

SUMMARY
Pyramidal neurons in the anterior cingulate cortex (ACC), a prefrontal region involved in processing the affective components of pain, display hyperexcitability in chronic neuropathic pain conditions, and their silencing abolishes hyperalgesia. We show that dopamine, through D1 receptor (D1R) signaling, inhibits pyramidal neurons of mouse ACC by modulation of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Activation of Gs-coupled D1R by dopamine induces the opening of HCN channels at physiological membrane potentials, driving a significant decrease in input resistance and excitability. Systemic L-DOPA in chronic neuropathic mice rescues HCN channel activity, normalizes pyramidal excitability in ACC, and blocks mechanical and thermal allodynia. Moreover, microinjection of a selective D1R agonist in the ACC relieves the aversiveness of ongoing neuropathic pain, while an ACC D1R antagonist blocks gabapentin- and lidocaine-evoked antinociception. We conclude that dopaminergic inhibition via D1R in ACC plays an analgesic role in physiological conditions and is decreased in chronic pain.
In brief
Lançon et al. show that dopamine inhibits pyramidal neurons in the anterior cingulate cortex (ACC) via D1R signaling and subsequent opening of HCN channels. Activation of ACC D1R provides analgesia and is necessary for relief of ongoing pain. Supplementing supraspinal dopamine normalizes ACC pyramidal hyperexcitability and reduces neuropathic hyperalgesia.
Graphical Abstract
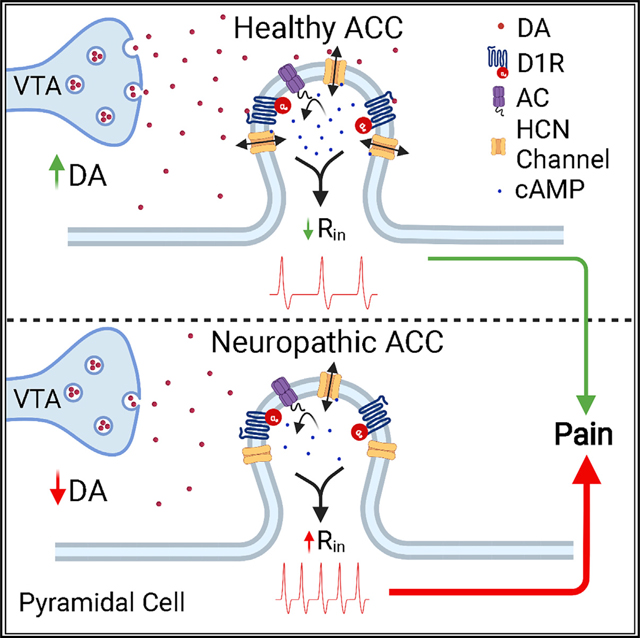
INTRODUCTION
Damage to peripheral nerves induces maladaptive changes in both peripheral and central nociceptive pathways that may increase the risk of chronic pain (Campbell and Meyer, 2006; Decosterd and Woolf, 2000). These maladaptive changes can elicit ongoing pain as well as allodynia, a pathological sensitivity to innocuous stimuli, and hyperalgesia, an increased sensitivity to painful stimuli (von Hehn et al., 2012). These hallmark symptoms of chronic pain are linked to an increase in pyramidal cell excitability in layer 2/3 of the anterior cingulate cortex (ACC), a region of the medial prefrontal cortex (mPFC) that is implicated in the affective components of pain in both human and animal studies (Cordeiro Matos et al., 2015; Eto et al., 2011; Kamiński et al., 2017; Mhalla et al., 2010; Rainville et al., 1997). A recent meta-analysis of brain imaging data showed that the ACC is the most consistently activated region in patients suffering from chronic pain, consistent with its critical role in higher order pain processing (Jensen et al., 2016). Additionally, in experimental rodent models of inflammatory and neuropathic pain, optogenetic inhibition of hyperexcitable pyramidal cells has been reported to produce analgesic effects, including decreased tactile allodynia and conditioned place preference, respectively (Kang et al., 2015; Sellmeijer et al., 2018). Collectively, these findings support the conclusion that the ACC is a region involved in regulatory top-down pain processing. Despite these findings, very little is known about how pyramidal neurons in the ACC are modulated in physiological or pathological states.
Hyperpolarization-activated cyclic-nucleotide-gated (HCN) channels control the excitability of many central neurons by regulating input resistance (Poolos et al., 2002). These cyclic AMP (cAMP)-gated cation channels are highly expressed in the mPFC and have decreased open-channel probabilities at physiological membrane potentials in chronic pain conditions (Cordeiro Matos et al., 2015; Notomi and Shigemoto, 2004; Santello et al., 2017). This reduction in HCN-mediated currents (Ih) results in increased input resistance and therefore excitability (Cordeiro Matos et al., 2015; Santello et al., 2017). Recent findings indicate that HCN channels are colocalized with Gs-coupled D1 dopamine (DA) receptors on dendritic spines of mPFC pyramidal neurons (Paspalas et al., 2013; Stoof and Kebabian, 1981).
DAergic modulation of the ACC is particularly interesting given the known role of the other major monoamines, including norepinephrine and serotonin, in prefrontal modulation in chronic pain states (Cordeiro Matos et al., 2015; Santello et al., 2017). In view of the well-documented effects that both norepinephrine and serotonin have on HCN channel function in the ACC and consequently on pathological cortical excitability, we hypothesized that DA, through postsynaptic D1 receptors and opening of HCN channels, can decrease pyramidal excitability and consequently influence top-down pain pathways by inhibition of pyramidal cells.
In the present study, we investigated the neuromodulatory effects of DA on intrinsic cellular excitability of pyramidal cells in the ACC, and we provide electrophysiological evidence of a functional interaction between D1 receptors and HCN channels. We also examined whether an increase in DAergic input to the ACC can reduce the pathological cellular dysfunction of the ACC observed in neuropathic conditions and alleviate the symptoms of chronic pain. We found that a tonic increase in cortical DA, via long-term systemic administration of L-DOPA and carbidopa, relieves neuropathy-induced allodynia, while acute intracortical injection of a selective D1 receptor (D1R) agonist drives conditioned place preference selectively in neuropathic animals, indicating the relief of the aversive state of ongoing pain. Furthermore, we provide evidence that pain relief evoked by gabapentin and lidocaine also requires D1R signaling in the ACC.
RESULTS
DAergic inhibition of pyramidal cells in ACC
Following application of DA (10–50 μM) by perfusion for 15 min, 50% (n = 9 of 18) and 54% (n = 7 of 13) of pyramidal cells in layer 2/3 of the ACC were inhibited relative to baseline (data not shown). The dichotomy in response to DA between responsive and non-responsive cells was obvious, as responsive cells showed a marked inhibition of −40.1% ± 10.1% in contrast to non-responsive cells, which expressed a +6.9% ± 4.5% excitation relative to baseline values following 50 μM DA application (Figure 1A; n = 18; unpaired t test: t(14) = 4.6; p = 0.0004). Criteria for responsiveness were based on the presence of a reversible decrease in excitability. Analysis of responsive cells showed a dose-dependent inhibition. We observed that 10 μM DA reduced excitability to 84.9% ± 6.4% of baseline (15% inhibition), while 50 μM DA inhibited cells to 59.7% ± 8.4% of baseline (40% inhibition; Figures 1B and 1C; n = 7–9; one-way ANOVA: F(2, 21) = 7.6; p = 0.0032). The inhibitory effects of DA were reversible following a 10-min washout in all responsive cells, with excitability values for both 10 μM and 50 μM DA treatments returning to within 1.6% of control values (Figure 1C).
Figure 1. DA inhibits pyramidal cells in layer 2/3 of the mouse ACC.

(A) Following application of 50 μM DA, a large subset of layer 2/3 pyramidal neurons in the ACC shows strong inhibition (n = 18).
(B) 10–50 μM DA reduces excitability in a dose-dependent and reversible fashion, compared to control conditions; electrophysiological current-clamp traces shown.
(C) Left panel: quantitative group analysis shows that 50 μM DA significantly inhibits pyramidal firing activity in the ACC in comparison to control conditions. Middle panel: input-output plot demonstrates interaction between stimulation intensity and firing frequency following treatment with DA. Right panel: application of 50 μM DA induces a significant increase in pyramidal rheobase (n = 7–9).
(D) Left panel: typical current-clamp recordings illustrating alterations in input resistance following treatment with 50 μM DA. Right panel: quantitative group analysis of input resistance of pyramidal cells treated with aCSF or 50 μM DA (n = 8) is shown.
Values represented as means ± SEM: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
The input-output plot illustrates the inhibitory effect of DA on pyramidal firing activity (Figure 1C). Interestingly, DA induces an increase in the threshold required to trigger an action potential at low stimulation intensities. This is represented in Figure 1C, where weak stimulations below 50 pA no longer elicited action potentials in cells treated with 50 μM DA (n = 7 and 9; two-way ANOVA: F(14, 270) = 56.0; p = 0.0001). This is further demonstrated by analyzing the current required to evoke an action potential. We found DA increased the current required to evoke an action potential at −60 mV from ≈40 pA to 56.7 ± 8.8 (10 μM) and 68.9 ± 10.6 pA (50 μM; n = 9; paired t test: t(8) = 3.5; p = 0.0101; Figure 1C). In pyramidal cells treated exclusively with artificial cerebrospinal fluid (aCSF) alone, we did not notice any significant change in excitability (+2.0% ± 6.1% relative to baseline values; n = 8; Figure 1C).
In concordance with the decreased excitability of layer 2/3 pyramidal cells of the ACC following DA application, we also observed a significant decrease in the input resistance of DA-responsive cells (Figure 1D). Input resistance for cells treated with aCSF alone was found to be 145.7 ± 8.7 MΩ, whereas in the presence of 50 μM DA, we measured an input resistance of 114.4 ± 10.2 MΩ (Figure 1D; n = 8; paired t test: t(7) = 2.3; p = 0.028). Similarly, 10 μM DA also induced a trend of reduction in input resistance; however, the difference was not found significant (154.8 ± 9.8 MΩ to 128.1 ± 8.7 MΩ; data not shown).
These electrophysiological results indicate that the neuromodulator DA induces a robust and reversible inhibition of pyramidal cells in the mouse ACC, by inducing a decrease in their input resistance.
DAergic inhibition of pyramidal cells mediated by D1Rs
To determine which receptor DA acts at on pyramidal cells, we tested the effects of the selective D1R agonist SKF81297. Application of 10 μM SKF81297 induced a strong, reversible inhibition of layer 2/3 pyramidal cells in the ACC (Figure 2A), similar to the effects produced by 50 μM DA (Figure 1C). SKF81297 elicited a significant decrease in excitability relative to control values (Figure 2B; n = 9; control: 98.0% ± 6.1%, SKF: 68.9% ± 3.2% relative to baseline; one-way ANOVA: F(2, 24) = 16.2; p = 0.0013). SKF81297 also evoked a significant decrease in input resistance (Figure 2C; n = 9; baseline: 151.2 ± 6.5 MΩ, SKF: 134.7 ± 9.2 MΩ; paired t test: t(8) = 2.17; p = 0.031).
Figure 2. DAergic inhibition of pyramidal cells in ACC is exclusively mediated by D1 receptors.

(A) Typical current-clamp recordings illustrating alterations in pyramidal firing activity following application of 10 μM SKF81297, 5 μM SCH23390 + 50 μM DA, and 10 μM quinpirole.
(B) Top panel: quantitative group analysis of cell excitability; SKF81297 induces a significant decrease in excitability, whereas 5 μM SCH23390 + 50 μM DA and 10 μM quinpirole did not have any inhibitory effect. Bottom panel: input-output plots recorded during each of 4 different treatments (n = 6–10) are shown.
(C) Left panel: typical traces illustrating the impact of various DAergic ligands on input resistance. Right panel: 10 μM SKF81297 induces a significant decrease in input resistance, whereas 5 μM SCH23390 + 50 μM DA and 10 μM quinpirole did not (n = 6–10).
(D) Left panels: electrophysiological current-clamp traces illustrating 10 μM DHPG-induced persistent firing along with the reversible inhibitory effects of 50 μM DA and 10 μM SKF81297. Right panel: quantitative group analysis illustrating the significant decrease in firing frequency during persistent firing following application of 50 μM DA or 10 μM SKF81297 in comparison to the control group (DHPG alone; n = 11–13) is shown.
Values represented as means ± SEM: *p < 0.05; ***p < 0.001; ****p < 0.0001.
We also investigated the effects of SCH23390, a selective D1R antagonist, on the inhibitory effects of DA. Addition of SCH23390 by pre-incubation and co-application blocks both the inhibitory effect of 50 μM DA (Figure 2A) and the DA-induced decrease in input resistance (Figure 2C). Group analysis further confirms this antagonism as SCH23390 application completely abolishes the effects of DA on excitability (n = 10; +7.6% ± 4.9% excitability relative to baseline) and on input resistance (n = 10; 166.0 ± 12.8 MΩ to 154.0 ± 13.9 MΩ following DA application; Figures 2B and 2C). Interestingly, directly blocking D1Rs and adenylate cyclase activation via the antagonist SCH23390 caused an increase in baseline excitability (Figure 2B; n = 10; p = 0.0004), suggesting the presence of basal dopamine tone in ACC.
We then investigated the effect of activating Gi-coupled D2-like receptors on pyramidal cell excitability (Tsu and Wong, 1996). We applied selective D2-like agonist quinpirole (10 μM) to pyramidal neurons in layer 2/3 of the ACC and observed that D2-like receptor activation increases their firing activity (Figure 2A). Following quinpirole application, we found a significant increase in excitability in comparison to aCSF controls (Figure 2B; n = 6 and 8; repeated-measure two-way ANOVA with Dunnett’s multiple comparison: F(3, 450) = 30.2; p = 0.0448). Altogether, these findings indicate that postsynaptic DAergic inhibition of pyramidal cells in the ACC exclusively depends on D1R and does not involve excitatory D2-like receptors.
DAergic modulation of persistent firing in ACC
Pyramidal cells in the ACC display persistent firing properties that make them capable of holding a working memory trace (Kamiński et al., 2017). Persistent firing was induced by coincident depolarization and stimulation with the mGluR1/5 agonist DHPG (10 μM). DHPG alone caused an increase of 91.5% ± 46.4% in persistent firing frequency relative to baseline (n = 11; Figure 2D). In a large majority of persistent firing cells, application of DA (10 μM) or the D1R agonist SKF81297 (10 μM) produced a significant decrease in firing frequency or even termination of the prolonged firing (n = 12 of 17 for DA; n = 13 of 18 for SKF81297; Figure 2D). SKF81297 and DA application induced a 61.8% ± 10.3% and 65.4% ± 6.9% reduction in persistent firing intensity, respectively, relative to baseline values (n = 12–13; one-way ANOVA: F(2, 30) = 15.3; p < 0.0001; Figure 2D).
HCN channels mediate DAergic modulation of ACC pyramidal cells
To determine whether HCN channels contribute to DAergic inhibitory mechanisms, we pre- and co-applied the HCN channel blocker ZD7288 alongside DA during recordings. Treatment with 10 μM ZD7288 completely abolishes DAergic inhibition in the ACC (Figure 3). Following DA application in presence of ZD7288, we noticed no decrease in either excitability (Figure 3A) or input resistance (Figures 3A and 3B). Whereas 50 μM DA alone induced a 40.3% ± 8.4% decrease in excitability (previously shown in Figure 1), co-application with ZD7288 evokes a 26.8% ± 12.4% increase in excitability (n = 8; one-way ANOVA: F(2, 22) = 13.4; p = 0.0002; Figure 3A). Blocking HCN channels suppresses the impact of DA on input resistance (n = 7; Figure 3B). These findings are in line with previous reports documenting the effect of ZD7288 on the input resistance of central neurons (Surges et al., 2004).
Figure 3. DAergic inhibition depends on HCN channels.

(A) Treatment with the HCN channel blocker 10 μM ZD7288 occludes the inhibitory effects of 50 μM DA. Representative current-clamp traces showing the effects of 10 μM ZD7288 on excitability (left) and group analysis (right; n = 8) are shown.
(B) 10 μM ZD7288 blocks the 50 μM DA-induced decrease in input resistance. Representative current-clamp traces showing the effects of 10 μM ZD7288 on input resistance (left) and quantitative group analysis (right; n = 7) are shown.
(C) Quantitative effects of 10 μM ZD7288 on firing frequency (left) and input resistance (right) at baseline (n = 8).
(D) Both 50 μM DA and 10 μM SKF81297 induce a significant increase in resting membrane potentials of layer 2/3 pyramidal neurons of the ACC. This effect is lost in presence of the D1R antagonist SCH23390 (5 μM) or when HCN channels are blocked with ZD7288 (10 μM; n = 7–17).
Values represented as means ± SEM: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To gauge the importance of HCN in maintaining the homeostatic excitability of pyramidal cells in the ACC, we tested the effect of ZD7288 alone, i.e., in the absence of DA. Application of ZD7288 dramatically increases both pyramidal excitability and input resistance to levels observed in neuropathic conditions (n = 8; unpaired t tests: t(7) = 3.3 and 2.5; p = 0.0053 and 0.0245; Figures 3C and 7A).
Figure 7. Role of dopamine and D1R signaling in the ACC in pain relief.

(A) Systemic L-DOPA and carbidopa prevents gabapentin-induced CPP in SNI mice. Saline-treated SNI mice spent significantly more time in the gabapentin (GBP) paired chamber after conditioning (post) compared to the time in the same chamber before conditioning (pre). L-Dopa-and-carbidopa-treated SNI mice did not show significant changes in chamber preference (left). CPP difference scores were calculated as the difference between post-conditioning and pre-conditioning time spent in the GBP-paired chamber (right; n = 10–16).
(B) D1R agonist in ACC produces CPP in SNI rats. Bilateral microinjection of SKF81297 in the ACC (0.5 μg/0.5 μL) had no effect on reflexive tactile responses in either SNI or sham rats (left). SNI rats spent significantly more time in the SKF81297 paired chamber after drug conditioning (post) compared to the time in the same chamber before conditioning (pre; middle). The difference score for the SKF81297-paired chamber is significantly larger in SNI rats than in sham rats (right; n = 15–17).
(C) D1R antagonist in ACC blocks gabapentin-evoked CPP in SNI rats. Bilateral microinjection of SCH23390 (10 μg/0.5 μL) in the ACC of SNI rats prevents GBP-evoked CPP (left). After drug conditioning (post), SNI rats microinjected with saline in the ACC spend significantly more time in the GBP-paired chamber than before conditioning (pre; left). Difference between post-conditioning and pre-conditioning time spent in the GBP-paired chamber (right; n = 11–15) is shown.
(D) D1R antagonist in ACC blocks CPP induced by lidocaine nerve block in a rat model of post-surgical pain. Bilateral microinjection of SCH23390 (10 μg/0.5 μL) in the ACC of rats with left hindpaw incision prevents CPP elicited by peripheral nerve blockade with injection of lidocaine in the popliteal fossa of the injured hindpaw (left). After lidocaine conditioning (post), injured rats microinjected with saline in the ACC spend significantly more time in the lidocaine-paired chamber than before conditioning (pre; left). The difference score for the lidocaine-paired chamber is significantly larger in saline and incision rats than in SCH and sham or SCH and incision rats (right; n = 6–8).
Bars represent means ± SEM; symbols show the values for individual animals: *p < 0.0001; **p < 0.01.
We found that application of DA or SKF81297 caused a measurable increase in the resting membrane potentials (RMPs) of pyramidal cells (n = 17 and 15; paired t test: t(16) = 6.9 and t(14) = 8.4; p < 0.0001 and p < 0.0001; Figure 3D). This significant shift in RMP was not present when either D1Rs or HCN channels were blocked (n = 9, 7, and 7; Figure 3D). Given the nature of HCN channels, the depolarizing shift in RMP could be due to increasing inward cationic currents.
Although these results indicate that HCN channels expression and availability are critical for DAergic inhibition, they do not prove a functional connection between DAergic modulation and HCN channel-mediated currents (Ih). To determine whether DA is involved in mediating Ih, we used voltage-clamp electrophysiology to record HCN currents, in presence of TTX and BaCl2 to block voltage-dependent Na and Kir channels.
As expected, our recordings indicate that application of 50 μM DA does not cause any significant changes in maximal Ih currents measured at −130 mV. However, we noticed a significant increase in Ih around physiological membrane potentials (e.g., −70 mV; Figure 4A). A large population (64.2%) of pyramidal neurons in the ACC showed an increase in Ih in response to DA (n = 9 of 14). At −70 mV, Ih increases from 18.4 ± 2.7 pA to 28.0 ± 3.1 pA following application of 50 μM DA (Figure 4A). This modulation of Ih was found reversible as washing out of DA with aCSF for 10 min brings back Ih to baseline values (n = 10; paired t test: t(9) = 3.8; p = 0.0022).
Figure 4. DA regulates HCN channel activity in pyramidal neurons of the ACC.

(A) Left panel: representative voltage-clamp traces showing HCN currents (holding voltage −130 mV to −40 mV; 10 steps) before and after 50 μM DA application in layer 2/3 ACC. Middle panel: no significant increase in max Ih (recorded at −130 mV) following DA application is shown. Right panel: 50 μM DA induces a strong and reversible increase in Ih around physiological membrane potentials (−70 mV; n = 9).
(B) Left panel: typical tail currents shown. Middle panel: group analysis fitted with Boltzmann function shows a significant increase in open HCN channels at physiological voltages (−90 mV to −50 mV). Right panel: significant depolarizing shift in half-activation potential of HCN channels after 50 μM DA application (n = 5) is shown.
Values represented as means ± SEM: *p < 0.05; **p < 0.01; ***p < 0.001.
A voltage-dependent increase in Ih is associated with an increase in fractional activation of HCN channels rather than in the number of HCN channels present at the cell surface (Biel et al., 2009). To check whether DA induces an increase in HCN open channel probability, we analyzed HCN tail currents before and after DA application (Figure 4B). We found a significant increase in fractional activation of Ih (n = 5; repeated-measure ANOVA: F(9, 40) = 64.7; p < 0.0001; Figure 4B). Consequently, DA induces a rightward depolarizing shift in the half-activation potential of HCN channels, from −89 ± 3 mV to −68 ± 8 mV (Figure 4B; n = 5; paired t test: t(8) = 2.5; p = 0.0390), indicating that DAergic inhibition is mediated by the modulation of HCN channel activity in pyramidal cells of the ACC.
To ascertain whether D1R activation also increased HCN activity in layer 5 pyramidal neurons, we analyzed sag ratios before and after 10 μM SKF81297 application. Despite limitations of somatic patching in large pyramidal cells, we found a modest but significant increase in the sag ratio following SKF81297 application at physiological relevant potentials, from 1.11 ± 0.01 to 1.13 ± 0.01 (n = 16; paired t test: t(15) = 2.4; p = 0.0317; data not shown). In line with previous results, we found no change in the max sag ratio recorded at −100 mV, from 1.10 ± 0.01 to 1.09 ± 0.01 (n = 16; paired t test: t(15) = 1.9; p = 0.0688).
DAergic inhibitory mechanisms remain functional in neuropathic conditions
To determine whether DA-mediated pyramidal inhibition is still effective in ACC in neuropathic conditions, we tested the effects of exogenous DA and SKF81297 on pyramidal excitability (following +100 pA pulse) in layers 2/3 and 5 in mice 4 weeks post-spared nerve injury (SNI) surgery. We observed 50% and 33% of layer 2/3 and layer 5 ACC pyramidal neurons responding to SKF81927, respectively, values similar to the percentage of responsive cells in sham animals. SNI mice with tactile and cold allodynia displayed significant inhibition in both layers 2/3 and layer 5 in response to 50 μM DA application, similar to naive mice (Figures 5A and 5C; n = 4–5; paired t test: t(4, 4) = 2.65 and 3.16; p = 0.038 and 0.025 for layer 2/3 and 5, respectively). Additionally, 10 μM SKF81297 also decreased pyramidal excitability in both SNI and sham mice (Figures 5B and 5D; n = 4–6; paired t test; t(4–5) = 3.33 and 5.00; p = 0.029 and 0.0077; for layers 2/3 and layer 5, respectively). At +40 pA stimulation, both 50 μM DA and 10 μM SKF81297 application resulted in full silencing of responsive pyramidal neurons (data not shown). Excitability of pyramidal neurons not exposed to DA or SKF81297 stayed within ~2% of baseline values in both SNI and sham mice (data not shown).
Figure 5. DAergic inhibition preserved in neuropathic mice across cortical layers.

(A) Typical electrophysiological traces and group analysis illustrating 50 μM DA induces significant inhibition of layer 2/3 ACC pyramidal neurons in sham and SNI mice (+100 pA stimulation; n = 4).
(B) Typical electrophysiological traces and group analysis illustrating 10 μM SKF81297 inhibits layer 2/3 ACC pyramidal neurons in both sham and SNI mice (+100 pA stimulation; n = 4).
(C) Typical electrophysiological traces and group analysis illustrating 50 μM DA induces significant inhibition of layer 5 ACC pyramidal neurons in sham and SNI mice (+100 pA stimulation; n = 5).
(D) Typical electrophysiological traces and group analysis illustrating 10 μM SKF81297 inhibits layer 5 ACC pyramidal neurons in both sham and SNI mice (+100 pA stimulation; n = 6).
Values represented as means: *p < 0.05; **p < 0.01.
L-DOPA and carbidopa administration rescues mechanical and cold sensitivity in SNI mice
Our electrophysiological results, showing that the DA inhibition mechanism is preserved in neuropathic conditions, suggest that DAergic input to the ACC could be diminished in neuropathic conditions. Therefore, we tested the effects of long-term administration of the DA precursor L-DOPA, with carbidopa to prevent its peripheral metabolism, in neuropathic mice. We measured sensory thresholds in neuropathic mice using the von Frey assay and acetone cold test to measure tactile and cold allodynia, two somatosensory hallmarks of chronic neuropathic pain. 2 weeks following SNI surgery, with continuous L-DOPA (75 mg/24 h/kg) and carbidopa (10 mg/24 h/kg) intake, we observed that treated mice did not display significant ipsilateral tactile hypersensitivity when compared to the control SNI group (n = 7–11; repeated-measure two-way ANOVA with Dunnett’s multiple comparisons: p = 0.0068; Figure 6A). Similarly, L-DOPA- and carbidopa-treated neuropathic mice did not display ipsilateral allodynic responses in the acetone cold test 3 weeks following SNI (Figure 6B).
Figure 6. L-DOPA and carbidopa treatment effective at reducing mechanical hypersensitivity in SNI mice.

(A) Mechanical withdrawal thresholds on ipsilateral paw (left) and contralateral paw (right) between treatment conditions. Groups: SNI, sham, SNI + L-DOPA, sham + L-DOPA, and SNI + delayed L-DOPA (supplementation starting 2 weeks post-SNI surgery rather than on the day of SNI surgery; n = 6–11).
(B) Mean scores of responses to cold in the acetone test on ipsi- and contralateral paws in several treatment conditions (n = 7–11).
(C) Input-output plots documenting L-DOPA-and-carbidopa-induced decrease in both pyramidal excitability (left) and input resistance (right; n = 13–27).
(D) Rescue of HCN channel activity in L-DOPA-and-carbidopa-treated SNI mice (left), with open channel probabilities on level with sham mice at resting membrane potential (right; n = 27–34).
Values represented as means ± SEM: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To test whether L-DOPA is effective at reducing hyperalgesia only when provided during a critical time window following SNI surgery, we delayed the start of L-DOPA treatment in one group of mice until 2 weeks following the surgery (SNI + delayed L-DOPA). Following 2 weeks on L-DOPA and 4 weeks post-SNI surgery, these mice were statistically identical to the sham, L-DOPA-treated sham, and L-DOPA-treated SNI groups and significantly more antiallodynic than the control SNI group (n = 8; repeated-measure two-way ANOVA with Dunnett’s multiple comparisons: p = 0.0120; Figure 6A). As a control, we added a group of mice who had undergone a sham surgery and supplemented their diet with L-DOPA and carbidopa. The L-DOPA-treated sham group did not differ significantly from the non-treated sham group (n = 6 and 10; Figure 6A). Consistent with established literature, mice supplemented with L-DOPA and carbidopa displayed significant motor side effects, including hyperactivity, starting 5 weeks following the onset of the treatment (Lundblad et al., 2005). No behavioral experiments were conducted on mice showing motor symptoms related to L-DOPA.
Daily L-DOPA and carbidopa administration rescues excitability and HCN function in neuropathic ACC
To assess how the behavioral characteristics of neuropathic mice translate to physiological properties of pyramidal neurons in the ACC, we analyzed the firing frequencies, input resistance, and HCN properties of layer 2/3 pyramidal neurons of SNI mice and SNI mice supplemented with L-DOPA and carbidopa. 4 weeks following SNI surgery, with continuous L-DOPA (75 mg/24 h/kg) and carbidopa (10 mg/24 h/kg) intake, we observed that neuropathic mice did not display cortical hyperexcitability (Figure 6C). ACC pyramidal cells of L-DOPA-treated SNI mice had firing frequencies on par with sham mice, significantly lower than SNI mice not supplemented with L-DOPA. At +100 pA stimulation, pyramidal cells from control SNI mice without L-DOPA fired at 18.0 ± 1.1 Hz, whereas pyramidal cells from L-DOPA-treated SNI mice fired at 13.8 ± 0.8 Hz (Figure 6C). This effect of the L-DOPA treatment on excitability is also illustrated by the input-output plot (Figure 6C; n = 13–27; repeated-measure two-way ANOVA with Dunnett’s multiple comparison: at 100 pA, p = 0.0274). The impact of the L-DOPA treatment of mice on excitability of pyramidal cells in the ACC is logically reflected in changes in their input resistance (Figure 6C) from 247.8 ± 7.9 MΩ in SNI to 199.0 ± 9.7 MΩ in SNI mice supplemented with L-DOPA and carbidopa (n = 9; repeated-measure two-way ANOVA with Dunnett’s multiple comparison: p = 0.0025).
As HCN channel dysfunction is a major driving force causing cortical hyperexcitability in SNI mice, we also measured Ih and the fractional activation of HCN channels in neuropathic conditions with or without treatment with L-DOPA. As we have previously reported (Cordeiro Matos et al., 2015), we measured a significant shunting of pyramidal Ih in SNI mice, whereas pyramidal cells of SNI mice supplemented with L-DOPA and carbidopa did not display this downward shift but rather displayed Ih values almost identical to those seen in sham mice across all voltages (Figure 6D; n = 27–34; repeated-measure two-way ANOVA with Dunnett’s multiple comparison). This shift in Ih is mirrored in the open channel probability of HCN at physiologically relevant membrane potentials. For example, at −70 mV, 16.9% ± 2.3% of HCN channels are open in the SNI + L-DOPA group, whereas only 8.8% ± 4.2% of HCN channels are open in the control SNI group (Figure 6D; n = 25–28; repeated-measure two-way ANOVA with Dunnett’s multiple comparison: p = 0.07).
Daily L-DOPA and carbidopa treatment relieves ongoing pain in SNI mice
Systemic L-DOPA and carbidopa significantly decreased tactile and thermal allodynia in SNI mice. To establish whether L-DOPA and carbidopa could also produce relief of pain aversiveness, we determined whether this treatment prevented gabapentin-induced conditioned place preference (CPP) in SNI mice. SNI mice were pretreated with L-DOPA and carbidopa (intraperitoneally [i.p.] 20 mg/10 mL/kg and 4 mg/10 mL/kg, per 24 h) for 12 days starting immediately after SNI surgery and then at 15 days after the surgery evaluated for conditioned place preference to acute administration of gabapentin (i.p. 30 mg/mL/kg). SNI mice pretreated with saline displayed significant conditioned place preference following gabapentin (Figure 7A; n = 16; paired t test; t(9) = 3.419; p = 0.0076), suggesting relief of ongoing pain. In contrast, SNI mice pretreated with L-DOPA and carbidopa did not display significant preference to the chamber paired with gabapentin, suggesting that increased dopaminergic signaling produced relief of ongoing pain revealed by decreased motivation to seek pain relief (Figure 7A; n = 10; paired t test; t(15) = 0.017; p = 0.987). The difference score between L-DOPA + carbidopa and saline pretreated mice was significantly different (Figure 7A; n = 10–16; unpaired t test; t(24) = 2.575; p = 0.0166).
Intra-ACC injection of D1R agonist SKF81297 produces conditioned place preference in neuropathic rats
Because DAergic inhibition of pyramidal cells is mediated by D1Rs, we investigated whether a single administration of SKF-81297, a dopamine D1R agonist, modulates allodynia and ongoing pain in animals with neuropathic pain. To allow site-specific SKF-81297 microinjection, we measured pain behavior in adult male Sprague Dawley rats with bilateral ACC cannulas and either SNI-induced chronic neuropathic pain or sham surgery. Bilateral microinjection of SKF-81297 in the ACC (0.5 μg/0.5 μL) had no effect on tactile responses in either SNI or sham rats (Figure 7B). ACC SKF81297 produced conditioned place preference only in SNI rats (Figure 7B; n = 15; paired t test; t(14) = 3.431; p = 0.0041). Intracortical injection of SKF81297 in sham-operated animals did not produce a chamber preference (Figure 7B; n = 17; paired t test; t(16) = 0.521; p = 0.609). As illustrated in Figure 7B, there is a significant difference between sham and SNI animals in CPP difference score (n = 15–17; unpaired t test: t(30) = 3.06; p = 0.0047).
Relief of ongoing neuropathic or incisional pain depends on D1R signaling in the ACC
Relief of pain is a reward that is associated with increased activity of VTA DAergic neurons and DA release in the mesolimbic pathway (Navratilova et al., 2012). As ventral tegmental area (VTA) DA neurons also project to the cortex, we sought to determine whether relief of pain in SNI rats elicited by gabapentin, a common treatment for neuropathic pain, is dependent on ACC D1R signaling. We therefore bilaterally microinjected the selective D1R antagonist SCH23390 (10 μg/0.5 μL) in the ACC before gabapentin conditioning and found that, while SNI rats pretreated with saline displayed a significant preference to the gabapentin paired chamber (Figure 7C; n = 11; paired t test; t(10) = 2.48; p = 0.033), no preference was observed in SNI rats pretreated with ACC SCH23390 (Figure 7C; n = 15; paired t test; t(14) = 0.123; p = 0.904).
To verify whether ACC D1R signaling is also necessary for relief of post-surgical pain, we produced relief of ongoing pain with lidocaine-induced nerve block in the incisional pain model. ACC saline pretreated rats that had undergone the incisional pain procedure showed significant preference to the chamber previously paired with lidocaine injected at the popliteal fossa (Figure 7D; n = 8; paired t test; t(7) = 3.14; p = 0.017). However, preference to lidocaine-induced nerve block was prevented by pretreatment of SCH23390 (10 μg/0.5 μL), bilaterally injected in the ACC before conditioning (Figure 7D; n = 6; paired t test; t(5) = 1.45; p = 0.206). SCH23390 had no effect in sham-operated rats. There was a significant effect of treatment group when comparing CPP difference score for SCH23390 and sham, saline and incision, and SCH23390 and incision animals (Figure 7D; n = 6–8; one-way ANOVA; p = 0.0114; Tukey’s multiple comparison test: SCH23390 and sham versus saline and incision p < 0.05; saline and incision versus SCH23390 and incision p < 0.05).
DISCUSSION
Understanding how chronic pain impacts cortical circuitry is key to the development of improved effective treatments for patients. Modulation of ACC neuronal activity by DA is of particular interest in this search, given the key role of ACC in the processing of affective components of pain perception, the presence of a dense mesocortical DAergic projection, and the expression of DA receptors across all cortical layers (Clarkson et al., 2017). Our findings indicate that DA is inhibitory to a large subpopulation of pyramidal neurons in layer 2/3 and layer 5 ACC, both in physiological and neuropathic pain states. DAergic inhibition of ACC neurons raised the rheobase by approximately 30 pA, a difference that could determine whether a neuron fires or not following pain-stimuli-evoked postsynaptic potentials in the ACC in vivo (Sikes and Vogt, 1992). We used selective D1R- and D2-like ligands to determine that only D1Rs are involved in this inhibition. The inhibition induced by the selective D1R agonist SKF81297 mimicked the inhibition induced by DA, while the selective D2-like agonist quinpirole evoked an increase in pyramidal excitability. Additionally, we show that the D1R antagonist SCH23390 antagonizes DA’s inhibitory effects. Although it is arguable that these DA-evoked excitatory changes are not present in vivo due to the upregulation of AMPA receptors via protein kinase A (PKA) phosphorylation, DA has been shown to decrease evoked postsynaptic potentials (EPSPs) in the ACC (Darvish-Ghane et al., 2016; Esteban et al., 2003; Sun et al., 2005).
The ACC plays a pivotal role in working memory, and patients suffering from chronic pain commonly report impairments in working memory performance (Phelps et al., 2021; Schnurr and MacDonald, 1995; Bradley et al., 1989). The property of persistent firing observed in pyramidal cells of the ACC provides a cellular substrate for working memory (Kamiński et al., 2017). We used a glutamatergic model of persistent firing evoked by the mGluR agonist DHPG to test the effects of both DA and SKF81297 on the frequency of action potentials during the persistent depolarization (plateau potential). Our results indicate that SKF81297 and DA consistently decrease the duration of plateau potential and the frequency of persistent firing in the ACC. Thus, the loss of working memory performance measured in chronic pain disorders and the overall effect of dopamine and D1R antagonists on working memory could be related to this strong DAergic modulation of persistent firing in the ACC (Cai and Arnsten, 1997; Bradley et al., 1989; Kamiński et al., 2017; Panegyres 2004; Sawaguchi and Goldman-Rakic, 1991).
The dependence of DAergic inhibition on Gs-coupled D1 receptors and not on Gi-coupled D2-like receptors led us to conclude that cAMP plays a pivotal role in controlling the excitability of ACC pyramidal neurons. Our group and others have shown that cAMP-gated HCN channel dysfunction plays a major role in promoting cortical hyperexcitability in neuropathic conditions (Cordeiro Matos et al., 2015; Santello et al., 2017). We conclude that the firing activity of pyramidal cells in the ACC is controlled by a balance of monoaminergic inputs through their Gs- and Gi-coupled receptors with opposite effects on HCN channel function: activation of Gi-coupled α2 adrenergic receptors shuts down HCN channels, while activation of Gs-coupled 5-HT7 and D1 receptors increase the opening of HCN channels (Santello et al., 2017; Zhang et al., 2013).
D1 receptors are colocalized with HCN channels on dendritic spines of pyramidal neurons, and we confirmed that DA modulates HCN channels through D1 receptors (Paspalas et al., 2013). Our results show D1 receptors induce an increase in resting membrane potential, which is prevented when HCN is blocked with ZD7288, likely due to inward cationic currents mediated by HCN channels. However, opening of HCN channels also induces a decrease in input resistance stemming from a reduction in charge insulation in dendritic spines (Surges et al., 2004). Therefore, the decrease in input resistance induced by D1R activation is likely overriding the slow depolarization, resulting in inhibition. Although DA does not increase the maximal amplitude of Ih, we noticed a significant increase in HCN-mediated currents at physiological membrane potentials. Accordingly, the half-activation potential, the membrane potential at which half of HCN channels are open, decreases drastically in the presence of DA. This suggests a change in internal cAMP levels rather than a change in the number of HCN channels at the cell surface. These results are also reflected in the fractional activation of Ih at physiological membrane potentials; at −60 mV, DA approximately triples Ih fractional activation.
We provide evidence that the signaling mechanisms underlying D1R- and HCN-mediated inhibition are still functional in the ACC of neuropathic animals, strongly suggesting hypoactive dopaminergic inputs. Interestingly, pathologies with hypoactive supraspinal DAergic pathways, such as Parkinson’s disease (PD) and major depression, show a high prevalence of non-myogenic chronic pain (Blanchet and Brefel-Courbon, 2018; Defazio et al., 2008; Djaldetti et al., 2004; Ford 2010). The VTA and the SNc, the main sources of mesocortical DA, display altered firing frequencies and cell death in depression and PD, respectively (Alexander 2004; Liu et al., 2018; McGeer et al., 1988). Furthermore, there is evidence linking a GABA-mediated reduction in dopaminergic activity in the VTA of rodents afflicted with chronic neuropathic pain (Huang et al., 2019; Ko et al., 2018). A reduction in cortical DA could be causing the high comorbidity of chronic pain in patients with PD, as fMRI studies display pathological hyperactivity in the ACC (Brefel-Courbon et al., 2005). Administration of L-DOPA, a DA precursor that increases DA release in the brain, and carbidopa, a peripheral DOPA decarboxylase inhibitor, alleviates symptoms of chronic pain and normalizes ACC hyperactivity in patients with PD (Antinori et al., 2018; Brefel-Courbon et al., 2005; Schestatsky et al., 2007). There is also increasing evidence linking other hypodopaminergic disorders, such as clinical depression, to chronic pain (Fishbain et al., 1997; Taylor et al., 2016).
To test this idea of a hypodopaminergic state in chronic pain conditions, we supplemented chronic neuropathic mice with systemic L-DOPA and carbidopa, a treatment that was successfully used to treat pain in patients with PD (Brefel-Courbon et al., 2005). 2 weeks following the start of L-DOPA and carbidopa administration, mice that had undergone SNI surgery did not show the typical hypersensitivity of animals in chronic pain. Thus, tactile and cold allodynia of SNI mice treated with L-DOPA and carbidopa did not differ from sham-operated controls. Additionally, delaying the L-DOPA treatment until 2 weeks post-SNI showed reversal of established tactile hypersensitivity. Daily L-DOPA and carbidopa treatment of SNI mice also prevented development of ongoing pain indicated by lack of CPP to gabapentin that was observed in saline-treated SNI mice. These data suggest that chronic neuropathic pain is associated with a hypodopaminergic state that persists for an extended period. Administration of L-DOPA both prevented and reversed established behavioral consequences of neuropathic pain, suggesting important therapeutic opportunities. In fact, clinical studies are already showing promising results using L-DOPA and carbidopa to treat chronic low back pain (Reckziegel et al., 2019). Although carbidopa was present to limit the peripheral effects of L-DOPA, we confirmed these behavioral changes are consistent with a reduction in hyperexcitability and input resistance of pyramidal cells in the ACC to levels on par with sham-operated mice and are not peripherally driven. We also measured a dramatic increase in Ih at relevant membrane potentials in L-DOPA-treated SNI mice, particularly at resting potentials where there are no HCN channels open in SNI conditions.
Consistent with the demonstration of hypodopaminergic state in animals with chronic pain and with the role of D1R on the electrophysiological properties of pyramidal neurons, we observed a CPP effect in SNI rats following acute activation of D1 receptors with SKF81297 in the ACC. As the CPP paradigm reflects learning resulting from reward and we did not observe SKF81297-paired chamber preference in sham animals, our findings indicate that D1R activation provides reward associated with pain relief (King et al., 2009; Porreca and Navratilova, 2017). Lack of SKF81297-paired chamber preference in sham animals is also in agreement with our findings of no hypoalgesia in the sham L-DOPA-treated group compared to the sham control group.
Furthermore, we show that the pain relief produced by gabapentin and lidocaine nerve block, treatments that are known to be analgesic in humans, depends on D1R signaling in the ACC. Gabapentin pain relief is associated with activation of VTA DAergic neurons and subsequent release of DA in the mesolimbic pathway (Navratilova et al., 2012). Bilateral injection of D1R antagonist SCH23390 in the ACC prevented the conditioned place preference induced by gabapentin in a model of neuropathic pain as well as the pain relief induced by a peripheral nerve block with lidocaine in a model of post-surgical pain. As L-DOPA and carbidopa treatment prevents gabapentin-induced CPP in SNI mice, we suggest that the pain relief evoked by opioids and other non-opioid analgesic drugs rely on increasing cortical DA levels (Johnson and North, 1992).
Although it remains unclear whether hyperexcitability in the ACC is a cause or effect of chronic pain state, increasing evidence suggests inhibiting pyramidal cells of the ACC induces analgesia (Kang et al., 2015; Kuner and Flor, 2016; Sellmeijer et al., 2018). Our findings, in two different species, strongly suggest that DA is a major inhibitory neuromodulator of pyramidal activity in the ACC in pathological pain states. We propose that dysregulation of this DAergic inhibition contributes to the expression of affective and somatosensory features typical of chronic pain, and normalization with D1R agonists may provide significant therapeutic benefit.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Philippe Séguéla (philippe.seguela@mcgill.ca).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This study did not generate code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Six-week old male C57BL/6 mice (Charles River Laboratories, QC, CA) were housed in the Montreal Neurological Institute (MNI) Animal Care Facility (ACF) and all procedures are following the Canadian Council on Animal Care guidelines. For CPP experiments, six-week old male C57BL/6J mice (Jackson Laboratories) were housed in the University of Arizona ACF. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Arizona and were in accordance with guidelines from the International Association for the Study of Pain. Mice were kept in a 12 hour light-dark cycle with free access to water and food.
Rats
Eight to nine-week old male Sprague-Dawley rats, 250–300 g (Harlan Laboratories, Indianapolis, IN), were housed in the University of Arizona ACF. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Arizona and were in accordance with guidelines from the International Association for the Study of Pain. Rats were kept in a 12 hour light-dark cycle with free access to water and food.
METHOD DETAILS
Spared Nerve Injury (SNI) and Incisional Injury Models
Isoflurane (2%) and ketamine/xylazine (80/12 mg/kg i.p.) were used to anesthetize mice and rats, respectively. Spared nerve injury (SNI) surgery to induce chronic neuropathic pain was performed on mice and rats 14 days prior to CPP and von Frey testing. For SNI, the tibial and peroneal nerves were ligated and cut, leaving the sural nerve intact. Incisional injury was performed according to the Brennan model (Brennan et al., 1996) as described previously (Navratilova et al., 2012).
Electrophysiological Recordings
Animals were anesthetized with an Avertin solution (2.5 g tribromoethanol in 5 mL amylene hydrate diluted in 100 mL ddH2O) (Sigma-Aldrich, St. Louis, MI). Then animals were transcardically perfused with a 4°C choline-chloride based cutting solution oxygenated with carbogen (O2 95%, CO2 5%) (Praxair). Brains were extracted and sliced into 300 μm sections using a vibratome (Leica VT1000). Slices were allowed to rest at room temperature for 1 hour in oxygenated (see above) artificial cerebrospinal fluid (aCSF) containing 124 mM NaCl, 2 mM KCl, 26 mM NaHCO3, 1.8 mM MgSO4, 1.25 mM NaH2PO4, 10 mM Glucose, 1.6 mM CaCl2, pH 7.4. Slices were submerged at 30–32°C on the stage of a Zeiss Axioskop microscope continuously perfused with oxygenated (see above) aCSF containing 1.8 mM Kynurenic Acid and 100 μM picrotoxin at a rate of 1 mL/minute. A near-infrared CCD camera coupled to a x63 water immersion objective was used during patch-clamp recordings (Sony XC-75). Cells were patched with BF150–75-10 glass pipettes (~6 MΩ) pulled with a Flaming Brown Micropipette Puller (Model P-97, Sutter Instruments, US). Pipettes were mounted on a MP-225 micromanipulator and filled with an intracellular solution containing 120mM K-gluconate, 10 mM HEPES, 0.2 mM EGTA, 20 mM KCl, 2 mM MgCl2, 7 mM diTrisPhosphate-Creatine, 4 mM Na2ATP, and 0.3 mM NaGTP (Sutter Instruments, Novato, CA). An Axopatch 200B amplifier and Digidata 1322A interface digitizer were used for data acquisition (Molecular Devices, San Jose, CA). Signals were low-pass filtered at 10 kHz for current-clamp recordings and at 2 kHz for voltage-clamp recordings, digitized at 20 kHz.
Neurons were patched in areas corresponding to layer 2/3 and layer 5 dACC according to stereotaxic coordinates (Paxinos and Franklin, 2008). Pyramidal cells were identified based on firing frequency, input resistance (MΩ), and spike adaptation to a 1 s 100 pA cuurent injection pulse. Cells with resting membrane potentials (RMP) above −50 mV or below −80 mV were excluded. Series resistance was compensated (≤35 MΩ).
Excitability was evaluated by bringing cells to −60 mV RMP and counting action potentials evoked by a 17-step protocol (current input ranging from −20 pA to +140 pA, 10 pA steps). Input-output was calculated based on action potentials evoked at each step. Input resistance (MΩ) was assessed by injecting cells with a 6-step protocol (current input ranging from −300 pA to 0 pA, 60 pA steps). Input resistance was averaged across steps and evaluated across treatments.
HCN induced sags were measured in layer 5 pyramidal neurons using whole-cell patch recording in current-clamp mode; two current steps of −300 pA and −120 pA were used to record max sag and sag at physiological relevant potentials, respectfully. sag ratios were calculated by measuring the sag amplitude and normalizing it to the steady-state voltage at the end of each current step, shown below.
HCN currents (Ih) were measured in voltage clamp mode in the additional presence of 1 μM TTX and 200 μM BaCl2 to block voltage-gated sodium channels and inward rectifying potassium channels (Kir), respectively. Cells were held at a holding potential of −40 mV followed by a 2.5 s step ranging from −40 mV to −130 mV holding voltage (10 step protocol, 10 mV steps). Following each respective step, cells were held at −130mV for 1 s to record tail currents. Current amplitudes of HCN channels were measured by calculating the difference between the current at the start of each step (during the peak) and at the end of each step during the plateau phase. Physiological Ih was measured at −70 mV and max Ih was determined at −130 mV holding voltage. Fractional activation of Ih was calculated by measuring the amplitude of the tail current after each step and normalized to the max tail current (see equation below). A Boltzmann function was fitted using GraphPad Prism 7 (GraphPad Software).
Ih amplitude was assessed by obtaining baseline values 5 minutes after patching and comparing them with values obtained following 10-minute treatment with 50 μM DA. Resting membrane potential (mV), temperature, series resistance (MΩ), and whole-cell capacitance were measured before each time point.
Intracranial ACC Cannulation
Stereotaxic cannulation surgeries were performed in animals anesthetized with a ketamine/xylazine combination (80/12 mg/kg, i.p.; Western Medical Supply/Sigma, Arcadia, CA). Bilateral cannulation of the ACC was performed as previously described (Johansen and Fields, 2004; Navratilova et al., 2015). A pair of 26-guage stainless steel guide cannulas cut 4 mm below the pedestal (Plastics One Inc., Roanoke, VA) were directed toward the following ACC injection site: AP: +2.6 mm from bregma; ML: ± 0.6 mm; DV: −1.8 mm from skull). Guide cannulas were cemented in place and secured to the skull by small stainless-steel machine screws. Stainless steel dummy cannulas were inserted into each guide to keep the guide free of debris. Rats then received a subcutaneous gentamycin (1 mg/ml) injection, were housed individually, and were allowed to recover for 7–10 days. Cannula locations were verified after the experiment; 10 rats were eliminated due to incorrect placement.
L-DOPA/Carbidopa Dosage and Administration for Electrophysiology and von Frey Assay
Mice were placed on a strict L-DOPA/Carbidopa regimen immediately following SNI surgery. Light-shielded L-DOPA and carbidopa were dissolved in their water supply and administered continually throughout the duration of the study (Blunt et al., 1993). Water intake per mouse was measured 3 times weekly and L-DOPA/carbidopa concentrations were adjusted so as each mouse received 75 mg/24 hr/kg L-DOPA and 10 mg/24 hr/kg carbidopa. In the case of delayed L-DOPA treatment group, administration of L-DOPA/carbidopa started after the first day of behavioral testing instead of immediately following SNI surgery.
Behavioral Tests
Von Frey
Animals were placed in suspended chambers with wire mesh floors for 30 minutes for habituation prior to testing. Mouse withdrawal thresholds were determined by stimulating the lateral planar surface of both the ipsi- and contralateral paws using an automatic von Frey apparatus (Ugo Basile, Italy). Rat withdrawal thresholds were calculated using a series of calibrated von Frey filaments (Stoelting, Wood Dale, IL). Logarithmically spaced increments ranging from 0.41 to 15 g (4–150 N) were applied perpendicular to the plantar surface of the ipsilateral hind paw until the filament buckled. Withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (“up and down” method), analyzed using a Dixon nonparametric test, and expressed as the mean withdrawal threshold (Dixon 1980).
Acetone cold test
Cold allodynia was measured by applying a drop of acetone to the lateral planar surface of the paw and measuring acute behavioral responses of the mice (0 = no response, 1 = hind paw flinching and stamping, 2 = hind paw licking, 3 = exaggerated response and/or vocalization).
Conditioned place preference (CPP)
Mice
SNI mice were pretreated daily with either saline or L-DOPA/carbidopa (20 mg/10ml/kg/ 24hr and 4 mg/10ml/kg/ 24 hr, i.p.) for 12 days starting on the day of SNI surgery. Mice were acclimated to CPP chambers on day 11 after surgery and baselines were assessed on day 12. Conditioning was performed on days 13 and 14 with morning injections of saline and confinement for 30 min in one chamber and gabapentin (30 mg/ml/kg, i.p.) injections followed by 30 min confinement in the opposite chamber in the afternoon. On test day (day 15), mice were placed in the middle chamber with free access to both conditioning chambers and the time spent in each chamber was recorded for 15 min. Ten mice were eliminated due to failed surgery or failed baselines.
Rats
A single trial conditioning protocol was used for CPP as previously described (King et al., 2009; Navratilova et al., 2013). On preconditioning day, rats were placed into the CPP boxes with free access to all three chambers. Anymaze software was used to determine the time spent in each chamber across 15 min. Animals spending more than 80% (720 s) or less than 20% (180 s) of the total time in either chamber were eliminated from further testing (19 rats).
Conditioning day - microinjection of SFK81297: SNI and sham rats with ACC cannulas received a saline injection into the ACC and were placed into a conditioning chamber for 30 min. Four hours later, rats received SKF81297 (0.5 μg/0.5 μl) into the ACC and were placed into the opposite chamber for 30 min.
Conditioning day- microinjection of SCH23390: SNI rats with ACC cannulas were pretreated with bilateral microinjections of either saline (0.5 μl) or SCH23390 (10 μg/0.5 μl) in the ACC. Following pretreatment, rats received a saline injection (i.p.) and placed into a conditioning chamber for 30 minutes. Four hours later, pretreated SNI rats received gabapentin (50 mg/ml/kg, i.p.) and placed into the opposite chamber for 30 minutes. The experiment with incision rats was performed in a similar way but in the afternoon, rats received injection of lidocaine (200 μl; 4%) into popliteal fossa under light isoflurane anesthesia to induce peripheral nerve blockade.
On test day, rodents were placed drug free into the middle CPP chamber and were allowed to explore all chambers for 15 minutes; the time in chambers was assessed using the Anymaze software to determine chamber preference. Difference scores were calculated as test time minus preconditioning time spent in the drug-paired chamber.
Drugs and Solutions
Reagents used for aCSF, cutting solution, internal pipette solution, DA, carbidopa, and xylazine were purchased from Sigma-Aldrich (St. Louis, Missouri). TTX, DHPG, SKF81297, ZD7288, quinpirole, and SCH23390 were obtained from Tocris (Bristol, UK). Levodopa was purchased from Santa Cruz Biotechnology (Dallas, Texas). Ketamine was obtained from Western Medical Supply (Arcadia, CA). All drugs were diluted and aliquoted into single-use samples stored at −20°C. Gabapentin was purchased from Spectrum Chemical MFG (Gardena, CA) and prepared fresh by dissolving in distilled water. Lidocaine was purchased from Qualitest Pharmaceuticals.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were analyzed using Clampfit 10 and statistical analysis was performed using GraphPad Prism 7. Values are represented as either raw or normalized means ± SEM with significance threshold set at p < 0.05. One-way ANOVA with Dunnett’s multiple comparison was used to compare multiple independent groups (aCSF versus 10 μM DA versus 50 μM DA). Unpaired t tests were used to compare two independent groups whereas the paired t test was used to compare two related groups (baseline versus treatment). Two-way ANOVA with Bonferroni multiple comparison was used to analyze significance between independent samples under multiple conditions. Significance for fractional activation of HCN channels was tested using repeated-measure ANOVA. CPP chamber preference was analyzed using the Anymaze software (Wood Dale, IL).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| SKF81297 | Tocris | Cat#1447 |
| Quinpirole | Tocris | Cat#1061 |
| DHPG | Tocris | 0805/5 |
| Dopamine | Sigma-Aldrich | Cat#H8502 |
| ZD7288 | Tocris | Cat#1000/10 |
| L-DOPA | Sigma-Aldrich | PHR1271 |
| Carbidopa | Sigma-Aldrich | PHR1655 |
| SCH23390 | Tocris | Cat#0925 |
| Gabapentin | Spectrum Chemical MFG Corp. | A4404 |
| Lidocaine | Qualitest Pharmaceuticals | #00603139447 |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Wild-Type C57Bl6 Mice | Charles River Canada | C57Bl6 |
| Wild-Type C57BL/6J mice | Jackson Laboratories | RRID:IMSR_JAX:000664 |
| Sprague-Dawley rats | Harlan Laboratories | #002 |
|
| ||
| Software and algorithms | ||
|
| ||
| pClamp | Molecular Devices | RRID:SCR_011323 |
| Prism 9 | Graphpad | RRID:SCR_002798 |
| ANY-maze | ANY-maze | RRID:SCR_014289 |
Highlights.
D1R activation is inhibitory to a subpopulation of pyramidal neurons in ACC
Inhibition via D1R is mediated by an increase in HCN channel fractional activation
Increasing cortical DA with L-DOPA reduces hyperalgesia in neuropathic mice
D1R signaling in ACC is required for relief of ongoing pain
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes for Health Research (PJT-1453098; to P.S.) and by NIH (R01 DA 041809; to F.P.). K.L. holds a studentship from the Louise and Alan Edwards Foundation for Pain Research. We thank our McGill colleagues Stephen Glasgow, Maria Zamfir, and Steven Cordeiro Matos for advice and technical support. Graphical abstract was created with BioRender.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alexander GE (2004). Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 6, 259–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori S, Fattore L, Saba P, Fratta W, Gessa GL, and Devoto P (2018). Levodopa prevents the reinstatement of cocaine self-administration in rats via potentiation of dopamine release in the medial prefrontal cortex. Addict. Biol. 23, 556–568. [DOI] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, and Zong X (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885. [DOI] [PubMed] [Google Scholar]
- Blanchet PJ, and Brefel-Courbon C (2018). Chronic pain and pain processing in Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 87 (Pt B), 200–206. [DOI] [PubMed] [Google Scholar]
- Blunt SB, Jenner P, and Marsden CD (1993). Suppressive effect of L-dopa on dopamine cells remaining in the ventral tegmental area of rats previously exposed to the neurotoxin 6-hydroxydopamine. Mov. Disord. 8, 129–133. [DOI] [PubMed] [Google Scholar]
- Bradley VA, Welch JL, and Dick DJ (1989). Visuospatial working memory in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 52, 1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brefel-Courbon C, Payoux P, Thalamas C, Ory F, Quelven I, Chollet F, Montastruc JL, and Rascol O (2005). Effect of levodopa on pain threshold in Parkinson’s disease: a clinical and positron emission tomography study. Mov. Disord. 20, 1557–1563. [DOI] [PubMed] [Google Scholar]
- Brennan TJ, Vandermeulen EP, and Gebhart GF (1996). Characterization of a rat model of incisional pain. Pain 64, 493–502. [DOI] [PubMed] [Google Scholar]
- Cai JX, and Arnsten AF (1997). Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys. J. Pharmacol. Exp. Ther. 283, 183–189. [PubMed] [Google Scholar]
- Campbell JN, and Meyer RA (2006). Mechanisms of neuropathic pain. Neuron 52, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson RL, Liptak AT, Gee SM, Sohal VS, and Bender KJ (2017). D3 receptors regulate excitability in a unique class of prefrontal pyramidal cells. J. Neurosci. 37, 5846–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro Matos S, Zhang Z, and Séguéla P (2015). Peripheral neuropathy induces HCN channel dysfunction in pyramidal neurons of the medial prefrontal cortex. J. Neurosci. 35, 13244–13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvish-Ghane S, Yamanaka M, and Zhuo M (2016). Dopaminergic modulation of excitatory transmission in the anterior cingulate cortex of adult mice. Mol. Pain 12, 1744806916648153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, and Woolf CJ (2000). Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158. [DOI] [PubMed] [Google Scholar]
- Defazio G, Berardelli A, Fabbrini G, Martino D, Fincati E, Fiaschi A, Moretto G, Abbruzzese G, Marchese R, Bonuccelli U, et al. (2008). Pain as a nonmotor symptom of Parkinson disease: evidence from a case-control study. Arch. Neurol. 65, 1191–1194. [DOI] [PubMed] [Google Scholar]
- Dixon WJ (1980). Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462. [DOI] [PubMed] [Google Scholar]
- Djaldetti R, Shifrin A, Rogowski Z, Sprecher E, Melamed E, and Yarnitsky D (2004). Quantitative measurement of pain sensation in patients with Parkinson disease. Neurology 62, 2171–2175. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, and Malinow R (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143. [DOI] [PubMed] [Google Scholar]
- Eto K, Wake H, Watanabe M, Ishibashi H, Noda M, Yanagawa Y, and Nabekura J (2011). Inter-regional contribution of enhanced activity of the primary somatosensory cortex to the anterior cingulate cortex accelerates chronic pain behavior. J. Neurosci. 31, 7631–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbain DA, Cutler R, Rosomoff HL, and Rosomoff RS (1997). Chronic pain-associated depression: antecedent or consequence of chronic pain? A review. Clin. J. Pain 13, 116–137. [DOI] [PubMed] [Google Scholar]
- Ford B (2010). Pain in Parkinson’s disease. Mov. Disord. 25 (Suppl 1), S98–S103. [DOI] [PubMed] [Google Scholar]
- Huang S, Borgland SL, and Zamponi GW (2019). Peripheral nerve injury-induced alterations in VTA neuron firing properties. Mol. Brain 12, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KB, Regenbogen C, Ohse MC, Frasnelli J, Freiherr J, and Lundström JN (2016). Brain activations during pain: a neuroimaging meta-analysis of patients with pain and healthy controls. Pain 157, 1279–1286. [DOI] [PubMed] [Google Scholar]
- Johansen JP, and Fields HL (2004). Glutamatergic activation of anterior cingulate cortex produces an aversive teaching signal. Nat. Neurosci. 7, 398–403. [DOI] [PubMed] [Google Scholar]
- Johnson SW, and North RA (1992). Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci. 12, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiński J, Sullivan S, Chung JM, Ross IB, Mamelak AN, and Rutishauser U (2017). Persistently active neurons in human medial frontal and medial temporal lobe support working memory. Nat. Neurosci. 20, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Kwak C, Lee J, Sim SE, Shim J, Choi T, Collingridge GL, Zhuo M, and Kaang BK (2015). Bidirectional modulation of hyperalgesia via the specific control of excitatory and inhibitory neuronal activity in the ACC. Mol. Brain 8, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, and Porreca F (2009). Unmasking the tonic-aversive state in neuropathic pain. Nat. Neurosci. 12, 1364–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MY, Jang EY, Lee JY, Kim SP, Whang SH, Lee BH, Kim HY, Yang CH, Cho HJ, and Gwak YS (2018). The role of ventral tegmental area gamma-aminobutyric acid in chronic neuropathic pain after spinal cord injury in rats. J. Neurotrauma 35, 1755–1764. [DOI] [PubMed] [Google Scholar]
- Kuner R, and Flor H (2016). Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 18, 20–30. [DOI] [PubMed] [Google Scholar]
- Liu D, Tang QQ, Yin C, Song Y, Liu Y, Yang JX, Liu H, Zhang YM, Wu SY, Song Y, et al. (2018). Brain-derived neurotrophic factor-mediated projection-specific regulation of depressive-like and nociceptive behaviors in the mesolimbic reward circuitry. Pain 159, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad M, Usiello A, Carta M, Håkansson K, Fisone G, and Cenci MA (2005). Pharmacological validation of a mouse model of l-DOPA-induced dyskinesia. Exp. Neurol. 194, 66–75. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Akiyama H, and McGeer EG (1988). Rate of cell death in parkinsonism indicates active neuropathological process. Ann. Neurol. 24, 574–576. [DOI] [PubMed] [Google Scholar]
- Mhalla A, de Andrade DC, Baudic S, Perrot S, and Bouhassira D (2010). Alteration of cortical excitability in patients with fibromyalgia. Pain 149, 495–500. [DOI] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Okun A, Qu C, Eyde N, Ci S, Ossipov MH, King T, Fields HL, and Porreca F (2012). Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc. Natl. Acad. Sci. USA 109, 20709–20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, King T, and Porreca F (2013). Evaluation of reward from pain relief. Ann. N Y Acad. Sci. 1282, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navratilova E, Xie JY, Meske D, Qu C, Morimura K, Okun A, Arakawa N, Ossipov M, Fields HL, and Porreca F (2015). Endogenous opioid activity in the anterior cingulate cortex is required for relief of pain. J. Neurosci. 35, 7264–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notomi T, and Shigemoto R (2004). Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J. Comp. Neurol. 471, 241–276. [DOI] [PubMed] [Google Scholar]
- Panegyres PK (2004). The contribution of the study of neurodegenerative disorders to the understanding of human memory. QJM 97, 555–567. [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Wang M, and Arnsten AF (2013). Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb. Cortex 23, 1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, and Franklin KB (2008). The Mouse Brain in Stereotaxic Coordinates (Gulf Professional; ). [Google Scholar]
- Phelps CE, Navratilova E, and Porreca F (2021). Cognition in the chronic pain experience: preclinical insights. Trends Cogn. Sci. 25, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, and Johnston D (2002). Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat. Neurosci. 5, 767–774. [DOI] [PubMed] [Google Scholar]
- Porreca F, and Navratilova E (2017). Reward, motivation, and emotion of pain and its relief. Pain 158 (Suppl 1), S43–S49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B, and Bushnell MC (1997). Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science 277, 968–971. [DOI] [PubMed] [Google Scholar]
- Reckziegel D, Tétreault P, Ghantous M, Wakaizumi K, Petre B, Huang L, Jabakhanji R, Abdullah T, Vachon-Presseau E, Berger S, et al. (2019). Gender dependent pharmacotherapy for blocking transition to chronic back pain: a proof of concept randomized trial. medRxiv. 10.1101/19006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Bisco A, Nevian NE, Lacivita E, Leopoldo M, and Nevian T (2017). The brain-penetrant 5-HT7 receptor agonist LP-211 reduces the sensory and affective components of neuropathic pain. Neurobiol. Dis. 106, 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaguchi T, and Goldman-Rakic PS (1991). D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science 251, 947–950. [DOI] [PubMed] [Google Scholar]
- Schestatsky P, Kumru H, Valls-Solé J, Valldeoriola F, Marti MJ, Tolosa E, and Chaves ML (2007). Neurophysiologic study of central pain in patients with Parkinson disease. Neurology 69, 2162–2169. [DOI] [PubMed] [Google Scholar]
- Schnurr RF, and MacDonald MR (1995). Memory complaints in chronic pain. Clin. J. Pain 11, 103–111. [DOI] [PubMed] [Google Scholar]
- Sellmeijer J, Mathis V, Hugel S, Li XH, Song Q, Chen QY, Barthas F, Lutz PE, Karatas M, Luthi A, et al. (2018). Hyperactivity of anterior cingulate cortex areas 24a/24b drives chronic pain-induced anxiodepressive-like consequences. J. Neurosci. 38, 3102–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikes RW, and Vogt BA (1992). Nociceptive neurons in area 24 of rabbit cingulate cortex. J. Neurophysiol. 68, 1720–1732. [DOI] [PubMed] [Google Scholar]
- Stoof JC, and Kebabian JW (1981). Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature 294, 366–368. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, and Wolf ME (2005). Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J. Neurosci. 25, 7342–7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surges R, Freiman TM, and Feuerstein TJ (2004). Input resistance is voltage dependent due to activation of Ih channels in rat CA1 pyramidal cells. J. Neurosci. Res. 76, 475–480. [DOI] [PubMed] [Google Scholar]
- Taylor AMW, Becker S, Schweinhardt P, and Cahill C (2016). Mesolimbic dopamine signaling in acute and chronic pain: implications for motivation, analgesia, and addiction. Pain 157, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsu RC, and Wong YH (1996). Gi-mediated stimulation of type II adenylyl cyclase is augmented by Gq-coupled receptor activation and phorbol ester treatment. J. Neurosci. 16, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Hehn CA, Baron R, and Woolf CJ (2012). Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73, 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cordeiro Matos S, Jego S, Adamantidis A, and Séguéla P (2013). Norepinephrine drives persistent activity in prefrontal cortex via synergistic α1 and α2 adrenoceptors. PLoS ONE 8, e66122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This study did not generate code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.