

Abstract
Background
Campylobacter is an animal and zoonotic pathogen of global importance, and a pressing need exists for effective vaccines, including those that make use of conserved polysaccharide antigens. To this end, we adapted Protein Glycan Coupling Technology (PGCT) to develop a versatile Escherichia coli strain capable of generating multiple glycoconjugate vaccine candidates against Campylobacter jejuni.
Results
We generated a glycoengineering E. coli strain containing the conserved C. jejuni heptasaccharide coding region integrated in its chromosome as a model glycan. This methodology confers three advantages: (i) reduction of plasmids and antibiotic markers used for PGCT, (ii) swift generation of many glycan-protein combinations and consequent rapid identification of the most antigenic proteins or peptides, and (iii) increased genetic stability of the polysaccharide coding-region. In this study, by using the model glycan expressing strain, we were able to test proteins from C. jejuni, Pseudomonas aeruginosa (both Gram-negative), and Clostridium perfringens (Gram-positive) as acceptors. Using this pgl integrant E. coli strain, four glycoconjugates were readily generated. Two glycoconjugates, where both protein and glycan are from C. jejuni (double-hit vaccines), and two glycoconjugates, where the glycan antigen is conjugated to a detoxified toxin from a different pathogen (single-hit vaccines). Because the downstream application of Live Attenuated Vaccine Strains (LAVS) against C. jejuni is to be used in poultry, which have a higher body temperature of 42 °C, we investigated the effect of temperature on protein expression and glycosylation in the E. coli pgl integrant strain.
Conclusions
We determined that glycosylation is temperature dependent and that for the combination of heptasaccharide and carriers used in this study, the level of PglB available for glycosylation is a step limiting factor in the glycosylation reaction. We also demonstrated that temperature affects the ability of PglB to glycosylate its substrates in an in vitro glycosylation assay independent of its transcriptional level.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12934-021-01728-7.
Keywords: Biological conjugation, PglB, Temperature, PGCT, One health, Poultry, Vaccine
Background
Protein Glycan Coupling Technology (PGCT) exploits genetically modified bacteria to produce glycoconjugates, in essence transforming bacterial cells into vaccine production factories [1]. The technology obviates the need to handle pathogens and eliminates expensive and time-consuming purification steps as well as simplifying glycan-acceptor protein coupling [2]. Its conception stemmed from the discovery that protein glycosylation is present in all domains of life [3, 4] and is the most common post-translational modification of proteins. In the late 1990s an N-linked protein glycosylation locus (pgl) was identified in the bacterium C. jejuni [5]. This was followed by the functional transfer of the locus into a laboratory strain of E. coli with the first glycoprotein generated heterologously [6]. Subsequent transfer of the pgl locus demonstrated that in E. coli, the oligosaccharyltransferase (OST) PglB could transfer different polysaccharide structures to an acceptor protein [7]. Concomitantly to these findings, it was demonstrated that PglB preferred a consensus sequence (sequon) D/E-X1-N-X2-S/T, (where X1 and X2 cannot be a proline) [8]. The discovery of the consensus sequon combined with the relaxed specificity of PglB towards different polysaccharide regions has contributed to burgeoning advancements in the field of glycoengineering. In 2011 Fisher et al., demonstrated that the addition of a sequence consensus ‘glycotag’ at either the N-or C-termini of a protein was sufficient for glycosylation [9]. This discovery meant that double-hit approach vaccines (where the protein and glycan are from the same pathogen) are a possibility, as any protein can become an acceptor. Furthermore, it allows new glycoconjugate vaccines to overcome the use of the already overly utilised CRM197, tetanus toxoid (TT) and Haemophilus influenzae protein D [10] carrier proteins. The use of new carrier proteins contributes towards avoiding possible interference between different vaccines [11], carrier-induced immune suppression [12], as well as increasing vaccine specificity and coverage.
Despite recent technical developments, the need to improve PGCT as an alternative glycoconjugate vaccine production platform remains. In this work, we generated a versatile glycoengineering strain (E. coli SDB1pgl) for the rapid production of glycoconjugates to be used recombinantly as well as to investigate the usefulness of such strains as part of a live attenuated vaccine strategy. The E. coli SDB1pgl was used to investigate glycosylation in vivo in a bid to understand the lack of protection observed in our previous study where poultry was vaccinated using avian pathogenic Escherichia coli (APEC) strain Chi7122pgl [13]. By creating a strain that can chromosomally express the conserved Campylobacter heptasaccharide glycan we introduced greater flexibility to the PGCT system, enabling the rapid testing of preferred carrier proteins, obviating the need for antibiotics for plasmid maintenance, minimising the possible spread of plasmids and increasing stability of the polysaccharide coding region as well as contributing to greater efficiency by decreasing the metabolic burden to the E. coli host [14–17]. Here we show that with appropriate modifications, Gram-positive or Gram-negative bacterial proteins could be modified with C. jejuni glycan in E. coli SDB1 cells [18]. The pgl locus is an optimal starting point as it contains both the necessary genes for the assembly of the C. jejuni heptasaccharide as well as the PglB OST. We demonstrated that by using an E. coli pgl integrant strain, we can rapidly generate different recombinant glycoconjugates as well as potentially produce glycoconjugates within the bacteria that, after the introduction of mutations to reduce toxicity, could be used as a delivery method for vaccines. Due to results we have observed previously, where no protection was seen against C. jejuni when using Chi7122pgl, we set out to determine factors that affect glycosylation when the pgl locus is inserted in the chromosome. As the live attenuated vaccines (LAVs) created in our previous study [13] were intended to be used in poultry which have a higher body temperature than humans or other farm animals [19], we investigated the effect of temperature on protein expression and glycosylation. An unequivocal relationship between temperature and glycosylation efficiency was demonstrated within bacterial cells as well as when carrying out the reaction using an in vitro glycosylation assay. This alteration in glycoconjugate production was shown to be independent of gene expression at the mRNA level. Finally, we determined that the addition of extra copies of pglB per cell improves glycosylation efficiency under the conditions tested.
Results
Generation of the SDB1pgl integrant
To establish a flexible system for the rapid generation of glycoconjugate vaccines against Campylobacter, we decided to insert the C. jejuni pgl locus into the chromosome of the E. coli SDB1 strain via allelic exchange. E. coli SDB1 [18] is a W3110-derivative strain lacking the O-antigen with the additional deletion of waaL and wecA genes encoding the O-antigen ligase and the initiating glycosyltransferase involved in O-antigen and enterobacterial common antigen (ECA) biosynthesis, respectively. This glycoengineering strain was chosen to maximise the amount of glycan substrate available for PglB (owing to the absence of competition with the WaaL ligase) and to ensure the faithful synthesis of C. jejuni heptasaccharide starting with the correct reducing end glycan, bacillosamine, transferred by the PglC initiating transferase instead of a mixture of GlcNAc-starting glycans initiated by the E. coli endogenous initiating transferase WecA.
To achieve chromosomal integration of the pgl locus, we used the suicide vector, pSECpgl [13] based on the pCVD442 plasmid [20] containing a pir-dependent R6K replicon and the sacB gene of Bacillus subtilis conferring sucrose sensitivity, which enables positive selection of integrants for the loss of backbone sequence. pSECpgl contains the pgl locus from C. jejuni 81116 C-terminally tagged with a kanamycin resistance (KanR) cassette and surrounded by homology arms for insertion between E. coli gidB and atpI genes (Fig. 1A).
Fig. 1.

Generation of functional SDB1pgl integrants. A Schematic diagram depicting the pSECpgl suicide plasmid. B Strategy for integration of the C. jejuni pgl locus in the SDB1 genome via allelic exchange. C Confirmation of correct integration of the pgl locus via PCR in five SDB1pgl clones. Oligos 1 and 6 align outside of the chosen homology arms, while oligos 2 and 5 align to the pgl locus and the KanR tag, respectively. Formation of amplicons 1&2 and 5&6 is possible only in case of successful integration in the correct location. D SDB1pgl integrants can glycosylate a model carrier protein (G-NetB). SDB1 transformed with a plasmid encoding pgl locus (pACYCpgl) as well as a plasmid encoding pgl locus with a non-functional PglB (pACYCpgl::pglB) were used as positive and negative controls, respectively. His-pulldown followed by western blotting confirms glycoprotein production
pSECpgl was delivered to recipient SDB1 cells via overnight conjugation with DAP-dependent MFD donor cells (Fig. 1B). Integrants were identified by growth on selective agar and confirmed by PCR. A successful first recombination event was identified by kanamycin-resistant and DAP-sensitive conjugants that were positive for a PCR amplifying the middle of the pgl locus with oligonucleotides 3&4 resulting in a ∼1.5 kb amplicon (step 3 in Fig. 1B).
To test whether the SDB1pgl integrants possessed a functional pgl locus, two clones were transformed with an IPTG-inducible plasmid encoding a hyperglycosylatable version of C. perfringens necrotic enteritis toxin (G-NetB). Both clones of SDB1pgl integrants tested were functional as they clearly glycosylated G-NetB (Fig. 1D). Six bands (green) corresponding to G-NetB glycosylated at one to six sites are visible by fluorescent western blotting when stained with SBA lectin, which recognises the terminal GalNAc residue of the C. jejuni heptasaccharide. In this specific example, the integrants outperformed the very same strain transformed with a plasmid encoding the pgl locus (pACYCpgl, ∼10–12 copies per cell), suggesting that, at least in this case, a single chromosomal copy of pgl in concert with the expression of G-NetB from a pEXT20 backbone provides more efficient glycoconjugate production. No glycosylation could be observed when SDB1 was transformed with a plasmid encoding a version of the pgl with a mutation in pglB (pACYCpgl::pglB), which served as a negative control.
Generation of vaccine candidates using SDB1pgl and SDB1pACYCpgl host strains
Most proteins are not natural substrates for the OST PglB, however with modifications a protein can be decorated with a PglB-compatible polysaccharide (Fig. 2). For this purpose, we modified G-NetB, G-ExoA, G-SodB and G-FlpA by adding glycotags, a signal peptide, either PelB or DsbA, for localisation to the periplasmic compartment and a polyhistidine tag for affinity purification of the glycoconjugates. All protein modifications are described in “Materials and methods” and are summarised in Fig. 2.
Fig. 2.

Depiction of the modifications performed in the carrier proteins to become suitable PglB substrates (addition of different numbers of non-natural glycotags (2, 8 and 10), PelB or DsbA leaders for co-localisation in the periplasm, and poly-histidine tag for purification as well as detoxifying mutations on both G-ExoA and G-NetB)
Comparison of glycosylation of carrier proteins between SDB1pgl and SDB1 pACYCpgl at 30 °C and 37 °C
The need to understand how different factors such as temperature, media and co-factors affect glycosylation and to determine optimal conditions to standardise PGCT has been an imperative goal. However, glycosylation variability has been observed and reported, depending on the glycan or the carrier protein used. To determine the effects that different copy numbers of the pgl glycosylation locus and different temperatures have on glycoconjugate formation, we compared the newly generated E. coli SDB1pgl strain with a single chromosomal copy of the pgl locus, to an E. coli SDB1 expressing the pgl locus from plasmid pACYCpgl (SDB1 pACYCpgl∼10–12 copies per cell) and assessed qualitatively the variability of glycosylation of carriers G-SodB, G-FlpA, G-NetB and G-ExoA at 30 °C and 37 °C (Fig. 3).
Fig. 3.

Comparison of glycosylation of four different carriers NetB and G-ExoA (A) and G-SodB, G-FlpA (B), by SDB1pgl, which expresses the C. jejuni heptasaccharide from its chromosome and SDB1 pACYCpgl, which expresses the C. jejuni heptasaccharide from a pACYC184-based plasmid, at two different temperatures (30 °C and 37 °C). In red is the anti-His signal and in green the anti-glycan signal
We observed that for three of the four proteins tested (G-FlpA, G-ExoA and G-SodB) glycosylation was evidently higher with the pgl locus residing on a plasmid (Fig. 3). Protein production for G-FlpA was similar for SDB1pgl and for SDB1 pACYCpgl at 30 °C (Table 1) however, its expression was markedly different at 37 °C (SDB1pgl_pEXT20-FlpA 37 °C—1.09 mg/ml and SDB1 pACYCpgl_pEXT20-FlpA 0.41 mg/ml). Even though, protein quantity produced in the E. coli cells containing the pgl locus in the chromosome (SDB1pgl) was 2.65 times higher than the protein produced in the E. coli cells expressing the pgl locus from the plasmid, the glycosylation level was markedly different, with only one sequon occupied in SDB1pgl_pEXT20-FlpA at 37 °C as opposed to two in SDB1pACYCpgl_pEXT20-FlpA at the same temperature.
Table 1.
Acceptor proteins quantities produced in the same E. coli SDB1 background but with the pgl locus in plasmid (SDB1 pACYCpgl) or inserted in the chromosome of SDB1 (SDB1pgl) at 30 °C and 37 °C (X indicates ORF inserted coding for G-SodB/G-FlpA/G-NetB/G-ExoA)
| Temperature (°C) | E. coli background | Acceptor proteins | |||
|---|---|---|---|---|---|
| G-SodB | G-FlpA | G-NetB | G-ExoA | ||
| 30 | SDB1 pACYCpgl pEXT20_X | 0.4 | 0.92 | 0.40 | 1.79 |
| SDB1pgl pEXT20_X | 0.24 | 0.90 | 0.28 | 0.83 | |
| 37 | SDB1 pACYCpgl pEXT20_X | 0.54 | 0.41 | 0.28 | 0.71 |
| SDB1pgl pEXT20_X | 0.45 | 1.09 | 0.28 | 0.55 | |
G-SodB was expressed similarly at both temperatures and by both bacterial hosts (Table 1). However, glycosylation was visibly better for the plasmid-based system suggesting that protein quantity is not directly related to glycosylation efficiency (Fig. 3). There was greater protein production for SDB1pACYCpgl_pMAD-G-ExoA than SDB1pgl_pMAD-G-ExoA at both temperatures (Table 1). There was a clear difference on the glycosylation pattern observed for SDB1pACYCpgl_pMAD-G-ExoA when compared with SDB1pgl_pMAD-G-ExoA. Glycosylation was clearly more efficient when the pgl locus was in a plasmid (SDB1pACYCpgl_pMAD-G-ExoA) as opposed to inserted in the chromosome (SDB1pgl_pMAD-G-ExoA).
The fourth protein tested G-NetB was unique in its phenotype since it was the only one modified more efficiently when the pgl locus was inserted in the chromosome. At 30 °C, six out of its ten sequons were glycosylated as opposed to two at 37 °C. Interestingly there were small differences in protein quantities produced using the different hosts, SDB1pACYCpgl_pEXT20-G-NetB (0.40 mg/ml) and SDB1pgl_pEXT20-G-NetB (0.28 mg/ml) at 30 °C (Table 1). SDB1pACYCpgl_pEXT20-G-NetB and SDB1pgl_pEXT20-G-NetB both produced same quantity of protein at 37 °C (0.28 mg/ml) (Table 1). In this instance there were fine differences between protein quantities, however SDB1pACYCpgl pEXT20-G-NetB produced a higher amount of protein at 30 °C and this did not correlate positively with more efficient glycosylation (Fig. 3, panel A). The glycosylation deficiency observed for three out of the four proteins tested in the SDB1pgl strains led to the hypothesis that PglB might be limiting the glycosylation reaction since pglB was being expressed constitutively from a single genomic copy as opposed to the expected 10–12 copies per bacterial cell from the pACYCpgl plasmid.
Generation of glycoconjugates using SDB1pgl with additional inducible PglB
To test if PglB and not the available heptasaccharide was the limiting factor on three out of four of the glycosylation reactions using the chromosomally inserted pgl locus, an additional IPTG inducible plasmid-borne pglB was electroporated into strains SDB1pgl pEXT20-SodB, SDB1pgl pEXT20-FlpA, SDB1pgl_pEXT20-G-NetB and SDB1pgl_pG-ExoA generating strains SDB1pgl_pEXT21_pglB_pEXT20-SodB, SDB1pgl_pEXT21pglB_pEXT20-FlpA, SDB1pgl_pEXT21_pglB_pEXT20-G-NetB and SDB1pgl_pEXT21pglB_p_G-ExoA. The plasmid backbone encoding PglB is pEXT21, which has an estimated copy number of ∼3 per cell [23], thereby increasing the total pglB copy number to 4 (one constitutively expressed from the endogenous promoter of the pgl locus and three from a Ptac IPTG-inducible promoter). Glycosylation efficiency was tested in these strains at 30 °C, the temperature previously identified as most favourable for glycosylation. The glycosylation efficiency improved considerably for carriers G-SodB (lane 1 and lane 5), where it is clearly visible that both sequons are occupied, as well as for G-NetB where all 10 sequons are occupied (lane 7), i.e., 3 more than without the extra inducible PglB (Fig. 4, lane 3). A ladder pattern as well as more intense bands could be observed for G-ExoA when we probed with SBA lectin (Fig. 4, lane 8), which is an improvement from the result obtained without the addition of the extra inducible PglB, lane 4). Furthermore, with the addition of inducible PglB we could observe an increase in glycosylation of FlpA with both sequons occupied (lane 5) as opposed to just one (lane 2). These observations led to the conclusion that efficient glycosylation of the protein carriers tested need more than one functional copy of pglB per cell, suggesting that once the polysaccharide coding region is integrated in the chromosome, pglB can be a limiting factor for the glycosylation reaction for these acceptors and not glycan production.
Fig. 4.

Comparison between glycosylation of carrier proteins at 30 °C in the presence and absence of additional inducible PglB. Glycosylation without additional inducible PglB of carriers G-SodB (lane 1), G-FlpA (lane2), G-NetB (lane 3) and G-ExoA (lane 4), followed by the effect of addition of inducible PglB in glycosylation of the same carriers at 30 °C, G-SodB (lane 5), G-FlpA (lane 6), G-NetB (lane 7) and G-ExoA (lane 8). 2 µg of each protein was loaded per well. In red is the anti-His and in green the anti-SBA signals
Glycosylation of all four carriers at 42 °C using SDB1pgl and SDB1pgl_pEXT21pglB
The idea of using bacterial cells as a vector for glycoconjugate delivery is a concept that is gaining traction in the veterinary field, particularly if the bacterial cells producing the vaccine can be added to the water or the feed of the target animal for ease of administration at scale.
Due to the glycosylation temperature dependency that we report in this manuscript, and the fact that we have been working towards a C. jejuni vaccine for chickens to reduce downstream campylobacteriosis in humans, we decided to investigate glycosylation efficiency at 42 °C, the approximate body temperature of chickens (Fig. 5). We observed a substantial decrease in glycosylation at 42 °C even when plasmid-borne inducible pglB was added when compared to glycosylation at 30 °C (Fig. 4). However, the effect of adding inducible PglB was still visible (Fig. 5, lanes 5, 6, 7 and 8) when compared to the chromosomally integrated pgl locus (SDB1pgl) only (Fig. 5, lanes 1 to 4). Interestingly, G-FlpA was the only protein to register an improvement in its glycosylation pattern. At this temperature and with the extra inducible PglB, both sequons were occupied (Fig. 5, lane 6).
Fig. 5.

Glycosylation of carrier proteins at 42 °C in SDB1pgl and SDB1pgl _pEXT21pglB backgrounds. Lanes, 5, 6, 7 and 8 show the increase in glycosylation potentiated by the addition of extra inducible PglB. Lanes 1, 2, 3 and 4 show lack of detectable glycosylation at 42 °C in SDB1pgl background. In red is the anti-His signal and in green the anti-glycan signal
pglB expression at 30 °C, 37 °C and 42 °C
To understand if the decrease in glycosylation observed at higher temperatures was a consequence of altered gene expression, transcription of pglB was analysed. Using quantitative RT-PCR and the Δ∆Ct [22] method we assessed the transcription of pglB compared to the housekeeping genes gapA and rpoS at the same three temperatures. Transcription of pglB at 30 °C was determined as the control condition since this is the temperature where the highest glycosylation of the carriers is observed. We observed an increase in the transcription of pglB at both 37 °C and 42 °C, of approximately sixfold and twofold (n = 9) respectively. However, this apparent increase in transcription of pglB does not appear to correlate with higher glycosylation of the carrier proteins (Fig. 6).
Fig. 6.
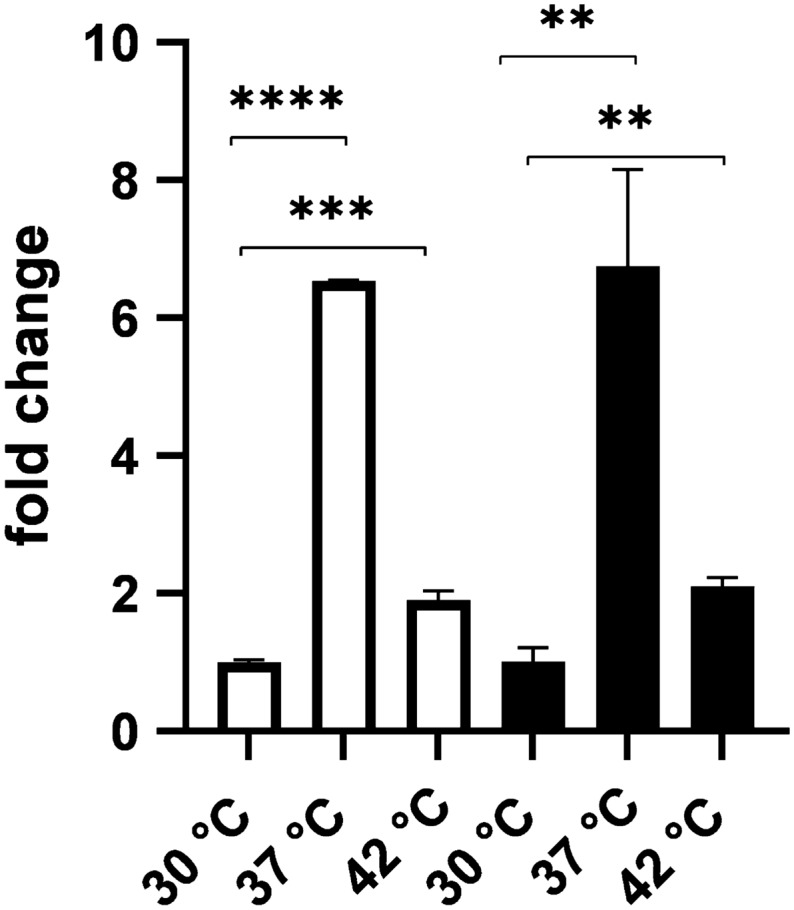
Transcription of pglB in SDB1pgl grown at 30 °C, 37 °C and 42 °C. In white pglB expression in SDB1pgl when compared to housekeeping gene gapA. In black pglB expression in SDB1pgl when compared to the housekeeping gene rpoS. Data was obtained using 3 biological replicates each with 3 technical replicates for each temperature (n = 9), p < 0.001
In vitro glycosylation at 30 °C, 37 °C and 42 °C
To try and understand the decrease in glycosylation at higher temperatures despite the increase in pglB expression, an in vitro glycosylation reaction was performed. For this assay all bacterial cell extracts were prepared at 30 °C, all the SDB1pEXT21pglB expressing E. coli was prepared in a single batch culture, ensuring the same level of PglB per reaction. The incubation of the reaction was then performed at 30 °C, 37 °C and 42 °C. In vitro glycosylation reactions had varied degrees of success depending on the carrier protein used. Using carrier protein FlpA we were able to determine a clear effect of temperature on the function of PglB. It was clear that the reactions at the higher temperatures of 37 °C and 42 °C had significantly less glycan being detected by SBA lectin (Fig. 7, lanes 6 and 9). G-NetB degraded when incubated in the in vitro reaction conditions while G-SodB presented a similar phenotype to FlpA, with glycosylation decreasing as the reaction temperature increases (Additional file 1: Fig. S1). G-ExoA was modified with the heptasaccharide to a very low level at 30 °C and below the detection level at the higher temperatures (Additional file 1: Fig. S2).
Fig. 7.

In vitro glycosylation reaction using carrier protein G-FlpA. L-Protein ladder PageRuler plus (Bioline, UK) Lane 1, 4 and 7—negative control, G-FlpA, no glycan donor incubated at 30 °C, 37 °C and 42 °C respectively; lane 2,5 and 8—G-FlpA combined with glycan donor from SDB1pACYCpgl and SDB1pEXT21pglB, incubated at 30 °C, 37 °C and 42 °C respectively; lane 3, 6 and 9—G-FlpA combined with glycan donor from SDB1pgl and SDB1pEXT21pglB incubated at 30 °C, 37 °C and 42 °C respectively
Discussion
Understanding glycosylation variability is central for increasing the applicability of PGCT for vaccine development. Currently, most glycoconjugates generated using PGCT, also referred to as biological conjugation, use a plasmid-based system [23–31]. Despite it being successful when the goal is to generate recombinant glycoconjugates, reducing the number of plasmids in the system and increasing genetic stability of PGCT elements is desirable for vaccine process development. There are clear advantages to have the polysaccharide coding region integrated in the bacterial chromosome. Firstly, issues regarding plasmid incompatibilities and plasmid stability are avoided. Secondly, chromosomal integration of the glycan-encoding locus reduces the need for antibiotic use in the cultures to maintain extrachromosomal copies of plasmids. Importantly, flexibility is introduced in the system. By keeping the polysaccharide region constant one can readily assess multiple carrier proteins in terms of glycosylation efficiency and yield, overall enabling the more rapid production and testing of candidate vaccines in either a recombinant form or to be used as a future LAV. Introduction of large polysaccharide coding regions in the chromosome of bacteria has been shown as enabling stable and metabolically efficient glycan production [14, 15, 32]. Furthermore, having multiple copies of two or three plasmids performing complex metabolic functions can elevate the metabolic burden arresting cell growth [13, 33], having a deleterious effect on glycoconjugate production.
In this study we aimed to compare the expression and glycosylation of G-SodB, G-FlpA, G-NetB and G-ExoA when the polysaccharide is expressed from the chromosome versus expression of the same polysaccharide from plasmid pACYCpgl at different temperatures. Despite the metabolic burden resulting from expressing the entire pgl locus in the medium copy plasmid pACYCpgl (10–12 copies), three out of four of the protein carriers tested exhibit better glycosylation in this combination (G-SodB, G-FlpA, G-ExoA). We did observe, however, that for both methods a lower culture temperature (30 °C) promoted more efficient glycosylation for three out of the four acceptors tested (G-SodB, G-NetB and G-ExoA). Interestingly, it was only G-FlpA, a C. jejuni native protein, that showed better glycosylation at 37 °C with the plasmid system, even if the protein level produced was approximately 3 times inferior to the protein produced by SDB1pgl. In this case a higher protein yield did not correlate with higher glycosylation, which could even indicate a potential feedback inhibitory effect. There was also variability of sequon occupation. It was noted that both sequons added to G-FlpA were occupied when the protein was glycosylated in the presence of more than one copy of pglB at 30 °C, 37 °C and 42 °C. On the other hand, for protein G-NetB the opposite was true, higher temperature led to lower sequon occupancy in SDB1pgl. No noticeable glycosylation was observed when G-NetB was in the presence of multiple copies of the C. jejuni heptasaccharide. Therefore, for this carrier protein, extra availability of donor glycan and OST did not positively correlate with higher yield of glycosylated protein. Furthermore, we concluded based on the G-NetB result that using the plasmid system for screening for glycosylated proteins may not always be suitable for highly and efficiently glycosylated proteins. Why such pronounced effects were observed for G-FlpA and G-NetB needs further investigation. However, one can speculate that it could be related to the coupling rate of PglB/ carrier/ glycan and the availability of substrates at a given time. Low glycosylation efficiency in SDB1pgl was intriguing and led us to hypothesise that one copy of PglB per cell may not be sufficient for efficient glycosylation. We tested our hypothesis by adding an inducible low copy plasmid (3 copies per cell ± 0.3) [23] expressing PglB to SDB1pgl. We observed a substantial increase on occupation of sequons and therefore glycosylation, on all carrier proteins tested at 30 °C. The addition of only extra pglB copy but not all the other necessary components for the expression of the C. jejuni heptasaccharide, indicated that enough C. jejuni heptasaccharide was being produced in SDB1pgl, but that one pglB copy per cell was insufficient to efficiently transfer it across to the different acceptor proteins.
Because of our interest in applying PGCT to LAVS and of the temperature dependence of glycosylation reported in this manuscript, we have also assessed glycosylation of all four carriers at 42 °C, as it is the reported avian body temperature [19]. We have observed that glycosylation still happens at 42 °C but it is not efficient (Fig. 5). To ensure that the decrease seen in glycosylation was not due to a decrease in pglB expression we investigated the fold-change of pglB expression at 30 °C, 37 °C and 42 °C and observed a fold-increase of approximately 6 and 2 respectively. However, an increase in the expression of pglB did not result in higher glycosylation of the carrier proteins. These results were followed by the investigation of the effect of the temperature in the function of PglB by using a modified version of the in vitro glycosylation reaction described by Jaroentmeechai et al., [21]. In this instance, and due to the reaction conditions, different levels of glycosylation success were observed for the protein carriers studied. It is important to understand that this assay is performed using lysed bacterial cell cultures with no addition of protease inhibitors and at temperatures that potentiate protein degradation. For example, G-NetB was unable to withstand these conditions and degraded in all three temperatures tested.
The different glycosylation efficiencies observed in the in vitro glycosylation reaction for G-SodB, G-FlpA G-NetB and G-ExoA were not surprising as even in vivo glycosylation of these four proteins shows variation. Successful G-FlpA glycosylation using the in vitro glycosylation reaction best illustrates the effect of temperature on PglB function (Fig. 7). G-FlpA was modified at all three temperatures, however the fluorescence detected by the LICOR imaging system clearly shows a decrease as the reactions were incubated at higher temperatures.
In this study we have established a correlation between temperature and glycosylation yield for 3 out of the 4 proteins tested using the plasmid system, whereby glycosylation is more efficient at 30 °C than at 37 °C even when the protein level produced is similar. We also determined that glycosylation decreases for all proteins tested at 42 °C, and that this is independent of pglB expression. Thus, indicating an effect of temperature on PglB function. The observed temperature dependence of glycosylation could have more significant implications on the development of LAVS expressing glycoconjugates. It would mean the determination of a minimum amount of glycoconjugate that confers protection and ensuring this could be achieved per bacterial cell. Indeed, it may explain the lack of protection observed with an APEC pgl integrant expressing G-NetB in our recent work [13]. Alternatively, LAVS could be produced at 30 °C, and repeatedly introduced to chickens over time. Because the temperature effect seems to be on PglB function and independent of impacts on pglB transcription, adding a pglB chromosomal copy, possibly under the control of a constitutive promoter, would not be expected to increase glycosylation to the level necessary per bacterial cell.
It was surprising to observe such direct correlation between temperature and PglB function, as this protein is native to C. jejuni—a bacterium well adapted to the chicken’s higher body temperature. This temperature effect was observed both in vitro and in vivo in E. coli suggesting that perhaps there are other unknown factors that help stabilise PglB function in C. jejuni. Questions that remain unanswered and are under investigation include whether PglB requires additional help from a chaperone, for instance, to perform its function; and whether protein folding and sequon accessibility are affected by the temperatures at which cultures are grown.
Overall, it was observed that glycosylation is more efficient when there are more copies of pglB available and at lower temperatures. However, pglB does not have to be expressed from a high-copy number plasmid, instead the addition of an inducible pglB on a low copy number plasmid was sufficient. Furthermore, there was no positive correlation between the amount of protein produced by the bacterial cell and its glycosylation level, in fact the opposite was consistently observed for G-FlpA. A fundamental understanding of PglB when expressed in E. coli in the context of biological conjugation is central to the vaccine manufacturing process and its increased applicability.
Conclusions
In this study we have established a correlation between temperature and glycosylation yield when we used the plasmid system, whereby glycosylation is more efficient at 30 °C than at 37 °C even when the protein level produced is similar. Furthermore, we have determined that for more efficient glycosylation when the pgl locus is integrated in the chromosome of SDB1 and at 30 °C, there is a need to have more than one copy of PglB available. This was true for all carrier proteins tested. Additionally, we determined that glycosylation decreases for all proteins tested at 42 °C, and that this is independent of pglB expression. Thus, indicating an effect of temperature on PglB function.
Materials and methods
Bacterial strains and growth conditions
Escherichia coli SDB1 (W3110, ΔwaaL ligase, ΔwecA GlcNAc transferase), acquired from the Feldman laboratory [18] was used in this study. Consistently, E. coli was grown in Luria Bertani Lennox (LB) broth or LB agar plates at 30 °C, 37 °C and 42 °C. Where appropriate, kanamycin (50 μg/ml), ampicillin (100 μg/ml), chloramphenicol (30 μg/ml) and spectinomycin (100 μg/ml) were added to the culture medium.
Generation of SDB1pgl integrants
SDB1 pgl integrants were generated via allelic exchange mediated by pSECpgl suicide plasmid (Table 2) [13]. The pSECpgl suicide vector (AmpR) was first transformed into electrocompetent diaminopimelic acid (DAP)-dependent E. coli pir + MFD donor cells [34] suitable for conjugation. Successful transformants were selected on LB agar plates supplemented with 0.3 mM DAP and 100 µg/ml Amp. 5 ml cultures of donor (MFD) and recipient (SDB1) strains were grown overnight at 37 °C and 180 rpm. The donor strain was grown in the presence of DAP (0.3 mM) and ampicillin (100 μg/ml) to maintain the suicide plasmid. Overnight cultures were diluted 1:100 in fresh LB medium with appropriate antibiotics. When cultures reached OD600nm of 0.6–0.8 they were centrifuged for 2 min at 10,000×g. The bacterial pellets were then washed in PBS three times followed by resuspension in 100 ml fresh LB medium without antibiotics. The bacterial cells were then combined in a 1:3 ratio of donor to recipient bacterial cells. 10 µl of this mix was spotted on a dry LB agar plate without antibiotics nor DAP for overnight conjugation at 37 °C. The next day, the lawn of conjugation mixture was resuspended in 2 ml of PBS. 100 µl of this mixture and 100 µl of tenfold dilutions were plated onto selective agar plates containing 50 μg/ml kanamycin, the antibiotic marker tagging the pgl locus and incubated overnight at 37 °C.
Table 2.
List of plasmids used in this study
Single colonies were picked and grown in selective media (LB with 50 μg/ml Kan) overnight at 37 °C. Kanamycin resistant colonies were confirmed for presence of the plasmid by PCR amplification of the pgl insert. Colonies that were positive by PCR were picked and grown overnight at 37 °C, 180 rpm in 5 ml LB medium with 50 μg/ml Kan. Cultures that grew overnight were centrifuged for 2 min at 10,000×g. The pellets were washed 3 × in PBS and resuspended in 2 ml of PBS. 50 μl of the resuspension was spotted or streaked onto selective sucrose plates (15 g/l bacterial agar, 10 g/l tryptone, 5 g/l yeast, 15% v/v sucrose) with 50 µg/ml Kan, but in the absence of ampicillin to permit loss of the vector but retention of the kanR-marked pgl region. Kanamycin and sucrose resistant, ampicillin sensitive clones, were analysed for genomic insertion at the correct site by PCR. Double recombination events that resulted in kanamycin-resistant, sucrose-resistant, and ampicillin-sensitive colonies were verified by PCR with oligonucleotides 1&2, 3&4 and 5&6 described in Table 3 (Fig. 1C). Oligonucleotides 1 and 6 align to regions of the SDB1 genome upstream and downstream of the left and right homology arms used, respectively. Oligonucleotide 2 aligns to the beginning of the pgl locus, while oligonucleotide 5 aligns to the Kan cassette that tags the locus, thus amplicons obtained with primers 1&2 and 5&6 (∼1 kb) will only be present in the case of successful integration of the pgl locus at the chosen site. Oligonucleotides 3&4, instead, amplify the middle of the pgl locus and were used as a proxy to confirm integration of an intact pgl locus.
Table 3.
List of primers used in this study
| Primer name | Sequence (5′–3′) | |
|---|---|---|
| 1 | fw | TGAGATCGAAAAGCAGCTGC |
| 2 | rev | TCCAAAGTGCCGTGGTTTTG |
| 3 | fw | GCCGCAAGATGAATACACGC |
| 4 | rev | CAAGCCCATGACCACTAGCA |
| 5 | fw | TGCCTGCTTGCCGAATATCA |
| 6 | rev | CCCAGTACATGTTCAGCAATG |
| pglBFw | fw | AAGAATTCATGTTGAAAAAAGAGTATTTAAAAAACCC |
| pglBRev | rev | AAGGATCCTTAAATTTTAAGTTTAAAAACCTTAGC |
| FwPglB | fw | ACCACTCCGTTGCTAAGATAAA |
| RevPglB | rev | GCCCGCTAGAATGTCTTTGA |
| gapAFw | fw | CTCCGCTGGCTAAAGTTATCA |
| gapARev | rev | CAGTCAGTTTGCCATTCAGTTC |
| rpoSFw | fw | GATGTGAATCGGCAAACGAATAG |
| rpoSRev | rev | CCGGATGATCGAGAGTAACTTG |
Restriction sites are underlined
Modification of carriers as acceptors for glycosylation
The proteins, superoxide dismutase (SodB) and fibronectin-binding protein (FlpA) from C. jejuni, detoxified necrotic enteritis toxin β (NetB) from Clostridium perfringens, and detoxified Pseudomonas aeruginosa exotoxin A (ExoA) were chosen as glycan acceptors. SodB and FlpA have been reported to reduce C. jejuni colonisation in poultry in previous studies [35, 36], NetB was shown to reduce necrotic enteritis in chickens [28], and ExoA, has been used previously in a successful Francisella tularensis vaccine study [29, 37]. Proteins G-SodB and G-FlpA were engineered to be PglB substrates as described in Vohra et al. [26]. All proteins were modified to ensure transport to the periplasmic compartment of the cell by addition of an N-terminal PelB signal peptide, apart from G-ExoA which contains a DsbA signal. All proteins contain a polyhistidine tag for affinity purification. G-NetB was modified to contain 5 DQNAT glycotags at both the C-and N-terminus [13] and detoxified by introducing the mutation W262A [38]. Finally, G-ExoA, a detoxified protein (L552V, ΔE553) contains two internal modifications that allow glycosylation of the protein by PglB, as well as containing four glycotags at the N-terminus and an additional 4 glycotags at the C-terminus [37].
Transformation of SDB1 with carriers pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB, pMAD-G-ExoA and with pACYCpgl containing the pgl locus
Escherichia coli strain SDB1, which lacks the function of wecA (ensuring the polysaccharide is built on undecaprenol with a bacillosamine instead of the preferential N-acetylglucosamine) and waaL (ensuring that no polysaccharide is transferred to lipid A) was used as the host strain for protein expression and glycoconjugate production. SDB1 was made electrocompetent and transformed with plasmids pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB and pMAD-G-ExoA (all ampicillin resistant) encoding for the glycosylatable proteins, as well as pACYCpgl encoding for the pgl locus and pACYCpglB::km, encoding the pgl locus where PglB is non-functional owing to insertion of a kanamycin resistance cassette (chloramphenicol resistant and chloramphenicol and kanamycin respectively) [39]. These constructs enabled the expression of the glycoconjugates and their unglycosylated versions. The transformants were selected on LB agar supplemented with 100 µg/ml ampicillin and 30 μg/ml chloramphenicol for the glycosylatable proteins, and for the unglycosylated proteins at 100 µg/ml ampicillin, 30 μg/ml chloramphenicol and 50 μg/ml kanamycin and incubated at 37 °C overnight [29].
Transformation of SDB1pgl with carriers pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB and pMAD-G-ExoA
Strain SDB1pgl, was transformed with carriers pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB and pMAD-G-ExoA to assess and compare the glycosylation potential of the pgl locus on the chromosome versus the pgl locus on a plasmid. SDB1pgl strain was made electrocompetent and transformed with 200 ng of pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB or pMAD-G-ExoA. The transformants were selected on LB agar supplemented with 100 µg/ml ampicillin and incubated at 37 °C overnight.
Generation of glycoconjugates using SDB1pgl, SDB1pACYCpgl and SDB1pACYCpglB::km
For all experiments, E. coli SDB1pgl was cultured in Luria–Bertani (LB) broth (Fisher Scientific, UK) supplemented with ampicillin (100 μg/ml) and kanamycin (50 μg/ml). Briefly, SDB1pgl pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB or pMAD-G-ExoA strains were grown overnight at 30 °C, 37 °C and 42 °C. The next day, the cultures were diluted 1/100 and allowed to grow at 30 °C, 37 °C and 42 °C until an OD600nm of 0.5 was reached. At that point 0.2% l-arabinose was added to SDB1pgl pMAD-G-ExoA, with a 0.2% l-arabinose supplementing addition 4 h later. 1 mM IPTG was added to SDB1pgl pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB. All cultures were grown for a further 16 h.
For all experiments, E. coli SDB1 pACYCpgl and SDB1 SDB1pACYCpglB::km [39] were cultured in LB broth supplemented with ampicillin and chloramphenicol at 100 μg/ml and 30 μg/ml, respectively. Briefly, transformants of E. coli SDB1 pACYCpgl or SDB1 pACYCpglB::km carrying pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB or pMAD-G-ExoA were grown overnight at 30 and 37 °C. The next day, the cultures were diluted 1/100 and allowed to grow at 30 and 37 °C until an OD600nm of 0.5 was reached. At that point 1 mM IPTG was added to SDB1 pACYCpgl pEXT20-SodB, SDB1 pACYCpgl pEXT20-FlpA, SDB1 pACYCpgl pEXT20-G-NetB for induction of glycosylatable carriers. To SDB1 pACYCpgl pMAD-G-ExoA 0.2% L-arabinose was added for induction of the acceptor protein. All cultures were grown for a further 16 h at either 30 or 37 °C.
Generation of glycoconjugates using SDB1pgl with additional inducible PglB
The strains SDB1pgl containing pEXT20-SodB, pEXT20-FlpA, pEXT20-G-NetB or pMAD-G-ExoA were transformed with pEXT21pglB to increase the copy number of the OST-encoding gene and discern if PglB was the limiting factor in the reaction. pEXT21pglB was generated by amplifying pglB from pACYCpgl using primers PglBFW and PglBrev (Table 3) and cloned into pEXT21 at EcoRI and BamHI restriction enzyme sites (Timothy Scott, personal communication).
Glycoconjugate purification for comparison and quantification
Bacterial cells from cultures induced for 24 h were harvested via centrifugation at 5300×g, 4 °C for 30 min and were resuspended in ice-cold lysis buffer (50 mM NaH2PO4, 300 mM NaCl, and 10 mM imidazole). Lysis, wash, and elution buffer pH’s were adjusted to 8 by addition of NaOH. Resuspended bacterial cells were subjected to five rounds of mechanical lysis using a prechilled Stansted High Pressure Cell Disruptor (Stansted Fluid Power Ltd., UK) under 60,000 psi (410 MPa) in continuous mode. The lysate was then centrifuged at 10,000×g at 4 °C for 60 min and the supernatant combined with Ni–NTA (Qiagen, Japan) resin for 1 h at 4 °C. This was followed by washing the column with 200 ml of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole pH 8.0), subsequently, 2 ml of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole pH 8.0) was used to elute the proteins of interest. The collected fractions were visualised by western blot, and the glycosylated protein fractions were pooled and concentrated using buffer exchange columns Vivaspin 2 (Vivaproducts, UK) into PBS, prior to quantification using a NanoDrop (ThermoFisher, UK). Quantification of both glycosylated and unglycosylated proteins was determined using extinction coefficient and the molecular weight calculated by ProtParam (Expasy); G-FlpA extinction coefficient 54,780 M−1 cm−1, Molecular weight (MW) = 49,042.09 Da, and G-SodB extinction coefficient 47,120 M−1 cm−1, Molecular weight (MW) = 28,878.43 Da, G-ExoA extinction coefficient 98,320 M−1 cm−1, Molecular weight (MW) = 76,042.02 Da and G-NetB extinction coefficient 52,830 M−1 cm−1, Molecular weight (MW) = 42,409.22 Da.
Assessment of glycoconjugate production by western blotting
To assess protein expression and glycosylation levels, a simultaneous two-channel fluorescence-based western blot (Odyssey LI-COR, LI-COR Biosciences, Hamburg Germany) was used to analyse the purified elution fractions. Freshly eluted samples were resuspended in 2 × Laemmli buffer and heated at 95 °C for 6 min. Heated samples, and a PageRuler Plus Prestained Protein Ladder (Bioline, UK) were separated on a NuPAGE 10% Bis–Tris Gel Novex®, then transferred using the Invitrogen TM iBlot TM2 gel transfer device to a Hybond™-C Extra nitrocellulose membrane (Amersham Biosciences, UK).
The membrane was then incubated with PBS 0.1% tween for 1 h at room temperature. This was followed by 3 × 10 min PBS washes. The following step was the incubation of the membrane with the primary antibody (mouse anti-His antibody (1:10,000)) and biotinylated soybean agglutinin (SBA) lectin (1:4000) for 1 h, a lectin specific for the GalNAc motif of the C. jejuni heptasaccharide. Following incubation, the membrane was washed for 30 min with PBS. The membrane was then incubated with the secondary antibodies used at a concentration of 1:10,000. IRDye® 680RD Goat anti-mouse for detection of His-tagged recombinant proteins and IRDye® 800CW Strepavidin to detect glycosylation (Odyssey® LI-COR Biosciences, UK). Detection of fluorescent signal was carried out using a LI-COR imaging system.
qRT-PCR of pglB at 30 °C, 37 °C and 42 °C in SDB1pgl
Overnight cultures of SDB1pgl were grown in LB medium supplemented with 50 μg/ml of kanamycin, at 30 °C, 37 °C and 42 °C. RNA was extracted as previously described [40], with the small modification that 2 µg of total RNA from each sample was treated with TURBO DNase (Invitrogen) according to the manufacturer’s instructions.
cDNA was generated from DNase treated RNA using superscript III kit (Invitrogen) using random hexamers and following the manufacturer’s instructions. 2 µl of each sample were used as template in a RT-PCR using SYBRGreen dye-based PCR amplification and detection system using ABI7500 Fast instrument (Applied Biosystems). Comparative expression of pglB in SDB1pgl was analysed against the expression of housekeeping genes gapA and rpoS and using the ΔΔCt method [22]. Amplification was carried out using the following primers at a final concentration of 500 nM FwPglB and RvPglB for pglB, gapAFw and gapARev for gapA, and rpoSFw and rpoSRev for rpoS (Table 3). Triplicate samples from three separate biological replicates were analysed (n = 9), statistical analysis was performed using a t-test.
In vitro glycosylation reaction
An in vitro glycosylation reaction was undertaken to assess the effect of temperature on PglB function. The assay was based on Jaroentomeechai et al. [21], modified in house (Elizabeth Atkins personal communication, manuscript in preparation). Briefly, cultures of SDB1pgl, SDB1 pACYCpgl, SDB1 pEXT20-SodB, SDB1 pEXT20-FlpA, SDB1 pEXT20-G-NetB or SDB1 pMAD-G-ExoA were grown in LB at 30 °C, supplemented with 50 µg/ml kanamycin, 50 µg/ml chloramphenicol and 100 µg/ml ampicillin, respectively. These cultures were then lysed using a high-pressure cell disruptor as above and chilled to be used as heptasaccharide donor, as well as acceptor proteins. To all reactions (described in Additional file 1: Table S1) extra PglB produced from SDB1 pEXT21 pglB was also added. The reactions were prepared and incubated at 30 °C, 37 °C and 42 °C overnight and the ability to transfer C. jejuni heptasaccharide to the acceptor proteins at different temperatures was assessed by western blot (as described above).
Supplementary Information
Additional file 1. Additional Figures and Tables.
Acknowledgements
The authors acknowledge funding from the BBSRC (BB/N001591/1 and BB/R008124/1) and the Engineering and Physical Sciences Research Board (EP/R013756/1). All figures were created with BioRender.com. We would like to thank Dr. Timothy Scott for the kind donation of pEXT21_pglB.
Abbreviations
- PGCT
Protein Glycan coupling technology
- LAVs
Live attenuated vaccines
- Bp
Base pairs
- Kda
Kilodalton
- GlcNAc
N-acetylglucosamine
- GalNAc
N-acetylgalactosamine
- Glc
Glucose
- OD
Optical density
- SBA
Soybean agglutinin
Authors' contributions
VST: Designed the study, performed the experimental work and wrote the manuscript. MM: Made the SDB1pgl integrants, Fig. 1, edited, read and approved the manuscript. AAS, MPS, AJG, BWW: Read, edited and approved the manuscript, THS: Constructed the pSECpgl vector, helped editing and approved the manuscript. JC: Helped designed the study, edited and approved the manuscript. All authors read and approved the final manuscript.
Funding
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results. This work was funded by BBSRC BB/N001591/1.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its additional files].
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Vanessa S. Terra, Email: Vanessa.Terra@lshtm.ac.uk
Marta Mauri, Email: Marta.Mauri@lshtm.ac.uk.
Mark P. Stevens, Email: Mark.Stevens@roslin.ed.ac.uk
Andrew J. Grant, Email: ajg60@cam.ac.uk
Brendan W. Wren, Email: Brendan.Wren@lshtm.ac.uk
Jon Cuccui, Email: Jon.Cuccui@lshtm.ac.uk.
References
- 1.Terra VS, Mills DC, Yates LE, Abouelhadid S, Cuccui J, Wren BW. Recent developments in bacterial protein glycan coupling technology and glycoconjugate vaccine design. J Med Microbiol. 2012;61:919–26. doi: 10.1099/jmm.0.039438-0. [DOI] [PubMed] [Google Scholar]
- 2.Dow JM, Mauri M, Scott TA, Wren BW. Improving protein glycan coupling technology (PGCT) for glycoconjugate vaccine production. Expert Rev Vaccines. 2020;19:507–27. doi: 10.1080/14760584.2020.1775077. [DOI] [PubMed] [Google Scholar]
- 3.Szymanski CM, Wren BW. Protein glycosylation in bacterial mucosal pathogens. Nat Rev Microbiol. 2005;3:225–37. doi: 10.1038/nrmicro1100. [DOI] [PubMed] [Google Scholar]
- 4.Eichler J. Extreme sweetness: protein glycosylation in archaea. Nat Rev. 2013;11:151–156. doi: 10.1038/nrmicro2957. [DOI] [PubMed] [Google Scholar]
- 5.Szymanski CM, Ruijin Y, Ewing CP, Trust TJ, Guerry P. Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol. 1999;32:1022–1030. doi: 10.1046/j.1365-2958.1999.01415.x. [DOI] [PubMed] [Google Scholar]
- 6.Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, et al. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–3. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 7.Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, et al. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci USA. 2005;102:3016–21. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kowarik M, Young NM, Numao S, Schulz BL, Hug I, Callewaert N, et al. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 2006;25:1957–66. doi: 10.1038/sj.emboj.7601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher AC, Haitjema CH, Guarino C, Çelik E, Endicott CE, Reading CA, et al. Production of secretory and extracellular N-linked glycoproteins in Escherichia coli. Appl Environ Microbiol. 2011;77:871–881. doi: 10.1128/AEM.01901-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palmu AA, Jokinen J, Borys D, Nieminen H, Ruokokoski E, Siira L, et al. Effectiveness of the ten-valent pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) against invasive pneumococcal disease: a cluster randomised trial. Lancet. 2013;381:214–22. doi: 10.1016/S0140-6736(12)61854-6. [DOI] [PubMed] [Google Scholar]
- 11.Findlow H, Borrow R. Interactions of conjugate vaccines and co-administered vaccines. Hum Vaccin Immunother. 2016;12:226–30. doi: 10.1080/21645515.2015.1091908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pichichero ME. Protein carriers of conjugate vaccines characteristics, development, and clinical trials. Hum Vaccin Immunother. 2013;9:2505–23. doi: 10.4161/hv.26109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauri M, Sannasiddappa TH, Vohra P, Corona-Torres R, Smith AA, Chintoan-Uta C, et al. Multivalent poultry vaccine development using Protein Glycan Coupling Technology. Microbl Cell Fact. 2021;20:193. doi: 10.1186/s12934-021-01682-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS. Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng. 1990;35:668–81. doi: 10.1002/bit.260350704. [DOI] [PubMed] [Google Scholar]
- 15.Tyo KEJ, Ajikumar PK, Stephanopoulos G. Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat Biotechnol. 2009;27:760–5. doi: 10.1038/nbt.1555. [DOI] [PubMed] [Google Scholar]
- 16.Englaender JA, Jones JA, Cress BF, Kuhlman TE, Linhardt RJ, Koffas MAG. Effect of genomic integration location on heterologous protein expression and metabolic engineering in E. coli. ACS Synth Biol. 2017;6:710–20. doi: 10.1021/acssynbio.6b00350. [DOI] [PubMed] [Google Scholar]
- 17.Yates LE, Natarajan A, Li M, Hale ME, Mills DC, DeLisa MP. Glyco-recoded Escherichia coli: recombineering-based genome editing of native polysaccharide biosynthesis gene clusters. Metab Eng. 2019;53:59–68. doi: 10.1016/j.ymben.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Quintanilla F, Iwashkiw JA, Price NL, Stratilo C, Feldman MF. Production of a recombinant vaccine candidate against Burkholderia pseudomallei exploiting the bacterial N-glycosylation machinery. Front Microbiol. 2014;5:381. doi: 10.3389/fmicb.2014.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giloh M, Shinder D, Yahav S. Skin surface temperature of broiler chickens is correlated to body core temperature and is indicative of their thermoregulatory status. Poult Sci. 2012;91:175–188. doi: 10.3382/ps.2011-01497. [DOI] [PubMed] [Google Scholar]
- 20.Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun. 1991;59:4310. doi: 10.1128/iai.59.12.4310-4317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaroentomeechai T, Stark JC, Natarajan A, Glasscock CJ, Yates LE, Hsu KJ, et al. Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat Commun. 2018;9(1):2686. doi: 10.1038/s41467-018-05110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Dykxhoorn DM, St Pierre R, Linn T. A set of compatible tac promoter expression vectors. Gene. 1996;177:133–136. doi: 10.1016/0378-1119(96)00289-2. [DOI] [PubMed] [Google Scholar]
- 24.Linton D, Allan E, Karlyshev A, Cronshaw AD, Wren BW. Identification of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in Campylobacter jejuni. Mol Microbiol. 2002;43:497–508. doi: 10.1046/j.1365-2958.2002.02762.x. [DOI] [PubMed] [Google Scholar]
- 25.Wacker M, Wang L, Kowarik M, Dowd M, Lipowsky G, Faridmoayer A, et al. Prevention of Staphylococcus aureus infections by glycoprotein vaccines synthesized in Escherichia coli. J Infect Dis. 2014;209:1551–61. doi: 10.1093/infdis/jit800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vohra P, Chintoan-Uta C, Terra VS, Bremner A, Cuccui J, Wren BW, et al. Evaluation of glycosylated FlpA and SodB as subunit vaccines against Campylobacter jejuni colonisation in chickens. Vaccines. 2020;8:1–14. doi: 10.3390/vaccines8030520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riddle MS, Kaminski RW, di Paolo C, Porter CK, Gutierrez RL, Clarkson KA, et al. Safety and immunogenicity of a candidate bioconjugate vaccine against Shigella flexneri 2a administered to healthy adults: a single-blind, randomized phase i study. Clin Vaccine Immunol. 2016;23:908–17. doi: 10.1128/CVI.00224-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandes da Costa SP, Mot D, Bokori-Brown M, Savva CG, Basak AK, van Immerseel F, et al. Protection against avian necrotic enteritis after immunisation with NetB genetic or formaldehyde toxoids. Vaccine. 2013;31:4003–8. doi: 10.1016/j.vaccine.2013.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuccui J, Thomas RM, Moule MG, D’Elia R, Laws TR, Mills DC, et al. Exploitation of bacterial N-linked glycosylation to develop a novel recombinant glycoconjugate vaccine against Francisella tularensis. Open Biol. 2013;3:130002. doi: 10.1098/rsob.130002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huttner A, Gambillara V. The development and early clinical testing of the ExPEC4V conjugate vaccine against uropathogenic Escherichia coli. Clin Microbiol Infect. 2018;24:1046–50. doi: 10.1016/j.cmi.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Reglinski M, Ercoli G, Plumptre C, Kay E, Petersen FC, Paton JC, et al. A recombinant conjugated pneumococcal vaccine that protects against murine infections with a similar efficacy to Prevnar-13. npj Vaccines. 2018;3:1–11. doi: 10.1038/s41541-018-0090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatz CFR, Bally B, Rohrer S, Steffen R, Kramme S, Siegrist CA, et al. Safety and immunogenicity of a candidate bioconjugate vaccine against Shigella dysenteriae type 1 administered to healthy adults: a single blind, partially randomized Phase I study. Vaccine. 2015;33:4594–601. doi: 10.1016/j.vaccine.2015.06.102. [DOI] [PubMed] [Google Scholar]
- 33.Bassalo MC, Garst AD, Halweg-Edwards AL, Grau WC, Domaille DW, Mutalik VK, et al. Rapid and efficient one-step metabolic pathway integration in E. coli. ACS Synth Biol. 2016;5:561–8. doi: 10.1021/acssynbio.5b00187. [DOI] [PubMed] [Google Scholar]
- 34.Ferrieres L, Hemery G, Nham T, Guerout AM, Mazel D, Beloin C, et al. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol. 2010;192:6418–6427. doi: 10.1128/JB.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chintoan-Uta C, Cassady-Cain RL, Al-Haideri H, Watson E, Kelly DJ, Smith DGE, et al. Superoxide dismutase SodB is a protective antigen against Campylobacter jejuni colonisation in chickens. Vaccine. 2015;33:6206–11. doi: 10.1016/j.vaccine.2015.09.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neal-McKinney JM, Samuelson DR, Eucker TP, Nissen MS, Crespo R, Konkel ME. Reducing Campylobacter jejuni colonization of poultry via vaccination. PLoS ONE. 2014;9:e114254. doi: 10.1371/journal.pone.0114254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall LE, Nelson M, Davies CH, Whelan AO, Jenner DC, Moule MG, et al. An O-Antigen glycoconjugate vaccine produced using protein glycan coupling technology is protective in an inhalational rat model of tularemia. J Immunol Res. 2018;2018:8087916. doi: 10.1155/2018/8087916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Savva CG, da Costa SPF, Bokori-Brown M, Naylor CE, Cole AR, Moss DS, et al. Molecular architecture and functional analysis of NetB, a pore-forming toxin from Clostridium perfringens. J Biol Chem. 2013;288:3512–22. doi: 10.1074/jbc.M112.430223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linton D, Dorrell N, Hitchen PG, Amber S, Karlyshev A, Morris HR, et al. Functional analysis of the Campylobacter jejuni N-linked protein glycosylation pathway. Mol Microbiol. 2005;55:1695–703. doi: 10.1111/j.1365-2958.2005.04519.x. [DOI] [PubMed] [Google Scholar]
- 40.Pumirat P, Cuccui J, Stabler RA, Stevens JM, Muangsombut V, Singsuksawat E, et al. Global transcriptional profiling of Burkholderia pseudomallei under salt stress reveals differential effects on the Bsa type III secretion system. BMC Microbiol. 2010;10:171. doi: 10.1186/1471-2180-10-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Additional Figures and Tables.
Data Availability Statement
All data generated or analysed during this study are included in this published article [and its additional files].