

Abstract
Here, we investigate the impact of hypoxia on the hepatic response of glucocorticoid receptor (GR) to dexamethasone (DEX) in mice via RNA‐sequencing. Hypoxia causes three types of reprogramming of GR: (i) much weaker induction of classical GR‐responsive genes by DEX in hypoxia, (ii) a number of genes is induced by DEX specifically in hypoxia, and (iii) hypoxia induces a group of genes via activation of the hypothalamic–pituitary–adrenal (HPA) axis. Transcriptional profiles are reflected by changed GR DNA‐binding as measured by ChIP sequencing. The HPA axis is induced by hypothalamic HIF1α and HIF2α activation and leads to GR‐dependent lipolysis and ketogenesis. Acute inflammation, induced by lipopolysaccharide, is prevented by DEX in normoxia but not during hypoxia, and this is attributed to HPA axis activation by hypoxia. We unfold new physiological pathways that have consequences for patients suffering from GC resistance.
Keywords: crosstalk, hypoxia, inflammation, mechanism, metabolism
Subject Categories: Chromatin, Transcription & Genomics; Metabolism
This study investigates the impact of hypoxia on hepatic GR function and unfolds new physiological pathways that might have implications for patients suffering from GC resistance.

Introduction
Glucocorticoids (GCs) are the major stress hormones, produced by the hypothalamic–pituitary–adrenal (HPA) axis. The production of GCs is initiated by stress, which activates the expression of corticotropin‐releasing hormone (CRH) in the hypothalamus, leading to the secretion of adrenocorticotropic hormone (ACTH) by the pituitary in the blood, which stimulates the adrenals (Spiga et al, 2014). GCs are mainly generated by the adrenal glands and regulate carbohydrate, lipid, and protein metabolism (Vegiopoulos & Herzig, 2007; Rose et al, 2010). They bind to the intracellular GC receptor (GR), a versatile transcription factor involved in a range of physiological responses. GR can directly regulate the expression of hundreds of genes, many of which have strong anti‐inflammatory effects, via DNA binding, e.g., in hepatocytes and white blood cells. GR additionally interacts with and prevents the activity of other transcription factors, such as nuclear factor κB (NF‐κB) (Timmermans et al, 2019).
In animal models, inhibition of GC production by surgical adrenalectomy, or inhibition of GR using RU486 (Dejager et al, 2010) or via genetic ablation (Kleiman et al, 2012) leads to extreme sensitivity for inflammatory stimuli, e.g., in lipopolysaccharide (LPS)‐induced endotoxemia. Synthetic GCs, such as dexamethasone (DEX), are widely used in inflammatory diseases like rheumatoid arthritis (RA) (Vandewalle et al, 2018) and recently shown to reduce mortality in patients seriously ill with COVID‐19 (Sterne et al, 2020). We and others have found that the transcriptional induction of genes with anti‐inflammatory effects (e.g., Tsc22d3 encoding Glucocorticoid‐Induced Leucine Zipper, GILZ, or Dusp1 encoding Map Kinase Phosphatase 1, MKP1) by GR in organs, such as liver, can become compromised by a mechanism that still needs to be unfolded. In acute, lethal inflammation, like septic shock, a functional decline of GR has been observed and has been called GC resistance (GCR) (Van Bogaert et al, 2011; Dendoncker et al, 2019; Vandewalle et al, 2021). Since the reduction of GR function is possibly the result of interference between transcription factors, certain authors have focused their attention on NF‐κB as a factor responsible for GCR (Dendoncker et al, 2019). However, under drastic inflammatory conditions, also hypoxia‐induced factors (HIFs) are activated in tissues (Cummins et al, 2016). Next to its anti‐inflammatory function, GR has important metabolic functions. In fat tissue, GR can activate lipolysis, leading to the release of free fatty acids (FFAs) and glycerol (de Guia & Herzig, 2015). GR also stimulates gluconeogenesis in hepatocytes (Rose & Herzig, 2013). Moreover, GR can play a role in the maturation of the skin barrier (Sevilla & Pérez, 2018) and the lung (Daniel Bird et al, 2015) during development.
Hypoxia has an important physiological role during normal life, and the liver is essential for the physiological regulation of gene expression during hypoxia (Jungermann & Kietzmann, 2000). The major pathway regulating the response to low oxygen (O2) concentrations involves the activation of HIFs, heterodimeric factors composed of HIFα (mainly HIF1α and HIF2α), and HIFβ. While the β‐subunit is constitutively expressed, HIFα subunits are hydroxylated by the O2 sensors prolyl‐4‐hydroxylases (PHDs) in normoxia (Wielockx & Meneses, 2016; Nakayama & Kataoka, 2019). This leads to the binding of the von Hippel‐Lindau protein (pVHL) and degradation by the 26S proteasome (Cockman et al, 2000; Schofield & Ratcliffe, 2004). In low O2 concentrations, PHDs lose their enzymatic activity, and HIFα is stabilized and translocates to the nucleus. Together with HIFβ and other co‐factors, they will bind to hypoxia responsive elements (HREs) in the promoter/enhancer of a selection of genes and regulate their expression, mostly by inducing transcription (Wielockx & Meneses, 2016). This is important for the activation of metabolic pathways involved in cellular glucose (Semenza, 2011), lipid metabolism (Mylonis et al, 2019), and erythropoiesis (Franke et al, 2013). Sensory systems have developed to detect environmental O2 concentrations and to adapt to hypoxia (Chang et al, 2015; Bleymehl et al, 2016). When rats are exposed to perinatal hypoxia, Crh mRNA levels are significantly higher in the paraventricular nucleus (PVN) of hypothalamic cells (Raff et al, 2007) and an age‐dependent ACTH and GC response is induced (Bruder et al, 2008). The administration of DMOG (dimethyloxalylglycine), a PHD inhibitor leading to increased HIF1α protein levels, elevates plasma GC levels in rats following an acute stressor such as air puffs (Harrell et al, 2015). The cortisol awakening response (CAR) in humans, a marker for the HPA axis function, is significantly higher when ascending to an altitude > 3,000 m compared to natives residing permanently at high altitude (Park et al, 2014). Altogether, hypoxia is able to activate the HPA axis and induce the production of GCs.
Several in vitro studies present an interaction between GR and HIF. Kodama et al (2003) have identified that a ligand‐dependent activation of GR increases hypoxia‐dependent gene expression and HRE activity in HeLa cells. Next, hypoxia causes a transcriptional upregulation of Nr3c1 in human renal proximal tubular epithelial cells (Leonard et al, 2005) and mouse pituitary AtT‐20 cells (Zhang et al, 2015). In contrast to the previous results, the expression of HIF1α target genes is decreased in hypoxic HEPG2 cells when stimulated with DEX (Wagner et al, 2008). These contradictory findings illustrate highly dynamic interactions between O2 concentrations and GR function mediated through HIF1α. Recently, it has been shown that hypoxia is involved in the rewiring of the GR cistrome, thereby indicating a reprogramming of the GR to changes in O2 concentrations (Yang et al, 2020). Furthermore, the GR function is essential in O2‐deprived conditions for the expansion of immature erythroid cells during stress erythropoiesis by increasing the erythrocyte count, hematocrit, and hemoglobin content of the peripheral blood (Bauer et al, 1999). Also in vivo studies in zebrafish larvae have found a crosstalk between HIF and GR (Vettori et al, 2017; Marchi et al, 2020). Although these papers are very valuable, we are convinced that we need to understand the impact of hypoxia on GR in a pathophysiological context in vivo.
Severe inflammatory conditions, such as systemic inflammatory response syndrome (SIRS) and sepsis, are associated with increased blood lactate levels (Garcia‐alvarez et al, 2014) and lead to GCR (Van Bogaert et al, 2011; Dendoncker et al, 2019; Vandewalle et al, 2021). Since HIFs are also activated during inflammation (Cummins et al, 2016), we investigated whether hypoxia has an impact on the transcriptional activity of GR, and if it accounts for (part of) the GCR. To study this, we compared the response of mice under normoxic conditions (21% O2) with mice under hypoxia (7% O2) and studied the genome‐wide transcriptional response to DEX and GR DNA‐binding profile, in liver. Also, the effect of hypoxia on the HPA axis activation and the metabolite profile in the blood was studied.
Results
Hypoxia modifies the GR response to DEX in the liver
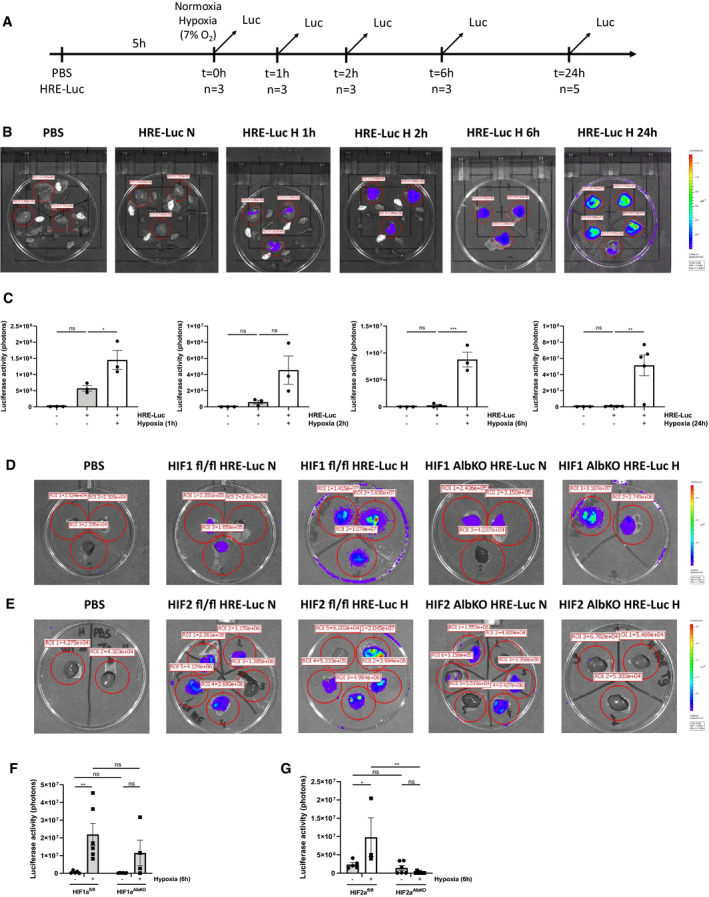
To study the induction of HIF activity in mouse liver during hypoxia, mice were hydrodynamically injected in the tail vein with PBS or with a HRE‐driven luciferase reporter plasmid (HRE‐luc) and were randomly assigned to normoxia (21% O2) or hypoxia (7% O2) (Fig 1A). Luciferase activity was measured. In normoxia, we detected a low signal in livers of mice injected with the reporter plasmid. Hypoxia strongly increased the luciferase activity, especially at the 6 and 24 h time point (Fig 1B and C). A transfection efficiency of about 50% of the HRE‐luc reporter plasmid was observed (Appendix Fig S1A and B). To investigate the role of HIF1α and HIF2α in the regulation of HRE‐luc during hypoxia (6 h), the reporter activity was measured in HIF1aAlbKO and HIF2aAlbKO mice and was found to be reduced in HIF1aAlbKO mice and absent in HIF2aAlbKO mice, when subjected to hypoxia (Fig 1D–G).
Figure 1. HIFs are activated in mouse liver during hypoxia.
-
A–CThe effect of hypoxia was estimated in C57BL/6J mice (n = 3) by a HRE‐luciferase (HRE‐luc) reporter plasmid at indicated time points. All mice were injected according to body weight. (A) Experimental setup. (B) Imaging of luciferase activity (purple signal) in the liver of control mice and mice under normoxic and hypoxic conditions at indicated time points. Red circles indicate the region of interest selected for the measurement of the luciferase activity. (C) Bioluminescent photon counts normalized to the PBS control group in liver of mice in normoxia and hypoxia at indicated time points. P‐values were calculated using one‐way ANOVA followed by post‐hoc Tukey’s test to correct for multiple testing during the pairwise multiple comparisons.
-
D, EImaging of luciferase activity in the liver of HIF1aAlbKO (D) and HIF2aAlbKO (E) mice after a hydrodynamic tail vein injection with a HRE‐luc reporter plasmid or control (PBS) according to body weight followed by 6‐h normoxia or hypoxia.
-
F, GBioluminescent photon counts normalized to the PBS control group in liver of (F) HIF1aAlbKO and (G) HIF2aALbKO mice and their wild‐type littermates 6 h after normoxia of hypoxia.
Data information: All bars represent mean ± SEM. Each individual data point represents individual mice. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ***P < 0.001; **P ≤ 0.01; *P ≤ 0.05. N = normoxia, H = hypoxia. Signal around the perimeter of the Petri dishes represents a specific luciferase signal.
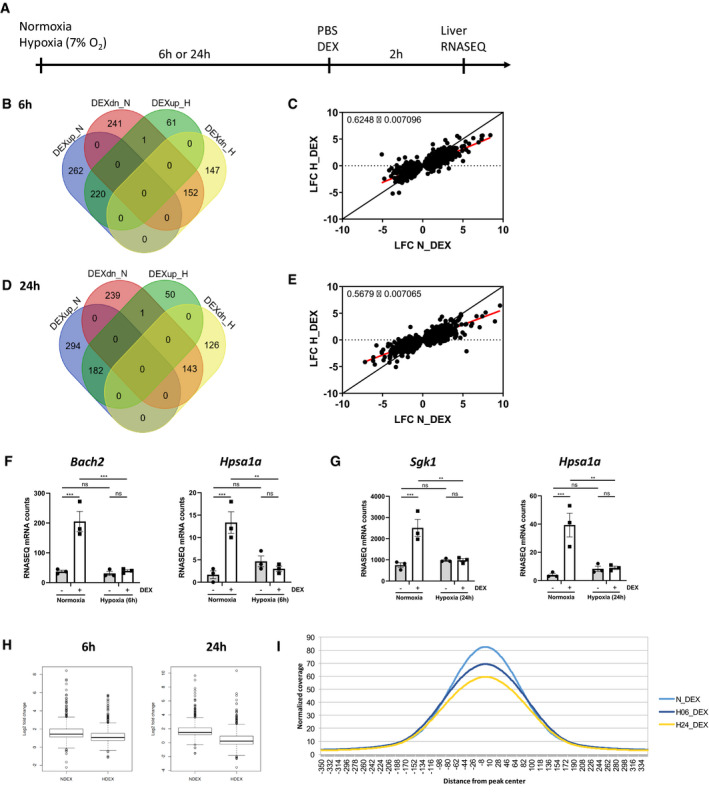
We investigated the effect of hypoxia on GR activity by injecting mice i.p. with PBS or DEX (10 mg/kg) after 6 or 24 h of normoxia or hypoxia. Since the liver is essential in the physiological regulation of gene expression during hypoxia (Jungermann & Kietzmann, 2000) and since the GR has important metabolic and anti‐inflammatory functions in this organ (Rose & Herzig, 2013; de Guia & Herzig, 2015), we isolated livers and performed genome‐wide transcriptomics analysis by RNA‐seq 2 h after PBS or DEX injection (Fig 2A). Of the 462 genes that were significantly upregulated by DEX in normoxia (log fold change [LFC] > 1 and P ≤ 0.05), only 220 were upregulated 6 h after hypoxia. Also, in normoxia, 394 genes were significantly downregulated by DEX (LFC < −1 and P ≤ 0.05), while only 152 genes were downregulated by DEX after 6‐h hypoxia (Fig 2B). Similarly, only 182 genes were upregulated by DEX 24 h after hypoxia, while 476 genes were upregulated by DEX in normoxia. 383 genes were significantly downregulated by DEX in normoxia, while only 143 genes were downregulated by DEX after 24 h of hypoxia (Fig 2D). When plotting the LFC of all DEX‐responsive genes (LFC > 1 and P ≤ 0.05 and LFC < −1 and P ≤ 0.05, compared to PBS) in normoxia and hypoxia, a clear reduction of the GR response was observed under hypoxic conditions, both after 6 h (Fig 2C, slope ± standard error of linear regression curve (LRC) = 0.6248 ± 0.007096) and 24 h (Fig 2E, slope of LRC = 0.5679 ± 0.007065). Representative examples of GR‐responsive genes are shown after DEX stimulation in normoxia and hypoxia based on the RNA‐seq data (Fig 2F and G), so under hypoxic conditions less genes are differentially expressed by DEX, and the fold induction of genes induced/repressed by DEX in hypoxia is less outspoken. Using Enrichr, we compared the pathways controlled by the genes induced by DEX in normoxia alone [6 h (n = 262) and 24 h (n = 294)] with those induced by DEX in both normoxia and hypoxia [6 h (n = 220) and 24 h (n = 182)]. Based on the “Wiki Pathways 2019 Mouse” function in Enrichr, we suggest a shift from inflammation control to metabolic control (Table 1).
Figure 2. Hypoxia modifies the GR response to DEX.
-
AFemale C57BL/6J mice were put in normoxia or hypoxia for 6 or 24 h, injected i.p. with PBS or DEX (10 mg/kg) and livers were isolated after 2 h for genome‐wide transcriptomics via RNA‐seq. N = 3 per group for a single RNA‐seq.
-
BVenn diagram depicting the number of genes upregulated (up, LFC > 1 and P ≤ 0.05) and downregulation (dn, LFC < −1 and P ≤ 0.05) by DEX in normoxia and hypoxia after 6 h.
-
CScatter plot showing log fold change (LFC) of all DEX‐upregulated genes (LFC > 1 and P ≤ 0.05) and DEX‐downregulated genes (LFC < −1 and P ≤ 0.05) in normoxia versus hypoxia after 6 h. The black line represents the diagonal, and the red line represents the real slope ± standard error of the data as analyzed by linear regression.
-
DVenn diagram depicting the number of genes upregulated (up, LFC > 1 and P ≤ 0.05) and downregulation (dn, LFC < −1 and P ≤ 0.05) by DEX in normoxia and hypoxia after 24 h.
-
EScatter plot showing log fold change (LFC) of all DEX‐upregulated genes (LFC > 1 and P ≤ 0.05) and DEX‐downregulated genes (LFC < −1 and P ≤ 0.05) in normoxia versus hypoxia after 24 h. The black line represents the diagonal, and the red line represents the real slope ± standard error of the data as analyzed by linear regression.
-
F, GExamples of GR‐responsive genes based on the RNA‐seq data 6 h (F) and 24 h (G) after normoxia or hypoxia and DEX stimulation.
-
H, IChIP‐seq on liver derived from mice, which were subjected to hypoxia (6 and 24 h), followed by DEX (10 mg/kg) injection and 2 h later sacrificed for liver isolation. (H) Box plots showing the LFC of genes responsive to DEX in both normoxia and hypoxia (6 and 24 h). The central band represents the median. The box ranges from the first quartile (Q1) to the third quartile (Q3), which represents the interquartile range (IQR = Q3 – Q1) and covers the central 50% of the data. The whiskers illustrate the minimum (Q1 – 1.5*IQR) and maximum (Q3 + 1.5*IQR) of the data. Outliers are shown as dots. N = 3 biological replicates per condition. (I) Histogram with coverage per position in a region 350 bp up‐ and downstream of the peaks found in normoxia and hypoxia after DEX stimulation (N_DEX, H06_DEX, and H24_DEX).
Data information: All bars represent mean ± SEM. N = 3 per group. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ***P < 0.001, **P < 0.01.
Table 1.
Enrichr pathway analysis based on genes induced by DEX in normoxia alone and genes induced by DEX in both normoxia and hypoxia.
| N_DEX only | N_DEX_H_DEX | ||
|---|---|---|---|
| Name | P‐value | Name | P‐value |
| Regulation of Cardiac Hypertrophy by miR‐208 WP1526 | 0.00729 | p53 signaling WP2902 | 0.00048 |
| MAPK signaling pathway WP493 | 0.0107 | Adipogenesis genes WP447 | 0.00089 |
| Signal Transduction of S1P Receptor WP57 | 0.01306 | White fat cell differentiation WP2872 | 0.01170 |
| Irinotecan Pathway WP475 | 0.02059 | Calcium Regulation in the Cardiac Cell WP553 | 0.022662 |
| Macrophage markers WP2271 | 0.02059 | Myometrial Relaxation and Contraction Pathways WP385 | 0.02691 |
| Gene regulatory network modeling somitogenesis WP2852 | 0.02480 | ErbB signaling pathway WP1261 | 0.03079 |
| Exercise‐induced Circadian Regulation WP544 | 0.02504 | Notch Signaling Pathway WP29 | 0.03079 |
| IL‐6 signaling Pathway WP387 | 0.02555 | Insulin Signaling WP65 | 0.03165 |
| Neural Crest Differentiation WP2074 | 0.02784 | MAPK signaling pathway WP493 | 0.03165 |
| Adipogenesis genes WP447 | 0.03372 | Apoptosis WP1254 | 0.03253 |
Wiki Pathways 2019 Mouse analysis of the 6 h + 24 h genes that are induced by DEX in normoxia only (N_DEX) or in normoxia and hypoxia (N_DEX_H_DEX). Remark that GR pathways controlling inflammation are lost when GR is induced in hypoxic conditions and are replaced by metabolic pathways.
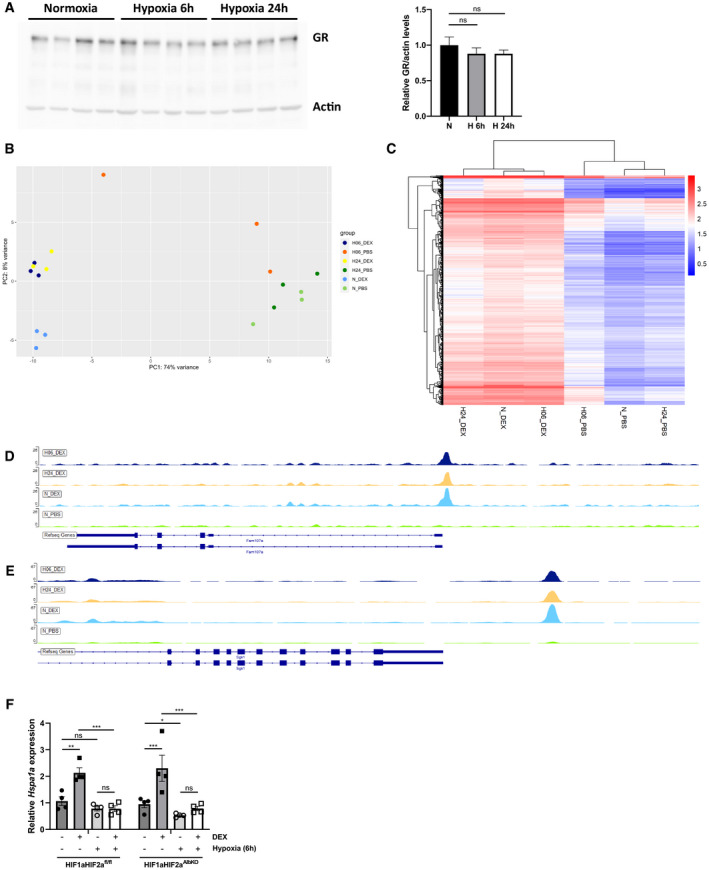
To explore the mechanism by which hypoxia changes the response of the GR to DEX, we investigated key aspects of the GR signaling in hypoxic conditions. Both after 6 and 24 h of hypoxia, the GR protein levels were not declined in the liver (Fig EV1A). Subsequently, we studied the effect of hypoxia on GR chromatin‐binding by means of genome‐wide ChIP‐seq. Liver samples were isolated as in RNA‐seq (Fig 2A): 2 h after PBS or DEX stimulation, 6 or 24 h after normoxia and hypoxia. Biological triplicates were used for all conditions, given a total of 18 analyzed samples (3 N_PBS, 3 N_DEX, 3 H_PBS_6 h, 3 H_DEX_6 h, 3 H_PBS_24 h and 3 H_DEX_24 h) and input controls. In Fig EV1B, the PCA plot clarifies the variance within samples per condition, based on the peaks found in the ChIP‐seq after normalization and scaling of the data. All samples are clustered together per group, except for one sample within the H_PBS_6 h group. However, this inconsistency is not problematic since most of the comparisons are based on the DEX stimulated conditions. When plotting the LFC of genes that responded to DEX regardless of their O2 concentration, we observed a clear DEX effect in normoxia. A reduced DEX effect on GR chromatin‐binding was detected both after 6 and 24 h of hypoxia (Fig 2H). We found that the coverage of the GR DNA‐binding peaks, as found after DEX stimulation in normoxia, was generally lower in hypoxia after DEX stimulation, both at the 6 and 24 h time points (Fig 2I). By means of illustration, GR peaks found at the promoter region of Fam107a and Sgk1 are shown (Fig EV1D and E). Figure EV1C represents a heatmap based on the GR DNA‐binding peaks after normalization and scaling of the ChIP‐seq data found in normoxia and hypoxia after DEX stimulation compared to input samples of this group. Log10 of the total area under the peak is displayed in this heatmap. Overall, we observed that the total area under the peak was higher after DEX stimulation compared to PBS both in normoxia and hypoxia (6 and 24 h). Additionally, the coverage of GR DNA‐binding peaks after DEX stimulation in hypoxia is highly correlated with the peak coverage in the liver of DEX‐stimulated mice in normoxia [Pearson correlation coefficient (PCC): N_DEX versus H_DEX_6 h = 0.9238885 and N_DEX versus H_DEX_24 h = 0.914772]. When we only compare the PBS treated groups in normoxia and hypoxia, the GR DNA binding profile is mostly altered after 6‐h hypoxia (PCC: N_PBS versus H_PBS_6 h = 0.8191357 and N_PBS versus H_PBS_24 h = 0.9431441).
Figure EV1. Hypoxia modifies the GR response to DEX in the liver.
-
AFemale C57BL/6J mice were put in normoxia or hypoxia for 6 and 24 h, liver was isolated for further analysis. GR protein levels were analyzed via Western blot using ACTIN as a loading control. GR protein levels were quantified using FIJI and normalized to ACTIN levels. P‐values were calculated using one‐way ANOVA. N = 4 biological replicates per group.
-
BPCA plot visualizing the variance within samples per conditions based on the peaks found in the ChIP‐seq experiment after normalization and scaling of the data.
-
CHeatmap based on the GR DNA‐binding peaks after normalization and scaling of the ChIP‐seq data found in normoxia and hypoxia after DEX stimulation compared to input samples of this group. Log10 of the total area under the peak is shown.
-
D, EExamples of specific GR DNA‐binding peaks, associated with the GR‐responsive genes induced by DEX Fam107a (D) and Sgk1 (E) in normoxia and hypoxia (6 and 24 h).
-
FHIF1aHIF2afl/fl and HIF1aHIF2aAlbKO mice were put in hypoxia for 6 h and stimulated with DEX. Hspa1a expression was measured in the liver via RT–qPCR. N = 4 per group, one experiment.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ***P < 0.001, **P < 0.01, *P ≤ 0.05.
Source data are available online for this figure.
To gain insight into the role of HIF in the expression of these GR‐responsive genes during hypoxia, we examined the expression of Hspa1a in the liver of HIF1aHIF2aAlbKO mice and wild‐type littermates after 6‐h hypoxia and DEX stimulation via RT–qPCR. The expression pattern of Hspa1a is similar in HIF1aHIF2afl/fl and HIF1aHIF2aAlbKO mice (Fig EV1F). Taken together, these data demonstrate that the reduced GR response to DEX in hypoxia can be linked to a reduced GR DNA‐binding (Fig 2I), but the changes are largely locus‐specific and not observed to the same degree at every GR peak and gene detected via ChIP‐seq. Furthermore, it seems that HIF1α and HIF2α in the liver are not involved in the reduced GR response to DEX upon hypoxia.
Expression of specific DEX‐responsive genes only in hypoxia
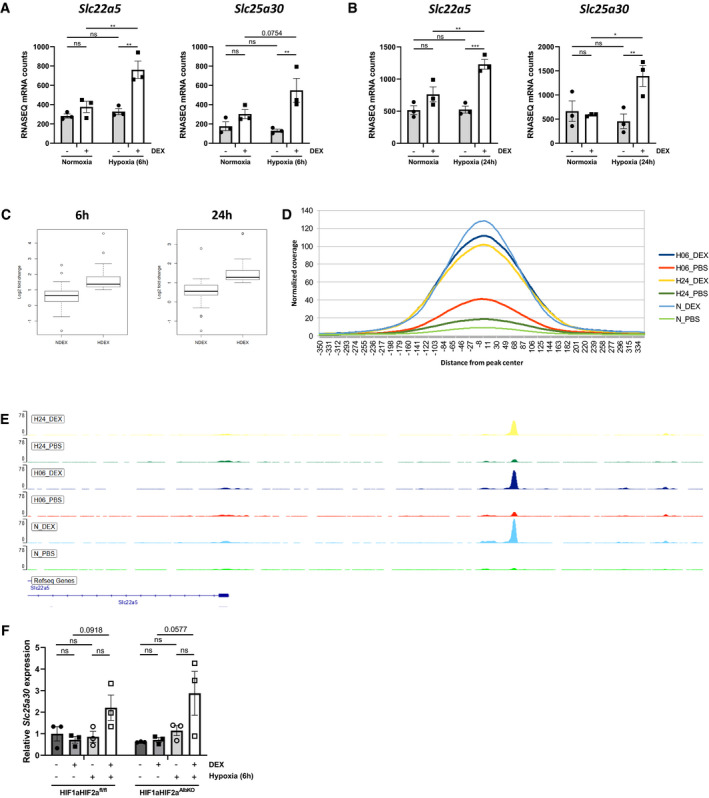
Both in mice that had been 6 and 24 h in hypoxia, we found expression of a set of DEX‐responsive genes that were not induced by DEX in normoxia. It concerns 61 genes at 6 h and 50 genes at 24 h (Fig 2B and D), showing an overlap of only 6 genes. An overview of these genes can be found in Dataset EV1 (6 h) and Dataset EV2 (24 h). Examples are shown in Fig 3A and B. The number of these newly acquired DEX‐responsive genes is considerably less compared to the amount of DEX‐responsive genes that are lost by hypoxia (262 genes at 6 h, 294 genes at 24 h). The general expression pattern of these genes was also evaluated in normoxia and hypoxia alone. Boxplots were used based on the median of the LFCs of these genes after 6‐h and 24‐h hypoxia (Appendix Fig S2A and B). First, when comparing the LFCs directly in normoxia and hypoxia, a median LFC of around 0 is observed. This indicates that hypoxia alone does not have an influence on the expression level of these genes. Second, DEX stimulation in normoxia does not lead to a significant induction of these genes but shows a fairly large non‐significant fold change. The individual LFC values tend toward a lower expression in hypoxia compared to normoxia. Overall, when hypoxia is combined with DEX treatment, DEX counteracts the initial downregulation that is observed in hypoxia alone. In hypoxia, the DEX effect is enhanced indicating that both hypoxia and DEX stimulation are necessary to induce the expression of this subset of genes. Since these genes are expressed by GR in hypoxic conditions, a crosstalk between GR and HIF may form the mechanism of this induction. By studying the pathways induced by the union of these DEX‐induced genes in hypoxia via Enrichr, we found that peroxisome proliferator‐activated receptor alpha (PPARα) is the top‐activated transcription factor. Also, GR and HIFs were found. When analyzing pathways that are induced by these unique DEX‐responsive genes in hypoxia, Enrichr revealed exclusively metabolic pathways, including fatty acid β‐oxidation. Finally, the top GO associated biological response found via Enrichr, was the response to GCs. Together, these data suggest that in hypoxia DEX is sensed, GR, PPARα, HIF1α, and HIF2α are activated, and metabolic reprogramming is induced.
Figure 3. Induction of specific DEX‐responsive genes in hypoxia.
-
A, BExamples of DEX‐responsive genes in hypoxia 6 h (A) and 24 h (B), which were not induced by DEX in normoxia.
-
C–EChIP‐seq on liver derived from mice which were subjected to hypoxia (6 and 24 h), followed by DEX (10 mg/kg) injection and 2 h later sacrificed for liver isolation. (C) Box plots showing the LFC of genes responsive to DEX in hypoxia, but not in normoxia (6 and 24 h). The central band represents the median. The box ranges from Q1 to Q3, which represents the interquartile range (IQR = Q3 – Q1) and covers the central 50% of the data. The whiskers illustrate the minimum (Q1 – 1.5*IQR) and maximum (Q3 + 1.5*IQR) of the data. Outliers are shown as dots. N = 3 biological replicates per condition. (D) Histogram showing the coverage per position in a region 350 bp up‐ and downstream of the GR DNA‐binding peaks found in hypoxia after PBS or DEX stimulation. (E) Examples of specific GR DNA‐binding peaks, associated with the DEX‐induced gene Slc22a5 in hypoxia (6 and 24 h).
-
FHIF1aHIF2afl/fl and HIF1aHIF2aAlbKO mice were put in hypoxia for 6 h and stimulated with DEX. Slc25a30 expression was measured in the liver via RT–qPCR. N = 3 per group, one experiment.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ***P < 0.001, **P < 0.01, *P ≤ 0.05.
We then studied the GR DNA‐binding profile of these specific DEX‐responsive genes in hypoxia. A clear DEX effect in hypoxia was observed when plotting the LFC of these genes induced by DEX in hypoxic conditions compared to the expression of these genes after DEX in normoxia (Fig 3C). Based on the GR DNA‐binding sites associated with DEX‐responsive genes in hypoxia, an increase in the coverage of these GR DNA‐binding peaks was observed after DEX in hypoxia compared to PBS (Fig 3D). However, the increase in peak coverage was still lower after DEX stimulation in hypoxia compared to normoxia, indicating that other mechanisms could be involved next to the alterations in the GR DNA‐binding profile responsible for the differences that we observed in the transcriptomics data. The GR DNA‐binding peaks of Slc22a5 6 and 24 h after hypoxia and DEX stimulation are shown in Fig 3E. Using HOMER de novo motif search, we could not detect a clear link between GR and HIF which could be responsible for the induction of the DEX‐responsive genes in hypoxia alone. We also investigated the expression levels of Slc25a30 in the liver of HIF1aHIF2afl/fl and HIF1aHIF2aAlbKO mice after 6 h hypoxia stimulated or not with DEX. The absence of HIF1α and HIF2α in the liver did not alter the expression pattern of Slc25a30 (Fig 3F). In conclusion, we identified DEX‐responsive genes in hypoxia linked with metabolic pathways and higher GR DNA‐binding after DEX stimulation in hypoxia. Further investigation will be necessary to identify other mechanisms involved in the regulation of the expression of these specific DEX‐responsive genes in hypoxia.
Hypoxia causes activation of GR by HPA axis stimulation
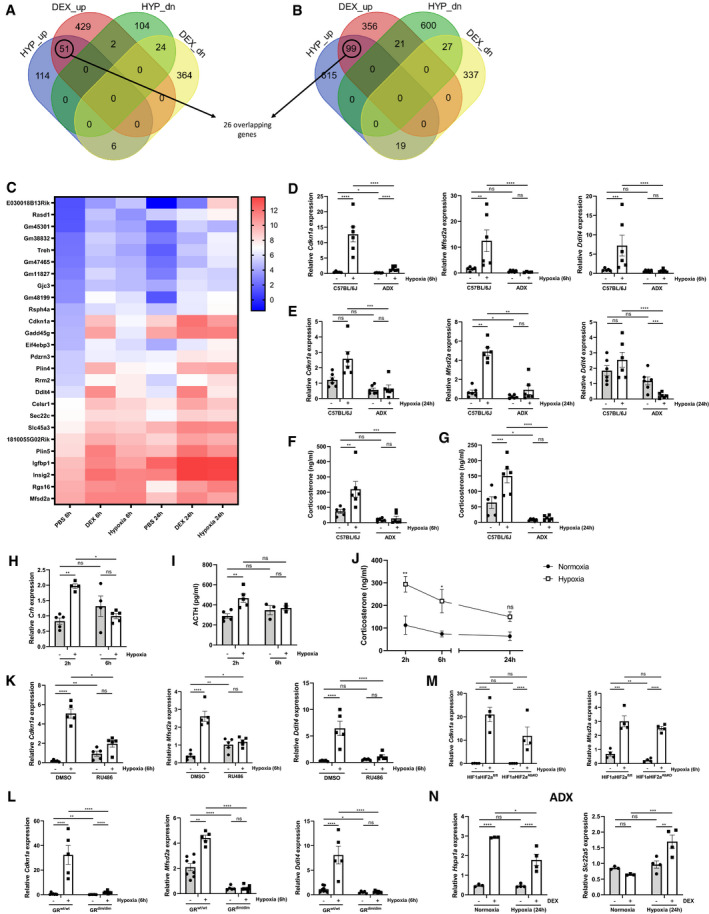
Since we have shown that the response to DEX under hypoxia is less pronounced, we wanted to investigate whether the GR is already activated upon exposure to hypoxia and therefore GR is not able to respond to DEX. Based on the RNA‐seq data, we found that hypoxia induced the expression of typical GR‐responsive genes in the liver (LFC > 1 and P ≤ 0.05): 51 genes after 6‐h hypoxia (Fig 4A) and 99 genes after 24‐h hypoxia (Fig 4B), with 26 genes overlapping. These genes are known GR‐stimulated genes, as they were also induced by DEX in normoxia in the RNA‐seq data, so these genes will be referred to as “stress GRE genes”. A heatmap of these stress GRE genes is displayed in Fig 4C. Figure EV2 shows heatmaps of the GR‐responsive genes after 6 h (Fig EV2A) and 24 h (Fig EV2B) of hypoxia. A functional survey via Enrichr of the union of the 6‐h and 24‐h genes (124 genes) reveals that transcription factors such as GR, PPARα, and HIFs, but also c‐Myc, Nanog, and Smad3, are associated with the expression of these genes. We studied whether the induction of these stress GRE genes in hypoxia can be linked with an increase in GR DNA‐binding. The coverage of GR DNA‐binding peaks was clearly higher after DEX stimulation in normoxia (Fig EV3A and B) but, when comparing the peak coverage in normoxia and hypoxia only, we observed an increase in GR DNA‐binding peaks in hypoxia for both time points, although less obvious after 24 h. The GR DNA‐binding peaks for Cdkn1a (Fig EV3C), Mfsd2a (Fig EV3D), Ddit4 (Fig EV3E), and Igfbp1 (Fig EV3F) are shown as examples.
Figure 4. Hypoxia causes activation of GR by HPA axis stimulation.
-
A, BThe effect of hypoxia on the GR response to DEX stimulation was studied via RNA‐seq, n = 3 per group. Venn diagram depicting the number of genes upregulated (up) or downregulated (dn) by DEX in normoxia and by hypoxia after 6 h (A) or 24 h (B) (LFC > 1 or LFC < −1 and P ≤ 0.05).
-
CHeatmap representing log2 values of shared stress GRE genes (counts) induced by hypoxia 6 and 24 h and induced by DEX in normoxia (LFC > 1 and P ≤ 0.05).
-
D–GFemale C57BL/6J and ADX mice were put in normoxia or hypoxia for 6 or 24 h, n = 6/group, two independent experiments. (D, E) Confirmation of RNA‐seq data via RT–qPCR [6 h (D) or 24 h (E)]. (F, G) Plasma GC concentration [6 h (F) or 24 h (G)].
-
H–JFemale C57BL/6J mice were put in normoxia or hypoxia for the indicated time points (2, 6, and 24 h). (H) Hypothalamic Crh mRNA expression levels were determined via RT–qPCR. (I) ACTH levels were measured in the plasma. N = 3–5 per group, one experiment. (J) Plasma GC concentration, mice in normoxia are depicted as black circles, mice in hypoxia are depicted as white squares. N = 5–6 per group.
-
KFemale C57BL/6J mice were put in normoxia or hypoxia for 6 h and injected i.p. with 5 mg RU486 or vehicle (DMSO). Liver was isolated and stress GRE gene expression was measured via RT–qPCR. N = 5 per group, one experiment.
-
LFemale WT and GRdim/dim mice were put in normoxia or hypoxia for 6 h, and liver was isolated. Stress GRE gene expression was measured via RT–qPCR. N = 5–9 per group, two independent experiments.
-
MHIF1aHIF2afl/fl and HIF1aHIF2aAlbKO mice were put in hypoxia for 6 h. Stress GRE genes were measured in the liver via RT–qPCR. N = 4 per group, one experiment.
-
NExpression levels of DEX‐responsive genes in normoxia and unique DEX‐responsive genes induced by hypoxia were measured in the liver of ADX mice after DEX stimulation during hypoxia (24 h) via RT–qPCR. N = 3–4 per group, one experiment.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P ≤ 0.05.
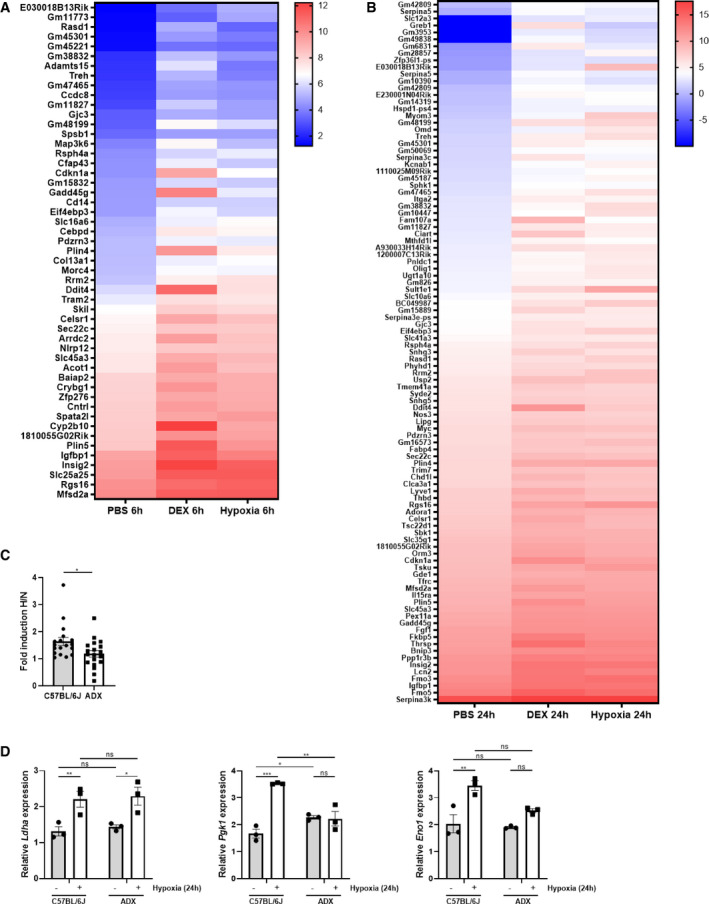
Figure EV2. Hypoxia itself causes the induction of GR‐responsive genes.
-
A, BFemale C57BL/6J mice were put in normoxia or hypoxia for 6 h (A) or 24 h (B), injected with PBS or DEX (10 mg/kg) and 2 h later, liver was isolated for analyses. N = 3 per group for a single RNA‐seq. (A) Heatmap representing stress GRE genes induced by hypoxia (6 h) and DEX in normoxia. Log2 values are shown of the counts of the stress GRE genes based on the RNA‐seq data (LFC > 1 and P ≤ 0.05). (B) Heatmap representing stress GRE genes induced by hypoxia (24 h) and DEX in normoxia. Log2 values are shown of the counts of the stress GRE genes based on the RNA‐seq data (LFC > 1 and P ≤ 0.05).
-
C, DFemales C57BL/6J and ADX mice were put in normoxia (N) and hypoxia (H, 6 and 24 h) and the expression of HIF target genes were evaluated in the liver via RT–qPCR. (C) Fold inductions (H/N) of HIF target genes in C57BL/6J and ADX mice. P‐value was calculated using a two‐way unpaired Student’s t‐test. (D) Examples of the expression levels of HIF target genes. N = 3 per group.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ***P < 0.001, **P < 0.01, *P ≤ 0.05.
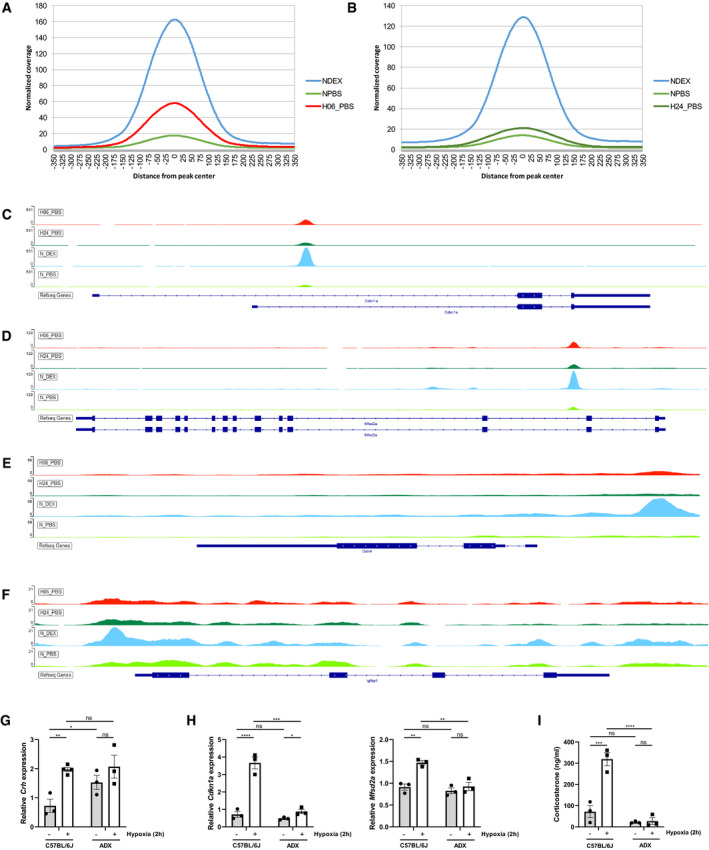
Figure EV3. Hypoxia causes activation of GR by HPA axis stimulation.
-
A, BHistogram showing the coverage per position in a region 350 bp up‐ and downstream of the peaks found in normoxia after PBS or DEX stimulation and in hypoxia 6 h (A) and 24 h (B).
-
C–FExamples of specific GR peaks, associated with the DEX‐induced and hypoxia‐induced genes Cdkn1a (C), Mfsd2a (D), Ddit4 (E), and Igfbp1 (F) in normoxia with and without DEX stimulation and in hypoxia (6 and 24 h).
-
G–IFemale C57BL/6J and ADX mice were put in normoxia and hypoxia (2 h) and hypothalamus and blood was collected. (G) Hypothalamic Crh mRNA levels measured via RT–qPCR. (H) Stress GRE gene expression. (I) Plasma GC levels. N = 3 per group.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P ≤ 0.05.
To investigate whether the induction of the stress GRE genes during hypoxia in the liver can be attributed to the activation of the HPA axis, we validated the RNA‐seq data via RT–qPCR in C57BL/6J and ADX mice, of which both adrenal glands are surgically removed. The expression levels of Cdkn1a, Mfsd2a, and Ddit4 were significantly increased in hypoxic conditions in C57BL/6J mice, both after 6 h (Fig 4D) and 24 h (Fig 4E), but not in ADX mice (Fig 4D and E), and the plasma GC levels were significantly higher after 6 and 24 h of hypoxia in C57BL/6J mice (Fig 4F and G). Since hypoxia increases GC production by the adrenal glands, we considered if hypoxia leads to the activation of the entire HPA axis by studying Crh mRNA levels in the hypothalamus and plasma ACTH levels during hypoxia. Indeed, both Crh (Fig 4H) and ACTH (Fig 4I) showed a transient increase. Also, plasma GC levels were significantly elevated after hypoxia (Fig 4J). To rule out whether the caging of the mice has an influence on the GC production, C57BL/6J mice were housed in normal cages and in a normoxic chamber and blood was collected after 2, 6, and 24 h. No differences in GC levels were observed (Appendix Fig S3). Furthermore, we also investigated the hypothalamic response in ADX mice in hypoxia. Hypothalamic Crh mRNA levels were increased after 2‐h hypoxia in C57BL/6J mice. It is known that Crh mRNA levels are higher in ADX mice (Dallman et al, 1994) because of a lack of GC control. Similar Crh mRNA levels were observed in the hypothalamus of C57BL/6J and ADX mice in hypoxia (Fig EV3G); however, due to high basal Crh mRNA levels in ADX mice, no significant difference was detected in ADX mice. The expression level of stress GRE genes was also measured in the hypothalamus of C57BL/6J and ADX mice after 2‐h hypoxia. A significant increase was observed after 2‐h hypoxia in C57BL/6J mice; however, these levels were significantly reduced in the hypothalamus of ADX mice (Fig EV3H). To confirm that the GC levels were reduced in ADX mice, GC levels were measured in the plasma of these mice. In C57BL/6J mice, GC levels were significantly increased in hypoxia (2 h), while this was not the case in ADX mice (Fig EV3I). Based on these data, it is likely that hypoxia activates the entire HPA axis, starting with Crh transcription in the hypothalamus, leading to ACTH and the induction of stress GRE genes in a GC‐dependent way.
Since ADX mice fail to activate GR, it could be possible that the reduced GC production also has an effect on HIF activity in general, as was shown in GR‐deficient zebrafish larvae (Marchi et al, 2020). Therefore, we have investigated the expression pattern of HIF target genes in the liver of C57BL/6J and ADX mice in normoxia and hypoxia (6 and 24 h). Fold inductions (H/N) were calculated between C57BL/6J and ADX mice for these genes. The mean fold induction in C57BL/6J mice was generally around 1.6, indicating an increase in the expression of HIF target genes in hypoxia. However, a significant reduction of the fold induction (1.2) in ADX mice was observed (Fig EV2C), suggesting that the absence of GC production influences the expression of HIF target genes. The expression pattern of a number of HIF target genes is shown in Fig EV2D. A future study is necessary to further investigate the impact of GR on HIF function.
GR is critical during the hypoxia‐induced stress response
We further investigated the role of GR in the stress response during hypoxia (6 h) by inhibiting GR using RU486, and by using GRdim/dim mice, which express a GR with a reduced dimerization potential and therefore are unable to induce dimer‐dependent GRE genes. RU486 pre‐treated mice (Fig 4K) and GRdim/dim mice (Fig 4L) were no longer able to increase the expression of hypoxia‐induced stress GRE genes in the liver. We further investigated whether the presence of HIF1α and HIF2α in the liver is essential for the expression of these stress GRE genes. Both in HIF1aHIF2aAlbKO mice and wild‐type littermates, Cdkn1a, Mfsd2a (Fig 4M), Ddit4, and Igfbp1 (Appendix Fig S4A) were significantly upregulated 6 h after hypoxia, suggesting that the induction of the stress GRE genes depends exclusively on GCs and GR. As a proof of concept, the expression of HIF target genes was also studied in the liver of HIF1aHIF2afl/fl and HIF1aHIF2aAlbKO mice in hypoxia (6 h) and was significantly reduced upon hypoxic conditions in the absence of liver HIF1α and HIF2α (Appendix Fig S4B).
Finally, we examined the response of ADX mice to GR‐responsive genes induced by DEX in normoxia but not in hypoxia in C57BL/6J mice (Fig 2, e.g. Hspa1a). In hypoxic conditions (24 h), when ADX mice were stimulated with DEX, typical GRE genes like Hspa1a were significantly upregulated both in normoxia and hypoxia. Also, unique DEX‐responsive genes induced by hypoxia (Fig 3) were expressed in ADX mice (Fig 4N). Based on these results, we conclude that the chronic GC production by the adrenals in hypoxia is responsible for the altered DEX response in the liver of hypoxic mice and is independent of liver HIF1α and HIF2α.
Involvement of hypothalamic HIF1α and HIF2α in activation of the HPA axis
To investigate the influence of hypoxia on gene expression in the hypothalamus, we performed an RNA‐seq analysis 2 h after hypoxia and found significant increase of 313 genes at the level of the hypothalamus (LFC > 0.3 and P < 0.05). HOMER motif enrichment analysis based on known motifs (vertebrates) identified 80 genes containing an HIF1β motif in their promoter (Fig 5A), and also, the HIF1α and HIF2α motif (Fig 5B) as well as the GRE motif were found. Based on the expression levels of the stress GRE genes and the appearance of HREs in the HOMER analysis, it is obvious that the hypothalamus reacts on HIF transcription factors as well as on the GR to hypoxia. Enrichr Pathway analysis coupled the 313 upregulated genes to a HIF signaling response, but it also showed that the transcription factor Hey2, which is a HIF1α/HIF2α‐induced gene, is associated with the observed differential expression in the hypothalamus. Hey2 binds the E‐box motif CACGTG (Steidl et al, 2000), the same motif which is bound by other transcription factors such as NPAS, NPAS2, c‐Myc, CLOCK, BMAL, and others, all of which were retrieved via HOMER motif enrichment analysis. The expression of stress GRE genes at the level of the hypothalamus was validated using RT–qPCR (Fig 5C). In addition, Western blot and immunohistochemistry (IHC) confirmed HIF1α and HIF2α protein stability in hypothalamus 2 h after hypoxia (Fig 5D). In normoxia, HIF1α and HIF2α were mainly found in the cytoplasm, while a clear nuclear enrichment was observed during hypoxia (Fig 5E). In conclusion, hypoxia stimulates the HPA axis, leading to GC production and the induction of stress GRE genes in liver and hypothalamus. Additionally, both HIF1α and HIF2α are stabilized and clear enrichment in the nuclei of hypothalamic cells during hypoxia was detected.
Figure 5. Involvement of hypothalamic HIF1α and HIF2α in activation of the HPA axis during hypoxia.
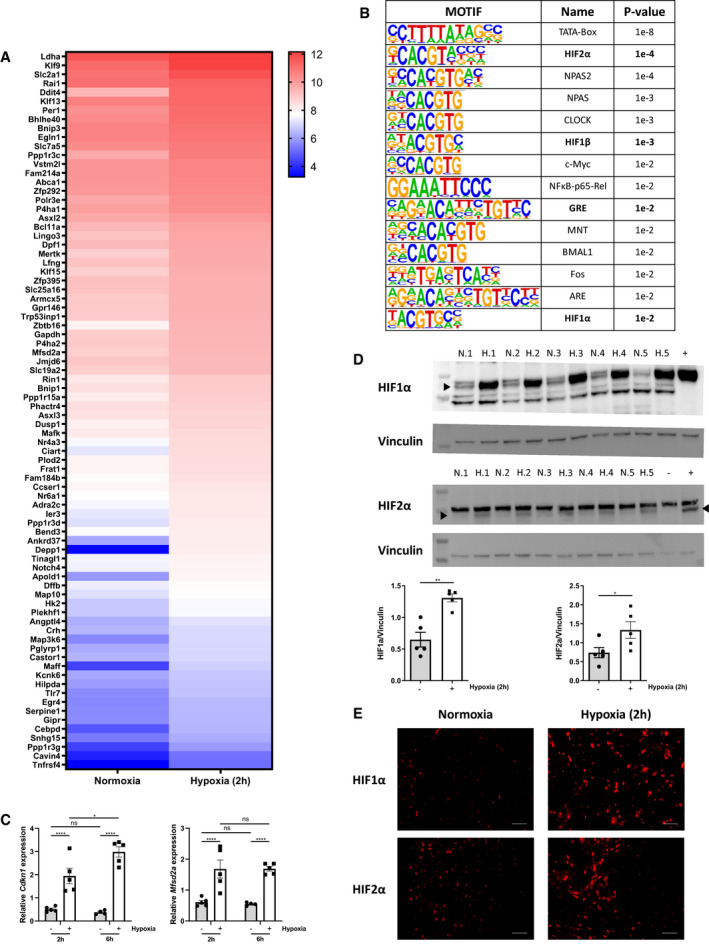
- Female C57BL/6J mice were put in normoxia or hypoxia for 2 h, hypothalamus was isolated for analyses. N = 3–4 per group for a single RNA‐seq. Heatmap represents genes upregulated by hypoxia (2 h) (LFC > 0.3 and P ≤ 0.05).
- HOMER motif analysis of hypoxia‐induced genes (start offset: −1 kb, end offset: 50 bp downstream of TSS). Enriched motifs with their name and P‐value are displayed.
- Female C57BL/6J mice were put in normoxia or hypoxia for the indicated time points. Hypothalamus was isolated and stress GRE genes were measured via RT–qPCR. N = 4–5 per group, one experiment.
- HIF1α and HIF2α protein levels were analyzed via Western blot using VINCULIN as a loading control. HIF1α and HIF2α protein levels were quantified using FIJI and normalized to VINCULIN levels. P‐values were calculated using Mann‐Whitney test. N = 5 biological replicates per group. N = normoxia, H = hypoxia.
- HIF1α and HIF2α expression was detected in hypothalamus samples via IHC, scale bar 50 µm.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ****P < 0.0001, **P < 0.01, *P ≤ 0.05.
Source data are available online for this figure.
Hypoxia causes lipolysis and liver ketogenesis in a GR‐dependent manner
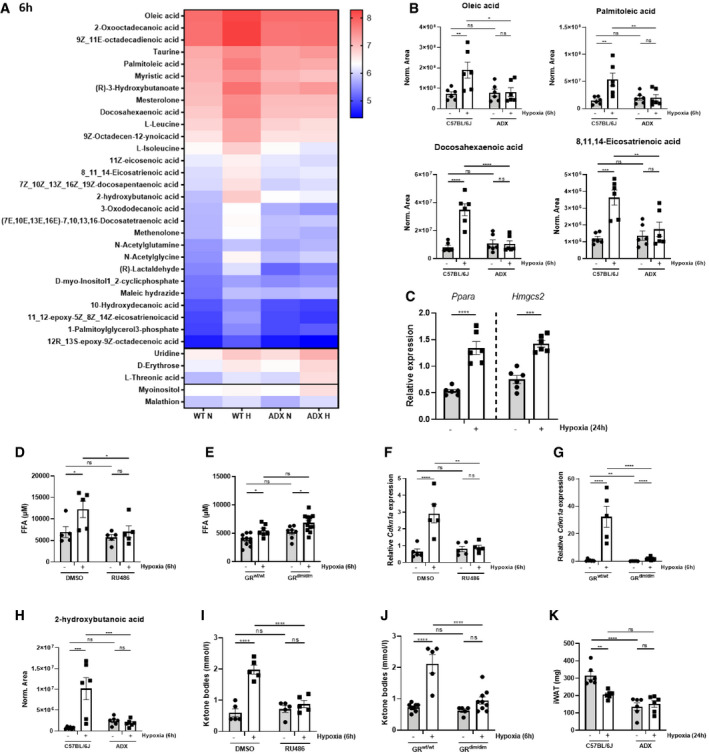
The transcriptomics data in the liver and Enrichr pathway analyses suggest that hypoxia instructs GR to regulate metabolic rather than inflammatory pathways. PPARα (encoded by the Nr1c1 gene) is one of the major sensors of the nutritional status, and it is highly expressed in the liver and primarily controls the oxidation of FFAs and ketogenesis (Grabacka et al, 2016; Wang et al, 2020). We investigated the metabolic profile of mice under hypoxic conditions and the effect on liver PPARα activity. The plasma of C57BL/6J and ADX mice was collected 6 h after normoxia or hypoxia for metabolic profile analysis. In the plasma of C57BL/6J control mice, the levels of numerous different FFAs were significantly increased after 6 h of hypoxia, while no differences were detected in the plasma of ADX mice (Fig 6A). Figure 6B depicts several examples. A complete overview of the metabolic profile is shown in Fig EV4A. Since FFAs are endogenous ligands and activators of PPARα, we investigated the PPARα gene response to hypoxia by measuring PPARα‐responsive genes (Ppara itself and Hmgcs2) in the liver. During early hypoxia (6 h), no significant changes were observed (Fig EV4B), but the PPARα response was significantly increased after 24‐h hypoxia (Fig 6C). To support that hepatic PPARα is activated, the expression level of PPARα‐dependent genes was measured via RT–qPCR in liver‐specific PPARa knock‐out (PPARaAlbKO) mice. The expression of these genes was significantly upregulated after 24 h in wild‐type mice, while the expression levels were significantly reduced in the liver of PPARaAlbKO mice upon hypoxia (Fig EV4C). To exclude whether DEX itself is able to induce the expression of Ppara and PPARα‐responsive genes (Cd36, Cpt1a, Cpt2, Slc25a20, Hadha, Acat1, Acox1, Acox2, Ehhadh, and Hmgcs2) in normoxia and hypoxia (6 and 24 h), we calculated the fold inductions (DEX/PBS) of these genes (Fig EV4D). We were unable to detect significant differences between the fold inductions in normoxia and hypoxia. Since the fold inductions are mainly around 1, we can assume that DEX is not sufficient to induce Ppara and PPARα‐responsive genes.
Figure 6. Hypoxia causes white adipose tissue lipolysis leading to FFA and liver ketone body production in a GR‐dependent manner.
-
AFemale C57BL/6J and ADX mice were put in normoxia and hypoxia for 6 and 24 h. Plasma was isolated for metabolomics analysis. The heatmap represents log10 of metabolites, which are significantly increased of the plasma of C57BL/6J mice (C57BL/6J P ≤ 0.05 and ADX P > 0.05, LFC > 1), significantly increased in the plasma of C57BL/6J and ADX mice (C57BL/6J P ≤ 0.05 and ADX P ≤ 0.05, LFC > 1), and significantly increased in the plasma of ADX mice only (C57BL/6J P > 0.05 and ADX P ≤ 0.05, LFC > 1).
-
BMetabolomics analysis identifying the presence of FFAs in the plasma of C57BL/6J and ADX mice 6 h after hypoxia. N = 6 per group, two independent experiments.
-
CPPARα gene response in the liver of C57BL/6J mice 24 h after hypoxia via RT–qPCR. P‐values were calculated using a two‐way unpaired Student’s t‐test. N = 6 per group, two independent experiments.
-
D–GFFA levels and stress GRE genes were determined in the plasma and iWAT of female C57BL/6J mice injected with 5 mg RU486 or vehicle (DMSO) (D, F), and in GRdim/dim mice and their wild‐type littermates (E, G) after 6 h of normoxia and hypoxia. N = 4–9 per group.
-
H–JBlood ketone body levels 6 h after normoxia and hypoxia in female C57BL/6J and ADX mice (H), in female C57BL/6J mice injected with 5 mg RU486 or vehicle (DMSO) (I), and in GRdim/dim mice and their wild‐type littermates (J). N = 5–9 per group.
-
KiWAT weight of C57BL/6J and ADX mice 6 h after normoxia or hypoxia. N = 5–6 per group.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P ≤ 0.05.
Figure EV4. Hypoxia causes white adipose tissue lipolysis leading to FFA and liver ketone body production in a GR‐dependent manner.
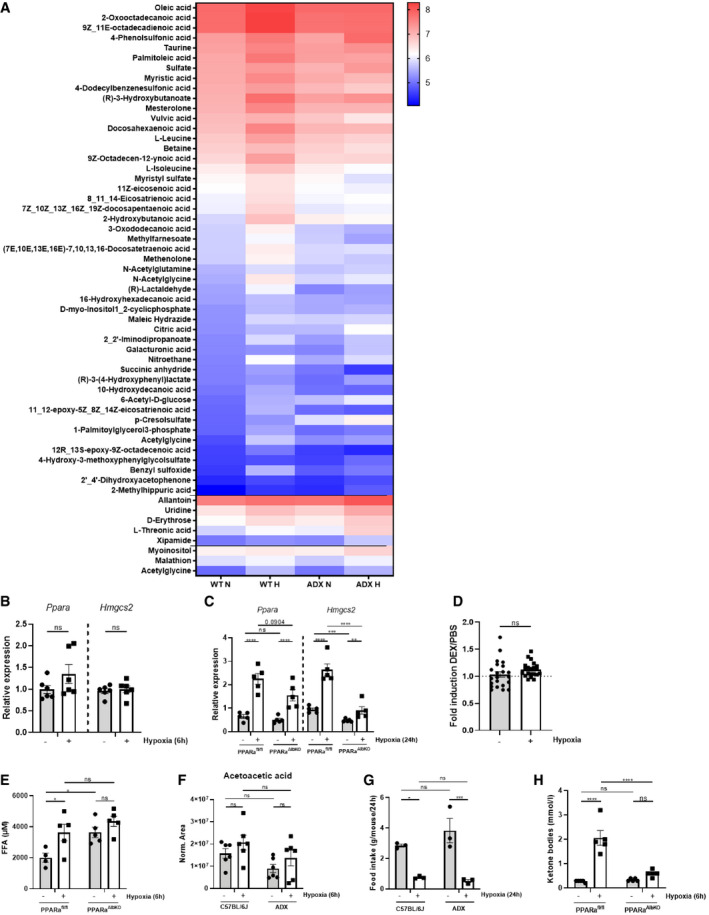
- Female C57BL/6J and ADX mice were put in normoxia and hypoxia for 6 h. Plasma was isolated for metabolomics analysis. The heatmap represents log10 of metabolites, which are significantly increased of the plasma of C57BL/6J mice (C57BL/6J P ≤ 0.05 and ADX P > 0.05), significantly increased in the plasma of C57BL/6J and ADX mice (C57BL/6J P ≤ 0.05 and ADX P ≤ 0.05), and significantly increased in the plasma of ADX mice only (C57BL/6J P > 0.05 and ADX P ≤ 0.05).
- Female C57BL/6J mice were put in normoxia and hypoxia for 6 h and liver was isolated for analyses. PPARα gene response in the liver of C57BL/6J mice 6 h after hypoxia via RT–qPCR. P‐values were calculated using a two‐way unpaired Student’s t‐test. N = 6 per group pooled from two independent experiments.
- PPARα gene response in the liver of PPARaAlbKO mice and their wild‐type littermates 24 h after hypoxia via RT–qPCR. N = 5 per group, one experiment.
- Fold inductions (DEX/PBS) of Ppara and PPARα‐responsive genes in normoxia and hypoxia (6 and 24 h) based on the RNA‐seq mRNA counts. N = 3 biological replicates for RNA‐seq data. P‐value was calculated using a two‐way unpaired Student’s t‐test. Fold inductions of 22 genes are depicted.
- FFA levels in the plasma of PPARafl/fl and PPARaAlbKO mice after 6 h hypoxia. N = 4–5 per group, one experiment.
- Metabolomics analysis identifying the presence of acetoacetic acid in the plasma of C57BL/6J and ADX mice 6 h after hypoxia. N = 6 per group, pooled from two independent experiments.
- The average food intake of C57BL/6J and ADX mice was measured after 24 h normoxia or hypoxia. Pooled data of 3 independent experiments.
- Blood ketone body levels 6 h after normoxia and hypoxia in PPARafl/fl and PPARaAlbKO mice. N = 5 per group, one experiment.
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P ≤ 0.05.
We further examined the role of GR in the release of FFAs in the plasma of mice during hypoxia. Inhibition of GR by RU486 prevented the release of FFAs in the plasma (Fig 6D), while in GRdim/dim mice, FFA levels were still increased by hypoxia, suggesting that the increased FFA production in white adipose tissue (WAT) is not affected by the reduced dimerization potential of GRdim/dim mice (Fig 6E). FFA levels were also measured in the plasma of PPARaAlbKO mice and wild‐type littermates after 6‐h hypoxia. In basal conditions, the FFA levels were already significantly higher in the plasma of PPARaAlbKO mice. These results are in line with the increased hepatic and plasma FFA levels in full PPARa knock‐out mice (Kersten et al, 1999; Leone et al, 1999). As expected, hypoxia significantly increased FFA levels in PPARafl/fl mice. However, no difference could be observed in PPARaAlbKO mice due to the high basal levels (Fig EV4E). Since the WAT is the most essential organ for lipolysis (Bolsoni‐Lopes & Alonso‐Vale, 2015), and GR can activate lipolysis (de Guia & Herzig, 2015), we studied if the stress GRE response is induced by hypoxia (6 h) in inguinal WAT (iWAT) and whether this response is GR‐dependent. Both in mice pre‐treated with RU486 (Fig 6F) and GRdim/dim mice (Fig 6G), the expression of stress GRE genes was absent during hypoxia.
Because the expression of Hmgcs2, the rate‐limiting enzyme in the ketone body (KB) biosynthesis (McGarry & Foster, 1980) is significantly increased after 24 h of hypoxia, we investigated the effect of hypoxia on the production of KBs. Three endogenous KBs, namely, β‐hydroxybutyrate (BHB), acetoacetate and acetone, are produced via ketogenesis in the liver, starting from acetyl‐CoA (Van Wyngene et al, 2018). The metabolomics analysis showed a significant increase of BHB in the plasma of C57BL/6J mice 6 h after hypoxia, but not in ADX mice (Fig 6H). Acetoacetic acid also tended to increase upon hypoxia, while these levels were lower in ADX mice (Fig EV4F). The last KB acetone was not detected via the metabolomics analysis. Furthermore, to investigate whether a reduced food intake might be responsible for the differences in KB production between C57BL/6J and ADX mice, the average food intake of C57BL/6J and ADX mice after 24‐h normoxia or hypoxia was determined. We did not observe a difference in food intake between C57BL/6J and ADX mice in normoxia and hypoxia; however, a significant reduction in food intake was observed in both C57BL/6J and ADX mice during hypoxic conditions (Fig EV4G). When GR was inhibited via RU486, KBs were not elevated in the plasma of these mice (Fig 6I). Interestingly, the increase in KBs was absent in GRdim/dim mice (Fig 6J), suggesting that intact GR dimerization is a necessary companion of PPARα (as shown earlier in starvation studies (Ratman et al, 2016)) for ketogenesis in hypoxia. We also confirmed the involvement of PPARα in the KB production, since KB levels were significantly increased in PPARafl/fl mice, while this was absent in PPARaAlbKO mice (Fig EV4H). To support that FFA production in hypoxia, is due to lipolysis, we investigated the weight of iWAT in C57BL/6J and ADX mice and found significant decrease in C57BL/6J mice, while in ADX mice, although their iWAT weight was already lower compared to C57BL/6J control mice, no effect was observed (Fig 6K). In conclusion, hypoxia leads to the activation of the HPA axis, which activates GR in the liver, hypothalamus, and iWAT. In the latter, hypoxia has GR dimer regulated stress effects as well as lipolytic effects, causing FFA release, and hepatic PPARα activation and KB production.
GR shows attenuated anti‐inflammatory capacity under hypoxic conditions
As we have demonstrated that liver GR is less responsive to DEX in hypoxia (Fig 2) and hypoxia activates the HPA axis, leading to higher GC production (Fig 4), we studied if hypoxia attenuates the anti‐inflammatory actions of DEX and if DEX is still able to protect against LPS‐induced lethal endotoxic shock (Figs 7 and EV5). Nfkbia, Dusp1, and others such as Fam107a, Tsc22d3, Vdr, and Serpina3c were significantly upregulated by DEX in normoxia, however less induced by DEX in hypoxia. Also, genes with known pro‐inflammatory capacities are repressed by DEX in normoxia, but are repressed to lesser extent by DEX in hypoxia (Fig EV5). These effects are most pronounced in the liver 24 h after hypoxia (Fig EV5B) and are less clear after 6 h (Fig EV5A). We measured the expression levels of Dusp1 and Nfkbia via RT–qPCR and found that they can no longer be induced by DEX 24 h after hypoxia in the liver, lung, kidney, and spleen (Fig 7A). We then studied whether DEX‐induced protection against LPS‐induced endotoxemia (Dejager et al, 2010; Kleiman et al, 2012), a very well‐known acute lethal inflammation model, is different in hypoxia compared to normoxia. First, we compared the LPS sensitivity of mice kept for 24 h in hypoxia or normoxia by increasing the dose of i.p. injected LPS (2.5, 5, and 14.5 mg/kg LPS) and recording lethal response. Mice in hypoxia displayed a higher sensitivity to LPS (Fig 7B). In order to confirm that the LD100 LPS dose in normoxia and hypoxia is comparable in terms of organ damage, parameters (lactate dehydrogenase (LDH), aspartate transaminase (AST), creatine kinase, urea) were measured in the plasma of mice 6 h after LPS injection. The increase of LDH, AST, creatine kinase, and urea was similar in the plasma of mice in normoxia and hypoxia in LPS‐induced endotoxemia (Fig 7C). It is known that LPS causes fast activation of the HPA axis leading to increased GC levels and repression of the pro‐inflammatory response of LPS (Beishuizen & Thijs, 2003). Since hypoxia induces chronic GC production, and since we hypothesize that the anti‐inflammatory function of GCs is compromised, GC levels were measured in the plasma of mice in normoxia and hypoxia 2 h after LPS injection. In normoxia, LPS causes a significant increase in GC levels, while in mice in hypoxia, no further increase could be observed when LPS is injected (Fig 7D). Next, the effect of LPS on the expression of genes with a known anti‐inflammatory function was measured via RT–qPCR and these genes (Fam107a, Dusp1, Tsc22d3, Vdr, and Serpina3c) were significantly induced after LPS injection in normoxia, but no difference in gene expression was detected in hypoxic conditions (Fig 7E). Furthermore, the expression of these genes was significantly increased in ADX mice by DEX stimulation (Fig 7F) in normoxia as well as in hypoxia, indicating that the chronic GC production, via the activation of the HPA axis by hypoxia, might be responsible for a reduced anti‐inflammatory induction by LPS or by injection of DEX.
Figure 7. GR shows attenuated anti‐inflammatory capacity under hypoxic conditions.
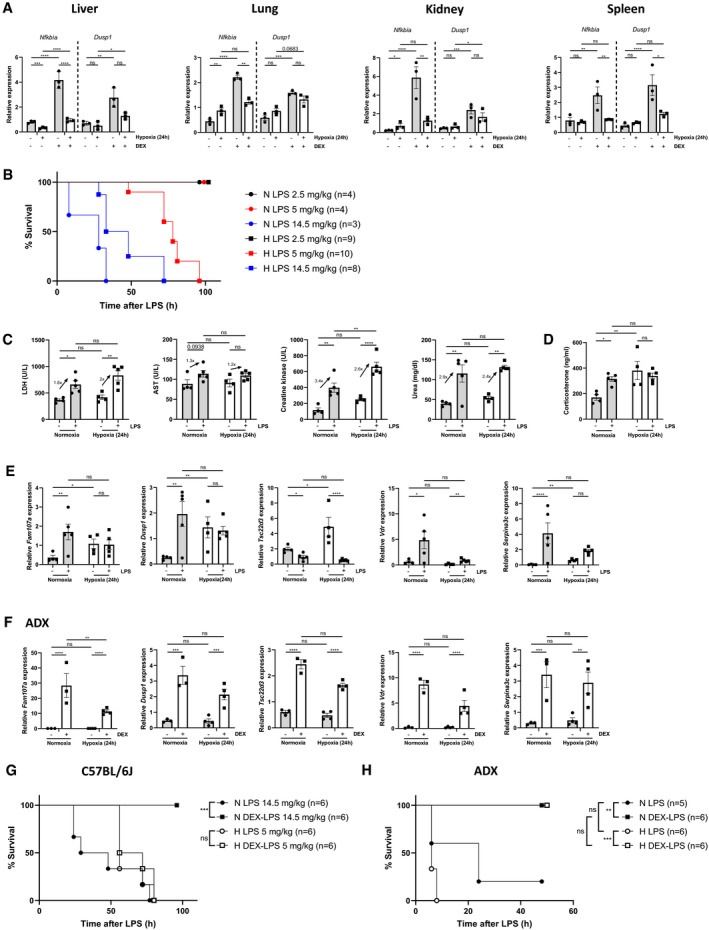
-
AFemale C57BL/6J mice (n = 3 per group) were put in normoxia or hypoxia for 24 h, injected with PBS or DEX (10 mg/kg) and 2 h later, indicated organs were isolated and gene expression was measured via RT–qPCR.
-
BLPS LD100 dose‐response in female C57BL/6J mice injected with indicated LPS doses after 24 h hypoxia. During the follow‐up of lethality, mice remained under hypoxic conditions. N‐values are indicated in the legend.
-
C, DOrgan damage parameters (C) and GC levels (D) were determined in the plasma of female C57BL/6J mice injected with 14.5 mg/kg LPS (normoxia) or 5 mg/kg LPS (hypoxia) 6 and 2 h after LPS injection, respectively. N = 4–5 per group.
-
EFemale C57BL/6J mice were injected with 14.5 mg/kg LPS (normoxia) or 5 mg/kg LPS (hypoxia) after 24 h normoxia or hypoxia. Liver was isolated 6 h after injected and typical GRE genes were measured via RT–qPCR. N = 4–5 per group.
-
FFemale ADX mice were put in normoxia or hypoxia for 24 h, injected i.p. with PBS or DEX (10 mg/kg) and 2 h later, liver was isolated for RT–qPCR analyses of typical GRE genes. N = 3–4 per group.
-
GFemale C57BL/6J mice were injected with 14.5 mg/kg LPS (normoxia) or 5 mg/kg LPS (hypoxia) after 24 h normoxia or hypoxia, with or without pre‐treatment with 10 mg/kg DEX 1 h before LPS injection. During the follow‐up of lethality, mice remained under normoxic or hypoxic conditions. Mice in normoxia: black circles (LPS), black squares (DEX‐LPS); mice in hypoxia: white circles (LPS), white squares (DEX‐LPS).
-
HFemale ADX mice were injected with 0.05 mg/kg LPS after 24‐h normoxia or hypoxia, with or without 10 mg/kg DEX pre‐treatment 1 h before LPS injection. During the follow‐up of lethality, mice remained under normoxic or hypoxic conditions. ADX mice in normoxia: black circles (LPS), black squares (DEX‐LPS); ADX mice in hypoxia: white circles (LPS), white squares (DEX‐LPS).
Data information: All bars represent mean ± SEM. P‐values were calculated using two‐way ANOVA followed by post‐hoc Šídák’s multiple comparisons test to correct for multiple testing during the pairwise multiple comparisons, except if otherwise stated. Survival curves were analyzed with a Log‐Rank (Mantel‐Cox) test. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P ≤ 0.05.
Figure EV5. GR shows attenuated anti‐inflammatory capacity under hypoxic conditions.

-
A, BHeatmap representing the DEX effect on the expression of pro‐ and anti‐inflammatory genes in normoxia and hypoxia after 6 h (A) and 24 h (B). Log2 values are shown of the counts of these genes on the RNA‐seq data. Genes with known pro‐inflammatory function are categorized below the black line, genes with known anti‐inflammatory function are shown above the black line.
-
CLPS LD100 dose‐response was determined in female ADX mice injected with indicated LPS doses after 24 h hypoxia. During the follow‐up of lethality, mice remained under hypoxic conditions. N‐values are indicated in the legend.
-
D–GFemale C57BL/6J mice were put in normoxia (D, F) or hypoxia (E, G) for 24 h, injected with PBS or DEX (10 mg/kg) and 1 h later injected with 14.5 mg/kg LPS (normoxia) or 5 mg/kg (hypoxia). Organ damage parameters (D, E) were determined in the plasma of the mice 24 h after LPS injection. N = 11–12 per group. Glucose levels (F, G) were measured via the tail vein. N = 6 per group.
Data information: All bars represent mean ± SEM. P‐value were analyzed with one‐way ANOVA. Survival curves were analyzed with a Log‐Rank (Mantel‐Cox) test. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P ≤ 0.05.
C57BL/6J mice were treated with DEX (10 mg/kg) or PBS after 24‐h normoxia or hypoxia, followed 1 h later by a single lethal LPS dose (normoxia 14.5 mg/kg LPS, hypoxia 5 mg/kg LPS). In normoxia, DEX was found to protect mice against LPS‐induced lethality, while no DEX protection was observed in mice under hypoxic conditions (Fig 7G). Also, a clear DEX effect was detected on organ damage parameters 24 h after LPS injection in normoxia (Fig EV5D), but not in hypoxia (Fig EV5E). It is known that GCs are important in preserving glucose levels during inflammatory conditions such as sepsis, since glucose is the primary energy source of the brain and it is important to maintain the maximal brain function upon inflammation (Kuo et al, 2015; Van Wyngene et al, 2018; Vandewalle & Libert, 2020). Based on our results, hypoxia activates the HPA axis, thereby increasing GC levels, which are not further increased upon an inflammatory stimulus such as LPS (Fig 7D). To investigate whether the activation of the HPA axis and increased GC levels in hypoxia has an influence on the glucose levels during inflammation, we performed an experiment in which female C57BL/6J mice were put in normoxia or hypoxia for 24 h followed by an intraperitoneal injection with PBS or DEX (10 mg/kg). 1 h later, these mice were injected intraperitoneally with 14.5 mg/kg LPS (normoxia) or 5 mg/kg (hypoxia). As expected, blood glucose levels were decreased 24 h after LPS‐induced endotoxemia in normoxia, DEX was not able to increase this (Fig EV5F). Similar results were obtained when mice were in hypoxic conditions (Fig EV5G), thereby indicating that the activation of the HPA axis and increased GC levels do not alter the glucose response to inflammation. To confirm that chronic GC production is responsible for the lack of DEX protection in hypoxia, we investigated whether DEX remained able to protect against a lethal LPS dose (0.05 mg/kg LPS) in ADX mice in hypoxia. First, LD100 LPS was checked in ADX mice both in normoxia and hypoxia. No difference in LPS sensitivity could be observed (Fig EV5C). In ADX mice, DEX was still able to induce a significant protection against a lethal LPS dose, independent of the O2 concentration in which the ADX mice were kept (Fig 7H). In conclusion, the anti‐inflammatory function of DEX is compromised under hypoxic conditions. The activation of the HPA axis followed by a chronic GC production by hypoxia might be responsible for this compromised function of DEX.
Discussion
The cellular responses to stress and inflammation are tightly regulated by the GC‐induced and hypoxia‐induced transcriptional responses. Several in vitro and in vivo studies using zebrafish larvae have illustrated the presence of a crosstalk between HIF and GR (Vettori et al, 2017; Marchi et al, 2020; Vanderhaeghen et al, 2021). Although these papers are very valuable, more convincing data are necessary to understand the impact of hypoxia on GR in a pathophysiological context in vivo. Therefore, we have investigated the in vivo impact of hypoxia on GR function in mice by genome‐wide transcriptional analysis in liver and by means of deep hypoxia. During these experiments, mice have been put under extreme hypoxic conditions [we applied 7% O2 based on previous studies (Bruder et al, 2008; Wang, Ma, et al, 2012)]; however, no mortality was detected after 6‐h or 24‐h hypoxia. Hypoxia stabilizes hypothalamic HIF1α and HIF2α and causes a complete, strong activation of the HPA axis and chronic production of GCs by the adrenals. As it is difficult to obtain blood from mice in a non‐stressed manner (Kim et al, 2018), the GC levels observed in normoxia might be potentially stress levels. In the absence of GC production via adrenalectomy, the negative feedback signal is removed, thereby increasing CRH and ACTH levels (Dallman et al, 1987, 1994). The importance of the HPA axis activation and GC production regarding the stress GRE response was confirmed in ADX mice upon hypoxia. As expected, basal Crh mRNA levels were increased in ADX mice, hypoxia tended to increase the Crh mRNA levels in ADX mice although not significant and to similar extent as C57BL/6J mice. The effect of reduced GC production was also clear on the expression levels of the stress GRE genes. In GR‐deficient zebrafish larvae, the reduced GC production also affects HIF activity in general (Marchi et al, 2020). We have observed a significant reduction in the fold induction of HIF target gene expression levels in ADX mice upon hypoxia, suggesting that GCs also influences the expression of HIF target genes. Further research is needed to determine the details of the contribution of HIF1α and/or HIF2α in the activation of the HPA axis upon hypoxia and the GC/GR stress response, and how GC/GR regulate these HIF effects.
Since the activation of the HPA axis is an essential component of how GR is engaged to a more metabolic program during hypoxia, we have studied this in more detail. Our data suggest that hypoxia (i) activates several GRE genes by means of HPA axis activation and GC production, (ii) causes DEX to induce genes that are not induced by DEX in normoxia in which hypoxia and DEX exert a synergistic effect, and (iii) strongly reduces the effect of DEX on canonical DEX‐induced genes in the liver. Based on RT–qPCR analysis in ADX mice, this last effect appears to be due to the HPA axis activation, although we do not have RNA‐seq data of ADX mice in normoxia and hypoxia combined with DEX stimulation. Furthermore, the effects of hypoxia on the gene expression patterns are independent on the presence of HIF1α and HIF2α in the liver. The mechanisms of action of GCs include binding of GC to the cytoplasmic GR, GR translocation to the nucleus, followed by DNA binding and regulation of gene expression (Timmermans et al, 2019). In our study, we found that hypoxia had no significant effects on Nr3c1 mRNA levels. Although it has been shown that increased GC levels can reduce GR protein levels, a phenomenon called homologous downregulation (Dong et al, 1988; Bellingham et al, 1992), liver GR protein levels were not decreased in hypoxic conditions. GR homologous downregulation is proposed as one of the mechanisms causing GCR (Van Bogaert et al, 2011; Dendoncker et al, 2019); however, GR is still active in hypoxic conditions, but its priorities are clearly altered. The reprogramming of GR induced during hypoxia is, in part, regulated by differences in the GR DNA‐binding profiles. Based on ChIP‐seq analysis, we found that (i) the induction of stress GRE genes by hypoxia can be linked with increases in GR DNA‐binding; (ii) the DEX‐responsive genes in hypoxia, which are not induced by DEX in normoxia, are associated with a higher coverage of GR DNA‐binding peaks after DEX in hypoxia compared to PBS; however, the overall peak coverage is still lower in comparison to DEX stimulation in normoxia; and (iii) the strongly reduced DEX effect on canonical DEX‐induced genes by hypoxia can be correlated with reduced GR DNA‐binding after DEX stimulation in the liver. We were not able to find evidence for a direct interaction between GR and HIF, which is in line with the results of Yang et al (2020) They also found that hypoxia depletes GR target genes involved in inflammatory responses and that hypoxia increases the expression of genes involved in O2 regulation, which we can confirm based on our RNA‐seq data. Alterations in GR DNA‐binding are thus partly responsible for the changes in DEX response in hypoxia.
Next to alterations in the GR DNA‐binding profiles, hypoxia can also induce changes in the chromatin structure via histone methylation, acetylation, and DNA methylation (Batie et al, 2018). Histone acetylation is correlated with transcriptional activation, independent of the acetylation site. Acetylation of histone H3 on lysine 27 (H3K27ac) is the most studied histone acetylation and is known as a marker for active enhancers (Calo & Wysocka, 2013). Recently, Yang et al (2020) have identified a differential acetylation of H3K27 in response to DEX in hypoxia. These changes in GR activation chromatin state might also be responsible for changes in the GR recruitment to DNA (Yang et al, 2020). The p300 co‐activator is also known as a powerful mediator of GR transactivation and brings histone acetylation enzymatic activity to the GR bound sites (Guo et al, 2017; Dendoncker et al, 2019). More than half of the GR DNA‐binding site have shown enrichment of the p300 co‐activator and thus an active histone mark (Yang et al, 2020), indicating that competition between GR and HIF for the p300 co‐activator might also be responsible for the alterations in the gene expression profile. Finally, hypoxia drives the expression of both miRNA 103 and 107, which causes a decreased expression of its known target KLF4 followed by the inhibition of GR co‐modulators such as CARM1 and NCOA2 (Chen et al, 2012; Yang et al, 2020). Further investigation will be necessary to determine whether posttranslational modifications such as histone acetylation contribute to the GR response during hypoxic conditions and/or whether the induction of miRNAs are involved in the altered GR DNA‐binding and transcriptomics profile upon hypoxia.
As mentioned, endogenous GCs promote lipolysis under a normal, basal physiologic state via the induction of lipase activity in the adipose tissue of mice (de Guia & Herzig, 2015). The mode of action of GR is thought to be (i) via gene induction of the Angiopoietin Like 4 gene (Angptl4) (ii) as well as by inhibition of the phosphodiesterase 3 gene (Enpp3 gene), both leading to increased phosphorylation and activation of hormone‐sensitive lipase by PKA, but also via (iii) increased gene expression of the adipose tissue triacylglycerol lipase‐coding gene (Pnpla2) (Wang, Gray, et al, 2012). In hypoxia, the production of GCs is dependent on the adrenals, since GC levels are significantly lower in the plasma of ADX mice. Of note, an important remark in the use of ADX mice is the fact that not only GCs, but also catecholamines like epinephrine and norepinephrine are no longer produced by the adrenal medulla (Kanczkowski et al, 2016). Since hypoxia‐induced FFA release is also blocked by RU486, it is clear, however, that this hypoxia effect is a GR effect and not a catecholamine response. The fact that GRdim/dim mice still respond to hypoxia by increased FFA levels argues for a requirement of intact GR dimerization in this regard in WAT. The stress GRE response is also induced in the iWAT and is intact GR dimerization‐dependent, showing that the GR reprogramming is also present in this tissue.
In Peruvian populations living at high altitude, a unique dyslipidemia pattern with high frequency of triglyceride levels have been observed (Gonzales & Tapia, 2013). Since FFAs are endogenous ligands of PPARα (Wang et al, 2020), the effect of hypoxia on β‐oxidation was further investigated. We reveal that hypoxia increases plasma FFA levels and PPARα‐mediated β‐oxidation followed by enhanced KB production (ketogenesis). Upon fasting, PPARα full knock‐out mice fail to induce mitochondrial and peroxisomal fatty acid oxidation genes (Lee & Gonzalez, 1996), which leads to increased levels of plasma and hepatic fatty acids, hypoketonemia, and hypothermia (Kersten et al, 1999; Leone et al, 1999). In PPARaAlbKO mice, plasma FFA levels were higher during basal conditions. Although we expected an increase of FFAs upon hypoxia in PPARaAlbKO mice, no significant differences were observed in the plasma of these mice. This might be associated with an increased fatty acid uptake and lipid accumulation in the liver (Yasuhara et al, 1991; Kersten et al, 1999). Since the production of KBs is also dependent on intact GR dimerization, a close interaction between GR and PPARα during ketogenesis in hypoxia appears essential. GR‐PPARα physical interaction during ketogenesis in starvation has already been shown in vitro by Ratman et al (2016). Furthermore, PPARα agonists are able to inhibit the expression of GC‐responsive GRE‐driven genes in a PPARα dependent manner (Bougarne et al, 2009). Also, the anti‐inflammatory activity of DEX is lower in PPARα knock‐out mice compared to wild‐type littermates (Cuzzocrea et al, 2008), corroborating the crosstalk between PPARα and GR. We also evaluated whether DEX itself is sufficient to induce PPARα mediated fatty acid oxidation pathways. However, no effect of DEX on the expression levels of genes involved in β‐oxidation was detected.
Several studies have investigated the function of HIF1α and HIF2α in lipid metabolism. Although we did not observe an increase in lipid biosynthesis, Li et al (2006) showed that HIF1α upregulates lipid biosynthesis in the liver by stimulating sterol regulatory element binding protein (SREBP)‐1 activity via SREBP cleavage‐activating protein (SCAP) during intermittent hypoxia. However, a protective role for HIF1α in the development of alcoholic fatty liver is proven by Nishiyama et al (2012). The absence of HIF1α in the liver leads to increased lipid biosynthesis and steatosis. Although we observed an increase in β‐oxidation during hypoxia, Liu et al found that inhibition of the HIF1α or HIF2α in the liver attenuates hypoxia‐reduced FFA β‐oxidation leading to improved hepatic fat metabolism. However, a dominant role for HIF1α in the decrease in β‐oxidation is stated by Belanger et al (2007), while Rankin et al suggest an important role for HIF2α in attenuating β‐oxidation (Rankin et al, 2009). We also detected higher KB levels in hypoxia and their dependency on the intact GR dimerization. The absence of HIF1β in mouse livers leads to decreased KB levels, suggesting a role for HIF1α and/or HIF2α in ketogenesis (Wang et al, 2009). Further investigation will be necessary to uncover the exact role of HIF1α and/or HIF2α and their link with PPARα in lipid metabolism and ketogenesis during hypoxia.
Hypoxia and inflammation are two closely linked phenomena in many pathological processes such as critical illness, sepsis, and inflammatory bowel diseases (IBDs). Critically ill patients often experience systemic inflammation in combination with hypoxia. Kiers et al (2018) found that mice, exposed to 9% O2 1 h before systemic inflammation, display an attenuated inflammatory cytokine response (Kiers et al, 2018). This is in contrast with the results obtained in our study where prolonged hypoxia (24 h, 7% O2) causes an increased sensitivity to a lethal LPS‐induced endotoxemia. Hypoxia is also present in tumors and is recognized as an important deleterious factor in cancer therapies. In tumors, enhanced angiogenesis is present; however, O2 levels are significantly lower ranging from 0.3 to 4.2% O2 (McKeown, 2014). In acute inflammation under normal O2 concentrations, when mice are injected with tumor necrosis factor (TNF) or LPS, endogenous GCs are important protective molecules against this SIRS (De Bosscher et al, 2016). They are well known for their anti‐inflammatory properties, primarily by counteracting the production of pro‐inflammatory cytokines, such as IL‐1β, IL‐6, and IFNβ (Libert et al, 1990, 1991; De Bosscher et al, 2003). Adrenalectomy causes a clear sensitization of mice to acute lethal SIRS induced by LPS (Dejager et al, 2010), illustrating that LPS causes HPA axis activation and GC production (Beishuizen & Thijs, 2003), thereby inducing the expression of genes with anti‐inflammatory functions in a negative feedback mechanism. We found that the LPS sensitivity in ADX mice is not altered in the presence of low O2 concentrations, and DEX can still protect these mice. The chronic GC production by hypoxia is thus responsible for the increased sensitivity for LPS and for the lack of DEX protection, since genes with anti‐inflammatory functions can no longer be upregulated by LPS, because no additional GCs can be induced. Previously, we have shown that in acute, lethal inflammation like septic shock, a functional decline of GR is observed, called GCR (Van Bogaert et al, 2011; Dendoncker et al, 2019). Despite GR is still functional in a condition of pure, deep hypoxia, it seems that the anti‐inflammatory priority of GR has changed to a more metabolic function. Since GR DNA‐binding is required for its anti‐inflammatory function (Uhlenhaut et al, 2013; Escoter‐Torres et al, 2020), the altered DEX response and the changes in the anti‐inflammatory profile of GR in hypoxia could be correlated with the differences in the GR DNA‐binding profile after DEX stimulation, when mice are in hypoxic conditions. The use of GCs attenuates the pro‐ and anti‐inflammatory responses present during sepsis. The first large‐scale clinical trials considering the use of GCs in sepsis patients involved high doses of GCs in the management of septic shock. Bolus injection of GCs significantly reduced the mortality rates (Minneci et al, 2009). However, more recent clinical trials recommend the use of a supraphysiological dose of hydrocortisone (200–300 mg/day) (Annane et al, 2002). In contrast to the supraphysiological dose of GCs, we used a rather high dose of DEX (10 mg/kg) because this concentration protects prophylactically against a lethal LPS‐induced endotoxemia (Van Looveren et al, 2020) and we wanted to investigate the effect of hypoxia on the pro‐ and anti‐inflammatory functioning of the GR. However, we have to take into account that other nuclear receptors such as the pregnane X receptor (PXR) might also be activated when using such a high dose of DEX (Pavek, 2016).
Since inflammation is able to induce the stabilization and activation of HIF (Cummins et al, 2016), it would be of great interest to know whether HIF might play a role in the GCR during sepsis. The protective effects of exogenous GCs like DEX against LPS‐induced endotoxemia are frequently associated with the inhibition of TNF production in white blood cells, such as macrophages (Bhattacharyya et al, 2007; Kleiman et al, 2012; Van Looveren et al, 2020). Nevertheless, hepatic GR is also crucial for the GC homeostasis and the protection against SIRS and sepsis (Van Bogaert et al, 2011; Jenniskens et al, 2018). When ascending to high altitude, factors like immunomodulation, hypoxia, environmental stressors, and physiologic adaptations may lead to increased susceptibility to pathogens (Basnyat & Starling, 2015). The most common syndrome starting within a few hours of ascent is acute mountain sickness (AMS). It is generally associated with headache, nausea, vomiting, anorexia, lassitude, and sleep disturbances (Taylor, 2011). Several studies have shown that prophylactic treatment of humans with synthetic GCs like DEX (Rock et al, 1989; Basu et al, 2002a; Kitsteiner et al, 2011), prednisolone (Basu et al, 2002b), or budesonide (Zheng et al, 2014; Berger et al, 2017; Zhu et al, 2020) prevents the development of AMS or reduce its symptoms. In contrast, our study shows that a few hours after hypoxia, the anti‐inflammatory GR response to DEX is strongly reduced compared to normoxia, probably due to the change in more metabolic priorities of the GR. We could speculate that DEX will first influence the metabolic functions of GR during hypoxia, while the anti‐inflammatory functions are less important. When people are pre‐treated with an exogenous GC like DEX before ascending to high altitude, the main anti‐inflammatory function of GR is not altered and will be able to prevent the development of AMS. In this regard, the actual outcome of the interplay between GR and hypoxia may depend on a “first come” principle. However, it would be of great interest to identify the effect of less severe hypoxia on the GR function, if the GC/GR response would be different. Also, adaptation to long‐term hypoxia could alter the metabolic and inflammatory effects linked to GR we observe during acute, deep hypoxia.
In summary, hypoxia activates the HPA axis, leading to GC production by the adrenal glands. The precise role of HIF1α and/or HIF2α in this activation requires further investigation. GCs are responsible for increased lipolysis and FFA levels in the bloodstream independent of intact GR dimerization. FFAs induce PPARα activity in the liver, leading to the production of KBs, which is GR dimerization‐dependent. Next to GR‐mediated metabolic changes, hypoxia sensitizes mice to LPS‐induced endotoxemia, which is caused by the HPA axis activation and GC production. Furthermore, DEX is no longer able to protect mice against LPS‐induced lethal shock and is probably linked with the changes in the GR DNA‐binding profile in hypoxia. Our data unfold new physiological pathways that may have consequences for patients suffering from GCR, when ascending to high altitudes or when hypoxia is present in critically ill patients.
Materials and Methods
Reagents and Tools table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Experimental models: Organisms/strains | ||
| C57BL/6J | Janvier | N/A |
| ADX C57BL/6J | Janvier | N/A |
| Hif1atm3Rsjo/Hif1atm3Rsjo Mus musculus (in manuscript referred to as HIF1aflox/flox) | Ben Wielockx (B6 background) | MGI Cat# 2386679 |
| Epas1tm1Mcs/ Epas1tm1Mcs Mus musculus (in manuscript referred to as HIF2aflox/flox) | Ben Wielockx (B6 background) | MGI Cat# 3526731 |
| B6. Cg‐Speer6‐ps1Tg(Alb‐cre)21Mgn/J Mus musculus (in manuscript referred to as Albumin Cre) | Dirk Elewaut |
IMSR Cat# JAX:003574 RRID: IMSR_JAX:003574 |
| Nr3c1tm3Gsc/Nr3c1tm3Gsc Mus musculus (in manuscript referred to as GRdim/dim) | Jan Tuckermann (FVB background) |
MGI Cat# 4455054 RRID: MGI:4455054 |
| Pparatm1a(EUCOMM)Wtsi/ Pparatm1a(EUCOMM)Wtsi Mus musculus (in manuscript referred to as PPARaflox/flox) | Karolien De Bosscher (B6 background) | MGI Cat# 4432239 |
| Recombinant DNA | ||
| HRE‐luciferase plasmid | Addgene | Cat# 26731 |
| Antibodies | ||
| Mouse anti‐human GR antibody (G‐5) (for western blot, 1:1,000) | Santa‐Cruz Biotechnology | Cat# Sc‐3932; RRID:AB_2687823 |
| Rabbit anti‐mouse polyclonal GR antibody (H300) (for ChIP‐seq, 5 µg per reaction) | Santa‐Cruz Biotechnology | Cat# sc‐8992; RRID:AB_2155784 |
| Mouse anti‐mouse monoclonal beta‐actin antibody (BA3R, 1:5,000) | ThermoFisher Scientific | Cat# MA5‐15739; RRID:AB_10979409 |
| Rabbit anti‐mouse polyclonal HIF1α antibody (1:1,000) | Cayman chemical | Cat# 10006421; RRID:AB_409037 |
| Rabbit anti‐mouse polyclonal HIF2α antibody (1:3,000) | Abcam | Cat# ab109616; RRID:AB_11156727 |
| Rabbit anti‐mouse polyclonal Vinculin antibody (1:1,000) | Cell signalling | Cat# 4650S; RRID:AB_10559207 |
| Mouse monoclonal luciferase antibody (1:1,000) | ThermoFisher Scientific | Cat# MA1‐16880; RRID:AB_568609 |
| Goat anti‐mouse polyclonal HRP antibody (1:5) | Vector Laboratories | Cat# MP‐7452; RRID:AB_2744550 |
| Oligonucleotides | ||
| qPCR primers (Table 2) |
This MS | N/A |
| Chemicals, enzymes and other reagents | ||
| LPS | Sigma Aldrich | Cat# L5886 |
| Rapidexon 2 mg/ml (DEX) | Dechra | NA |
| RU486 | Sigma Aldrich | Cat# M‐8046 |
| XenoLight™ D‐Luciferin ‐ K+ Salt | Caliper Life Sciences | Cat# 122799 |
| Software | ||
| GraphPad Prism v.8 | GraphPad Software | GraphPad Prism; RRID:SCR_002798 |
| HOMER | (Heinz et al, 2010) | HOMER; RRID:SCR_010881 |
| HISAT2 | (Kim et al, 2019) | HISAT2; RRID: SCR_015530 |
| FeatureCounts | (Liao et al, 2014) | FeatureCounts; RRID:SCR_012919 |
| DESeq2 | (Love et al, 2014) | DESeq2; RRID:SCR_015687 |
| Other | ||
| Aurum total RNA mini kit | Bio‐rad | Cat# 732‐6820 |
| SensiFast cDNA synthesis kit | GC Biotech BV | Cat# BIO‐650504 |
| SensiFast Sybr no‐ROX mice | GC Biotech BV | Cat# CSA‐01190 |
| Roche Light Cycler 480 instrument | Roche | RRID :S CR_020502 |
| Glucose meetstrips FreeStyle Lite | Abbott | N/A |
| FreeStyle Frredom Lite glucose meter | Abbott | N/A |
| Precision Xtra ß‐Ketone teststrips | Abbott | N/A |
| Freestyle Precision Neo meter | Abbott | N/A |
| Free Fatty Acid Assay Kit | Abnova | Cat# KA1667 |
| ACTH ELISA kit | Antibodies‐online | Cat# ABIN1113286 |
| Corticosterone ELISA kit | Arbor assays | Cat# K014‐H1; RRID:AB_2877626 |
Table 2.
Primer sequences used for RT–qPCR.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| Hprt | AGTGTTGGATACAGGCCAGAC | CGTGATTCAAATCCCTGAAGT |
| Rpl | CCTGCTGCTCTCAAGGTT | TGGTTGTCACTGCCTCGTACTT |
| Tbp | GAAGCTGCGGTACAATTCCAG | CCCCTTGTACCCTTCACCAAT |
| Fam107a | CAGACCAGAGTACAGAGAGTGG | GTGGTTCATAAGCAGCTCACG |
| Ddit4 | CAAGGCAAGAGCTGCCATAG | CCGGTACTTAGCGTCAGGG |
| Cdkn1a | CCTGGTGATGTCCGACCTG | CCATGAGCGCATCGCAATC |
| Mfsd2a | AGAAGCAGCAACTGTCCATTT | CTCGGCCCACAAAAAGGATAAT |
| Crh | CCTCAGCCGGTTCTGATCC | AGCAACACGCGGAAAAAGTTA |
| Ppara | AGAGCCCCATCTGTCCTCTC | ACTGGTAGTCTGCAAAACCAAA |
| Hmgcs2 | GAAGAGAGCGATGCAGGAAAC | GTCCACATATTGGGCTGGAAA |
| Nfkbia | TGAAGGACGAGGAGTACGAGC | TTCGTGGATGATTGCCAAGTG |
| Dusp1 | GTTGTTGGATTGTCGCTCCTT | TTGGGCACGATATGCTCCAG |
| Tsc22d3 | CCAGTGTGCTCCAGAAAGTGTAAG | AGAAGGCTCATTTGGCTCAATCTC |
| Vdr | ACCCTGGTGACTTTGACCG | GGCAATCTCCATTGAAGGGG |
| Serpina3c | CTGGGGCTCGTGATAACTGG | TCGAGTTGTGTCCCATTTTCTTT |
| Hspa1a | TGGTGCAGTCCGACATGAAG | GCTGAGAGTCGTTGAAGTAGGC |
| Slc22a5 | ACTGTGCCAGGGGTGCTAT | GCAACTGAGGCTTCGTAGAAT |
Methods and Protocols
Mice
Female C57BL/6J mice as well as bilaterally adrenalectomized (ADX) C57BL/6J mice were purchased from Janvier (Le Genest‐St. Isle, France). ADX mice were ordered at the age of 7 weeks and bilateral adrenalectomy was performed 2 weeks before delivery. HIF1afl/fl , HIF2afl/fl (provided by Dr. Ben Wielockx), and PPARafl/fl (provided by Prof. Dr. Karolien De Bosscher) were crossed with Albumin Cre transgenic mice, and the offspring was intercrossed to generate HIF1afl/fl Albumin CreTg/+ (HIF1aAlbKO), HIF2afl/fl Albumin CreTg/+ (HIF2aAlbKO), HIF1aHIF2afl/fl Albumin CreTg/+ (HIF1aHIF2aAlbKO), and PPARafl/fl Albumin CreTg/+ (PPARaAlbKO) mice, all in a C57BL/6J background. GRdim/dim mice were generated by Reichardt et al (1998) and kept on a FVB/N background (generously provided by Dr. Jan Tuckermann, Ulm, Germany). Heterozygous GRdim/wt mice were intercrossed to generate GRwt/wt and GRdim/dim homozygous mutant mice. All offspring was genotyped by PCR on genomic DNA isolated from toe biopsies. Mice were housed in a temperature‐controlled, specific pathogen free (SPF) air‐conditioned animal house with 14‐ and 10‐h light/dark cycles and received food and water ad libitum. The drinking water of ADX mice was supplemented with 0.9% NaCl. All mice were used at the age of 8–12 weeks, and all experiments were approved by the institutional ethics committee for animal welfare of the Faculty of Sciences, Ghent University, Belgium.
Reagents
LPS from Salmonella abortus equi was purchased from Sigma‐Aldrich N.V. (L‐5886). For in vivo DEX injection, Rapidexon (Medini N.V.) was used. LPS and DEX were diluted in pyrogen‐free phosphate‐buffered saline (PBS). RU486 (Mifepristone, Sigma‐Aldrich N.V.), a GR antagonist, was diluted in DMSO. Luciferin (XenoLight™ D‐Luciferin ‐ K+ Salt) was purchased from Caliper Life Sciences.
Injections and sampling
All injections were given intraperitoneally (i.p.), except for the hydrodynamic intravenous (i.v.) tail injection of the DNA plasmid. Injection volumes were always adapted to the bodyweight of the mice. In lethality experiments, mice were monitored by monitoring rectal body temperature. Mice with body temperature below 28°C were euthanized using cervical dislocation. Blood was taken via cardiac puncture after sedation of the mice with a ketamine/xylazine solution (Sigma‐Aldrich N.V.) or via retro‐orbital eye bleeding after sedation with isoflurane. To obtain mouse plasma, samples blood was collected in EDTA‐coated tubes, and samples were centrifuged at 960 g for 15 min at 4°C. Plasma samples were stored at −80°C for metabolomics analysis or at −20°C for biochemical analysis. For sampling of liver and white adipose tissue, mice were killed by cervical dislocation at indicated time points.
Hypoxia treatment
Mice were randomly assigned to the normoxia group and hypoxia group. The normoxia group was exposed to room air (21% O2), whereas the hypoxia group was placed in a ventilated hypoxic chamber with 7% O2 and 93% N2 for the indicated time points. The oxygen levels were monitored with a Greisinger GOX 100 oxygen sensor (Conrad).
Detection of HIF activity
Mice were injected in the tail vein over 5 s with a HRE‐luc reporter plasmid solution (Addgene, #26731; 10 µg/ml in sterile, endotoxin‐free PBS) or PBS (control) in a volume equivalent to 10% of the body weight, as described by Van Bogaert et al (2011). The HRE‐luc plasmid contains three hypoxia response elements (24‐mers, TGTCACGTCCTGCACGACTCTAGT) from the Pgk1 gene upstream of firefly luciferase. Five hours after transfection (HRE‐Luc 0 h), mice were placed in normoxic or hypoxic conditions, and visualized after indicated time points. Briefly, mice were injected with 200 μl of a 15 mg/ml potassium salt luciferin solution. Ten minutes after injection, livers were isolated and visualized via the imaging chamber of the IVIS Spectrum In vivo Imaging System (Caliper Life Sciences). Photon emission was integrated over a period of 2 min and recorded as pseudo‐color images. Living Image (Caliper Life Sciences) was used for image analysis. The regions of interest (ROI, red circles) were selected based on the luciferase signal (purple) detected over all images. To confirm the specificity of the technique used for the injection of the HRE‐luc reporter plasmid, liver and other organs were also visualized (Fig 1B; PBS, HRE‐Luc N, HRE‐Luc H 1 h, HRE‐Luc H 2 h). For the PBS‐injected mice and mice injected with the HRE‐luc plasmid in normoxia, only 1 picture is shown representing the luciferase signal at the different time points when mice were put in normoxia or hypoxia. Data were acquired as photons/cm2/s, and results are normalized to the PBS control group. N = normoxia, H = hypoxia.
Food intake experiment
To determine whether hypoxia has an influence on the food intake during hypoxia, C57BL/6J or ADX mice were placed in normoxia or hypoxia for 24 h with access to a limited amount of food. The food was weighed at the start of the experiment, and 2, 6, and 24 h after the start of the experiment. Alternating experiments were performed with C57BL/6J and ADX mice in normoxia and hypoxia with 4–5 mice per group. The amount of food eaten during the experiment was calculated and divided by the amount of mice present per group to calculate the average amount of food (in grams) each mouse had eaten over 24 h. The experiment with C57BL/6J or ADX mice was performed in triplicate.
RNA sequencing
Liver
Total RNA was isolated with Aurum total RNA mini kit (Bio‐Rad) according to the manufacturer’s instructions. RNA concentration was measured, and RNA quality was checked with the Agilent RNA 6000 Pico Kit (Agilent Technologies). The 6‐h library was constructed using the TruSeq Stranded mRNA Library Prep kit (Illumina) according to the manufacturer’s instructions and sequenced paired‐end on an Illumina NextSeq500 instrument. The 24‐h library was constructed using a Stranded NextSeq high‐output mRNA Library Prep kit (2x150p) according to the manufacturer’s instructions and sequenced single‐end on an Illumina NextSeq500 instrument. The data were quality checked and pre‐processed (Illumina adapter removal) with Trimmomatic v 0.39 (Bolger et al, 2014). Reads were mapped to the mouse (mm10) reference genome using known splice junctions from the Ensemble 95 version of the annotation with HISAT2 (Kim et al, 2019). Gene level reads counts were obtained with featureCounts (Liao et al, 2014), and differential expressed genes were found by the DESeq2 R package (Love et al, 2014) with the false discovery rate (FDR) set at 5%. Motif finding for multiple motifs or de novo motif finding was performed using the HOMER software. We used the promoter region (start offset: −1 kb, end offset: 50 bp downstream of transcription start site (TSS)) to search for known motif enrichment and de novo motifs (Heinz et al, 2010). Visualizations were made using the R software. Gene ontology (GO) term enrichment on selected gene groups was performed via the Enrichr tool (Chen et al, 2013).
Hypothalamus
Total RNA was isolated with Aurum total RNA mini kit (Bio‐Rad) according to the manufacturer’s instructions. RNA concentration was measured, and RNA quality was checked with the Agilent RNA 6000 Pico Kit (Agilent Technologies). The 2‐h library was constructed using the TruSeq Stranded mRNA Library Prep kit (Illumina) according to the manufacturer’s instructions and sequenced paired‐end on an Illumina NextSeq500 instrument. The data were quality checked and pre‐processed (Illumina adapter removal) with Trimmomatic v 0.39 (Bolger et al, 2014). Reads were mapped to the mouse (mm10) reference genome using known splice junctions from the Ensemble 95 version of the annotation with HISAT2 (Kim et al, 2019). Gene level reads counts were obtained with featureCounts (Liao et al, 2014), and differential expressed genes were found by the DESeq2 R package (Love et al, 2014) with the false discovery rate (FDR) set at 5%. %. Motif finding for multiple motifs or de novo motif finding was performed using the HOMER software. We used the promoter region (start offset: −1 kb, end offset: 50 bp downstream of TSS) to search for known motif enrichment and de novo motifs (Heinz et al, 2010). Visualizations were made using the R software. Gene ontology (GO) term enrichment on selected gene groups was performed via the Enrichr tool (Chen et al, 2013).
Real‐time quantitative PCR
Liver was isolated and stored in RNA later (Life Technologies Europe), and white adipose tissue was snap‐frozen and stored at −20°C before RNA was isolated. Total RNA was isolated with the Aurum total RNA mini kit (Bio‐Rad) according to manufacturer’s instructions. RNA concentration was measured with the Nanodrop 8000 (Thermo Fisher Scientific), and 1,000 ng RNA was used to prepare cDNA with Sensifast cDNA Synthesis Kit (Bioline). cDNA was diluted 20 times in ultrapure water for use in RT–PCRs. RT–PCR primers for used targets are listed in Table 2. RT–PCR was performed with sensiFast Sybr no‐ROX mix (Bioline) and was performed in duplicate in a Roche LightCycler480 system (Applied Biosystems). The stability of the housekeeping genes (HKGs) was determined by Genorm. Results are given as relative expression values normalized to the geometric mean of the HKGs, calculated in the qBase+ software (Biogazelle).
Metabolomics experiments
Plasma metabolites were extracted by adding 10 µl of plasma to 990 µl of an 80% methanol (in water) extraction buffer containing 2 µM of deuterated (d27) myristic acid as internal standard) following extraction overnight at −80°C. Precipitated proteins and insolubilities were removed by centrifugation at 20,000× g for 20 min at 4°C. The supernatant was transferred to the appropriate mass spectrometer vials. Measurements were performed using a Vanquish LC System (Thermo Scientific) in‐line connected to a Lumos Orbitrap mass spectrometer (Thermo Scientific). 2 µl of sample was injected and concentrated on a Hilicon iHILIC‐Fusion(P) SS precolumn after which it was loaded onto a Hilicon iHILIC‐Fusion(P) SS column SS (Achrom). A linear gradient was carried out starting with 90% solvent A (LC‐MS grade acetonitrile) and 10% solvent B (10 mM ammonium acetate pH 9.3). From 2 to 20 min, the gradient changed to 80% B and was kept at 80% until 23 min. Next, a decrease to 40% B was carried out to 25 min, further decreasing to 10% B at 27 min. Finally, 10% B was maintained until 35 min. The solvent was used at a flow rate of 200 µl/min, and the columns temperature was kept constant at 25°C. The mass spectrometer operated in negative ion mode, and settings of the HESI probe were as follows: sheath gas flow rate at 35, auxiliary gas flow rate at 14. Spray voltage was set at 2.9 kV, temperature of the capillary at 300°C and S‐lens RF level at 50. A full scan (resolution of 240,000 and scan range of m/z 70–750) was applied. For the data analysis, we used Compound Discoverer 3.0 (Thermo Scientific) a software platform for analyzing metabolites. Identification of metabolites was done with in‐house libraries as well as with third party databases.
Endotoxemia experiments
24 h before LPS injections, mice were put under normoxic or hypoxic conditions. Female C57BL/6J mice were injected i.p. with PBS or DEX (10 mg/kg) followed by a lethal dose of LPS (LD100 14.5 mg/kg in normoxia, 5 mg/kg in hypoxia) dissolved in sterile PBS to induce endotoxin shock. Female ADX mice were injected i.p. with a lethal LPS dose of 0.05 mg/kg. For survival experiments, rectal body temperature and lethality were monitored. For experiments aimed to isolate blood and organ samples, mice were injected i.p. with LPS or PBS in control mice. For biochemical analysis, mice were euthanized via cervical dislocation at indicated time points and plasma and organs were isolated.
Biochemical analysis
Analysis of mouse plasma aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatinine, troponin, creatine kinase (CK), and lactate dehydrogenase (LDH) levels were kindly provided to us by the University Hospital of Ghent. Blood glucose and ketone body levels were measured in tail blood with the use of OneTouch Verio glucose meter (LifeScan) and Freestyle Precision Neo meter (Abbott), respectively. ACTH (Antibodies‐online GmbH), corticosterone (Tebu‐Bio), and free fatty acid (Abnova) were measured in mouse plasma with the use of colorimetric assays according to manufacturer’s instructions.
Western blot analysis
Liver
For the detection of GR, total protein was isolated out of snap frozen liver with RIPA lysis buffer, supplemented with protease inhibitor cocktail (Roche). Protein samples containing 50 µg of protein were separated by electrophoresis on an 8% gradient SDS‐polyacrylamide gel and transferred to nitrocellulose filters (pore size 0.45 µm). After blocking the membranes with a ½ dilution of Starting Block/PBST 0.1% (Thermo Fisher Scientific), membranes were incubated overnight at 4°C with primary antibodies against GRα/β (1:1,000, G5, sc‐393232; Santa Cruz), and β‐actin (1:5000; Life Technologies Europe) as an internal control. Blots were washed with PBST 0.1% and then incubated for 1 h at room temperature with Amersham ECL anti‐mouse antibody (1:2,000, NA931, GE Healthcare Life Sciences). Immunoreactive bands were visualized and quantified using an Amersham Imager 600 (GE Healthcare Life Sciences).
Hypothalamus
For Western blot, hypothalamus tissue samples were lysed for protein extraction using RIPA buffer with inhibitors: PMSF, protease inhibitors, sodium orthovanadate (all from Chem Cruz). The tissue samples were lysed under hypoxic conditions (3% O2) in the whitley hypoxia chamber. For lysis, samples were incubated for 30 min on ice and with periodic vortexing followed by centrifugation for removal of debris. The samples were aliquoted and then stored at −80°C until further processing. The protein concentrations were measured using the Bicinchoninic acid assay (BCA) (Thermo Fisher). 25 µg of protein from each sample was mixed with LDS sample buffer and 5% mercaptoethanol and denaturation was performed at 99°C for 5 min. Following denaturation, proteins were separated on a 4–12% Bis‐Tris protein gels (Thermo Fisher) and transferred to the membrane. The membranes were blocked with 5% Milk with TBST. After washing, the membranes were incubated with primary antibodies overnight at 4°C. The primary antibodies used were as follows: HIF‐1α (Cayman chemical) or HIF‐2α (Abcam), or later with Vinculin (Cell signaling) after stripping in 5% Milk with TBST. After overnight incubation with primary antibodies, the membranes were washed followed by incubated with secondary Rabbit IgG HRP (R&D systems) for 1 h at room temperatures. The membranes were then subjected to imaging using Fusion Fx (Peqlab, VWR). The quantifications were performed using Fiji (ImageJ distribution 1.52K).
Immunohistochemistry
Liver
For the evaluation of the transfection efficiency of the HRE‐luciferase reporter plasmid, excised liver from PBS and HRE‐Luc‐injected mice were fixed in 4% paraformaldehyde overnight at 4°C, dehydrated, and embedded in paraffin. Tissue sections of 5 µm were cut. For the luciferase staining, tissue sections were dewaxed, incubated with antigen retrieval buffer (1x citrate buffer pH6, Vector H‐3300) for 20 min at boiling temperature in a PickCell electric pressure cooker and cooled down. Peroxidase blocking was done with 3% H2O2 in methanol for 10 min. Blocking buffer (5% goat serum in 1% bovine serum albumin (BSA) in PBS) was added to the slides for 30 min at room temperature. Primary antibody against luciferase (MA1‐16880, Thermo Fisher Scientific) was diluted 1:1,000 in 1% BSA in PBS and incubated overnight at 4°C. Then, slides were incubated with the goat anti‐mouse secondary HRP antibody (MP‐7452, Vector Laboratories, 1:5 in PBS) for 30 min. To signal of the staining was increased via incubating the slides with the Tyramide Signal Amplification (TSA) kit (1:75, Perkin Elmer) for 10 min, followed by 30 min incubation with ABC (PK‐6100, Vector Laboratories). Counterstaining was done with Hoechst reagent (Sigma‐Aldrich NV, 1:1,000 in PBS). Pictures were taken with an Olympus BX51 Discussion Microscope.
Hypothalamus
For immunofluorescence staining of HIF1α and HIF2α on hypothalamus samples, OCT embedded hypothalamus tissue was cut into 7‐µm sections at −20°C using a cryotome and stored at −20°C until further processing. For staining, the sections were dried for 20 min at room temperature. Fixed with paraformaldehyde, washed with phosphate‐buffered saline (PBS), and with PBS containing 0.1% Tween‐20, blocked with 5% normal goat serum followed by primary antibody staining with HIF1α (Cayman chemical) or HIF2α (Novus biologicals) for 3 days at 4°C and subsequent secondary antibody staining. Slides were mounted in fluorescent mounting medium and stored at 4°C until analysis. Fluorescent images were acquired on an ApoTome II Colibri (Carl Zeiss, Jena, Germany), scale bar (50 µm). Images were analyzed using either Zen software (Carl Zeiss, Jena, Germany) or Fiji (ImageJ distribution 1.52K).
ChIP‐sequencing
ChIP‐seq was used to investigate the GR DNA‐binding in conditions of normoxia and hypoxia using biological triplicates for all conditions, for a total of 18 analyzed samples (3 N_PBS, 3 N_DEX, 3 H_PBS_6 h, 3 H_DEX_6 h, 3 H_PBS_24 h, and 3 H_DEX_24 h) and input controls. All conditions were processed in one run, which included the immunoprecipitation (IP) and sequencing. Therefore, all comparisons listed in the manuscript are considered “within experiment” comparisons. Snap frozen liver samples were derived from mice that were injected with PBS or 10 mg/kg DEX in normoxic conditions and 6 or 24 h after hypoxia. 200 mg of tissue was homogenized in PBS and then crosslinked with 2% formaldehyde in PBS for 20 min at RT while rotating. This reaction was stopped by adding 125 mM glycine for 10 min at RT. Tissue was then collected in ice‐cold PBS and subsequently lysed in lysis buffer (0.1% SDS, 1% Triton X‐100, 0.15 M NaCl, 1 mM EDTA and 20 mM Tris pH8, supplemented with protease inhibitors). Lysates were sonicated at 4°C to yield 200–800 bp DNA fragments. IP was performed on 100 µl lysates diluted 1:3 in incubation buffer (0.15% SDS, 1% Triton X‐100, 0.15 M NaCl, 1 mM EDTA, and 20 mM HEPES) with 5 µg of rabbit anti‐GR antibody (H300, sc‐8992; Santa Cruz) or normal rabbit IgG (10500C; Invitrogen) as negative control at 4°C overnight. After 2 h, BSA‐blocked nProtein sepharose beads (GE Healthcare) were added to the lysates. The following day, beads were washed in WBI, WBII, LiCl buffer and twice in EDTA/HEPES buffer (0.5 M EDTA and 1 M HEPES). Chromatin was eluted in 200 µl elution buffer (0.1 M NaHCO3 and 10% SDS) supplemented with proteinase K. Next, proteins were decrosslinked by raising the incubation temperature to 65°C for 16 h and DNA purified using the PCR purification kit (Qiagen), and eluting in 50 µl of Qiagen Elution Buffer. Fifteen ng of purified DNA was used to generate ChIP‐seq libraries according to manufacturer’s protocol using NEBNext Ultra II DNA Library Prep Kit (E7645L, NEB). Libraries were single‐end sequencing by Illumina NextSeq 500. The ChIP‐seq experiment was performed in collaboration with the group of Jorma J. Palvimo (Finland), where the IP pulldown, library preparation and sequencing were executed. Spike‐in samples were not used based on their experience and the fact that all samples were processed within one experiment. Samples were normalized for sequencing depth, background signal, and scaled to 1e7 reads per sample per condition. These scaled data were used for all visualizations. The obtained reads were mapped to the mm10 reference genome using the bwa software (bwa mem). Peak calling was done using HOMER v4.11 (Heinz et al, 2010) requiring fourfold enrichment over input and local background and at least 50 tags per peak. To increase our data reliability and counter variability, only bound regions found in all 3 replicates of a condition were considered in this work. Motif finding, annotation, visualizations, and detection of differentially bound regions were also done with HOMER (find motifs genome, annotate peaks, and find differential peak replicates). Peak region motif finding was done 1 kb upstream to 50 bp downstream of the TSS.
Statistics
Data were expressed as means ± standard errors of the means (SEM). Statistical significance was evaluated with two‐way unpaired Student’s t‐test and one‐way or two‐way ANOVA in GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA). If applicable, one‐way and two‐way ANOVA analysis were followed by post‐hoc analysis to correct for multiple testing during the pairwise multiple comparisons using the Tukey’s test or the Šídák’s multiple comparisons test for one‐way and two‐way ANOVA, respectively. The results of the two‐way ANOVA tests can be found in Dataset EV3. Fold changes or ratios were log (Y) transformed before statistical analysis. Survival curves were subjected to the Log‐Rank (Mantel‐Cox) test to investigate whether statistical significance could be observed during different groups. The Pearson correlation coefficient (PCC) was determined using the R statistics software and the correlation coefficient function.
Author contributions
TV conceived and performed the experiments and co‐wrote the manuscript. ST performed all bio‐informatics analysis of RNA sequencing and ChIP‐sequencing data. LVW, KVL, JV, CW, LD, GC, KDB, and LN performed experiments. ME, SD, JDB, and JVB provided general technical assistance. WVB provided a hypoxia chamber and performed part of the hypoxia experiments. BG did the metabolomics experiments. BW and DW did Western blot analysis and immunohistochemistry for HIF1a and HIF2a on hypothalamus samples. KL performed the immunohistochemistry for luciferase on the liver samples. JJP and VP performed the processing of the samples for ChIP sequencing. RB and CL supervised the research and co‐wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View
Source Data for Figure 5D
Acknowledgements
The authors wish to thank Jan Tuckermann, Ben Wielockx, and Karolien De Bosscher for providing GRdim/dim mice, HIF1afl/fl and HIF2afl/fl mice, and PPARafl/fl mice, respectively. We thank Joke Vanden Berghe and animal house caretakers for animal care. We acknowledge the VIB Nucleomics Core for RNA sequencing. The EMBL GeneCore is acknowledged for ChIP sequencing. Research in the author’s laboratories was funded by the Agency for Innovation of Science and Technology in Flanders (IWT), the Research Council of Ghent University (GOA grant BOF19‐GOA‐004 and Methusalem grant BOF.MET.2021.0001.0), the Research Foundation Flanders (FWO‐Vlaanderen Research grants G025220N and G014921N and SBO‐grant S002721N and S003122N), and Flanders Institute for Biotechnology (VIB).
EMBO reports (2022) 23: e53083.
Data availability
RNA‐seq data: Gene expression. Deposited at the National Center for Biotechnology Information Gene Expression Omnibus public database (http://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE162100 and GSE162155 for RNA‐seq data 6 and 24 h after hypoxia and DEX stimulation in liver, respectively, accession number GSE162673 for RNA‐seq data of hypothalamus samples (2 h hypoxia). ChIP‐seq data: accession number GSE163242.
References
- Annane D, Sébille V, Charpentier C, Bollaert PE, François B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troché G et al (2002) Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. J Am Med Assoc 288: 862–871 [DOI] [PubMed] [Google Scholar]
- Basnyat B, Starling JM (2015) Infectious diseases at high altitude. Microbiol Spectr 3: 1–7 [DOI] [PubMed] [Google Scholar]
- Basu M, Sawhney RC, Kumar S, Pal K, Prasad R, Selvamurthy W (2002a) Glucocorticoids as prophylaxis against acute mountain sickness. Clin Endocrinol 57: 761–767 [DOI] [PubMed] [Google Scholar]
- Basu M, Sawhney RC, Kumar S, Pal K, Prasad R, Selvamurthy W (2002b) Hypothalamic‐pituitary‐adrenal axis following glucocorticoid prophylaxis against acute mountain sickness. Horm Metab Res 34: 318–324 [DOI] [PubMed] [Google Scholar]
- Batie M, del Peso L, Rocha S (2018) Hypoxia and chromatin: a focus on transcriptional repression mechanisms. Biomedicines 6: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schütz G, Beug H (1999) The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev 13: 2996–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beishuizen A, Thijs LG (2003) Endotoxin and the hypothalamo‐pituitary‐adrenal (HPA) axis. J Endotoxin Res 9: 3–24 [DOI] [PubMed] [Google Scholar]
- Belanger AJ, Luo Z, Vincent KA, Akita GY, Cheng SH, Gregory RJ, Jiang C (2007) Hypoxia‐inducible factor 1 mediates hypoxia‐induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator‐activated receptor α/retinoid X receptor. Biochem Biophys Res Commun 364: 567–572 [DOI] [PubMed] [Google Scholar]
- Bellingham DL, Sar M, Cidlowski JA (1992) Ligand‐dependent down‐regulation of stably transfected human glucocorticoid receptors is associated with the loss of functional glucocorticoid responsiveness. Mol Endocrinol 6: 2090–2102 [DOI] [PubMed] [Google Scholar]
- Berger MM, Macholz F, Sareban M, Schmidt P, Fried S, Dankl D, Niebauer J, Bärtsch P, Mairbäurl H (2017) Inhaled budesonide does not prevent acute mountain sickness after rapid ascent to 4559 m. Eur Respir J 50: 1–4 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, Muglia LJ (2007) Macrophage glucocorticoid receptors regulate Toll‐like receptor 4‐mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 109: 4313–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleymehl K, Pérez‐Gómez A, Omura M, Moreno‐Pérez A, Macías D, Bai Z, Johnson RS, Leinders‐Zufall T, Zufall F, Mombaerts P (2016) A sensor for low environmental oxygen in the mouse main olfactory epithelium. Neuron 92: 1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolsoni‐Lopes A, Alonso‐Vale MIC (2015) Lipolysis and lipases in white adipose tissue – An update. Arch Endocrinol Metab 59: 335–342 [DOI] [PubMed] [Google Scholar]
- Bougarne N, Paumelle R, Caron S, Hennuyer N, Mansouri R, Gervois P, Staels B, Haegeman G, De Bosscher K (2009) PPARα blocks glucocorticoid receptor α‐mediated transactivation but cooperates with the activated glucocorticoid receptor α for transrepression on NF‐κB. Proc Natl Acad Sci USA 106: 7397–7402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruder ED, Taylor JK, Kamer KJ, Raff H (2008) Development of the ACTH and corticosterone response to acute hypoxia in the neonatal rat. Am J Physiol Regul Integr Comp Physiol 295: 1195–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J (2013) Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AJ, Ortega FE, Riegler J, Madison DV, Krasnow MA (2015) Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 527: 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H‐Y, Lin Y‐M, Chung H‐C, Lang Y‐D, Lin C‐J, Huang J, Wang W‐C, Lin F‐M, Chen Z, Huang H‐D et al (2012) MiR‐103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res 72: 3631–3641 [DOI] [PubMed] [Google Scholar]
- Cockman ME, Masson N, Mole DR, Jaakkola P, Chang G, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH (2000) Hypoxia inducible factor‐a binding and ubiquitylation by the von hippel‐lindau tumor suppressor protein *. J Biol Chem 275: 25733–25741 [DOI] [PubMed] [Google Scholar]
- Cummins EP, Keogh CE, Crean D, Taylor CT (2016) The role of HIF in immunity and inflammation. Mol Aspects Med 47‐48: 24–34 [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Bruscoli S, Mazzon E, Crisafulli C, Donato V, Di Paola R, Velardi E, Esposito E, Nocentini G, Riccardi C (2008) Peroxisome proliferator‐activated receptor‐α contributes to the anti‐inflammatory activity of glucocorticoids. Mol Pharmacol 73: 323–337 [DOI] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Jacobson L, Levin N, Cascio CS, Shinsako J (1987) Characterization of corticosterone feedback regulation of ACTH secretion. Ann N Y Acad Sci 512: 402–414 [DOI] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Levin N, Walker CD, Bradbury MJ, Suemaru S, Scribner KS (1994) Corticosteroids and the control of function in, the hypothalamo‐pituitary‐adrenal (HPA) axis. Ann N Y Acad Sci 746: 22–31 [DOI] [PubMed] [Google Scholar]
- Daniel Bird A, McDougall ARA, Seow B, Hooper SB, Cole TJ (2015) Minireview: glucocorticoid regulation of lung development: lessons learned from conditional GR knockout mice. Mol Endocrinol 29: 158–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Beck IM, Ratman D, Vanden BW, Libert C (2016) Activation of the glucocorticoid receptor in acute inflammation: the SEDIGRAM concept. Trends Pharmacol Sci 37: 4–16 [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G (2003) The interplay between the glucocorticoid receptor and nuclear factor‐κB or activator protein‐1: molecular mechanisms for gene repression. Endocr Rev 24: 488–522 [DOI] [PubMed] [Google Scholar]
- Dejager L, Pinheiro I, Puimège L, Fan YD, Gremeaux L, Vankelecom H, Libert C (2010) Increased glucocorticoid receptor expression and activity mediate the LPS resistance of SPRET/EI mice. J Biol Chem 285: 31073–31086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendoncker K, Timmermans S, Vandewalle J, Eggermont M, Lempiäinen J, Van Hamme E, Dewaele S, Vandevyver S, Ballegeer M, Souffriau J et al (2019) TNF‐α inhibits glucocorticoid receptor‐induced gene expression by reshaping the GR nuclear cofactor profile. Proc Natl Acad Sci USA 116: 12942–12951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Poellinger L, Gustafsson JÅ, Okret S (1988) Regulation of glucocorticoid receptor expression: evidence for transcriptional and posttranslational mechanisms. Mol Endocrinol 2: 1256–1264 [DOI] [PubMed] [Google Scholar]
- Escoter‐Torres L, Greulich F, Quagliarini F, Wierer M, Uhlenhaut NH (2020) Anti‐inflammatory functions of the glucocorticoid receptor require DNA binding. Nucleic Acids Res 48: 8393–8407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke K, Gassmann M, Wielockx B (2013) Erythrocytosis: the HIF pathway in control. Blood 122: 1122–1128 [DOI] [PubMed] [Google Scholar]
- Garcia‐alvarez M, Marik P, Bellomo R (2014) Sepsis‐associated hyperlactatemia. Crit Care 18: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales GF, Tapia V (2013) Association of high altitude‐induced hypoxemia to lipid profile and glycemia in men and women living at 4100m in the Peruvian Central Andes. Endocrinol Y Nutr 60: 79–86 [DOI] [PubMed] [Google Scholar]
- Grabacka M, Pierzchalska M, Dean M, Reiss K (2016) Regulation of ketone body metabolism and the role of PPARα. Int J Mol Sci 17: 2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guia RM, Herzig S (2015) How do glucocorticoids regulate lipid metabolism? Adv Exp Med Biol 872: 127–144 [DOI] [PubMed] [Google Scholar]
- Guo B, Huang X, Cooper S, Broxmeyer HE (2017) Glucocorticoid hormone‐induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nat Med 23: 424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell CS, Rowson SA, Neigh GN (2015) Pharmacological stimulation of hypoxia inducible factor‐1α facilitates the corticosterone response to a mild acute stressor. Neurosci Lett 600: 75–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple Combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenniskens M, Weckx R, Dufour T, Vander PS, Pauwels L, Derde S, Téblick A, Güiza F, Van Den Berghe G, Langouche L (2018) The hepatic glucocorticoid receptor is crucial for cortisol homeostasis and sepsis survival in humans and male mice. Endocrinology 159: 2790–2802 [DOI] [PubMed] [Google Scholar]
- Jungermann K, Kietzmann T (2000) Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology 31: 255–260 [DOI] [PubMed] [Google Scholar]
- Kanczkowski W, Sue M, Bornstein SR (2016) Adrenal gland microenvironment and its involvement in the regulation of stress‐induced hormone secretion during sepsis. Front Endocrinol 7: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W (1999) Peroxisome proliferator‐activated receptor alpha mediates the adaptive response to fasting. J Clin Invest 103: 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiers D, Wielockx B, Peters E, van Eijk LT, Gerretsen J, John A, Janssen E, Groeneveld R, Peters M, Damen L et al (2018) Short‐term hypoxia dampens inflammation in vivo via enhanced adenosine release and adenosine 2B receptor stimulation. EBioMedicine 33: 144–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Foong D, Cooper MS, Seibel MJ, Zhou H (2018) Comparison of blood sampling methods for plasma corticosterone measurements in mice associated with minimal stress‐related artefacts. Steroids 135: 69–72 [DOI] [PubMed] [Google Scholar]
- Kitsteiner JM, Whitworth JD, Nashelsky J (2011) Preventing acute mountain sickness. Am Fam Physician 84: 398–400 [PubMed] [Google Scholar]
- Kleiman A, Hubner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, Bauer M, Schmid W, Schutz G, Libert C et al (2012) Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin‐1 in macrophages. FASEB J 26: 722–729 [DOI] [PubMed] [Google Scholar]
- Kodama T, Shimizu N, Yoshikawa N, Makino Y, Ouchida R, Okamoto K, Hisada T, Nakamura H, Morimoto C, Tanaka H (2003) Role of the glucocorticoid receptor for regulation of hypoxia‐dependent gene expression *. J Biol Chem 278: 33384–33391 [DOI] [PubMed] [Google Scholar]
- Kuo T, McQueen A, Chen T‐C, Wang J‐C (2015) Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol 872: 99–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SST, Gonzalez FJ (1996) Targeted disruption of the peroxisome proliferator‐activated receptor alpha gene, PPARalpha. Ann N Y Acad Sci 804: 524–529 [DOI] [PubMed] [Google Scholar]
- Leonard MO, Godson C, Brady HR, Taylor CT (2005) Potentiation of glucocorticoid activity in hypoxia through induction of the glucocorticoid receptor. J Immunol 174: 2250–2257 [DOI] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP (1999) A critical role for the peroxisome proliferator‐activated receptor α (PPARα) in the cellular fasting response: the PPARα‐null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA 96: 7473–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Bosch‐Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, Polotsky VY (2006) Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia‐inducible factor‐1α. Physiol Genomics 25: 450–457 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Libert C, Brouckaert P, Shaw A, Fiers W (1990) Induction of interleukin 6 by human and murine recombinant interleukin 1 in mice. Eur J Immunol 20: 691–694 [DOI] [PubMed] [Google Scholar]
- Libert C, Van BS, Brouckaert P, Shaw A, Fiers W, Libert C, Bladel VAN, Brouckaert P, Shaw A (1991) Involvement of the liver, but not of IL‐6, in IL‐1‐induced desensitization to the lethal effects of tumor necrosis factor. This information is current as Why The JI ? Submit online. • Rapid Reviews ! 30 days * from submission to initial decision • No. J Immunol 146: 2625–2632 [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi D, Santhakumar K, Markham E, Li N, Storbeck KH, Krone N, Cunliffe VT, Van Eeden FJM (2020) Bidirectional crosstalk between hypoxia‐inducible factor and glucocorticoid signalling in zebrafish larvae. PLoS Genet 16: 1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry JD, Foster DW (1980) Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem 49: 395–420 [DOI] [PubMed] [Google Scholar]
- McKeown SR (2014) Defining normoxia, physoxia and hypoxia in tumours ‐ Implications for treatment response. Br J Radiol 87: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minneci PC, Deans KJ, Eichacker PQ, Natanson C (2009) The effects of steroids during sepsis depend on dose and severity of illness: an updated meta‐analysis. Clin Microbiol Infect 15: 308–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylonis I, Simos G, Paraskeva E (2019) Hypoxia‐inducible factors and the regulation of lipid metabolism. Cells 8: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Kataoka N (2019) Regulation of gene expression under hypoxic conditions. Int J Mol Sci 20: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama Y, Goda N, Kanai M, Niwa D, Osanai K, Yamamoto YU, Senoo‐Matsuda N, Johnson RS, Miura S, Kabe Y et al (2012) HIF‐1α induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J Hepatol 56: 441–447 [DOI] [PubMed] [Google Scholar]
- Park JY, Hwang TK, Park HK, Ahn RS (2014) Differences in cardiovascular and hypothalamic‐pituitary‐adrenal axis functions between high‐altitude visitors and natives during a trek on the Annapurna circuit. Neuroendocrinology 99: 130–138 [DOI] [PubMed] [Google Scholar]
- Pavek P (2016) Pregnane X receptor (PXR)‐mediated gene repression and cross‐talk of PXR with other nuclear receptors via coactivator interactions. Front Pharmacol 7: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff H, Jacobson L, Cullinan WE (2007) Augmented hypothalamic corticotrophin‐releasing hormone mRNA and corticosterone responses to stress in adult rats exposed to perinatal hypoxia. J Neuroendocr 19: 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin EB, Rha J, Selak MA, Unger TL, Keith B, Liu Q, Haase VH (2009) Hypoxia‐inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol 29: 4527–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratman D, Mylka V, Bougarne N, Pawlak M, Caron S, Hennuyer N, Paumelle R, De Cauwer L, Thommis J, Rider MH et al (2016) Chromatin recruitment of activated AMPK drives fasting response genes co‐controlled by GR and PPARα. Nucleic Acids Res 44: 10539–10553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P et al (1998) DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93: 531–541 [DOI] [PubMed] [Google Scholar]
- Rock PB, Johnson TS, Larsen RF, Fulco CS, Trad LA, Cymerman A (1989) Dexamethasone as prophylaxis for acute mountain sickness. Chest 95: 568–573 [DOI] [PubMed] [Google Scholar]
- Rose AJ, Herzig S (2013) Metabolic control through glucocorticoid hormones: an update. Mol Cell Endocrinol 380: 65–78 [DOI] [PubMed] [Google Scholar]
- Rose AJ, Vegiopoulos A, Herzig S (2010) Role of glucocorticoids and the glucocorticoid receptor in metabolism: insights from genetic manipulations. J Steroid Biochem Mol Biol 122: 10–20 [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5: 343–354 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2011) Regulation of metabolism by hypoxia‐inducible factor 1. Cold Spring Harb Symp Quant Biol 76: 347–353 [DOI] [PubMed] [Google Scholar]
- Sevilla LM, Pérez P (2018) Glucocorticoid receptor signaling in skin barrier function. Intech 13: 23–44 [Google Scholar]
- Spiga F, Walker JJ, Terry JR, Lightman SL (2014) HPA axis‐rhythms. Compr Physiol 4: 1273–1298 [DOI] [PubMed] [Google Scholar]
- Steidl C, Leimeister C, Klamt B, Maier M, Nanda I, Dixon M, Clarke R, Schmid M, Gessler M (2000) Characterization of the human and mouse HEY1, HEY2, and HEYL genes: cloning, mapping, and mutation screening of a new bHLH gene family. Genomics 66: 195–203 [DOI] [PubMed] [Google Scholar]
- Sterne JAC, Murthy S, Diaz JV, Slutsky AS, Villar J, Angus DC, Annane D, Azevedo LCP, Berwanger O, Cavalcanti AB et al (2020) Association between administration of systemic corticosteroids and mortality among critically Ill patients with COVID‐19: a meta‐analysis. J Am Med Assoc 324: 1330–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A (2011) High‐altitude illnesses: physiology, risk factors, prevention, and treatment. Rambam Maimonides Med J 2: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans S, Souffriau J, Libert C (2019) A general introduction to glucocorticoid biology. Front Immunol 10: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hübner N, Evans RM (2013) Insights into negative regulation by the glucocorticoid receptor from genome‐wide profiling of inflammatory cistromes. Mol Cell 49: 158–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bogaert T, Vandevyver S, Dejager L, Van Hauwermeiren F, Pinheiro I, Petta I, Engblom D, Kleyman A, Schütz G, Tuckermann J et al (2011) Tumor necrosis factor inhibits glucocorticoid receptor function in mice: a strong signal toward lethal shock. J Biol Chem 286: 26555–26567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Looveren K, Timmermans S, Vanderhaeghen T, Wallaeys C, Ballegeer M, Souffriau J, Eggermont M, Vandewalle J, Van WL, De BK et al (2020) Glucocorticoids limit lipopolysaccharide‐induced lethal inflammation by a double control system. EMBO Rep 32: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wyngene L, Vandewalle J, Libert C (2018) Reprogramming of basic metabolic pathways in microbial sepsis: therapeutic targets at last? EMBO Mol Med 10: e8712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderhaeghen T, Beyaert R, Libert C (2021) Bidirectional crosstalk between hypoxia inducible factors and glucocorticoid signalling in health and disease. Front Immunol 12: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle J, Libert C (2020) Glucocorticoids in sepsis: to be or not to be. Front Immunol 11: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle J, Luypaert A, De BK, Libert C (2018) Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab 29: 42–54 [DOI] [PubMed] [Google Scholar]
- Vandewalle J, Timmermans S, Paakinaho V, Vancraeynest L, Dewyse L, Vanderhaeghen T, Wallaeys C, Van Wyngene L, Van Looveren K, Nuyttens L et al (2021) Combined glucocorticoid resistance and hyperlactatemia contributes to lethal shock in sepsis. Cell Metab 33: 1–14 [DOI] [PubMed] [Google Scholar]
- Vegiopoulos A, Herzig S (2007) Glucocorticoids, metabolism and metabolic diseases. Mol Cell Endocrinol 275: 43–61 [DOI] [PubMed] [Google Scholar]
- Vettori A, Greenald D, Wilson GK, Peron M, Facchinello N, Markham E, Sinnakaruppan M, Matthews LC, McKeating JA, Argenton F et al (2017) Glucocorticoids promote Von Hippel Lindau degradation and Hif‐1α stabilization. Proc Natl Acad Sci USA 114: 9948–9953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AE, Huck G, Stiehl DP, Jelkmann W, Hellwig‐Bürgel T (2008) Dexamethasone impairs hypoxia‐inducible factor‐1 function. Biochem Biophys Res Commun 372: 336–340 [DOI] [PubMed] [Google Scholar]
- Wang JC, Gray NE, Kuo T, Harris CA (2012) Regulation of triglyceride metabolism by glucocorticoid receptor. Cell Biosci 2: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XL, Suzuki R, Lee K, Tran T, Gunton JE, Saha AK, Patti M‐E, Goldfine A, Ruderman NB, Gonzalez FJ et al (2009) Ablation of ARNT/HIF1β in liver alters gluconeogenesis, lipogenic gene expression, and serum ketones. Cell Metab 9: 428–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ma Y‐Y, Song X‐L, Cai H‐Y, Chen J‐C, Song L‐N, Yang R, Lu J (2012) Upregulations of glucocorticoid‐induced leucine zipper by hypoxia and glucocorticoid inhibit proinflammatory cytokines under hypoxic conditions in macrophages. J Immunol 188: 222–229 [DOI] [PubMed] [Google Scholar]
- Wang Y, Nakajima T, Gonzalez FJ, Tanaka N (2020) PPARs as metabolic regulators in the liver: lessons from liver‐specific PPAR‐null mice. Int J Mol Sci 21: 2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielockx B, Meneses A (2016) PHD2: from hypoxia regulation to disease progression. Hypoxia 4: 53–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Berry A, Sauer C, Baxter M, Donaldson IJ, Forbes K, Donn R, Matthews L, Ray D (2020) Hypoxia regulates GR function through multiple mechanisms involving microRNAs 103 and 107. Mol Cell Endocrinol 518: 111007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuhara M, Ohama T, Matsuki N, Saito H, Shiga J, Inoue K, Kurokawa K, Teramoto T (1991) Induction of fatty liver by fasting in suncus. J Lipid Res 32: 887–891 [PubMed] [Google Scholar]
- Zhang C, Qiang Q, Jiang Y, Hu L, Ding X, Lu Y, Hu G (2015) Effects of hypoxia inducible factor‐1α on apoptotic inhibition and glucocorticoid receptor downregulation by dexamethasone in AtT‐20 cells. BMC Endocr Disord 15: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C‐R, Chen G‐Z, Yu J, Qin J, Song P, Bian S‐Z, Xu B‐D, Tang X‐G, Huang Y‐T, Liang X et al (2014) Inhaled budesonide and oral dexamethasone prevent acute mountain sickness. Am J Med 127: 1001–1009.e2 [DOI] [PubMed] [Google Scholar]
- Zhu X, Liu Y, Li N, He Q (2020) Inhaled budesonide for the prevention of acute mountain sickness: a meta‐analysis of randomized controlled trials. Am J Emerg Med 38: 1627–1634 [DOI] [PubMed] [Google Scholar]