

Abstract
More and more evidence suggests that microRNA (miRNA) and RNA editing play key roles in the development and progression of tumor. However, the influence of miRNA-mediated RNA editing on tumor stem cells remains unclear. In this study, the results demonstrated that miR-17, which was downregulated in melanoma stem cells, acted as a tumor inhibitor by suppressing the stemness of melanoma stem cells and promoting cell differentiation. MiR-17 targeted ADAR2 (adenosine deaminase acting on RNA 2), a gene encoding an editing enzyme required for the maintenance of melanoma stem cell stemness. In melanoma stem cells, ADAR2 was responsible for DOCK2 mRNA editing, which was able to increase the stability of DOCK2 mRNA. The in vitro and in vivo data demonstrated that DOCK2 mRNA editing upregulated the expressions of stemness and anti-apoptotic genes by activating Rac1 and then phosphorylating Akt and NF-κB, thus leading to oncogenesis of melanoma stem cells. Our findings contribute new perspectives to miRNA-regulated RNA editing in tumor progression.
Keywords: melanoma stem cells, stemness, RNA editing, miR-17, Rac1/Akt/NF-κB pathway
Graphical abstract
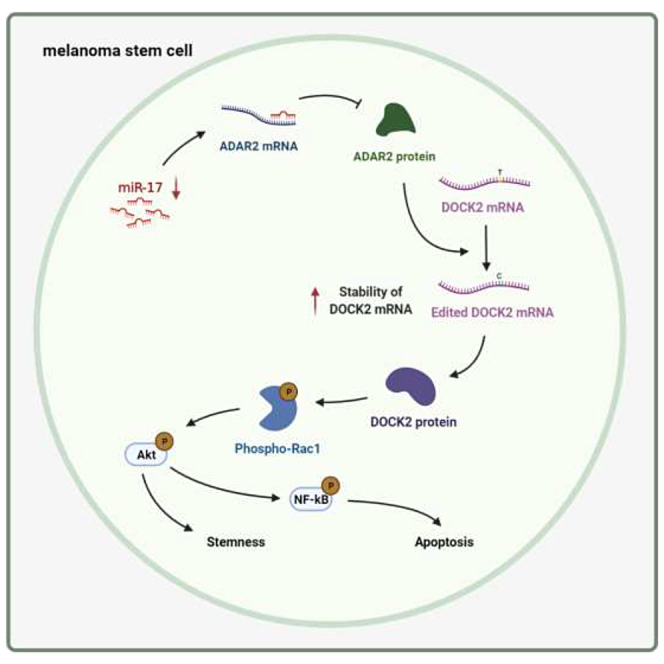
MiR-17 inhibits RNA editing to achieve post-transcriptional regulation of DOCK2, leading to the suppression of the stemness of melanoma stem cells. Our findings indicate that miRNA-regulated RNA editing plays important roles in tumor progression and that miR-17, a tumor suppressor, may be a potential therapeutic target for the treatment of melanoma.
Introduction
Malignant tumors are one of the most serious diseases that threaten human health. In recent years, the incidence of malignant tumors has shown a significant increase. Among malignant tumors, melanoma has the highest degree of malignancy and is more likely to invade nearby tissues and spread to other parts of the body than other types of skin cancer.1 Owing to the lack of specific therapeutic targets, chemotherapy and biotherapy are commonly used strategies for treating cancer.2 However, clinical treatment often results in resistance of cancer cells to these treatments, causing tumor recurrence.3,4 Increasingly, studies have shown that cancer stem cells are closely associated with chemotherapy resistance and tumor recurrence.5 Cancer stem cells have the features of self-renewal, high proliferation, and drug resistance.6,7 These specific characteristics of cancer stem cells may be caused by a series of regulatory mechanisms of gene expression. It is well known that post-transcriptional modification is an important mode of gene expression regulation,8 in which microRNA (miRNA) and RNA editing are two important post-transcriptional regulatory mechanisms.9,10
miRNAs, a class of non-coding small RNAs of ∼22 nt in length, are involved in the regulation of various physiological processes by specifically targeting the 3′ untranslated regions (3′ UTRs) of mRNAs.11 To date, many studies have shown that miRNAs are dysregulated in various cancers, revealing that miRNAs may have the potential to act as tumor-suppressor genes or oncogenes in the development of tumors.12 As reported, RNA editing enzymes can selectively edit miRNA precursors, thereby affecting the maturation process of miRNAs and making the mechanisms of miRNA regulation more complicated.13, 14, 15 However, whether miRNAs can affect the level of RNA editing by regulating the expression of RNA-editing genes remains unclear, as does the role of miRNA-mediated regulation of RNA editing in tumorigenesis.
The ubiquitous RNA-editing mode in animals is adenosine deaminase acting on RNA (ADAR) editing enzyme-catalyzed A (adenosine) to I (inosine) RNA editing.16 In cancers, a prominent feature is angiogenesis. Cancer cells can get into the blood, travel through the blood vessels, and form metastatic tumors in other parts of the body.17 Previous studies have found that arteries are the type of tissue with the highest RNA-editing level,18 suggesting that RNA editing plays a key role in the development of tumors. In tumorigenesis, ADAR2-induced RNA editing of different genes can cause functional changes in tumor cells to eliminate tumorigenicity. ADAR2-mediated editing of an oncogene PODXL, which encodes podocyte marker-like protein (PODXL), leads to the inhibition of tumor progression.19 The edited GABA receptor α3 can suppress breast cancer cell invasion and metastasis.20
Documented findings reveal that miRNAs and RNA editing play vital roles in tumorigenesis. Therefore, it becomes critical to clarify the effects of miRNA-mediated regulation of RNA editing on cancer stem cells. To address this issue, miR-17, which was predicted to target ADAR2, was characterized in our study. The results demonstrated that miR-17 inhibited the ADAR2-mediated dedicator of cytokinesis 2 (DOCK2) mRNA editing, thus resulting in the suppression of the stemness of melanoma stem cells.
Results
Downregulation of miR-17 in melanoma stem cells
To characterize the expression of miR-17 in melanoma stem cells, the melanoma stem cells and the corresponding melanoma non-stem cells were sorted from two types of melanoma cells (i.e., MDA-MB-435 and A375). MDA-MB-435 stem cells were obtained from a previous study in our laboratory.21 To sort the stem cells from A375 cells, the aldehyde dehydrogenase 1 (ALDH1)-positive cells were regarded as cancer stem cells (Figure 1A, P4 region) and the ALDH1-negative cells were classified as cancer non-stem cells (Figure 1A, P5 region). At the same time, the results of in vitro tumorsphere formation assays showed that the ALDH1-positive cells could form large suspension cell spheres (Figure 1B), demonstrating that these cells might be cancer stem cells.
Figure 1.

Downregulation of miR-17 in melanoma stem cells
(A) Sorting of melanoma stem cells. Based on the detection of ALDH1 activity, the ALDH1 fluorescent substrate BODIPY-aminoacetate (BAAA) was used to conduct fluorescence-activated cell sorting. As a control group, the ALDH1 activity was inhibited by DEAB. The ALDH1-negative cells shown at the P5 region were cancer non-stem cells and the ALDH1-positive cells shown at the P4 region were potential melanoma stem cells. (B) In vitro tumorsphere formation assay. Representative images showing the sphere formation of the ALDH1-positive cells are indicated. Scale bars, 100 μm. (C) Tumorigenicity of cancer stem cells in vivo. The ALDH1-positive cells were subcutaneously injected into BALB/C nude mice. As a control, ALDH1-negative cells were included in the injection. Forty days later, the tumors were examined. Arrows indicate the tumors. (D) Differential expressions of stemness genes in ALDH1-negative cells and ALDH1-positive cells. The mRNA levels of stemness genes were evaluated by quantitative real-time PCR (∗∗p < 0.01). (E) Differential expression of miR-17 in melanoma stem cells, gastric cancer stem cells, and cancer non-stem cells. Quantitative real-time PCR was used to detect the level of miR-17 (∗∗p < 0.01). (F) Northern blot analysis of miR-17 expression in melanoma stem cells and non-stem cells. The probes are indicated on the right. U6 was used as a control. The quantified data are also indicated (∗∗p < 0.01). (G) Genomic localization of miR-17. The co-expressing genes related to miR-17 are indicated by blue boxes. (H) Expression profiles of the co-expressing genes associated with miR-17. The expression levels of LOC105370313, LOC105370314, miR-17, and GPC5 were determined using quantitative real-time PCR in melanoma stem cells (MDA-MB-435) and melanoma non-stem cells (∗p < 0.05). (I) Gene set enrichment analysis of miR-17 and stemness genes of melanoma. The expression data were collected from TCGA-SKCM.
To determine the tumor formation ability of the ALDH1-positive cells in vivo, immunodeficient mice were subcutaneously injected with 500 cells isolated from the spheres of tumorsphere formation assays. The results showed subcutaneous tumor generated in mice inoculated with the ALDH1-positive cells, whereas subcutaneous tumors were not observed in mice inoculated with the ALDH1-negative cells (Figure 1C). Furthermore, the expression levels of ALDH1, Nanog, c-myc, and klf4, the stemness genes of melanoma stem cells,22, 23, 24 were assessed in the ALDH1-positive cells and the corresponding negative cells. As expected, the stemness genes were notably upregulated in the ALDH1-positive cells compared with the ALDH1-negative cells (Figure 1D). These cumulative data suggested that the ALDH1-positive cells from A375 cells indeed were melanoma stem cells.
Quantitative real-time PCR data indicated that miR-17 was markedly downregulated in the melanoma stem cells by comparison with the melanoma non-stem cells (Figure 1E). The results showed that miR-17 was significantly downregulated in gastric cancer stem cells sorted from MGC-803 cells, while miR-17 was significantly upregulated in gastric cancer stem cells sorted from HGC-27 and MKN-45 cells (Figure 1E). MiR-17 presented different profiles in different gastric cancer stem cells. Therefore, the miR-17 expression was further characterized in melanoma stem cells. Northern blot confirmed the downregulation of miR-17 in melanoma stem cells (Figure 1F). Our findings suggested that miR-17 played important roles in melanoma stem cells.
The analysis showed that miR-17 was located in a region of 800 bp of the non-protein-coding gene C13orf25 at 13q31.3 (Figure 1G). In the genome, the co-expressed genes related to miR-17 included LOC105370313, LOC105370314, and GPC5. The results of quantitative real-time PCR revealed that the expression profiles of LOC105370313, LOC105370314, and GPC5 in melanoma stem cells and non-stem cells were comparable with that of miR-17 (Figure 1H), indicating the co-expression of LOC105370313, LOC105370314, GPC5, and miR-17.
To explore the relationship between the expression levels of miR-17 and stemness genes in the clinic, gene set enrichment analysis (GSEA) was performed using data from The Cancer Genome Atlas-Skin Cutaneous Melanoma (TCGA-SKCM). The results demonstrated that the miR-17 expression level was significantly negatively correlated with the expression levels of stemness genes in melanoma (Figure 1I), showing that the low expression of miR-17 was clinically associated with melanoma stem cells.
Collectively, these findings demonstrated that miR-17 was downregulated in melanoma stem cells, suggesting its essential role in tumorigenesis of melanoma stem cells.
Anti-tumor activity of miR-17 in melanoma stem cells
To explore the impact of miR-17 on tumorigenesis, miR-17 was overexpressed in melanoma stem and non-stem cells, followed by evaluation of the growth and stemness of cancer cells. The results of quantitative real-time PCR demonstrated that miR-17 content was markedly increased when the synthesized miR-17 was transfected into cells (Figure 2A). miR-17 overexpression led to a significant decrease of melanoma stem cell viability by comparison with the control (Figure 2B). However, miR-17 overexpression had no impact on the proliferation of melanoma non-stem cells (Figure 2B). Flow cytometry analysis indicated that the cell cycle of miR-17-overexpressed cancer stem cells was arrested in the G1 phase compared with the control (Figure 2C), thus contributing to the inhibition of melanoma stem cell proliferation mediated by miR-17 overexpression.
Figure 2.

Anti-tumor activity of miR-17 in melanoma stem cells
(A) Overexpression of miR-17 in melanoma stem cells and non-stem cells. Cancer stem cells were transfected with the synthesized miR-17 or the control (miR-17-scrambled). The miR-17 expression level was evaluated by quantitative real-time PCR at 36 h after transfection (∗∗p < 0.01). (B) Impact of miR-17 overexpression on the proliferation of melanoma stem cells. The viability of melanoma cells was determined at 36 h after transfection of miRNA (∗∗p < 0.01). (C) Effect of miR-17 on the cell cycle of melanoma stem cells. The percentage distribution in the cell-cycle phases of melanoma stem cells and non-stem cells was analyzed at 36 h after transfection with miR-17 or miR-17-scrambled (∗∗p < 0.01). (D) Effects of miR-17 overexpression on apoptosis of melanoma stem cells. At 48 h after transfection, the caspase-3/7 activity of the miRNA-transfected cells was detected (∗∗p < 0.01). (E) Detection of apoptosis by flow cytometry. Annexin V assays were performed to detect apoptosis of cancer cells at 48 h after transfection of miRNA (∗∗p < 0.01). (F and G) Effects of miR-17 overexpression on the expression of stemness genes (F) and differentiation genes (G) in melanoma stem cells. The expression levels of genes were evaluated by quantitative real-time PCR at 36 h after transfection of synthesized miR-17 in melanoma cells (∗∗p < 0.01). (H) Detection of ALDH1-positive cells in miR-17-silenced melanoma cells. MDA-MB-435 cells were transfected with AMO-miR-17. Thirty-six hours later, the cells were subjected to flow cytometry analysis. Based on the detection of ALDH1 activity, the ALDH1 fluorescent substrate BODIPY-aminoacetate (BAAA) was used to detect the fluorescence-activated cell population. As a control group, the ALDH1 activity was inhibited by DEAB. The ALDH1-negative cells shown at the P3 region were cancer non-stem cells and the ALDH1-positive cells shown at the P4 region were potential melanoma stem cells. AMO-miR-17-scrambled served as a control (∗∗p < 0.01). (I) Percentage of tumorsphere formation of miR-17-silenced melanoma cells. MDA-MB-435 cells were transfected with AMO-miR-17-scrambled or AMO-miR-17 and cultured for 7 days, and the percentage of tumorsphere formation of melanoma cells was examined (∗∗p < 0.01). (J) Impact of miR-17 on tumorigenesis of melanoma stem cells in vivo. Nude mice were injected with melanoma stem cells (MDA-MB-435), followed by the injection of miR-17, miR-17-scrambled, AMO-miR-17, or AMO-miR-17-scrambled. The tumor volume of mice was then measured once every 3 days for 1 month. The numerical data were the mean of four mice (∗p < 0.05; ∗∗p < 0.01). (K) Influence of miR-17 on tumor size. The solid tumor images were obtained after sacrifice of mice. Scale bar, 1 cm. (L) Immunohistochemical analysis of stemness markers in solid tumors of the mice injected with miR-17-overexpressed or -silenced melanoma stem cells. Brown represents ALDH1 or Sox2 protein. Nuclei were stained with hematoxylin (blue). Scale bars, 50 μm. (M) Influence of miR-17 silencing on tumorigenesis of MDA-MB-435 cells. The numerical data were the means of four mice. The tumor volumes were measured every 3 days for 1 month (∗p < 0.05; ∗∗p < 0.01). (N) Impact of miR-17 silencing on tumor size in the mice injected with MDA-MB-435 cells. The tumor images were obtained 30 days after AMO injection. Scale bar, 1 cm. (O) Immunohistochemical analysis of stemness markers in solid tumors of the mice injected with miR-17-silenced MDA-MB-435 cells. Arrows indicate the positive signals. Scale bars, 50 μm. (P) Effects of miR-17 silencing on the expressions of stemness genes in normal tissues. Nude mice were injected with melanoma stem cells (MDA-MB-435), followed by the injection of AMO-miR-17 or AMO-miR-17-scrambled. The normal tissues close to the solid tumors were subjected to immunohistochemical analysis to examine the stemness markers. Representative immunohistochemical images are shown. Arrows indicate the positive signals. Scale bars, 50 μm.
To investigate whether the melanoma cell-cycle arrest led to apoptosis, the apoptotic pathway activity of melanoma cells transfected with miR-17 was characterized. The results indicated that miR-17 overexpression could cause melanoma stem cell apoptosis by comparison with the control (Figure 2D), showing that miR-17 overexpression triggered cancer stem cell-cycle arrest, thereby promoting cell apoptosis. Furthermore, the data of Annexin V assays revealed that following the transfection of miR-17, the percentage of apoptotic cells (Annexin V-positive) in melanoma stem cells, but not in melanoma non-stem cells, was markedly increased (Figure 2E). These observations suggested that miR-17 has a positive effect on the apoptosis of melanoma stem cells.
Furthermore, to explore the impact of miR-17 overexpression on the stemness of melanoma stem cells, the expression levels of stemness genes in melanoma stem cells transfected with miR-17 were determined. Our results found that overexpression of miR-17 led to significant downregulation of stemness genes (Nanog, Oct-4, Sox2, and c-fos) (Figure 2F) and upregulation of some differentiation genes compared with the controls (Figure 2G). Thus, these findings revealed that miR-17 could suppress the stemness of melanoma stem cells by promoting cell differentiation.
To explore the influence of miR-17 silencing on the stemness of melanoma cells, miR-17 was knocked down in melanoma cells (MDA-MB-435), followed by the examination of ALDH1-positive cells. Flow cytometry results demonstrated that miR-17 silencing led to a significant increase in the number of melanoma stem cells (Figure 2H). At the same time, the tumorsphere formation percentage of miR-17-silenced melanoma cells was significantly increased compared with the control (Figure 2I). These results showed that miR-17 promoted the stemness of melanoma cells.
To further evaluate the influence of miR-17 on tumorigenesis in vivo, melanoma stem cells (MDA-MB-435) were injected into nude mice, followed by the injection of miR-17, miR-17-scrambled, anti-microRNA oligonucleotide (AMO)-miR-17, or AMO-miR-17-scrambled. The results showed that the tumor growth rate in the mice treated with miR-17 was significantly decreased compared with the control (miR-17-scrambled) (Figure 2J). miR-17 silencing significantly increased the tumor growth rate (Figure 2J). Solid tumor sizes generated similar results (Figure 2K). Downregulation or upregulation of ALDH1 and Sox2 were also observed in miR-17-overexpressed or miR-17-silenced mice using immunohistochemical staining (Figure 2L). These data indicated that miR-17 could suppress tumorigenesis of melanoma stem cells in vivo.
To evaluate the impact of miR-17 silencing on tumorigenesis of MDA-MB-435 cells in vivo, MDA-MB-435 cells were injected into nude mice, followed by the injection of AMO-miR-17 or AMO-miR-17-scrambled. The results showed that the tumor growth rate in AMO-miR-17-injected mice was higher than that in the control mice (Figure 2M). miR-17 silencing significantly increased tumor sizes (Figure 2N). Compared with AMO-miR-17-scrambled, immunohistochemical staining revealed that miR-17 silencing promoted the expression of stemness genes (Figure 2O).
To explore the effects of miR-17 silencing on normal tissues, the mice were injected with melanoma stem cells and then AMO-miR-17. The results of immunohistochemical analysis of normal tissues close to the solid tumors of mice demonstrated that the expression profile of stemness genes in the mice treated with AMO-miR-17 was similar to that of the control mice (Figure 2P). These data indicated the safety of miR-17 as a therapeutic target.
Taken the above data together, miR-17 acted as a tumor suppressor by inhibiting the stemness of melanoma stem cells and promoting cell differentiation.
Anti-tumor mechanism of miR-17 in melanoma stem cells
To reveal the anti-tumor mechanism of miR-17, the target genes of miR-17 were predicted. Based on the target prediction, miR-17 might target 18 genes (HBP1, NFAT5, TBC1D15, PSD3, DCBLD2, BMPR2, ENPP5, PLAG1, E2F3, BTN2A1, Rab5b, CCND1, ADAR2, PKD2, NPAT, ZFYVE26, EIF4G2, and ZNFX1) (Figure 3A). Among the 18 genes, miR-17 overexpression significantly decreased the expression levels of ADAR2, BC1D15, ENPP5, and Rab5b genes in melanoma stem cells (MDA-MB-435) (Figure 3B), showing that ADAR2, BC1D15, ENPP5, and Rab5b might be targeted by miR-17. To explore whether the potential targets were involved in melanoma stem cells, ADAR2, BC1D15, ENPP5, or Rab5b was knocked down by gene-specific small interfering RNA (siRNA) in melanoma stem cells (MDA-MB-435), followed by examination of cell viability. The results indicated that ADAR2 silencing led to a significant decrease of cell viability compared with the control, while BC1D15, ENPP5, or Rab5b silencing had no effect on cell viability (Figure 3C). In this context, miR-17 might target ADAR2 gene (Figure 3D). However, ADAR1 and ADAR3 were not target genes of miR-17 (Figure 3D).
Figure 3.

Anti-tumor mechanism of miR-17 in melanoma stem cells
(A) Number of the potential target genes of miR-17. The target genes of miR-17 were predicted using five algorithms, namely RNA22, TarBase, TargetRank, miRDB, and miRTarBase. Based on the target gene prediction, the potential targets of miR-17 included 18 genes (HBP1, NFAT5, TBC1D15, PSD3, DCBLD2, BMPR2, ENPP5, PLAG1, E2F3, BTN2A1, Rab5b, CCND1, ADARB1, PKD2, NPAT, ZFYVE26, EIF4G2, and ZNFX1). (B) Influence of miR-17 overexpression on the expressions of 18 potential target genes in melanoma stem cells. Melanoma stem cells (MDA-MB-435) were transfected with miR-17 or the control (miR-17-scrambled). At 36 h after transfection, the expression levels of 18 potential target genes were examined by quantitative real-time PCR (∗p < 0.05; ∗∗p < 0.01). HBP, HMG-box transcription factor 1; NFAT5, nuclear factor of activated T cells 5; TBC1D15, TBC1 domain family member 15; PSD3, pleckstrin and sec7 domain containing 3; DCBLD2, discoidin, CUB, and LCCL domain containing 2; BMPR2, bone morphogenetic protein receptor type 2; ENPP5, ectonucleotide pyrophosphatase/phosphodiesterase family member 5; PLAG1, pleomorphic adenoma gene 1; E2F3, E2F transcription factor 3; BTN2A1, butyrophilin subfamily 3 member A1; Rab5b, Ras oncogene family; CCND1, cyclin D1; ADARB1, adenosine deaminase RNA specific B1; PKD2, polycystin 2, transient receptor potential cation channel; NPAT, nuclear protein, co-activator of histone transcription; ZFYVE26, zinc finger FYVE-type containing 26; EIF4G2, eukaryotic translation initiation factor 4 gamma 2; ZNFX1, zinc finger NFX1-type containing 1. (C) Impact of TBC1D15, ENPP5, Rab5b, or ADAR2 silencing on the viability of melanoma stem cells. Melanoma stem cells (MDA-MB-435) were transfected with gene-specific siRNA. As a control, siRNA-scrambled was included in the transfection. At 36 h after transfection, cell viability was determined (∗p < 0.05). (D) Prediction of genes targeted by miR-17. The underlining shows the seed sequence of miR-17. (E) Direct target of ADAR2 3′ UTR by miR-17. The direct interaction between miR-17 and ADAR2 3′ UTR was examined by luciferase reporter gene assays as described in materials and methods. Luciferase activity was normalized to the ratio of firefly and Renilla luciferase activities (∗∗p < 0.01). (F) Direct interaction between miR-17 and ADAR2 mRNA. Biotinylated miR-17 pull-down assays using the total RNAs extracted from melanoma stem cells were conducted. The pulled-down RNAs were subjected to quantitative real-time PCR to detect ADAR2. As a control, miR-17-scrambled was included in the assays (∗∗p < 0.01). (G and H) Interaction between miR-17 and ADAR2 gene in melanoma stem cells. AMO-miR-17 (G) or miR-17 (H) was transfected into melanoma stem cells. As a control, miR-17-scrambled or AMO-miR-17-scrambled was included in the transfection. Quantitative real-time PCR was used to evaluate the level of ADAR2 mRNA at 36 h after transfection (∗∗p < 0.01).
To characterize the interaction between miR-17 and ADAR2, dual-luciferase reporter assays were carried out in the melanoma cells. A plasmid containing the ADAR2 3′ UTR and synthesized miR-17 were co-transfected into MDA-MB-435 cells. It was found that following transfection with miR-17 and ADAR2 3′ UTR, the luciferase activity of the cells was markedly reduced by comparison with the controls (Figure 3E), revealing that miR-17 directly targeted the ADAR2 gene. To further confirm the direct interaction between miR-17 and ADAR2 mRNA, biotinylated miR-17 pull-down assays using the total RNAs extracted from melanoma stem cells were performed. The results of quantitative real-time PCR demonstrated that miR-17 pulled down the ADAR2 mRNA from the extracted RNAs of melanoma stem cells, while there was little ADAR2 mRNA for the control biotin-miRNA-17-scrambled (Figure 3F), confirming the direct interaction between miR-17 and ADAR2 mRNA.
To examine whether ADAR2 could be regulated by miR-17 in cells, the expression of miR-17 was overexpressed or silenced in melanoma stem cells, and the ADAR2 expression level was assessed. The results showed that overexpressing or silencing of miR-17 in melanoma stem cells led to markedly decreased or increased ADAR2 expression level (Figures 3G and 3H). These cumulative data indicated that ADAR2 was a target gene of miR-17.
Role of ADAR2 in melanoma stem cells
To clarify the role of ADAR2 in the regulation of proliferation and stemness of melanoma stem cells, ADAR2 was knocked out in melanoma stem cells. The specific guide RNA (gRNA) targeting ADAR2 was synthesized (Figure 4A) to knock out ADAR2 in melanoma stem cells (MDA-MB-435). The sequencing data revealed that the ADAR2 gene was knocked out in melanoma stem cells (Figure 4B). Western blotting showed that ADAR2 protein was not detected in ADAR2-knockout melanoma stem cells (Figure 4C). These data indicated that the ADAR2 gene was knocked out in melanoma stem cells.
Figure 4.

Role of ADAR2 in melanoma stem cells
(A) Schematic diagram of the gRNA sequence targeting ADAR2. The target site was located in the second exon of ADAR2. (B) Sequence of ADAR2 in ADAR2-knockout (mutant) and wild-type melanoma stem cells (MDA-MB-435). Protospacer adjacent motif (PAM) is boxed. (C) Detection of ADAR2 protein in ADAR2-knockout melanoma stem cells. The ADAR2 protein was examined by western blot in mutant and wild-type melanoma stem cells (MDA-MB-435). β-Tubulin was used as a control. (D) Impact of ADAR2 knockout on the viability of melanoma stem cells. ADAR2-wild-type (WT) or ADAR2-knockout (mutant) MDA-MB-435 cells (1 × 104) were cultured for 36 h and the cell viability evaluated (∗∗p < 0.01). (E) Effects of ADAR2 knockout on the tumorsphere formation capacity of melanoma stem cells. The mutant or WT of melanoma stem cells (MDA-MB-435) were cultured for 10–14 days, and the tumorsphere was then examined (∗∗p < 0.01). Scale bars, 50 μm. (F) Tumorsphere formation capacity of a single melanoma stem cell (MDA-MB-435). Scale bars, 10 μm. (G) Impact of ADAR2 knockout on the stemness of melanoma stem cells. Quantitative real-time PCR was used to measure the expression levels of stemness genes in mutant or wild-type melanoma stem cells (MDA-MB-435) (∗∗p < 0.01). (H) Impact of ADAR2 knockout on the differentiation of melanoma stem cells. Quantitative real-time PCR was used to evaluate the expression levels of differentiation genes (∗∗p < 0.01).
To investigate the effects of ADAR2 deletion on melanoma stem cells, the proliferation, tumorsphere formation capacity, and stemness gene expression of ADAR2-knockout melanoma stem cells (MDA-MB-435) were evaluated. The data from 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assays revealed that ADAR2 knockout significantly reduced the viability of cancer stem cells (Figure 4D). The tumorsphere formation capacity of ADAR2-knockout melanoma stem cells was significantly decreased compared with that of normal melanoma stem cells (Figure 4E). A single ADAR2-knockout melanoma stem cell could not form tumorsphere (Figure 4F), showing that ADAR2 knockout decreased the stem cell characteristics of melanoma stem cells. At the same time, the results indicated that ADAR2 knockout significantly downregulated the expressions of the stemness genes of melanoma stem cells (Figure 4G), while the expression levels of the differentiation genes were significantly upregulated in the ADAR2-knockout melanoma stem cells (Figure 4H). These results demonstrated that ADAR2 was required for the maintenance of melanoma stem cell stemness.
Editing of mRNAs mediated by ADAR2 in melanoma stem cells
To reveal the underlying mechanism of ADAR2 in melanoma cells, the mRNAs edited by ADAR2 were characterized. Western blot analysis indicated that the ADAR2 complex was obtained (Figure 5A). Data from RNA immunoprecipitation demonstrated that 50 mRNAs with higher content were bound to ADAR2 (Figure 5B), suggesting that these mRNAs might be the editing substrates of ADAR2. To reveal the mRNAs edited by ADAR2, the 50 mRNAs were amplified and then sequenced. The results showed that there existed RNA editing in the 3′ UTR of AP2A2 (adapter-related protein complex 2 subunit alpha 2) and in the open reading frame (ORF) of DOCK2 (Figure 5C). Three distinct editing sites were located in the 3′ UTR of AP2A2, while an obvious editing site was located in the ORF of DOCK2 (Figure 5C).
Figure 5.

Editing of mRNAs mediated by ADAR2 in melanoma stem cells
(A) Immunoprecipitation of the ADAR2 complex. The cell lysate was incubated with the ADAR2-specific antibody, followed by western blot analysis with the antibody against ADAR2. IgG alone was used as a control. Arrow indicates the immunoprecipitated ADAR2 protein. M, protein marker. (B) mRNAs with higher content in the ADAR2 complex. RNAs extracted from the immunoprecipitated product using ADAR2-specific antibody were sequenced. (C) Schematic diagram of RNA editing. The 50 mRNAs with higher content in the ADAR2 complex were subjected to sequencing. Among them, RNA-editing sites were found in 3′ UTR of AP2A2 and ORF of DOCK2. The numbers indicate the locations.
Role of DOCK2 mRNA editing in melanoma stem cells
To reveal the roles of the editing of AP2A2 or DOCK2 in melanoma stem cells, the expression profile of AP2A2 or DOCK2 in stem cells and non-stem cells was examined. Data from quantitative real-time PCR indicated that DOCK2 was markedly upregulated in melanoma stem cells by comparison with melanoma non-stem cells, while the expression level of AP2A2 was not differentially expressed in two types of cells (Figure 6A). Western blots generated similar results (Figure 6B). Overall, these results showed that DOCK2 played an important role in melanoma stem cells.
Figure 6.

Role of DOCK2 mRNA editing in melanoma stem cells
(A) Expression profiles of AP2A2 and DOCK2 in melanoma stem cells (MDA-MB-435) and melanoma non-stem cells. The levels of gene expression were quantified by quantitative real-time PCR. (B) Western blot analysis of AP2A2 and DOCK2 in melanoma non-stem cells and melanoma stem cells (MDA-MB-435). β-Tubulin was used as a control. (C) Expression of the edited DOCK2 mRNA in ADAR2-knockout melanoma stem cells. Quantitative real-time PCR and western blot were used to examine the expression of DOCK2 in ADAR2-knockout melanoma stem cells (MDA-MB-435). β-Tubulin was used as a control. (D) Influence of DOCK2 mRNA editing on the proliferation of ADAR2-knockout melanoma stem cells. ADAR2-knockout melanoma stem cells were co-transfected with DOCK2-specific shRNA and the plasmid expressing the edited DOCK2 mRNA. At 48 h after co-transfection, cell viability was evaluated. (E) Effects of DOCK2 mRNA editing on apoptosis of DOCK2-edited cells. Caspase-3/7 activity was evaluated at 48 h after co-transfection of DOCK2-specific shRNA and the plasmid expressing the edited DOCK2 mRNA. (F) Impact of DOCK2 mRNA editing on apoptosis of cells using Annexin V assays. At 48 h after co-transfection of DOCK2-specific shRNA and the plasmid expressing the edited DOCK2 mRNA, the cells were subjected to Annexin V assays. (G) Effects of DOCK2 mRNA editing on the expression levels of stemness genes in melanoma stem cells. Quantitative real-time PCR and western blot were used to examine the expressions of stemness genes. (H) Influence of DOCK2 mRNA editing on the expressions of differentiation genes in melanoma stem cells. Gene expression was determined by quantitative real-time PCR and western blot. β-Tubulin was used as a control. (I) Impact of DOCK2 mRNA editing on tumorsphere formation capacity of melanoma stem cells. ADAR2-knockout melanoma stem cells were co-transfected with DOCK2-specific shRNA and the plasmid expressing the edited DOCK2 mRNA. Ten days later, the percentage of tumorsphere formation was examined. (J) Stability of edited or unedited DOCK2 mRNA. mRNA levels of edited or unedited DOCK2 mRNA were quantified by quantitative real-time PCR. (K) Impact of DOCK2 mRNA editing on activation of Rac1 and phosphorylation of Akt and the p65 subunit of NF-κB. Western blot was used to detect the proteins. β-Tubulin was used as a control. (L) Influence of DOCK2 mRNA editing on the expression of Bcl-2 in melanoma stem cells. The expression level of Bcl-2 was determined by quantitative real-time PCR and western blot. β-Tubulin was used as a control. (M) Model for the role of miR-17-mediated DOCK2 mRNA editing in melanoma stem cells. In all panels, asterisks indicate the statistical significance of the difference between treatments (∗∗p < 0.01).
To evaluate the influence of DOCK2 mRNA editing on melanoma stem cells, the edited and unedited DOCK2 mRNA was characterized. In the ADAR2-knockout melanoma stem cells (MDA-MB-435), the sequencing data revealed that DOCK2 was not edited. Thus, the ADAR2-knockout melanoma stem cells were co-transfected with DOCK2-specific short hairpin RNA (shRNA), which could knock down the endogenous DOCK2, and the plasmid expressing the edited DOCK2 mRNA. The results indicated that the edited DOCK2 mRNA was expressed in the ADAR2-knockout melanoma stem cells (Figure 6C). MTS data revealed that the expression of DOCK2 mRNA editing significantly increased the viability of ADAR2-knockout melanoma stem cells (Figure 6D). Simultaneously, caspase-3/7 activity detection showed that DOCK2 mRNA editing significantly decreased the apoptotic activity of DOCK2-edited cells (Figure 6E). Annexin V assays essentially generated similar results (Figure 6F). These data showed that DOCK2 mRNA editing suppressed apoptosis of melanoma stem cells.
Further results indicated that DOCK2 mRNA editing significantly increased the expression levels of stemness genes (Oct-3/4, Sox2, ALDH1, and c-myc) in melanoma stem cells (Figure 6G). Furthermore, the differentiation genes (MDA-5, CK18, MUC1, and ZO-1) were significantly downregulated in DOCK2-edited melanoma stem cells (Figure 6H). At the same time, compared with unedited DOCK2 mRNA, the tumorsphere formation capacity of DOCK2 mRNA editing was significantly increased (Figure 6I). These data revealed that DOCK2 mRNA editing played a positive role in the maintenance of the stemness of melanoma stem cells.
To further evaluate the influence of DOCK2 mRNA editing on DOCK2 mRNA stability, the mRNA levels of edited and unedited DOCK2 mRNAs were examined. The results showed that the stability of the edited DOCK2 mRNA became worse than that of the unedited DOCK2 mRNA (Figure 6J).
As reported, DOCK2, a guanine nucleotide exchange factor (GEF), specifically activates the isoforms of small G protein Rac.25,26 Therefore, the influence of DOCK2 mRNA editing on the activation of Rac1 was characterized. The results showed that DOCK2 mRNA editing significantly increased the amount of activated Rac1 compared with unedited DOCK2 mRNA (Figure 6K), indicating that DOCK2 mRNA editing promoted Rac1 activation. In DOCK2-edited melanoma stem cells, the content of phosphorylated Akt was greatly increased compared with the control (Figure 6K), showing that Rac1 activation led to the phosphorylation of Akt. At the same time, Rac1 activation promoted the phosphorylation of the p65 subunit of nuclear factor κB (NF-κB) (Figure 6K). Phosphorylation of Akt upregulated the expressions of stemness genes Sox2, ALDH1, Oct3/4, and c-myc (Figure 6G), consistent with previous reports.27, 28, 29, 30 Activation of the NF-κB signaling pathway promoted the expression of anti-apoptotic factor Bcl-2 (Figure 6L), consistent with the reported data.31
Collectively, these cumulative findings proved that miR-17 suppressed the expression of ADAR2 by targeting its 3′ UTR, leading to the inhibition of DOCK mRNA editing in melanoma stem cells (Figure 6M). DOCK2 mRNA editing, which increased the stability of DOCK2 mRNA, upregulated the expression of stemness and anti-apoptotic genes by activating Rac1 and then phosphorylating Akt and NF-κB, thus contributing to the maintenance of stemness and the inhibition of apoptosis of melanoma stem cells (Figure 6M).
Influence of DOCK2 mRNA editing on tumorigenesis of melanoma stem cells in vivo
To explore the influence of DOCK2 mRNA editing on tumorigenesis of melanoma stem cells in vivo, DOCK2-edited cells and DOCK2-unedited cells were injected into nude mice (Figure 7A). The results showed that DOCK2 mRNA editing significantly promoted tumor growth in mice compared with that of DOCK2-unedited treatment (Figure 7B). Besides, the sizes and weights of the solid tumors from the mice treated with DOCK2-edited cells were significantly increased (Figures 7C and 7D). Immunohistochemical results revealed that DOCK2 mRNA editing promoted the expression of stemness genes in solid tumors (Figure 7E). These data demonstrated that DOCK2 mRNA editing promoted in vivo tumorigenesis of melanoma stem cells.
Figure 7.

Influence of DOCK2 mRNA editing on tumorigenesis of melanoma stem cells in vivo
(A) Flow chart of the in vivo experiments. Tumor growth was examined every 3 days. At 45 days after cell injection, the mice were sacrificed for tumor examination. (B) Effects of DOCK2 mRNA editing on tumor growth in mice. The tumor volume of mice with different treatments was examined every 3 days. Asterisks indicate the statistical significance of difference between treatments (∗∗p < 0.01). (C) Impact of DOCK2 mRNA editing on tumor size. Scale bar, 1 cm. (D) Influence of DOCK2 mRNA editing on tumor weight (∗∗p < 0.01). (E) Immunohistochemical analysis of stemness markers in solid tumors of the mice injected with DOCK2-edited or DOCK2-unedited melanoma stem cells. Brown represents ALDH1 or Sox2 protein. Nuclei were stained with hematoxylin (blue). Scale bars, 50 μm. (F) Western blot analysis of the DOCK2 protein, the activated Rac1, the phosphorylated Akt, and the p65 subunit of NF-κB in solid tumors of mice injected with DOCK2-edited or DOCK2-unedited melanoma stem cells. β-Tubulin was used as a control. (G) Expressions of stemness genes and Bcl-2 in the solid tumors of mice injected with DOCK2 mRNA-edited melanoma stem cells. Western blot was used to determine the protein level. β-Tubulin was used as a control.
To assess the effects of DOCK2 mRNA editing on the signaling pathway related to stemness and apoptosis of melanoma stem cells in vivo, the content of DOCK2, activated Rac1, phosphorylated Akt, and p65 subunit of NF-κB, and the expression levels of Bcl2 and stemness genes in solid tumors were determined. Western blot analysis demonstrated that the DOCK2 content in solid tumors of mice injected with DOCK2 mRNA-edited melanoma stem cells was much higher than that in the control (Figure 7F), showing that DOCK2 mRNA editing increased DOCK2 mRNA stability. The results demonstrated that DOCK2 mRNA editing promoted the activation of Rac1 and the phosphorylation of Akt and the p65 subunit of NF-κB by comparison with the controls (Figure 7F). Besides, the stemness genes (Oct-3/4, Sox2, ALDH1, and c-myc) and Bcl-2 gene were upregulated in the solid tumors of mice injected with DOCK2 mRNA-edited cells (Figure 7G). These findings indicated that DOCK2 mRNA editing activated Rac1 and then phosphorylated Akt and NF-κB, leading to the upregulation of stemness and anti-apoptotic genes in vivo.
Taken together, these findings revealed that DOCK2 mRNA editing can promote tumorigenesis of melanoma stem cells in vivo.
Discussion
It is well known that RNA editing and miRNAs, two important components of gene expression regulation networks, play essential roles in the maintenance and differentiation processes of cancer stem cells. As reported, miRNAs can regulate the self-renewal character of cancer stem cells by targeting stemness-related genes.32 A-to-I RNA editing, a molecular process that has been recognized as an important mechanism in post-transcriptional modifications of mammalian transcripts, is associated with human cancers.33 It has been found that some genes undergoing RNA editing can acquire oncogenic properties and enhance the stem-like characteristics of cancer cells, suggesting that RNA editing has major effects on the regulation of the stemness of cancer stem cells.20,34 ADAR1-mediated miR-200b editing, as a tumor suppressor, reduces cancer cell aggressiveness and viability.35 To date, however, the role of miRNA-mediated regulation of RNA editing in cancer stem cells remains unclear. In this study, the findings revealed that miR-17 overexpression reduced the stemness of melanoma stem cells by inhibiting the expression of ADAR2, which could edit DOCK2 mRNA to ensure its stability. Our data demonstrated that miR-17 was significantly downregulated in two types of melanoma stem cells compared with melanoma non-stem cells. However, the expression profile of miR-17 was different in different types of gastric cancer stem cells. MiR-17 was significantly downregulated in gastric cancer stem cells sorted from MGC-803 cells, while it was significantly upregulated in gastric cancer stem cells sorted from HGC-27 and MKN-45 cells. Therefore, miR-17 was characterized in melanoma stem cells. In this context, our findings contributed a novel clue to reveal the role of miRNA-regulated RNA editing in the stemness maintenance of melanoma stem cells.
RNA editing is involved in tumorigenesis.36 However, the underlying mechanism of miRNA-regulated RNA editing in cancer stem cells has not been extensively explored. In this study, the cumulative results showed that miR-17 downregulated ADAR2 to suppress DOCK2 mRNA editing in melanoma stem cells. DOCK2 mRNA editing increased the stability of DOCK2 mRNA, thus inhibiting apoptosis of melanoma stem cells by activating Rac1. In humans, the ADAR family contains ADAR1, ADAR2, and ADAR3.37,38 ADAR1 and ADAR2 are responsible for A-to-I editing events of pre-mRNA, predominantly in non-coding regions such as introns or UTRs of mRNAs and long non-coding RNAs.39 Our findings showed that ADAR2 underwent RNA editing in the ORF of DOCK2 mRNA. As reported, ADAR2 was able to enhance RNA stability by restricting the interaction between RNA-destabilizing proteins and their cognate substrates, thereby increasing the abundance of target RNA.40 In our study, DOCK2 mRNA editing enhanced DOCK2 mRNA stability in melanoma stem cells. DOCK2, an atypical form of GEF,41 activated Rac1, thereby triggering the Rac1/AKT/NF-κB signaling pathway. Our findings revealed that miRNA-regulated RNA editing plays a crucial role in tumor progression. Therefore, miRNA-regulated RNA editing may be a promising therapeutic target in diverse tumor contexts.42
Materials and methods
Cell cultures
Melanoma cell lines (MDA-MB-435 and A375) and gastric cancer cell lines (HGC-27, MKN-45, and MGC-803) were purchased from the American Type Culture Collection. MDA-MB-435 non-stem cells were cultured in Leibovitz's L-15 medium (MilliporeSigma, USA) supplemented with 10% fetal bovine serum (FBS). A375 and MGC-803 non-stem cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA) supplemented with 10% FBS. HGC-27 and MKN-45 non-stem cells were cultured in RIPM-1640 medium (Gibco) supplemented with 10% FBS. Melanoma stem cells and gastric cancer stem cells were cultured in DMEM/F12 medium (Invitrogen, USA) supplemented with 10 ng/mL basic fibroblast growth factor (Beyotime Biotechnology, Shanghai, China), 20 ng/mL epidermal growth factor (Beyotime), 5 μg/mL insulin (Beyotime), and 2% B-27 (MilliporeSigma). Melanoma stem cells, gastric cancer stem cells, gastric non-stem cells, and A375 non-stem cells were cultured at 37°C in a humidified atmosphere with 5% CO2. MDA-MB-435 non-stem cells were cultured in a 100% humidified atmosphere at 37°C.
Sorting of cancer stem cells and cancer non-stem cells
The sorting of melanoma stem cells and gastric stem cells was conducted using an ALDEFLUOR kit according to the manufacturer’s protocol (Cyagen Biosciences, USA). To detect aldehyde dehydrogenase 1 (ALDH1), a marker of cancer stem cells, the melanoma and gastric cancer cells were suspended in ALDEFLUOR assay buffer containing ALDH1 fluorescent substrate BODIPY-aminoacetate (BAAA) at 1 mM and incubated for 40 min at 37°C. As a negative control, cancer cells were treated with 50 mM diethylaminobenzaldehyde (DEAB), a specific ALDH1 inhibitor. Following incubation, the cells were centrifuged at 300 × g for 5 min. After removing the supernatant, the cell pellet was resuspended in 0.5 mL of ALDEFLUOR assay buffer and stored at 4°C for fluorescence-activated cell sorting (FACS). FACS was conducted with a flow cytometer at an excitation of 575 nm. The sorted melanoma stem cells were used for the subsequent assays. As reported, cancer stem cells can be cultured for up to 5 months.43, 44, 45, 46, 47
In vitro tumorsphere formation assay
In vitro tumorsphere formation assay proceeded under serum-free and non-adherent conditions. In an ultralow adherent 96-well plate (Corning, USA), a single ALDH1-positive cell was seeded per well containing DMEM/F12 medium supplemented with 10 ng/mL basic fibroblast growth factor (Beyotime), 20 ng/mL epidermal growth factor (Beyotime), 5 μg/mL insulin (Beyotime), and 2% B-27 (MilliporeSigma). Cells were cultured for 2 weeks, after which tumorspheres were examined under inverted phase-contrast microscopy. The forming tumorspheres were scattered in DMEM/F12 medium supplemented with the aforementioned elements. Subsequently, a single cell of these was used for the in vitro tumorsphere formation assay. The in vitro tumorsphere formation assay was conducted three times.
Quantifications of miR-17 and mRNAs by real-time PCR
To detect the expressions of miR-17 and mRNAs, a mirVana miRNA Isolation Kit (Ambion, USA) was utilized to extract total RNAs from cells and tissues. The extracted RNAs were treated with DNase I, and the cDNA was then reversely transcribed from the total RNAs using an miR17-specific or mRNA-specific primer with a TaqMan microRNA reverse transcription kit (Applied Biosystems, USA). The total volume of the real-time PCR reaction was 10 μL, which contained 0.5 μL of RT product, 1 μL of TaqMan miRNA Assay reagent (Applied Biosystems), and 5 μL of TaqMan 2× Universal PCR Master Mix (Applied Biosystems). PCR was performed at 95°C for 10 min, followed by 50 cycles at 95°C for 15 s and 60°C for 1 min. The 2−ΔΔCt method was used to determine relative individual miRNA quantities, and U6 (Applied Biosystems) was used as an internal standard for normalization. To assess the mRNA levels of Nanog homeobox (Nanog), c-fos, Octamer-binding transcription factor-3/4 (Oct-3/4), sex-determining region Y-box 2 (Sox2), cytokeratin 18 (CK18), Mucin 1 (MUC1), α-smooth muscle actin (α-SMA), cyclin-dependent kinase inhibitor 1A (CDKN1A), and Zonula occludens 1 (ZO-1), quantitative real-time PCR was conducted with sequence-specific primers (Nanog, 5′-GCT TGC CTT GCT TTG AAG CA-3′ and 5′-TTC TTG ACT GGG ACC TTG TC-3′; c-fos, 5′-CCG GGG ATA GCC TCT CTT ACT-3′ and 5′-CCA GGT CCG TGC AGA AGT C-3′; Oct-3/4, 5′-GAG CAA AAC CCG GAG GAG T-3′ and 5′-TTC TCT TTC GGG CCT GCA C-3′; Sox2, 5′-GCC GAG TGG AAA CTT TTG TCG-3′ and 5′-GGC AGC GTG TAC TTA TCC TTC T-3′; CK18, 5′-GTT GAC CGT GGA GGT AGA TGC-3′ and 5′-GAG CCA GCT CGT CAT ATT GGG-3′; MUC1, 5′-TGC CGC CGA AAG AAC TAC G-3′ and 5′-TGG GGT ACT CGC TCA TAG GAT-3′; α-SMA, 5′-CTA TGA GGG CTA TGC CTT GCC-3′ and 5′-GCT CAG CAG TAG TAA CGA AGG A-3′; CDKN1A, 5′-TGT CCG TCA GAA CCC ATG C-3′ and 5′-AAA GTC GAA GTT CCA TCG CTC-3′; ZO-1, 5′-CAA CAT ACA GTG ACG CTT CAC A-3′ and 5′-CAC TAT TGA CGT TTC CCC ACT C-3′; GAPDH, 5′-GGT ATC GTG GAA GGA CTC ATG AC-3′ and 5′-ATG CCA GTG AGC TTC CCG TTC AG-3′; ADAR2, 5′-CTG ACA CGC TCT TCA ATG GTT-3′ and 5′-GGC GCA GTT CGT TCA AGA T-3′). The human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as a control.
Northern blot
Total RNAs were loaded to the wells of a denaturing 15% polyacrylamide gel using 1× TBE buffer (90 mM Tris-boric acid, 2 mM ethylenediaminetetraacetic acid [EDTA] [pH 8.0]). Following electrophoresis, RNA was transferred to a nylon membrane (Amersham Biosciences, UK). The membrane was hybridized overnight at 55°C with a digoxin (DIG)-labeled DNA probe (miR-17, 5′-CTA CCT GCA CTG TAA GCA CTT TG-3′; U6, 5′-GGG CCA TGC TAA TCT TCT CTG TAT CGT T-3′) after UV crosslinking. The membrane was subsequently detected using Detection Starter Kit II (Roche, Germany) and DIG High Prime DNA Labeling.
Silencing and overexpression of miR-17 in melanoma stem cells
To overexpress miR-17, melanoma cells were transfected with 50 nM of the synthesized miR-17 or miR-17-scrambled using Lipofectamine 2000 (Life Technologies, USA). The miR-17 sequence was 5′-CAA AGU GCU UAC AGU GCA GGU AG-3′. The miR-17 sequence was randomly scrambled to generate miR-17-scrambled (5′-UUC UCC GAA CGU GUC ACG UTT-3′) as a control group. MiR-17 and miR-17-scrambled were synthesized by Shanghai GenePharma (Shanghai, China). After transfection, the cells were harvested at different time points for subsequent use.
The expression of miR-17 was silenced in melanoma cells using an AMO. The melanoma cells were transfected with 50 nM of AMO-miR-17 (5′-ACT GTA AGC ACT TTG-3′) or AMO-miR-17-scrambled (5′-CAC TGG CAT GAC GCA-3′). AMOs were synthesized with a phosphorothioate backbone and a 2′-O-methyl modification at nucleotides 2 and 8 (Sangon Biotech, Shanghai, China). After transfection, the cells were collected at different time points.
Cell viability and proliferation analysis
Cell viability and proliferation analysis were conducted using MTS assays (Promega, USA). Cells were incubated in a 96-well plate (100 μL of culture medium and 20 μL of MTS reagent per well). Following incubation at 37°C in 5% CO2 for 1.5 h, the absorbance was measured at 450 nm. All experiments were repeated three times.
Cell-cycle analysis
Flow cytometry was utilized to perform cell-cycle analysis. Cell samples were fixed in ice-cold ethanol overnight and incubated with DNase-free RNase A (20 mg/mL) for 30 min. Following centrifugation at 300 × g for 5 min, propidium iodide (PI) was used to stain the cells. A flow cytometer was used to measure the fluorescence intensity of 1 × 104 cells at an excitation wavelength of 488 nm.
Analysis of caspase-3/7 activity
To detect caspase-3/7 activity, the caspase-Glo 3/7 assay (Promega, USA) was utilized. According to the manufacturer’s protocol, cells were seeded in a 96-well plate at a concentration of 1 × 104/well. After incubation at 37°C in 5% CO2 for 48 h, caspase-3/7 activity was measured by exposing the cells to medium with 100 μL of caspase-Glo 3/7 reagent for 30 min in darkness. The luminescence of the cells was measured.
Apoptosis assay
Cells were collected and stained with the FITC (fluorescein isothiocyanate) Annexin V Apoptosis Detection Kit I (Becton Dickinson, USA) according to the manufacturer’s protocol. In brief, the cells were harvested and rinsed with cold phosphate-buffered saline (PBS) and resuspended in 1× Annexin binding buffer at 1 × 106 cells/mL. Subsequently 5 μL of Alexa Fluor 488 Annexin V and 1 μL of PI were added to the cells, followed by incubation at room temperature for 15 min in darkness, and 400 μL of 1× Annexin binding buffer was then added to the sample. The specimen was measured using a flow cytometer at an excitation of 575 nm.
Target gene prediction of miRNA
Five computational target gene prediction algorithms, including RNA22 (https://cm.jefferson.edu/rna22/Interactive/), TarBase (https://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=tarbasev8%2Findex), TargetRank (http://hollywood.mit.edu/targetrank/), miRDB (http://mirdb.org/), and miRTarBase (https://mirtarbase.cuhk.edu.cn/∼miRTarBase/miRTarBase_2019/php/index.php), were used to predict the genes targeted by miR-17. The overlapping genes predicted by the algorithms were the potential targets of miRNA.
Dual-luciferase reporter assay
The sequence of miR-17 binding site of ADAR2 3′ UTR (5′-GCACUUU-3′) was mutated, generating ADAR2 3′ UTR mutant (5′-…AUGAGCG…-3′). The pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega) was used to construct recombinant plasmids of ADAR2 3′ UTR and ADAR2 3′ UTR mutant. The cloning was confirmed by DNA sequencing. Subsequently, melanoma cells were seeded into a 96-well plate and co-transfected with 0.2 nM ADAR2 3′ UTR or ADAR2 3′ UTR mutant and 50 nM of synthesized miR-17 or miR-17-scrambled using Lipofectamine 2000, followed by incubation for 36 h. The dual-luciferase reporter assay (Promega) was used to measure the relative luciferase activity of specimens according to the manufacturer’s protocol.
miRNA pull-down assay
Melanoma stem cells (MDA-MB-435) were transfected with 50 nM of the synthesized biotin-miR-17 or biotin-miR-17-scrambled. At 36 h after transfection, the cells were incubated in lysis buffer (25 mM Tris-HCl, 100 mM KCl, 5 mM MgCl2, 0.3% NP-40, 50 U of RNasin Plus Ribonuclease Inhibitor [Promega], 1× Halt protease inhibitor cocktail [Thermo Fisher Scientific, USA]) for 20 min on ice. Dynabeads M-280 Streptavidin beads (Thermo Fisher Scientific) were activated according to the manufacturer’s protocol and blocked for 2 h at 4°C in lysis buffer containing 10 mg/mL RNase-free bovine serum albumin and yeast tRNA (Sigma-Aldrich, USA). After washing with lysis buffer, the beads were incubated with the lysate for 4 h at 4°C, followed by RNA extraction. The extracted RNAs were subjected to quantitative real-time PCR using miR-17-specific primers.
Construction of ADAR2 knockout mutant of melanoma stem cells
A sequence-specific guide RNA (gRNA) (5′-TGT GAA GGA AAA CCG CAA TC-3′) cloned into lentiCRISPRv2 plasmid (Hanheng Biotechnology, Shanghai, China) was used to knock out ADAR2 gene in melanoma stem cells. Melanoma stem cells were then infected with lentivirus isolated from 293T cells transfected with the recombinant gRNA-lentiCRISPRv2 plasmid. To assess the gene-editing activity of this gRNA, the genomic DNA of lentivirus-transfected melanoma stem cells was extracted and the ADAR2 gene was amplified using sequence-specific primers (5′-AGA TTA CTT GTT TAG TTG AGG CTG ATA G-3′ and 5′-CGG TGG GAA TGG TGG TAA GAC A-3′), followed by digestion with T7 endonuclease 1 (New England Biolabs, USA) at 37°C for 15 min. The digested products were analyzed with agarose gel electrophoresis. Subsequently the lentivirus-transfected melanoma stem cells were cultured in stem cell medium containing 1 μg/mL puromycin (Thermo Fisher Scientific) for 48 h. A single colony was selected, passaged, and genotyped. After puromycin screening, the ADAR2 knockout mutant of melanoma stem cells (MDA-MB-435) was confirmed by DNA sequencing and western blot with ADAR2-specific antibody.
Western blot analysis
Proteins were isolated from cells or tissues. Subsequently, the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by electrotransfer to a polyvinylidene fluoride membrane (Millipore, USA). The membrane was incubated with a primary antibody at 4°C overnight and then incubated in blocking solution TBST (Tris-buffered saline with Tween 20, supplemented with 5% skim milk) for 2 h at room temperature. The membrane was then incubated with horseradish peroxidase-conjugated secondary antibody (Sigma-Aldrich) for 2 h at room temperature. Finally, the membrane was ready for scanning to detect protein signals using Western Lightning Plus-ECL Oxidizing Reagent Plus (PerkinElmer, USA).
RNA immunoprecipitation
Cells were crosslinked under UV. The cells were then resuspended with hypotonic buffer (10 mM N-(2-hydroxyethyl)piperazine-N-(2-ethanesulfonic acid) [HEPES], 1.5 mM MgCl2, 10 mM KCl [pH 7.9]), followed by incubation for 2 min on ice. The cell suspension was added to NP-40 (Sigma, USA) at 0.4% final concentration and then centrifuged at 3,000 × g for 7 min at 4°C. The nuclei were rinsed two times with hypotonic buffer. After centrifugation at 3,000 × g for 2 min at 4°C, the nuclei were resuspended with buffer C (20 mM HEPES, 0.33 M NaCl, 1.5 mM MgCl2, 25% glycerol, 1 mM dithiothreitol, 0.2 mM EDTA [pH 7.9]) and incubated for 3 h at 4°C. The nuclei were incubated with rProtein-A beads (Smart-Life Sciences Biotechnology, Changzhou, China) coated with ADAR2-specific antibody (Santa Cruz Biotechnology, USA) or with the control immunoglobulin G (IgG) (Santa Cruz Biotechnology) overnight at 4°C. After washes with buffer C, the mixture was incubated with RNase-free DNase I (Vazyme Biotech, Nanjing, China) for15 min at 37°C and further incubated with proteinase K for 15 min at 55°C. Subsequently the RNAs were extracted with acidic phenol (Ambion, USA).
Expression of the edited DOCK2 mRNA in ADAR2-knockout melanoma stem cells
In the ADAR2-knockout melanoma stem cells (MDA-MB-435), DOCK2 was unedited based on sequencing analysis. To express the edited DOCK2 mRNA in the ADAR2-knockout melanoma stem cells, the endogenous DOCK2 was knocked down by DOCK2-specific shRNA (5′-GGT GCA GTC CAT GAT GTA CGA-3′). In brief, DOCK2-specific shRNA was cloned into pLenti-U6-Puro plasmid (Vigene Biosciences, China). Subsequently the recombinant plasmid, the psPAX2 plasmid (Vigene Biosciences), and PMD2.G plasmid (Vigene Biosciences) were co-transfected into 293T cells. After co-transfection, viral particles were harvested at 48 h and infected with the ADAR2-knockout melanoma stem cells. The cells were cultured in DMEM/F12 medium (Invitrogen) supplemented with 10 μg/mL puromycin (Sigma-Aldrich). After culture for 3 weeks, the cells with puromycin resistance were harvested to examine the knockdown efficiency of DOCK2 by western blot and quantitative real-time PCR. To express the edited DOCK2 mRNA in DOCK2-silenced and ADAR2-knockout melanoma stem cells, the edited DOCK2 mRNA was amplified by PCR using sequence-specific primers (5′-AGC CAC CCC CTG ACG GCT TC-3′ and 5′-TTA TTC GAA CCA AGA GAA CA-3′) and then cloned into pcDNA3.1 plasmid (Invitrogen). Lipofectamine 2000 (Invitrogen) was used to transfect the recombinant plasmid into the cells. After transfection, the cells were harvested at different time points for later use.
DOCK2 mRNA stability assay
To examine the mRNA stability of DOCK2, actinomycin D (Sigma-Aldrich) was added into the cell culture medium to block cell transcription. At different times after cell culture, total RNAs were extracted with TRIzol reagent (TransGen Biotech, Beijing, China). The cDNA was then synthesized using HiScriptII Q RT SuperMix for qRCR reagent kit (Vazyme Biotech). The RNAs were subjected to quantitative real-time PCR with sequence-specific primers (DOCK2, 5′-CAT CCG TGA TAT GTG GTA-3′ and 5′-AAG TGT CTC TAA TAT ACG T-3′; GAPDH, 5′-GGT ATC GTG GAA GGA CTC ATG AC-3′ and 5′-ATG CCA GTG AGC TTC CCG TTC AG-3′) using Hieff qPCR SYBR Green Master Mix (Yeasen Biotech, Shanghai, China). GAPDH was used as an internal standard for normalization.
Detection of activated Rac1
A Rac1 activation assay kit was used to examine the activated Rac1 protein according to the manufacturer’s protocol (Cell Biolabs, USA). In brief, cells were treated with cell lysis buffer. Subsequently the lysate was added with the agarose beads coated by p21-binding domain of p21 activated kinase protein (Cell Biolabs), which specifically bound to the activated Rac1. After incubation at 4°C for 1 h, the mixture was centrifuged at 14,000 × g for 10 s. Finally, western blot analysis was used to detect the supernatant using Rac1-specific monoclonal antibody.
Tumorigenicity assay in vivo
Cancer stem cells (1 × 104 cells/mL) or cancer non-stem cells (1 × 104 cells/mL) were mixed with Corning Matrigel matrix (Corning, USA) at a ratio of 1:1. Subsequently, 100 μL of cell suspension was subcutaneously injected into BALB/C nude mice (4 weeks old) to induce tumor growth. The tumor volume was measured once every 3 days. Forty days later, the mice were sacrificed to determine the size and weight of solid tumors.
The DOCK2 edited cells (106 cells/mL) and the DOCK2 unedited cells (106 cells/mL) were mixed with Corning Matrigel matrix (Corning) at a ratio of 1:1, respectively. Subsequently, 100 μL of cell suspension was subcutaneously injected into BALB/C nude mice (4 weeks old) to induce tumor growth. The tumor volume was measured once every 3 days. Forty-five days later, the nude mice were sacrificed. The sizes and weights of solid tumors were determined.
To evaluate the role of miR-17 in tumorigenesis in vivo, MDA-MB-435 stem cells (106 cells/mL) were subcutaneously injected into BALB/C nude mice. Three days later, the mice were subcutaneously and intravenously injected with miR-17, miR-17-scrambled, AMO-miR-17, or AMO-miR-17-scrambled at 80 mg/kg once every 3 days. The tumor volume was examined every 3 days. Thirty days later the mice were sacrificed, followed by examination of tumor sizes and weights of solid tumors.
Animal experiments were approved by The Animal Experiment Center of Zhejiang University, China. All methods were conducted in accordance with the approved guidelines.
Gene set enrichment analysis
GSEA was conducted to evaluate the relationship between the expression profiles of miRNAs and mRNAs using data from the TCGA-SKCM cohort. GSEA was carried out using GSEA version 3.10.1 (https://www.omicstudio.cn/tool/29).
Immunohistochemical analysis
Solid tumors were incubated in 10% buffered formalin (pH 7.4) for 24 h. The tumors were cut into 3- to 5-μm pieces and mounted on a slide. The slide was dewaxed and hydrated in 100%, 95%, and 80% ethanol for 5 min each. The primary antibody was then incubated with the slide in a humidity chamber. After washes with PBS, the slide was covered with the secondary antibody and incubated for 10 min at room temperature. Streptavidin peroxidase was added, followed by incubation for 10 min at room temperature. AEC buffer and AEC chromogen (Santa Cruz Biotechnology) were mixed and added to the slide. After incubating for 10 min at room temperature, the slide was examined under a fluorescence microscope.
Statistical analysis
Statistical analysis was performed with one-way analysis of variance. The significance of the difference between two groups was examined using Student’s t test.
Acknowledgments
This work was financially supported by the National Key Research and Development Program of China (2018YFD0900504) and Innovation Group Project of Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (no. 311021006).
Author contributions
X.Z.: conceptualization, funding acquisition, project administration, supervision, writing. and editing. Y.Z.: data curation, validation, formal analysis, methodology, validation, resources, software, and writing. X.Y. and Y.C.: data curation, methodology, validation, and resources.
Declaration of interests
The authors declare no competing interests.
References
- 1.Lo J.A., Fisher D.E. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science. 2014;346:945–949. doi: 10.1126/science.1253735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Hollander P., Savage M.I., Brown P.H. Targeted therapy for breast cancer prevention. Front. Oncol. 2013;3:250. doi: 10.3389/fonc.2013.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Song J., Shih Ie M., Salani R., Chan D.W., Zhang Z. Annexin XI is associated with cisplatin resistance and related to tumor recurrence in ovarian cancer patients. Clin. Cancer Res. 2007;13:6842–6849. doi: 10.1158/1078-0432.CCR-07-0569. [DOI] [PubMed] [Google Scholar]
- 4.Colak S., Medema J.P. Cancer stem cells—important players in tumor therapy resistance. FEBS J. 2014;281:4779–4791. doi: 10.1111/febs.13023. [DOI] [PubMed] [Google Scholar]
- 5.Alvero A.B., Chen R., Fu H.H., Montagna M., Schwartz P.E., Rutherford T., Silasi D.A., Steffensen K.D., Waldstrom M., Visintin I., Mor G. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009;8:158–166. doi: 10.4161/cc.8.1.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean M., Fojo T., Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 7.Hurt E.M., Kawasaki B.T., Klarmann G.J., Thomas S.B., Farrar W.L. CD44+ CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer. 2008;98:756–765. doi: 10.1038/sj.bjc.6604242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006;34:721–733. doi: 10.1093/nar/gkj471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Lopez J., Hourcade Jde D., Del Mazo J. Reprogramming of microRNAs by adenosine-to-inosine editing and the selective elimination of edited microRNA precursors in mouse oocytes and preimplantation embryos. Nucleic Acids Res. 2013;41:5483–5493. doi: 10.1093/nar/gkt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ha M., Kim V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 11.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 12.Iorio M.V., Croce C.M. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012;4:143–159. doi: 10.1002/emmm.201100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawahara Y., Zinshteyn B., Chendrimada T.P., Shiekhattar R., Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8:763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawahara Y., Zinshteyn B., Sethupathy P., Iizasa H., Hatzigeorgiou A.G., Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W., Chendrimada T.P., Wang Q., Higuchi M., Seeburg P.H., Shiekhattar R., Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y., Wondimu A., Yan S., Bobb D., Ladisch S. Tumor gangliosides accelerate murine tumor angiogenesis. Angiogenesis. 2014;17:563–571. doi: 10.1007/s10456-013-9403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan M.H., Li Q., Shanmugam R., Piskol R., Kohler J., Young A.N., Liu K.I., Zhang R., Ramaswami G., Ariyoshi K., et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550:249–254. doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan T.H., Qamra A., Tan K.T., Guo J., Yang H., Qi L., Lin J.S., Ng V.H., Song Y., Hong H., et al. ADAR-mediated RNA editing predicts progression and prognosis of gastric cancer. Gastroenterology. 2016;151:637–650.e10. doi: 10.1053/j.gastro.2016.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gumireddy K., Li A., Kossenkov A.V., Sakurai M., Yan J., Li Y., Xu H., Wang J., Zhang P.J., Zhang L., et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat. Commun. 2016;7:10715. doi: 10.1038/ncomms10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang F., Wei J., Zhang S., Zhang X. Shrimp miR-S8 suppresses the stemness of human melanoma stem-like cells by targeting the transcription factor YB-1. Cancer Res. 2017;77:5543–5553. doi: 10.1158/0008-5472.CAN-17-1375. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 23.Silva J., Nichols J., Theunissen T.W., Guo G., van Oosten A.L., Barrandon O., Wray J., Yamanaka S., Chambers I., Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gawlik-Rzemieniewska N., Bednarek I. The role of NANOG transcriptional factor in the development of malignant phenotype of cancer cells. Cancer Biol. Ther. 2016;17:1–10. doi: 10.1080/15384047.2015.1121348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunisaki Y., Nishikimi A., Tanaka Y., Takii R., Noda M., Inayoshi A., Watanabe K., Sanematsu F., Sasazuki T., Sasaki T., Fukui Y. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J. Cell Biol. 2006;174:647–652. doi: 10.1083/jcb.200602142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang L., Yang J., Jo C.H., Boland A., Zhang Z., McLaughlin S.H., Abu-Thuraia A., Killoran R.C., Smith M.J., Cote J.F., Barford D. Structure of the DOCK2-ELMO1 complex provides insights into regulation of the auto-inhibited state. Nat. Commun. 2020;11:3464. doi: 10.1038/s41467-020-17271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L., Ge C., Zhao F., Zhang Y., Wang X., Yao M., Li J. NRBP2 overexpression increases the chemosensitivity of hepatocellular carcinoma cells via Akt signaling. Cancer Res. 2016;76:7059–7071. doi: 10.1158/0008-5472.CAN-16-0937. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z., Kang L., Zhang H., Huang Y., Fang L., Li M., Brown P.J., Arrowsmith C.H., Li J., Wong J. AKT drives SOX2 overexpression and cancer cell stemness in esophageal cancer by protecting SOX2 from UBR5-mediated degradation. Oncogene. 2019;38:5250–5264. doi: 10.1038/s41388-019-0790-x. [DOI] [PubMed] [Google Scholar]
- 29.Zhang F., Li K., Yao X., Wang H., Li W., Wu J., Li M., Zhou R., Xu L., Zhao L. A miR-567-PIK3AP1-PI3K/AKT-c-Myc feedback loop regulates tumour growth and chemoresistance in gastric cancer. EBioMedicine. 2019;44:311–321. doi: 10.1016/j.ebiom.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bamodu O.A., Chang H.L., Ong J.R., Lee W.H., Yeh C.T., Tsai J.T. Elevated PDK1 expression drives PI3K/AKT/MTOR signaling promotes radiation-resistant and dedifferentiated phenotype of hepatocellular carcinoma. Cells. 2020;9:746. doi: 10.3390/cells9030746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Heijden M., Zimberlin C.D., Nicholson A.M., Colak S., Kemp R., Meijer S.L., Medema J.P., Greten F.R., Jansen M., Winton D.J., Vermeulen L. Bcl-2 is a critical mediator of intestinal transformation. Nat. Commun. 2016;7:10916. doi: 10.1038/ncomms10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samaeekia R., Adorno-Cruz V., Bockhorn J., Chang Y.F., Huang S., Prat A., Ha N., Kibria G., Huo D., Zheng H., et al. miR-206 inhibits stemness and metastasis of breast cancer by targeting MKL1/IL11 pathway. Clin. Cancer Res. 2017;23:1091–1103. doi: 10.1158/1078-0432.CCR-16-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maas S., Kawahara Y., Tamburro K.M., Nishikura K. A-to-I RNA editing and human disease. RNA Biol. 2006;3:1–9. doi: 10.4161/rna.3.1.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shigeyasu K., Okugawa Y., Toden S., Miyoshi J., Toiyama Y., Nagasaka T., Takahashi N., Kusunoki M., Takayama T., Yamada Y., et al. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight. 2018;3:e99976. doi: 10.1172/jci.insight.99976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramirez-Moya J., Baker A.R., Slack F.J., Santisteban P. ADAR1-mediated RNA editing is a novel oncogenic process in thyroid cancer and regulates miR-200 activity. Oncogene. 2020;39:3738–3753. doi: 10.1038/s41388-020-1248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan T.W., Fu T., Bahn J.H., Jun H.I., Lee J.H., Quinones-Valdez G., Cheng C., Xiao X. RNA editing in cancer impacts mRNA abundance in immune response pathways. Genome Biol. 2020;21:268. doi: 10.1186/s13059-020-02171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higuchi M., Maas S., Single F.N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 38.Lehmann K.A., Bass B.L. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 39.Bazak L., Haviv A., Barak M., Jacob-Hirsch J., Deng P., Zhang R., Isaacs F.J., Rechavi G., Li J.B., Eisenberg E., Levanon E.Y. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014;24:365–376. doi: 10.1101/gr.164749.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anantharaman A., Tripathi V., Khan A., Yoon J.H., Singh D.K., Gholamalamdari O., Guang S., Ohlson J., Wahlstedt H., Ohman M., et al. ADAR2 regulates RNA stability by modifying access of decay-promoting RNA-binding proteins. Nucleic Acids Res. 2017;45:4189–4201. doi: 10.1093/nar/gkw1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo X., Chen S.Y. Dedicator of cytokinesis 2 in cell signaling regulation and disease development. J. Cell Physiol. 2017;232:1931–1940. doi: 10.1002/jcp.25512. [DOI] [PubMed] [Google Scholar]
- 42.Xu X., Wang Y., Mojumdar K., Zhou Z., Jeong K.J., Mangala L.S., Yu S., Tsang Y.H., Rodriguez-Aguayo C., Lu Y., et al. A-to-I-edited miRNA-379-5p inhibits cancer cell proliferation through CD97-induced apoptosis. J. Clin. Invest. 2019;129:5343–5356. doi: 10.1172/JCI123396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimono Y., Zabala M., Cho R.W., Lobo N., Dalerba P., Qian D., Diehn M., Liu H., Panula S.P., Chiao E., et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., He L., Du Y., Zhu P., Huang G., Luo J., Yan X., Ye B., Li C., Xia P., et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell. 2015;16:413–425. doi: 10.1016/j.stem.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Shah M., Cardenas R., Wang B., Persson J., Mongan N.P., Grabowska A., Allegrucci C. HOXC8 regulates self-renewal, differentiation and transformation of breast cancer stem cells. Mol. Cancer. 2017;16:38. doi: 10.1186/s12943-017-0605-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhan Y., Chen Z., He S., Gong Y., He A., Li Y., Zhang L., Zhang X., Fang D., Li X., Zhou L. Long non-coding RNA SOX2OT promotes the stemness phenotype of bladder cancer cells by modulating SOX2. Mol. Cancer. 2020;19:25. doi: 10.1186/s12943-020-1143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin Y., Hou Y., Liu S., Zhu P., Wan X., Zhao M., Peng M., Zeng H., Li Q., Jin T., et al. A novel long non-coding RNA lnc030 maintains breast cancer stem cell stemness by stabilizing SQLE mRNA and increasing cholesterol synthesis. Adv. Sci. (Weinh) 2021;8:2002232. doi: 10.1002/advs.202002232. [DOI] [PMC free article] [PubMed] [Google Scholar]