

Abstract
High‐salt diets have recently been implicated in hypertension, cardiovascular disease, and autoimmune disease. However, whether and how dietary salt affects host antiviral response remain elusive. Here, we report that high salt induces an instant reduction in host antiviral immunity, although this effect is compromised during a long‐term high‐salt diet. Further studies reveal that high salt stimulates the acetylation at Lys663 of p97, which promotes the recruitment of ubiquitinated proteins for proteasome‐dependent degradation. p97‐mediated degradation of the deubiquitinase USP33 results in a deficiency of Viperin protein expression during viral infection, which substantially attenuates host antiviral ability. Importantly, switching to a low‐salt diet during viral infection significantly enhances Viperin expression and improves host antiviral ability. These findings uncover dietary salt‐induced regulation of ubiquitinated cellular proteins and host antiviral immunity, and could offer insight into the daily consumption of salt‐containing diets during virus epidemics.
Keywords: p97, salt, USP33, Viperin, virus
Subject Categories: Metabolism; Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications & Proteolysis
High salt levels in the food promote the degradation of the Viperin deubiquitinase USP33, thus interfering with Viperin stability and weakening antiviral responses. A low‐salt diet enhances Viperin expression and promotes antiviral activity.
Introduction
Sodium chloride (salt, NaCl) is essential for the maintenance of normal physiological reactions in the body, while excess salt intake, which remains prevalent in both adult and children populations around the world, leads to a marked increase in the incidence of numerous disorders (Brown et al, 2009). High salt consumption has been implicated in hypertension and cardiovascular disease (Kotchen et al, 2013; O'Donnell et al, 2014). In addition, recent studies have clearly demonstrated that high salt promotes inflammatory responses and drives autoimmune diseases (Kleinewietfeld et al, 2013; Wu et al, 2013). However, whether and how high salt affects host antiviral ability remain ambiguous, which results in a lack of guidance about dietary salt intake during viral infection.
High salt has been reported to regulate some types of immune cells. High salt can induce pathogenic TH17 cells via the p38/MAPK pathway (Kleinewietfeld et al, 2013), and promote TH17 cell differentiation dependently on serum glucocorticoid kinase 1 (SGK1) (Wu et al, 2013). In addition, high salt enhances lipopolysaccharide‐induced, but inhibits IL‐4‐induced, activation of macrophages (Zhang et al, 2015), and leads to an overall imbalance of immune homeostasis (Binger et al, 2015). The complex and dynamic regulation of immune cells by high salt could result in uncertain changes in biological function. In addition, high salt locally enhances antibacterial ability. High salt reduces intestinal survival of Lactobacillus and a gut‐initiated TH17 response (Wilck et al, 2017; Faraco et al, 2018), and accumulated Na+ at the site of bacterial skin infections promotes cutaneous antibacterial defense (Binger et al, 2015). Compared with bacterial infection, viral infection can induce the opposed responses of the body to nutrition metabolism (Wang et al, 2016). Interestingly, our preliminary data suggested that high salt increased the viral load in mouse blood. Thus, it is quite valuable to clearly clarify how dietary salt affects viral infection. In addition, exploring the detailed mechanisms by which high salt regulates cellular proteins and signaling will promote an understanding of the roles of high salt in affecting various human diseases.
Protein ubiquitination, which is a reversible process controlled by ubiquitin E3 ligases and deubiquitinases, has been recognized to be essential for regulating cellular protein levels and signaling pathways. Ubiquitinated proteins could be delivered to the proteasome or lysosome for degradation. In addition, p97, also referred to as valosin‐containing protein (VCP), plays an essential role in regulating endoplasmic reticulum‐associated degradation (ERAD), and can recruit ubiquitinated protein for further degradation to maintain cellular homeostasis (Meyer et al, 2012). Among the deubiquitinases, ubiquitin‐specific protease 33 (USP33) has recently been reported to be involved in the pathogenesis of many cancers (Yuasa‐Kawada et al, 2009; Huang et al, 2015; Liu et al, 2016). USP33 can also promote the replication of centrosomes by deubiquitinating CP110 proteins (Li et al, 2013). Thus far, whether and how high salt regulates the process of protein ubiquitination remains unknown.
Here, we unexpectedly found that a short‐term high‐salt diet (HSD) promoted viral infection in mice, while a relative long‐term HSD did not significantly increase mouse infection of viruses. Further studies revealed that high salt inhibits the virus‐ and interferon‐induced expression of antiviral Viperin proteins, which in turn substantially attenuates cellular antiviral ability. Mechanistically, high salt stimulates the acetylation of p97, which recruits USP33, a key deubiquitinase of Viperin, for proteasome‐dependent degradation, thus attenuating Viperin protein stability and lowering cellular Viperin levels. Interestingly, we revealed that a low‐salt diet (LSD) during viral infection enhances in vivo antiviral immunity in virus‐infected mice. This study uncovers a previously undescribed activity of high salt by which high salt induces acetylation‐ and ubiquitination‐mediated degradation of cellular proteins, and, more importantly, elucidates the role of dietary salt in regulating host antiviral ability.
Results
High salt is an instant promotion factor of viral infection
To determine whether a HSD affects host antiviral ability, we fed mice a normal salt diet (NSD, 0.45% NaCl) or a HSD (4% NaCl) for different durations according to the reported protocol (Wilck et al, 2017; Faraco et al, 2018), and then infected the mice with vesicular stomatitis virus (VSV), a commonly used virus model (Guo et al, 2019; Zuo et al, 2020). Interestingly, we noticed that as compared with an NSD, a continuous 30‐day HSD seemed to slightly upregulate virus titers and viral RNA levels in the blood and spleens of mice challenged with viruses, but the difference was not significant (Figs 1A and EV1A and B). However, a short 7‐day HSD markedly increased the VSV titers and viral RNA levels in the mouse blood (Figs 1B and EV1C). Consistently, a 7‐day HSD did result in an increase in the VSV RNA levels in the mouse spleen, lung, liver, and kidney tissues (Fig EV1D). These results suggested that a HSD could impair host antiviral ability in an acute manner. Thus, we further injected mice with a high‐salt solution (3% NaCl) and then infected these mice with VSV either immediately or 12 h post injection. The results showed that the injection of a high‐salt solution intravenously can immediately promote viral infection (Fig 1C), which obviously is not due to a gut‐initiated immune response. Similarly, at 12 h after the high‐salt injection the mouse antiviral ability was still lower than the group without high‐salt injection (Fig 1C).
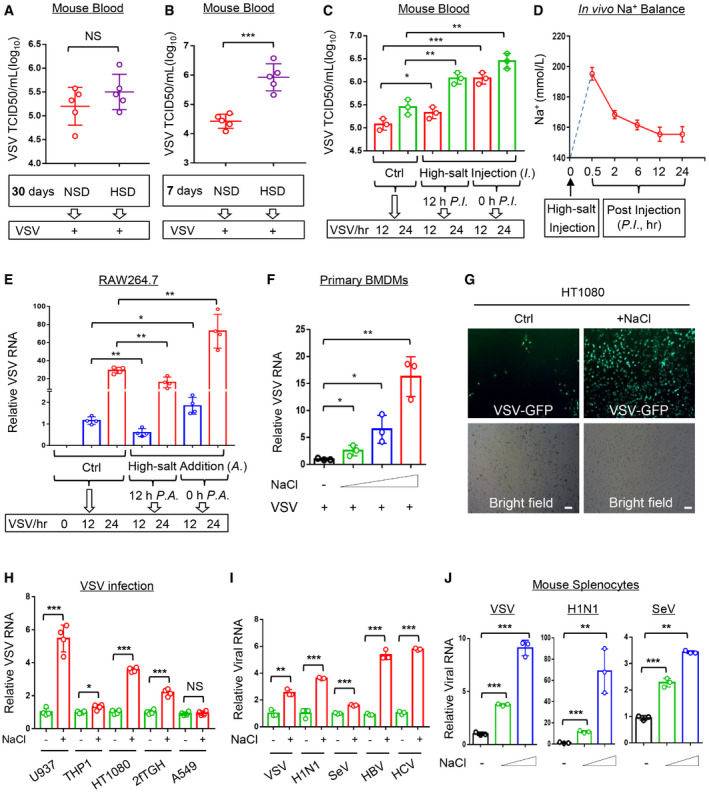
Figure 1. High salt is an instant promotion factor of viral infection.
-
A, BVirus titers in the blood from mice (n = 5) fed an NSD (0.45% NaCl) or HSD (4% NaCl) for 30 days (A) or 7 days (B) and then intraperitoneally infected with VSV (1 × 108 PFU per gram body, 48 h) were analyzed by the TCID50 assay.
-
CMice (n = 3) were intravenously injected with the high‐salt solution (3% NaCl) or control salt solution (0.9% NaCl, Ctrl). Then mice were intraperitoneally infected with VSV (1 × 109 PFU per gram body, 12 or 24 h) either 12 h post injection (P.I.) or immediately (0 h P.I.). Virus titers in the blood were analyzed by the TCID50 assay.
-
DMice (n = 3) were intravenously injected with the high salt solution (3% NaCl). Na+ concentration in sera was analyzed at different time points by a Micro Blood Sodium Concentration Assay Kit. Data were shown as mean and SEM of three biological replicates.
-
ERAW264.7 cells were treated with additional NaCl (+34 mM: the final NaCl concentration increased by 34 mM) or control ddH2O (Ctrl). Then cells were infected with VSV (MOI = 1.0, 12 or 24 h) either 12 h post addition (P.A.) of NaCl or immediately (0 h P.A.). Viral RNA levels were analyzed by RT–qPCR.
-
FRT–qPCR analysis of viral RNA levels in mouse primary BMDMs infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+17, 34 and 51 mM).
-
GHT1080 cells were infected with VSV‐GFP (MOI = 0.5, 24 h) immediately after addition of NaCl (+34 mM). VSV‐GFP viruses were observed by fluorescence. Scale bars, 100 μm.
-
HRT–qPCR analysis of viral RNA levels in U937, THP1, HT1080, 2fTGH and A549 cells infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
-
IRAW264.7 cells were infected with VSV/H1N1/SeV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM), or were transfected with HBV‐replicon or HCV‐replicon and then treated with additional NaCl (+34 mM) for 12 h. Viral RNA levels were analyzed by RT‐qPCR.
-
JRT–qPCR analysis of viral RNA levels in mouse primary splenocytes infected with VSV/H1N1/SeV (MOI = 1.0, 24 h) immediately after addition of NaCl (+17 and 34 mM).
Data information: Data (A, B, C, D) represent mean and SEM of five (A, B) or three (C, D) biological replicates; Data (E, F, H, I, J) show the mean and SD of four (E, H) or three (F, I, J) biological replicates. For all statistical testing, P‐values were calculated using two‐tailed unpaired Student’s t‐test. NS, not significant (P > 0.05). *P < 0.05, **P < 0.01 and ***P < 0.001.
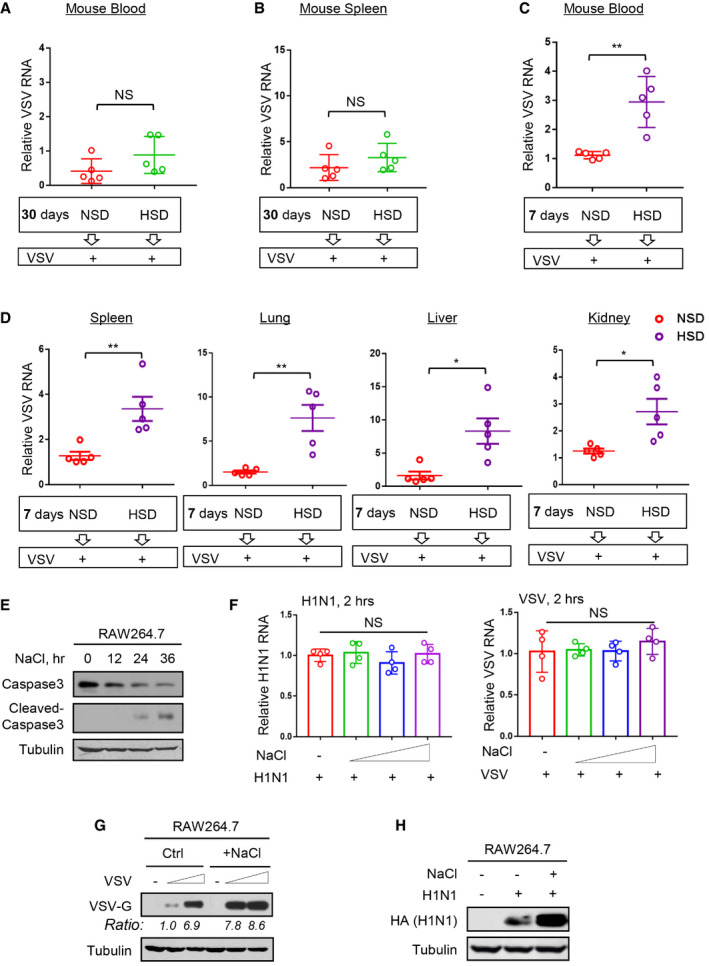
Figure EV1. High salt is an instant promotion factor of viral infection.
-
A, BMice (n = 5) were fed an NSD (0.45% NaCl) or HSD (4% NaCl) for 30 days and then intraperitoneally infected with VSV (1 × 108 PFU per gram body, 48 h). VSV RNA levels in mouse blood (A) or spleen (B) were detected by RT‐qPCR.
-
C, DMice (n = 5) were fed an NSD (0.45% NaCl) or HSD (4% NaCl) for 7 days and then intraperitoneally infected with VSV (1 × 108 PFU per gram body, 48 h). VSV RNA levels in mouse blood (C) or different tissues (spleen, lung, liver, and kidney) (D) were detected by RT‐qPCR.
-
EWestern blot analysis of Caspase 3 and cleaved‐Caspase 3 in RAW264.7 cells treated with additional NaCl (+51 mM) for 12, 24, and 36 h.
-
FRT–qPCR analysis of viral RNA levels in RAW264.7 cells infected with H1N1 or VSV (MOI = 1.0) for only 2 h immediately after addition of NaCl (+17, 34, and 51 mM).
-
GRAW264.7 cells were infected with VSV (MOI = 1.0 and 2.0, 24 h) immediately after addition of NaCl (+34 mM). VSV‐G protein levels were analyzed by western blot.
-
HRAW264.7 cells were infected with H1N1 (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM). H1N1‐encoded HA protein levels were analyzed by western blot.
Data information: Data (A–D) show mean and SEM of five biological replicates; Data (E, G, H) are representative of at least two biological replicates; Data (F) represent mean and SD of four biological replicates. For all statistical testing: P‐values were calculated using two‐tailed unpaired Student’s t‐test. N.S, not significant (P > 0.05). *P < 0.05 and **P < 0.01.
Source data are available online for this figure.
We next observed high‐salt‐mediated regulation of viral infection in cell models. Considering that high salt concentrations are rapidly balanced in vivo, as also shown in our analyses (Fig 1D), we speculated that a continuous long‐time treatment of cells with high salt could not represent the actual salt situation in vivo under normal physiological conditions. One of the cell status analyses demonstrated that the continuous treatment of cells with high salt for over 24 h led to a strong induction of Caspase 3 activation (Fig EV1E), indicating that these cells were undergoing apoptosis. To establish high‐salt experimental cell models that approximately mimic the conditions of high‐salt treatment in vivo, we tried to shorten the duration of the high‐salt treatment in cells. Thus, we infected RAW264.7 cells with viruses immediately or 12 h after high‐salt treatment, since high salt enters cells very rapidly. We observed that compared with the group without high‐salt treatment, the group pretreated with high salt for 12 h followed by 24 h of VSV infection, which actually resulted in a final 36 h of high‐salt treatment in the cells, exhibited decreased virus levels to some extent (Fig 1E). In contrast, the group infected with VSV immediately after the addition of high salt showed higher levels of viruses than the control group (Fig 1E); this was consistent with the results of viral infection after short‐term treatment of mice with high salt. We further demonstrated that the co‐existence of high salt and viruses did not affect virus entry, as shown by the comparable intracellular virus levels between the high‐salt and control groups within 2 h of infection (Fig EV1F). Based on these observations from mice and in vitro cell models, we postulated that the presence of high salt in cells within a certain period of time could promote viral infection.
Further analysis demonstrated that the presence of high salt promoted VSV infection in a dose‐dependent manner (Figs 1F and EV1G). This effect occurred in different types of cells, including macrophages and fibroblasts (Fig 1G and H). Interestingly, we noticed that the presence of high salt did not promote VSV infection in A549 lung epithelium cells (Fig 1H). Next, we found that infection of cells by different viruses, including RNA viruses (VSV, H1N1, SeV, and HCV) and one DNA virus (HBV), can be enhanced by the presence of high salt (Figs 1I and EV1H). Similarly, high salt promoted viral infection of mouse primary splenocytes in a dose‐dependent manner (Fig 1J). Taken together, these findings suggested that high salt is a factor that instantly promotes viral infection in cells.
High salt restricts IFN‐I‐induced antiviral activity but not IFN‐I signaling
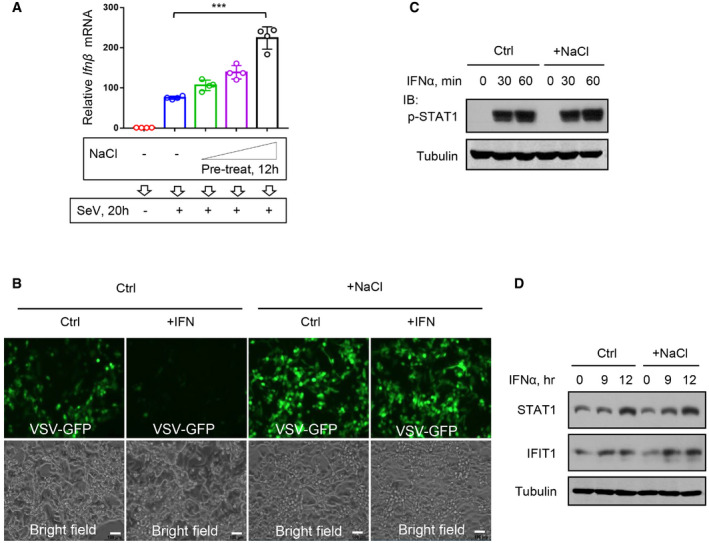
To understand the regulation of viral infection by high salt, we further explored the underlying mechanisms. We noticed that under conditions of viral infection, type‐I interferon (IFN‐I) receptor 1 (Ifnar1) deficiency resulted in higher viral load, which was not further increased by high salt in Ifnar1 −/− cells (Fig 2A), suggesting a role of IFN‐I in high‐salt‐mediated regulation of viral infection. However, this high‐salt treatment with instant viral infection did not result in higher Ifnβ production than that induced by viral infection alone (Fig 2B). We also found that long‐time (12 h) high‐salt pretreatment plus subsequent virus infection with high salt (20 h) can lead to the induction of higher levels of Ifnβ (Fig EV2A), which could explain the above results showing that 12 h of high‐salt pretreatment eventually resulted in fewer viruses in the cells (Fig 1E). Further studies revealed that the presence of high salt inhibited IFN‐I‐induced antiviral activity in RAW264.7 cells in a dose‐dependent manner (Fig 2C). Moreover, the results from 2fTGH cells confirmed the effect of high salt on inhibition of IFN‐I‐induced antiviral activity (Fig EV2B). In line with these observations, STAT1 deficiency also resulted in higher viral load, and the viral load was not further upregulated by high salt in STAT1‐deficient cells (Fig 2D). These findings suggest that high salt inhibits IFN‐I‐induced antiviral activity.
Figure 2. High salt restricts Viperin expression and IFN‐I‐induced antiviral activity.
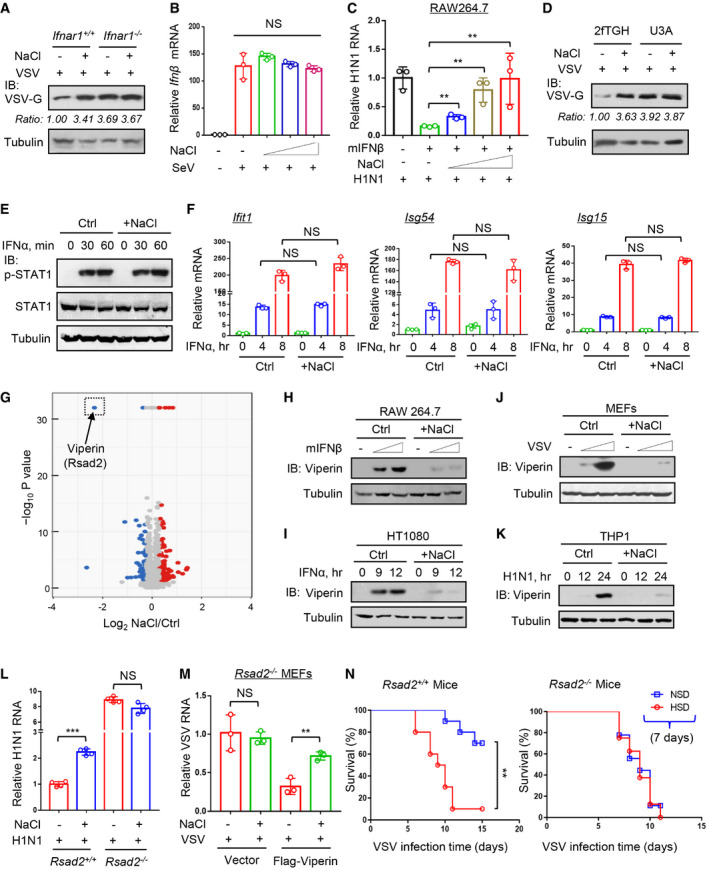
- Western blot analysis of VSV‐G protein levels in Ifnar1+/+ or Ifnar1 −/− MEF cells infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
- RT–qPCR analysis of Ifnβ mRNA levels in HT1080 cells infected with SeV (MOI = 1.0, 10 h) immediately after addition of NaCl (+17, 34 and 51 mM).
- RAW264.7 cells were treated with mIFNβ (50 IU/ml, 20 h) immediately after addition of NaCl (+17, 34, and 51 mM). After washing, cells were infected with H1N1 (MOI = 1.0, 24 h) and viral RNA levels were analyzed by RT–qPCR.
- Western blot analysis of VSV‐G protein levels in U3A (STAT1‐deficient) and its parental cells 2fTGH infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
- Western blot analysis of STAT1‐Tyr701 phosphorylation (p‐STAT1) in HEK293T pretreated with additional NaCl (+34 mM) or control ddH2O (Ctrl) for 12 h and then treated with IFNα (1,000 IU/ml) as indicated.
- RT–qPCR analysis of Ifit1, Isg54, and Isg15 mRNA levels in HEK293T pretreated with additional NaCl (+34 mM) for 12 h and then treated with IFNα (1,000 IU/ml) as indicated.
- RAW264.7 cells were treated with mIFNβ (500 IU/ml) for 12 h immediately after addition of NaCl (+34 mM) or control ddH2O (Ctrl). Whole cell lysates were subjected to a quantitative proteomic analysis with a TMT2‐plex method to identify the differentially expressed proteins. The upregulated and downregulated proteins were analyzed in the volcano plot. The most dramatically downregulated ISG protein, Viperin (Rsad2), was marked in the dotted frame.
- Western blot analysis of Viperin protein in RAW264.7 treated with mIFNβ (100 and 500 IU/ml, 12 h) immediately after addition of NaCl (+34 mM).
- Western blot analysis of Viperin protein in HT1080 treated with IFNα (1,000 IU/ml) and NaCl (+34 mM) as (H).
- Western blot analysis of Viperin protein in MEF cells infected with VSV (MOI = 0.5 and 1.0, 24 h) immediately after addition of NaCl (+34 mM).
- Western blot analysis of Viperin protein in THP1 infected with H1N1 (MOI = 1.0) immediately after addition of NaCl (+34 mM).
- RT–qPCR analysis of viral RNA levels in Rsad2+/+ or Rsad2 −/− MEF cells infected with H1N1 (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
- Rsad2 MEF cells were transfected with empty vectors or Flag‐Viperin, and then infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM). Viral RNA levels were analyzed by RT‐qPCR.
- Survival curves of 8‐week‐old Rsad2 +/+ or Rsad2 −/− mice administrated with an NSD or HSD for 7 days and then intraperitoneally infected with VSV (1 × 108 PFU per gram body mouse, n = 10).
Data information: Data (B, C, F, L, M) represent mean and SD of three (B, C, F, M) or four (L) biological replicates, and P‐values were calculated using two‐tailed unpaired Student’s t‐test; Data (A, D, E, H‐K) are representative of at least two biological replicates; Data (N) show the mean and SEM of 10 biological replicates, and P‐value was calculated using logrank (Mantel–Cox) tests. For all statistical testing: NS, not significant (P > 0.05), **P < 0.01 and ***P < 0.001.
Source data are available online for this figure.
Figure EV2. High salt does not affect IFN‐I‐induced signaling.
- RT–qPCR analysis of IFNβ mRNA levels in HT1080 cells pretreated with additional NaCl (+17, 34, and 51 mM) for 12 h and then infected with SeV (MOI = 1.0) for 20 h.
- 2fTGH cells were treated with IFNα (50 IU/ml, 20 h) immediately after addition of NaCl (+34 mM). After washing, cells were infected with VSV‐GFP (MOI = 0.1, 24 h) and then viruses were observed by fluorescence. Scale bars, 10 μm.
- Western blot analysis of STAT1‐Y701 phosphorylation (p‐STAT1) in HEK293T cells treated with additional NaCl (+34 mM, 12 h) and then stimulated with IFNα (1,000 IU/ml) for 30 and 60 min.
- Western blot analysis of STAT1 and IFIT1 levels in HEK293T cells stimulated with IFNα (1,000 IU/ml) immediately after addition of NaCl (+34 mM) as indicated.
Data information: Data (A) represent mean and SD of four biological replicates; Data (C, D) are representative of at least two biological replicates. For all statistical testing: P‐values were calculated using two‐tailed unpaired Student’s t‐test. ***P < 0.001.
Source data are available online for this figure.
To our surprise, this high‐salt treatment did not inhibit IFN‐I‐induced activation of STAT1 in different types of cells (Figs 2E and EV2C). Furthermore, we found that high salt did not downregulate the mRNA expression of several representative IFN‐I‐stimulated genes (ISGs), including Ifit1, Isg54, and Isg15 (Fig 2F). Consistently, IFN‐I‐induced expression of ISG proteins was not inhibited by short‐time high salt treatment (Fig EV2D), suggesting that short‐time treatment of cells with high salt did not affect IFN‐I‐induced signaling pathway. Based on these findings, we speculated that there is a possibility that high salt could affect certain key antiviral effectors induced by IFN‐I.
High salt exacerbates viral infection by reducing antiviral Viperin protein
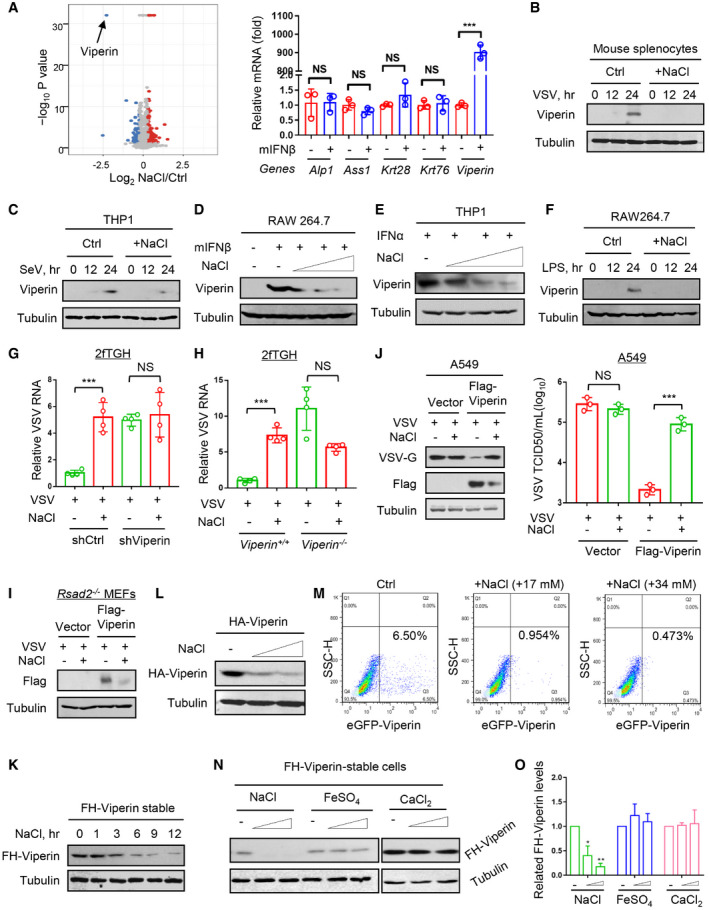
To find out the detailed mechanisms, we next performed quantitative proteomic analyses to determine the influence of high salt on IFN‐I‐induced antiviral proteins. To this end, RAW264.7 cells were treated with mIFNβ (500 IU/ml) for 12 h immediately after the addition of NaCl (+34 mM), and then whole cell lysates were subjected to a quantitative proteomic analysis with a TMT2‐plex method to identify the differentially expressed proteins. We analyzed the differential expression genes with more than two‐fold changes. Interestingly, among these main differential genes, Viperin (Rsad2), as an IFN‐induced antiviral protein, was dramatically downregulated by high‐salt treatment in two quantitative proteomic analyses (Figs 2G and EV3A, left), whereas the other genes were shown not to be IFN‐I‐induced genes (Fig EV3A, right). Given that we have confirmed that high‐salt‐mediated regulation of viral infection is dependent on IFN‐I (Figs 2A,C,D), we think that those genes that cannot respond to IFN‐I could not be the main contributor to this regulation. Thus, we further observed whether Viperin is a main contributor to high‐salt‐mediated promotion of viral infection. We firstly confirmed that the Viperin protein induction by both IFNα and IFNβ can be inhibited in the presence of high salt in either RAW264.7 or HT1080 cells (Fig 2H and I). Consistently, high salt strongly blocked different virus‐induced Viperin protein production (Figs 2J and K, and EV3B and C). The high‐salt‐mediated downregulation of Viperin proteins was dose dependent (Fig EV3D and E). In addition, high salt can also restrict lipopolysaccharide (LPS)‐induced Viperin protein production (Fig EV3F), suggesting that high salt is a broad‐spectrum inhibitor of Viperin protein induction.
Figure EV3. High salt exacerbates viral infection by decreasing antiviral protein Viperin.
-
ARAW264.7 cells were treated with mIFNβ (500 IU/ml) for 12 h immediately after addition of NaCl (+34 mM) or control ddH2O (Ctrl). Differentially expressed proteins were analyzed by a TMT2‐plex Mass analysis (left). RT–qPCR was used to analyze mRNA levels of several top differential genes in RAW264.7 cells treated with mIFNβ (500 IU/ml) for 12 h (right). Data were shown as mean and SD of three biological replicates.
-
B, CWestern blot analysis of Viperin in mouse splenocytes infected with VSV (B) or in THP1 cells infected with SeV (C) (MOI = 1.0, 12 and 24 h) immediately after addition of NaCl (+34 mM).
-
D, EWestern blot analysis of Viperin in RAW264.7 treated with mIFNβ (300 IU/ml) (D) or in THP1 treated with IFNα (1,000 IU/ml) (E) for 12 h immediately after addition of NaCl (+17, 34, and 51 mM).
-
FWestern blot analysis of Viperin in RAW264.7 treated with LPS (2.5 μg/ml, 12 and 24 h) immediately after addition of NaCl (+34 mM).
-
GRT–qPCR analysis of VSV RNA levels in 2fTGH cells transfected with control shRNAs (shCtrl) or shRNAs against Viperin (shViperin) and then infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
-
HRT–qPCR analysis of VSV RNA levels in Viperin+/+ or Viperin −/− 2fTGH cells infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
-
IWestern blot analysis of Flag‐Viperin in Rsad2 −/− MEF cells transfected with vectors or Flag‐Viperin and then infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
-
JWestern blot analysis of VSV‐G in A549 cells transfected with vectors or Flag‐Viperin and then infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM). VSV titers in culture supernatants were analyzed by the TCID 50 assay.
-
KWestern blot analysis of Flag‐HA‐tagged (FH)‐Viperin levels in stable FH‐Viperin‐expressing HeLa cells treated with additional NaCl (+34 mM) as indicated.
-
LWestern blot analysis of HA‐Viperin in HEK293T cells transfected with HA‐Viperin and then treated with additional NaCl (+17, 34 mM) for 12 h.
-
MFlow cytometry analysis of eGFP‐Viperin in HEK293T cells transfected eGFP‐Viperin and then treated with additional NaCl (+17, 34 mM) for 12 h.
-
NWestern blot analysis of FH‐Viperin levels in stable FH‐Viperin‐expressing HeLa cells treated with additional NaCl (+17 and 34 mM), or FeSO4 (20 and 40 μM), or CaCl2 (20 and 40 μM) for 12 h.
-
OThe densities of FH‐Viperin protein bands in (N) were quantitated with the Image J. Data represent mean and SD of three biological replicates.
Data information: Data (A, G, H, J) represent mean and SD of three (A, J) or four (G, H) biological replicates; Data (B–F, I–L, N) are representative of at least two biological replicates. For all statistical testing, P‐values were calculated using two‐tailed unpaired Student’s t‐test. NS, not significant (P > 0.05). *P < 0.05, **P < 0.01 and ***P < 0.001.
Source data are available online for this figure.
Next, to examine whether high salt regulates viral infection through Viperin, we knocked down Viperin in fibroblast cells. The results showed that Viperin knockdown restricted the high‐salt‐mediated promotion of viral infection (Fig EV3G). Consistently, knockout of human Viperin or mouse orthologous gene Rsad2 abolished the effects of high salt on both VSV and H1N1 infection (Figs 2L and EV3H). Furthermore, re‐expression of Viperin in Rsad2 −/− MEFs rescued high‐salt‐mediated regulation of viral infection (Figs 2M and EV3I). In addition, given that we have demonstrated that the epithelial cell line A549 is deficient in the production of Viperin proteins during viral infection (Yuan et al, 2020), we utilized A549 cells to observe the effect of high salt. The results showed that high salt cannot promote viral infection in A549 cells (Fig EV3J), while transfection of A549 cells with Flag‐Viperin reconstructed high‐salt‐mediated enhancement of viral infection (Fig EV3J). Importantly, mice fed a short‐term (7‐day) HSD showed significantly higher mortality rates when challenged with viruses than control mice (Fig 2N, left). However, a short‐term HSD cannot increase the mortality rates of Rsad2 −/− mice challenged with viruses (Fig 2N, right). Collectively, we demonstrated that high salt reduces Viperin protein levels during viral infection, which substantially exacerbates viral infection.
Identification of USP33 as a key deubiquitinase controlling Viperin protein stability
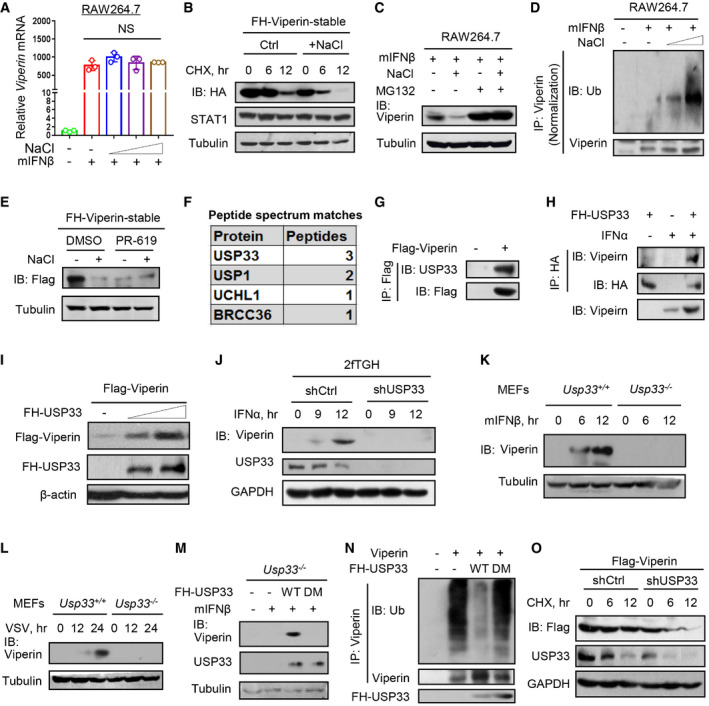
We observed that short‐time high‐salt treatment did not downregulate the IFN‐I‐induced Viperin mRNA levels (Fig 3A), but promoted the degradation of Viperin proteins (Fig 3B), suggesting that high salt regulates Viperin at the protein level. In line with this observation, a short‐time high‐salt treatment decreased exogenously expressed Viperin protein in a time‐ and dose‐dependent manner (Fig EV3K–M). Interestingly, we found that two additional metal salt compounds, FeSO4 and CaCl2, did not noticeably downregulate the Viperin protein levels (Fig EV3N and O). We further found that high‐salt‐mediated downregulation of Viperin proteins can be blocked by a proteasome inhibitor MG132 (Fig 3C). Consistently, high salt promoted Viperin ubiquitination in a dose‐dependent manner (Figs 3D and EV4A). However, high salt did not increase the level of UBE4A (Fig EV4B), a recently identified ubiquitin E3 ligase of Viperin (Yuan et al, 2020). Furthermore, PR‐619, a pan‐deubiquitinase inhibitor (Ritorto et al, 2014), decreased the Viperin protein levels and blocked the high‐salt‐mediated downregulation of Viperin (Fig 3E), suggesting that high salt could target certain deubiquitinases to regulate Viperin proteins.
Figure 3. Identification of USP33 as a key deubiquitinase controlling Viperin protein stability.
-
ART–qPCR analysis of Viperin mRNA in RAW264.7 treated with mIFNβ (300 IU/ml, 12 h) immediately after addition of NaCl (+17, 34 and 51 mM).
-
BWestern blot analysis of FH‐Viperin protein in stable FH‐Viperin‐expressing HeLa cells pretreated with additional NaCl (+34 mM) and then treated with CHX (50 μM) for 6 and 12 h.
-
CRAW264.7 were treated with MG132 (10 μM) and additional NaCl (+34 mM), followed by mIFNβ (300 IU/ml) treatment for 12 h. Viperin protein levels were analyzed by immunoblotting.
-
DImmunoprecipitation analysis of Viperin ubiquitination in RAW264.7 treated with mIFNβ (300 IU/ml) for 12 h immediately after addition of NaCl (+34 mM).
-
EWestern blot analysis of FH‐Viperin protein in stable FH‐Viperin‐expressing HeLa cells treated with PR‐619 (10 μM) together with additional NaCl (+34 mM) for 12 h.
-
FMass spectrometry analysis of potential Viperin‐binding deubiquitinases in HEK293T cells transfected with Flag‐Viperin.
-
GImmunoprecipitation analysis of the interaction between USP33 and Flag‐Viperin in HEK293T cells transfected with Flag‐Viperin.
-
HImmunoprecipitation analysis of the interaction between Viperin and FH‐USP33 in 2fTGH cells transfected with FH‐USP33 and then treated with IFNα (1,000 IU/ml) for 12 h.
-
IWestern blot analysis of Flag‐Viperin levels in HEK293T cells co‐transfected with Flag‐Viperin and increasing amount of Flag‐HA (FH)‐USP33.
-
JWestern blot analysis of Viperin in 2fTGH cells transfected with control shRNAs (shCtrl) or shRNAs against USP33 (shUSP33) and then treated with IFNα (1,000 IU/ml) for different times.
-
K, LWestern blot analysis of Viperin protein in Usp33+/+ or Usp33 −/− MEF cells treated with IFNβ (500 IU/ml) (K) or infected with VSV (MOI = 1.0) (L) as indicated.
-
MWestern blot analysis of Viperin protein in Usp33 −/− MEF cells transfected with FH‐USP33 (wild type, WT; C194S and H673Q, DM) and then stimulated with mIFNβ (500 IU/ml) for 12 h.
-
NImmunoprecipitation analysis of Viperin ubiquitination in HEK293T cells co‐transfected with Viperin and FH‐USP33 (WT or DM).
-
OWestern blot analysis of Flag‐Viperin in stable Flag‐Viperin‐expressing HEK293T cells transfected with shCtrl or shUSP33 and then treated with CHX (50 μM) as indicated.
Data information: Data (A) represent mean and SD of three biological replicates; Data (B–E, G–O) are representative of at least two biological replicates. For all statistical testing, P values were calculated using two‐tailed unpaired Student’s t‐test. NS, not significant (P > 0.05).
Source data are available online for this figure.
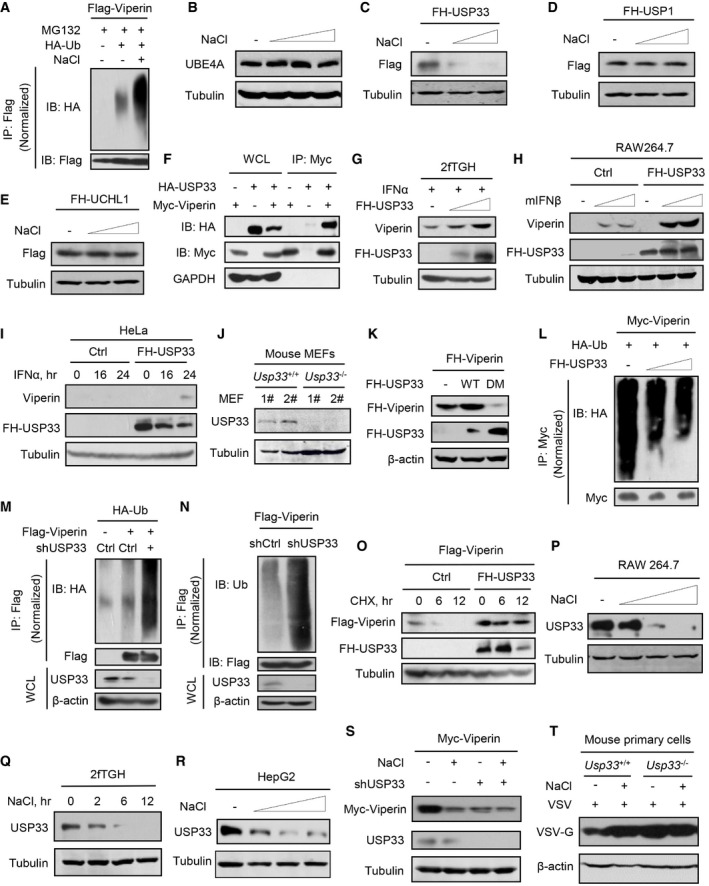
Figure EV4. High salt downregulates USP33, which is a key deubiquitinase of Viperin.
-
AImmunoprecipitation analysis of ubiquitination of Flag‐Viperin in HEK293T cells co‐transfected with Flag‐Viperin and HA‐Ub, and then treated with MG132 (10 μM) and additional NaCl (+34 mM) for 12 h.
-
BWestern blot analysis of UBE4A in RAW264.7 cells treated with additional NaCl (+17, 34, and 51 mM) for 12 h.
-
C–EWestern blot analysis of Flag‐HA (FH)‐USP33 (C), FH‐USP1 (D), FH‐UCHL1 (E) in HEK293T cells transfected with the corresponding plasmids and then treated with additional NaCl (+17 and 34 mM) for 12 h.
-
FImmunoprecipitation analysis of the interaction between Myc‐Viperin and HA‐USP33 in HEK293T cells co‐transfected with Myc‐Viperin and HA‐USP33.
-
GWestern blot analysis of Viperin in 2fTGH cells transfected with increasing amount of FH‐USP33 and then treated with IFNα (5,000 IU/ml) for 15 h.
-
HWestern blot analysis of Viperin in RAW264.7 transfected with empty vectors (Ctrl) or FH‐USP33 and then treated with mIFNβ (500 and 1,000 IU/ml) for 12 h.
-
IWestern blot analysis of Viperin in HeLa cells transfected with FH‐USP33 and then treated with IFNα (1,000 IU/ml) for different times.
-
JWestern blot analysis of USP33 protein levels in two clones (1# and 2#) of Usp33+/+ and Usp33 −/− MEF cells.
-
KWestern blot analysis of FH‐Viperin in HEK293T cells co‐transfected FH‐Viperin and FH‐USP33 (wild type, WT; C194S and H673Q, DM).
-
LImmunoprecipitation analysis of Myc‐Viperin ubiquitination in HEK293T cells co‐transfected with Myc‐Viperin, HA‐Ub, and increasing amount of FH‐USP33.
-
MImmunoprecipitation analysis of Flag‐Viperin ubiquitination in HEK293T cells co‐transfected with Flag‐Viperin, HA‐Ub, and shCtrl or shUSP33.
-
NImmunoprecipitation analysis of Flag‐Viperin ubiquitination in HEK293T cells co‐transfected with Flag‐Viperin and shCtrl or shUSP33.
-
OWestern blot analysis of Flag‐Viperin levels in HEK293T cells co‐transfected Flag‐Viperin and FH‐USP33, and then treated with CHX (50 μg/ml) as indicated.
-
PWestern blot analysis of USP33 in RAW264.7 cells treated with additional NaCl (+17, 34, and 51 mM) for 12 h.
-
QWestern blot analysis of USP33 in 2fTGH cells treated with additional NaCl (+34 mM) for different times.
-
RWestern blot analysis of USP33 in HepG2 cells treated with additional NaCl (+17, 34, and 51 mM) for 12 h.
-
SWestern blot analysis of Myc‐Viperin in HEK293T cells co‐transfected with Myc‐Viperin and shCtrl (−) or shUSP33, then treated with additional NaCl (+34 mM) for 12 h.
-
TWestern blot analysis of VSV‐G in Usp33+/+ and Usp33 −/− mouse liver cells infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
Data information: Data (A–T) are representative of at least two biological replicates.
Source data are available online for this figure.
Next, we performed a mass spectrometry analysis to identify the possible deubiquitinases that target Viperin. The results showed several potential Viperin‐interacting deubiquitinases, including USP33, USP1, UCHL1, and BRCC36 (Fig 3F). Among them, only USP33 was notably downregulated by high salt (Fig EV4C–E). Further studies revealed that USP33 was able to interact with Viperin (Figs 3G and H, and EV4F), and dramatically upregulated both exogenously expressed Viperin and IFN‐I‐induced Viperin (Figs 3I and EV4G–I). Knockdown or knockout of USP33 blocked Viperin protein induction by either IFN‐I (Figs 3J and K, and EV4J) or viruses (Fig 3L). Re‐expression of wild‐type (WT) USP33 but not a mutant lacking deubiquitinase activity (C194S and H673Q, DM; Berthouze et al, 2009) in Usp33 −/− MEFs rescued IFN‐I‐induced Viperin protein production (Fig 3M). Consistently, USP33‐WT but not USP33‐DM increased Viperin protein levels (Fig EV4K) and decreased ubiquitination levels of Viperin (Figs 3N and EV4L). USP33 knockdown resulted in a strong increase in Viperin ubiquitination levels (Fig EV4M and N). In line with these findings, USP33 knockdown markedly accelerated Viperin protein degradation (Fig 3O), while USP33 overexpression improved Viperin protein stability (Fig EV4O). These findings suggested that USP33 is a key deubiquitinase that controls Viperin protein stability and levels in cells.
High salt reduces cellular USP33 and, therefore, decreases Viperin protein stability
We further observed how high salt regulates USP33. We noticed that high salt downregulated the USP33 protein levels in a dose‐dependent manner in different types of cells (Figs 4A and B, and EV4P–R). Importantly, a 7‐day HSD resulted in a significant decrease in the USP33 protein levels in mice, while mice fed a 30‐day HSD had comparable or slightly decreased levels of USP33, compared with mice fed a control diet (Fig 4C), suggesting a possible balance and adaptation of the body to high salt during a long‐term HSD.
Figure 4. High salt reduces cellular USP33 and, therefore, decreases Viperin protein stability.
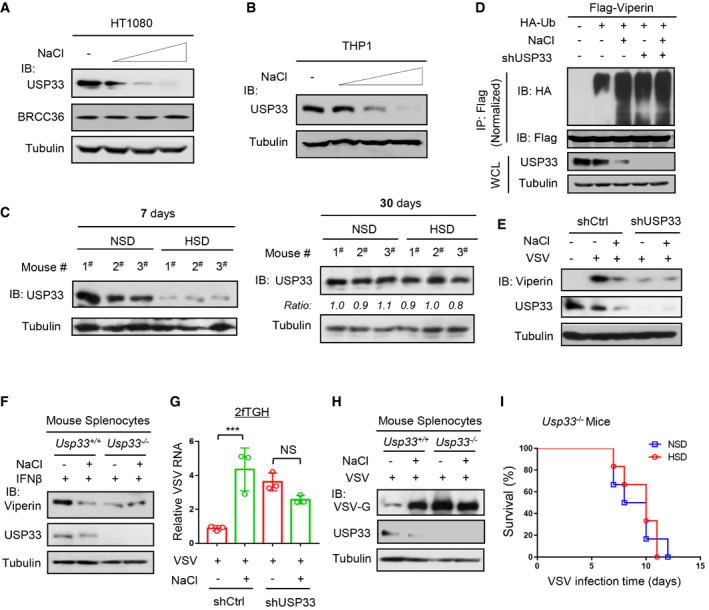
-
A, BWestern blot analysis of USP33 proteins in HT1080 (A) or THP1 (B) cells treated with additional NaCl (+17, 34 and 51 mM) for 12 h.
-
CWestern blot analysis of USP33 levels in the spleen tissues of mice fed an NSD or HSD for 7 days or 30 days as indicated.
-
DImmunoprecipitation analysis of Viperin ubiquitination in HEK293T cells co‐transfected with Flag‐Viperin, HA‐Ub, and shUSP33, then treated with additional NaCl (+34 mM) for 12 h.
-
EWestern blot analysis of Viperin levels in 2fTGH cells transfected with shCtrl or shUSP33 and then infected with VSV (MOI = 1.0, 12 h) immediately after addition of NaCl (+34 mM).
-
FWestern blot analysis of Viperin in the splenocytes of Usp33 +/+ or Usp33 −/− mice treated with mIFNβ (500 IU/ml, 12 h) immediately after addition of NaCl (+34 mM).
-
GRT‐qPCR analysis of VSV RNA levels in 2fTGH cells transfected with shCtrl or shUSP33 and then infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM).
-
HMouse splenocytes of Usp33 +/+ or Usp33 −/− mice were infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM). VSV‐G protein levels were analyzed by Western blot.
-
ISurvival curves of 8‐week‐old Usp33 −/− mice fed an NSD (0.5% NaCl) or HSD (4% NaCl) for 7 days and then infected with VSV (1 × 108 PFU per gram body mouse, n = 10).
Data information: Data (A–F, H) are representative of at least two biological replicates; Data (G) represent mean and SD of three biological replicates, and P‐values were calculated using two‐tailed unpaired Student’s t‐test; Data (I) show mean and SEM of 10 biological replicates, and P‐value was calculated using logrank (Mantel–Cox) tests. For all statistical testing: NS, not significant (P > 0.05). ***P < 0.001.
Source data are available online for this figure.
Furthermore, we found that USP33 knockdown inhibited the high‐salt‐mediated increase in Viperin ubiquitination and decrease in Viperin protein levels (Figs 4D and EV4S). USP33 deficiency also abolished the high‐salt‐mediated downregulation of Viperin proteins induced by viruses (Fig 4E) or IFN‐I (Fig 4F). As a consequence, knockout or knockdown of USP33 blocked the high‐salt‐mediated promotion of viral infection (Figs 4G and H, and EV4T). Consistently, high salt lost the ability to increase the mortality rates of Usp33 −/− mice when challenged with viruses (Fig 4I). Together, these findings suggested that high salt reduces USP33 levels to lower Viperin protein stability, thus attenuating host antiviral ability.
High salt stimulates degradation of ubiquitinated proteins via p97
We noticed that short‐time high‐salt treatment did not affect USP33 mRNA levels (Fig 5A), but regulated proteasome‐dependent degradation of USP33 proteins (Fig 5B). Surprisingly, we found that high salt actually reduced USP33 ubiquitination levels (Figs 5C and EV5A). Based on the contradiction between the protein and ubiquitination levels of USP33, we postulated that high salt may specifically promote the degradation of ubiquitinated proteins. Thus, we immunoprecipitated ubiquitinated USP33 proteins from Flag‐USP33‐overexpressing cells and found a dramatic decrease in ubiquitinated Flag‐USP33 when the cells were treated with high salt (Fig 5D, left). However, the non‐ubiquitinated USP33 protein levels remained relatively stable before and after high‐salt treatment (Fig 5D, right). Considering that p97 has been well documented to be an important adaptor that recruits ubiquitinated proteins and promotes the delivery of ubiquitinated proteins to the proteasome for degradation, we determined the role of p97 in the high‐salt‐mediated regulation of USP33. We noticed that a p97 inhibitor, NMS873, largely inhibited USP33 downregulation by high salt (Fig 5E). Furthermore, p97 knockout abolished the effects of high salt on USP33 protein levels (Fig 5F) and ubiquitination (Fig 5G), suggesting that high salt regulates the ubiquitination and protein levels of USP33 through p97.
Figure 5. High salt stimulates degradation of ubiquitinated proteins via p97.
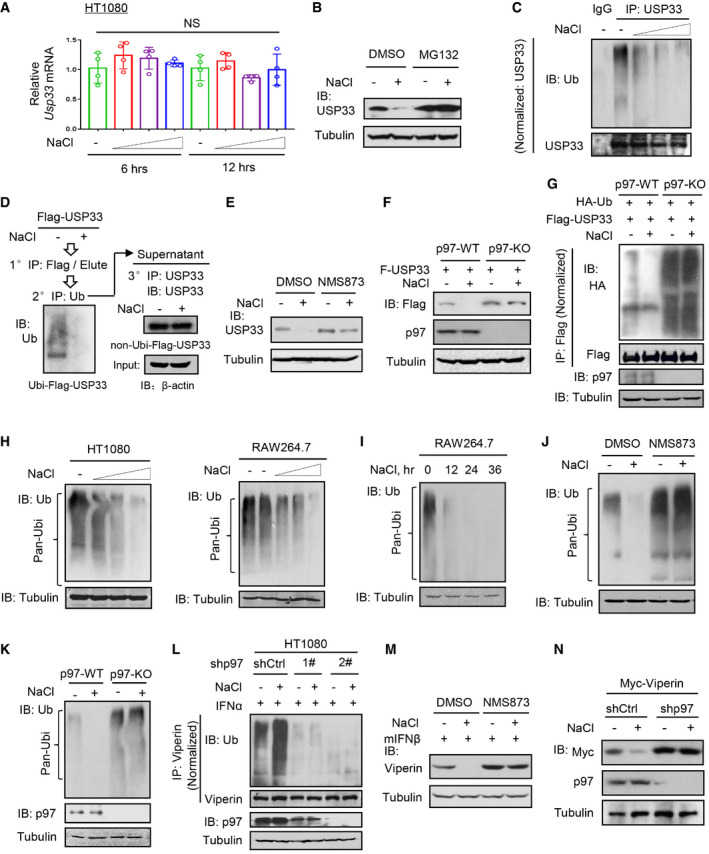
-
ART–qPCR analysis of USP33 mRNA levels in HT1080 cells treated with additional NaCl (+17, 34 and 51 mM) for 6 and 12 h.
-
BWestern blot analysis of USP33 protein in RAW264.7 treated with MG132 (10 μM) and additional NaCl (+34 mM) for 12 h.
-
CImmunoprecipitation analysis of USP33 ubiquitination in RAW264.7 treated with additional NaCl (+17, 34 and 51 mM) for 12 h.
-
DHEK293T cells were transfected with Flag‐USP33. Flag‐USP33 proteins were immunoprecipitated with Flag beads (1° IP) and then eluted by Flag peptides. The eluted Flag‐USP33 proteins were further immunoprecipitated by an anti‐Ub antibody (2° IP). The immunoprecipitates were subjected to immunoblotting by an anti‐Ub antibody (left), and the supernatant were further immunoprecipitated (3° IP) and immunoblotted by a USP33 antibody (right).
-
EWestern blot analysis of USP33 in HT1080 cells treated with NMS873 (10 μM) and additional NaCl (+34 mM) for 12 h.
-
FWestern blot analysis of USP33 protein in p97‐sufficient (WT) or p97‐knockout (KO) 2fTGH cells transfected with Flag‐USP33 (F‐USP33) and then treated with additional NaCl (+34 mM) for 12 h.
-
GImmunoprecipitation analysis of ubiquitination of Flag‐USP33 in p97‐WT or p97‐KO 2fTGH cells co‐transfected with Flag‐USP33 and HA‐Ub and then treated with NaCl (+34 mM) for 12 h.
-
H, IWestern blot analysis of ubiquitination of cellular proteins (Pan‐Ubi) in HT1080 and RAW264.7 cells treated with additional NaCl (+17, 34 and 51 mM) for 12 h (H), or in RAW264.7 treated with additional NaCl (+34 mM) for different times (I).
-
JWestern blot analysis of ubiquitination of cellular proteins (Pan‐Ubi) in HT1080 cells treated with NMS873 (10 μM) and additional NaCl (+34 mM) for 12 h.
-
KWestern blot analysis of ubiquitination of cellular proteins (Pan‐Ubi) in p97‐WT or p97‐KO HEK293T cells treated with additional NaCl (+34 mM) for 12 h.
-
LImmunoprecipitation analysis of Viperin ubiquitination in HT1080 cells transfected with shCtrl or shp97 (1# or 2#) and then treated with IFNα (1,000 IU/ml, 12 h) immediately after addition of NaCl (+34 mM).
-
MWestern blot analysis of Viperin in RAW264.7 treated with NMS873 (10 μM), together with additional NaCl (+34 mM), and then treated with mIFNβ (500 IU/ml) for 12 h.
-
NWestern blot analysis of Myc‐Viperin in HEK293T cells co‐transfected with Myc‐Viperin and shCtrl or shp97, and then treated with additional NaCl (+34 mM) for 12 h.
Data information: Data (A) represent mean and SD of four biological replicates; Data (B–N) are representative of at least two biological replicates. For all statistical testing, P‐values were calculated using two‐tailed unpaired Student’s t‐test. NS, not significant (P > 0.05).
Source data are available online for this figure.
Figure EV5. High salt stimulates degradation of ubiquitinated proteins via p97.
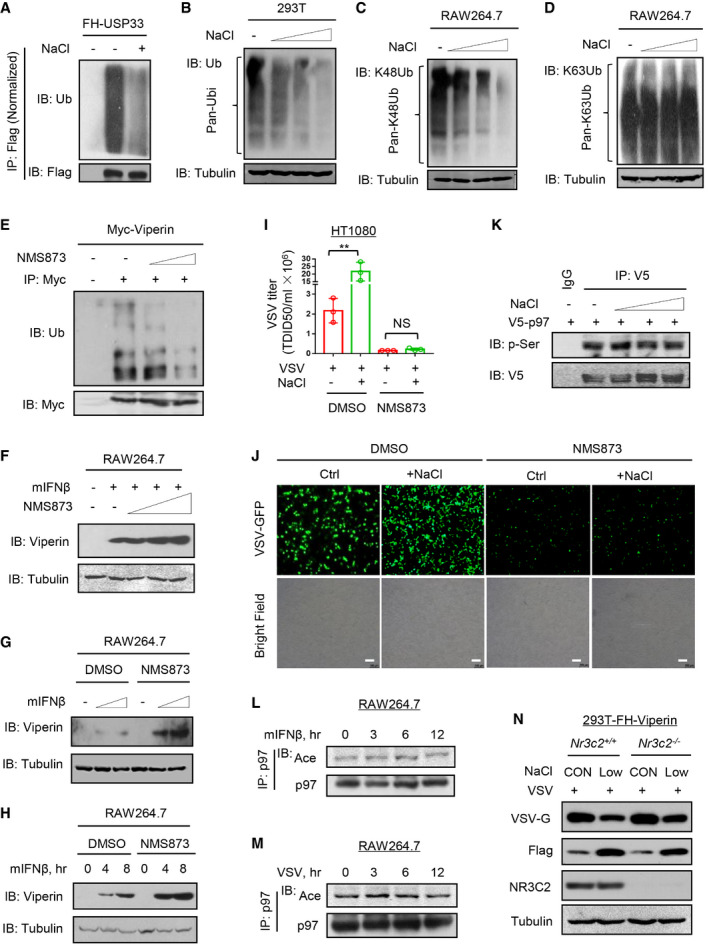
-
AImmunoprecipitation analysis of FH‐USP33 ubiquitination in HEK293T cells transfected with FH‐USP33 and then treated with additional NaCl (+34 mM) for 12 h.
-
BImmunoprecipitation analysis of ubiquitination (pan‐Ubi) of cellular proteins in HEK293T cells treated with additional NaCl (+17, 34, and 51 mM) for 12 h.
-
C, DImmunoprecipitation analysis of K48‐linked (C) or K63‐linked (D) ubiquitination of cellular proteins in RAW264.7 cells treated with additional NaCl (+17, 34, and 51 mM) for 12 h using a specific anti‐K48‐Ub chain or anti‐K63‐Ub chain antibody.
-
EImmunoprecipitation analysis of Myc‐Viperin ubiquitination in HEK293T cells transfected with Myc‐Viperin and then treated with NMS873 (3 and 5 μM) for 6 h.
-
FWestern blot analysis of Viperin in RAW264.7 pretreated with NMS873 (1, 3 and 6 μM) for 2 h and then treated with mIFNβ (500 IU/ml) for 6 h.
-
G, HWestern blot analysis of Viperin in RAW264.7 pretreated with NMS873 (10 μM) for 2 h and then treated with mIFNβ (500 and 1,000 IU/ml) for 6 h (G) or with mIFNβ (500 IU/ml) for 4 and 8 h (H).
-
ITCID50 assay of VSV titers in culture supernatants from HT1080 cells infected with VSV (MOI = 1.0, 24 h) immediately after addition of NaCl (+34 mM) and NMS873 (10 μM). Data represent mean and SD of three biological replicates. NS, not significant (P > 0.05). **P < 0.01 (two‐tailed unpaired Student’s t‐test).
-
JFluorescence microscopy of VSV with a green fluorescent protein gene (GFP) in HT1080 cells infected with VSV‐GFP (MOI = 0.5, 24 h) immediately after addition of NaCl (+34 mM) and NMS873 (10 μM). Scale bars, 100 µm.
-
KImmunoprecipitation analysis of pan‐serine phosphorylation (p‐Ser) of V5‐tagged p97 (V5‐p97) in RAW264.7 cells transfected with V5‐p97 and then treated with additional NaCl (+17, 34 and 51 mM) for 12 h using an anti‐pan‐serine phosphorylation antibody.
-
L, MImmunoprecipitation analysis of pan‐acetylation (Ace) of p97 in RAW264.7 cells treated with mIFNβ (500 IU/ml) (L) or VSV (MOI = 1.0) (M) for indicated times.
-
NHEK293T cells with stable expression of FH‐Viperin were used to make Nr3c2 +/+ and Nr3c2 −/− cells using CRISPR‐Cas9. Then cells were infected with VSV (MOI = 1.0) in media containing normal (CON) or reduced (Low, −34 mM) concentration of NaCl for 12 h. VSV‐G and FH‐Viperin levels were analyzed by western blot.
Data information: Data (A–H, K–N) are representative of at least two biological replicates; Data (I) represent mean and SD of three biological replicates. For all statistical testing, P‐values were calculated using two‐tailed unpaired Student’s t‐test. NS, not significant (P > 0.05). **P < 0.01.
Source data are available online for this figure.
Given that p97 widely regulates the degradation of ubiquitinated proteins, we further determined the effects of high salt on ubiquitinated proteins in the whole cells. Interestingly, high salt strongly promoted the downregulation of cellular ubiquitinated proteins in a dose‐ and time‐dependent manner in different cells (Figs 5H and I, and EV5B). Further studies revealed that high salt induced a decrease in the Lys48‐ but not Lys63‐linked ubiquitination of proteins (Fig EV5C and D). Moreover, both the p97 inhibitor NMS873 and p97 knockout abolished the high‐salt‐mediated decrease in the ubiquitination of cellular proteins (Fig 5J and K), suggesting that the effect of high salt on cellular ubiquitinated proteins is dependent on p97.
Consistent with these observations, the p97 inhibitor reduced Viperin ubiquitination levels (Fig EV5E). Furthermore, the high‐salt‐mediated upregulation of Viperin ubiquitination was inhibited by p97 knockdown (Fig 5L). Consequently, both NMS873 and p97 knockdown restricted the high‐salt‐mediated downregulation of Viperin protein levels (Fig 5M and N). Additionally, the p97 inhibitor increased the IFN‐I‐induced expression of Viperin proteins (Fig EV5F–H), and restricted the high‐salt‐mediated promotion of viral infection (Fig EV5I and J). Collectively, we uncovered a high‐salt‐stimulated signaling mechanism by which high salt promotes the degradation of ubiquitinated proteins in cells through p97.
High salt promotes p97 acetylation to recruit USP33 for degradation
We further explored the molecular mechanism by which high salt regulates p97 activity. High salt did not change the p97 protein levels in either RAW264.7 or HT1080 cells (Fig 6A and B). However, high salt markedly promoted the interaction of p97 with USP33 (Fig 6C). Our previous studies have revealed the roles of several different post‐translational modifications of cellular proteins in regulating IFN‐I antiviral signaling (Guo et al, 2019; Yuan et al, 2020; Zuo et al, 2020). Thus, we further analyzed the potential post‐translational modifications of p97. The results showed that high salt robustly stimulated p97 acetylation but not serine phosphorylation (Figs 6D and EV5K). In addition, neither viral infection nor IFN‐I treatment significantly induced p97 acetylation (Figs EV5L and M). We then performed mass spectrometry analyses and found two potential acetylation sites of p97, Lys663 and Lys651, which could be stimulated by high salt (Fig 6E and F). We found that mutating Lys663 but not Lys651 of p97 to arginine (R) abolished the high‐salt‐induced upregulation of p97 acetylation (Fig 6G), suggesting that high salt could stimulate the acetylation of p97 at Lys663.
Figure 6. High salt promotes p97 acetylation to recruit USP33 for degradation.
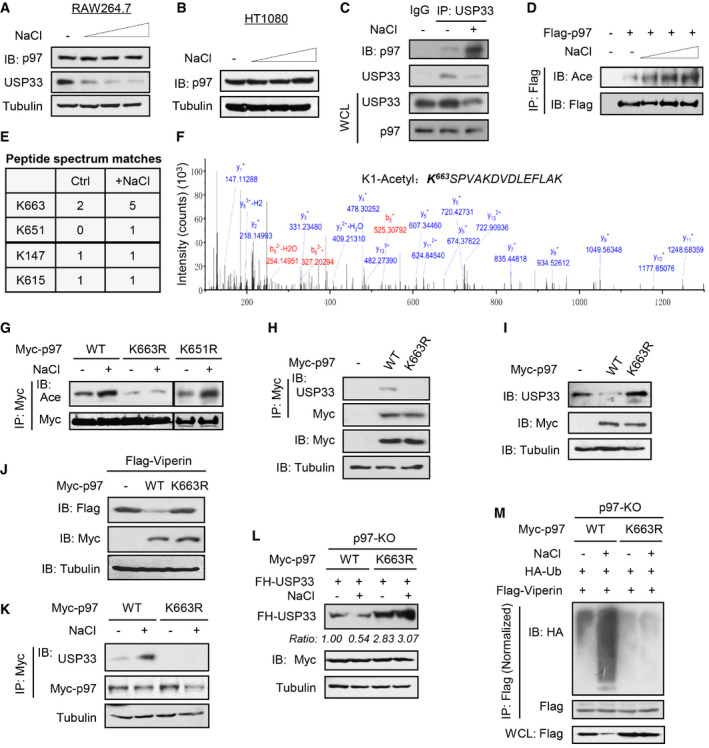
-
A, BWestern blot analysis of p97 levels in RAW264.7 (A) or HT1080 (B) cells treated with additional NaCl (+17, 34 and 51 mM) for 12 h.
-
CImmunoprecipitation analysis of the interaction between USP33 and p97 in RAW264.7 treated with additional NaCl (+34 mM) for 12 h.
-
DImmunoprecipitation analysis of acetylation of Flag‐p97 in HEK293T cells transfected with Flag‐p97 and then treated with additional NaCl (+17, 34 and 51 mM) for 12 h.
-
E, FMass spectrometry analysis of acetylation of Flag‐p97 (E) in RAW264.7 transfected with Flag‐p97 and then treated with additional NaCl (+34 mM) or control ddH2O (Ctrl) for 12 h. The intensity of p97‐K663 site was shown in (F).
-
GImmunoprecipitation analysis of acetylation of Myc‐p97 in HEK293T cells transfected with Myc‐p97 (WT, K663R or K651R) and then treated with additional NaCl (+34 mM) for 12 h.
-
HImmunoprecipitation analysis of the interaction between USP33 and Myc‐p97 in HEK293T cells transfected with Myc‐p97 (WT or K663R).
-
IWestern blot analysis of USP33 levels in HEK293T cells transfected with empty vectors (−) or Myc‐p97 (WT or K663R).
-
JWestern blot analysis of Flag‐Viperin levels in HEK293T cells co‐transfected with Flag‐Viperin and Myc‐p97 (WT or K663R).
-
KImmunoprecipitation analysis of the interaction between USP33 and Myc‐p97 in HEK293T cells transfected with Myc‐p97 (WT or K663R) and then treated with additional NaCl (+34 mM) for 12 h.
-
LWestern blot analysis of FH‐USP33 levels in p97‐KO HEK293T cells co‐transfected with Myc‐p97 (WT or K663R) and Flag‐USP33, and then treated with additional NaCl (+34 mM) for 12 h.
-
MImmunoprecipitation analysis of Viperin ubiquitination in p97‐KO 2fTGH cells co‐transfected with Myc‐p97 (WT or K663R), Flag‐Viperin, and HA‐Ub as indicated, and then treated with additional NaCl (+34 mM) for 12 h.
Data information: Data (A–D, G–M) are representative of at least two biological replicates.
Source data are available online for this figure.
Importantly, mutating Lys663 blocked the binding of p97 to USP33 (Fig 6H) and the subsequent downregulation of USP33 proteins (Fig 6I). Consistently, overexpression of p97, but not its K663R mutant, reduced Viperin protein levels (Fig 6J). We further explored the role of Lys663 of p97 in the high‐salt‐mediated regulation of USP33 and Viperin. The results showed that mutating Lys663 of p97 abolished the high‐salt‐stimulated interaction between p97 and USP33 (Fig 6K). In p97‐KO cells, in which high salt cannot reduce USP33 (Fig 5F), re‐expressing wild‐type p97 but not its K663R mutant rescued the high‐salt‐mediated downregulation of USP33 proteins (Fig 6L). Moreover, mutating Lys663 of p97 inhibited the high‐salt‐mediated increase in Viperin ubiquitination and decrease in Viperin protein levels (Fig 6M). Taken all together, our findings revealed that high salt stimulates p97 acetylation at Lys663, which promotes the proteasome‐dependent degradation of USP33 and in turn lowers the level of Viperin protein induced by viruses or IFN‐I, and ultimately attenuates host antiviral ability.
A low‐salt diet during viral infection improves host antiviral ability
Interestingly, we noticed that lowering the NaCl concentration in the cell culture media attenuated p97 acetylation (Fig 7A). Consistently, lowering the salt concentration upregulated the levels of USP33 protein (Fig 7B). When the salt concentration in the cell culture media was reduced, IFN‐I can induce higher levels of Viperin proteins (Fig 7C), and substantially inhibited viral infection in a dose‐dependent manner (Fig 7D). In addition, given that aldosterone‐mineralocorticoid receptor system is readily activated by the treatment of low salt, we analyzed whether the aldosterone‐mineralocorticoid receptor NR3C2 plays a role in regulating low‐salt‐mediated inhibition of viral infection. The results showed that knockout of NR3C2 did not significantly affect the effect of low salt on viral infection (Fig EV5N).
Figure 7. A low‐salt diet during viral infection improves Viperin expression and host antiviral ability.
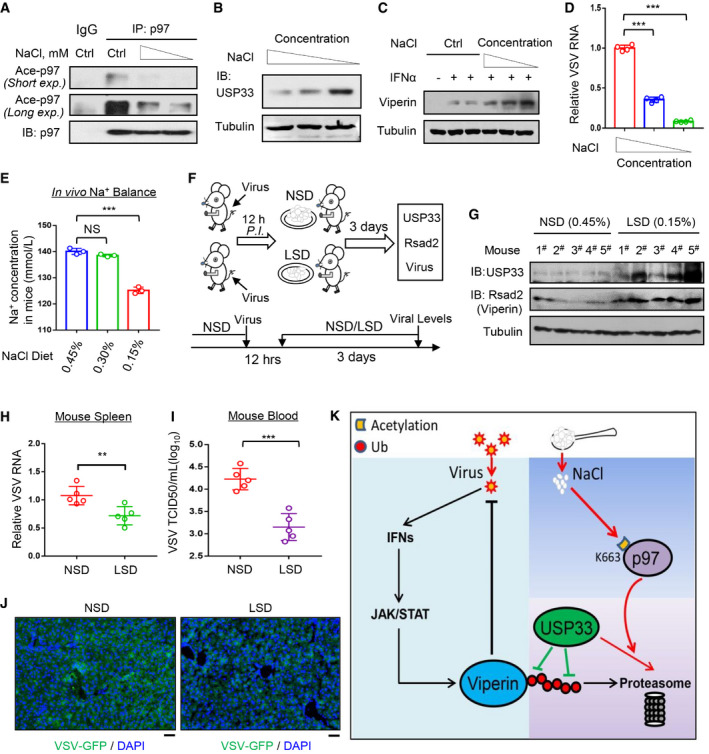
- Immunoprecipitation analysis of acetylation of p97 in RAW264.7 cells cultured in media containing reduced concentration of NaCl (−17 and −34 mM) for 12 h.
- Western blot analysis of USP33 in RAW264.7 cells cultured in media containing reduced concentration of NaCl (−17 and −34 mM) for 12 h.
- Western blot analysis of Viperin in HT1080 cells treated with IFNα (1,000 IU/ml, 12 h) in media containing reduced concentration of NaCl (−17, −34 and −51 mM).
- RAW264.7 cells were infected with VSV (MOI = 1.0, 12 h) in media containing reduced concentration of NaCl (−17 and −34 mM). Viral RNA levels were analyzed by RT‐qPCR.
- Mice (n = 3) were fed an NSD (0.45% NaCl) or LSD (0.30% or 0.15% NaCl) for 3 days. Na+ concentration in mouse serum was analyzed by a Micro Blood Sodium Concentration Assay Kit.
- Schematic diagram of analysis of in vivo viral levels in VSV‐infected mice fed either an NSD (0.45% NaCl) or LSD (0.15% NaCl).
- Western blot analysis of USP33 and Viperin protein levels in the spleen tissues of mice (n = 5) fed an NSD (0.45% NaCl) or LSD (0.15% NaCl) for 3 days as indicated.
- Mice (n = 5) were intraperitoneally infected with VSV (1 × 108 PFU per gram body). Twelve hours post infection, mice were fed an NSD (0.45% NaCl) or LSD (0.15% NaCl) for 3 days. Then VSV RNA levels in mouse spleen tissues were detected by RT‐qPCR.
- Virus titers in the mouse blood (n = 5) from (H) were analyzed by the TCID50 assay.
- Immunofluorescence analysis of the amount of VSV‐GFP in liver tissues of mice intraperitoneally infected with VSV‐GFP (1 × 107 PFU per gram body) and then fed an NSD/LSD as (H). Scale bars, 20 µm.
- A proposed model of high‐salt‐mediated restriction of host antiviral ability by activating p97 acetylation and subsequent degradation of the deubiquitinase USP33 and antiviral protein Viperin.
Data information: Data (A–C, G) are representative of at least two biological replicates; Data (D) represent mean and SD of four biological replicates; Data (E, H, I) show mean and SEM of three (E) or five (G–I) biological replicates. For all statistical testing: P‐values were calculated using two‐tailed unpaired Student’s t‐test. N.S, not significant (P > 0.05) and **P < 0.01, ***P < 0.001 (two‐tailed unpaired Student’s t‐test).
Source data are available online for this figure.
Thus, we further explored the effects of a low‐salt diet (LSD) on regulating host antiviral ability. By testing the diets with different levels of salt in mice, we found that a diet containing 0.15% NaCl can result in decreased NaCl concentrations in vivo (Fig 7E). Hence, mice were fed an NSD (0.45% NaCl) or LSD (0.15% NaCl) 12 h post infection with viruses. After 3 days, USP33 and Viperin protein levels, as well as viral levels, were measured in mouse tissues and blood (Fig 7F). We found that an LSD improved both USP33 and Rsad2 protein levels in mouse spleen tissues (Fig 7G). In line with these increased antiviral proteins, switching to an LSD during viral infection significantly reduced viral RNA levels in mouse spleen tissues (Fig 7H), and lowered viral titers in blood (Fig 7I). The immunofluorescence analysis of VSV‐GFP in mouse liver tissues confirmed that an LSD inhibited viral infection in vivo (Fig 7J). Taken together, these findings demonstrated that an LSD during viral infection enhances host antiviral ability.
Discussion
High salt has been implicated in numerous disorders. The impacts of a long‐term HSD have been observed in animal models and in the clinic, which revealed the association between high salt and hypertension, cardiovascular disease, and autoimmune diseases (Kleinewietfeld et al, 2013; Manzel et al, 2014; O'Donnell et al, 2014). Here, we uncovered the role of high salt in regulating antiviral responses. On the whole, high salt stimulates acetylation of p97 at Lys663, which activates p97 to induce degradation of certain ubiquitinated proteins. USP33, whose levels are critically controlled by p97‐mediated degradation (Chan et al, 2014), thus undertook a substantial decrease in protein levels under treatment of high salt. USP33 is an important deubiquitinase of antiviral protein Viperin and, therefore, its deficiency results in increased ubiquitination of Viperin protein and decreased protein levels of Viperin. As a potent antiviral protein induced by IFN‐I signaling, Viperin deficiency in cells largely attenuates IFN‐I antiviral potency upon viral infection, which consequently leads to a promotion of viral infection by high salt.
In this study, we found that a short‐term (7 days) HSD promoted viral infection in mice. However, this effect was largely compromised during the relatively long‐term (30 days) consumption of the HSD. Using high‐salt cell models, we further revealed that short‐time high‐salt treatment stimulates the p97‐dependent degradation of USP33, a key stabilizer of antiviral Viperin protein produced during viral infection, which leads to a marked decrease in cellular antiviral ability. In line with this cell model, we noticed that a short‐term (7 days) HSD strongly reduced the USP33 protein levels in mice. However, USP33 protein levels were not markedly downregulated during the relatively long‐term (30 days) HSD (Fig 4C), which suggests a homeostasis mechanism by which the host counters USP33 reduction. With these findings, we further revealed that switching to an LSD during viral infection substantially upregulated USP33 and Viperin protein levels in mice and eventually improved host antiviral immunity. Thus, this study could provide insight into the consumption of a diet containing salt during a period of virus epidemic.
It is understandable that it is difficult to study the actual effects of high salt in a perfect cell model. The conditions of cells treated with high salt for a long time does not represent the real response of the body to high salt in vivo, since the body has many regulators, including arterial pressure, renal sympathetic tone, and the renin–angiotensin–aldosterone system, to rapidly control sodium balance (Bie, 2018). In addition, as observed in our studies, continuous high‐salt treatment of cells for over 24 h could have led to cell apoptosis to some extent, which will never happen in normal bodies. Actually, long‐time treatment of cells with high salt could induce more destructive cell responses. To reduce the destructive effects on cell conditions caused by long‐time high‐salt treatment and be as close as possible to the time length of in vivo regulation of high salt concentration by the salt balance mechanisms, we harvested cells within 12 h of high‐salt treatment in those experiments with NaCl treatment alone, and in other cases, we treated cells with other stimulators (such as viruses or IFN‐I) immediately after the addition of high salt, in order to decrease the duration of high‐salt treatment in cells. We noticed that these cell models showed consistent antiviral phenotypes with an in vivo HSD mouse model. Importantly, with these cell models we clearly demonstrated that high salt strongly blocks virus‐ and IFN‐I‐induced Viperin expression and ultimately leads to a marked reduction in cellular antiviral ability.
High‐salt‐mediated inhibition of Viperin protein expression is capable of greatly lowering cellular antiviral ability, which was clearly demonstrated by the observations that high salt lost the ability to affect antiviral activity in Viperin‐knockdown, Viperin −/− or Rsad2 −/− cells and Rsad2 −/− mice (Figs 2L–N and EV3G–J). These facts indicate the important role of Viperin proteins in regulating cellular antiviral response. In line with this idea, Viperin has been demonstrated to be a powerful antiviral effector against a broad array of viruses (Helbig et al, 2005; Wang et al, 2007; Seo et al, 2011), and Viperin can even directly block viral RNA synthesis by generating the ribonucleotide ddhCTP (Gizzi et al, 2018). In fact, a recent study has reported that RSAD2/Viperin is one of the top ISGs that undergo the most dramatic induction by IFN‐I (Liu et al, 2012). Thus, we speculated that viral infection activates cellular antiviral immune response, such as type‐I interferon signaling, which produces various antiviral proteins, while high salt restricts some of these antiviral proteins upregulated by viruses to different extents, leading to a reduction in cellular antiviral ability. Among these inhibited antiviral proteins, Viperin shows a very dramatic downregulation by high salt and, therefore, plays a major role in affecting host antiviral ability.
We previously elucidated that Viperin protein is quite unstable in cells and undergoes rapid degradation upon expression via HAT1‐UBE4A signaling (Yuan et al, 2020). Here, we revealed the deubiquitinase USP33 as another key regulator of Viperin protein stability. Cellular deficiency of USP33 blocked Viperin protein induction by both viruses and IFN‐I (Fig 3K and L). Thus, we think that high‐salt‐mediated reduction of USP33 largely restricts Viperin protein induction during viral infection. In addition, this study for the first time uncovered not only acetylation‐mediated regulation of p97 activity but also p97‐mediated regulation of cellular antiviral response. Although p97 has been reported to promote replication of several viruses, including poliovirus (Arita et al, 2012) and human cytomegalovirus (Lin et al, 2017), the roles of p97 in regulating cellular antiviral response remain unexplored. We disclosed that acetylation of p97 by high salt damages Viperin‐mediated antiviral activity, and inhibition of p97 strongly blocks viral infection in cells (Fig EV5I and J). In addition, an interesting observation is that high salt affects the universal ubiquitination via p97 in macrophages. Thus, it will be attractive to study how the regulation of protein ubiquitination by high salt affects other physiological and pathological functions of macrophages in the future. Together, this study revealed USP33 and p97 as two important host factors for antiviral immunity.
In conclusion, we reveal that high salt stimulates the acetylation of p97 that recruits USP33 for rapid degradation, which, in turn, inhibits Viperin protein induction during viral infection, leading to a reduction in host antiviral ability (Fig 7K). These findings promote an understanding of the dietary salt‐mediated regulation of cellular signaling proteins, uncover the instant detrimental effect of high salt on antiviral immunity, and highlight the benefit of a low‐salt diet to host antiviral ability, which could provide valuable guidance for the consumption a salt‐containing diet during viral infection.
Materials and Methods
Mice
Rsad2 −/− and Usp33 −/− mice on a C57BL/6 background were generated by the Cyagen Biosciences Inc. (Guangzhou, China). Ifnar1 −/− mice were nice gifts from Prof. Chunsheng Dong (Soochow University). Wild‐type C57BL/6 mice were purchased from the Laboratory Animal Center of Soochow University. Mice were maintained and bred in special pathogen‐free (SPF) conditions in the Experimental Animal Center of Soochow University. Six‐ to eight‐week‐old mice were used for most experiments. No specific randomization procedures were used. Animals were distributed into groups by animal caretakers unaware of the study design. Animal care and use protocol adhered to the National Regulations for the Administration of Affairs Concerning Experimental Animals. All protocols and procedures for mouse studies were performed in accordance with the Laboratory Animal Management Regulations with approval of the Scientific Investigation Board of Soochow University.
High‐salt diet, low‐salt diet, and high‐salt injection
Mice received normal chow (0.45% NaCl) and tap water ad libitum (NSD), or NaCl‐rich chow (4% NaCl) and tap water containing 1% NaCl ad libitum (HSD), or low‐salt chow (0.30% or 0.15% NaCl) and tap water ad libitum (LSD). The chows containing different concentrations of NaCl are purchased from the Jiangsu Xietong Bioengineering Company (Nanjing, China). For high‐salt injection, mice were intravenously injected via tail vein with the high‐salt solution (3% NaCl in ddH2O) or control salt solution (0.9% NaCl in ddH2O).
Cells isolation from mice
The primary mouse splenocytes were isolated from mouse spleen tissues, which were cut into small pieces and digested by the erythrocyte lysis buffers. After centrifugation, the cells were collected and then cultured in RPMI medium until further experiments. For the mouse embryonic fibroblasts (MEFs), Rsad2 −/−, Ifnar1 −/−, and Usp33 −/− embryos were obtained from the pregnant 13‐day‐old mice. Briefly, the embryos were cut into pieces and digested in 0.05% trypsin/EDTA with 100 units of DNase I, and then cultured in the DMEM medium containing 10% FBS, 200 mM l‐glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. For the BMDMs, mouse bone marrows were prepared from the 8‐week‐old adult mice. Then the cells were cultured in RPMI medium supplemented with GM‐CSF (50 ng/ml) for 7 days.
Expression constructs and reagents
HA‐tagged mouse Viperin was a nice gift from Dr. Peter Cresswell (Yale University). Myc‐tagged mouse Viperin was a nice gift from Dr. Zhenghong Yuan (Fudan University, China). Flag‐tagged human Viperin was a nice gift from Dr. Chunfu Zheng (Fujian Medical University, China). Myc‐tagged human Viperin and Flag‐HA (FH)‐tagged Viperin were generated using PCR amplification from Flag‐tagged human Viperin. HA‐ubiquitin (HA‐Ub) were gifts from Dr. Lingqiang Zhang (State Key Laboratory of Proteomics, Beijing). FH‐USP33, FH‐USP1, and FH‐UCHL1 were purchased from Addgene. Myc‐p97 and V5‐p97 were purchased from the Vigene Biosciences. All shRNAs against Viperin (shViperin), USP33 (shUSP33) or p97 (shp97) were purchased from GENECHEM (Shanghai, China). HBV‐replicon and HCV‐replicon were nice gifts from Dr. Serge Y. Fuchs (University of Pennsylvania). All mutations were generated by the QuickChange Lightning site‐Directed Mutagenesis Kit (Stratagene, 210518). All plasmids were confirmed by DNA sequencing. Recombinant human IFNα and IFNβ were purchased from PEPROTECH. Recombinant mouse IFNβ (mIFNβ) was purchased from the R&D Systems. Flag peptide (F3290) and LPS (L2630) were purchased from Sigma. NMS873 and PR‐619 were purchased from the Selleck. Cycloheximide (CHX), MG132, puromycin, Polybrene, and other chemicals were purchased from Sigma.
Cell culture and maintenance, transfection
HEK293T, A549, RAW264.7, HeLa, HT1080, 2fTGH, HepG2, HCT116, and Vero cells were obtained from the American Type Culture Collection. U3A cells were kindly provided by Guo‐Qiang Chen (Shanghai Jiaotong University, China). The cells above were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone) supplemented with 10% FBS (GIBCO, Life Technologies), 100 units/ml penicillin, and 100 μg/ml streptomycin. U937 and THP1 cells were obtained from ATCC and were cultured in RPMI 1640 medium (HyClone). All cells were cultured at 37°C under 5% CO2. All transient transfections were carried out using either Lipofectamine 2000 (Thermo Fisher), or LongTrans (Ucallm), or PEI (Polyetherimide) according to manufacturer’s instructions.
For short‐time (12 h) treatment of cells in media containing reduced concentration of sodium chloride, the media were made by 1× PBS sterile buffer supplemented with 4.0 mM l‐Glutamine, 4,500 mg/l Glucose, 110 mg/l Sodium Pyruvate, and different concentrations of NaCl (final concentration: 154 mM, 137 mM and 120 mM), together with 10% FBS (GIBCO, Life Technologies), 100 units/ml penicillin, and 100 μg/ml streptomycin.
Lentivirus preparation
HEK293T cells were co‐transfected with shRNAs (shViperin, or shUSP33, or shp97, or control shRNAs) and three helper vectors (pMDL, pVSVG, and pREV). After 48 h, the culture supernatant containing lentiviruses was collected. Then the lentiviral particles were purified by PEG‐8000 and collected in cold 1× PBS. The aliquot lentiviruses were stored at −80°C until further experiments.
Mass spectrometry analysis
For analysis of Viperin‐binding proteins, HEK293T cells were transfected with empty vectors or Flag‐Viperin. 48 h after transfection, cells were harvested by Nonidet P‐40 lysis buffer containing 150 mM NaCl, 20 mM Tris‐HCl (PH7.4), 1% NP‐40, 0.5 mM EDTA, PMSF (50 μg/ml), and protease inhibitors mixtures (Sigma). For analysis of the acetylation of p97, RAW264.7 cells were transfected with Flag‐p97 and then treated with or without NaCl (+34 mM) for 12 h. Next, the M2 affinity gel was used to pull down Flag‐Viperin from the whole cell lysates. SDS‐PAGE gels were stained with the Silver Staining kits (Beyotime, P0017S). The gel bands from control and experimental samples were carefully excised, and then were digested with trypsin. The resulting tryptic peptides were purified using C18 Zip‐Tip and analyzed by an Orbitrap Elite hybrid mass spectrometer (Thermo Fisher) coupled with a Dionex LC. The peptide spectrum matches (PSMs) for Viperin were obtained after database search using Proteome Discoverer 1.4 against a UniProt protein database containing 89,796 entries with the addition of 247 common contaminants. False discovery rate for peptide identification was set as 1%.
Quantitative proteomic analysis with a TMT2‐plex method
RAW264.7 cells were treated with mIFNβ (500 IU/ml) for 12 h immediately after the addition of NaCl (+34 mM) or control ddH2O (Ctrl). Protein–peptide identification and quantitation was performed by the PTM BioLab Inc. (Hangzhou, China). Sample was sonicated three times on ice using a high‐intensity ultrasonic processor (Scientz) in lysis buffer (8 M urea, 1% Protease Inhibitor Cocktail). The remaining debris was removed by centrifugation at 12,000 g at 4°C for 10 min. Finally, the supernatant was collected and the protein concentration was determined with BCA kit according to the manufacturer’s instructions. For digestion, the protein solution was reduced with 5 mM dithiothreitol for 30 min at 56°C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in darkness. The protein sample was then diluted by adding 100 mM TEAB to urea concentration < 2 M. Finally, trypsin was added at 1:50 trypsin‐to‐protein mass ratio for the first digestion overnight and 1:100 trypsin‐to‐protein mass ratio for a second 4‐h digestion. After trypsin digestion, peptide was desalted by Strata X C18 SPE column (Phenomenex) and vacuum‐dried. Peptide was reconstituted in 0.5 M TEAB and processed according to the manufacturer’s protocol for TMT kit/iTRAQ kit. Briefly, one unit of TMT/iTRAQ reagent was thawed and reconstituted in acetonitrile. The peptide mixtures were then incubated for 2 h at room temperature and pooled, desalted, and dried by vacuum centrifugation. The tryptic peptides were dissolved in 0.1% formic acid (solvent A) and directly loaded onto a homemade reversed‐phase analytical column (15‐cm length, 75 μm i.d.). The gradient was comprised of an increase from 6% to 23% solvent B (0.1% formic acid in 98% acetonitrile) over 26 min, 23% to 35% in 8 min and climbing to 80% in 3 min then holding at 80% for the last 3 min, all at a constant flow rate of 400 nl/min on an EASY‐nLC 1000 UPLC system. The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus (Thermo) coupled online to the UPLC. The electrospray voltage applied was 2.0 kV. The m/z scan range was 350–1,800 for full scan, and intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were then selected for MS/MS using NCE setting as 28 and the fragments were detected in the Orbitrap at a resolution of 17,500. A data‐dependent procedure that alternated between 1 MS scan followed by 20 MS/MS scans with 15.0‐s dynamic exclusion was carried out. Automatic gain control (AGC) was set at 5E4. Fixed first mass was set as 100 m/z. The resulting MS/MS data were processed using Maxquant search engine (v.1.5.2.8). Tandem mass spectra were searched against human uniprot database concatenated with reverse decoy database.
CRISPR‐Cas9‐mediated genome editing
The lentiCRISPRv2 vector was a nice gift from Dr. Fangfang Zhou (Soochow University, China). Briefly, small‐guide RNAs with either Viperin, or USP33, or p97 gene targets were cloned into the lentiCRISPRv2 vector and then were transfected into HEK293T cells. Forty‐eight hours after transfection, the supernatant containing lentiviruses was collected. Furthermore, HEK293T or 2fTGH cells were infected by the supernatant. After 48 h, these cells were split into 96‐well plates and were cultured under puromycin selection until further experiments. gRNA for Viperin: ATTGCTCACGATGCTCACGC; USP33: TCCAGACATGCCC AAAGATT; p97: GCGTATCGACCCATCCGGAA. NR3C2: CATCACTTCTTCTA GACGAC.
Viral infection in vitro
Vesicular stomatitis virus (VSV) and Sendai virus (SeV) were gifts from Dr. Chen Wang (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences). Influenza A virus (H1N1, PR/8/34) was a gift from Dr. Jianfeng Dai (Soochow University, China). VSV‐GFP was a gift from Dr. Chunsheng Dong (Soochow University, China). Cells were firstly treated with IFN‐I for 18–20 h. After washing twice by 1× PBS, cells were cultured in serum‐free medium and then infected with different viruses diluted with DMEM serially for 1.5 h. Then the supernatant was removed and cells were further cultured in DMEM medium with10% FBS until harvest.
Viral infection in vivo
Eight weeks old mice were injected intraperitoneally (i.p.) with VSV (1 × 108 PFU per gram body mouse). After 3 days, mouse lung, liver, spleen, and kidney tissues were gained and grinded into small pieces for further analysis by western blot, RT–qPCR, or ELISA. Immunofluorescence pictures of VSV‐GFP and cell nucleus (DAPI) in mouse liver tissues were captured by the Servicebio company (Wuhan, China). Mouse survival observation continued until 15 days.
TCID50 assay
Vesicular stomatitis virus viral titers were determined by a standard 50% tissue culture‐infective dose (TCID50) assay. Briefly, mouse serum containing viruses or the cell culture supernatants containing viruses were collected and diluted with DMEM serially. Then the diluted supernatants were put on the monolayer of Vero cells in 96‐well plates. The TCID50 was calculated by the Spearman–Karber algorithm.
Cycloheximide (CHX) pulse chase assay
The half‐life of Viperin protein was determined by a cycloheximide (CHX) pulse chase assay. Cells were transfected with Flag‐Viperin and shRNA plasmids. Seventy‐two hours after transfection, cells were treated with DMSO or CHX (50 μg/ml) for 0, 6, and 12 h. Cells were then harvested and the equal amounts of boiled lysates were analyzed by western blot.
In vivo ubiquitination assay
Cells were transfected with the corresponding plasmids, together with or without HA‐ubiquitin (Ub). Forty‐eight or 72 h after transfection, cells were harvested in the RIPA lysis buffer with N‐ethylmaleimide (10 mM). To analyze the ubiquitination of either Viperin or USP33, proteins were immunoprecipitated by a specific antibody (Ab) on a rotor at 4°C. After washing three times with the high‐salt (500 mM NaCl) washing buffer and subsequently twice with the normal salt (150 mM NaCl) washing buffer, the immunoprecipitates were analyzed by western blot using the anti‐HA or anti‐Ub Ab.
Total RNA extraction
Cell lines or mouse primary cells were harvested with the TRIzol reagent (Invitrogen). The cDNA was produced by reverse transcription using oligo (dT) or random primer according to the manufacturer's instructions (Invitrogen) using 1 μg of total RNAs. Then the RNA levels were analyzed by real‐time quantitative PCR (RT–qPCR) assay.
Real‐time quantitative PCR (RT–qPCR)
Real‐time qPCR was conducted with the SYBR Green (Selleck). The relative gene expression levels were calculated using the change‐in‐cycling threshold (2−ΔΔ C t) method. Quantitation of all target genes was normalized to the control gene β‐actin, and all data are shown as fold change normalized to that in either unstimulated or uninfected cells accordingly. The results were analyzed from three independent experiments and are shown as the average mean ± SD. The primer sequences are as following:
SeV:
Forward: 5'‐GATGACGATGCCGCAGCAGTAG‐3'
Reverse: 5'‐CCTCCGATGTCAGTTGGTTCACTC‐3'
VSV:
Forward: 5’‐ACGGCGTACTTCCAGATGG‐3’
Reverse: 5’‐CTCGGTTCAAGATCCAGGT‐3’
HBV:
Forward: 5'‐GAGTGTGGATTCGCACTCC‐3'
Reverse: 5'‐GAGGCGAGGGAGTTCTTCT‐3'
HCV:
Forward: 5'‐ TCTTCACGCAGAAAGCGTCTAG‐3'
Reverse: 5'‐ GGTTCCGCAGACCACTATGG‐3'
Mouse‐Ifnβ:
Forward: 5'‐CTTCGTGTTTGGTAGTGATGGT‐3'
Reverse: 5'‐GGGGATGATTTCCAGCCGA‐3'
Human Viperin:
Forward: 5'‐TGCTTAAGGAAGCTGGTATGGAG‐3'
Reverse: 5'‐TCACCAACTTGCCCAGGTAT‐3'
Mouse Rsad2:
Forward: 5'‐CAGGCTGGTTTGGAGA AGATCAAC‐3'
Reverse: 5'‐TACTCCCCATAGTCCTTG AACCATC‐3'
Human Ifit1:
Forward: 5'‐CACAAGCCATTTTCTTTGCT‐3'
Reverse: 5′‐ACTTGGCTGCATATCGAAAG‐3 ′
Human Isg15:
Forward: 5'‐GGGACCTGACGGTGAAGATG‐3'
Reverse: 5'‐CGCCGATCTTCTGGGTGAT‐3'
Human Isg54:
Forward: 5'‐CACCTCTGGACTGGCAATAGC‐3'
Reverse: 5'‐GTCAGGATTCAGCCGAATGG‐3'
Alpl:
Forward: 5'‐CCAGAAAGACACCTTGACTGTGG‐3'
Reverse: 5'‐ TCTTGTCCGTGTCGCTCACCAT‐3'
Ass1:
Forward: 5'‐ CACTCTACGAGGACCGCTATCT‐3'
Reverse: 5'‐ CTCAAAGCGGACCTGGTCATTC‐3'
Krt28:
Forward: 5'‐ AGACCTACTGCCGCCTCATAGA‐3'
Reverse: 5'‐ CACCGTCTTCACCAGTGTGGTT‐3'
Krt76:
Forward: 5'‐ GCAGGCTAAAGTGGACAGTCTG‐3'
Reverse: 5'‐GGACCACAGATGTGTCACTGAC‐3'
Usp33:
Forward: 5'‐CAGTCTGCATCTCCAAAGAGAA‐3'
Reverse: 5'‐ACTACTGGACCCCAAAACCA‐3'
β‐actin:
Forward: 5'‐ACCAACTGGGACGACATGGAGAAA‐3'
Reverse: 5'‐ATAGCACAGCCTGGATAGCAACG‐3'
Western blot
Equivalent amount of proteins was subjected to SDS–PAGE and then were transferred to PVDF membranes (Millipore). Membranes were blocked with either 5% nonfat milk or 5% BSA for 0.5 h at room temperature, and then incubated with the corresponding primary antibodies (Abs). After washing three times with PBST (1× PBS and Tween 20), the membranes were subjected to the secondary Abs (HRP‐conjugated goat anti‐rabbit or goat anti‐mouse [Bioworld]) in 5% nonfat milk for 1 h. After washing three times with PBST, the membranes were visualized with the ECL Prime (Thermo Scientific). The antibodies with indicated dilutions were as follows: USP33 (Proteintech, 20445‐1‐Ap, 1:1,000), p97 (Santa Cruz, sc‐133211, 1:500), p‐STAT1(Y701) (Cell Signaling Technology, 9167, 1:1,000), Phospho‐(Ser/Thr) Phe antibody (Abcam, ab17464, 1:500), Caspase3 (Cell Signaling Technology, 7272, 1:1,000), Viperin (Abcam, ab107359, 1:1,000), UBE4A (Santa Cruz, sc‐365904, 1:1,000), Flag (Sigma, F7425, 1:5,000), HA (Sigma, ab9110, 1:5,000), Ubiquitin (Ub) (Santa Cruz, sc‐8017, 1:500), K48‐Ub (Cell Signaling, #4289, 1:500), K63‐Ub (Millipore, #05‐1308, 1:500), STAT1 (Cell Signaling Technology, 8826, 1:5,000), HA (H1N1) (Sino Biological, 11684‐T56, 1:5,000), VSV‐G (sc‐66180, 1:1,000), Myc (Abmart, M20002, 1:5,000), Lysine‐acetylation (Santa Cruz, sc‐81623, 1:1,000), BRCC36 (Abcam, ab115172, 1:1,000), NR3C2 (Proteintech,21854‐1‐AP), β‐Actin (Proteintech, 66009, 1:5,000), Tubulin (Proteintech, 66031‐1‐Ig, 1:5,000), GAPDH (Proteintech, 10494‐1‐AP, 1:5,000), and IFIT1 (Santa Cruz, sc‐134948, 1:1,000).
Immunoprecipitation
Cells were harvested in lysis buffer containing 150 mM NaCl, 20 mM Tris‐HCl (pH 7.4), 1% Nonidet P‐40, 0.5 mM EDTA, PMSF (50 μg/ml), and protease inhibitor mixtures (Sigma). When protein ubiquitination was examined, RIPA buffer (Beyotime) was used and N‐ethylmaleimide (10 mM) was added into the lysis buffer. The cell lysates were incubated with specific Abs on a rotor at 4°C. Protein G‐agarose beads (Millipore; 16‐266) were firstly washed twice and then were added into the supernatant. The mixture was incubated for 2–3 h on a rotor at 4°C. For immunoprecipitation of Flag/Myc/HA‐tagged proteins, either M2 affinity gel (A2220; Sigma‐Aldrich), or Myc, or HA magnetic beads (Selleck) were added to the cell lysates. Then the lysates were rotated for 2–3 h on a rotor at 4°C. After washing three times with the washing buffer containing 150 mM NaCl, the immunoprecipitates were analyzed by western blot.
Construction of Viperin‐stable cell lines
HEK293T cells were transfected with pOZ‐FH‐C‐puro (empty vectors; Addgene) or Flag‐HA‐Viperin, together with PCL Ampho plasmids. Thirty‐six hours after transfection, the supernatant was collected and used to infect either HeLa, or HEK293T, or A549 cells. The infected cells were cultured for 24 h with polybrene (8 μg/ml). After removing the culture medium, the cells were cultured in 10% FBS without polybrene for 24 h. Then cells were cultured in 10% FBS with puromycin (1.5 μg/ml) for 2 weeks’ selection. The stable Viperin‐expressing cells were maintained in 10% FBS medium with puromycin (1.5 μg/ml) until 2 days before further experiments.
Micro blood sodium concentration assay
Eight‐week‐old mice were injected intraperitoneally (i.p.) with the high‐salt solution (3% NaCl in ddH2O) or control salt solution (0.9% NaCl in ddH2O). At different time points, the concentration of Na+ in mouse sera was analyzed by the Micro blood sodium concentration assay kit (BC2805, Solarbio).
Statistical analysis
Comparison between different groups was analyzed using the two‐tailed unpaired Student’s t‐test. Values of P < 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001; NS, not significant. Kaplan–Meier survival curves were generated and analyzed for mouse survival study performed in the Graph Pad Prism 7.0.
Author contributions
YY, YM, TR, FH, LQ, XC, YZ, H‐GZ, and JH performed the experiments. H‐GZ, YM, TR, and WS assisted with mouse experiments, tissue processing, and analysis. GX helped the mass spectrometry analysis. WZ helped the acetylation analysis. TR, JH, CQ, QD, and QW assisted with the transfections and RT‐qPCR analysis. CZ, YX, DW, WS, and JJ discussed the data and manuscript. HZ and YY designed experiments, analyzed data and wrote the paper. HZ was responsible for research supervision, coordination, and strategy.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgments
We thank Dr. Peter Cresswell (Yale University School of Medicine), Dr. Serge Y. Fuchs (University of Pennsylvania), Dr. Guo‐Qiang Chen (Shanghai Jiaotong University), Dr. Zhenghong Yuan (Fudan University), Dr. Chen Wang (China Pharmaceutical University) and Dr. Lingqiang Zhang (National Center of Protein Sciences, China) for important reagents. We also thank Dr. Serge Y. Fuchs (University of Pennsylvania) for valuable discussion. This work is supported by grants from the National Key R&D Program of China (2018YFC1705500): 2018YFC1705505, the National Natural Science Foundation of China (31770177, 31970846 and 32000540), the national program of 1,000 Young Talents (2014), Jiangsu Provincial Distinguished Young Scholars (BK20130004), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
EMBO reports (2022) 23: e53466.
See also: S Ghosh & ENG Marsh (January 2022)
Data availability
The data for quantitative proteomic analysis generated in this study have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD028645 (http://www.ebi.ac.uk/pride/archive/projects/PXD028645).
References
- Arita M, Wakita T, Shimizu H (2012) Valosin‐containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J Virol 86: 5541–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthouze M, Venkataramanan V, Li Y, Shenoy SK (2009) The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. EMBO J 28: 1684–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie P (2018) Mechanisms of sodium balance: total body sodium, surrogate variables, and renal sodium excretion. Am J Physiol Regul Integr Comp Physiol 315: R945–R962 [DOI] [PubMed] [Google Scholar]
- Binger KJ, Gebhardt M, Heinig M, Rintisch C, Schroeder A, Neuhofer W, Hilgers K, Manzel A, Schwartz C, Kleinewietfeld M et al (2015) High salt reduces the activation of IL‐4‐ and IL‐13‐stimulated macrophages. J Clin Invest 125: 4223–4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown IJ, Tzoulaki I, Candeias V, Elliott P (2009) Salt intakes around the world: implications for public health. Int J Epidemiol 38: 791–813 [DOI] [PubMed] [Google Scholar]
- Chan NC, den Besten W, Sweredoski MJ, Hess S, Deshaies RJ, Chan DC (2014) Degradation of the deubiquitinating enzyme USP33 is mediated by p97 and the ubiquitin ligase HERC2. J Biol Chem 289: 19789–19798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraco G, Brea D, Garcia‐Bonilla L, Wang G, Racchumi G, Chang H, Buendia I, Santisteban MM, Segarra SG, Koizumi K et al (2018) Dietary salt promotes neurovascular and cognitive dysfunction through a gut‐initiated TH17 response. Nat Neurosci 21: 240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizzi AS, Grove TL, Arnold JJ, Jose J, Jangra RK, Garforth SJ, Du Q, Cahill SM, Dulyaninova NG, Love JD et al (2018) A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558: 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Zuo Y, Qian L, Liu J, Yuan Y, Xu K, Miao Y, Feng Q, Chen X, Jin L et al (2019) ADP‐ribosyltransferase PARP11 modulates the interferon antiviral response by mono‐ADP‐ribosylating the ubiquitin E3 ligase β‐TrCP. Nat Microbiol 4: 1872–1884 [DOI] [PubMed] [Google Scholar]
- Helbig KJ, Lau DT, Semendric L, Harley HA, Beard MR (2005) Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 42: 702–710 [DOI] [PubMed] [Google Scholar]
- Huang Z, Wen P, Kong R, Cheng H, Zhang B, Quan C, Bian Z, Chen M, Zhang Z, Chen X et al (2015) USP33 mediates Slit‐Robo signaling in inhibiting colorectal cancer cell migration. Int J Cancer 136: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA (2013) Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496: 518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotchen TA, Cowley AW Jr, Frohlich ED (2013) Salt in health and disease–a delicate balance. N Engl J Med 368: 1229–1237 [DOI] [PubMed] [Google Scholar]
- Li J, D'Angiolella V, Seeley ES, Kim S, Kobayashi T, Fu W, Campos EI, Pagano M, Dynlacht BD (2013) USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 495: 255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Prendergast J, Grey F (2017) The host ubiquitin‐dependent segregase VCP/p97 is required for the onset of human cytomegalovirus replication. PLoS Pathog 13: e1006329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang Q, Li K, Gong Z, Liu Z, Xu Y, Swaney MH, Xiao K, Chen Y (2016) Prognostic significance of USP33 in advanced colorectal cancer patients: new insights into β‐arrestin‐dependent ERK signaling. Oncotarget 7: 81223–81240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G (2012) Systematic identification of type I and type II interferon‐induced antiviral factors. Proc Natl Acad Sci USA 109: 4239–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M (2014) Role of "Western diet" in inflammatory autoimmune diseases. Curr Allergy Asthma Rep 14: 404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol 14: 117–123 [DOI] [PubMed] [Google Scholar]
- O'Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, Yan H, Lee SF, Mony P, Devanath A et al (2014) Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med 371: 612–623 [DOI] [PubMed] [Google Scholar]
- Ritorto MS, Ewan R, Perez‐Oliva AB, Knebel A, Buhrlage SJ, Wightman M, Kelly SM, Wood NT, Virdee S, Gray NS et al (2014) Screening of DUB activity and specificity by MALDI‐TOF mass spectrometry. Nat Commun 5: 4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JY, Yaneva R, Cresswell P (2011) Viperin: a multifunctional, interferon‐inducible protein that regulates virus replication. Cell Host Microbe 10: 534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Huen SC, Luan HH, Yu S, Zhang C, Gallezot JD, Booth CJ, Medzhitov R (2016) Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell 166: 1512–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hinson ER, Cresswell P (2007) The interferon‐inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2: 96–105 [DOI] [PubMed] [Google Scholar]
- Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, Haase S, Mähler A, Balogh A, Markó L et al (2017) Salt‐responsive gut commensal modulates T(H)17 axis and disease. Nature 551: 585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK (2013) Induction of pathogenic TH17 cells by inducible salt‐sensing kinase SGK1. Nature 496: 513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Miao Y, Qian L, Zhang Y, Liu C, Liu J, Zuo Y, Feng Q, Guo T, Zhang L et al (2020) Targeting UBE4A revives viperin protein in epithelium to enhance host antiviral defense. Mol Cell 77: 734–747 [DOI] [PubMed] [Google Scholar]
- Yuasa‐Kawada J, Kinoshita‐Kawada M, Rao Y, Wu JY (2009) Deubiquitinating enzyme USP33/VDU1 is required for Slit signaling in inhibiting breast cancer cell migration. Proc Natl Acad Sci USA 106: 14530–14535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W‐C, Zheng X‐J, Du L‐J, Sun J‐Y, Shen Z‐X, Shi C, Sun S, Zhang Z, Chen X‐Q, Qin MU et al (2015) High salt primes a specific activation state of macrophages, M(Na). Cell Res 25: 893–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Feng Q, Jin L, Huang F, Miao Y, Liu J, Xu Y, Chen X, Zhang H, Guo T et al (2020) Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat Commun 11: 1146 [DOI] [PMC free article] [PubMed] [Google Scholar]