blotting, immunostaining, and in vitro experiments. Then, a global sequence alignment was performed between the collected peptides and identified ncPEPs. ncPEPs that matched the reported peptides were marked as validated in SPENCER.
Immunogenic analysis
To assess the neoantigen potential of ncPEPs, we constructed an immunogenicity analysis based on the current model of epitope immunogenicity, which proposes that a peptide must first be presented by MHC-I and then recognized as foreign by T cells to elicit an antitumor T cell response (42). According to this model, predicting the immunogenicity of a peptide requires a comprehensive evaluation of both antigen presentation and T cell recognition. Hence, previous research (43) identified three key parameters related to these two aspects, including MHC-I binding affinity, MHC-I binding stability and T cell recognition probability, which were systematically selected based on data from a global consortium. Therefore, to improve the prediction accuracy, we constructed a comprehensive pipeline for predicting immunogenicity by integrating the methods suggested by the above research.
Since it is generally accepted that MHC-I molecules bind peptides 8–14 aa in length (44), we first divided each ncPEP into 8–14-mer segments using a sliding window algorithm. Specifically, peptides shorter than 8 aa were considered nonimmunogenic. Then, the scores of the three parameters for all fragments were calculated. Notably, human MHC-I is also known as the human leukocyte antigen I (HLA-I) complex and can be designated into 12 supertype groups (45). Since different supertypes of HLA-I complexes can recognize totally different antigen sequences, patient-specific HLA-I genotypes should be provided when predicting the immunogenicity of each peptide. However, due to the lack of genome or exon sequencing data, we could not infer the accurate HLA-I genotype of each patient. Therefore, we calculated the three parameter scores of each ncPEP under all known HLA genotypes to comprehensively assess their immunogenicity. The detailed methodology is described below:
First, the MHC-I binding affinity was inferred as the IC50 value for each peptide sequence under a specific HLA-I genotype. With the default parameters, we used NetMHCpan 4.0 (44) to calculate the binding affinity score between peptide fragments and each HLA-I genotype. In addition to strong binding affinity, sufficient MHC-I binding stability is also required to enable the presentation of the peptide-MHC complex on the cell membrane through a series of transport processes and, ultimately, recognition by CD8+ T cells. Accordingly, netMHCstabpan (46) was used to predict the binding stability score between peptide fragments and each HLA-I genotype. In addition, the TCR recognition probability of ncPEPs was calculated using the multistate thermodynamic model (Supplementary methods) described by Łuksza et al. (47).
Finally, using the thresholds reported in a previous study (43), ncPEPs that contained a fragment with MHC binding affinity <34 nM, MHC binding stability >1.4 h, and T cell recognition probability >10–11 were predicted to be immunogenic.
Database and web interface implementation
All the metadata in SPENCER are stored and managed in MySQL tables. The server-backend was developed based on Java and the web-frontend interfaces were implemented in HyperText Markup Language (HTML), Cascading Style Sheets (CSS), and JavaScript (JS). To provide visualization of all analysis results, multiple statistical diagrams were generated by EChars on the website. In addition, the interactive heat maps showing the detailed expression levels of tumor-specific and upregulated peptides were constructed by Element UI toolkits. Furthermore, to intuitively visualize the secondary structure of ncPEPs, the ViennaRNA package (48) was integrated into the website interface. IBS (49) was also applied to present the domain organization of sORFs. Particularly, the Lorikeet spectral annotator (https://uwpr.github.io/Lorikeet/) was implemented to display the MS evidence for each identified ncPEP.
RESULTS
Database content
SPENCER has collected 2806 cancer-related MS data points from cancer patients in 55 studies across 15 cancer types (Figure 2A and Supplementary Table S1). MaxQuant was applied to search for ncPEPs in each collected MS dataset, and identified a total of 29 526 ncPEPs from 1726 tissue samples across 15 different types of cancer (Supplementary Table S3). The identified small peptides were mainly translated from five types of ncRNAs, including lncRNA, misc-RNA, sRNA, snRNA and rRNA (Figure 2B). Specifically, 19 831 peptides were translated from 6803 lncRNAs, 9688 from 34 misc-RNAs, 3 from 2 sRNAs, 2 from 1 snRNAs and 2 from 2 rRNAs. As a result, we found that 1.53% (6842/448 331) of the ncRNAs could be translated into small peptides, which was consistent with the findings of previous studies (50). According to differential expression analysis, ncPEPs in each study were divided into four groups: tumor-specific, upregulated in cancer, downregulated in cancer, and others. As a result, we obtained 8060 (27.30%) tumor-specific small peptides, 446 (1.51%) significantly upregulated small peptides, 447 (1.51%) downregulated small peptides, and 20 573 (69.68%) other small peptides spanning 15 cancer types (Figure 2C, left side).
Figure 2.

Overview of the data in SPENCER. (A) Statistics on the number of cancer-related proteomic MS datasets published each year. The green line represents manually curated studies, and the pink line represents tumor patient samples. (B) The coding source of ncPEPs is shown in a bubble plot, presenting 5 different RNA types, including lncRNA (blue), misc-RNA (green), sRNA (pink), snRNA (red), and rRNA (yellow). (C) The number of total small peptides (left) and immunogenetic peptides (right) identified by SPENCER in different cancer types is shown in bar plots, with four groups: tumor-specific (deep green), upregulated in cancer (pink), downregulated in cancer (red), and others (light green). (D) The proportions of peptides with experimental evidence and immunogenicity are illustrated in a pie plot. (E) The occurrence frequency of tumor-specific and tumor-upregulated peptides in patient samples of different cancer types is shown in a dot plot.
To date, many ncRNA translation products have been reported in numerous other studies. To further verify the reliability of our identified ncPEPs, we collected a series of ncPEPs reported in other studies and aligned them against the identified ncPEPs. As a result, 74.71% (22 060/29 526) of the identified ncPEPs were reported in other studies and were marked as validated in SPENCER (Figure 2D). Furthermore, in consideration of the great potential of using ncPEPs as novel neoantigens in tumor immunotherapy, SPENCER has also performed an immunogenicity analysis of the identified ncPEPs. According to the previous pipeline proposed by Well et al. (43), we assessed the immunogenicity of all the identified ncPEPs using three feature scores: MHC-I binding affinity, MHC-I binding stability, and T cell recognition probability. We found that 15.23% (4497/29 526) of the ncPEPs were expected to have immunogenic potential. (Figure 2C, right side). Of these, 3269 small peptides were verified by experimental evidence, accounting for 11.07% of all the ncPEPs (Figure 2D). In addition, to further explore the potential of using these ncPEPs to develop novel cancer vaccines, the fractions of tumor-specific and tumor-upregulated peptides in patients with different cancer types were calculated. Strikingly, we found that 83.78% (24 736/29 526) of the ncPEPs were simultaneously expressed in multiple cancer types. Of these, 24.35% (6022/24 736) were tumor-specific, and 1.69% (418/24 736) were upregulated in cancer. In addition, among these ncPEPs, 77.87% (19 263/24 736) were experimentally validated, and 3603 were predicted to be immunogenic (Figure 2E).
Web interface and usage
SPENCER provides a user-friendly web interface that enables users to search, browse, and download all the cancer-associated ncPEPs in the database. The main features of the web interface are described in more detail in the following sections.
Search
First, SPENCER has developed a ‘QUICK SEARCH’ module on the ‘HOME’ page for providing users with a direct investigation of the curated data. In this module, a variety of options are provided for querying the database, such as cancer type and tissue type. Additionally, detailed information on ncPEPs, including peptide ID, transcript ID, gene ID, gene name and RNAcentral ID, can be searched in this module. In addition, to allow a user to quickly access ncPEPs of interest, SPENCER provides more detailed options on the ‘Search’ page, such as expression group, immunogenicity, status, and ncRNA location. Users can conduct an advanced search to restrict the output by inputting more accurate and detailed search conditions. Furthermore, we annotated ncPEPs with the associated ID in other external resources to provide more comprehensive information on the peptides. Accordingly, users can search by PubMed ID, Ensembl ID, GeneCard ID or another database ID to ascertain whether their interested research focus/ncRNA has translation products in SPENCER (Figure 3A). Moreover, to support sequence similarity searching for either nucleotides or peptides of interest, the BLAST suite is integrated into SPENCER on the ‘BLAST’ page (Figure 3B). A complete set of parameters for BLAST searching are supported for further adjustment.
Figure 3.

Basic functions of the SPENCER web interface. (A) The main patterns of the search interface of SPENCER. (B) The BLAST interface for sequence similarity searching in SPENCER. (C) The browsing interface for ncRNA-encoded small peptides in SPENCER. (D) Results in SPENCER are divided into three levels, including the gene, transcript and peptide levels. (E) The visualization tools in SPENCER.
Browse
All ncPEPs can be surveyed on the ‘BROWSE’ page (Figure 3C). When choosing a particular ‘Cancer’ and ‘Tissues’ catalog, SPENCER will provide a statistical table for the displayed ncPEPs and show histograms presenting the distribution of ncPEPs in different expression groups and RNA types. To reveal ncPEP expression differences between cancer and normal samples, we further constructed a section providing a differential expression overview for each study in SPENCER. This section contains a volcano map for presenting the expression level of all the identified peptides. In addition, an interactive expression heatmap was developed to visualize the detailed expression level of tumor-specific and upregulated peptides in each sample. If users are interested in detailed information on an ncPEP on the heatmap, they can easily access the ncPEP specific page by clicking on each cell in the expression heatmap. Moreover, this page has an interactive result table spread over multiple subpages displaying all ncPEPs in a certain ‘Cancer’ and ‘Tissues’ catalog. SPENCER also provides a filter option box for the results table at the bottom of the page, allowing the user to narrow an extensive set of results. Using this filter option box, users can filter the data based on various criteria, including expression groups, validation status, and immunogenicity.
Detail
To comprehensively display the source and structure of ncPEPs, detailed data in SPENCER are stored at three levels, including the gene, transcript, and peptide levels (Figure 3D). Users can access the relevant detail page for each level by clicking on the associated gene name, associated transcript ID, peptide ID in the browse results. The ‘Gene’ page provides a table of gene information and shows the gene ID, gene name, description, genome location, strand, and cross-reference with the target gene. In addition to the basic information, an overview of gene-associated ncRNA transcripts is provided on the ‘Gene’ page. On the ‘Transcript’ page, SPENCER provides basic information about the transcript and an overview of the peptides involved. Since several studies have shown that RNA secondary structure can affect RNA translation (51), ViennaRNA (48) was applied to visualize ncRNA secondary structures to facilitate further research on mechanisms (Figure 3D and E). In addition, to intuitively display the positions of sORFs and translated peptides in the transcript, a schematic diagram was constructed using IBS (49). Detailed information, including sequence, cancer type, tissue source, expression level, and the dataset from which this result was derived, can be assessed on the ‘Peptide’ page. In particular, to fully understand the MS evidence for the ncPEPs, a spectral visualization module is presented on the ‘Peptide’ page using the Lorikeet spectral annotator. This module shows the ion peaks and peptide sequence fragmentation for each annotated mass spectrum. To promote the study of ncPEPs in cancer immunotherapy, SPENCER displays the scores of three key immunogenicity parameters for each ncPEP under each HLA genotype on the ‘Peptide’ page. Furthermore, all curated data in SPENCER are well referenced, allowing investigators to independently verify findings in greater detail (Supplementary Table S2).
SUMMARY AND PERSPECTIVES
Progress in the research on ncPEPs has provided interesting avenues for deciphering the mysteries of life and may advance further exploration of cancer development (52). Recently, cancer-associated ncPEPs have shown considerable potential as novel targets in the development of antitumor drugs and therapy (53). Identifying and exploring ncPEPs in tumors will benefit studies of cancer pathogenesis, diagnosis, and immunotherapy. Thus, we developed SPENCER, a database of ncPEPs in cancer patients. SPENCER displays the expression profiles of ncPEPs at multiple levels and helps to visualize the expression patterns of small peptides in different cancer types.
At present, SPENCER contains 29 526 small peptides encoded by 6842 ncRNAs in 1,007 tumor patients across 15 types of cancer, and most of the associated sORFs were significantly enriched in lncRNAs (P = 0.0023, OR = 1.5172, hypergeometric test). Most noteworthy is that SPENCER has integrated an immunogenicity prediction pipeline based on the model proposed by Wells et al. (43) to further explore the potential of ncPEPs as targets for cancer immunotherapy. To date, the rules for predicting the immunogenicity of epitopes typically incorporate MHC-I binding affinity as well as filters and ranking criteria obtained from prior knowledge. Since the current immunogenicity prediction focuses solely on MHC-I binding capability, and ignores other mechanisms such as MHC-I binding stability and TCR recognition, false positives may be introduced into the prediction results. To more precisely evaluate the immunogenicity of ncPEPs, our immunogenicity prediction pipeline integrates peptide features associated with MHC-I presentation and TCR recognition. In addition, we implemented strict cutoffs derived from systematic testing across substantial patient samples from a global consortium (43), which could improve the reliability of the immunogenicity predictions. Among the prediction results, some of the tumor-specific peptides were predicted to contain known epitopes that have been validated in previous studies to stimulate an immune response. For instance, the potential epitope in the peptide STDTGVSLPSYEEDQGSK showed a high similarity with a known neoantigen that has been proven by a centralized set of verification experiments, including HLA binding and immunological analyses (43). In addition, two peptides (HVISYSLSPFEQR and MRHVISYSLSPFEQR) specifically expressed in colon cancer were found to contain epitopes that were previously detected as neoantigens in human primary tumors; these epitopes could be used to develop promising tumor vaccines (20). These results implied that the immunogenicity prediction by SPENCER is accurate and reliable for discovering potential tumor neoantigens. By integrating the expression and immunogenicity status of ncPEPs, SPENCER can provide promising targets for cancer immunotherapy research. Based on the curated dataset in SPENCER, we also found that many immunogenic peptides were expressed specifically in cancer tissues. Moreover, the majority were expressed in multiple cancer types and different patient samples. These widely expressed and immunogenic ncPEPs are potential promising actionable targets for tumor vaccines, which would benefit patients with various types of cancer.
In conclusion, SPENCER provides useful information on ncPEPs to help experimental biologists interpret cancer-related ncPEPs and explore molecular mechanisms involving ncRNA in cancer. We will continue to improve SPENCER in the following ways: (i) continuous collection and analysis of newly published MS datasets of different cancer types, (ii) refining of the functional and validated status of peptides and (iii) adding an analysis pipeline for proteomics data to identify ncPEPs and perform immunogenicity evaluations. We believe that with continuous improvement, SPENCER can become an effective tool to analyze the functional roles of ncPEPs, and can contribute to cancer diagnosis and treatment.
DATA AVAILABILITY
SPENCER is a comprehensive online database available at http://spencer.renlab.org.
Supplementary Material
Contributor Information
Xiaotong Luo, School of Life Sciences, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China; State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Yuantai Huang, School of Life Sciences, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China; State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Huiqin Li, School of Life Sciences, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China.
Yihai Luo, School of Life Sciences, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China.
Zhixiang Zuo, State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Jian Ren, School of Life Sciences, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China; State Key Laboratory of Oncology in South China, Cancer Center, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University, Guangzhou 510060, China.
Yubin Xie, School of Life Sciences, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31801105, 81772614, U1611261, 81802438, 31771462]; National Key R&D Program of China [2017YFA0106700]; Program for Guangdong Introducing Innovative and Entrepreneurial Teams [2017ZT07S096]; Guangdong Basic and Applied Basic Research Foundation [2020A1515010220, 2021B1515020108]. Funding for open access charge: National Natural Science Foundation of China [31801105, 81772614, U1611261, 81802438, 31771462]; National Key R&D Program of China [2017YFA0106700]; Program for Guangdong Introducing Innovative and Entrepreneurial Teams [2017ZT07S096]; Guangdong Basic and Applied Basic Research Foundation [2020A1515010220, 2021B1515020108].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dunham I., Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R.et al.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Houseley J., Rubbi L., Grunstein M., Tollervey D., Vogelauer M.. A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol. Cell. 2008; 32:685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li D., Tolleson W.H., Yu D., Chen S., Guo L., Xiao W., Tong W., Ning B.. Regulation of cytochrome P450 expression by microRNAs and long noncoding RNAs: Epigenetic mechanisms in environmental toxicology and carcinogenesis. J. Environ. Sci. Health C. 2019; 37:180–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Landgraf P., Rusu M., Sheridan R., Sewer A., Iovino N., Aravin A., Pfeffer S., Rice A., Kamphorst A.O., Landthaler M.et al.. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007; 129:1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang T., Alvarez A., Hu B., Cheng S.-Y.. Noncoding RNAs in cancer and cancer stem cells. Chin. J. Cancer. 2013; 32:582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang W.-T., Han C., Sun Y.-M., Chen T.-Q., Chen Y.-Q.. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019; 12:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xing J., Liu H., Jiang W., Wang L.. LncRNA-encoded peptide: functions and predicting methods. Front. Oncol. 2021; 10:3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J., Zhu S., Meng N., He Y., Lu R., Yan G.-R.. ncRNA-encoded peptides or proteins and cancer. Mol. Ther. 2019; 27:1718–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang J.-Z., Chen M., Chen D., Gao X.-C., Zhu S., Huang H., Hu M., Zhu H., Yan G.-R.. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol. Cell. 2017; 68:171–184. [DOI] [PubMed] [Google Scholar]
- 10. Polycarpou-Schwarz M., Groß M., Mestdagh P., Schott J., Grund S.E., Hildenbrand C., Rom J., Aulmann S., Sinn H.-P., Vandesompele J.et al.. The cancer-associated microprotein CASIMO1 controls cell proliferation and interacts with squalene epoxidase modulating lipid droplet formation. Oncogene. 2018; 37:4750–4768. [DOI] [PubMed] [Google Scholar]
- 11. Zheng X., Chen L., Zhou Y., Wang Q., Zheng Z., Xu B., Wu C., Zhou Q., Hu W., Wu C.et al.. A novel protein encoded by a circular RNA circPPP1R12A promotes tumor pathogenesis and metastasis of colon cancer via Hippo-YAP signaling. Mol. Cancer. 2019; 18:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farkona S., Diamandis E.P., Blasutig I.M.. Cancer immunotherapy: the beginning of the end of cancer. BMC Med. 2016; 14:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bethune M.T., Li X.-H., Yu J., McLaughlin J., Cheng D., Mathis C., Moreno B.H., Woods K., Knights A.J., Garcia-Diaz A.et al.. Isolation and characterization of NY-ESO-1–specific T cell receptors restricted on various MHC molecules. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang R.-F., Wang H.Y.. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017; 27:11–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peng M., Mo Y., Wang Y., Wu P., Zhang Y., Xiong F., Guo C., Wu X., Li Y., Li X.et al.. Neoantigen vaccine: an emerging tumor immunotherapy. Mol. Cancer. 2019; 18:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan C.-M., Wang J.-P., Tang Y.-Y., Zhao J., He S.-Y., Xiong F., Guo C., Xiang B., Zhou M., Li X.-L.et al.. circMAN1A2 could serve as a novel serum biomarker for malignant tumors. Cancer Sci. 2019; 110:2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Charpentier M., Croyal M., Carbonnelle D., Fortun A., Florenceau L., Rabu C., Krempf M., Labarrière N., Lang F.. IRES-dependent translation of the long non coding RNA meloe in melanoma cells produces the most immunogenic MELOE antigens. Oncotarget. 2016; 7:59704–59713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carbonnelle D., Vignard V., Sehedic D., Moreau-Aubry A., Florenceau L., Charpentier M., Mikulits W., Labarriere N., Lang F.. The melanoma antigens MELOE-1 and MELOE-2 are translated from a bona fide polycistronic mRNA containing functional IRES sequences. PLoS One. 2013; 8:e75233–e75233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Godet Y., Desfrançois J., Vignard V., Schadendorf D., Khammari A., Dreno B., Jotereau F., Labarrière N.. Frequent occurrence of high affinity T cells against MELOE-1 makes this antigen an attractive target for melanoma immunotherapy. Eur. J. Immunol. 2010; 40:1786–1794. [DOI] [PubMed] [Google Scholar]
- 20. Laumont C.M., Vincent K., Hesnard L., Audemard É., Bonneil É., Laverdure J.-P., Gendron P., Courcelles M., Hardy M.-P., Côté C.et al.. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018; 10:eaau5516. [DOI] [PubMed] [Google Scholar]
- 21. Liu H., Zhou X., Yuan M., Zhou S., Huang Y.E., Hou F., Song X., Wang L., Jiang W.. ncEP: a manually curated database for experimentally validated ncRNA-encoded proteins or peptides. J. Mol. Biol. 2020; 432:3364–3368. [DOI] [PubMed] [Google Scholar]
- 22. Dragomir M.P., Manyam G.C., Ott L.F., Berland L., Knutsen E., Ivan C., Lipovich L., Broom B.M., Calin G.A.. FuncPEP: a database of functional peptides encoded by Non-Coding RNAs. Non-coding RNA. 2020; 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hazarika R.R., De Coninck B., Yamamoto L.R., Martin L.R., Cammue B.P., van Noort V.. ARA-PEPs: a repository of putative sORF-encoded peptides in Arabidopsis thaliana. BMC Bioinformatics. 2017; 18:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang Y., Wang J., Zhao Y., Wang H., Liu T., Li Y., Cui T., Li W., Feng Y., Luo J.et al.. cncRNAdb: a manually curated resource of experimentally supported RNAs with both protein-coding and noncoding function. Nucleic Acids Res. 2021; 49:D65–D70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olexiouk V., Van Criekinge W., Menschaert G.. An update on sORFs.org: a repository of small ORFs identified by ribosome profiling. Nucleic Acids Res. 2018; 46:D497–D502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hao Y., Zhang L., Niu Y., Cai T., Luo J., He S., Zhang B., Zhang D., Qin Y., Yang F.et al.. SmProt: a database of small proteins encoded by annotated coding and non-coding RNA loci. Brief. Bioinform. 2018; 19:636–643. [DOI] [PubMed] [Google Scholar]
- 27. Choteau S.A., Wagner A., Pierre P., Spinelli L., Brun C.. MetamORF: a repository of unique short open reading frames identified by both experimental and computational approaches for gene and metagene analyses. Database. 2021; 2021:baab032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen Y., Li D., Fan W., Zheng X., Zhou Y., Ye H., Liang X., Du W., Zhou Y., Wang K.. PsORF: a database of small ORFs in plants. Plant Biotechnol. J. 2020; 18:2158–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tyanova S., Temu T., Cox J.. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016; 11:2301–2319. [DOI] [PubMed] [Google Scholar]
- 30. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M.et al.. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019; 47:D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi M., Carver J., Chiva C., Tzouros M., Huang T., Tsai T.H., Pullman B., Bernhardt O.M., Hüttenhain R., Teo G.C.et al.. MassIVE.quant: a community resource of quantitative mass spectrometry-based proteomics datasets. Nat. Methods. 2020; 17:981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moriya Y., Kawano S., Okuda S., Watanabe Y., Matsumoto M., Takami T., Kobayashi D., Yamanouchi Y., Araki N., Yoshizawa A.C.et al.. The jPOST environment: an integrated proteomics data repository and database. Nucleic Acids Res. 2019; 47:D1218–D1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sharma V., Eckels J., Taylor G.K., Shulman N.J., Stergachis A.B., Joyner S.A., Yan P., Whiteaker J.R., Halusa G.N., Schilling B.et al.. Panorama: a targeted proteomics knowledge base. J. Proteome Res. 2014; 13:4205–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schwenk J.M., Omenn G.S., Sun Z., Campbell D.S., Baker M.S., Overall C.M., Aebersold R., Moritz R.L., Deutsch E.W.. The human plasma proteome draft of 2017: Building on the human plasma peptideatlas from mass spectrometry and complementary assays. J. Proteome Res. 2017; 16:4299–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma J., Chen T., Wu S., Yang C., Bai M., Shu K., Li K., Zhang G., Jin Z., He F.et al.. iProX: an integrated proteome resource. Nucleic Acids Res. 2019; 47:D1211–D1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. The Rnacentral Consortium. RNAcentral: a hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019; 47:D221–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sayers E.W., Barrett T., Benson D.A., Bolton E., Bryant S.H., Canese K., Chetvernin V., Church D.M., Dicuccio M., Federhen S.et al.. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2010; 38:D5–D16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. UniProt, T., C. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M.. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics : MCP. 2014; 13:2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W.. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gruber A.R., Lorenz R., Bernhart S.H., Neuböck R., Hofacker I.L.. The Vienna RNA websuite. Nucleic Acids Res. 2008; 36:W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schreiber R.D., Old L.J., Smyth M.J.. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011; 331:1565–1570. [DOI] [PubMed] [Google Scholar]
- 43. Wells D.K., van Buuren M.M., Dang K.K., Hubbard-Lucey V.M., Sheehan K.C.F., Campbell K.M., Lamb A., Ward J.P., Sidney J., Blazquez A.B.et al.. Key parameters of tumor epitope immunogenicity revealed through a consortium approach improve neoantigen prediction. Cell. 2020; 183:818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jurtz V., Paul S., Andreatta M., Marcatili P., Peters B., Nielsen M.. NetMHCpan-4.0: improved peptide–MHC Class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol. 2017; 199:3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang M., Claesson M.H.. Classification of human leukocyte antigen (HLA) supertypes. Methods Mol. Biol. 2014; 1184:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rasmussen M., Fenoy E., Harndahl M., Kristensen A.B., Nielsen I.K., Nielsen M., Buus S.. Pan-specific prediction of peptide-MHC Class I complex stability, a correlate of T cell immunogenicity. J. Immunol. 2016; 197:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Łuksza M., Riaz N., Makarov V., Balachandran V.P., Hellmann M.D., Solovyov A., Rizvi N.A., Merghoub T., Levine A.J., Chan T.A.et al.. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017; 551:517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gruber A.R., Bernhart S.H., Lorenz R.. The ViennaRNA web services. Methods Mol. Biol. 2015; 1269:307–326. [DOI] [PubMed] [Google Scholar]
- 49. Liu W., Xie Y., Ma J., Luo X., Nie P., Zuo Z., Lahrmann U., Zhao Q., Zheng Y., Zhao Y.et al.. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics. 2015; 31:3359–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verheggen K., Volders P.J., Mestdagh P., Menschaert G., Van Damme P., Gevaert K., Martens L., Vandesompele J.. Noncoding after all: Biases in proteomics data do not explain observed absence of lncRNA translation products. J. Proteome Res. 2017; 16:2508–2515. [DOI] [PubMed] [Google Scholar]
- 51. Kawaguchi D., Shimizu S., Abe N., Hashiya F., Tomoike F., Kimura Y., Abe H.. Translational control by secondary-structure formation in mRNA in a eukaryotic system. Nucleosides Nucleotides Nucleic Acids. 2020; 39:195–203. [DOI] [PubMed] [Google Scholar]
- 52. Wang S., Mao C., Liu S.. Peptides encoded by noncoding genes: challenges and perspectives. Signal Transduct. Targeted Ther. 2019; 4:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu S., Wang J., He Y., Meng N., Yan G.-R.. Peptides/proteins encoded by non-coding RNA: a novel resource bank for drug targets and biomarkers. Front. Pharmacol. 2018; 9:1295–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SPENCER is a comprehensive online database available at http://spencer.renlab.org.