

Abstract
Clostridioides difficile infections have emerged as the leading cause of healthcare‐associated infectious diarrhea. Disease symptoms are mainly caused by the virulence factors, TcdA and TcdB, which are large homologous multidomain proteins. Here, we report a 2.8 Å resolution cryo‐EM structure of native TcdA, unveiling its conformation at neutral pH. The structure uncovers the dynamic movement of the CROPs domain which is induced in response to environmental acidification. Furthermore, the structure reveals detailed information about the interaction area between the CROPs domain and the tip of the delivery and receptor‐binding domain, which likely serves to shield the C‐terminal part of the hydrophobic pore‐forming region from solvent exposure. Similarly, extensive interactions between the globular subdomain and the N‐terminal part of the pore‐forming region suggest that the globular subdomain shields the upper part of the pore‐forming region from exposure to the surrounding solvent. Hence, the TcdA structure provides insights into the mechanism of preventing premature unfolding of the pore‐forming region at neutral pH, as well as the pH‐induced inter‐domain dynamics.
Keywords: Clostridioides difficile, cryo‐EM, native structure, TcdA, Toxin A
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Structural Biology
TcdA and TcdB are major Clostridioides difficile multidomain virulence factors. This study presents the high‐resolution cryo‐EM structure of the native TcdA toxin at neutral pH and provides insight into pH‐induced inter‐domain dynamics.

Introduction
Clostridioides difficile (C. difficile) is the leading cause of healthcare‐associated infectious diarrhea and classified as one of the top three urgent threats by the Center for Disease Control and Prevention (Lessa et al, 2015; CDC, 2019). In the United States alone the infection is responsible for close to half a million disease incidences and almost 30,000 deaths annually (Lessa et al, 2015). C. difficile infections (CDI) particularly affects elderly patients above 65 years undergoing antibiotic therapy, and the clinical outcomes range from mild diarrhea to pseudomembranous colitis, toxic megacolon and death (Songer, 2004; Lessa et al, 2015). Standard‐of‐care treatment of CDI is comprised of antibiotics such as metronidazole and vancomycin, however, 20–30% of patients experience recurrent infections (Johnson, 2009). Prolonged hospital stays, expensive treatments, and strict guidelines for preventing spread of the infection at the hospitals make CDI a substantial financial burden in many countries (Balsells et al, 2019).
Pathogenicity of C. difficile is mainly mediated by two large homologous exotoxins, TcdA and TcdB, with molecular weights of 308 and 270 kDa, respectively (Aktories et al, 2017). Both toxins consist of four known functional domains (Fig 1A) that contribute to a multistep intoxication mechanism of epithelial host cells (Fig 1B). The C‐terminal combined repetitive oligopeptides (CROPs) domain, which is comprised of a series of continuous short repeats (SRs) interspersed with long repeats (LRs), and the central delivery and receptor‐binding domain (DRBD) have been shown to facilitate binding to cell surface receptors (LaFrance et al, 2015; Yuan et al, 2015; Tao et al, 2016, 2019; Chen et al, 2018, 2021). The toxins then enter the cells via receptor‐mediated endocytosis, where the acidic environment of the endosomes triggers conformational changes in the toxins, which is predicted to facilitate pore formation (Florin & Thelestam, 1986; Qa’dan et al, 2000; Barth et al, 2001; Frisch et al, 2003; Giesemann et al, 2006). The mechanism and structure of the pore is still unknown; however, pore formation is thought to be mediated by the central DRBD which consists of a globular subdomain (GSD) and an elongated hydrophobic stretch of four α‐helices stretched across a scaffold of β‐sheets (Aktories et al, 2017; Chandrasekaran & Lacy, 2017). It has been suggested that the DRBD is involved in pore formation by unfolding and inserting the hydrophobic stretch into the endosomal membrane (Genisyuerek et al, 2011; Zhang et al, 2014; Orrell et al, 2017, 2020). The N‐terminal glucosyltransferase domain (GTD) and the cysteine protease domain (CPD) are believed to translocate across the endosomal membrane through the formed pore and into the cytosol. Once inside the cytosol, the CPD activity is induced by intracellular inositol hexakisphosphate (InsP6) where it proteolytically cleaves and releases the GTD from the rest of the toxin (Egerer et al, 2007; Reineke et al, 2007; Aktories et al, 2017). The free GTD in the cytosol then targets small GTPases of the Rho family, including Rho, Rac, and Cdc42, and inactivates their function by transferring a glucose moiety from UDP‐glucose onto a conserved residue of the GTPases (Just et al, 1995a, 1995b; Sehr et al, 1998). While TcdA and TcdB share the same mode of action, various in vivo studies have shown contradicting results regarding the individual roles of each toxin during disease. Earlier studies proposed that TcdA was the key virulence factor during disease, negating the role of TcdB (Lyerly et al, 1988; Warny et al, 1994; Kyne et al, 2001; Giannasca & Warny, 2004). However, in recent years, it has been shown that both toxins cause the same spectrum of clinical disease symptoms in humans, although TcdB is around 10 times more potent at disrupting epithelial integrity (Sambol et al, 2000; Drudy et al, 2007; King et al, 2015). Overall, it has been accepted that while both TcdA and TcdB are essential in C. difficile pathogenesis, TcdB may play a more significant role in the advanced and severe aspects of the disease (Chandrasekaran & Lacy, 2017).
Figure 1. Overall structure of native TcdA.
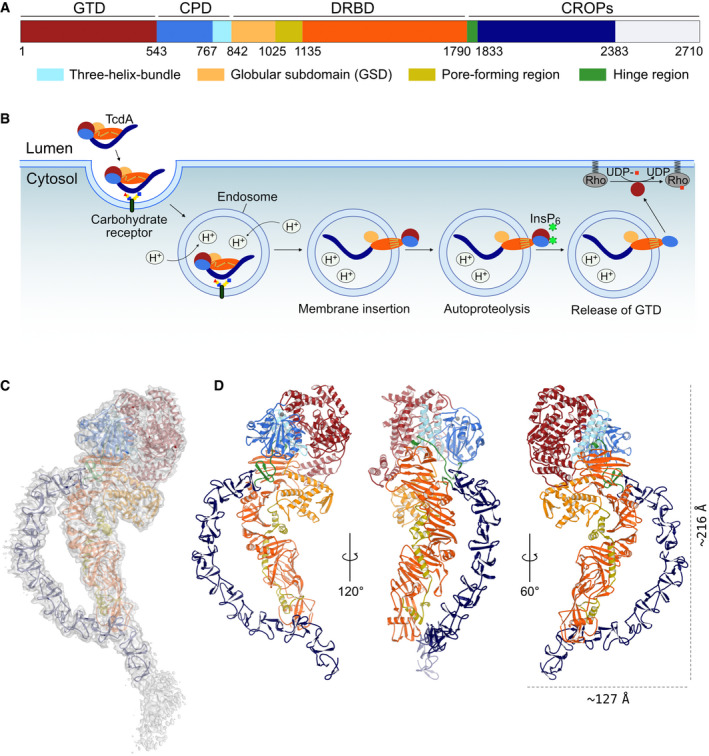
- A schematic representation of TcdA, which shows the domain organization including identified subdomains and functional regions. GTD, glycosyltransferase domain (dark red); CPD, cysteine protease domain (blue) containing the three‐helix bundle (3HB) (light blue); DRBD, delivery and receptor‐binding domain (orange) containing the globular subdomain (GSD) (yellow), the pore‐forming region (olive), and the hinge region (green); CROPs, combined repetitive oligopeptides (dark blue). The white area of the C‐terminal part of the CROPs domain represents a part of the structure that could not be modeled in the TcdA structure due to deteriorating cryo‐EM map quality.
- The multistep mechanism of intoxication. Various carbohydrate moieties (and gp96 or LPLR) are receptors for TcdA. The toxin is internalized by endocytosis and acidification of the endosome, which triggers a conformational change in the DRBD and results in pore formation and translocation of the GTD, and likely also CPD, into the cytosol. Next, inositol hexakisphosphate (InsP6) binds and activates the CPD which cleaves and releases the GTD. The GTD translocates to the cell membrane and glycosylates the Rho family of GTPases causing pathogenic cell rounding and apoptotic cell death.
- Cryo‐EM map of native TcdA contoured at 7 σ with a cartoon representation of the structure placed inside. The individual domains are colored as in panel A.
- Cartoon representation of TcdA from three different angles with zinc shown as a gray sphere.
Structural insights into different fragments of TcdA and TcdB have been reported and have allowed a better understanding of the function of the individual toxin domains (Pruitt & Lacy, 2012; Chandrasekaran & Lacy, 2017). Recently, the full‐length structure of TcdB, crystallized at pH 5.2 (endosomal pH) and with the help of three VHH antibodies (nanobodies), was determined to 3.9 Å resolution (Chen et al, 2019). This structure displays an “open” conformation of the CROPs domain of TcdB, which is distinct from a prior model presented by negative stain electron microscopy (EM) at neutral pH (Pruitt et al, 2010), further demonstrating pH‐dependent flexibility of the toxin structure. Furthermore, the structure of a truncated TcdA construct, consisting of the GTD, the CPD, and the DRBD (TcdA3‐1832, referred to as TcdA1832) crystallized at pH 6.0, has been determined to a resolution of 3.3 Å (Chumbler et al, 2016). However, this structure did not contain the CROPs domain, which comprises one‐third of the full‐length structure. The authors docked the TcdA1832 structure into a 3D map generated by negative stain EM, which indicated a possible interaction between the CROPs domain and the distal tip of the DRBD (Chumbler et al, 2016). Attempts at crystallizing the full‐length TcdA have proven difficult, likely due to the elongated structure of the CROPs domain being structurally flexible relative to the rest of the protein (Pruitt et al, 2010).
Due to the lack of high‐resolution full‐length structures of TcdA and TcdB at neutral pH, there are still plenty of unknowns related to the dynamics of the individual domains and how they interact with each other within the tertiary structure of these multidomain proteins. Furthermore, structural and mechanistic understanding of these toxins is a significant priority in order to comprehend the mode of host cell entry, which can serve as an important guide to successfully develop new anti‐toxin therapeutics.
Here, we present a 2.8 Å resolution structure of native TcdA at pH 7.5, obtained by single‐particle cryogenic electron microscopy (cryo‐EM). This high‐resolution structure of TcdA provides new important insight into the structural and functional dynamics of the toxin. Whereas the previous full‐length TcdB structure provides an important snapshot of the structural conformation at endosomal pH, the TcdA structure represents the mature host cell entry‐ready conformation at neutral pH prior to receptor binding and endocytosis. Together, the two structures demonstrate a large conformational rearrangement of the CROPs domain which is primarily promoted by nine residues in the hinge region linking the DRBD with the CROPs domain. Furthermore, when comparing TcdB and TcdA1832 with TcdA, there are regional movements occurring inside the DRBD, which may be attributed to the pH‐triggered movement of the CROPs domain. Importantly, the TcdA structure reveals detailed information about the interaction area between the CROPs domain and the tip of the DRBD, where the CROPs domain interacts closely with a highly conserved β‐hairpin loop in the C‐terminal part of the pore‐forming region. Additionally, a loop inside the GSD, which historically has been included as a part of the DRBD, also closely interacts with two α‐helices in the N‐terminal part of the pore‐forming region. This interaction between the loop of the GSD and the two α‐helices is not seen in the TcdA1832 and TcdB structures where the loop is highly destabilized. Interestingly, both the CROPs domain and the GSD interact with areas of the pore‐forming region that contain residues which previously have been shown to be important for pore formation (Genisyuerek et al, 2011; Zhang et al, 2014). Hence, we provide compelling evidence that the hydrophobic pore‐forming region is not only shielded by the β‐scaffold of the DRBD but also by extensive interactions with the CROPs domain and GSD. This likely serves to stabilize the pore‐forming region and to keep it in a fixed position until endosomal acidification triggers its membrane insertion.
Results
Cryo‐EM structure of native TcdA
Because of the known difficulties crystallizing full‐length TcdA, likely due to its large size and structural flexibility, we decided to use single‐particle cryo‐EM to examine the structure of native TcdA from the hypervirulent C. difficile strain R20291. A solution of highly purified TcdA at pH 7.5 was applied to carbon grids and was plunge frozen in liquid ethane. Two cryo‐EM data sets were collected, and the data were merged into a large data set consisting of 13,758 raw movies. Data processing led to an electrostatic potential map at a resolution of 2.8 Å based on the Fourier shell correlation 0.143 cutoff, with local resolutions of the EM map reaching ~2 Å (Fig EV1A). Attempts were also made to obtain an electrostatic potential map of full‐length TcdA at pH 4.5. Unfortunately, the quality of the particles in the images collected was not sufficiently high to proceed with further processing. This was probably due to non‐optimal sample conditions and protein aggregation (data not shown). The obtained cryo‐EM map of TcdA at neutral pH (Fig 1C) made it possible to build and refine a continuous structure from residues 2 to 2,383 of the 2,710 residues constituting the full‐length TcdA. Following residue 2,383 in the C‐terminal part of the CROPs domain, the electrostatic potential density of the cryo‐EM map quickly deteriorates, which is likely due to the highly flexible nature of the remaining CROPs domain protruding from the DRBD. Figure EV1B shows 2D classes of TcdA, which confirm that the entire CROPs domain is present in the particles but becomes averaged out in the final 3D map. The final structure was validated using the MolProbity validation tool in Phenix (Williams et al, 2018) with good statistics for the geometry restraints and a Ramachandran plot with 92.35/7.56/0.08% of favored, allowed, and outliers, respectively (Table EV1).
Figure EV1. Road map for obtaining a 2.8 Å resolution cryo‐EM map of TcdA.
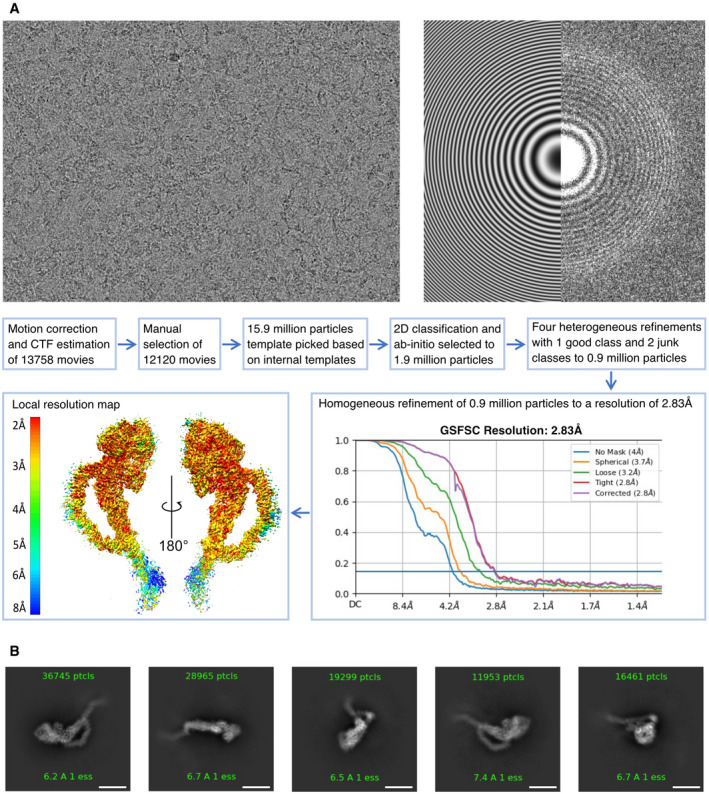
- Workflow from cryoSPARC processing. Top images show examples of raw micrograph (left) and corresponding contrast transfer function (CTF) fit in Fourier space (right) with theoretical fit shown on the left side and experimental data on the right side. In total, 13,758 movies were processed using patch motion correction and CTF estimation including data curation based on defocus, CTF fit, and motion correction values. The particles were picked using a template based on initial 2D classes from a subset of the data. Good particles were selected using 2D classification and ab initio refinements followed by multiple heterogeneous refinement runs and resulted in 0.9 million good particles. A final homogeneous refinement run yielded a map at 2.8 Å resolution based on an FSC threshold of 0.143 (graph bottom right). The resulting map (bottom left) is colored according to local resolution (contoured at 6.7 σ).
- Representative 2D classes of TcdA particles showing side, back, and top views. The region extending from residue 2,383 of the CROPs domain of TcdA is visible in the 2D classes but averaged out in the final 3D map because of high flexibility in this region. Scale bars, 100 Å.
The cryo‐EM structure of TcdA (Fig 1D) reveals the three main components of the toxin as previously described (Chumbler et al, 2016; Chen et al, 2019). The GTD and CPD form the superior “head” of the toxin, displaying a large inter‐domain interaction surface and the DRBD, containing the GSD, the pore‐forming region, and part of the hinge region, forms the central “body”, interacting with the GTD and CPD domains in one end. The CROPs domain at neutral pH extends from the hinge region of the DRBD, curves around the section of the DRBD that interacts with the distal tip, and continues onwards into a kinked and elongated C‐terminal “tail.” This conformation of the TcdA CROPs domain supports the previously reported ~25 Å negative stain EM structure of TcdA (Fig EV2A; Pruitt et al, 2010), which showed similar overall conformations of TcdA and TcdB at pH 7.0. Likewise, the CROPs domain in TcdA at pH 4.5 adopts a conformation (Pruitt et al, 2010), which is strikingly similar to the conformation of the TcdB crystal structure (Chen et al, 2019; Fig EV2B), indicating that TcdA and TcdB have similar functional dynamics in response to acidification.
Figure EV2. Comparison of TcdA and TcdB structures.
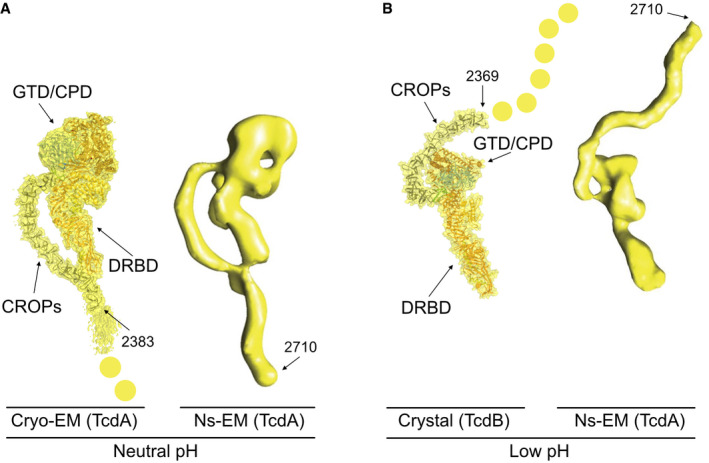
- Comparison of TcdA and TcdB structures to negative staining EM (Ns‐EM) of TcdA. The 2.8 Å cryo‐EM map with structure of TcdA modeled into the map next to a negative stain EM map of TcdA at neutral pH (Pruitt et al, 2010). Yellow dots extending from the cryo‐EM map represent the C‐terminal part of the CROPs which is averaged out in the final 3D map.
- Crystal structure of TcdB at pH 5.2 with a reoriented CROPs domain (Chen et al, 2019) compared to a negative stain EM map of TcdA at pH 4.5 (Pruitt et al, 2010). Yellow dots represent the C‐terminal part of the CROPs domain, which is present in TcdA but not in TcdB. Negative stain EM images from Pruitt et al, 2010, are used with permission from PNAS.
The individual domains in the cryo‐EM TcdA structure show overall similarity to the previously determined crystal structures of TcdA1832 at pH 6.0 (Chumbler et al, 2016) and the full‐length TcdB at pH 5.2 (Chen et al, 2019) with calculated Cα r.m.s.d values as shown in Table 1. However, the orientation of the DRBD in the cryo‐EM structure of TcdA is somewhat different compared to the crystal structures of TcdA1832 and TcdB, as indicated by the slightly elevated Cα r.m.s.d values. Also, the CROPs domain in the TcdA structure only shows similarity to the TcdB CROPs domain in the region between residues 1,811 and 2,187, whereafter the similarity decreases dramatically.
Table 1.
Inter‐individual domain superpositions.
| TcdA | TcdA1832 | TcdB | ||
|---|---|---|---|---|
| r.m.s.d (Å) | No. Cα atoms | r.m.s.d (Å) | No. Cα atoms | |
| GTD (1–543) | 0.785 | 483 | 1.409 | 490 |
| CPD (544–842) | 0.722 | 279 | 1.165 | 260 |
| GSD (843–1,025) | 0.759 | 126 | 1.773 | 138 |
| DRBD (1,026–1,802) | 2.123 | 711 | 2.822 | 652 |
| CROPs (1,811–2,383) | n.a. | – | 3.776 | 419 |
| CROPs (1,811–2,187) | n.a. | – | 1.708 | 319 |
| CROPs (2,188–2,383) | n.a. | – | 8.934 | 130 |
Cα r.m.s.d values of domain superpositions using the “align” command in PyMol.
The distinct conformation of the CROPs domain and the DRBD at neutral pH
The structure of TcdA at neutral pH including almost two‐thirds of the CROPs domain is the first to be determined at high resolution. To visualize the conformational differences between the cryo‐EM structure of TcdA, the crystal structure of TcdA1832 without the CROPs domain at moderately acidic pH and the crystal structure of full‐length TcdB at acidic pH, we have superimposed the GTD and CPD domains of the three structures, respectively (Fig 2A and B). Despite the interdomain similarity of TcdA and TcdA1832, the position of the DRBDs in the two structures is rotated by ~10° relative to the GTD and CPD domains (calculated by DynDom; Lee et al, 2003), and thus the DRBD of TcdA1832 has moved away from the position where it interacts with the CROPs domain in the TcdA structure (Fig 2A). Interestingly, we see a comparable rotation of the DRBD between TcdB at acidic pH and TcdA at neutral pH, relative to the GTD and CPD domains (Fig 2B). Furthermore, the same superposition of TcdA with TcdA1832 reveals a ~32° rotation of the GSD (residues 842–1,025 in TcdA) compared to the GSD in TcdA1832 (Fig 2A). A similar rotation of the GSD is also seen in TcdB (Fig 2B). Hence, the TcdA1832 structure (pH 6.0) mimics the conformation of TcdB structure (pH 5.2) to some degree, which may be due to the acidic crystallization conditions. The observed rotation of the GSD is in the opposite direction of the rotation of the DRBD and therefore is not likely the result of a multidomain motion in one direction. These findings indicate that the movements of the DRBD and the GSD are related to a decrease in pH and potentially to the movement of the CROPs domain, since the CROPs domain in the TcdA structure at neutral pH is in a closed conformation when interacting with the DRBD.
Figure 2. Comparison of the cryo‐EM structure of TcdA (pH 7.5) to the crystal structure of TcdA1832 (pH 6.0) and full‐length TcdB (pH 5.2).
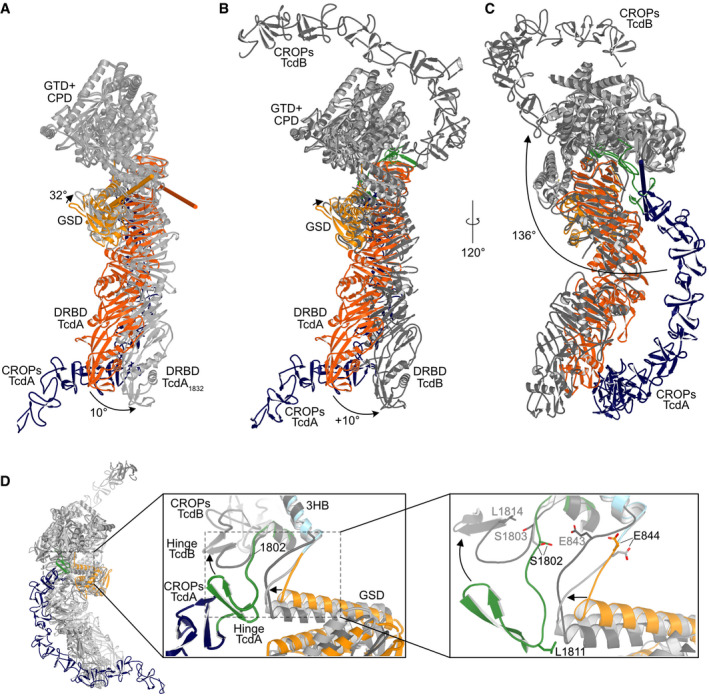
- Superposition of TcdA and TcdA1832. The GTD and CPD of TcdA are superimposed on the corresponding domains of TcdA1832 (all in light gray). The remaining domains of TcdA are colored as in Fig 1. In the TcdA1832 structure, the DRBD (light gray) swings away from the CROPs domain present in TcdA with a ~10° motion around the screw axis described by the orange rod, while the GSD of TcdA1832 tilts ~32° in the opposite direction around the axis illustrated by the yellow rod.
- Superposition of TcdA and TcdB. The GTD and CPD of TcdA are superimposed on the corresponding domains of TcdB (all in dark gray). The remaining domains of TcdA are colored as in Fig 1. The DRBD and GSD of TcdB move in a fashion similar to the corresponding domains of TcdA1832.
- The same superposition of TcdA and TcdB as in B but rotated to illustrate the ~136° rotation of the CROPs domain around the screw axis illustrated by the dark blue rod.
- Close‐up view of the hinge region in a superposition of TcdA (colored as in Fig 1), TcdA1832 (light gray), and TcdB (dark gray). Residues in TcdB are written in gray and in black for TcdA/TcdA1832.
Figure 2C illustrates a dramatic conformational difference in the CROPs domain when comparing the TcdA and TcdB structures. When superimposing the GTD and CPD domains of the two structures, there is a ~136° rotation of the CROPs domains of TcdA at neutral pH and TcdB at acidic pH. This movement is facilitated by the hinge (residues 1,790–1,833), which was previously defined in TcdB as a β‐hairpin, similar to the short repeats (SR) found in the CROPs domain, attached to a short α‐helix interspersed by a loop that connects the CROPs domain to the DRBD (Chen et al, 2019). In the TcdB structure, the hinge was shown to interact directly with the β‐flap, which is important for CPD activation, and the three‐helix bundle (3HB) (residues 767–842) located in a cleft among the GTD, CPD, DRBD, and CROPs domains. In the cryo‐EM map of the TcdA structure, the hinge region is also well defined (Fig EV3A). However, in the TcdA structure, where the CROPs domain is interacting with the distal tip of the DRBD, the entire β‐hairpin (SR) part of the hinge is positioned in a cleft among the CPD, GSD, and DRBD (Fig 2D). In this position, the hinge forms no interactions with either the 3HB or other residues in the CPD. Instead, the β‐hairpin of the hinge is occupying space where the GSD is otherwise positioned in both the TcdB and TcdA1832 structures. This position of the hinge region in the interdomain cleft of the TcdA structure results in a displacement of the linker between the 3HB and the GSD (residues 843–849) causing the GSD to tilt away from the hinge region. The rotation of the hinge primarily occurs through substantial changes in the backbone conformation of residues Ser1802 to His1810 (Tyr1805‐Glu1813 in TcdB) (Figs 2D and EV4), while the reorientation of the GSD is promoted by changes in residues Glu844 to Val846.
Figure EV3. The 2.8 Å cryo‐EM map presented in different regions of the TcdA structure.
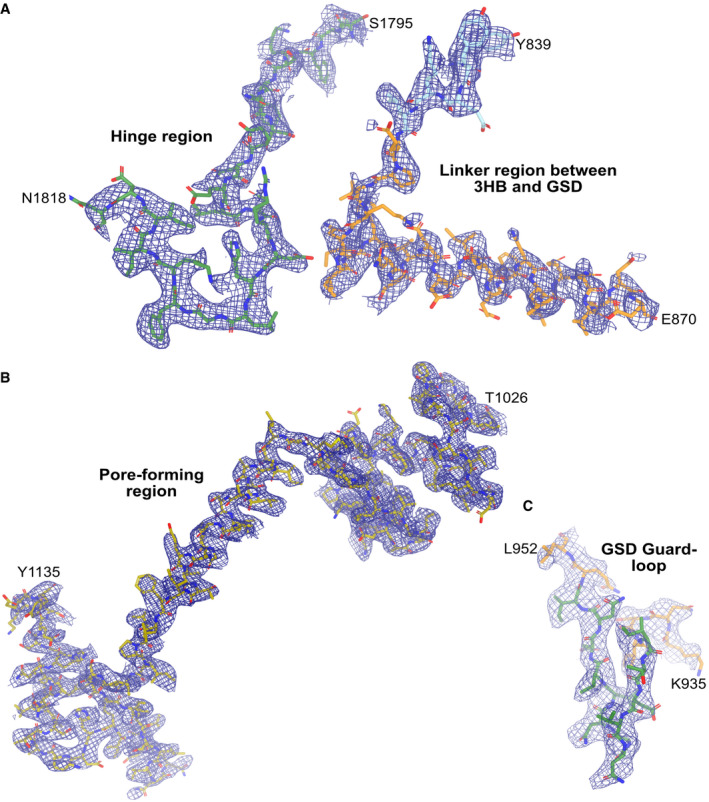
- Map of the hinge region (residues 1,795–1,818) as well as the linker region between the 3HB and the GSD (residues 839–870).
- Map of the entire pore‐forming region from residues 1,026–1,135.
- Map of the guard loop in GSD from residues 936–952. All maps are contoured at 8 σ.
Figure EV4. Movement of the hinge SR and CROPs SR1 during acidification.
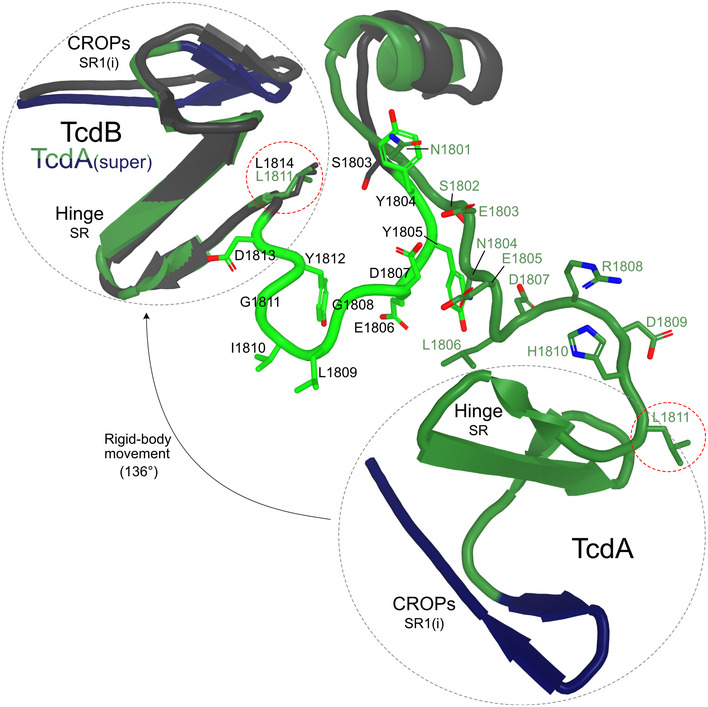
Close‐up view showing the rigid body movement of the TcdA hinge SR (dark green) and the SR1 of the CROPs domain (dark blue) in a superposition of the TcdA and TcdB CROPs domains. The ~136° rotation of the hinge region and CROPs region relative to the GTD and the CPD is displayed by the superimposed hinge SR and SR1 of the CROPs domain of TcdA on the corresponding regions of TcdB at low pH (dark gray). This rotation is primarily facilitated by changes in the backbone conformations of residues Ser1802 to His1810 of the hinge loop in TcdA (Tyr1805‐Asp1813 in TcdB (light green)). The red circle highlights the movement of Leu1811 during acidification, while the gray circle highlights movement of hinge SR and CROPs SR1.
Protection of the pore‐forming region by the CROPs domain and the GSD
The elongated α‐helical hydrophobic pore‐forming region (residues 1,025–1,135) lies in a continuous groove stretching across the surface of the β‐scaffold comprising the majority of the DRBD. The region consists of four α‐helices (α1, α2, α3, and α4) and three β‐strands (β3, β4, and β5). It was previously proposed that the DRBD serves to support and protect the hydrophobic residues in the pore‐forming region by being dormant at neutral pH. Then, during the acidification of the endosome, the DRBD undergoes structural changes to create a transmembrane passageway to the cytosol (Zhang et al, 2014; Chen et al, 2019). However, not much is known in detail about this protection mechanism, and the structural elements important for the activation of endosomal pore formation are unknown as well. In the TcdA structure, the entire pore‐forming region is well defined in the cryo‐EM map (Fig EV3B) and the C‐terminal half of the region is structurally similar to the TcdA1832 and TcdB structures. However, the N‐terminal part (including α1 and α2) of TcdA shows interesting differences when compared to TcdB, while the α1 and α2 helices of the TcdB structure are highly flexible with no electron density in the map; in TcdA, this part of the pore‐forming region is more ordered and buried into the DRBD (Figs 3A and 4).
Figure 3. Protection of the pore‐forming region in TcdA.
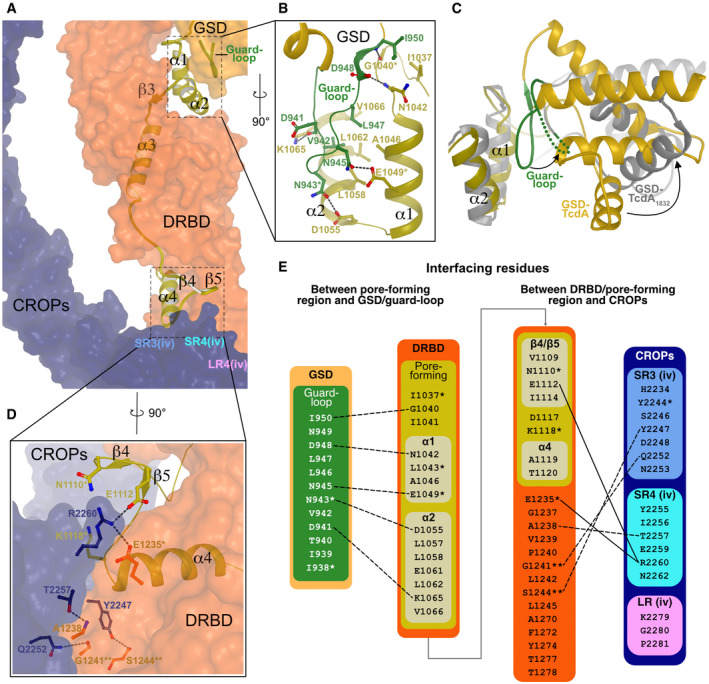
- Surface representation of the DRBD and the CROPs domain with cartoon illustration of the pore‐forming region and the GSD guard loop. Both the GSD of the DRBD and part of the CROPs domain interact with parts of the pore‐forming region. The estimated position of the repeat units SR3 (iv), SR4 (iv), and LR (iv) interacting with the DRBD and pore‐forming region is written in light blue, cyan, and pink, respectively.
- The GSD of the TcdA structure is positioned so that a loop closely interacts with the α1 and α2 helices of the pore‐forming region. This “guard loop” (green) in GSD, residues Ile939 to Ile950, forms several H‐bonds and hydrophobic interactions with residues in both the α1 and α2 helices.
- In a superposition of α1 and α2 of both TcdA and TcdA1832 (gray), the GSD of the TcdA1832 structure has rotated away from α1 and α2 of the pore‐forming region and the guard loop is disordered leaving the α1 and α2 helices exposed to the solvent.
- Hydrogen‐bond interactions between SR3 and SR4 in CROPs unit iv with the tip of the DRBD and β4, β5, and α4 of the pore‐forming region. The interaction area among the SR3 (iv), SR4 (iv), and LR (iv) in the CROPs domain and the DRBD includes three H‐bonds and Arg2260 forming two salt bridges to Glu1112 and Glu1235. The CROPs domain has several residues interfacing with residues in the DRBD and the pore‐forming region that are not shown in this panel.
- Interface residues between the pore‐forming region and the GSD guard loop as well as the DRBD/pore‐forming region and the CROPs domain as determined by the PISA webtool from the European Bioinformatics Institute (Krissinel & Henrick, 2007). Only interacting residues are listed for each domain. Inter‐domain hydrogen bonds and salt bridges are illustrated with dashed and solid lines, respectively. *strictly conserved in the family of LCTs. **strictly conserved in the family of LCTs except for TpeL, which does not contain a CROPs domain.
Figure 4. GSD movements relative to the pore‐forming region.
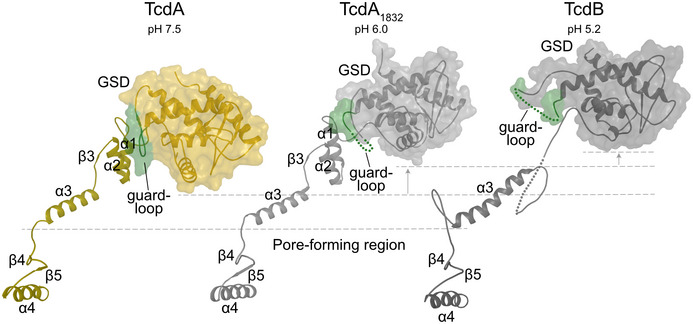
The α3, β4, β5, and α4 of the pore‐forming region in TcdA at pH 7.5 (olive), TcdA1832 at pH 6.0 (light gray) and TcdB at pH 5.2 (dark gray) are superimposed. The GSD and pore‐forming region are illustrated as a cartoon while the GSD is additionally shown with a transparent surface. The GSD of TcdA1832 and TcdB is rotated relative to the GSD of TcdA (orange) in a progressive manner corresponding to the decreasing pH, and the guard loop (green) goes from a well‐ordered loop interacting with the α1 and α2 helices of the pore‐forming region in TcdA to being disordered and dislocated from α1 to α2 and disordered in TcdA1832 and TcdB.
In addition to being shielded by the DRBD, the pore‐forming region is also elegantly kept in place by the GSD and the CROPs domain in conjunction, protecting residues important for pore formation in the N‐ and C‐terminal regions, respectively (Fig 3A). In TcdA, the N‐terminal helices, α1 and α2, are guarded by a well‐defined and conserved loop (residues Ile939–Ile950) in the GSD (Figs EV3C and EV5). This “guard‐loop” is positioned so that it closely interacts with both α1 and α2 helices, forming several hydrogen bonds and hydrophobic interactions (Fig 3A, B, and E). Residues Asp941, Asn943, and Asn945 in the guard loop show hydrogen bonding to conserved residues Lys1065, Asp1055, and Glu1049, respectively, in α2 and α1, while residues Asp948 and Ile950 in the guard loop and residues Asn1042 and Gly1040 N‐terminal of α1 are also forming H‐bonds. In addition, residues Val942, Leu947, and Ile950 in the guard loop are forming direct hydrophobic interactions with residues Ile1037, Ala1046, Leu1058, Leu1062, and Val1066, in the two helices. Figure 3E shows a list of the residues forming the entire interface area between the guard loop and the N‐terminal part of the pore‐forming region.
Figure EV5. Alignment of large clostridial cytotoxins.
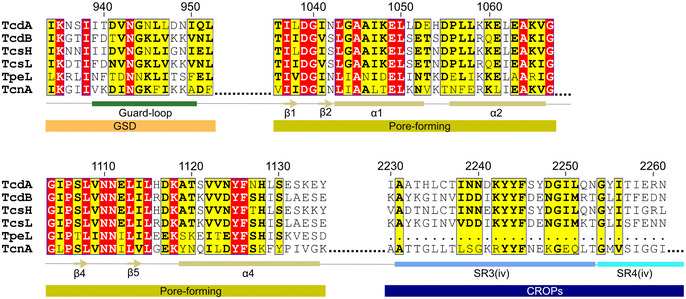
Amino acid sequence alignment of the guard loop in the GSD, part of the pore‐forming region, and SR3 (iv) and SR4 (iv) of the CROPs domain among the large clostridial cytotoxins. TcdA and TcdB originate from C. difficile strain R20291, TcsH and TcsL are both from C. sordellii strain VPI 9048, TpeL originates from C. perfringens strain CP4, and TcnA is from C. novyi B strain NCTC 9691.
Interestingly, when superimposing the α1 and α2 helices from TcdA onto TcdA1832, the GSD of TcdA1832 tilts away from the two helices, which disrupts the interactions between α1/α2 and the guard loop and leaves the loop disordered (Figs 3C and 4). This withdrawal of the guard loop in TcdA1832 is even more prominent when superimposed onto the TcdB structure, which results in the uncovering of the two helices and exposes them to the surrounding solvent (Fig 4).
At the distal end of the DRBD, there is a large interaction area between the tip of the domain and the two short repeats, SR3 and SR4, and long repeat, LR, in unit iv of the CROPs domain (Fig 3A, D, and E). This interaction area includes three H‐bonds among residues Tyr2247, Gln2252, and Thr2257 in the SRs with residues Ser1244, Gly1241, and Ala1238, respectively, in the DRBD (Fig 3D). Furthermore, Arg2260 in SR4 forms a salt bridge to the Glu1235 in the DRBD, which is strictly conserved in the family of LCTs. Arg2260 also forms another salt bridge to the highly conserved residue, Glu1112, which resides in the loop between β4 and β5 of the pore‐forming region. Besides these H‐bond interactions, the SR3/4 and LR from unit iv of the CROPs domain have several residues interfacing directly with residues in the DRBD and the pore‐forming region including highly conserved residues in the β4/β5 β‐hairpin known to be important for pore formation (Fig 3E).
The CROPs domain
While the full structure of the TcdB CROPs domain has been determined previously at low pH (Chen et al, 2019), only small 127‐aa and 255‐aa structures of the C‐terminal fragment of the CROPs domain of TcdA have been determined (Ho et al, 2005; Greco et al, 2006). Using these fragments, the authors constructed a putative model of the complete CROPs domain of TcdA that showed an extended S‐shaped structure, which was later confirmed by low‐resolution EM (Pruitt et al, 2010). The CROPs domains of TcdA (residues 1,833–2,710) and TcdB (residues 1,834–2368) have a unique structure as they are made up of two types of repetitive sequences, SRs of 15–21 residues interspersed with LRs of 30 residues. The CROPs domain of TcdA is composed of 32 SRs and 7 LRs (Ho et al, 2005), while the much shorter CROPs domain of TcdB is composed of only 20 SRs and 4 LRs (Chen et al, 2019). Each SR consists of a β‐hairpin connected to a flexible loop, while the LR is composed of three β‐strands that form a twisted anti‐parallel β‐sheet structure in tandem with the β‐hairpin of the previous SR. Adjacent SRs are packed together with a ~120° rotation in relation to the previous SR, forming straight rod‐like segments of β‐solenoid structure. As the repetitive stacking of the SRs is interrupted by a LR, it causes a ~132–146° kink in the rod‐like segment creating the curved shape of the CROPs domain. Due to the presence of seven LRs in the CROPs domain of TcdA, it can be divided into eight structurally equivalent units (i–viii), while TcdB is composed of five units (i–v) (Ho et al, 2005; Chen et al, 2019).
From the cryo‐EM map of TcdA, we were able to build the structure of the N‐terminal part of the CROPs domain to residue 2383 (SR4 in unit v) (Fig 5A). The hinge, which connects the CROPs domain to the DRBD, contains an SR‐like sequence N‐terminal of the first SR1 in unit i of the CROPs domain. To compare the structures of the CROPs domains of TcdA and TcdB, we superimposed the two CROPs domains and calculated a Cα r.m.s.d of 1.71 Å (Table 1) from residue Leu1811 in the hinge region to residue Ile2187 in LR (iii) of TcdA (Val2187 in TcdB; Fig 5B). Beyond residue 2,187, the similarity of the two CROPs domains decreases. Structurally, the CROPs domains of TcdA and TcdB are structurally highly similar from the beginning of the hinge SR until LR (iii), suggesting a highly rigid structure. After the hinge SR until LR (iii) region, the CROPs domains curve in different directions with the CROPs domain of TcdB rotating ~50° relative to TcdA. The CROPs domain of TcdB passes the tip of the DRBD on the opposite side, whereas the CROPs domain of TcdA extends downwards and away from the DRBD. Both TcdA and TcdB CROPs domains are composed of a similar number of SRs in each unit until LR (iii). Following LR (iii), the CROPs of TcdB consist of five SRs in unit iv, compared to only four SRs in the corresponding CROPs unit of TcdA. This difference may be the origin of the differing rotation angles in the latter part of the C terminus, although alternatively, it could also be attributed to the low pH of the TcdB crystal, crystal packing, or perhaps the interaction between the DRBD and the CROPs domain in TcdA. From the superposition of the CROPs domains in TcdA and TcdB (Fig 5B), it is clear that the SR in the hinge forms a rigid unit together with the CROPs domain. Hence, the repeat unit of the CROPs and the hinge SR jointly undergo a rigid body movement during the pH‐induced rotation of the CROPs domain with only residues 1,802–1,810 comprising the actual hinge movement (Fig EV4).
Figure 5. The CROPs domain.
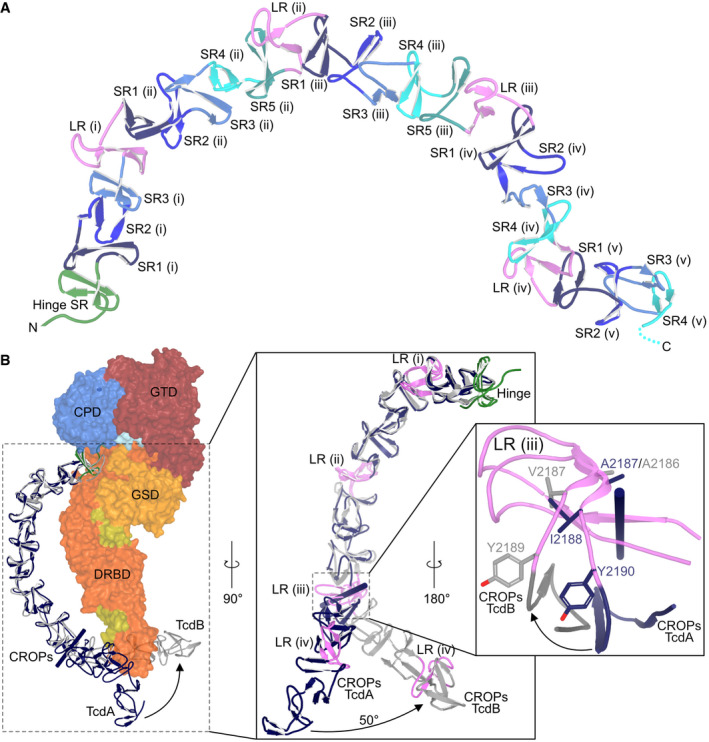
- Structure of the CROPs domain of TcdA. As shown for TcdB (Chen et al, 2019), the link between the DRBD and the CROPs domain is separated by the hinge region, which contains its own SR (green). This is followed by a series of SRs (blue/green colors) that are separated by LRs (pink). The CROPs domain was modeled until residue 2,383 in the SR4 (unit v).
- Structural comparison of the CROPs domains of TcdA (blue) and TcdB (gray). The CROPs domain of TcdB (residues 1,802–2,368) is superimposed onto the partial CROPs domain of TcdA (residues 1,833–2,383). The two domains superimpose with a Cα r.m.s.d. of 1.71 Å (Table 1) from residue 1,810 in the hinge region to residue 2,187 in LR (iii) of TcdA. Beyond that, the two domains follow different paths where CROPs domain of TcdB rotates ~50° relative to that of TcdA.
Discussion
Numerous studies of different fragments of TcdA and TcdB have provided important insights into the structure and function of the toxins which is summarized in a recent review (Aktories et al, 2017). In addition, the recent full‐length crystal structure of TcdB at acidic pH revealed a distinct structure of the CROPs domain in an open conformation (Chen et al, 2019), confirming the previously demonstrated pH‐induced structural dynamics of the toxins at endosomal conditions (Florin & Thelestam, 1986; Qa’dan et al, 2000; Barth et al, 2001; Giesemann et al, 2006; Pruitt et al, 2010). Despite these significant advances, we are still missing key structural insights into the interactions and dynamics between the central DRBD and the CROPs domain in the host cell entry‐ready toxins at neutral pH prior to receptor binding and endocytosis. To answer some of these questions, we have determined a high‐resolution cryo‐EM structure of native TcdA at neutral pH. The structure provides a snapshot of the toxin from residues 2–2,383, showing interactions of the CROPs domain with a highly conserved part of the pore‐forming region in the DRBD. This TcdA structure provides the first view of the toxin at neutral pH and its comparison with the previous low pH TcdA1832 and TcdB structures reveals important functional dynamics in response to acidification.
The regional movements seen in the DRBD and the GSD in TcdA1832 (Fig 2A) and TcdB (Fig 2B) relative to TcdA are potentially a result of the change in pH or the lack of interaction between the DRBD and CROPs domain in the former structures. This could be an important mechanism of the LCTs in response to acidic pH, which facilitates the bending of the hinge region (Fig 2D) and thereby promotes the dramatic rearrangement of the CROPs domain (Fig 2C). The extension of the hinge from the multidomain cleft has been suggested to mediate inter‐domain structural communication. This is supported by earlier studies showing drastic reduction in toxicity of TcdB when the hinge region is deleted (Zhang et al, 2013). The conformational changes in the hinge results in a rigid body rotation of the hinge SR β‐hairpin together with the CROPs domain (Fig EV4). This brings the hinge SR and the N‐terminal part of the CROPs domain from a position distant from the β‐flap and the 3HB at neutral pH into a position that is proximal to this functionally important region, which allows direct interactions with the CPD at acidic pH (Fig 2D). Hence, the observed flexibility of the hinge region supports a potential function in the movement of the CROPs domain and the hinge SR during acidification. Furthermore, the similar conformations of the DRBD and GSD of TcdA1832 (Fig 2A) and TcdB (Fig 2B) suggest that the TcdA1832 and TcdB structures being exposed to moderately acidic and acidic pH, respectively, mimic the toxins’ structural rearrangements in the acidified endosome to some extent.
The TcdA structure unveils the first detailed information about the residue interactions among SR3, SR4, and LR in unit iv of the CROPs domain and the distal tip of the DRBD, revealing multiple H‐bonds and hydrophobic interactions between the two domains (Fig 3D and E). Interestingly, the positively charged Arg2260 is elegantly positioned in a pocket consisting of several negatively charged residues, where it forms H‐bonds to both Glu1235 of the DRBD, which is strictly conserved in the family of LCTs, and Glu1112 in the highly conserved β‐harpin loop (β4 and β5) in the C‐terminal part of the pore‐forming region (Fig 3D). The extensive interaction surface between the CROPs domain and the DRBD including the conserved pore‐forming region likely has a role in shielding of α4, β4, and β5 from solvent exposure and thereby prevents premature conformational changes in the DRBD until it reaches the endosomal compartment. Premature exposure of the hydrophobic pore‐forming region prior to an accessible endosomal membrane will likely lead to aggregate formation and prevent translocation and pore formation. Notably, the importance of residues Gly1241 and Ser1244 in the DRBD forming hydrogen bonds to residues Thr2252 and Tyr2247 in the CROPs domain is emphasized by being strictly conserved in the family of LCTs (except for TpeL, which does not contain a CROPs domain). The reason why the C‐terminal part of the pore‐forming region in the TcdB structure showed no pH‐triggered conformational alterations is likely because β4 and β5 are kept in place by direct interactions with one of the nanobodies used for crystallization (Chen et al, 2019). The binding of this nanobody sterically interfered with an interaction that is similar to the observed interaction between the CROPs domain and the DRBD in the TcdA structure (Fig 3D). This is further supported by the FRET analysis performed on TcdB by the same authors, finding no significant differences in FRET between the position of the CROPs domain at acidic and neutral pH. In this experiment, they used the same nanobody for immobilization of TcdB, which most likely prevented adoption of the closed conformation of the CROPs domain. On the other hand, the authors found a pH‐dependent conformational change in the CROPs domain into a closed conformation at neutral pH, when they performed SAXS studies in the absence of the nanobody.
The N‐terminal helices, α1 and α2, in the pore‐forming region are likewise shielded from solvent exposure by a highly conserved loop (residues Ile939–Ile950) in the GSD, which we refer to as the “guard‐loop” (Fig 3B and E). This guard loop interacts with α1 and α2 through multiple H‐bonds and hydrophobic interactions, elegantly protecting the two helices inside the hydrophobic groove positioned between the DRBD and GSD. Superposition of the α1 and α2 helices of TcdA and TcdA1832 clearly shows that the guard loop of the latter is disordered and the protection of α1 and α2 is disrupted due to the rotation of GSD, likely due to a lower than neutral pH as described previously (Fig 3C). This protecting mechanism may be an important function of the GSD, where it uses the guard loop to alternately protect and expose α1 and α2 to the surrounding solvent, depending on the environmental pH. This is further supported by the TcdB structure (Chen et al, 2019), which at an even more acidic pH than TcdA1832 is almost completely missing electron density for both the guard loop and the α1 and α2 helices, strongly indicating a high degree of flexibility in this region when the environmental pH is decreasing (Fig EV4). Due to these novel insights into the important role of the GSD in stabilizing the pore‐forming region, we argue that this subdomain, instead of being included as a part of the DRBD, may be considered its own functional domain. Furthermore, the crucial role of the hinge region is highlighted. In addition to facilitating movement of the CROPs domain during acidification, the reposition of the hinge leads to rotation of the GSD (Fig 2D), likely resulting in the exposure of the pore‐forming region (Fig 3C). These findings demonstrate a coherent inter‐domain mechanism mediated by the hinge, as the linker between all four domains of the toxin. The hinge has also been suggested to be involved in regulation of TcdA and TcdB autoprocessing, as the hinge interacts with the 3HB and the β‐flap in the TcdB structure (Chen et al, 2019). Also, TcdA1‐1795 (Chumbler et al, 2016) and TcdB1‐1805 (Chen et al, 2019), shorter fragments of the toxins without the hinge, are much more efficient in InsP6‐induced autocleavage of the GTD compared to the full‐length structures.
The hydrophobic α‐helices in the pore‐forming region (Fig 3) have been experimentally shown to be important for pore formation in TcdB (Genisyuerek et al, 2011; Zhang et al, 2014). These hydrophobic helical elements resemble similar motifs in the pore‐forming region of diphtheria toxin (Zhang et al, 2014), which suggests a “double‐dagger” model for the TcdB pore formation similar in principle to the model originally suggested for the diphtheria toxin pore (Choe et al, 1992; Wang & London, 2009). However, it is still not clear how these pores are formed and whether a single protein molecule alone can establish translocation. The hydrophobic α‐helices in TcdA, likely to be inserted into the endosomal lipid bilayer as two helical hairpins, were initially revealed by Chumbler et al (2016). It was proposed that the elongated β‐sheet structure of the DRBD provides a protecting scaffold around these hydrophobic segments, maintaining them in a shielded, yet readily accessible conformation until pore formation. Insights from our TcdA structure support this mechanism and additionally provide detailed information about the important role of the GSD and CROPs domain, which in conjunction contribute to the shielding of the pore‐forming region. The importance of the DRBD translocation machinery is further supported in a recent publication reporting that the DRBD including the GSD and the pore‐forming region are evolutionary conserved in hundreds of bacteria not related to clostridia and the conventional LCT domain architecture (Orrell et al, 2020). The authors also demonstrated that a TcdB fragment consisting of residues 851–1,473, which contains the intact GSD, comprises all the components needed for pore formation and translocation, whereas a similar fragment starting from residue 881 with a truncated GSD was unable to facilitate translocation, highlighting the critical function of the GSD during intoxication.
Furthermore, we display that the structures of the CROPs domains of TcdA and TcdB are highly similar from the first SR in the hinge region until LR (iii), after which the domains change direction and rotate ~50° relative to each other (Fig 5B). The hinge, connecting the CROPs domain to the DRBD, was first described by Chen et al (2019), where they also recognized this SR‐like structure N‐terminal of the first SR1 of the CROPs domain. Superimposing the CROPs domain of TcdA at neutral pH onto the CROPs domain of the acidic TcdB structure reveals that the CROPs SR1 and hinge SR move as a rigid body during acidification (Fig EV4). Therefore, since the hinge SR has a similar sequence motif, is structurally similar to the other CROPs SRs, and moves in conjunction with the CROPs domain as a unit, we propose to expand the CROPs domain to also include this hinge SR, starting from residue Leu1811. Including this SR in the CROPs domain was also suggested in a recent study that investigated the binding of TcdB to the cell surface receptor chondroitin sulfate proteoglycan 4 (CSPG4; Gupta et al, 2017).
The TcdA structure also explains why TcdA is unable to bind to the frizzled protein and CSPG4 receptors, which are previously described structurally in crystal structure complexes of TcdB and the two receptors (Chen et al, 2018, 2021). Frizzled protein is shown to bind to a pocket in DRBD of TcdB, which is formed by residues Leu1433, Met1437, Ser1486, Leu1493, Ser1495, and Phe1597. The corresponding residues in TcdA show very little conservation and the pocket is occluded by bulky residues such as Lys1434, Tyr1485, Glu1494, and Tyr1496. CSPG4 primarily interacts with residues in the hinge region (residues 1,809–1,825) and the CPD (residues Ser573, Arg575, Pro602, Tyr603, Tyr621, and Leu661–Ser667), which are also poorly conserved in TcdA. In addition, the conformation of the CROPs domain in the TcdA structure sterically interferes with binding of both receptors and would therefore have to be displaced for receptor binding to take place.
In conclusion, the findings in this study provide new important details in elucidating the pH‐induced structural and functional dynamics of the large clostridial toxins, as well as structural insights into the shielding of the pore‐forming region at neutral pH.
Materials and Methods
Protein purification
Native TcdA was purified from C. difficile strain R20291 (NCTC13366) as described in Aminzadeh and Jørgensen (2021). Briefly, the first‐stage seed culture was prepared by inoculating sterilized tryptone‐yeast extract (TY) (Formedium, Hunstanton, NK, UK) medium with C. difficile glycerol stock and incubating unagitated for 24 h at 37°C under anaerobic conditions. The second‐stage seed culture was prepared by inoculating sterilized TY medium with the first‐stage seed culture (1%, v/v), and likewise incubating unagitated for 24 h at 37°C under anaerobic conditions. To prepare a large‐scale culture for toxin purification, a Spectra/Por® 1.6–8 kDa dialysis tube (Repligen, Rancho Dominguez, CA, USA) was filled with 1.5 l of PBS, pH 7.5, and immersed in 15 l of TY, 5 mM ZnCl2, and 10 mM glucose culture medium in a 15 l Nalgene™ culture vessel (Thermo Fisher Scientific, Waltham, MA, USA) and sterilized. After sterilization and creation of anaerobiosis, the medium was equilibrated overnight at 37°C prior to inoculation with second‐stage seed culture (1%, v/v) into the dialysis tube and incubated unagitated for 3 days at 37°C. After 3 days, the bacterial culture in the dialysis tube was centrifuged at 18,500 g for 20 min at 4°C, filtered at 0.22 μm, and dialyzed using a Quattro 1000 Ultrafiltration/Diafiltration pump with Pellicon® 2 Biomax 50 kDa membrane cassettes (Merck Millipore Ltd., Carrigtwohill, CO, Ireland) in 50 mM Tris‐HCl, pH 7.5. Hereafter, TcdA was purified by Q Sepharose anion‐exchange chromatography, followed by MonoQ anion‐exchange chromatography, and finally by gel filtration chromatography into 50 mM Tris‐HCl, pH 7.5 for cryo‐EM grid preparation. Purified protein was aliquoted and stored with 20% glycerol at −80°C until use. Samples for cryo‐EM were prepared by thawing and diluting the protein from the frozen samples in 50 mM Tris‐HCl, pH 7.5, before applying to EM grids. In addition, samples were also diluted in 100 mM sodium acetate, pH 4.5, immediately before applying to EM grids.
Cryo‐EM data collection
C‐Flat 2/2–3Cu 300 mesh holey carbon grids (Protochips, Morrisville, NC, USA) were glow discharged for 45 s at 15 mA using a Quorum GloQube® Plus (Quorom Technologies Ltd, Laughton, ES, UK). Three μl of TcdA (0.5–1 mg/ml) was applied to grids, blotted for 4–5 s, and plunge frozen in liquid ethane using a Leica EM GP2 plunge freezer (Leica Microsystems GmbH, Wetzlar, HE, Germany) maintained at 10°C and 99% humidity. Frozen grids were stored in liquid nitrogen until imaging. Data collection was performed on the Titan Krios G3i (Thermo Fisher Scientific, Waltham, MA, USA) at the Danish National cryo‐EM Facility—EMBION (Aarhus node), operated at 300 kV with a Bioquantum/K3 setup (Ametek/Gatan, Pleasanton, CA, USA) automated with the EPU2.7 software. A nominal magnification of 130,000× (pixel size of 0.64 Å) and an underfocus range between 0.5 and 1.5 μm were used for data collection. Exposures were collected as movies of 56 dose fractions with an exposure rate of ~17.5 electrons pixel−1 s−1 and a total exposure of ~60 electrons Å−2.
Image processing
A total of 13,758 raw movies were obtained for the TcdA sample at neutral pH from two rounds of data collection, and image processing was performed using cryoSPARC v3.2 software (Punjani et al, 2017; Structura Biotechnology, Toronto, ON, Canada). Patch motion correction was performed with the Alignparts algorithm (Rubinstein & Brubaker, 2015) and patch CTF estimation was performed using the L‐BFGS algorithm (Zivanov et al, 2020) as used in the cryoSPARC implementation. The exposures were curated based on ice thickness, CTF fit resolution, and total motion. A template picker was used to select 15.9 million particle images based on internally generated volumes from selected 2D classes based on an initial blob picking round. The particles were 2D classified to remove contamination and bad particles resulting in an initial set of 2.6 million good particles. An initial round of ab initio structure determination followed by several rounds of heterogeneous refinement resulted in a final selection of 0.9 million good particles. These particles were used for a final homogeneous refinement which resulted in a 2.8 Å map of TcdA at neutral pH (Fig EV1A). Furthermore, a total of ~8,000 raw movies were obtained for the TcdA sample prepared at acidic pH but the quality of the particles obtained was not sufficient for further processing.
Model building
An initial model for TcdA was built using the Coot version 0.9.5 EL software (Emsley et al, 2010) by using the previously determined crystal structure of TcdA (TcdA1832) (PDB: 4R04) as a reference. The missing CROPs domain in TcdA1832 was built using selected repetitive units from the previously determined structure of the C‐terminal part of the TcdA CROPs domain (PDB: 2QJ6) for guidance. After the first round of model building, the model was subjected to automated molecular dynamics flexible fitting using Namdinator (Kidmose et al, 2019) to improve the geometric parameters and the real space fit to the electrostatic potential map. The Namdinator model was used for subsequent iterative rebuilding using Coot followed by refinement using the Real_Space_Refine tool in the Phenix software package version 1.19.2 (Afonine et al, 2018). The final model was evaluated using MolProbity (Williams et al, 2018). A Ramachandran plot according shows that 99.94% of the residues are within the allowed region. The statistics of the map reconstruction and the model refinement are presented in Table EV1.
Author contributions
RJ conceived the idea for using cryo‐EM on TcdA. AA purified the protein and prepared samples for cryo‐EM. AA, RJ, and TB prepared the grids for cryo‐EM imaging. TB conceived the cryo‐EM optimization and data collection strategies, executed multiple cryo‐EM data collection runs, and performed initial image processing and 3D reconstructions. CEL continued the image processing generating the final 2.8 Å cryo‐EM map under supervision of TB. RJ determined the structure of TcdA and performed all real‐space refinements in Phenix. TB and CEL did multiple flexible fitting runs using Namdinator. RJ made all the figures and tables and analyzed all data. CEL and TB contributed to Fig EV1. AA wrote the manuscript and contributed to data analysis. RJ revised the manuscript and contributed with intellectual content to the manuscript. CEL and TB wrote the details regarding cryo‐EM data collection and image processing. All authors have read, revised, and approved the final version of the manuscript to be published.
Conflict of interest
RJ and AA are co‐founders of Proxi Biotech ApS, which had no role in this study.
Supporting information
Expanded View Figures PDF
Table EV1
Acknowledgements
We thank Jesper L. Karlsen at Dept. of Mol. Biol. & Gen. at Aarhus University for excellent support on matters related to scientific computing. Furthermore, we thank Jill Cornell at Department of Biological Sciences at Purdue University for careful reading of the manuscript. We gratefully acknowledge financial support from the Novo Nordisk Fonden (Grant no.: NNF17OC0029548 to RJ) and we acknowledge the EMBION Cryo‐EM Facility at iNANO, Aarhus University, for providing access to high‐end cryo‐EM microscopes.
EMBO reports (2022) 23: e53597.
Contributor Information
Thomas Boesen, Email: thb@inano.au.dk.
René Jørgensen, Email: renejoe@ruc.dk.
Data availability
The cryo‐EM map of TcdA has been deposited to the EMDB (https://www.ebi.ac.uk/emdb/) under accession number EMD‐13574, and the TcdA structure to the PDB (https://www.rcsb.org) under accession number 7POG.
References
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD (2018) Real‐space refinement in PHENIX for cryo‐EM and crystallography. Acta Crystallogr Sect D Struct Biol 74: 531–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktories K, Schwan C, Jank T (2017) Clostridium difficile toxin biology. Annu Rev Microbiol 71: 281–307 [DOI] [PubMed] [Google Scholar]
- Aminzadeh A, Jørgensen R (2021) Systematic evaluation of parameters important for production of native toxin A and toxin B from Clostridioides difficile . Toxins (Basel) 13: 240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsells E, Shi T, Leese C, Lyell I, Burrows J, Wiuff C, Campbell H, Kyaw MH, Nair H (2019) Global burden of Clostridium difficile infections: a systematic review and meta‐analysis. J Glob Health 9: 10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K (2001) Low ph‐induced formation of ion channels by Clostridium difficile toxin B in target cells. J Biol Chem 276: 10670–10676 [DOI] [PubMed] [Google Scholar]
- CDC (2019) Antibiotic resistance threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services, CDC. www.cdc.gov/DrugResistance/Biggest‐Threats.html [Google Scholar]
- Chandrasekaran R, Lacy DB (2017) The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev 41: 723–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Lam K‐H, Liu Z, Mindlin FA, Chen B, Gutierrez CB, Huang L, Zhang Y, Hamza T, Feng H et al (2019) Structure of the full‐length Clostridium difficile toxin B. Nat Struct Mol Biol 26: 712–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Tao L, Wang T, Zhang J, He A, Lam K, Liu Z, He X, Perry K, Dong M et al (2018) Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360: 664–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zeng JI, Liu Z, Thaker H, Wang S, Tian S, Zhang J, Tao L, Gutierrez CB, Xing LI et al (2021) Structural basis for CSPG4 as a receptor for TcdB and a therapeutic target in Clostridioides difficile infection. Nat Commun 12: 3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S, Bennett MJ, Fujii G, Curmi PMG, Kantardjieff KA, Collier RJ, Eisenberg D (1992) The crystal structure of diphtheria toxin. Nature 357: 216–222 [DOI] [PubMed] [Google Scholar]
- Chumbler NM, Rutherford SA, Zhang Z, Farrow MA, Lisher JP, Farquhar E, Giedroc DP, Spiller BW, Melnyk RA, Lacy DB (2016) Crystal structure of Clostridium difficile toxin A. Nat Microbiol 1: 1–15 [DOI] [PubMed] [Google Scholar]
- Drudy D, Fanning S, Kyne L (2007) Toxin A‐negative, toxin B‐positive Clostridium difficile . Int J Infect Dis 11: 5–10 [DOI] [PubMed] [Google Scholar]
- Egerer M, Giesemann T, Jank T, Fullner Satchell KJ, Aktories K (2007) Auto‐catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J Biol Chem 282: 25314–25321 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florin I, Thelestam M (1986) Lysosomal involvement in cellular intoxication with Clostridium difficile toxin B. Microb Pathog 1: 373–385 [DOI] [PubMed] [Google Scholar]
- Frisch C, Gerhard R, Aktories K, Hofmann F, Just I (2003) The complete receptor‐binding domain of Clostridium difficile toxin A is required for endocytosis. Biochem Biophys Res Commun 300: 706–711 [DOI] [PubMed] [Google Scholar]
- Genisyuerek S, Papatheodorou P, Guttenberg G, Schubert R, Benz R, Aktories K (2011) Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol Microbiol 79: 1643–1654 [DOI] [PubMed] [Google Scholar]
- Giannasca PJ, Warny M (2004) Active and passive immunization against Clostridium difficile diarrhea and colitis. Vaccine 22: 848–856 [DOI] [PubMed] [Google Scholar]
- Giesemann T, Jank T, Gerhard R, Maier E, Just I, Benz R, Aktories K (2006) Cholesterol‐dependent pore formation of Clostridium difficile toxin A. J Biol Chem 281: 10808–10815 [DOI] [PubMed] [Google Scholar]
- Greco A, Ho JGS, Lin SJ, Palcic MM, Rupnik M, Ng KKS (2006) Carbohydrate recognition by Clostridium difficile toxin A. Nat Struct Mol Biol 13: 460–461 [DOI] [PubMed] [Google Scholar]
- Gupta P, Zhang Z, Sugiman‐Marangos SN, Tam J, Raman S, Julien J‐P, Kroh HK, Lacy DB, Murgolo N, Bekkari K et al (2017) Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor‐binding determinants. J Biol Chem 292: 17290–17301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JGS, Greco A, Rupnik M, Ng KKS (2005) Crystal structure of receptor‐binding C‐terminal repeats from Clostridium difficile toxin A. Proc Natl Acad Sci USA 102: 18373–18378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S (2009) Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcomes. J Infect 58: 403–410 [DOI] [PubMed] [Google Scholar]
- Just I, Selzer J, Wilm M, von Eichel‐Streiber C, Mann M, Aktories K (1995a) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375: 500–503 [DOI] [PubMed] [Google Scholar]
- Just I, Wilm M, Selzer J, Rex G, Von Eichel‐Streiber C, Mann M, Aktories K (1995b) The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270: 13932–13936 [DOI] [PubMed] [Google Scholar]
- Kidmose RT, Juhl J, Nissen P, Boesen T, Karlsen JL, Pedersen BP (2019) Namdinator ‐ Automatic molecular dynamics flexible fitting of structural models into cryo‐EM and crystallography experimental maps. IUCrJ 6: 526–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AM, Mackin KE, Lyras D (2015) Emergence of toxin A‐negative, toxin B‐positive Clostridium difficile strains: Epidemiological and clinical considerations. Future Microbiol 10: 1–4 [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Kyne L, Warny M, Qamar A, Kelly CP (2001) Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357: 189–193 [DOI] [PubMed] [Google Scholar]
- LaFrance ME, Farrow MA, Chandrasekaran R, Sheng J, Rubin DH, Lacy DB (2015) Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB‐induced cytotoxicity. Proc Natl Acad Sci USA 112: 7073–7078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RA, Razaz M, Hayward S (2003) The DynDom database of protein domain motions. Bioinformatics 19: 1290–1291 [DOI] [PubMed] [Google Scholar]
- Lessa F, Mu Y, Bamberg W, Beldavs Z, Dumyati G, Dunn J, Farley M, Holzbauer S, Meek J, Phipps E et al (2015) Burden of Clostridium difficile infection in the United States. N Engl J Med 372: 825–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyerly DM, Krivan HC, Wilkins TD (1988) Clostridium difficile: its disease and toxins. Clin Microbiol Rev 1: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrell KE, Mansfield MJ, Doxey AC, Melnyk RA (2020) The C. difficile toxin B membrane translocation machinery is an evolutionarily conserved protein delivery apparatus. Nat Commun 11: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrell KE, Zhang Z, Sugiman‐Marangos SN, Melnyk RA (2017) Clostridium difficile toxins A and B: Receptors, pores, and translocation into cells. Crit Rev Biochem Mol Biol 52: 461–473 [DOI] [PubMed] [Google Scholar]
- Pruitt RN, Chambers MG, Ng KK‐S, Ohi MD, Lacy DB (2010) Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci 107: 13467–13472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt RN, Lacy DB (2012) Toward a structural understanding of Clostridium difficile toxins A and B. Front Cell Infect Microbiol 2: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) CryoSPARC: Algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Qa’dan M, Spyres LM, Ballard JD (2000) pH‐induced conformational changes in Clostridium difficile toxin B. Infect Immun 68: 2470–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reineke J, Tenzer S, Rupnik M, Koschinski A, Hasselmayer O, Schrattenholz A, Schild H, von Eichel‐Streiber C (2007) Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446: 415–419 [DOI] [PubMed] [Google Scholar]
- Rubinstein JL, Brubaker MA (2015) Alignment of cryo‐EM movies of individual particles by optimization of image translations. J Struct Biol 192: 188–195 [DOI] [PubMed] [Google Scholar]
- Sambol SP, Merrigan MM, Lyerly D, Gerding DN, Johnson S (2000) Toxin gene analysis of a variant strain of Clostridium difficile that causes human clinical disease. Infect Immun 68: 5480–5487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K (1998) Glucosylation and ADP ribosylation of Rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry 37: 5296–5304 [DOI] [PubMed] [Google Scholar]
- Songer JG (2004) The emergence of Clostridium difficile as a pathogen of food animals. Anim Heal Res Rev 5: 321–326 [DOI] [PubMed] [Google Scholar]
- Tao L, Tian S, Zhang J, Liu Z, Robinson‐McCarthy L, Miyashita S‐I, Breault DT, Gerhard R, Oottamasathien S, Whelan SPJ et al (2019) Sulfated glycosaminoglycans and low‐density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat Microbiol 4: 1760–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, Zhang X, Stallcup WB, Miao J, He X et al (2016) Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538: 350–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, London E (2009) The membrane topography of the diphtheria toxin t domain linked to the a chain reveals a transient transmembrane hairpin and potential translocation mechanisms. Biochemistry 48: 10446–10456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warny M, Vaerman JP, Avesani V, Delmee M (1994) Human antibody response to Clostridium difficile toxin A in relation to clinical course of infection. Infect Immun 62: 384–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB et al (2018) MolProbity: More and better reference data for improved all‐atom structure validation. Protein Sci 27: 293–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Zhang H, Cai C, Zhu S, Zhou Y, Yang X, He R, Li C, Guo S, Li S et al (2015) Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 25: 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shi L, Li S, Yang Z, Standley C, Yang Z, ZhuGe R, Savidge T, Wang X, Feng H (2013) A segment of 97 amino acids within the translocation domain of Clostridium difficile toxin B is essential for toxicity. PLoS One 8: e58634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Park M, Tam J, Auger A, Beilhartz GL, Lacy DB, Melnyk RA (2014) Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc Natl Acad Sci USA 111: 3721–3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Scheres SHW (2020) Estimation of high‐order aberrations and anisotropic magnification from cryo‐EM data sets in RELION‐3.1. IUCrJ 7: 253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]