

Abstract
Purpose:
CD8+ T lymphocytes can kill autologous melanoma cells, but their activity is impaired when poorly immunogenic tumor phenotypes evolve in the course of disease progression. Here, we analyzed three consecutive melanoma lesions obtained within one year of developing stage IV disease for their recognition by autologous T cells.
Experimental Design:
One skin (Ma-Mel-48a) and two lymph node (Ma-Mel-48b, Ma-Mel-48c) metastases were analyzed for T-cell infiltration. Melanoma cell lines established from the respective lesions were characterized, determining the T-cell–stimulatory capacity, expression of surface molecules involved in T-cell activation, and specific genetic alterations affecting the tumor–T-cell interaction.
Results:
Metastases Ma-Mel-48a and Ma-Mel-48b, in contrast with Ma-Mel-48c, were infiltrated by T cells. The T-cell–stimulatory capacity was found to be strong for Ma-Mel-48a, lower for Ma-Mel-48b, and completely abrogated for Ma-Mel-48c cells. The latter proved to be HLA class I–negative due to an inactivating mutation in one allele of the beta-2-microglobulin (B2M) gene and concomitant loss of the other allele by a deletion on chromosome 15q. The same deletion was already present in Ma-Mel-48a and Ma-Mel-48b cells, pointing to an early acquired genetic event predisposing to development of β2m deficiency. Notably, the same chronology of genetic alterations was also observed in a second β2m-deficient melanoma model.
Conclusion:
Our study reveals a progressive loss in melanoma immunogenicity during the course of metastatic disease. The genetic evolvement of T-cell resistance suggests screening tumors for genetic alterations affecting immunogenicity could be clinically relevant in terms of predicting patient responses to T-cell–based immunotherapy.
Introduction
CD8+ T lymphocytes (CTL) can exert potent in vivo cytotoxicity against autologous melanoma cells. This is demonstrated by the remarkable clinical responses observed in adoptive cellular therapy with autologous tumor-reactive T cells and therapy with immune-modulatory antibodies that release T cells from suppressive signals (1-5). The efficacy of these treatment regimens is based on the high intrinsic immunogenicity of melanoma cells that allows CTL to respond to multiple HLA class I–restricted tumor antigens (6). However, only a subgroup of patients receiving immunotherapy experiences clinical benefit, whereas others do not respond at all or after initial responses show progressive disease. In particular, the coexistence of therapy-responding and nonresponding metastases in individual patients suggests heterogeneous T-cell responsiveness of the different lesions (1). The underlying mechanisms therefore are most likely multifactorial. An immunosuppressive tumor microenvironment generated by regulatory T cells, myeloid-derived suppressor cells, or other immune regulators can restrict T-cell effectiveness (7-9). Furthermore T-cell activity can be hampered by specific genetic alterations that affect tumor immunogenicity. Interestingly, the antitumor activity of the T cells is considered a driving force that selectively favors the outgrowth of low-immunogenic melanoma phenotypes. Thus, besides eliminating tumor cells, T cells shape the immunogenicity of malignant cells, thereby supporting the escape of tumors from immune surveillance. This so-called T-cell–mediated immunoediting of tumors, initially defined by Schreiber and colleagues (10), has been observed in several mouse model studies, and evidences that this process also takes place in patients with cancer have been provided. By investigating metastatic cell lines and in some cases tissue samples, several genetic alterations affecting the tumor cells’ immunogenicity have been defined, including the loss of single or multiple HLA alleles and HLA haplotypes as well as the total lack of HLA class I antigen expression (11-15). So far, only a few studies followed the course of melanoma immunogenicity during disease progression due to the poor availability of tissue from sequential tumor metastases, corresponding cell lines, and autologous peripheral blood T lymphocytes. Coulie and colleagues (6) were the first to examine T-cell recognition of autologous tumor cells derived from two sequential metastatic melanoma lesions. They observed that T-cell responses toward melanoma cells from the second metastases were impaired as these cells expressed only one of the six HLA class I alleles (16). In 2005, Yamshchikov and colleagues (14) demonstrated that recognition of tumor cells from two consecutive melanoma lesions by autologous differentiation antigen-specific CD8+ T cells was compromised by an HLA haplotype loss in tumor cells from the first metastasis and by a mixed HLA-low and HLA-negative tumor cell phenotype in the second metastasis. Hence, as a basis for improving melanoma immunotherapy, more information is needed about the type and the sequence of genetic alterations in the tumor cells with potential impact on their recognition by cognate T cells.
Here, we monitored the immunogenicity of three consecutive melanoma metastases from patient Ma-Mel-48 obtained within one year of developing stage IV disease. We observed a gradual loss in immunogenicity culminating in complete T-cell resistance of the tumor cells caused by an irreversible HLA class I–negative phenotype. This originated from two different types of genetic alterations, a deletion on chromosome 15q where the B2M gene maps and an inactivating B2M gene mutation. Notably, tumor cells from all metastases of patient Ma-Mel-48 carried the same deletion on chromosome 15q, identifying this alteration as an early predisposing genetic event in the development of β2m deficiency. The same chronology of genetic alterations was also observed in a second patient model of β2m-deficient melanoma.
Materials and Methods
Patient samples
Patient Ma-Mel-48 presented with melanoma stage II in December 2000 at the age of 85 years. Progression to stages III and IV was diagnosed in October 2001 and June 2002, respectively. The patient was treated with temozolomide at the beginning of 2003 but never received immunotherapy. In September 2003, the patient died. Patient Ma-Mel-100 presented with melanoma stage II in January 1998 at the age of 72 years. Progression to stage III was diagnosed in September 2002. From February 1998 until September 2002, the patient received adjuvant IFNα therapy. In September 2005, the patient died.
Samples including tumor tissues and peripheral blood mononuclear cells (PBMC) were collected after approval by the institutional review board and patient informed written consent. Tissues were mechanically divided for cryopreservation and generation of the corresponding cell lines. Small tissue pieces were distributed in cell culture dishes, and outgrowing cells were split for the first time at 90% cell confluence. Melanoma cell lines were cultured in RPMI-1640 medium supplemented with glutamine, 10% FCS, and penicillin/streptomycin. Cells were cultured at 37°C in a 5% CO2 atmosphere. Cell lines were authenticated by genetic profiling at the Institute for Forensic Medicine (University Hospital Essen) using the AmpFLSTR-Profiler Plus Kit (Applied Biosystems) and routinely tested every 6 months.
Antibodies
The following murine mAbs were used for IHC: W6/32 to detect HLA class I antigens (Dianova); bbm.1 to stain for β2m (kindly provided by G. Moldenhauer, German Cancer Research Center, Heidelberg, Germany); anti–HMB-45 (Dako) to detect melanoma cells; anti-CD3 (BD Pharmingen) to stain for T cells.
For flow cytometry, the mouse mAbs were as follows: anti–HLA-ABC-APC (eBiosciences), anti–CD54-PE and anti–HLA-DR-PECy7 (Beckmann Coulter), anti–PD-L1-PE and anti–PD-L2 (Biolegend), anti–B7-H3 (R&D Systems), anti–B7-H4 (eBiosciences); L243 was used for detection of HLA-DR molecules (17) and HC10 for labelling of β2m-free HLA heavy chains (18, 19).
The following antibodies were used for Western blot analysis: mouse anti–Melan-A/MART-1 (Zytomed), mouse anti-Tyrosinase and anti-MITF (Santa Cruz Biotechnology), rabbit anti-DCT/TRP2 (kindly provided by V. Hearing, National Cancer Institute, NIH, Bethesda); mouse anti-STAT1, rabbit anti-phospho(p)STAT1, rabbit anti-JAK1, rabbit anti-GAPDH (Cell Signaling Technology); rabbit anti-IRF1 and mouse anti-β2m (Santa Cruz Biotechnology); rabbit anti-β2m (Sigma); mouse anti-TAP1 (NOB-1) and mouse anti-tapasin (TO-3; ref. 20); mouse mAb HC10 (18, 19).
Mixed lymphocyte-tumor cell culture
Anti–CD8-Microbeads (Miltenyi Biotec) were used for positive selection of CD8+ T cells from PBMC. Selected T cells were cocultured in 24-well plates at 106 cells per well with irradiated (100 Gray) autologous tumor cells at 105 cells per well in 2 mL AIM-V medium (GIBCO-BRL) supplemented with 10% human AB serum (complete medium). IL2 was added on day 3 at 250 IU/mL (Chiron Corporation). Lymphocytes (106 cells/well) were restimulated weekly with 105 irradiated tumor cells in IL2-supplemented complete medium.
Intracellular cytokine staining
For detection of intracellular IFNγ and TNFα, lymphocytes were stimulated for 4 hours with the indicated tumor cells (effector-to-target ratio of 1:1 or 1:2) in AIM-V complete medium containing 10 μg/mL Brefeldin A (Sigma-Aldrich). Then cells were stained with antihuman CD3-PE/Cy7 and CD8-APC-Alexafluor700 antibodies (Beckman Coulter) followed by fixation and permeabilization using the Fixation/Permeabilization Concentrate and Diluent Kit (eBioscience) and addition of an anti-IFNγ–FITC antibody (Beckman Coulter) or anti-TNFα–Pacific Blue antibody (Biolegend). Cells were analyzed in a Gallios flow cytometer, and the Kaluza software was used for data analysis (Beckman Coulter). Where indicated, antibody W6/32 (50 μg/mL; purified from hybridoma supernatant, kindly provided by M. Fatho, Mainz) and anti–PD-L1 (10 μg/mL; Biolegend) were added to block the TCR/HLA class I and PD-1/PD-L1 interactions, respectively. Mouse monoclonal IgG1 (mIgG; R&D Systems) was used as control antibody.
SNP array analysis
Genomic DNA was isolated from melanoma cell lines of patients Ma-Ma-48 and Ma-Mel-100 and from nonfixed peripheral blood cells (available only for patient Ma-Mel-48) using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. SNP arrays were performed using CytoScan HD Array from Affymetrix. Hybridization was done according to the manufacturer’s protocol, and data analysis was performed applying the program Chromosome Analysis Suite from Affymetrix. SNP array data files are accessible at the NCBI GEO database (accession number: GSE60218).
To infer the phylogenetic relationship of Ma-Mel-48 cells, genotype calls were extracted from the CytoScan HD Array data using the linux shell and R (21). As an outgroup genotype, calls from the autologous blood sample (166) were used. We identified 111,029 variable sites among the cell lines and blood sample. Of those, 64,023 were parsimony informative. We then used the maximum parsimony criterion, minimizing the total number of evolutionary steps required to explain the relationship of the tested samples. We ran maximum parsimony in MEGA (22) using 500 bootstrap replications and complete deletion of missing data, as well as subtree pruning–regrafting as a search method.
Additional information for Materials and Methods is provided in Supplementary Methods.
Results
Consecutive metastases from patient Ma-Mel-48 show heterogeneous T-cell infiltration and HLA class I antigen expression
To determine the development of melanoma immunogenicity during disease progression, we collected tumor tissues from three consecutive metastases of patient Ma-Mel-48. The first Ma-Mel-48a, a skin metastasis, was excised in July 2002, a few weeks after diagnosis of stage IV disease. The second and the third lesions Ma-Mel-48b and Ma-Mel-48c, lymph node metastases, were surgically removed in January 2003 and July 2003, respectively (Fig. 1A).
Figure 1.

Heterogeneous T-cell infiltration and HLA class I antigen expression in different metastases from patient Ma-Mel-48. A, localization and excision date of three sequential metastases from patient Ma-Mel-48. B, serial sections from cryopreserved Ma-Mel-48a and −48c metastatic lesions were analyzed for the expression of HMB-45 (melanoma cells), CD3 (T cells), and HLA class I antigens by IHC. Red staining indicates positive cells. C, flow cytometric analysis of total HLA class I antigen expression on melanoma cell lines established from the different metastases of patient Ma-Mel-48. Cells were stained with mAb W6/32. Histograms from one representative of three independent experiments are shown. Numbers indicate mean fluorescence intensity (MFI).
Serial tissue sections of the three metastases were first analyzed by IHC for their infiltration by CD3+ T lymphocytes. T cells accumulated in the periphery and center of metastases Ma-Mel-48a and Ma-Mel-48b pointing to a close tumor-T cell interaction (Fig. 1B and Supplementary Fig. S1). In contrast, no T-cell infiltrate was detected in metastasis Ma-Mel-48c (Fig. 1B). The presence of T cells correlated with the expression of HLA class I molecules, stained for with the antibody W6/32 binding a structural epitope formed by complexes of the β2m light chain and the HLA heavy chains. HMB-45–positive melanoma cells expressed HLA class I molecules in metastasis Ma-Mel-48a but not in Ma-Mel-48c (Fig. 1B).
Next, cell lines established from the respective metastatic lesions were analyzed for their HLA class I surface expression by flow cytometry. Ma-Mel-48a as well as Ma-Mel-48b cells were MHC class I–positive, whereas Ma-Mel-48c cells proved to be negative (Fig. 1C). Thus, the data generated by flow cytometry were in agreement with the results obtained by IHC.
Progressive loss in T-cell recognition of the different melanoma cell lines
On the basis of the HLA class I expression pattern, we analyzed the T-cell–stimulatory capacity of Ma-Mel-48a and Ma-Mel-48b cells. Mixed lymphocyte-tumor cell cultures (MLTC) were set up, consisting of purified autologous peripheral blood CD8+ T cells and Ma-Mel-48a or Ma-Mel-48b tumor cells. CD8+ T cells were stimulated in weekly intervals with the indicated irradiated tumor cells to increase the frequency of tumor-reactive T lymphocytes. After two stimulations, CD8+ T cells were harvested and analyzed for their responsiveness toward the tumor cells. As shown in Fig. 2A, approximately 9% of CD8+ T cells from MLTC with Ma-Mel-48a cells (MLTC-48a) responded to Ma-Mel-48a cells with IFNγ production, whereas only around 2% of the CD8+ T cells reacted against Ma-Mel-48b cells. Interestingly, CD8+ T cells harvested from MLTC-48b showed limited responsiveness to Ma-Mel-48b as well as Ma-Mel-48a cells (Fig. 2B). Approximately 2% of the T cells recognized Ma-Mel-48b cells, and comparable reactivity toward Ma-Mel-48a cells was observed. These results pointed to a lower T-cell–stimulatory capacity of Ma-Mel-48b cells as compared with Ma-Mel-48a cells. In either case, production of CD8+ T-cell cytokines proved to be HLA class I–dependent (Fig. 2C), thus no T-cell reactivity toward the HLA class I–negative Ma-Mel-48c cells was observed (Fig. 2A and B).
Figure 2.
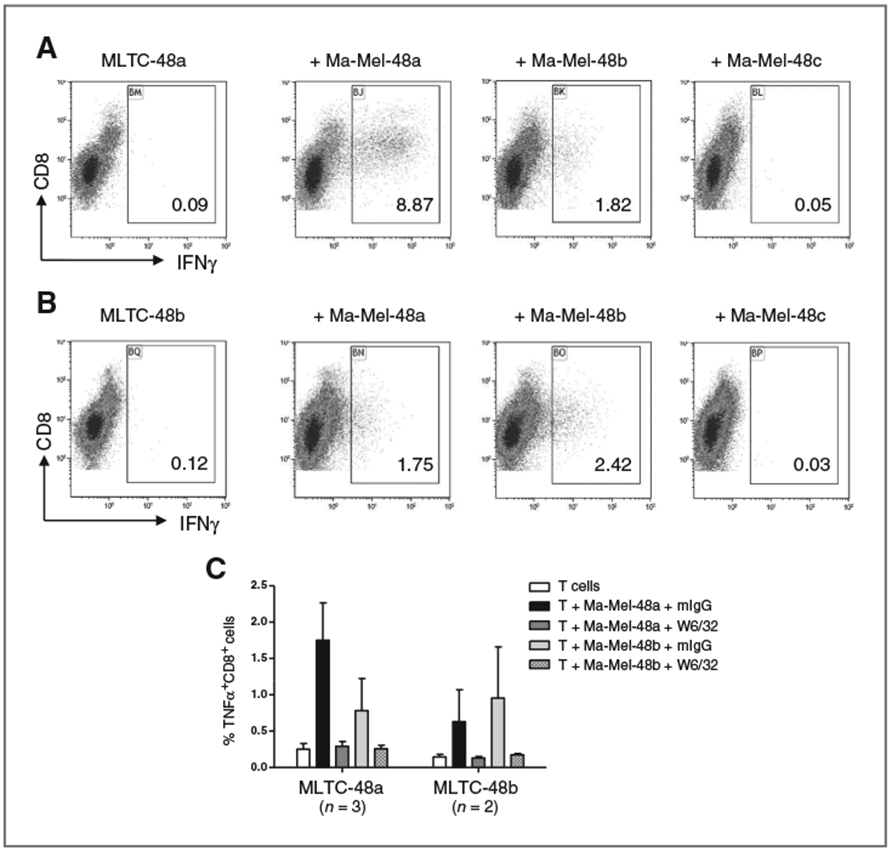
Tumor cells from sequential lesions differ in their T-cell–stimulatory capacity. A–C, analysis of the T-cell–stimulatory capacity of the different Ma-Mel-48 tumor cell lines. In autologous MLTC, isolated CD8+ T cells were stimulated twice with Ma-Mel-48a (MLTC-48a; A, C) or Ma-Mel-48b (MLTC-48b; B, C) cells and subsequently analyzed for their response toward different target cells by intracellular staining for IFNγ or TNFα. A, left, first dot plot: spontaneous IFNγ production by CD8+ T cells from MLTC-48a; second to fourth dot plot: production of IFNγ in response to the different target cells indicated above. B, left, first dot plot: spontaneous IFNγ production by CD8+ T cells from MLTC-48b; second to fourth dot plot: production of IFNγ in response to the different target cells indicated above. Results from one representative of two independent experiments are presented, numbers in dot plots indicate % IFNγ+CD8+ T cells within CD8+CD3+ T cells. C, to demonstrate HLA class I–dependent production of T-cell cytokines, tumor cells were incubated with blocking mAb W6/32 before addition of T cells. Control cells were incubated with mouse IgG1 (mIgG). The mean % TNFα+CD8+ T cells within CD8+CD3+ T cells of n = 2 or n = 3 independent experiments is presented.
In contrast with CD8+ T cells, autologous CD4+ T cells did not react toward any of the Ma-Mel-48 cell lines that lacked HLA class II expression even in the presence of IFNγ, due to epigenetic silencing of the transcriptional regulator CIITA (Supplementary Fig. S2 and data not shown).
Low immunogenic phenotype of Ma-Mel-48b cells is not due to altered expression of HLA alleles or immunomodulatory B7 molecules
To define the molecular mechanisms underlying differential T-cell recognition, we first analyzed Ma-Mel-48a and Ma-Mel-48b cells for potential alterations in HLA allele expression. HLA genotype analysis demonstrated that the HLA-A*0101, -B*0801, -C*0701 homozygous haplotype of autologous peripheral blood cells was conserved in the cell lines, both expressing considerable amounts of HLA-A−, HLA-B−, and HLA-C–specific mRNA (Fig. 3A). Data generated by qRT-PCR pointed to elevated HLA-B mRNA levels in Ma-Mel-48b cells as compared with Ma-Mel-48a cells. Accordingly, staining of Ma-Mel-48b cells for surface expression of HLA class I antigens and β2m-free HLA heavy chains was more intense compared with Ma-Mel-48a cells (Figs. 1C and 3A). Not expecting this shift in HLA class I allele expression to be responsible for the lower immunogenic phenotype of Ma-Mel-48b cells, we studied the surface expression of additional molecules involved in tumor–T-cell interaction. As shown in Fig. 3B, both cell lines expressed similar levels of the immunomodulatory B7 molecules PD-L1 (B7-H1), PD-L2 (B7-DC), and B7-H3, whereas no expression of B7-H4 was noted in either case. Though inhibitory PD-L1 was clearly detectable on Ma-Mel-48a and Ma-Mel-48b cells, its blockade did not enhance T-cell activities in our in vitro settings (Supplementary Fig. S3). Both melanoma cell lines also expressed similar levels of CD54 (ICAM-1), of importance for T-cell adhesion to the tumor cells (Fig. 3C).
Figure 3.
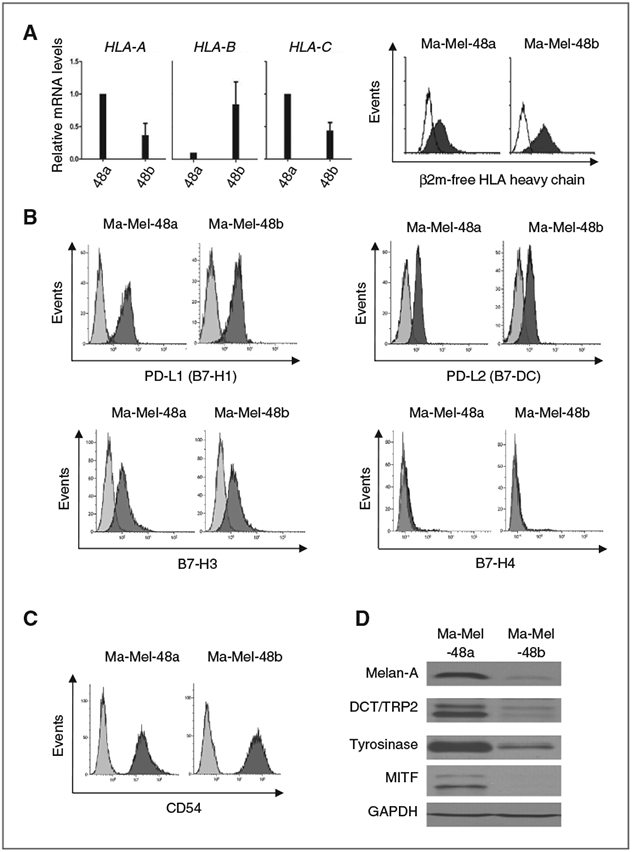
Conserved HLA phenotype but altered antigen expression in Ma-Mel-48b cells. A, left, mRNA levels of HLA-A, HLA-B, HLA-C in Ma-Mel-48a and Ma-Mel-48b cells were quantified by qRT-PCR and normalized to endogenous GAPDH mRNA. Expression levels, given as mean (+ SEM) of three independent experiments, are depicted relative to the expression in Ma-Mel-48a cells. A, right, flow cytometric analysis of β2m-free HLA heavy chain expression on Ma-Mel-48a and Ma-Mel-48b cells with mAb HC10. B, C, analysis of PD-L1 (B7-H1), PD-L2 (B7-DC), B7-H3, B7-H4, and CD54 surface expression on melanoma cell lines Ma-Mel-48a and Ma-Mel-48b by flow cytometry. Histograms from one representative of three independent experiments are shown. D, expression of MDA (Melan-A, Tyrosinase, DCT/TRP2) and its transcriptional regulator MITF in Ma-Mel-48a and Ma-Mel-48b cells was determined by Western blot. GAPDH served as a loading control. Data from one representative of three independent experiments are presented.
Although the analysis of specific surface molecules did not reveal significant differences between Ma-Mel-48a and Ma-Mel-48b cells, we postulated that variations in antigen expression levels could be responsible for the differential immunogenicity. Although the specificity of tumor-reactive T cells could not be studied due to limited sample material, we investigated the expression of melanoma differentiation antigens (MDA) in the tumor cells. As shown in Fig. 3D, Melan-A, Tyrosinase, and DCT/TRP2 as well as their transcriptional regulator MITF were decreased in Ma-Mel-48b as compared with Ma-Mel-48a cells, suggesting MDA down-regulation in Ma-Mel-48b cells could contribute to their lower T-cell–stimulatory capacity. Interestingly, Ma-Mel-48b dedifferentiation was not associated with a slow proliferative phenotype, as frequently observed for MITF-low melanoma cells (Supplementary Fig. S4; ref. 23).
The HLA class I–negative phenotype of Ma-Mel-48c cells is caused by β2m deficiency
To determine whether the HLA class I–negative phenotype of Ma-Mel-48c cells was reversible, cells were treated with type I and type II IFN. As shown in Fig. 4A, surface levels of HLA class I antigens were enhanced on Ma-Mel-48a and Ma-Mel-48b cells in response to IFN treatment. However, HLA class I antigen expression on Ma-Mel-48c cells could not be restored (Fig. 4A), although basic expression of interferon pathway components in Ma-Mel-48c cells was similar to the other cell lines and also the upregulation of STAT1, pSTAT1, and IRF1 in response to interferon treatment was comparable (Fig. 4B; Supplementary Fig. S5A). Analysis of several components of the antigen processing and presentation machinery (APM) by Western blot revealed expression of HLA heavy chains, TAP1, and tapasin in all cell lines, whereas β2m protein was expressed in Ma-Mel-48a and Ma-Mel-48b but not in Ma-Mel-48c cells, even when cells were treated with IFN (Fig. 4B). Also, mRNA expression of specific APM components was comparable between the three melanoma cell lines and was detectable in similar amounts also in melanocytes, but only Ma-Mel-48c cells lacked B2M mRNA (Fig. 4C; Supplementary Fig. S5B). These findings suggested the HLA class I–negative phenotype of Ma-Mel-48c cells was caused by the lack of B2M gene expression. Although B2M mRNA was not detectable by qRT-PCR, we obtained a shortened B2M-specific product by RT-PCR only for Ma-Mel-48c cells with primers amplifying the coding region from the start to the stop codon (Fig. 5A). Sequence analysis of this PCR product identified a 60-bp deletion starting in codon 96 (Exon II) of the B2M gene, leading to a C-terminally altered gene product (Fig. 5B). Notably, this B2M deletion variant could not be detected by qRT-PCR (Fig. 4C), as the assay primers were located in the B2M Exon II–III boundary. To confirm that the loss in HLA class I antigen expression was solely due to β2m deficiency, Ma-Mel-48c cells were transfected with a B2M expression plasmid. As shown in Fig. 5C, induction of HLA class I antigen expression was detectable on a population of transiently B2M-transfected cells. In agreement with the results obtained for the cell line, β2m was not detected in tumor cells in the corresponding metastatic lesion while it was expressed by normal cells (Fig. 5D).
Figure 4.
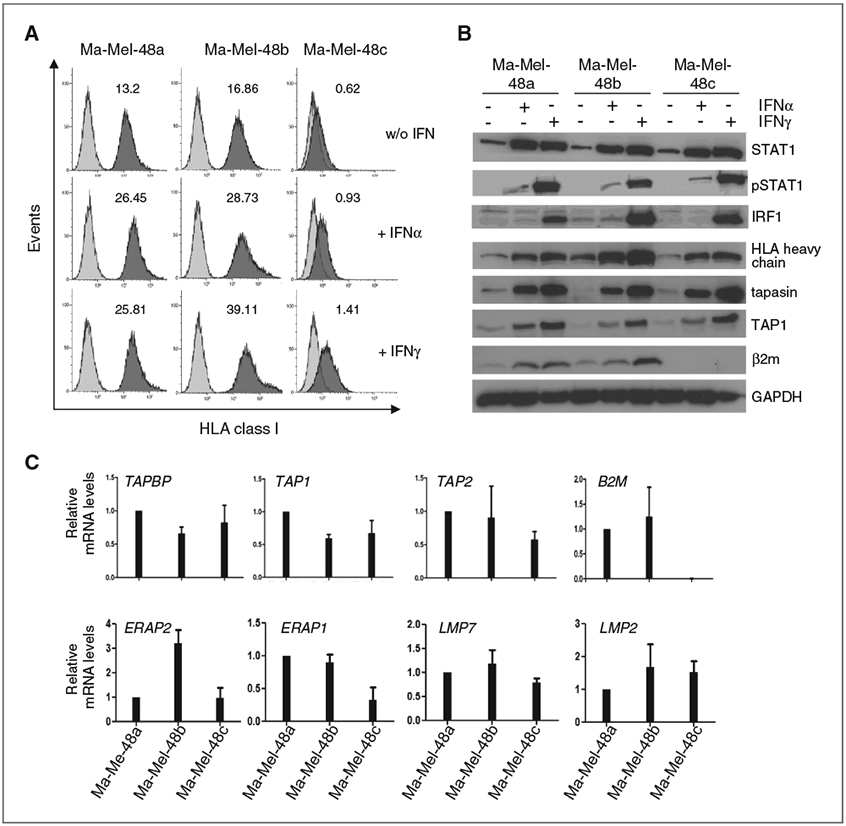
Interferon treatment of Ma-Mel-48c cells does not restore HLA class I antigen surface expression. The different Ma-Mel-48 cell lines were treated with IFNα (1000 U/mL) or IFNγ (500 U/mL) for 48 hours, controls were left untreated. A, cells were stained with mAb W6/32 for detection of HLA class I antigen expression. Data from one representative of three independent experiments are depicted, numbers indicate MFI values. B, cell lysates were analyzed by Western blot for the protein levels of interferon pathway components (STAT1, pSTAT1, IRF1) or APM components (HLA heavy chains, tapasin, TAP1, β2m). GAPDH served as loading control. One representative of three independent experiments is depicted. C, mRNA levels of different APM components were quantified by qRT-PCR and normalized to endogenous GAPDH mRNA. Expression levels, given as mean (+SEM) of three independent experiments, are depicted relative to the expression in Ma-Mel-48a cells.
Figure 5.

β2m deficiency of Ma-Mel-48c cells is caused by B2M gene mutation subsequent to B2M allele loss. A, semiquantitative analysis of B2M mRNA expression by RT-PCR. Ma-Mel-48c cells express a shortened B2M mRNA, a PCR product in the size of wild-type (wt) mRNA was only detectable in Ma-Mel-48a and Ma-Mel-48b cells. B, comparison of the C-terminal amino acid sequence (aa) of mutant β2m, as expressed in Ma-Mel-48c cells, and wt β2m. Capital letters indicate the aa sequence. The first 95 aa are identical in wt and mutant proteins. Stars indicate stop codons. C, Ma-Mel-48c cells were transfected with a B2M expression plasmid. Expression of HLA class I antigens on transient transfectants was determined by flow cytometry. D, analysis of β2m expression in metastatic lesion Ma-Mel-48c by IHC. Tumor cells were negative for β2m and did not stain red. E, SNP results given as allelic distribution of chromosome 15q are shown for DNA obtained from autologous PBMC and melanoma cells. All Ma-Mel-48 cell lines show loss of one chromosomal allele in the region 15q13.3 to 15q21.2 (Chr.15: 33,045,756-50,579,508; hg19). The location of B2M at 15q21.1 (Chr.15: 45,003,675-45,011,075) is shown by the dashed line. F, maximum parsimony tree showing the phylogenetic relationship of the melanoma cell lines and the blood sample (166) used as outgroup. Within the patient, a melanoma lineage leading to the studied cell lines evolved and was genetically divergent from the blood sample. A B2M loss evolved on this lineage. Then, a lineage leading to Ma-Mel-48a diverged from the melanoma ancestor and cumulated genotypic differences specific to this lineage. Later the lineages of Ma-Mel-48b and Ma-Mel-48c diverged and cumulated their specific genotypic differences. Hundred percent of the bootstrap replicates showed this grouping.
B2M allele loss is an early predisposing genetic event in the development of β2m deficiency
Previously, we reported that β2m deficiency is caused by the coincidence of a B2M gene mutation and allelic B2M loss. The B2M gene maps to chromosome 15q21.1 and can be lost as a result of chromosome 15q aberrations (12, 24). To define whether this was also the case in Ma-Mel-48c cells, SNP array analyses were performed on DNA obtained from the various tumor cell lines and autologous peripheral blood cells as a constitutive, normal control. Indeed, a partial deletion on chromosome 15q was observed in Ma-Mel-48c cells, encompassing the region 15q13.3 to 15q21.2 (Fig. 5E). Interestingly, the same portion of chromosome 15q was also found deleted in Ma-Mel-48a and Ma-Mel-48b cells.
From the genome-wide SNP genotype data, we therefore inferred the phylogenetic relationship of the melanoma cell lines and the corresponding autologous blood sample. A phylogenetic tree based on maximum parsimony showed that Ma-Mel-48b and Ma-Mel-48c cells were more closely related to each other than to Ma-Mel-48a cells, indicating that a lineage leading to Ma-Mel-48a evolved first. The grouping was present in all 500 bootstrap replicates of the analysis (Fig. 5F). Overall, these results demonstrated that the allelic loss of B2M occurred early in the course of disease and that a subsequent 60-bp deletion in the remaining B2M allele resulted in β2m deficiency in Ma-Mel-48c cells.
β2m-deficient melanoma cells from patient Ma-Mel-100 show the same chronology of genetic alterations
In addition to patient Ma-Mel-48, we observed loss of HLA class I antigen expression on melanoma cells from patient Ma-Mel-100 (Fig. 6A). Tumor cells from the two regional lymph node metastases Ma-Mel-100a and Ma-Mel-100b excised in April 2004 and May 2005, respectively, could not be stained with mAb W6/32 (Fig. 6B; Supplementary Fig. S6A). Consistent with the lack of HLA class I expression, T cells did not infiltrate the tumor lesion but were localized in the periphery (Fig. 6B). Also, cell lines established from the metastases showed an HLA class I–negative phenotype, which remained stable in the presence of type I and II interferons (Fig. 6C, data not shown). But HLA class I surface expression on Ma-Mel-100a and Ma-Mel-100b cells could be induced upon transfection of a B2M expression plasmid (Fig. 6C). Both cell lines also expressed similar amounts of B2M-specific mRNA (Fig. 6D); however, only Ma-Mel-100a cells lacked β2m expression in situ and in vitro (Fig. 6B and E), while the protein was detected in lysates from Ma-Mel-100b cells (Fig. 6E).
Figure 6.

B2M allele loss is also the initial genetic alteration in β2m-deficient melanoma cells from patient Ma-Mel-100. A, location and excision date of two sequential metastases from patient Ma-Mel-100. B, serial sections from cryopreserved metastasis Ma-Mel-100a were analyzed for the expression of HMB-45 (melanoma cells), CD3 (T cells), HLA class I antigens, and β2m by IHC. Red staining indicates positive cells. C, flow cytometric analysis of total HLA class I antigens on cell lines established from metastases Ma-Mel-100a and Ma-Mel-100b before and after transient transfection with a B2M expression plasmid. Cells were stained with mAb W6/32. Representative histograms from one of three independent experiments are shown. D, B2M mRNA levels were quantified by qRT-PCR and normalized to endogenous GAPDH mRNA. Expression levels, given as mean (+ SEM) of three independent experiments, are depicted relative to the expression in Ma-Mel-100b cells. E, β2m protein levels in cell lysates were determined by Western blot. GAPDH served as loading control. Representative results from one of three independent experiments are depicted. F, SNP array results showing the allelic distribution of chromosome 15q from DNA obtained from Ma-Mel-100a and Ma-Mel-100b cells. One chromosomal allele in the region 15q12 to 15q22.2 (Chr.15: 26,840,997-61,436,881;hg19) is lost in both Ma-Mel-100 cell lines. The location of B2M at 15q21.1 is shown by the dashed line. A subsequent B2M gene mutation was only detected in Ma-Mel-100b cells, identified as 12-bp deletion in Exon II.
Screening of the cells by SNP array for the underlying molecular alterations revealed deletion of the same region on chromosome 15q in Ma-Mel-100a and Ma-Mel-100b cells, ranging from 15q12 to 15q22.2 (Fig. 6F). Thus, the shared loss of one B2M allele was the initial genetic alteration in the development of β2m deficiency. Additional alterations, acquired subsequently, were different in both cell lines. Sequence analysis of the coding region revealed a 12-bp deletion in Exon II of the B2M gene in Ma-Mel-100b cells. In contrast, only wild-type B2M mRNA was detected in Ma-Mel-100a cells, pointing to an unknown posttranscriptional defect in B2M expression (Fig. 6F; Supplementary Fig. S6B).
Discussion
During disease progression melanoma cells acquire genetic alterations that can affect their recognition by cytotoxic T cells. Although some mutations increase the T-cell sensitivity of melanoma cells (25-27), others decrease the tumor cell’s immunogenicity and interfere with the effectiveness of immunotherapies. In terms of predicting responses to T-cell–based immunotherapy, it is important to understand the type and the sequence of genetic alterations that hamper the recognition of melanoma cells by cognate T cells.
Here, we studied the T-cell–stimulatory capacity of melanoma cells from three consecutive metastases of patient Ma-Mel-48 obtained at different times within one year of progressive stage IV disease. Although melanoma cells from skin metastasis Ma-Mel-48a strongly stimulated autologous CD8+ T cells, tumor cells from the lymph node metastasis Ma-Mel-48b cells, excised half a year later, did not. The lower T–cell-stimulatory capacity of Ma-Mel-48b cells could not be explained by an altered expression of immunomodulatory B7 molecules or the adhesion molecule CD54. In addition, there was no expression of HLA-DR molecules, described as ligands of the regulatory receptor LAG3 (28-31). Furthermore, HLA allele alterations, known to protect melanoma cells from certain T-cell specificities, were not detected in Ma-Mel-48b cells (13, 15, 32). We therefore assume the lower immunogenicity of Ma-Mel-48b cells was more likely due to differences in the antigen expression pattern. Specific T-cell antigens could be downregulated in Ma-Mel-48b in comparison with Ma-Mel-48a cells, as observed for the MDA (33). Furthermore, alterations in proteasome subunits and peptidases involved in the processing and presentation of tumor antigens could have contributed to the differential immunogenicity of the tumor cells (34-36).
The gradual decrease of melanoma immunogenicity in patient Ma-Mel-48 culminated in the complete T-cell resistance of Ma-Mel-48c cells, as these cells acquired an irreversible HLA class I–negative phenotype. This was caused by a lack in β2m expression due to an inactivating mutation in one allele of the B2M gene and concomitant loss of the second allele. We previously reported that β2m-deficient melanoma cells are characterized by deletions on chromosome 15q including the B2M gene mapping to 15q21.1 (24). Notably, loss of chromosome 15q material in melanoma is not a very rare event (37, 38). Of 70 metastatic melanoma samples analyzed, 16% were positive for loss of heterozygosity in chromosome region 15q21-22 (39). In the near future, exome studies will provide more detailed information about the frequency of B2M gene mutations in melanoma metastases, which we expect to be lower than B2M allelic losses.
By SNP array analysis on DNA from Ma-Mel-48c cells, we could clearly narrow down the deletion on chromosome 15q including the B2M gene. Notably, exactly the same deletion on chromosome 15q was already present in Ma-Mel-48a and Ma-Mel-48b cells. A phylogenetic tree inferred from SNP array data demonstrated that in patient Ma-Mel-48 a lineage leading to metastasis Ma-Mel-48a cells evolved first, followed by the lineages leading to Ma-Mel-48b and Ma-Mel-48c. This branching order is consistent with the timing of the excision of the metastases. The localized deletion on chromosome 15q, including one B2M allele, detected in all metastatic cell lines is thus an early event on the lineage leading to all three studied melanoma metastases. To our knowledge, this is the first study demonstrating that allelic B2M loss is the initial genetic alteration in the development of β2m deficiency. The same chronology of genetic alterations was also observed in tumor cells of patient Ma-Mel-100. Here, two metastases shared a common deletion on chromosome 15q associated with loss of a B2M allele. Remarkably, both metastases were found to be β2m deficient due to different subsequent alterations. A specific B2M gene mutation was identified in Ma-Mel-100b cells, whereas Ma-Mel-100a cells expressed significant amounts of only wild-type B2M mRNA. We assume that an unknown posttranscriptional mechanism blocks B2M mRNA translation in Ma-Mel-100a cells that requires further investigations.
Although our data suggest deletion of a B2M allele can be an early event in the course of melanoma progression, most studies detected β2m-deficient melanoma cells in late-stage disease (12, 14). Interestingly, Del Campo and colleagues (40) recently found nests of β2m-negative tumor cells to be present in a very early lesion from a patient with melanoma, who later developed a completely HLA class I–negative metastasis. Although the first β2m-negative tumor cells were detectable before immunotherapy, the completely HLA class I–negative lesion occurred under dendritic cell vaccination, leading to the assumption that vaccine-induced T-cell activity enriched β2m-deficient tumor cells. This was proposed also by previous studies that detected β2m loss in several recurrent metastases from patients receiving immunotherapy, such as patient Ma-Mel-100 who was treated with IFNα (13, 41). Because patient Ma-Mel-48 did not receive immunotherapy, this suggests that spontaneous antitumor T-cell responses led to the outgrowth of the HLA class I–negative melanoma immunophenotype. Besides, T-cell–independent mechanisms should also be taken into consideration. Recently, Garrido and colleagues demonstrated that HLA class I–negative melanoma cells showed enhanced proliferation, migration, and invasion in comparison with their HLA class I–positive counterparts, pointing to HLA class I molecules as tumor suppressors (42). Irrespective of the driving force, HLA class I alterations, in particular HLA class I loss, impede T-cell recognition of melanoma and will negatively influence all T-cell–based immunotherapies.
The infiltration of melanoma metastases by T cells is currently discussed as a biomarker that predicts responsiveness to immunotherapy, in which patients with T-cell–inflamed metastases more likely respond to treatment (43-45). Different mechanisms determine the migration of T cells into a metastatic lesion, such as the release of chemokines by tumor cells (45, 46). Furthermore, it has been described that the intensity of the T-cell infiltrate correlates with the level of HLA class I antigens expressed in the tumor (47). In accordance, CD3+ T cells were not detected in the center of metastasis Ma-Mel-48c or Ma-Mel-100a. T cells were rather located at the periphery, an observation also noted in other HLA class I–deficient melanoma lesions (unpublished data). This suggests that within the group of patients with melanoma with non T-cell–inflamed tumors a subgroup might exhibit HLA abnormalities. Thus, we propose that biopsies from patients with melanoma should be screened for genetic HLA alterations, including losses on chromosome 15q21.1, as this could prove valuable for immunotherapeutic treatment decisions.
Supplementary Material
Translational Relevance.
Metastatic melanoma is an aggressive, frequently deadly disease. In recent years, immunotherapies with anti–CTLA-4 and anti–PD-1 antibodies, exploiting the capacity of CD8+ T cells to kill immunogenic tumor cells, have shown great clinical promise. However, not all patients benefit from immunotherapy and currently biomarkers predicting responses to treatment are lacking. In our study, a progressive loss in immunogenicity was observed for melanoma cells derived from three consecutive patient metastases. This culminated in complete T-cell resistance of the tumor cells due to loss of HLA class I expression. Genetic analysis revealed an early acquired chromosomal deletion and subsequent inactivating gene mutation leading to β2m deficiency and the observed HLA class I–negative phenotype. Our findings suggest that tumors can genetically evolve to avoid being recognized by the immune system. Detecting genetic alterations affecting tumor immunogenicity could be of significant value in determining which patients are likely to benefit from melanoma immunotherapy.
Grant Support
This study was supported in part by the Helmholtz Alliance on Cancer Immunotherapy and by the NRW/EU-Ziel2-Programm 2007–2013 (005-1006-0057; to A. Paschen and D. Schadendorf); the DFG TRR77 and Integrated Research and Treatment Centre Transplantation (IFB-Tx, BMBF 01EO1302; to C.S. Falk); and PHS grants RO1CA138188 and RO1CA110249 awarded by the National Cancer Institute (to S. Ferrone).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Reprints and Subscriptions To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at pubs@aacr.org.
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 2005; 23:2346–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014;14:135–46. [DOI] [PubMed] [Google Scholar]
- 7.Becker JC, Andersen MH, Schrama D, Thor Straten P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol Immunother 2013;62:1137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umansky V, Sevko A. Melanoma-induced immunosuppression and its neutralization. Semin Cancer Biol 2012;22:319–26. [DOI] [PubMed] [Google Scholar]
- 9.Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol 2012;22:327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565–70. [DOI] [PubMed] [Google Scholar]
- 11.D'Urso CM, Wang ZG, Cao Y, Tatake R, Zeff RA, Ferrone S. Lack of HLA class I antigen expression by cultured melanoma cells FO-1 due to a defect in B2m gene expression. J Clin Invest 1991;87:284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paschen A, Méndez RM, Jimenez P, Sucker A, Ruiz-Cabello F, Song M, et al. Complete loss of HLA class I antigen expression on melanoma cells: a result of successive mutational events. Int J Cancer 2003; 103:759–67. [DOI] [PubMed] [Google Scholar]
- 13.Chang CC, Campoli M, Restifo NP, Wang X, Ferrone S. beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component down-regulation in melanoma cells derived from recurrent metastases following immunotherapy. J Immunol 2005;174:1462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamshchikov GV, Mullins DW, Chang CC, Ogino T, Thompson L, Presley J, et al. Sequential immune escape and shifting of T cell responses in a long-term survivor of melanoma. J Immunol 2005; 174:6863–71. [DOI] [PubMed] [Google Scholar]
- 15.Méndez R, Ruiz-Cabello F, Rodríguez T, Del Campo A, Paschen A, Schadendorf D, et al. Identification of different tumor escape mechanisms in several metastases from a melanoma patient undergoing immunotherapy. Cancer Immunol Immunother 2007;56:88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda H, Lethé B, Lehmann F, van Baren N, Baurain JF, de Smet C, et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997;6:199–208. [DOI] [PubMed] [Google Scholar]
- 17.Röhn TA, Reitz A, Paschen A, Nguyen XD, Schadendorf D, Vogt AB, et al. A novel strategy for the discovery of MHC class II-restricted tumor antigens: identification of a melanotransferrin helper T-cell epitope. Cancer Res 2005;65:10068–78. [DOI] [PubMed] [Google Scholar]
- 18.Stam NJ, Vroom TM, Peters PJ, Pastoors EB, Ploegh HL. HLA-A- and HLA-B-specific monoclonal antibodies reactive with free heavy chains in western blots, in formalin-fixed, paraffin-embedded tissue sections and in cryo-immuno-electron microscopy. Int Immunol 1990;2: 113–25. [DOI] [PubMed] [Google Scholar]
- 19.Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F. Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J Immunol 2003;171: 1918–26. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Campoli M, Cho HS, Ogino T, Bandoh N, Shen J, et al. A method to generate antigen-specific mAb capable of staining formalin-fixed, paraffin-embedded tissue sections. J Immunol Methods 2005;299:139–51. [DOI] [PubMed] [Google Scholar]
- 21.R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2010. [Google Scholar]
- 22.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 2013;30:2725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res 2008;68:650–6. [DOI] [PubMed] [Google Scholar]
- 24.Paschen A, Arens N, Sucker A, Greulich-Bode KM, Fonsatti E, Gloghini A et al. The coincidence of chromosome 15 aberrations and beta2-microglobulin gene mutations is causative for the total loss of human leukocyte antigen class I expression in melanoma. Clin Cancer Res 2006;12:3297–305. [DOI] [PubMed] [Google Scholar]
- 25.Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, et al. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci U S A 2005;102:16013–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013;19: 747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Rooij N,van Buuren MM, Philips D,Velds A,Toebes M, Heemskerk B et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 2013; 31:e439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer 2006;119:317–27. [DOI] [PubMed] [Google Scholar]
- 29.Quandt D, Fiedler E, Boettcher D, Marsch WCh, Seliger B. B7-h4 expression in human melanoma: its association with patients' survival and antitumor immune response. Clin Cancer Res 2011;17:3100–11. [DOI] [PubMed] [Google Scholar]
- 30.Hamaï A, Meslin F, Benlalam H, Jalil A, Mehrpour M, Faure F, et al. ICAM-1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res 2008;68:9854–64. [DOI] [PubMed] [Google Scholar]
- 31.Do JS, Valujskikh A, Vignali DA, Fairchild RL, Min B. Unexpected role for MHC II-peptide complexes in shaping CD8 T-cell expansion and differentiation in vivo. Proc Natl Acad Sci U S A 2012;109: 12698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiménez P, Cantón J, Collado A, Cabrera T, Serrano A, Real LM, et al. Chromosome loss is the most frequent mechanism contributing to HLA haplotype loss in human tumors. Int J Cancer 1999;83:91–7. [DOI] [PubMed] [Google Scholar]
- 33.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A 2002;99:16168–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Y, Sijts AJ, Song M, Janek K, Nussbaum AK, Kral S, et al. Expression of the proteasome activator PA28 rescues the presentation of a cytotoxic T lymphocyte epitope on melanoma cells. Cancer Res 2002;62:2875–82. [PubMed] [Google Scholar]
- 35.Kamphausen E, Kellert C, Abbas T, Akkad N, Tenzer S, Pawelec G, et al. Distinct molecular mechanisms leading to deficient expression of ER-resident aminopeptidases in melanoma. Cancer Immunol Immunother 2010;59:1273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vigneron N, Van den Eynde BJ. Proteasome subtypes and the processing of tumor antigens: increasing antigenic diversity. Curr Opin Immunol 2012;24:84–91. [DOI] [PubMed] [Google Scholar]
- 37.Höglund M, Gisselsson D, Hansen GB, White VA, Säll T, Mitelman F, et al. Dissecting karyotypic patterns in malignant melanomas: temporal clustering of losses and gains in melanoma karyotypic evolution. Int J Cancer 2004;108:57–65. [DOI] [PubMed] [Google Scholar]
- 38.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med 2005;353:2135–47. [DOI] [PubMed] [Google Scholar]
- 39.Maleno I, Aptsiauri N, Cabrera T, Gallego A, Paschen A, Loepez-Nevot MA, et al. Frequent loss of heterozygosity in the β2-microglobulin region of chromosome 15 in primary human tumors. Immunogenetics 2011;63:65–71. [DOI] [PubMed] [Google Scholar]
- 40.Del Campo AB, Kyte JA, Carretero J, Zinchencko S, Méndez R, González-Aseguinolaza G, et al. Immune escape of cancer cells with beta2-microglobulin loss over the course of metastatic melanoma. Int J Cancer 2014;134:102–13. [DOI] [PubMed] [Google Scholar]
- 41.Benitez R, Godelaine D, López-Nevot MA, Brasseur F, Jiménez P, Marchand M, et al. Mutations of the beta2-microglobulin gene result in a lack of HLA class I molecules on melanoma cells of two patients immunized with MAGE peptides. Tissue Antigens 1998;52: 520–9. [DOI] [PubMed] [Google Scholar]
- 42.Garrido C, Paco L, Romero I, Berruguilla E, Stefansky J, Collado A, et al. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis 2012;33:687–93. [DOI] [PubMed] [Google Scholar]
- 43.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14:1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 2012;72:1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine gene signature identifies lymph node-like structures in melanoma: potential for patient selection for immunotherapy? Sci Rep 2012;2:765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009;69:3077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al-Batran SE, Rafiyan MR, Atmaca A, Neumann A, Karbach J, Bender A, et al. Intratumoral T-cell infiltrates and MHC class I expression in patients with stage IV melanoma. Cancer Res 2005; 65:3937–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.