

Abstract
The exon junction complex controls the translation, degradation, and localization of spliced mRNAs, and three of its core subunits also play a role in splicing. Here, we show that a fourth subunit, Barentsz, has distinct functions within and separate from the exon junction complex in Drosophila neuromuscular development. The distribution of mitochondria in larval muscles requires Barentsz as well as other exon junction complex subunits and is not rescued by a Barentsz transgene in which residues required for binding to the core subunit eIF4AIII are mutated. In contrast, interactions with the exon junction complex are not required for Barentsz to promote the growth of neuromuscular synapses. We find that the Activin ligand Dawdle shows reduced expression in barentsz mutants and acts downstream of Barentsz to control synapse growth. Both barentsz and dawdle are required in motor neurons, muscles, and glia for normal synapse growth, and exogenous Dawdle can rescue synapse growth in the absence of barentsz. These results identify a biological function for Barentsz that is independent of the exon junction complex.
Keywords: Barentsz, exon junction complex, Dawdle, neuromuscular junction, synapse
Subject Categories: Development, Neuroscience, RNA Biology
Barentsz promotes the growth of Drosophila neuromuscular synapses by increasing Activin signaling independent of its known role in the exon junction complex.
Introduction
Post‐transcriptional regulation enables rapid and localized changes in gene expression, making it an important regulatory mechanism in the nervous system (Goldie & Cairns, 2012). The exon junction complex (EJC) links distinct modes of post‐transcriptional regulation by marking properly spliced mRNAs for preferential translation, efficient nonsense‐mediated decay (NMD), or specific subcellular localization (Nott et al, 2004; Palacios et al, 2004; Chang et al, 2007; Ghosh et al, 2012; Chazal et al, 2013; Metze et al, 2013; Boehm et al, 2014; Choe et al, 2014; Schlautmann & Gehring, 2020). During splicing, the core EJC protein eIF4AIII (EIF4A3 in humans) is recruited by the spliceosomal protein CWC22 to mRNA 20‐24 nucleotides upstream of exon junctions (Gehring et al, 2009; Alexandrov et al, 2012; Barbosa et al, 2012; Steckelberg et al, 2012). A dimer of Mago nashi (Mago)/MAGOH and Y14/RBM8A then associates with eIF4AIII (Gehring et al, 2009; Herold et al, 2009). These proteins together with the accessory subunit RnpS1/RNPS1 facilitate the splicing of a subset of introns in genes that include MAP kinase, piwi, and pro‐apoptotic transcripts (Ashton‐Beaucage et al, 2010; Roignant & Treisman, 2010; Haremaki & Weinstein, 2012; Michelle et al, 2012; Hayashi et al, 2014; Malone et al, 2014). In addition, this complex prevents the splicing of cryptic splice sites within introns and blocks resplicing of previously spliced transcripts (Blazquez et al, 2018; Boehm et al, 2018; Gehring & Roignant, 2020; Joseph & Lai, 2021).
A fourth subunit, known as Barentsz (Btz), Cancer susceptibility candidate gene 3 (CASC3), or Metastatic lymph node 51 (MLN51), associates with the complex following the completion of splicing and plays a role in cytoplasmic EJC‐mediated processes such as translation, NMD, and mRNA localization (van Eeden et al, 2001; Palacios et al, 2004; Shibuya et al, 2006; Chazal et al, 2013). The transition between RNPS1 and CASC3‐containing forms of the EJC alters the set of transcripts subject to NMD (Mabin et al, 2018; Gerbracht et al, 2020). In the mouse brain, haploinsufficiency for Magoh, Rbm8a, or Eif4a3 causes severe microcephaly, but complete loss of Casc3 does not reduce brain size relative to body size, and its effects on the brain can be attributed to developmental delay (Silver et al, 2010; Mao et al, 2015, 2016, 2017). Btz/CASC3 shuttles between the cytoplasm and the nucleus and can localize to stress granules, P‐bodies, and neuronal RNP granules independently of other EJC subunits (Macchi et al, 2003; Barbee et al, 2006; Baguet et al, 2007; Fritzsche et al, 2013; Cougot et al, 2014). However, a biological function for Btz/CASC3 acting outside the EJC has not yet been reported.
During Drosophila development, the synapses formed by larval motor neurons on their target muscles, known as neuromuscular junctions (NMJs), add new branches and synaptic boutons as the muscles increase in size (Schuster et al, 1996; Menon et al, 2013). This coordinated growth requires multiple signals to pass between the two cell types. Motor neurons secrete Wingless (Wg), which acts on the nerve terminals through its canonical signaling pathway, and on the muscle through a mechanism involving nuclear import of the Frizzled receptor (Mathew et al, 2005; Miech et al, 2008; Mosca & Schwarz, 2010). Muscles secrete the retrograde signal Glass‐bottom boat (Gbb), a bone morphogenetic protein (BMP) family member that acts on motor neurons through the Wishful thinking receptor, leading to expression of the exchange factor Trio and enhanced function of the receptor protein tyrosine phosphatase Lar (Keshishian & Kim, 2004; Ball et al, 2010; Berke et al, 2013). Gbb production by muscles is dependent on two Activin‐like ligands, Dawdle (Daw) and Maverick (Mav), which are produced by glial cells surrounding the nerve terminal as well as by the muscles themselves (Parker et al, 2006; Serpe & O'Connor, 2006; Ellis et al, 2010; Fuentes‐Medel et al, 2012). However, the canonical Activin signaling pathway does not appear to be necessary for NMJ growth (Kim & O'Connor, 2014). Additional signals that contribute to the control of synapse growth include neurotrophins, stress response mechanisms, and a pathway that transmits information about muscle size through the extent of postsynaptic differentiation (Xiong et al, 2010; Milton et al, 2011; Chen & Ganetzky, 2012; Ho & Treisman, 2020).
Here, we show that Btz has both EJC‐dependent and EJC‐independent functions in neuromuscular development. It acts in muscles to control the distribution of mitochondria, a function that requires physical and functional interactions with other EJC subunits. Btz also acts in motor neurons, muscles, and glia to promote NMJ growth, and interaction with other EJC subunits is dispensable for this process. Analysis of transcripts that show altered levels in neurons or muscles in btz mutants identified changes in expression of the Activin family member daw, as well as of a potential regulator of Activin signaling. daw acts in the same tissues as btz to promote NMJ growth, and exogenous daw can rescue NMJ size in btz mutants. These results indicate that Btz acts independently of the EJC to stimulate neuromuscular synapse growth and that this function is mediated by Activin signaling.
Results and Discussion
A tool for the analysis of EJC‐dependent and EJC‐independent functions of Btz
We and others have previously demonstrated that the Mago, Y14, and eIF4AIII subunits of the EJC have a function in splicing that is independent of Btz (Ashton‐Beaucage et al, 2010; Roignant & Treisman, 2010; Haremaki & Weinstein, 2012; Michelle et al, 2012; Hayashi et al, 2014; Malone et al, 2014). To test whether Btz can also act independently of the other EJC subunits, we generated two rescue transgenes that express GFP‐tagged Btz with 5′ and 3′ endogenous regulatory sequences that encompass the entire intergenic region between btz and the adjacent genes (Fig 1A). One transgene expresses the wild‐type Btz protein (Btz‐WT), while the other has mutations in two conserved residues (H215A and D216A) in the SELOR (Speckle localizer and RNA‐binding) module (Btz‐HD). These changes correspond to the H220A and D221A mutations that have been shown to disrupt the association of human MLN51 with eIF4AIII and the EJC without preventing it from interacting with RNA (Ballut et al, 2005; Bono et al, 2006; Gehring et al, 2009; Daguenet et al, 2012). We confirmed that HA‐tagged Btz‐WT protein, but not Btz‐HD, co‐immunoprecipitated with Myc‐tagged eIF4AIII when expressed in cultured S2R+ cells (Fig 1B).
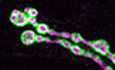
Figure 1. Interaction with the EJC is required for Btz function in the ovary.
-
ADiagram of the genomic rescue constructs in which wild‐type Btz (Btz‐WT) or Btz with the H215A and D216A mutations (Btz‐HD) is C‐terminally tagged with GFP and driven by its endogenous regulatory sequences.
-
BCo‐immunoprecipitation of HA‐tagged Btz‐WT or Btz‐HD with Myc‐tagged GFP or eIF4AIII from transfected S2R+ cells using Myc‐Trap Magnetic Agarose beads. The upper Western blot is blotted with anti‐Myc and the lower with anti‐HA. Input samples are shown on the left and immunoprecipitates on the right. Btz‐WT co‐immunoprecipitates with eIF4AIII, but Btz‐HD does not, although the protein is equally stable. The asterisk represents a likely cleavage product of Btz.
-
C–FStage 10 egg chambers stained with anti‐Vasa (C‐F) or anti‐GFP (E’, F’). (C) wild‐type; (D) germline clones homozygous for the null allele btz2 ; (E) btz2 germline clones rescued with Btz‐WT; (F) btz2 germline clones rescued with Btz‐HD. Posterior is to the right. Scale bars, 80 μm. Vasa localization to the posterior pole of the oocyte is lost in btz mutants and is rescued by the wild‐type but not the EJC interaction‐defective transgene. Wild‐type Btz‐GFP, but not the mutant form, is also slightly enriched at the posterior pole.
Source data are available online for this figure.
The full EJC, including Btz, is required to localize oskar mRNA to the posterior of the oocyte, leading to the assembly of germ plasm there (van Eeden et al, 2001; Palacios et al, 2004). Therefore, we tested the ability of the wild‐type and mutant transgenes to rescue posterior germ plasm formation in btz mutants, using Vasa protein as a readout for germ plasm (Breitwieser et al, 1996). As predicted, the Btz‐WT transgene rescued the loss of posteriorly localized Vasa in oocytes derived from btz mutant germline clones, and the encoded Btz‐WT‐GFP protein was enriched at the posterior pole (Fig 1C–E). However, Btz‐HD‐GFP failed to localize to the posterior pole or to rescue Vasa localization (Fig 1F), confirming that H215 and D216 are necessary for Btz to function in the context of the EJC. These transgenes thus enable us to distinguish EJC‐dependent from possible EJC‐independent functions of Btz.
Btz acts through the EJC to control the distribution of mitochondria in muscle
In addition to its effect on oskar mRNA localization in the developing oocyte (van Eeden et al, 2001; Palacios et al, 2004), btz has an essential role during development: most flies transheterozygous for a null allele of btz (van Eeden et al, 2001) and a deficiency covering the region (Df(3R)BSC497) die as late pupae. Our wild‐type transgene fully rescued the lethality of these transheterozygous btz mutants, while the mutant transgene provided a partial rescue (Table 1), suggesting that some of the functions of Btz that are required for viability may be EJC‐independent. We found that expressing GFP‐tagged Btz with a muscle‐specific GAL4 driver fully rescued the mutant flies to viability, while expressing it with a motor neuron driver or a glial driver partially rescued the lethality (Table 1). These results indicate that the most critical requirement for Btz is in muscle, but it also functions in motor neurons and glia.
Table 1.
Rescue of viability in btz mutants.
| Genotype | % of eclosed pupae | n |
|---|---|---|
| btz/+ | 100 | 186 |
| btz/Df | 8.5 | 176 |
| btz/Df, Btz‐WT | 94.0 | 133 |
| btz/Df, Btz‐HD | 40.9 | 115 |
| btz/Df, UAS‐BtzGFP | 6.5 | 92 |
| btz/Df, BG380‐GAL4> | 7.4 | 95 |
| btz/Df, BG380‐GAL4>UAS‐BtzGFP | 24.3 | 103 |
| btz/Df, repo‐GAL4> | 1.0 | 100 |
| btz/Df, repo‐GAL4>UAS‐BtzGFP | 24.1 | 83 |
| btz/Df, mhc‐Gal4> | 6.9 | 131 |
| btz/Df, mhc‐Gal4>UAS‐BtzGFP | 88.6 | 220 |
| btz/Df, mhc‐Gal4>UAS‐dawdle | 28.5 | 165 |
| btz/Df, da‐GAL4> | 5.4 | 74 |
| btz/Df, da‐GAL4>UAS‐BtzGFP | 97.4 | 78 |
| btz/Df, da‐GAL4>UAS‐dawdle | 23.5 | 132 |
The table shows the percentage of pupae from which adult flies eclosed and the total number of pupae counted for the indicated genotypes. Adult viability of btz2 /Df(3R)BSC497 is substantially rescued by the wild‐type btz transgene and by UAS‐btz expression ubiquitously or in muscles and partially rescued by the mutant transgene, by UAS‐btz expression in motor neurons or glia, and by ubiquitous or muscle‐specific UAS‐daw expression.
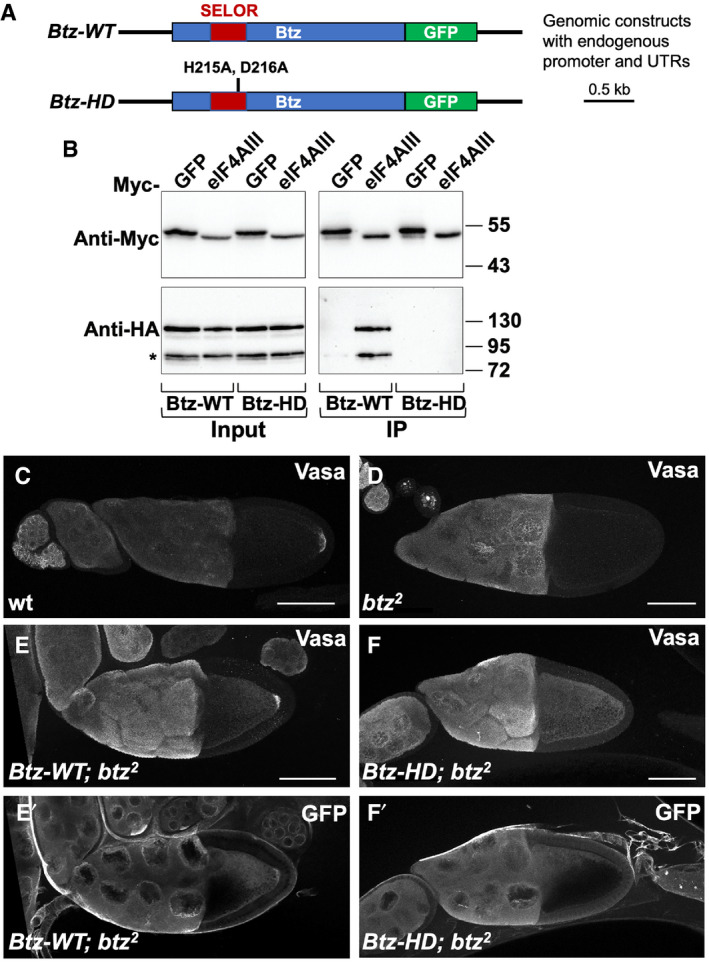
We observed that one striking effect of loss of btz in larval muscles was a change in the distribution of mitochondria. In wild‐type muscles, mitochondria detected either by staining for the ATP5A subunit of the ATP synthase complex or by electron microscopy were enriched at the dorsal surface, but also distributed between the muscle fibers (Fig 2A, B, L and M). In btz mutants, almost all mitochondria were concentrated at the dorsal surface (Fig 2C and N). We quantified these differences by measuring the ratio of anti‐ATP5A staining intensity in the dorsal peak relative to the next largest peak in a line scan through the muscle (Fig 2O). The normal distribution of ATP5A staining was restored by expressing UAS‐BtzGFP in muscle with G14‐GAL4 or by the presence of our Btz‐WT transgene (Fig 2D, E and O), but not by the Btz‐HD transgene (Fig 2F and O), suggesting that this function of Btz is mediated by the EJC. To confirm this, we used the muscle‐specific driver C57‐GAL4 to express UAS‐RNAi transgenes targeting Btz or the EJC subunits Mago and eIF4AIII and examined the localization of ATP5A staining. Depletion of each of these subunits caused mitochondria to concentrate at the dorsal surface (Fig 2G–J and O). The specificity of the ATP5A antibody was confirmed by the lack of staining in muscles in which bellwether (blw), which encodes ATP5A, was knocked down (Fig 2K). There was no change in the overall level of ATP5A in btz mutant muscles (Fig 2P), suggesting that the total number of mitochondria is not altered. These results indicate that Btz acts as a subunit of the EJC to promote the even distribution of mitochondria within muscles.
Figure 2. Btz acts as an EJC component to control the distribution of mitochondria in muscle.
-
A–KConfocal xz‐sections of larval muscle 6 in segment A3 stained with anti‐ATP5A to label mitochondria. The dorsal surface (D) is to the right and ventral (V) to the left. (A) white (w)1118 control; (B) btz2 /+ heterozygote; (C) btz2 / Df(3R)BSC497; (D) G14‐GAL4/UAS‐btz; btz2 / Df(3R)BSC497; (E) Btz‐WT; btz2 / Df(3R)BSC497; (F) Btz‐HD; btz2 / Df(3R)BSC497; (G) C57‐GAL4/+ control; (H) C57‐GAL4>UAS‐btz RNAi; (I) C57‐GAL4>UAS‐mago RNAi; (J) C57‐GAL4>UAS‐eIF4AIII RNAi; (K) C57‐GAL4>UAS‐blw RNAi. In wild‐type muscles, mitochondria are present at the dorsal surface (red arrow in A) and also distributed between the fibers (yellow arrow in A), while in btz mutant muscles and in muscles expressing btz, mago, or eIF4AIII RNAi, they are concentrated at the dorsal surface. Expression of UAS‐btz in muscle or the presence of the wild‐type, but not the HD mutant, Btz transgene restores the wild‐type distribution to btz mutants. The loss of ATP5A staining in muscles in which blw is knocked down confirms that the signal is specific. Scale bars, 10 μm.
-
L–NElectron micrographs showing cross‐sections of larval muscle 6 in segment A3, with the dorsal surface to the right. (L) btz2 /+; (M) Df(3R)BSC497/+; (N) btz2 / Df(3R)BSC497. Yellow arrows indicate examples of mitochondria interspersed between muscle fibers, and red arrows indicate mitochondria at the dorsal surface. Scale bars, 2 μm.
-
OQuantification of the ratio of the dorsal surface peak of ATP5 intensity to the second highest peak for the genotypes indicated. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant by Mann–Whitney test. n = 9 (btz/+), n = 11 (Df/+, btz/Df), n = 6 (btz/Df; G14>btz, C57‐GAL4/+), n = 4 (btz/Df; Btz‐WT, C57>eIF4AIII RNAi), n = 5 (btz/Df; Btz‐HD), n = 10 (C57>btz RNAi), or n = 3 (C57>mago RNAi). Error bars show mean ± SEM.
-
PWestern blot of wild type (Canton S) and btz2 /Df(3R)BSC497 larval carcasses with anti‐ATP5A and anti‐tubulin. Duplicate samples of 15 ng total protein are shown for each genotype. The overall level of ATP5A is not altered in btz mutant muscles.
Source data are available online for this figure.
Little is known about the regulation of this mitochondrial distribution, although it can be altered by mutations that disrupt mitochondrial fission or increase fusion (Han et al, 2012; Chao et al, 2016; Wang et al, 2016a, 2016b). Btz itself is not thought to be localized to mitochondria, but it has been found to interact with proteins that regulate the expression of mitochondrial RNAs (Bratic et al, 2011; Guruharsha et al, 2011; Baggio et al, 2014). Since the effect of Btz on mitochondria is EJC‐dependent, it may result from a change in the translation or stability of a transcript that regulates mitochondrial localization. Interestingly, oskar mRNA, which is localized by the EJC in the ovary (van Eeden et al, 2001; Palacios et al, 2004), encodes a protein that traps mitochondria at the posterior pole of the embryo through interactions with the actin cytoskeleton (Hurd et al, 2016).
Btz acts in motor neurons, muscles, and glia to promote normal NMJ growth
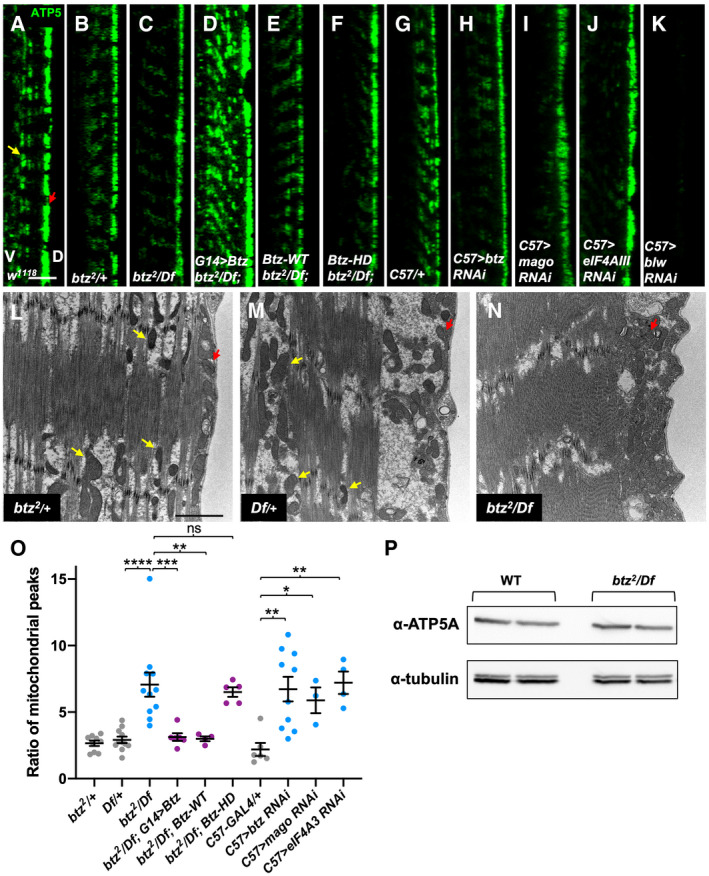
The requirement for btz in motor neurons and glia as well as muscles to promote viability suggested that it might be required for normal development of the NMJ, a structure that is formed by the conjunction of these three cell types and influenced by signaling interactions between them. Indeed, we found that btz mutant NMJs had fewer synaptic boutons than controls, and these tended to be more closely spaced along shorter axon branches (Fig 3A–C), although the size of individual boutons was not significantly altered (Fig EV1A). To measure synapse size, we quantified bouton numbers relative to muscle surface area (Fig 3K), as this has been shown to correlate well with other measures such as the extent and number of axon branches (Ho & Treisman, 2020). We confirmed that the reduction in bouton number was due to loss of btz function, because it could be rescued by ubiquitous expression of our UAS‐BtzGFP transgene with the daughterless (da)‐GAL4 driver (Fig 3D and K).
Figure 3. Btz acts in muscles, motor neurons, and glia to control NMJ size.
-
A–JConfocal images of the NMJ on larval muscles 6 and 7 in segment A3, stained with anti‐HRP (magenta) to label the nerve and anti‐Dlg (green) as a postsynaptic marker of synaptic boutons. (A) w1118 control; (B) btz2 /+; (C) btz2 /Df(3R)BSC497; (D) UAS‐btz/+; da‐GAL4, btz2 /Df(3R)BSC497; (E) BG380‐GAL4/+; UAS‐btz/+; btz2 /Df(3R)BSC497; (F) 24B‐GAL4/UAS‐btz; btz2 /Df(3R)BSC497; (G) BG380‐GAL4/+; 24B‐GAL4/UAS‐btz; btz2 /Df(3R)BSC497; (H) BG380‐GAL4/+; UAS‐btz RNAi/+; (I) mhc‐GAL4/UAS‐btz RNAi; (J) repo‐GAL4/UAS‐btz RNAi. Scale bar, 30 μm. Insets in (A, C) show enlargements of the boxed regions, illustrating the closer spacing of boutons in btz mutants.
-
K, LQuantifications of the number of boutons normalized to muscle surface area (×1,000) in the indicated genotypes. ****P < 0.0001; ***P < 0.001; **P < 0.01; ns, not significant by unpaired t‐test with Welch’s correction. n = 23 (w; da‐GAL4, btz/Df), n = 6 (btz/+), n = 10 (Df/+, UAS‐BtzGFP/+; btz/Df), n = 8 (btz/Df), n = 22 (da>btz‐GFP; btz/Df), n = 13 (BG380‐GAL4; btz/Df; BG380+24B>Btz‐GFP; btz/Df; da‐GAL4/+), n = 5 (BG380>Btz‐GFP; btz/Df), n = 11 (24B‐GAL4; btz/Df; da>btz RNAi), n = 12 (24B>Btz‐GFP; btz/Df; btz RNAi/+), n = 9 (BG380+24B‐GAL4; btz/Df), n = 15 (BG380‐GAL4/+), n = 14 (BG380>btz RNAi; repo>btz RNAi), n = 16 (mhc‐GAL4/+), n = 28 (mhc>btz RNAi), or n = 21 (repo‐GAL4/+). Error bars show mean ± SEM. NMJ size is reduced in btz mutants and rescued by expressing UAS‐btz with da‐GAL4 or with the combination of the BG380‐GAL4 and 24B‐GAL4 drivers, but not with any single driver. Expressing UAS‐btz RNAi with da‐GAL4 or in motor neurons with BG380‐GAL4, muscles with mhc‐GAL4, or glia with repo‐GAL4 also reduces NMJ size.
Source data are available online for this figure.
Figure EV1. Btz protein localization at the NMJ.
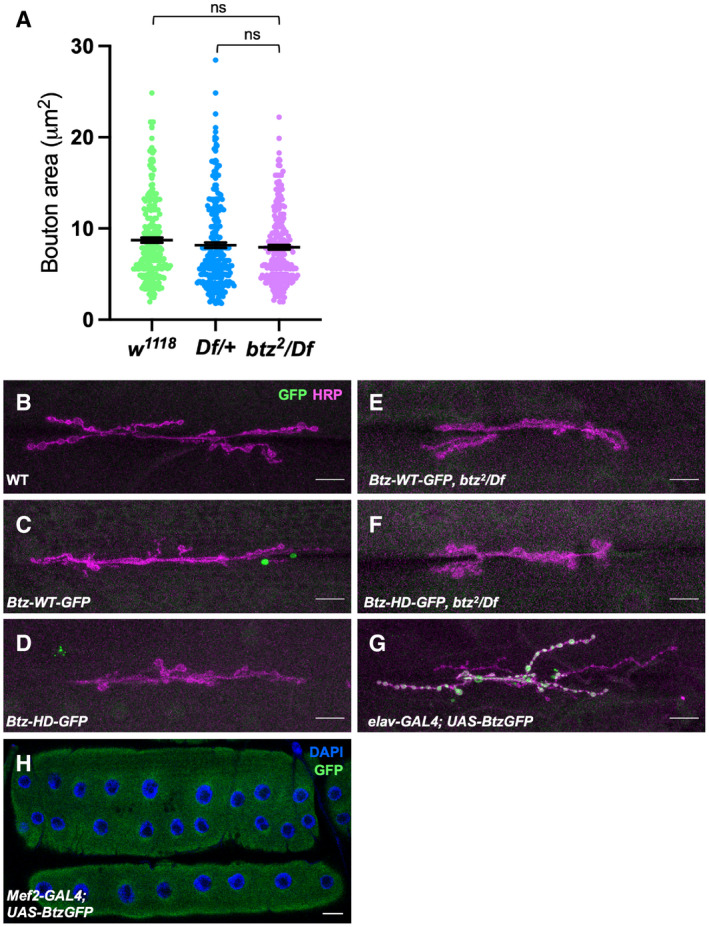
-
AQuantification of the area of individual synaptic boutons at the NMJ on muscles 6/7 in segment A3 in w1118 and Df(3R)BSC497/+ controls and in btz2 /Df(3R)BSC497 larvae. btz mutants show no significant reduction in bouton area compared to the controls. ns, not significant by Mann–Whitney test. n = 248 boutons from 6 NMJs (w1118 ), 262 boutons from 6 NMJs (Df/+), or 246 boutons from 6 NMJs (btz/Df). Error bars show mean ± SEM.
-
B–HLarval NMJs on muscle 6/7 in segment A3 stained with anti‐HRP (magenta in B‐G), DAPI (blue in H) and anti‐GFP (green). (B) Canton S control; (C) Btz‐WT‐GFP; (D) Btz‐HD‐GFP; (E) Btz‐WT‐GFP; btz2 /Df(3R)BSC497; (F) Btz‐HD‐GFP; btz2 /Df(3R)BSC497; (G) elav‐GAL4; UAS‐Btz‐GFP; (H) Mef2‐GAL4; UAS‐Btz‐GFP. Tagged Btz‐WT and Btz‐HD expressed from the btz promoter fell below our level of detection, even in the absence of endogenous btz. GFP‐tagged Btz overexpressed in neurons with elav‐GAL4 was localized to synaptic boutons, while GFP‐tagged Btz overexpressed in muscle with Mef2‐GAL4 was distributed throughout the muscle. Scale bars, 30 μm.
Source data are available online for this figure.
To determine in which cell types Btz functions to promote NMJ growth, we first attempted to look at the expression of our GFP‐tagged Btz transgenes. However, the level of expression of both the wild‐type and mutant proteins at the NMJ was too low for us to detect, even in the absence of endogenous btz (Fig EV1B–F). When overexpressed in neurons, BtzGFP localized to synaptic boutons, but when overexpressed in muscle, it was more diffusely distributed (Fig EV1G and H). As these expression results were inconclusive, we investigated the site of Btz function by using RNAi to knock it down in motor neurons, muscles, or glia. Depleting Btz from motor neurons with BG380‐GAL4 (an insertion in futsch) (Sanyal, 2009), from muscles with myosin heavy chain (mhc)‐GAL4 or from glia with reversed polarity (repo)‐GAL4 all caused a similar reduction in synaptic bouton number normalized to muscle surface area (Fig 3H–J and L), indicating that Btz is required in all three cell types. Consistent with this conclusion, we were unable to rescue NMJ growth in btz mutants by providing wild‐type Btz only to motor neurons, or to both muscles and glia using 24B‐GAL4 (an insertion in held out wings) (Zaffran et al, 1997) (Fig 3E, F and K). However, expressing UAS‐BtzGFP in all three cell types by combining 24B‐GAL4 with BG380‐GAL4 rescued NMJ size to the same extent as ubiquitous Btz expression (Fig 3G and K).
Btz controls NMJ size independently of the EJC
To test whether Btz regulates NMJ size through the EJC, we compared the effects of knocking down other EJC subunits in motor neurons to the effect of knocking down btz. In contrast to the reduction in bouton number caused by expressing btz RNAi with BG380‐GAL4 (Fig 4A, B and I), expressing mago RNAi or eIF4AIII RNAi with the same driver significantly increased the number of boutons relative to muscle surface area when compared to controls (Fig 4A, C, D and I). We also found that this function of Btz did not require the residues that interact with the EJC; the reduced NMJ size of btz mutants was rescued equally well by both our Btz‐WT and Btz‐HD transgenes (Fig 4E–I). These data indicate that unlike other known functions of Btz, its role in promoting normal synapse growth is not mediated by the EJC. The increase in NMJ size when mago or eIF4AIII is knocked down suggests that the EJC may even antagonize this function, perhaps by reducing the concentration of free Btz. Although it is clear that the other EJC subunits can perform important functions independently of Btz (Ashton‐Beaucage et al, 2010; Roignant & Treisman, 2010; Haremaki & Weinstein, 2012; Michelle et al, 2012; Hayashi et al, 2014; Malone et al, 2014; Mao et al, 2017), Btz functions independent of the EJC have not previously been reported.
Figure 4. Btz controls NMJ size independently of the EJC.
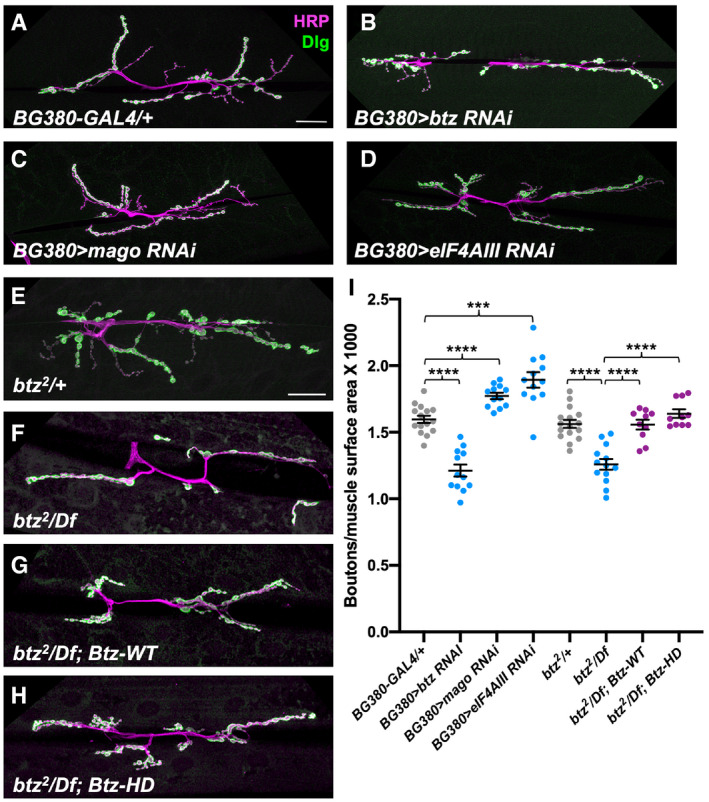
-
A–HConfocal images of the NMJ on larval muscles 6 and 7 in segment A3, stained with anti‐HRP (magenta) and anti‐Dlg (green). (A) BG380‐GAL4/+ control; (B) BG380‐GAL4/+; UAS‐btz RNAi/+; (C) BG380‐GAL4/+; UAS‐mago RNAi/+; (D) BG380‐GAL4/+; UAS‐eIF4AIII RNAi/+; (E) btz2 /+ control; (F) btz2 /Df(3R)BSC497; (G) Btz‐WT; btz2 /Df(3R)BSC497; (H) Btz‐HD; btz2 /Df(3R)BSC497. Scale bars, 30 μm (shown in A for A‐D and in E for E‐H).
-
IQuantifications of the number of boutons normalized to muscle surface area (×1,000) in the indicated genotypes. ****P < 0.0001; ***P < 0.001 by unpaired t‐test or Mann–Whitney test. n = 15 (BG380‐GAL4), n = 12 (BG380>btz RNAi; BG380>mago RNAi; BG380>eIF4AIII RNAi), n = 16 (btz/+), n = 13 (btz/Df), or n = 10 (Btz‐WT; btz/Df; Btz‐HD; btz/Df). Error bars show mean ± SEM. NMJ size is reduced by knocking down btz in motor neurons, but slightly increased by knocking down mago or eIF4AIII. The size deficit in btz mutants is equally well rescued by the wild type or EJC interaction‐defective btz transgenes.
Source data are available online for this figure.
Btz acts through Dawdle signaling to control NMJ size
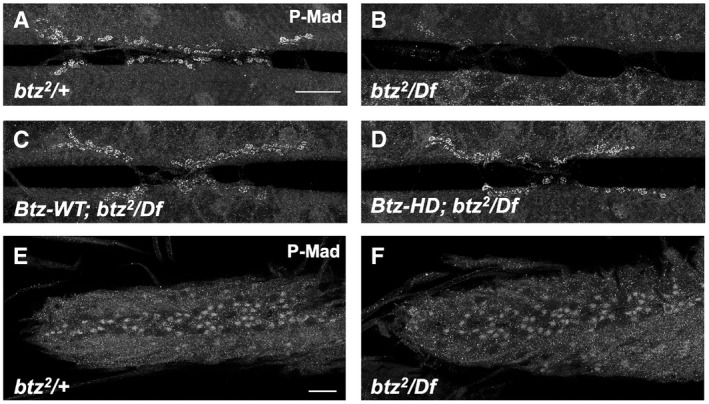
We next wished to identify targets of regulation by Btz that could explain its effect on NMJ size. We therefore examined gene expression in dissected central nervous systems or carcasses (consisting predominantly of body wall muscles) of btz2 /Df(3R)BSC497 third instar larvae compared to heterozygous btz2 /+ controls. From an RNA‐Seq analysis of three biological replicates, we found 220 genes that showed significantly altered expression (q < 0.05) in btz mutant larval carcasses and 103 that showed significantly altered expression in the central nervous system (Dataset EV1). Some of these expression changes are likely to be due to contamination by other tissues, as they are genes that are highly expressed in tissues such as the salivary gland, fat body, or testis. In addition, genes located within Df(3R)BSC497 showed an approximately 50% reduction in expression due to the absence of one wild‐type copy. Removing these genes from the list left 80 potential Btz targets in muscle and 33 in the CNS (Dataset EV1). One of the genes that was significantly downregulated in both the CNS and carcass samples in btz mutants was daw, which encodes an Activin homologue that has been implicated in the control of NMJ growth (Ellis et al, 2010; Fuentes‐Medel et al, 2012). In addition, two genes that were upregulated in btz mutant carcasses, larval translucida (ltl) and faulty attraction (frac), encode proteins that can regulate the activity of TGF‐β and BMP homologues in other contexts (Miller et al, 2011; Szuperak et al, 2011). We confirmed the changes in daw, ltl, and frac expression by quantitative RT–PCR (Fig 5A). daw was also strongly downregulated and frac upregulated in homozygous btz2 larval carcasses, but ltl was not significantly altered, suggesting that heterozygosity for Df(3R)BSC497 contributed to its upregulation (Fig EV2). Consistent with a reduction in signaling by members of the TGF‐β family (Fuentes‐Medel et al, 2012; Sulkowski et al, 2016), phosphorylation of the downstream factor Mothers against Dpp (Mad) was reduced at btz mutant NMJs (Figs 5B and C, and EV3A and B). Btz regulates TGF‐β signaling independently of the EJC, as Mad phosphorylation was rescued by both the Btz‐WT and Btz‐HD transgenes (Fig EV3C and D). Loss of btz did not affect Mad phosphorylation in the ventral nerve cord (Fig EV3E and F), which is regulated by muscle‐derived Gbb (Sulkowski et al, 2014, 2016), indicating that its effect is limited to local TGF‐β signaling at the synapse. Activin signaling regulates glutamate receptor expression and distribution at the synapse (Kim & O'Connor, 2014), which in turn can affect synaptic levels of pMad (Sulkowski et al, 2014), perhaps explaining the reduced synaptic pMad in btz mutants.
Figure 5. Daw signaling mediates the effects of Btz on NMJ size.
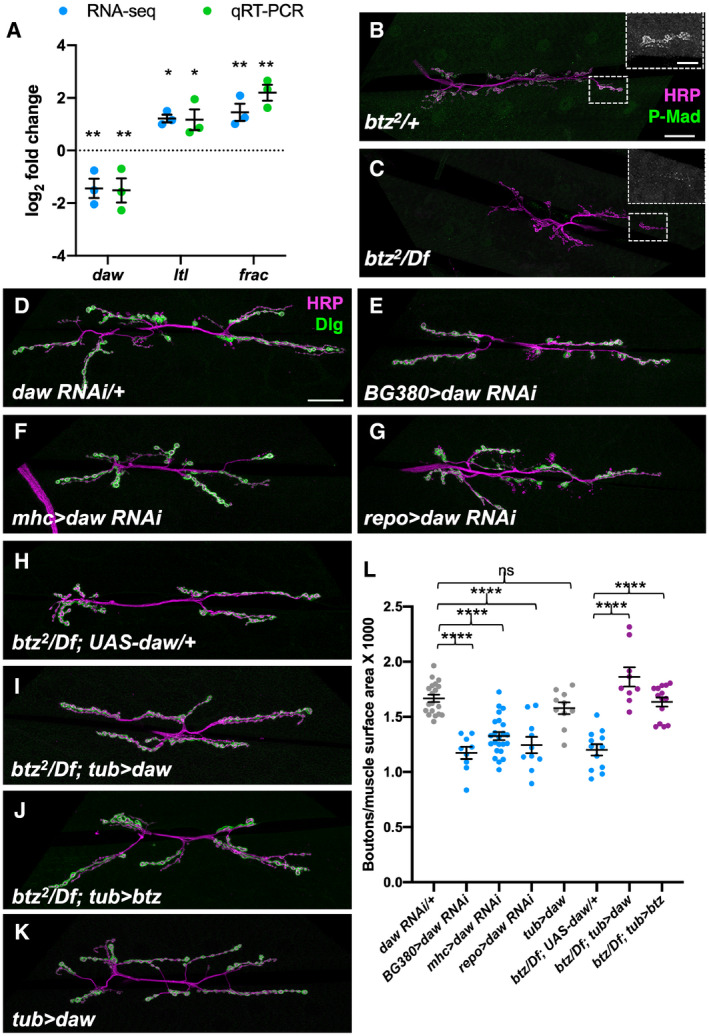
-
ALog2 fold changes in daw, ltl, and frac mRNA in btz2 /Df(3R)BSC497 larval carcasses compared to btz2 /+, measured by RNA‐Seq (blue) or qRT–PCR (green). **q < 0.01; *q < 0.05 by unpaired t‐test corrected for false discovery rate with the Benjamini–Hochberg procedure (RNA‐Seq) or **P < 0.01; *P < 0.05 by unpaired t‐test (qRT–PCR). n = 3 for each sample. Error bars show mean ± SD.
-
B–KConfocal images of the NMJ on larval muscles 6 and 7 in segment A3, stained with anti‐HRP (magenta) and anti‐P‐Mad (green in B, C) or anti‐Dlg (green in D‐K). (B) btz2 /+ control; (C) btz2 /Df(3R)BSC497. Insets show enlargements of the boxed regions stained with anti‐P‐Mad. Synaptic P‐Mad is lost in btz mutants. (D) UAS‐daw RNAi/+ control; (E) BG380‐GAL4/+; UAS‐daw RNAi/+; (F) mhc‐GAL4/UAS‐daw RNAi; (G) UAS‐daw RNAi/+; repo‐GAL4/+; (H) UAS‐daw/+; btz2 /Df(3R)BSC497; (I) tub‐GAL4/UAS‐daw; btz2 /Df(3R)BSC497; (J) tub‐GAL4/UAS‐btz; btz2 /Df(3R)BSC497; (K) tub‐GAL4/UAS‐daw. Scale bars, 30 μm (shown in B for B‐C and in D for D‐K). Scale bar for insets in (B, C), 10 μm (shown in B).
-
LQuantifications of the number of boutons normalized to muscle surface area (×1,000) in the indicated genotypes. ****P < 0.0001; ns, not significant by unpaired t‐test or Mann–Whitney test. n = 19 (UAS‐daw RNAi/+), n = 9 (BG380>daw RNAi; btz/Df; tub>daw), n = 23 (mhc>daw RNAi), n = 10 (repo>daw RNAi; tub>daw), n = 12 (btz/Df; UAS‐daw), or n = 14 (btz/Df; tub>btz). Error bars show mean ± SEM. Knocking down daw in motor neurons, muscles, or glia reduces NMJ size. Expressing UAS‐daw with tub‐GAL4 rescues NMJ size in btz mutants as efficiently as expressing UAS‐btz, but has no significant effect on NMJ size in a wild‐type background.
Source data are available online for this figure.
Figure EV2. Target gene expression in btz homozygotes.
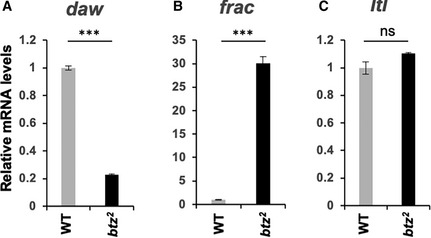
-
A–CRelative mRNA levels measured by qRT–PCR for daw (A), frac (B), and ltl (C) in btz2 homozygous larval carcasses compared to wild‐type (Canton S). daw and frac show changes consistent with those seen in btz2 /Df(3R)BSC497, but the change in ltl levels is not significant. ***P < 0.001; ns, not significant by unpaired t‐test. n = 3 for each sample. Error bars show mean ± SD.
Figure EV3. Btz regulates P‐Mad levels at the NMJ independently of the EJC.
-
A–FConfocal images of the NMJ on larval muscles 6 and 7 in segment A3 (A‐D) or the larval ventral nerve cord (E, F), stained with anti‐P‐Mad. (A, E) btz2 /+ control; (B, F) btz2 /Df(3R)BSC497; (C) Btz‐WT; btz2 /Df(3R)BSC497; (D) Btz‐HD; btz2 /Df(3R)BSC497. Scale bars, 20 μm. P‐Mad is lost from the synapse but not from neuronal cell bodies in btz mutants, and is rescued by both the wild type and EJC interaction‐defective btz transgenes.
daw was previously suggested to act in muscles and/or glia to regulate NMJ size (Ellis et al, 2010; Fuentes‐Medel et al, 2012). We used RNAi to knock it down in muscles, motor neurons, and glia and found that like Btz, it was required in all three cell types for normal synaptic bouton numbers (Fig 5D–G and L). To test whether Daw could mediate the effect of Btz on synapse growth, we expressed a UAS‐daw transgene (Serpe & O'Connor, 2006) in btz mutant larvae. Ubiquitous expression of daw with tubulin (tub)‐GAL4 rescued the btz mutant phenotype as effectively as ubiquitous expression of btz (Fig 5H–J and L). Importantly, overexpressing daw with tub‐GAL4 in wild‐type larvae did not increase synaptic bouton number (Fig 5K and L), indicating that it specifically restores a function that is lost in btz mutants. daw overexpression also partially rescued the lethality of btz mutants (Table 1). These results suggest that Btz regulates NMJ growth primarily by promoting Daw signaling.
Potential mechanisms for NMJ growth regulation by Btz and Activin
Additional studies will be required to understand the EJC‐independent mechanism by which Btz regulates the levels of daw and other mRNAs that contribute to NMJ growth. Although the crystal structure of the EJC shows that eIF4AIII forms most of the contacts with mRNA and Btz contacts only a single base (Bono et al, 2006), Btz has been reported to be capable of binding mRNA in vitro (Degot et al, 2004). Indeed, one of the few reports of a potentially EJC‐independent function for this subunit is that it regulates the translation of some mRNAs that lack introns (Chazal et al, 2013). Alternatively, another RNA‐binding protein might link Btz to the target mRNAs responsible for its function in NMJ growth. The Drosophila genome encodes 29 members of the DEAD‐box family in addition to eIF4AIII, including several that function in the nervous system. In a mass spectrometry screen, Btz was isolated in complexes with RNA‐binding proteins that include Syncrip, which regulates Gbb levels in muscle (Halstead et al, 2014), as well as CG4612, Spenito and Quaking‐related 58E‐3 (Guruharsha et al, 2011). We have not determined whether Btz directly interacts with the daw, ltl, and frac mRNAs or controls their levels through an indirect mechanism; for instance, daw expression in the fat body is sensitive to dietary and metabolic changes (Chng et al, 2014).
It is not immediately clear why both btz and daw are required in three distinct cell types for normal NMJ growth. It is possible that production of sufficient Daw requires its secretion from all three cell types. Growth does not appear to be proportional to Daw expression, as overexpressing Daw does not increase synapse size, but a threshold quantity of Daw may be required to trigger a fixed expansion of the NMJ. This would be consistent with the transient requirement for BMP signaling early in development for NMJs to reach their normal size (Berke et al, 2013). Alternatively, Daw might be differently processed or post‐translationally modified in each cell type, or secreted as a heterodimer with different TGF‐β family members or in a complex with different proteins. This type of mechanism has been reported for Gbb: presynaptically produced Gbb can be distinguished from the postsynaptically produced pool due to its trafficking by and release with the neuronal protein Crimpy (James et al, 2014). As ltl and frac encode ECM proteins that may be deposited by multiple cell types, regulation of their mRNAs by Btz could contribute to its multi‐site requirement. In developing wing discs and motor neurons, either a reduction or an increase of Frac or Ltl expression can negatively regulate TGF‐β signaling (Miller et al, 2011; Szuperak et al, 2011), indicating that the correct dosage of these proteins is critical. They could affect the activity of Daw, Gbb, or other ligands such Mav and Myoglianin, which are produced by glia and regulate NMJ growth (Fuentes‐Medel et al, 2012; Sulkowski et al, 2014; Augustin et al, 2017), or Activin‐β, which is released from motor neurons to regulate muscle size (Moss‐Taylor et al, 2019).
Activin signaling similarly promotes synaptogenesis and synaptic function in the mammalian hippocampus (Shoji‐Kasai et al, 2007; Hasegawa et al, 2014) and can boost axonal regeneration (Omura et al, 2015). Activin mRNA levels are regulated by neuronal activity (Andreasson & Worley, 1995; Dow et al, 2005), but the mechanism of this effect is not known. Our results raise the possibility that activity exerts its effects through a Btz‐dependent but EJC‐independent mechanism that impinges on Activin mRNA. The discovery that Btz can act separately from the EJC should prompt a broader exploration of its possible functions in neuronal granules (Fritzsche et al, 2013).
Materials and Methods
Fly stocks and genetics
To generate the Btz‐WT and Btz‐HD rescue transgenes, genomic DNA spanning from 27700111 to 27703445 on 3R was amplified by PCR and cloned into pattB‐QF‐hsp70. Mutations changing the H215 and D216 residues to A were introduced by PCR and Gibson cloning. The primers used to make Btz‐WT were Btz5UTRF (tagcggatccgggaattgggGTCCAGCCGTCGATACCGTTTTC), Btz5UTRR (cacccttttcTTCCTGCCCAGTGGCCG), eGFPlinkerF (tgggcaggaaGAAAAGGGTGGGCGCGC), eGFPlinkerR (acgcctgcacTCACGTGGACCGGTGCTTG), Btz3UTRF (gtccacgtgaGTGCAGGCGTACGCCGG), and Btz3UTRR (tagaggtaccctcgagccgcAACTTCGTGTTAAACGTTTATTTAAATTCAATGATCGATTAGTTTTTGCAAATACATTTCTCTC). To make Btz‐HD, Btz‐WT was used as a template with the primers Btz5UTRF, BtzHDAAR (cgtgctgcAGACCAGCGATCACCGCC), BtzHDAAF (gctggtctGCAGCACGATTCGAGGCC), and Btz3UTRR. The transgenes were injected by BestGene and integrated at the VK2 attP site. UAS‐BtzGFP was generated by amplifying the coding region of btz cDNA LD31454 by PCR and inserting it into pTWG (Drosophila Genomics Resource Center) by Gateway cloning. The plasmid was injected into embryos and integrated at random locations on the second and third chromosomes. Ovary phenotypes were examined in germline clones generated by crossing Btz‐WT or Btz‐HD; FRT82, btz2 /SM6‐TM6B females to hsFLP/Y; FRT82, ovoD /TM3 males and heat‐shocking the offspring for 1 h at 37°C during the pupal stage. Other stocks used were w1118 (BL#3605), Df(3R)BSC497 (BL#25001), G14‐GAL4 (Aberle et al, 2002), C57‐GAL4 (Budnik et al, 1996), da‐GAL4 (BL#55850), BG380‐GAL4 (BL#42736), 24B‐GAL4 (BL#1767), mhc‐GAL4 (BL#55133), repo‐GAL4 (BL#7415), tub‐GAL4 (Lee & Luo, 2001), elavc155‐GAL4 (BL#458), Mef2‐GAL4 (BL#27390), UAS‐btz RNAi (BL#30482), UAS‐mago RNAi (VDRC#28132), UAS‐eIF4AIII RNAi (VDRC#108580), UAS‐blw RNAi (VDRC#34664), UAS‐daw RNAi (BL#34974), and UAS‐daw (Parker et al, 2006).
Immunohistochemistry
To examine larval NMJs, 50 first instar larvae were collected on a grape juice agar plate and incubated at 25°C until they reached the third instar larval stage. Larval filets were prepared by pinning the larvae to silicone plates, dissecting them in ice‐cold Ca2+‐free HL3 saline (pH = 7.4), fixing in 4% formaldehyde in PBS for 15 min at room temperature, and permeabilizing with 0.2% Triton X‐100 in PBS. Adult ovaries were dissected in PBS, fixed in 4% formaldehyde in PBS for 30 min on ice, and permeabilized with 0.2% Triton X‐100 in PBS. Both tissue types were stained with primary antibody overnight at 4°C. The following antibodies were used: rabbit anti‐Vasa (1:5,000; (Gilboa & Lehmann, 2004)), chicken anti‐GFP (1:400; Life Technologies A10262), mouse anti‐Discs large (Dlg) (1:50; 4F3 from the Developmental Studies Hybridoma Bank (DSHB)), mouse anti‐ATP5A (1:500, Abcam 15H4C4), and rabbit anti‐P‐Mad (Persson et al, 1998; Sulkowski et al, 2014). Tissues stained with primary antibodies were washed three times for 5 min each with 0.2% Triton X‐100 in PBS and incubated with secondary antibodies conjugated to Alexa Fluor‐488 or Alexa Fluor‐633 (1:200, Jackson ImmunoResearch) for 2 h at room temperature. The neuronal membrane was visualized with Alexa Fluor‐488, Alexa Fluor‐633, or TRITC‐conjugated anti‐horseradish peroxidase (HRP) (1:200; Jackson ImmunoResearch). The muscle cells were visualized either using the background signal of the other antibodies used to stain the NMJ or with TRITC‐conjugated phalloidin (1:5,000; Abcam ab235138). The stained tissues were mounted in Fluoromount‐G (SouthernBiotech). Samples were imaged with a Leica SP5 or SP8 confocal microscope using a 63× oil objective. Images were captured with a resolution of 1,024 × 1,024 pixels and processed in ImageJ and Adobe Photoshop. The images shown are z projections of confocal stacks acquired from serial laser scanning or xz projections of these stacks (Fig 2).
Cell culture, co‐immunoprecipitation, and Western blotting
Btz‐WT and Btz‐HD were HA‐tagged and inserted into pPAC‐PL by Gibson cloning using the GFP‐tagged Btz‐WT and Btz‐HD plasmids as templates and the primers pPAC‐HA‐BtzF1 (atgtatccgtatgatgttccggattatgcaGCCGAAGTGGAGAAACCAA), pPAC‐HA‐BtzR1 (caatgtatcttatcatgtctttaTTCCTGCCCAGTGGC), pPAC‐vector‐F (AGACATGATAAGATACATTGATGAGTTT), and pPAC‐vector‐HA‐R (ggaacatcatacggatacatGGTGGCCCGATCC). eIF4AIII was Myc‐tagged and inserted into pPAC‐PL by Gibson cloning using eIF4AIII cDNA FI20117 as template and the primers pPAC‐cMyc‐eIF4A3‐F2 (ATGgaacaaaaacttattagcgaagaagatcttGCGCGCAAGAATGC), pAC‐eIF4A3‐R1 (caatgtatcttatcatgtctttaGATCAAGTCAGCCACGTTC), pPAC‐vector‐F and pPAC‐vector‐cMyc‐R (aagatcttcttcgctaataagtttttgttccatGGTGGCCCGATCC).
S2R+ cells were transfected with pPAC‐HA‐Btz‐WT or pPAC‐HA‐Btz‐HD and pPAC‐Myc‐eIF4AIII or pPAC‐Myc‐GFP (Bawankar et al, 2021) using Effectene (Qiagen) according to the manufacturer’s protocol. After 72 h, cells were harvested, washed once with PBS, and pelleted by centrifugation at 400× g for 10 min. The cell pellet was lysed in 0.5 ml of NET buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Triton, and 1 mM EDTA pH 8.0 supplemented with protease inhibitors and 10% glycerol) and sonicated for 3 cycles of 30 s at high power setting followed by incubation with RNase A at ice for 30 min. The lysate was centrifuged at 15,000× g for 10 min at 4°C. 10% of the volume was taken out as an input sample and the rest was incubated with Myc‐Trap Magnetic Agarose beads (Chromotek) on a head‐to‐toe rotary mixer at 4°C. The beads were washed three times for 15 min with NET buffer, and proteins were eluted by boiling the beads for 2 min at 95°C in SDS–PAGE loading dye supplemented with 100 mM DTT. For Western blots, the samples were run on a 10% SDS–PAGE gel. Gels were transferred to nitrocellulose membranes (Thermo Scientific) and were blocked for 1 h with 0.3% PBST supplemented with 5% low‐fat milk. Membranes were incubated with PBST + 5% milk supplemented with primary antibodies overnight at 4°C. Blots were washed with 0.3% PBST for three times 10 min and incubated with HRP‐conjugated secondary antibodies (1:10,000, Bio‐Rad) for 1 h. Blots were developed with enhanced chemiluminescence (Pierce). Antibodies used were mouse anti‐Myc (1: 5,000 anti‐c‐Myc clone 9E10) and mouse anti‐HA (1:5,000 anti‐HA clone 12CA5).
To measure mitochondrial abundance, body walls of third instar larvae (10 larvae/genotype) were dissected in ice‐cold PBS. Protein extraction was carried out by homogenizing body walls in 100 µl of lysis buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5% NP‐40) supplemented with protease inhibitors for 1 min at 30 oscillations/s using a TissueLyser (Qiagen). Lysates were centrifuged at 18,000 g for 10 min at 4°C to remove cell debris. Protein concentrations were determined using Bradford reagent (Bio‐Rad). Samples were boiled for 5 min at 95°C in Laemmli buffer, separated on a 10% SDS–PAGE gel, and transferred to a nitrocellulose membrane (Bio‐Rad). After blocking with 5% milk in 0.05% Tween in PBS for 1 h at RT, the membrane was incubated with primary antibody in blocking solution overnight at 4°C. Primary antibodies used were mouse anti‐ATP5A (1:1,000, Abcam ab14748) and mouse anti‐β‐tubulin (1:2,000, Biolegend #903401). The membrane was washed 3 times in PBST for 15 min and incubated 1 h at room temperature with HRP‐conjugated secondary antibody (1:10,000, Jackson Laboratories) in blocking solution. Protein bands were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Electron microscopy
Electron microscopy samples were processed as described (Ramachandran & Budnik, 2010) with some modifications. Briefly, body‐wall muscles of third instar larvae were dissected in Jan’s saline containing 128 mM NaCl, 2 mM KCl, 4 mM MgCl2, and 35.5 mM sucrose in 5 mM HEPES buffer (pH 7.2) with 0.1 M CaCl2. The body‐wall muscles were pinned on a silicone dish using insect pins and fixed with modified Trump’s fixative containing 4% paraformaldehyde, 1% glutaraldehyde, and 2 mM MgCl2 in 0.1 M sodium cacodylate buffer (pH 7.2) at room temperature for 30 min and then overnight at 4°C. Fixed muscles were rinsed with 0.1 M sodium cacodylate buffer and post‐fixed with 1% OsO4 in 0.1 M cacodylate buffer, followed by block staining with 1% uranyl acetate aqueous solution overnight at 4°C. The samples were rinsed in water, dehydrated in a graded ethanol series, infiltrated with propylene oxide/Spurr mixtures, and finally embedded in Spurr resin (Electron Microscopy Sciences, PA, USA). 70 nm ultra‐thin sections were cut and mounted on 200 mesh copper grids and stained with uranyl acetate and lead citrate. Imaging was performed on an electron microscope (CM12, FEI, Eindhoven, The Netherlands) at 120 kV and recorded digitally using a camera system (Gatan 4k × 2.7K) with Digital Micrograph software (Gatan Inc., Pleasanton, CA).
Quantification
The surface of muscles 6 and 7 was outlined and the enclosed area was quantified in ImageJ. The numbers of synaptic boutons were counted manually, and the area of individual boutons was quantified using the ellipse tool in ImageJ. To quantify mitochondrial localization, ImageJ was used to measure fluorescent intensity in random line scans taken from xz‐projections of ATP5A‐stained muscle. The highest peak, at the dorsal surface of the muscle, was normalized to the second‐highest peak within the muscle.
All quantifications were carried out blind. Statistical significance between each genotype and the controls was determined by two‐tailed Student’s t‐test, with Welch’s correction when the variances were significantly different. Each figure legend has details about sample sizes, precision measures, statistical analysis, and definitions of significance thresholds. No outliers were excluded.
RNA extraction
Third instar larvae were dissected in 4°C DEPC‐treated 0.1 M phosphate buffer (pH 7.4). Dissected central nervous systems and larval carcasses were mechanically homogenized with a plastic pestle in 200 µl TRIzol (Invitrogen). Total RNA was extracted from the samples using TRIzol/chloroform extraction: The tissues were incubated with a total volume of 450 µl TRIzol for 5 min and centrifuged at 17,000 g for 10 min at 4°C. The resultant supernatant was incubated with 107 µl chloroform, shaken vigorously by hand for 15 s, incubated at room temperature for 10 min, and centrifuged at 14,000 g for 10 min at 4°C. Approximately 250 µl of the upper aqueous phase was transferred to a new tube and incubated with 267 µl isopropanol at room temperature for 10 min. The RNA was pelleted by centrifugation at 17,000 g for 10 min at 4°C. The extracted RNA was washed 2× with 70% ethanol and purified using RNeasy Purification Kits (Qiagen). The RNA was eluted in 100 µl RNAse‐free water and further purified and concentrated by sodium acetate precipitation: 10 µl 3 M sodium acetate and 440 µl 100% ethanol were added to the RNA solution and it was incubated at −80°C overnight. The RNA was pelleted by centrifugation at 17,000 g for 30 min at 4°C, and the pellet was washed 2× with 70% ethanol and resuspended in water.
Quantitative reverse transcription polymerase chain reaction
The purified RNA was treated with RQ1 RNase‐Free DNase (Promega). Reverse transcription was performed from 1 μg of total RNA using SuperScript™ II Reverse Transcriptase (Thermo Fisher Scientific). Quantitative reverse transcription polymerase chain reaction (qPCR) was carried out using 10 ng of cDNA and 100 nM of each primer pair with a Roche LightCycler 480 machine and LightCycler 480 SYBR Green I Master 2X (Roche, 04887352001). The PCR program was: 10 min at 95°C, 45 cycles of 95°C for 15 s and 60°C for 1 min. The primers used were AGACCATAGCCATCCAGTCG and GCAGGTGATTTGGATGAGGT for daw, AGCTACGGATGCAGTGGAAC and GCCGATGCTTAGGTAGGTGA for ltl, and CACCACCGATGGAAAAACTT and TCAAAACTGATCTTGGGTTCG for frac.
RNA‐Seq
Total RNA was isolated from larval carcasses or central nervous systems from each genotype in triplicate using TRIzol (Invitrogen). RNA quality and quantity were assessed using the Bioanalyzer 2100 (Agilent Inc.). Library preparation and sequencing was carried out by the NYU Genome Technology Center. RNA‐Seq library preps were constructed using the Illumina TruSeq RNA sample Prep Kit v2 (Cat #RS‐122‐2002), using 500 ng of total RNA as input, amplified by 12 cycles of PCR, and run on an Illumina 2500 (v4 chemistry), as paired‐end reads of 50 bp. For each RNA‐seq sample, sequence quality was assessed with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and sequencing adapters were removed with Trimmomatic (Bolger et al, 2014). Cleaned reads were aligned to the Drosophila reference genome (dm3) with Tophat2 v2.1.1 1. The Picard CollectRnaSeqMetrics program (https://broadinstitute.github.io/picard/picard‐metric‐definitions.html#RnaSeqMetrics) was used to generate QC metrics including ribosomal RNA content, median per‐gene coverage, bases aligned to intergenic regions, 5’/3’ biases, and the distribution of the bases within exons, UTRs, and introns. Per sample gene expression profiles were computed using Cufflinks v2.2.1 1 and the RefSeq genome annotation for the Drosophila reference genome dm3 (Celniker et al, 2002). For multi‐sample comparison, principal component analysis and hierarchical clustering were used to verify that the expression profiles of the sequenced samples clustered as expected by sample tissue and genotype. Differential gene expression was computed for various contrasts between genotypes with the Cufflinks protocol (Trapnell et al, 2012) with default thresholds.
Author contributions
CHH, CP, PB, and J‐YR designed and carried out experiments, analyzed data, and edited the manuscript. ZT and SB analyzed data. JET designed and supervised experiments, analyzed data, obtained funding, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgments
We thank Kavita Arora, Thomas Hurd, Ruth Lehmann, Brian McCabe, Micaela Serpe, the Bloomington Drosophila Stock Center, the Vienna Drosophila Resource Center, the Drosophila Genomics Resource Center, and the Developmental Studies Hybridoma Bank for fly stocks and reagents. We thank NYULH DART Microscopy Laboratory members Alice Liang, Chris Petzold, and Kristen Dancel‐Manning for consultation and assistance with transmission electron microscopy; this core is partially funded by NYU Cancer Center support grant National Institutes of Health/National Cancer Institute P30CA016087. Flybase provided invaluable information for this study. We are grateful to Hui Hua Liu, DanQing He, and Ariel Hairston for technical assistance. The manuscript was improved by the critical comments of Maria Bustillo, Neha Ghosh, Ashley Jordan, and Hongsu Wang. This work was funded by National Science Foundation grant MCB‐1051022 to J.E.T.
EMBO reports (2022) 23: e53231.
Data availability
The RNA‐Seq dataset produced in this study is available in NCBI Gene Expression Omnibus, accession number GSE165971 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165971).
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS (2002) wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila . Neuron 33: 545–558 [DOI] [PubMed] [Google Scholar]
- Alexandrov A, Colognori D, Shu MD, Steitz JA (2012) Human spliceosomal protein CWC22 plays a role in coupling splicing to exon junction complex deposition and nonsense‐mediated decay. Proc Natl Acad Sci U S A 109: 21313–21318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson K, Worley PF (1995) Induction of beta‐A activin expression by synaptic activity and during neocortical development. Neuroscience 69: 781–796 [DOI] [PubMed] [Google Scholar]
- Ashton‐Beaucage D, Udell CM, Lavoie H, Baril C, Lefrançois M, Chagnon P, Gendron P, Caron‐Lizotte O, Bonneil É, Thibault P et al (2010) The exon junction complex controls the splicing of MAPK and other long intron‐containing transcripts in Drosophila . Cell 143: 251–262 [DOI] [PubMed] [Google Scholar]
- Augustin H, McGourty K, Steinert JR, Cochemé HM, Adcott J, Cabecinha M, Vincent A, Halff EF, Kittler JT, Boucrot E et al (2017) Myostatin‐like proteins regulate synaptic function and neuronal morphology. Development 144: 2445–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggio F, Bratic A, Mourier A, Kauppila TE, Tain LS, Kukat C, Habermann B, Partridge L, Larsson NG (2014) Drosophila melanogaster LRPPRC2 is involved in coordination of mitochondrial translation. Nucleic Acids Res 42: 13920–13938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baguet A, Degot S, Cougot N, Bertrand E, Chenard MP, Wendling C, Kessler P, Le Hir H, Rio MC, Tomasetto C (2007) The exon‐junction‐complex‐component metastatic lymph node 51 functions in stress‐granule assembly. J Cell Sci 120: 2774–2784 [DOI] [PubMed] [Google Scholar]
- Ball RW, Warren‐Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An BS, Wang TT, White JH, Haghighi AP (2010) Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron 66: 536–549 [DOI] [PubMed] [Google Scholar]
- Ballut L, Marchadier B, Baguet A, Tomasetto C, Seraphin B, Le Hir H (2005) The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat Struct Mol Biol 12: 861–869 [DOI] [PubMed] [Google Scholar]
- Barbee SA, Estes PS, Cziko A‐M, Hillebrand J, Luedeman RA, Coller JM, Johnson N, Howlett IC, Geng C, Ueda R et al (2006) Staufen‐ and FMRP‐containing neuronal RNPs are structurally and functionally related to somatic P bodies. Neuron 52: 997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa I, Haque N, Fiorini F, Barrandon C, Tomasetto C, Blanchette M, Le Hir H (2012) Human CWC22 escorts the helicase eIF4AIII to spliceosomes and promotes exon junction complex assembly. Nat Struct Mol Biol 19: 983–990 [DOI] [PubMed] [Google Scholar]
- Bawankar P, Lence T, Paolantoni C, Haussmann IU, Kazlauskiene M, Jacob D, Heidelberger JB, Richter FM, Nallasivan MP, Morin V et al (2021) Hakai is required for stabilization of core components of the m(6)A mRNA methylation machinery. Nat Commun 12: 3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H (2013) Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity‐dependent plasticity. J Neurosci 33: 17937–17950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez L, Emmett W, Faraway R, Pineda JMB, Bajew S, Gohr A, Haberman N, Sibley CR, Bradley RK, Irimia M et al (2018) Exon junction complex shapes the transcriptome by repressing recursive splicing. Mol Cell 72: 496–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm V, Britto‐Borges T, Steckelberg AL, Singh KK, Gerbracht JV, Gueney E, Blazquez L, Altmuller J, Dieterich C, Gehring NH (2018) Exon junction complexes suppress spurious splice sites to safeguard transcriptome integrity. Mol Cell 72: 482–495 [DOI] [PubMed] [Google Scholar]
- Boehm V, Haberman N, Ottens F, Ule J, Gehring NH (2014) 3' UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep 9: 555–568 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bono F, Ebert J, Lorentzen E, Conti E (2006) The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126: 713–725 [DOI] [PubMed] [Google Scholar]
- Bratic A, Wredenberg A, Grönke S, Stewart JB, Mourier A, Ruzzenente B, Kukat C, Wibom R, Habermann B, Partridge L et al (2011) The Bicoid stability factor controls polyadenylation and expression of specific mitochondrial mRNAs in Drosophila melanogaster . PLoS Genet 7: e1002324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitwieser W, Markussen FH, Horstmann H, Ephrussi A (1996) Oskar protein interaction with Vasa represents an essential step in polar granule assembly. Genes Dev 10: 2179–2188 [DOI] [PubMed] [Google Scholar]
- Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M (1996) Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg . Neuron 17: 627–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker SE, Wheeler DA, Kronmiller B, Carlson JW, Halpern A, Patel S, Adams M, Champe M, Dugan SP, Frise E et al (2002) Finishing a whole‐genome shotgun: release 3 of the Drosophila melanogaster euchromatic genome sequence. Genome Biol 3: research0079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF (2007) The nonsense‐mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74 [DOI] [PubMed] [Google Scholar]
- Chao YH, Robak LA, Xia F, Koenig MK, Adesina A, Bacino CA, Scaglia F, Bellen HJ, Wangler MF (2016) Missense variants in the middle domain of DNM1L in cases of infantile encephalopathy alter peroxisomes and mitochondria when assayed in Drosophila . Hum Mol Genet 25: 1846–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazal PE, Daguenet E, Wendling C, Ulryck N, Tomasetto C, Sargueil B, Le Hir H (2013) EJC core component MLN51 interacts with eIF3 and activates translation. Proc Natl Acad Sci U S A 110: 5903–5908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ganetzky B (2012) A neuropeptide signaling pathway regulates synaptic growth in Drosophila . J Cell Biol 196: 529–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng WB, Bou Sleiman MS, Schupfer F, Lemaitre B (2014) Transforming growth factor beta/activin signaling functions as a sugar‐sensing feedback loop to regulate digestive enzyme expression. Cell Rep 9: 336–348 [DOI] [PubMed] [Google Scholar]
- Choe J, Ryu I, Park OH, Park J, Cho H, Yoo JS, Chi SW, Kim MK, Song HK, Kim YK (2014) eIF4AIII enhances translation of nuclear cap‐binding complex‐bound mRNAs by promoting disruption of secondary structures in 5'UTR. Proc Natl Acad Sci U S A 111: E4577–E4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougot N, Daguenet É, Baguet A, Cavalier A, Thomas D, Bellaud P, Fautrel A, Godey F, Bertrand É, Tomasetto C et al (2014) MLN51 triggers P‐body disassembly and formation of a new type of RNA granules. J Cell Sci 127: 4692–4701 [DOI] [PubMed] [Google Scholar]
- Daguenet E, Baguet A, Degot S, Schmidt U, Alpy F, Wendling C, Spiegelhalter C, Kessler P, Rio M‐C, Le Hir H et al (2012) Perispeckles are major assembly sites for the exon junction core complex. Mol Biol Cell 23: 1765–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degot S, Le Hir H, Alpy F, Kedinger V, Stoll I, Wendling C, Seraphin B, Rio MC, Tomasetto C (2004) Association of the breast cancer protein MLN51 with the exon junction complex via its speckle localizer and RNA binding module. J Biol Chem 279: 33702–33715 [DOI] [PubMed] [Google Scholar]
- Dow AL, Russell DS, Duman RS (2005) Regulation of activin mRNA and Smad2 phosphorylation by antidepressant treatment in the rat brain: effects in behavioral models. J Neurosci 25: 4908–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eeden FJ, Palacios IM, Petronczki M, Weston MJ, St Johnston D (2001) Barentsz is essential for the posterior localization of oskar mRNA and colocalizes with it to the posterior pole. J Cell Biol 154: 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JE, Parker L, Cho J, Arora K (2010) Activin signaling functions upstream of Gbb to regulate synaptic growth at the Drosophila neuromuscular junction. Dev Biol 342: 121–133 [DOI] [PubMed] [Google Scholar]
- Fritzsche R, Karra D, Bennett K, Ang F, Heraud‐Farlow J, Tolino M, Doyle M, Bauer K, Thomas S, Planyavsky M et al (2013) Interactome of two diverse RNA granules links mRNA localization to translational repression in neurons. Cell Rep 5: 1749–1762 [DOI] [PubMed] [Google Scholar]
- Fuentes‐Medel Y, Ashley J, Barria R, Maloney R, Freeman M, Budnik V (2012) Integration of a retrograde signal during synapse formation by glia‐secreted TGF‐beta ligand. Curr Biol 22: 1831–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Lamprinaki S, Hentze MW, Kulozik AE (2009) The hierarchy of exon‐junction complex assembly by the spliceosome explains key features of mammalian nonsense‐mediated mRNA decay. PLoS Biol 7: e1000120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Roignant JY (2020) Anything but ordinary – emerging splicing mechanisms in eukaryotic gene regulation. Trends Genet 37: 355–372 [DOI] [PubMed] [Google Scholar]
- Gerbracht JV, Boehm V, Britto‐Borges T, Kallabis S, Wiederstein J, Ciriello S, Aschemeier DU, Krüger M, Frese CK, Altmüller J et al (2020) CASC3 promotes transcriptome‐wide activation of nonsense‐mediated decay by the exon junction complex. Nucleic Acids Res 48: 8626–8644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Marchand V, Gaspar I, Ephrussi A (2012) Control of RNP motility and localization by a splicing‐dependent structure in oskar mRNA. Nat Struct Mol Biol 19: 441–449 [DOI] [PubMed] [Google Scholar]
- Gilboa L, Lehmann R (2004) Repression of primordial germ cell differentiation parallels germ line stem cell maintenance. Curr Biol 14: 981–986 [DOI] [PubMed] [Google Scholar]
- Goldie BJ, Cairns MJ (2012) Post‐transcriptional trafficking and regulation of neuronal gene expression. Mol Neurobiol 45: 99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruharsha KG, Rual J‐F, Zhai BO, Mintseris J, Vaidya P, Vaidya N, Beekman C, Wong C, Rhee D, Cenaj O et al (2011) A protein complex network of Drosophila melanogaster . Cell 147: 690–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead JM, Lin YQ, Durraine L, Hamilton RS, Ball G, Neely GG, Bellen HJ, Davis I (2014) Syncrip/hnRNP Q influences synaptic transmission and regulates BMP signaling at the Drosophila neuromuscular synapse. Biol Open 3: 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SM, Tsuda H, Yang Y, Vibbert J, Cottee P, Lee SJ, Winek J, Haueter C, Bellen HJ, Miller MA (2012) Secreted VAPB/ALS8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Dev Cell 22: 348–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haremaki T, Weinstein DC (2012) Eif4a3 is required for accurate splicing of the Xenopus laevis ryanodine receptor pre‐mRNA. Dev Biol 372: 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Mukai H, Asashima M, Hojo Y, Ikeda M, Komatsuzaki Y, Ooishi Y, Kawato S (2014) Acute modulation of synaptic plasticity of pyramidal neurons by activin in adult hippocampus. Front Neural Circuits 8: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, Handler D, Ish‐Horowicz D, Brennecke J (2014) The exon junction complex is required for definition and excision of neighboring introns in Drosophila . Genes Dev 28: 1772–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold N, Will CL, Wolf E, Kastner B, Urlaub H, Luhrmann R (2009) Conservation of the protein composition and electron microscopy structure of Drosophila melanogaster and human spliceosomal complexes. Mol Cell Biol 29: 281–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CH, Treisman JE (2020) Specific isoforms of the guanine‐nucleotide exchange factor dPix couple neuromuscular synapse growth to muscle growth. Dev Cell 54: 117–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd TR, Herrmann B, Sauerwald J, Sanny J, Grosch M, Lehmann R (2016) Long Oskar controls mitochondrial inheritance in Drosophila melanogaster . Dev Cell 39: 560–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James RE, Hoover KM, Bulgari D, McLaughlin CN, Wilson CG, Wharton KA, Levitan ES, Broihier HT (2014) Crimpy enables discrimination of presynaptic and postsynaptic pools of a BMP at the Drosophila neuromuscular junction. Dev Cell 31: 586–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B, Lai EC (2021) The exon junction complex and intron removal prevent re‐splicing of mRNA. PLoS Genet 17: e1009563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshishian H, Kim YS (2004) Orchestrating development and function: retrograde BMP signaling in the Drosophila nervous system. Trends Neurosci 27: 143–147 [DOI] [PubMed] [Google Scholar]
- Kim MJ, O'Connor MB (2014) Anterograde Activin signaling regulates postsynaptic membrane potential and GluRIIA/B abundance at the Drosophila neuromuscular junction. PLoS One 9: e107443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Luo L (2001) Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 24: 251–254 [DOI] [PubMed] [Google Scholar]
- Mabin JW, Woodward LA, Patton RD, Yi Z, Jia M, Wysocki VH, Bundschuh R, Singh G (2018) The exon junction complex undergoes a compositional switch that alters mRNP structure and nonsense‐mediated mRNA decay activity. Cell Rep 25: 2431–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi P, Kroening S, Palacios IM, Baldassa S, Grunewald B, Ambrosino C, Goetze B, Lupas A, St Johnston D, Kiebler M (2003) Barentsz, a new component of the Staufen‐containing ribonucleoprotein particles in mammalian cells, interacts with Staufen in an RNA‐dependent manner. J Neurosci 23: 5778–5788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Mestdagh C, Akhtar J, Kreim N, Deinhard P, Sachidanandam R, Treisman J, Roignant JY (2014) The exon junction complex controls transposable element activity by ensuring faithful splicing of the piwi transcript. Genes Dev 28: 1786–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Brown HE, Silver DL (2017) Mouse models of Casc3 reveal developmental functions distinct from other components of the exon junction complex. RNA 23: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, McMahon JJ, Tsai YH, Wang Z, Silver DL (2016) Haploinsufficiency for core exon junction complex components disrupts embryonic neurogenesis and causes p53‐mediated microcephaly. PLoS Genet 12: e1006282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Pilaz LJ, McMahon JJ, Golzio C, Wu D, Shi L, Katsanis N, Silver DL (2015) Rbm8a haploinsufficiency disrupts embryonic cortical development resulting in microcephaly. J Neurosci 35: 7003–7018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew D, Ataman B, Chen J, Zhang Y, Cumberledge S, Budnik V (2005) Wingless signaling at synapses is through cleavage and nuclear import of receptor DFrizzled2. Science 310: 1344–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon KP, Carrillo RA, Zinn K (2013) Development and plasticity of the Drosophila larval neuromuscular junction. Wiley Interdiscip Rev Dev Biol 2: 647–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metze S, Herzog VA, Ruepp MD, Muhlemann O (2013) Comparison of EJC‐enhanced and EJC‐independent NMD in human cells reveals two partially redundant degradation pathways. RNA 19: 1432–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelle L, Cloutier A, Toutant J, Shkreta L, Thibault P, Durand M, Garneau D, Gendron D, Lapointe E, Couture S et al (2012) Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Mol Cell Biol 32: 954–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miech C, Pauer HU, He X, Schwarz TL (2008) Presynaptic local signaling by a canonical Wingless pathway regulates development of the Drosophila neuromuscular junction. J Neurosci 28: 10875–10884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CM, Liu N, Page‐McCaw A, Broihier HT (2011) Drosophila MMP2 regulates the matrix molecule Faulty attraction (Frac) to promote motor axon targeting in Drosophila . J Neurosci 31: 5335–5347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton VJ, Jarrett HE, Gowers K, Chalak S, Briggs L, Robinson IM, Sweeney ST (2011) Oxidative stress induces overgrowth of the Drosophila neuromuscular junction. Proc Natl Acad Sci U S A 108: 17521–17526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca TJ, Schwarz TL (2010) The nuclear import of Frizzled2‐C by Importins‐beta11 and alpha2 promotes postsynaptic development. Nat Neurosci 13: 935–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss‐Taylor L, Upadhyay A, Pan X, Kim MJ, O'Connor MB (2019) Body size and tissue‐scaling is regulated by motoneuron‐derived activins in Drosophila melanogaster . Genetics 213: 1447–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Le Hir H, Moore MJ (2004) Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev 18: 210–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Omura K, Tedeschi A, Riva P, Painter M, Rojas L, Martin J, Lisi V, Huebner E, Latremoliere A et al (2015) Robust axonal regeneration occurs in the injured CAST/Ei mouse CNS. Neuron 86: 1215–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios IM, Gatfield D, St Johnston D, Izaurralde E (2004) An eIF4AIII‐containing complex required for mRNA localization and nonsense‐mediated mRNA decay. Nature 427: 753–757 [DOI] [PubMed] [Google Scholar]
- Parker L, Ellis JE, Nguyen MQ, Arora K (2006) The divergent TGF‐beta ligand Dawdle utilizes an Activin pathway to influence axon guidance in Drosophila . Development 133: 4981–4991 [DOI] [PubMed] [Google Scholar]
- Persson U, Izumi H, Souchelnytskyi S, Itoh S, Grimsby S, Engstrom U, Heldin CH, Funa K, ten Dijke P (1998) The L45 loop in type I receptors for TGF‐beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett 434: 83–87 [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Budnik V (2010) Electron microscopy of Drosophila larval neuromuscular junctions. Cold Spring Harb Protoc 2010: pdb.prot5474 [DOI] [PubMed] [Google Scholar]
- Roignant JY, Treisman JE (2010) Exon junction complex subunits are required to splice Drosophila MAP kinase, a large heterochromatic gene. Cell 143: 238–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S (2009) Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila . Gene Exp Patterns 9: 371–380 [DOI] [PubMed] [Google Scholar]
- Schlautmann LP, Gehring NH (2020) A day in the life of the exon junction complex. Biomolecules 10: 866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS (1996) Genetic dissection of structural and functional components of synaptic plasticity. I. Fasciclin II controls synaptic stabilization and growth. Neuron 17: 641–654 [DOI] [PubMed] [Google Scholar]
- Serpe M, O'Connor MB (2006) The metalloprotease Tolloid‐related and its TGF‐beta‐like substrate Dawdle regulate Drosophila motoneuron axon guidance. Development 133: 4969–4979 [DOI] [PubMed] [Google Scholar]
- Shibuya T, Tange TO, Stroupe ME, Moore MJ (2006) Mutational analysis of human eIF4AIII identifies regions necessary for exon junction complex formation and nonsense‐mediated mRNA decay. RNA 12: 360–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji‐Kasai Y, Ageta H, Hasegawa Y, Tsuchida K, Sugino H, Inokuchi K (2007) Activin increases the number of synaptic contacts and the length of dendritic spine necks by modulating spinal actin dynamics. J Cell Sci 120: 3830–3837 [DOI] [PubMed] [Google Scholar]
- Silver DL, Watkins‐Chow DE, Schreck KC, Pierfelice TJ, Larson DM, Burnetti AJ, Liaw H‐J, Myung K, Walsh CA, Gaiano N et al (2010) The exon junction complex component Magoh controls brain size by regulating neural stem cell division. Nature Neurosci 13: 551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckelberg AL, Boehm V, Gromadzka AM, Gehring NH (2012) CWC22 connects pre‐mRNA splicing and exon junction complex assembly. Cell Rep 2: 454–461 [DOI] [PubMed] [Google Scholar]
- Sulkowski MJ, Han TH, Ott C, Wang Q, Verheyen EM, Lippincott‐Schwartz J, Serpe M (2016) A novel, noncanonical BMP pathway modulates synapse maturation at the Drosophila neuromuscular junction. PLoS Genet 12: e1005810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski M, Kim YJ, Serpe M (2014) Postsynaptic glutamate receptors regulate local BMP signaling at the Drosophila neuromuscular junction. Development 141: 436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuperak M, Salah S, Meyer EJ, Nagarajan U, Ikmi A, Gibson MC (2011) Feedback regulation of Drosophila BMP signaling by the novel extracellular protein Larval translucida. Development 138: 715–724 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Clark C, Geisbrecht ER (2016a) Analysis of mitochondrial structure and function in the Drosophila larval musculature. Mitochondrion 26: 33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Clark C, Geisbrecht ER (2016b) Drosophila clueless is involved in Parkin‐dependent mitophagy by promoting VCP‐mediated Marf degradation. Hum Mol Genet 25: 1946–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA (2010) Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol 191: 211–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffran S, Astier M, Gratecos D, Semeriva M (1997) The held out wings (how) Drosophila gene encodes a putative RNA‐binding protein involved in the control of muscular and cardiac activity. Development 124: 2087–2098 [DOI] [PubMed] [Google Scholar]