

Abstract
An increasing number of identified Parkinson’s disease (PD) risk loci contain genes highly expressed in innate immune cells, yet their role in pathology is not understood. We hypothesize that PD susceptibility genes modulate disease risk by influencing gene expression within immune cells. To address this, we have generated transcriptomic profiles of monocytes from 230 individuals with sporadic PD and healthy subjects. We observed a dysregulation of mitochondrial and proteasomal pathways. We also generated transcriptomic profiles of primary microglia from brains of 55 subjects and observed discordant transcriptomic signatures of mitochondrial genes in PD monocytes and microglia. We further identified 17 PD susceptibility genes whose expression, relative to each risk allele, is altered in monocytes. These findings reveal widespread transcriptomic alterations in PD monocytes, with some being distinct from microglia, and facilitate efforts to understand the roles of myeloid cells in PD as well as the development of biomarkers.
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder of aging that affects motor, cognitive and other functions 1. Familial cases of PD account for a minority of cases and are inherited in a mendelian form 2. Nevertheless, recent Genome Wide Association Studies (GWAS) have also established that sporadic forms of PD are highly heritable, and several variants have been identified that greatly increase one’s risk for developing sporadic PD 2. Understanding the function of these genes is a necessary step toward designing new and effective treatments. Recent genetic studies have identified over 78 PD risk loci 3, and many of these loci contain genes involved in immune function. While these and other findings suggest that the immune system plays an important role in PD, the underlying mechanisms of immune dysfunction are largely unknown. Genomic analysis has demonstrated that PD-associated susceptibility alleles alter the expression of nearby genes in peripheral monocytes 4-6 and that there is significant enrichment of PD-heritability in gene sets highly expressed in microglia 7. As of yet, it is unclear whether these peripheral monocytes, brain resident microglia, or both, influence disease risk and progression.
Several studies have identified altered myeloid functions in PD. Healthy microglia are essential for clearing of debris, such as α-synuclein 8,9 and for maintaining brain homeostasis. In addition, α-synuclein can activate microglia, releasing neurotoxic factors that may lead to death of dopaminergic neurons 10-13. Peripheral monocytes from PD patients have been shown to be hyperactive in response to α- synuclein stimulation 14. Monocytes have also been found to be capable of entering and interacting with the central nervous system (CNS) via the meninges 15-17 and may be involved in the phagocytosis of protein aggregates of debris from degenerating neurons 18,19. The Braak hypothesis, which proposes that α-synuclein pathology starts in the periphery 20, and the gut-origin hypothesis of PD 21-23 also postulates that peripheral immune cells might be exposed to PD-pathology early during the disease. Collectively, these studies support the importance of non-neuronal cell types including peripheral immune cells and brain resident glial cells in PD pathophysiology. However, there are critical gaps in our understanding of how these cells contribute to PD, in part due to the challenge of accessing patient-derived samples, and while some studies have characterized monocytes or microglia in PD 24,25, they were limited in sample size.
The Myeloid cells in Neurodegenerative Diseases (MyND) initiative is a collaborative effort with the goal of creating a multi-omic atlas of myeloid cells from the periphery and from autopsied brains of subjects with PD, Alzheimer’s disease (AD), and age-matched controls. This study reports the first phase of this initiative, which profiles the transcriptome of CD14+ monocytes and microglia from PD subjects and age-matched controls. Peripheral blood cells such as monocytes perform many of the fundamental cellular processes that are perturbed in PD 26, and we hypothesize that they may recapitulate some of the cellular pathology observed in the PD brain. As a source of patient tissue in which to study early disease processes, blood samples are easily accessible, cost- efficient, and can be obtained with minimal risk to the patients. Here, we performed large-scale, unbiased, systematic analysis incorporating genomic, and bulk and single-cell transcriptomic data to identify genes and co-expression networks that are dysregulated in PD myeloid cells. We observed that PD monocytes exhibited profound alterations for genes involved in mitochondrial and proteasomal function. Through single-cell RNA-sequencing (scRNA-seq) we found that these genes were highly expressed in the proinflammatory intermediate subpopulation. We further performed expression quantitative trait loci (eQTL) analysis of monocytes and identified 17 variants driving variation in mRNA abundance and PD susceptibility. Finally, we report that the mitochondrial transcriptome signature is discordant in the microglia and macrophages compared to monocytes, with the expression of mitochondrial genes downregulated in PD microglia and upregulated in PD monocytes.
Results
Participant recruitment and sample collection
Participants have been recruited from five clinical sites in New York City (see Methods). For each participant we have isolated CD14+ monocytes from 230 participants, including 135 with a diagnosis of sporadic idiopathic PD (“cases”) and 95 age-matched participants (“controls”) with no reported neurological or auto-immune diseases (Fig. 1; Extended Data Fig. 1A). Participants have a mean age of 67 years old. Sex, age and other demographic information can be found in supplemental material (Extended Data Fig. 1B, Table S1). The average age of onset (considered as age of diagnosis) in the PD group is 57.3 years old, with a disease duration of 8.3 years and Hoehn & Yahr (H&R)27 scale of 1.8 (see additional clinical information in Extended Data Fig. 1C, D).
Figure 1. Overview of the study design.

Parkinson’s disease and age-matched control subjects were recruited from five clinical sites: Movement Disorder Center at Mount Sinai Beth Israel (MSBI), Bendheim Parkinson and Movement Disorders Center at Mount Sinai (BPMD), Fresco Institute for Parkinson’s and Movement Disorders at New York University (NYUMD), and the Alzheimer’s Research Center (ADRC) and Center for Cognitive Health (CCH) at Mount Sinai Hospital. Fresh blood samples from PD and age-matched healthy subjects were collected following a rigorous, standardized set of procedures and used to isolate peripheral blood mononuclear cells (PBMCs). From the PBMCs, CD14+ monocytes were isolated using magnetic beads. Primary microglia were isolated from independent autopsied brains from two brain banks: Netherlands Brain Bank (NBB) and the Neuropathology Brain Bank and Research CoRE at Mount Sinai Hospital. Primary human microglia were isolated using CD11b+ beads. mRNAs from these cells were profiled using RNA-seq and single-cell RNA-Seq. Genome-wide genotyping was performed using DNA isolated from these samples. The data generated enabled to (from left to right) (i) description of the transcriptomic profiling of PD-monocytes at the gene, transcript and splicing levels (n=230), (ii) understanding of the contribution of the different monocyte subpopulation to the disease (n=10), (iii) integration of genomic and expression data to identify monocyte eQTLs (n=180) and (iv) comparison of the transcriptome signatures of PD peripheral monocytes to CNS microglia (n, samples=128, N, donors=55).
Primary human microglia have been isolated from postmortem brain tissues from independent donors. For this study, microglia from up to six different brain regions from 13 PD donors (22 samples) and 42 age-matched control donors (106 samples) have been used for RNA sequencing and downstream analysis (Table S2). The average age of death is 80.22 years old and 78.5 years old for control and PD cases, respectively. Disease duration in the PD group is 13.5 years, and sex is balanced (see Methods)
PD monocytes show mitochondrial and proteasomal alterations
We isolated human fresh monocytes from patient-derived blood using CD14+ beads. After rigorous quality control, we retained RNA-sequencing (RNA-seq) data from monocytes of 230 subjects for all downstream analyses. RNA-seq data were normalized and corrected to account for the effect of known biological and technical covariates (see methods; Fig. S1-S4). RNA-seq based quantifications enabled assessment of coding and non-coding differential gene expression, differential isoform expression, and differential splicing analyses (Fig. S1). A total of 300 differentially expressed genes (DEGs) were identified when comparing PD-derived monocytes to controls (False Discovery Rate [FDR] < 0.05). Of these, 162 identified DEGs were upregulated while 138 DEGs were downregulated (Fig. 2A, Table S3). The effect sizes for most of the DEGs were small (∣log2 fold change (FC)∣ < 0.5). The DEGs were not driven by LRRK2 or GBA mutation carriers, or by individuals of Ashkenazi Jewish (AJ) ancestry (Fig. S3C, D). Additionally, sex contributed small proportion of total variation in gene expression (mean variance 0.25% and standard deviation of 2.1%) (Fig. S3A). As the majority of PD cases were taking dopaminergic medication, Levodopa (L-dopa), we tested if gene expression was correlated with L-dopa equivalent daily dose (LEDD) on a subset of individuals (n=110). We found no significant correlation for any genes at FDR < 0.05, and of the DEGs, only four genes were significant at a nominal P-value < 0.05 threshold (Fig. S3E).
Figure 2. Transcriptomic analysis of PD-derived monocytes and age-matched controls.

(A) Volcano plot showing the fold-change (FC) of genes (log2 scale) between PD-monocytes (n=135) and controls (n=95) (x-axis) and their P-values significance (y-axis, −log10 scale). DEGs at FDR < 0.05 are highlighted in red (upregulated genes) and blue (downregulated genes). Moderated t-statistic (two-sided) is used for statistical test. (B) Pathway analysis for the upregulated (left panel) and downregulated (right panel) DEGs. Significance is represented in the x-axis (−log10 scale of the q-value). Only the 20 most significant pathways (FDR q-value < 0.05) with a minimum of 5 genes overlap are shown. Pathways are grouped and colored by biologically-related processes. n=230 independent samples (C) Examples of selected mitochondrial (top panel) and proteasomal (bottom panel) DEGs. Adjusted expression of the voom normalized counts after regressing covariates is shown. Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range. n=230 (D) Fold-change (log2 scale) correlation of DEGs between MyND monocytes (x-axis) and AMP-PD whole blood (y-axis). Genes are colored by significance, considering significant DEGs at FDR < 0.05. (E) Fold-change (log2 scale) correlation of DEGs between bulk monocytes (x-axis) and single-cell across-clusters analysis (y-axis). Four outlier genes were removed for easier visualization. Genes are colored by significance, considering significant DEGs at q-value < 0.05.
We performed gene set enrichment analysis (GSEA) to evaluate which biological processes were enriched for DEGs. The upregulated DEGs were significantly enriched for a number of Gene Ontology (GO) biological processes (BP) including mitochondrial function, antigen presentation, and RNA splicing (Fig. 2B, Table S4). The most significant BP were related to mitochondrial oxidative phosphorylation (OXPHOS), which includes essential components of respiratory chain complexes such as NADH dehydrogenase (NDUFA1 and B1) and Cytochrome C Oxidase (COX5A, 6B1, 7A2 and 7B) (FDR q-value < 0.05) (Fig. 2B, C, Table S4). Using a curated mitochondrial gene list 28, we found significant enrichment for OXPHOS genes (P-value = 0.00015, Fisher’s exact test), but not for other mitochondrial processes such as dynamics or mito-nuclear crosstalk (P-value = 0.80, Fisher’s exact test) and quality control (P-value = 0.66, Fisher’s exact test). The downregulated DEGs were overrepresented for functions including proteolysis, protein modification, differentiation and activation of innate immunity , and metabolic processes (FDR q-value < 0.05). Some of the genes in the proteolysis process are involved in proteasomal structure (PSMC5, PSMD5, and PSMD11) (Fig. 2B, C), ubiquitin (USP10), and autophagy-related function (GSK3B, PIK3R4, STAM). While most DEGs were part of a coherent biological function, we identified many that were not part of any processes including members encoding for the S100 proteins (S100A4, S100A6, S100P), which play an important role in inflammatory responses and function as damage-associated molecular pattern (DAMP) molecules 29,30. The S100 proteins have been shown to be upregulated in the substantia nigra and cerebrospinal fluid (CSF) of patients with PD as well as in a mouse model following MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), a toxin that causes parkinsonism in treated mice 31.
We next expanded these analyses to isoform transcript-level and local splicing (using intronic excision ratios) to identify transcriptomic dysregulation due to alternative splicing. We observed 1020 differentially expressed transcripts (DETs) and 161 differential splicing events (DS) at FDR < 0.05, corresponding to 939 and 158 unique genes, respectively (Extended Data Fig. 2 and 3, Table S5, S6). With the exception of mitochondrial function, the pathway analysis of DET and DS identified the same biological processes as the DEGs and expanded the list of genes involved in the protein degradation machinery including autophagy-related, proteasome, and lysosomal functions (Extended Data Fig. 2 and 3; Table S7, S8). These include the transmembrane protein 175 (TMEM175), which encodes a lysosomal K+ channel, and is in a PD GWAS locus that has been shown to play a critical role in lysosomal and mitochondrial function and PD pathogenesis 32. Also, two members (MTOR and RICTOR) of the rapamycin (mTOR) signaling pathway, a central regulator of the autophagy process 33, were identified in the DS analysis. Interestingly, some genes show significant alterations at the expression, transcript and splicing level (i.e.: NDUFV3, RICTOR). Together, these results highlight key genes involved in the machinery of protein degradation that have aberrant RNA splicing in PD monocytes.
With respect to the reproducibility of our results, we have performed two separate analyses to replicate our findings. First, we incorporated whole blood (WB) transcriptomic data from 780 PD cases and 504 controls from the Parkinson's Progression Markers Initiative (PPMI) (one of the cohorts of the Accelerating Medicines Partnership: Parkinson's Disease [AMP-PD]). Although not a direct replication since the AMP-PD transcriptome is from WB but given the large sample size, we expected to capture some of the monocyte-specific effects in blood. After QC (Fig. S5), we found 4484 genes at FDR < 0.01 (Table S9). We observed a significant overlap of the DEGs in the monocyte dataset and AMP-PD (P-Value = 0.041, Fisher-exact test). For the majority (6 out of 8 genes) of the mitochondrial DEGs in monocytes, we observed a concordant direction of effect in WB (Fig. 2D, 6C, S5F). However, the effect size in AMP-PD WB was weaker than in monocytes (mean FC = 0.09 in WB; mean FC = 0.27 in monocytes; for genes with FDR < 0.05; P-value < 2x10−16, independent-sample t-test) despite the large sample size (n = 1284) in AMP-PD. These results demonstrate the improved power of purified cell populations over mixtures of cell types such as whole blood, which may result in failure to properly capture the activity of cell-type-specific effects. Finally, we also validated our bulk RNA-seq findings in scRNA-seq of CD14+ monocytes by multiplexing 10 independent monocyte samples (seven PD, three controls; see below for further details). The effect size (normalized effect) 34 of DEGs from across-clusters gene expression from scRNA-seq were highly correlated with effect size from bulk RNA-seq DEGs (Spearman ρ =0.59, P-value = 5.04x10−6, Spearman rank correlation) (Fig. 2E, Table S10). For example, S100P and S100A6 were significant in both datasets (adjusted P-value < 0.05), and the majority (9 of 11) of the other members of the S100 gene family shared the same directionality in both datasets.
Figure 6. Parkinson’s disease susceptibility alleles alter gene expression in monocytes.

(A) Estimated proportion of heritability mediated by cis-genetic component of expression (h 2med/h 2g) in monocytes, DLPFC 46, and microglia 45 for AD 61, PD 3, Schizophrenia 62 and Height 63 GWAS. Bars indicate +/− SEM. (B) Colocalization of PD GWAS loci and monocyte cis expression or splicing QTLs. Shown in the bar plots are Posterior Probability (PPH4) from coloc 47 that supports the hypothesis (PPH4) that both eQTL (or sQTL) and PD GWAS share the same single variant. PD loci with suggestive colocalization (PPH4 > 0.5) are shown along with the eGene and the lead eQTL SNP (in LD with the lead GWAS SNP; r2 > 0.8). Genes in bold indicate reliable evidence in favor of a colocalized signal (defined as PPH3 + PPH4 > 0.8, PPH4/PPH3 > 2). n=180 independent samples. (C) Boxplot of selected eQTLs with gene expression (PEER adjusted) per individual stratified by genotype. The eQTL P-value and effect size (linear regression, see QTLtools) are listed on top. The PD GWAS effect allele is in bold. Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range. n=180 independent samples. (D) Fine-mapping of the BST1 locus. Colocalization of monocyte eQTL (top panel) and PD GWAS association (middle panel). Fine-mapping of BST1 using PolyFun 64 prioritizes two variants within the 95% credible set (bottom panel), one of which is a lead eQTL SNP (rs34559912). (E) Example of an sQTL within FAM49B showing intronic ratios stratified by genotypes (left panel). The PD effect allele and most significant intronic excision (chr8:129903350:129970943) within FAM49B are in bold. The red (bold) line represents the most significant junction. sQTL boxplot of chr8:129903350:129970943 intronic excision ratio (PEER adjusted) per individual stratified by genotype (right panel). Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range. n=180 independent samples.
To place the transcriptome changes in a systems-level framework, we performed Weighted Gene Co-expression Network Analysis (WGCNA) with the protein-coding genes of the 230 monocyte samples. We identified 65 modules of strongly co-expressed groups of genes, ranging from turquoise (largest, 2541 genes) to orangered3 (smallest, 30 genes), 6 of which were enriched for DEGs (Fig. S6, Extended Data Fig. 4, Table S11). We used LD score regression (LDSC) 35 to partition PD GWAS heritability into bins of correlated SNPs located within genes from each module. We found that 16 out of 65 modules showed enrichment for PD heritability (FDR < 0.05) including green, salmon, and red modules involving mitochondrial, lysosomal, and immune function, respectively (Extended Data Fig. 4A). Next, we correlated the module eigengene, the first principal component (PC) of the module gene expression level, with PD diagnosis. We observed three modules that were significantly correlated with PD diagnosis (FDR < 0.05, Two sided Wilcoxon signed-rank test), including turquoise (ubiquitin-related activity), antiquewhite4 (proteasome), and darkseagreen4 (Extended Data Fig. 4B). Given that multiple modules are associated with mitochondrial or lysosomal function, we considered taking the eigengene of all genes in either mitochondrial (n = 1302) or lysosomal (n = 526) GO categories. We found that the mitochondrial and lysosomal eigengenes were significantly upregulated and downregulated in PD, respectively (Fig. 3A, B). Taken together, these results illustrate that several co-expression gene modules in monocytes are enriched for PD heritability and further suggest subtle disruption of gene expression in specific biological networks including those with mitochondrial and proteo-lysosomal function.
Figure 3. Co-expression networks in monocytes capture PD-specific processes.

(A) Eigengene analysis of all genes in the “mitochondrial” GO category (n = 1302) between PD and controls (Left panel; Two sided Wilcoxon rank-signed test, P-value = 0.0012). Example of a module (green) enriched for PD heritability, mitochondrial genes, and upregulated DEGs (Right panel). Edges represent co-expression connectivity. Nodes in orange are upregulated DEGs at FDR < 0.05; yellow triangles are genes in PD GWAS loci. (B) Eigengene analysis of all genes in the “lysosome” GO category (n = 526) between PD and controls (Two sided Wilcoxon rank sum test, P-value = 0.0013) (Left panel). Example of a module (salmon) enriched for PD heritability, proteo-lysosomal genes, and downregulated DEGs. Nodes in orange are upregulated DEGs at FDR < 0.05; grey are selected proteo-lysosomal genes; and yellow triangles are genes in PD GWAS loci (Right panel). Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range.
scRNA-seq profiling of PD CD14+ monocytes
Human monocytes are subdivided into at least three different subpopulations (classical, intermediate, and non-classical) according to their surface expression of the receptor CD14 and the Fc receptor CD16 36. The three monocyte subsets are phenotypically and functionally different 37,38. To investigate whether the composition and gene expression profiles of monocyte subpopulations are altered in PD, we used flow cytometry (FACS) and scRNA-seq analysis to characterize different monocyte subpopulations from PD patients and age-matched healthy controls. First, we performed FACS analysis to assess differences in proportion of monocyte subsets between PD and controls. Using FACS, we did not observe any differences in the proportions of monocyte subpopulations in a subset of PD (n = 11) and control (n = 11) samples (P-value > 0.05, unpaired t-test) (Extended Data Fig. 5A), contrary to previous reports 39,40. Secondly, we performed scRNA-seq of CD14+ monocytes by multiplexing 10 individuals (seven PD, three controls, Table S12) on the 10x Chromium system with an expected yield of 20,000 single-cells. We identified six clusters including two main subpopulations that were detected corresponding to classical (CD14++/CD16−) and a CD16+ population that corresponds to intermediate (CD14++/CD16+) monocytes (Fig. 4A). The non-classical (CD14−/CD16++) monocyte subpopulation was not captured in the scRNA-seq due to the use of CD14+ selection method. Similar to our findings with FACS, we found no differences in proportions of monocyte subpopulations in PD vs controls (P-value > 0.05; Two sided Wilcoxon signed-rank test) (Extended Data Fig. 5B). After QC (Fig. S7, Extended Data Fig. 5C-F), we performed differential gene expression between the subpopulations (classical and intermediate, without considering diagnosis) and observed that 927 total genes were differentially expressed at FDR < 0.05 (Table S13). As expected many of the DEGs between clusters were marker genes for classical monocytes (CD14) or for intermediate populations (FCGR3A). We found that genes implicated in mitochondrial and proteasomal function, pathways enriched for DEGs in PD bulk monocytes, were highly expressed in the intermediate population relative to the classical population, showing that this subpopulation is enriched for genes related to PD pathophysiology. Specifically, genes that are members of the mitochondrial cytochrome c oxidase and NADH dehydrogenase families and proteasomal genes were highly expressed in the intermediate monocytes (Fig. 4B). We did observe some disease-related genes to be highly expressed in the classical subpopulation as well (e.g., S100A8). Finally, we performed differential expression analysis within each subpopulation and identified several DEGs that were only detected within the intermediate subpopulation but not in the bulk analysis. These included genes from several members of the complement component (C1QA), interferons (IFITM2), and chemokine (CXCL16) in the intermediate monocytes (Fig. 4C, Table S14). In summary, our scRNA-seq data enables the evaluation of molecular aspects of monocyte heterogeneity. Overall, these results suggest that intermediate monocytes, which comprise about ~8% of circulating monocytes and are involved in the production of reactive oxygen species (ROS) and inflammatory responses, are affected at the transcriptional level in PD.
Figure 4. Single-cell profiling of CD14+ monocytes from PD and control subjects.

(A) Generation of scRNA-seq from seven PD and three controls yielded 19,144 cells. Uniform Manifold Approximation and Projection (UMAP) visualization representing the six clusters including CD14++/CD16− classical monocytes (purple) CD14++/CD16+ intermediate cluster (green). (B) Comparison of the relative levels of expression of mitochondrial and proteasomal genes in the classical vs. intermediate monocytes using normalized effect (without considering diagnosis) 34,60. n=10 independent samples (C) Volcano plot showing the normalized effect within CD14++/CD16+ intermediate cluster of PD-monocytes and controls (x-axis) and their significance (y-axis, −log10 P-value). DEGs at q-value < 0.05 are highlighted in red (upregulated genes) and in blue (downregulated genes). Wald statistic is used for statistical test.
Monocyte signature in PD is distinct from microglia
We next sought to determine whether PD monocyte signatures are recapitulated in primary microglia. To address this question, we isolated CD11b+ primary microglia from fresh post mortem autopsied brains of 13 donors with PD and 42 controls (Extended Data Fig. 6A, B, Table S2). For comparison, we also isolated microglia from patients with other brain disorders, including depression (n = 72 samples), and neuropsychiatric disorders such as Schizophrenia [SCZ] and Bipolar disorder (n = 115 samples). The microglia samples were isolated from multiple brain regions based on the availability of high-quality tissues from the brain banks (Extended Data Fig. 6A, B) and subjected to RNA-seq. After rigorous QC and controlling for biological and technical covariates (Extended Data Fig. 6C-F), we performed differential gene expression between 22 PD and 106 control samples using a statistical method that accounts for repeated measures (in this case multiple brain regions from the same donor) while properly controlling the false discovery rate 41. Given the small sample size, we did not find any DEGs at FDR 0.05 in microglia but identified 222 DEGs at a suggestive threshold (FDR < 0.10) (Extended Data Fig. 6G, Table S16). In terms of reproducibility, we compared the results with an external dataset performed by single nuclei in sporadic cases of PD 42, where the authors described 29 genes significantly associated with microglial activation in PD. Comparing both datasets we found a significant overlap with our DEGs at FDR 0.1 (Fisher's exact test, P-value = 0.041, OR = 6.56).
A key finding is that OXPHOS gene signature is discordant compared to what we observed in peripheral monocytes, showing a downregulation in PD microglia (Fig. 5A, Extended Data Fig. 6H), signature specific to PD but not other brain disorders (Fig. 5B). In order to understand if the discordant gene expression is not due to cells being derived from the periphery and the CNS, we differentiated peripheral monocytes to monocyte-derived macrophages (MDMs) and performed qPCR of targeted OXPHOS genes. We observed that PD monocytes show higher expression of OXPHOS genes (COX7B, NDUFA1, PET100) in line with what we observed in Fig. 2. However, when monocytes from the same donors were differentiated to MDMs the directionality of OXPHOS genes was flipped (Fig 5C), opposite to the effect in monocytes from the same donors and the same directionality as microglia. These results demonstrate that directionality is cell-specific rather than CNS vs periphery, pointing to some alteration at the differentiation level.
Figure 5. Comparing the transcriptome profiles of PD monocytes and primary microglia.

(A) Effect size (log2[FC]) barplots of PD vs control differential expression in different datasets: substantia nigra (SN; light purple) 43, human microglia from MyND (dark purple), monocytes from MyND (dark green) and whole blood from AMP-PD (light green). Left panel: nuclear mitochondrial genes and proteasomal genes which are DEGs at FDR < 0.05 in monocytes from MyND. Right panel: All S100 genes tested across datasets. Bars indicate +/− SEM. Corrected P-value: *FDR < 0.05 in all datasets; *FDR < 0.15 for microglia MyND. Moderated t-statistic (two-sided) is used for statistical test. (B) Heatmap showing the fold-change (log2 scale) of disease vs controls of OXPHOS genes (y-axis) across different diseases (PD, Depression or Psychiatric disorders [Bipolar and Schizophrenia]). Blue represents log2(FC) < 0 (downregulated genes) and red represents log2(FC) > 0 (upregulated genes) when comparing disease vs. controls. Selected mitochondrial genes are shown. Nominal P-value: * P-value < 0.05; ** P-value < 0.01 for disease vs control differential expression. (C) qPCR validating the top differentially expressed OXPHOS genes (COXB, NDUFA1 and PET100) in monocytes and MDMs of controls (n = 11) and PD patients (n = 12). Graphs represent the fold change expression compared to controls. P-value was calculated via t-test. Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range.
Apart from the OXPHOS genes, we also identified genes involved in proteasomal function (PSMB5, PSMG2, PSMB2), complement component (C1QA, C1QB, and C1QC), and S100 calcium-binding proteins (S100A4 and S100A6) (nominal P-value < 0.05) in PD microglia. (Fig. 5A). The discordant gene expression signature between monocytes and microglia is consistent across most nuclear-encoded mitochondrial genes, whereas proteasomal genes and S100 family share a consistent directionality in monocytes and microglia (Fig. 5A). We accessed an independent dataset obtained from meta-analysis of 8 studies with gene expression profiles from bulk brain substantia nigra (SN) 43. 20 out of 151 OXPHOS genes were significant at FDR < 0.05 in the meta-analysis DEG of SN and all 20 genes were downregulated in PD. Although this is not a direct replication, it highlights the downregulation of OXPHOS genes in post-mortem PD brains (Fig. 5A). Taken together, our results show a reproducible discordant pattern of gene expression for mitochondrial OXPHOS genes in the monocytes and macrophages of PD subjects.
PD common variants alter gene expression in monocytes
The majority of PD risk-associated variants are located in non-coding regions of the genome. It is reasonable to hypothesize that a subset of these may cause disease by altering gene regulatory mechanisms as either expression (eQTL) or splicing (sQTL) quantitative trait loci. Here, we performed cis-eQTL analysis using monocytes from 180 subjects of European ancestry (Fig. S8) to systematically interrogate PD risk loci from the most recent GWAS 3 to uncover putative PD-dysregulated genes based on gene expression and splicing regulation. We identified 4,030 and 1,786 genes with cis-eQTLs and sQTLs at FDR < 0.05, respectively (Table S17, S18). Using a mediated expression score regression (MESC) 44, we estimate 26% (S.E.10%) of PD disease heritability is mediated by the cis-genetic component of monocyte gene expression levels (Fig. 6A). This estimate in monocytes is similar to what we observed in primary microglia (23%; S.E. 16%) 45 but lower than in prefrontal cortex (40%; S.E. 11%) 46, suggesting that a substantial proportion of PD heritability can be attributed to other CNS cells. Nevertheless, given that a large proportion of PD disease heritability is mediated by eQTLs in myeloid cells 4, we performed colocalization analysis 47 to determine whether a shared variant is responsible for both GWAS and QTL signal in a locus. We found that GWAS and eQTL signals colocalized in 15 out of 78 PD loci, suggesting that the disease-associated SNP (or one in very high LD to it) drove variation in expression in monocytes (Fig. 6B, Table S19). We observed suggestive levels of colocalization (PPH4 > 0.5) of GWAS and eQTL at three additional loci, including at the NOD2 locus (Fig. 6C), where the PD risk allele rs34559912-A decreases expression of NOD2. At the LRRK2 locus, we observe that the PD risk allele rs76904798-T increases LRRK2 expression in monocytes, consistent with what has previously been reported 4. We validate a previously identified eQTL at the cathepsin B (CTSB) locus, where the PD risk allele rs2740595-C decreases expression of CTSB (Fig. 6C). We found that the PD risk rs34559912-T allele, located within an intron of BST1, was strongly associated with lower expression of BST1 (Fig. 6C). The genetic analysis suggests that decreased expression of BST1 in monocytes is associated with increased risk for PD. Using a functional fine-mapping approach (see Methods), we found that the lead eQTL SNP (rs34559912) is also the top fine-mapped SNP (the SNP with the highest posterior probability of being causal within the 95% credible set) at the BST1 locus (Fig. 6D) and is within a monocyte-specific enhancer (Table S19). Notably, 60% (9 of 15 colocalized loci) of the lead eQTL SNP (or the lead GWAS SNP) are within CD14+ monocytes histone acetylation marks (H3K27ac) associated with enhancer activity, with one (rs34559912-BST1) specific to monocytes. Additionally, 27% (4 of 15 colocalized loci) of these lead eQTL SNPs (or the lead GWAS SNPs) are within microglia histone marks (H3K27ac), with two (rs6658353-B4GALT3 and rs1293298-CTSB) specific to microglia 48. Of these four, all are within chromatin accessibility (ATAC-seq) peaks in microglia 48, and of which three and one are within a PU.1 enhancer and promoter, respectively (Table S19). In a companion study, we have additionally fine-mapped all the PD GWAS loci and show that variants within the 95% credible sets for CTSB, LRRK2, RAB29, and GPNMB loci are located within microglia-specific enhancers (Schilder et al. in prep). In addition to expression, we performed sQTL analysis to identify local genetic effects that drive variation in RNA splicing in monocytes. We observed six PD risk alleles affecting the splicing of nearby genes (Fig. 6B). An example is PD risk allele rs2306528-T associated with an exon skipping event in FAM49B (Fig. 6E), a novel regulator of mitochondrial function 49. These results suggest that PD risk alleles modulate disease susceptibility by regulating the expression or splicing of genes in peripheral monocytes.
Discussion
Multiple lines of evidence implicate alterations in the immune system in PD 26, but the contribution of specific immune cells and their mechanisms in PD remains unclear. Here, we present a population-scale transcriptomic study of peripheral monocytes and primary microglia from subjects with PD. Our findings suggest widespread gene expression alterations in the PD myeloid cells, some of which are shared between the periphery and the CNS, while others have discordant effects. A key finding of our work is that peripheral monocytes derived from PD patients show widespread dysregulation in mitochondria and proteo-lysosomal pathways. We further show that mitochondrial genes are upregulated in peripheral monocytes but are downregulated in CNS microglia and peripheral MDMs, pointing to a cell specific effect. Our single-cell-resolution analysis further suggests that these transcriptional alterations are specific to the intermediate monocyte subpopulation. By intersecting transcriptomics and genetics, we also demonstrate a large proportion (~22%) of PD risk alleles alter the expression or splicing of genes in monocytes.
Although dysregulation of mitochondrial homeostasis in PD has been previously reported, these studies were mostly restricted to studying dopaminergic neurons, fibroblasts or whole blood from individuals with PD 40,50-56. Our work provides a unique view of monocyte transcriptome alterations associated with PD pathophysiology. We report that proteo-lysosomal signature is downregulated in PD monocytes, which could be in line with the accumulation of aberrant proteins associated with the disease. However, contrary to most published results, we also report an unexpected finding that OXPHOS genes are upregulated in peripheral monocytes from individuals with sporadic PD. The increased expression of OXPHOS genes in PD is a finding that has only been shown for two genes (SDHB and ATP5A) in lymphoblasts 57. A plausible explanation could be that PD monocytes reflect a hyperactive state with increased OXPHOS activity 57, and may be responsible for the elevated oxidative stress in PD. Another possibility is that the increased OXPHOS activity in PD monocytes is a compensatory effect of dysfunctional mitochondria due to the rapid turnover of monocytes. Due to monocyte heterogeneity, we further investigated the contribution of different subclusters at the single cell level and showed that OXPHOS and proteo-lysosomal genes are highly expressed in the proinflammatory intermediate CD14++/CD16+ subpopulation. We also generate the first (to our knowledge) unbiased transcriptomic dataset of freshly isolated microglia from PD and controls, where we observed the opposite effect, as the OXPHOS genes are downregulated in PD microglia. The data from microglia was replicated with SN data, although it is not a direct replication due to the absence of independent and sufficiently large microglia dataset from PD subjects. The results presented in this study are consistent and confirm previous observations showing reduction of OXPHOS gene expression in post-mortem brains of individuals with PD 50,51. When we differentiated peripheral monocytes (where OXPHOS genes are upregulated in PD) to MDMs upon M-CSF treatment we observed that the monocyte signature is discordant, showing that PD MDMs have lower expression of OXPHOS genes compared to controls. What these results reveal is a dysfunctional differentiation towards the macrophage phenotype in PD and points out that the discordant effects are due to cell specific differences. Taken all together, we hypothesize that the upregulation in OXPHOS genes in monocytes could be due to a compensatory effect to counteract the dysregulation observed in the macrophages. This work also improves our understanding of the PD-associated genetic risk factors influencing innate-immune mechanisms. Although a large proportion of PD heritability is mediated by the cis-genetic component of gene expression in neuronal tissues, our findings provide evidence that about ~25% of PD heritability is estimated to be mediated by myeloid cell-specific cis-eQTLs. This estimate is consistent with our observation that in at least 17 loci, the PD risk variants are likely to modify disease susceptibility, at least in part, by modulating gene expression or splicing in peripheral monocytes. However, given that many of the monocyte lead eQTL SNPs (or fine-mapped credible set of SNPs) are also within microglia enhancers, it is plausible that the observed genetic effect on monocyte gene expression may be a proxy for infiltrating macrophages and/or resident microglia found at the sites of neuropathology. Given the current data, it is difficult to discern the exact cellular context in which these variants may act. It is also plausible that many of these eQTL-PD GWAS colocalizations may be identified in other CNS cell types (e.g., neurons, astrocytes or oligodendrocytes), where some of these genes are also expressed. Future studies incorporating eQTL datasets from primary human microglia, or from scRNA-seq will be an important resource in pinpointing the cellular contexts in which PD-causing genetic variants affect gene expression.
Our work has major implications for discovering novel blood-based biomarkers for differentiating individuals with PD from control individuals. To date, biomarker studies in PD have largely focused on candidate approaches, with an emphasis on protein measures obtained in the CSF or brain imaging 1,58,59 which is considerably more difficult to obtain than blood. The use of monocyte gene expression to discover novel biomarkers of the disease state has several advantages. Firstly, monocytes isolated from peripheral blood are highly accessible human tissue, unlike the brain. While peripheral blood may be more easily obtained than primary monocytes, our results suggest that the magnitude of effects for DEGs was two-fold higher in monocytes compared to whole blood despite the smaller sample size (n = 230 in monocytes compared to 1,284 for whole blood from AMP-PD cohort). These results emphasize the power of purified cell populations that are not mixtures of cell types such as whole blood, which may result in the failure to properly capture the activity of cell-type-specific effects. Secondly, our study shows that peripheral monocytes from PD cases differ from those of control subjects. To this end, we identified several genes whose expression is altered not only in PD monocytes but also exhibit altered expression levels in microglia and SN of individuals with PD. For example, the S100 family of genes whose upregulation is reproducible in all four datasets that we have compared (monocytes, whole blood, microglia, and SN) and which have also been shown to be upregulated in CSF of patients with PD 31 are excellent candidates for potential blood-based biomarkers. Further longitudinal studies are necessary to assess whether transcriptional changes in monocytes are predictive of disease progression.
In summary, by defining the transcriptional signatures of peripheral monocytes from sporadic PD patients, we have uncovered PD-associated alterations of mitochondrial and proteo-lysosomal genes in peripheral cells. We demonstrate that although the same mitochondrial processes are altered in PD monocytes and microglia, the direction of effect of altered genes are distinct. Building on our data, future research should assess the functional bioenergetic properties of the CNS and peripheral cells in sporadic PD to unravel potential mechanisms leading to the dysregulation described here. Overall, these results provide support for the utility of monocyte gene expression profiles as potent tools for understanding molecular mechanisms, for the identification of novel therapeutic targets, and for the development of blood-based biomarkers.
Methods
See Supplementary information for demographics and clinical overview of study cohorts.
PBMC, monocyte isolation, and MDM differentiation
A maximum of 30 ml of blood was collected in Vacutainer blood collection tubes with acid citrate dextrose (ACD) (BD Biosciences). Fresh blood was shipped to the Raj laboratory and processed within 2-3 hours. First, blood was centrifuged at 1,500 g for 15 mins, and aliquots of whole blood and plasma were stored at −80 °C. Subsequently, blood was diluted in 2-fold PBS (Gibco) and PBMCs were isolated using SepMate tubes (StemCell Technologies) filled with 15 ml of Ficoll-Plaque PLUS (GE Healthcare) through a 15 mins centrifugation at 1,200 g. After washing with PBS, 5 million PBMCs were sorted to monocytes using CD14+ magnetic beads (Miltenyi) in the AutoMacs sorter and following manufacturer’s instructions. PBMCs and monocyte viability was assessed using Countess II Automated Cell counter (Thermo Fisher). After sorting, monocytes were stored in RLT buffer (Qiagen) + 1% 2-Mercaptoethanol (Sigma Aldrich) at −80 °C. Purity of the monocyte sorting was assessed via FACS and expression markers (RNA-seq). Remaining PBMCs were cryopreserved in 90% FBS (Germini) + 10% DMSO (Sigma Aldrich) at a concentration of 10 million cells/ml in Nalgene cryogenic vials (ThermoScientific). Vials were placed in NalGene CryoFreezing containers at −80 °C during 24-72 hours, and subsequently placed at liquid nitrogen for storage long-term.
For MDM differentiation, 0.5 million monocytes were plated at a concentration of 1 million cells/ml in 24 well/plate and differentiated to MDMs upon treatment with 50 ng/ml of M-CSF during 6 days, changing media every 3 days. Day 6 M-CSF was removed from media and cells were collected 24 h later in RLT.
DNA isolation and genotyping
DNA isolation and genotyping
When isolating DNA from blood, an aliquot of 1 ml was used. We used the QiAamp DNA Blood Midi kit (Qiagen) and followed the manufacturer's instructions. DNA quality and concentration was assessed using a Nanodrop. Samples were genotyped using the Illumina Infinium Global Screening Array (GSA), which contains a genome-wide backbone of 642,824 common variants plus custom disease SNP content (~ 60,000 SNPs). Additionally, we performed targeted genotyping for specific regions associated with neurodegenerative diseases (LRRK2, GBA and APOE). LRRK2 and GBA genotyping was outsourced to the Dr. William Nichols’ laboratory at the Cincinnati Children’s Hospital. SNP genotyping was performed for the G2019S variant in LRRK2 and the 11 most common variants in GBA (84GG, IVS2+1, E326K, T369M, N370S, V394L, D409G, L444P, A456P, RecNcil, R496H). The three major APOE isoforms (APOE 2, APOE 3, APOE 4) were assessed in the laboratory using Taqman assays for both rs429358 (C___3084793_20) and rs7412 (C___904973_10) from ThermoScientific following manufacturer’s instructions. 10 ng of DNA were added to the SNP reaction mix in a 96-well plate. Fluorescence reading of the Taqman assays was performed using QuantStudio 7 Flex (Applied Biosystems). See Supplementary information for genotype quality control and imputation.
Transcriptomic analysis
RNA isolation, library preparation and sequencing
RNA was isolated from monocyte samples stored in RLT buffer. After thawing on ice, RNA was isolated using RNeasy Mini kit (Qiagen) following the manufacturer's instructions including the DNase I optional step. Once RNA was isolated, samples were stored at −80 °C upon library preparation. Prior to library preparation, RNA concentration was assessed using Qubit and the RNA integrity number (RIN) by TapeStation using Agilent RNA ScreenTape System (Agilent Technologies). The median for the RIN values across the cohort is 9.7. Only 8 samples showed RIN < 5 and removing these samples did not alter results. Library preparations were done either in-house or at Genewiz Inc. using in both cases ribo-depletion strategy to remove rRNA. For in-house library preparation, we used the TruSeq Stranded Total RNA Sample Preparation kit (Illumina), with the Low Sample (LS) protocol and followed the manufacturer’s instructions. For samples prepared at Genewiz, we shipped the RNA and samples were processed using the Standard RNA-seq protocol. Samples were sequenced in 3 independent batches at Genewiz Inc. with a depth of 60 million 150-bp paired-end reads using Illumina HiSeq 4000 platform.
RNA-seq data processing, quality control, and normalization
To process FASTQ files, we utilized RAPiD-nf, an efficient RNA-seq processing pipeline implemented in the NextFlow framework 65. Following adapter trimming with trimmomatic (v0.36) 66, all samples were aligned to the hg38 build (GRCh38.primary_assembly) of the human reference genome using STAR (2.7.2a) 67 with indexes created from GENCODE (v30) 68. Gene expression was quantified using RSEM (1.3.1) 69; splice junction reads were extracted and quantified using Regtools (0.5.1) 70. Sequencing quality and technical metrics were assessed both before alignment with FASTQC (0.11.8) 71 and after alignment with Picard (2.20) 72 and Samtools (v1.9) 73.
As part of the RAPiD 3.0 pipeline, FASTQC was run for all samples and MultiQC was used to visualize and interpret the results. No samples were removed based on FASTQC metrics. Post alignment quality control of RNA-seq data was performed using Picard. Initial inclusion criteria consisted of at least 20 million passed reads, at least 20% of reads mapping to coding regions, and ribosomal rate < 30%. Additional QC was completed analyzing estimated counts, Transcripts Per Million (TPM), Counts Per Million (CPM), and TMM-voom (trimmed means of M-values) normalizations. Samples were removed if they were determined to be sex mismatches based on the expression of genes UTY and XIST compared to reported sex (Fig. S2D). Four samples were removed based on sex mismatches. Based on immune cell marker gene expression no samples were removed for having cell type contamination. Outliers were also removed after adjusting for covariates using dimensionality reduction through principal component analysis (PCA) and MDS that were selected to be used in differential analyses (Fig. S2E). Seven samples were removed after PCA and MDS analysis.
Individual gene and transcript level counts and TPM used for downstream analyses were generated using RSEM and assembled to a matrix via the tximport R package. Then, CPM were calculated using cpm() function from the edgeR packing in R. Lowly expressed genes were filtered out, which were defined as having less than one count per million in at least 30% of the samples leading to a total of 13,667 genes.
Understanding sources of expression variation and covariate selection
To understand major sources of variation in the gene expression data, we used the R package variancePartition 74, which uses a linear mixed model to attribute a percentage of variation in expression based on selected covariates on a per gene basis. This package was used with each of the datasets that were analyzed and treated separately. This way, we could identify the sources of variation for each dataset and thus regress out the specific covariates in each of the analyses. After extensive QC (see Supplementary information), we decided to use a design which includes those covariates that explained the most variance in gene expression (on average across genes) according to variancePartition results, which is as follows: expression ~ rna_batch + age + sex + RIN + PCT_USABLE_BASES + PCT_RIBOSOMAL_RNA + MDS1 + MDS2 + MDS3 + MDS4.
Differential Expression Analysis
Differential expression analysis was performed between PD cases and controls through a linear model using the R package limma version 3.38.3 75. These data were normalized using TMM values calculated from edgeR and voom transformed (Fig. S4A). Limma fits a linear model, and then runs a Bayesian moderated t-test which provides a P-value. P-values were then adjusted for multiple testing correction using the Benjamini-Hochberg FDR correction, which is implemented in the limma package. Differential isoform expression was performed following the same protocol.
Pathway and Gene Set Enrichment analysis
(i) Pathway analysis: we performed pathway analysis independently using the following input gene sets: upregulated DEGs (162), downregulated DEGs (138), differential splicing events (161) and DE transcripts (939) at FDR < 0.05. We used GSEA 76, focusing on Biological processes from Gene Ontology and limiting to gene sets between 10-500 genes. We show the 20 most significantly enriched pathways with at least five genes that overlap. (ii) Gene set enrichment analysis: to test specific pathways we used curated gene sets and tested statistical enrichment using Fisher exact test. The pathway lists were arranged as follows: (1) All gene ontology gene sets were downloaded from the amigo.geneontology.org resource searching for the specific pathways: mitochondria (315 genes), proteasome (450), lysosome (682), inflammatory response (694). (2) Mitochondrial curated list: 315 genes 28. From this gene list, we separated the different specific mitochondrial pathways (OXPHOS, Mitonuclear cross-talk and mitochondrial dynamics) following the paper specifications. (3) Proteasomal curated list: 39 genes 77. (4) Ubiquitin-related curated list: 428 genes combined from ubiquitin-like modifier activating enzymes (HGNC dataset), ubiquitin conjugating enzymes E2 (HGNC dataset) and ubiquitin ligase E3. (5) Lysosomal curated list: 435 genes from The Human Lysosome Gene Dataset.
qPCR validation
Briefly, RNA was reversed transcribed to cDNA and qPCR was performed using Taqman assays (ThermoScientific) for targeted genes. Fluorescence reading of the Taqman assays was performed using QuantStudio 7 Flex (Applied Biosystems). Results were analyzed using the comparative threshold cycle (Ct) and expressed as fold-change.
Splicing analysis
See Supplementary information for splicing analysis.
Parkinson’s Disease Progressive Marker Initiative (PPMI) RNA-seq data analysis
See Supplementary information for whole blood PPMI transcriptomic analysis.
Co-expression Network Analysis
Expressed genes were filtered by protein-coding according to GENCODE annotation version 30 (n = 11,475 protein-coding genes), and expression data was transformed using voom. To minimize the effect of confounders we used the “num.sv” function in the Bioconductor package sva embedded with the permutation-based approach algorithm “be” 78, 79, to get the number of surrogate variables (SVs) which estimated 12 SVs to be regressed from the whole matrix (n=230 samples). Then, using the sva_network package, we computed the SV loadings of the standardized expression matrix with singular value decomposition (SVD), and computed the residuals after regressing the top 12 SVs. Linear regression between the SVs and the covariates showed correlation mostly with technical covariates, including lane, batch, percentage of ribosomal bases and other sequencing metrics such as % of mRNA and intergenic bases (Fig. S6A).
The co-expression network analysis was performed using the R package of Weighted Gene Correlation Network Analysis (WGCNA) 80 following the standard pipeline to fit a scale-free topology (R2 > 0.8) and applying a Soft Threshold power of 5 into a signed network model (Fig. S6B). The adjacency matrices were constructed using the average linkage hierarchical clustering of the topological overlap dissimilarity matrix (1-TOM). Coexpression modules were defined using a dynamic tree cut method with minimum module size of 20 genes and deep split parameter of 4. Modules highly correlated with each other, corresponding to a module eigengene (ME) correlation of 0.75 were merged, resulting in a total of 65 modules (Fig S6D). The genes were prioritized based on their module membership value, also known as eigengene-based connectivity (kME). The top hub genes for each module are shown in Table S11. calculated the Pearson correlation between the MEs and disease diagnosis, and prioritized those modules with FDR adjusted P-value from a Wilcoxon rank-signed test. Network visualization was done using the “exportNetworkToCytoscape” function from WGCNA R package to export the lists of nodes and edges, and the ggraph R package 81 to create the figures. See extended information for Heritability analysis.
Single-cell RNA-seq data generation and processing
Using cryopreserved PBMCs, monocytes were isolated for scRNA-seq following the same protocol as previously described 82. scRNA-seq with multiplexed cell hashing 82 was performed at the New York Genome Center (NYGC) on purified monocytes from 10 donors, including three controls and seven PD patients (two with GBA mutations and one with a LRRK2 mutation).
See Supplementary information for extensive QC in scRNA-seq. After QC, we used the R package Seurat (v3.1.0) 83, 84 to remove non-protein-coding genes identified through biomaRt 85, keeping 14,827 protein-coding genes out of a total 24,914 genes. We also filtered-out low-quality cells that expressed less than 200 genes or over 2,500 genes, and cells that expressed greater than 5% mitochondrial genes using the “FilterCells” function in Seurat, reducing the total number of cells to 19,144. Lastly, expression counts were normalized using the “preprocess_cds” function in monocle3 with unique molecular identifier (UMI) count and % mitochondrial genes as covariates. Dimensionality reduction was then performed using Uniform Manifold Approximation and Projection (UMAP) 86, 87 via the “reduce_dimension” function in monocle3 34. To identify cell subpopulations, we applied Louvain clustering via the “cluster_cells” function in monocle3 using only the top 2000 most variable genes (identified with the “FindVariableGenes” function in Seurat) (Extended Data Fig. 5C). This yielded 6 discrete clusters, of which the largest two were identified as Classical (Cluster 1; CD14++/CD16−) and Intermediate (Cluster 2; CD14++/CD16+) monocytes based on the expression of cell-type markers. We performed differential expression between PD and controls without considering the independent subclusters (“across-clusters”) as replication of the bulk RNA-seq. We also identified differentially expressed genes between Classical and Intermediate monocyte clusters with the “fit_models” function in monocle3, which by default fits a generalized linear model for each gene with a quasi-Poisson expression response function, calculates coefficients under the Wald test, and corrects for multiple hypothesis testing using false discovery rate 88.
Human microglia isolation and transcriptome data generation
Fresh isolation of human microglia:
Post-mortem brain samples were obtained from the Netherlands Brain Bank (NBB) and the Neuropathology Brain Bank and Research CoRE at Mount Sinai Hospital. The permission to obtain brain material was obtained from the Ethical Committees and the project was approved by the IRB from both institutions. Written informed consent for autopsy and necessary clinical data was previously obtained. Controls were donors with non-neurological diagnoses and including various causes of death such as euthanasia, cardio-respiratory disease or cancer. For PD we included samples with confirmed clinical diagnosis without neuropathological confirmation. Brain tissue was stored in Hibernate media (Gibco) at 4 °C upon processing, which happened within 24 hours after autopsy (Extended Data Fig. 6). Microglia were isolated from the following regions, all of which have been linked to PD 89-92: corpus callosum (CC; 13 samples), medial frontal gyrus (MFG; 40 samples), superior temporal gyrus (STG; 30 samples), thalamus (THA; 23 samples), sub-ventricular zone (SVZ; 18 samples) and substantia nigra (SN; 1 sample). Microglia were isolated as previously described before 93 with minor modifications. In brief, tissue was first mechanically homogenized with the help of cell strainer and pipetting following enzymatic digestion with 0.33 mg/ml of DNase I (Sigma Aldrich) and 0.2% of Trypsin (Invitrogen) in a shaking incubator (140 rpm, 37 °C) for 30 minutes. After washing the tissue in GKN/BSA buffer (PBS + 2 g/L d-(1)-glucose + 0.3% bovine serum albumin (BSA), pH 7.4), cells were resuspended in 20 ml of GKN/BSA and 10 ml of Percoll (GE Healthcare) was added to the top drop-wise. The Percoll gradient was generated with 40 minutes of centrifugation at 4000 rpm 4 °C with no brake. The top myelin phase was discarded and the second layer, mainly containing astrocytes and microglia, was transferred to a new tube. Microglia were purified using human CD11b+ magnetic beads (Miltenyi) following the manufacturer’s instructions and the manual magnetic sorter. Microglia samples were stored in RLT buffer + 1% 2-Mercaptoethanol. RNA was isolated as previously described. Library preparation was performed at Genewiz using the Ultra-low input system which uses Poly-A selection to remove the rRNA. Purity of microglia was confirmed by qPCR comparing the homogenate, positive and negative fraction.
Microglia transcriptomic analysis
RNA-seq data were processed using the RAPiD pipeline, with the same configuration as MyND analysis. RNA-seq QC was performed by applying three filters to remove samples (considering the whole cohort): (i) less than 10 million reads aligned to the reference genome (GRCh38) using the STAR aligner; (ii) samples with more than 20% of the reads aligned to ribosomal regions; (iii) samples with less than 10% of the reads mapping to coding bases. Gene counts were generated by RSEM and tximport. Genes with more than 1 cpm in 30% of the samples were kept for downstream analysis. Differential expression was performed using the DREAM method 41 from variancePartition R package 74 to account for repeated measures. Since each donor can contribute multiple samples from different brain regions (Extended Data Fig. 6B), we modeled the donor as a random effect and added selected covariates to adjust for possible technical and biological confounders. In order to determine the covariates to add to the model we ran variancePartition (Extended Data Fig. 6E). The final model used was expression ~ donor_id + tissue + sex + age + fastqc_percent_gc + featurecounts_assigned + picard_pct_mrna_bases + picard_pct_pf_reads_aligned + picard_pct_ribosomal_bases + lane.
Quantitative Trait Loci Analysis
To perform eQTL mapping, following the GTEx pipeline 94 we converted gene expression matrices to BED format, performed TMM normalization, filtered for lowly expressed genes, removing any gene with less than one TPM in 20% of samples and at least 6 counts in 20% of samples, and inverse normal transformed each gene across samples. We tested 18,430 genes. Then, PEER 95 factors were calculated to estimate hidden confounders within our expression data. We created a combined covariate matrix that included the PEER factors and the first four genotyping ancestry MDS values. We used 15 PEER factors as covariates in our QTL model (Fig. S9A). To confirm that our DNA and RNA samples were from the same donor, we used mbv from QTLtools 96. Based on this, we removed 7 samples. To test for cis-eQTLs, linear regression was performed using the QTLtools nominal pass for each SNP-gene pair using a one megabase window within the transcription start site (TSS) of a gene. To test for association between gene expression and the top variant in cis, we used the QTLtools permutation pass which performs gene-based permutation with 1000 permutations. To identify eGenes, we performed FDR correction (using a threshold of ≤ 0.05) on the P-value of association adjusted for the number of variants tested in cis given by the fitted beta distribution. We estimated replication of MyND monocyte cis-eQTLs (discovery) using CD14+ eQTL data set from Fairfax et al. (replication)97 using the q-value R package to estimate π1 (Fig. S9C).
To perform splicing quantitative trait loci analysis (sQTLs), we used junction counts generated from regtools. All junction files were clustered using the Leafcutter script, specifying for each junction in a cluster a maximum length of 100kb. Following GTEx, introns without read counts in at least 50% of samples or with fewer than 10 read counts in at least 10% of samples were removed. Introns with insufficient variability across samples based on the thresholds provided by GTEx consortium 98 leaving us with a final set of 107,838 junctions within 35,056 clusters. Filtered counts were normalized using prepare_phenotype_table.py from leafcutter, merged, and converted to BED format, with the start/end positions from the gene to which an intron was mapped. We created a combined covariate matrix that includes 15 PEER factors and the first 4 genotyping ancestry MDS values as input to the analysis (Fig. S9B). QTL mapping was performed with QTLtools, testing all variants within 1 megabase of the transcription start site, with 1000 permutations, grouping SNPs by gene. Genes with splicing QTLs were identified by FDR correction (<0.05) of the permutation P-values.
Colocalization analysis
GWAS Data: we used the latest PD GWAS full summary statistics 3. Liftover of the full summary statistics from hg19 to hg38 was performed using GWAS harmonization 99. To perform the colocalization analysis between GWAS and eQTL data, we used the coloc.abf function from the coloc package 47 with default parameters. Our criteria for considering a signal to be colocalized was PPH3 + PPH4 > 0.8, PPH4/PPH3 > 2. All SNPs tested within 1 Mb either side of each GWAS locus were considered for colocalization analysis. To annotate SNPs, we incorporated CD14+ monocyte H3K27ac marks from HaploReg v4.1 100 and microglia H3K27ac marks, ATAC-seq peaks, and PU.1 annotations from Nott et al. 48 data on the UCSC genome browser.
Fine-mapping
The echolocatoR R-based pipeline101 was employed to functionally fine-map PD GWAS loci. Specifically, we used PolyFun+SuSiE 102,103 which computes SNP-wise heritability-derived prior probabilities using a L2-regularized extension of stratified LD SCore (S-LDSC) regression 35,104,105. A UK Biobank baseline model composed of 187 binarized epigenomic and genomic annotations was used as the annotation input 106. We applied PolyFun+SuSiE to PD GWAS summary statistics and LD reference generated from 337K UK Biobank individuals of white British ancestry.
Statistics and reproducibility
We have generated genotyping (GWAS chip) and RNA-seq (bulk and single-cell) data from human CD14+ monocytes, and bulk RNA-seq data from human CD11b+ microglia. Samples were age and sex matched between case and control groups. Statistical significance was determined by the statistical tests, and/or R packages, indicated in the methods section and/or figure and table legends corresponding to each analysis. Sample size for each analysis is indicated in both the methods section and figure and table legends. Data were excluded from each analysis based only on criteria that was outlined in the methods section corresponding to each analysis. Researchers were not blind to disease group when assigning samples to sequencing batches or when performing statistical analysis of sequencing data. Further information on statistics and reproducibility is available in the corresponding sections of the methods and in the Reporting Summary.
Data availability
Processed read counts and full eQTL summary statistics are available from Zenodo online data sharing portal at https://zenodo.org/record/4715907. Raw RNA-seq data and genotypes from the MyND cohort are made available via dbGAP (study accession ID: phs002400.v1.p1) at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002400.v1.p1. RNA-seq data and genotypes of Parkinson's Progression Markers Initiative (PPMI) cohort were obtained from the Accelerating Medicines Partnership program for Parkinson's disease (AMP-PD) Knowledge Platform. For up-to-date information on the study, https://www.amp-pd.org.
Code availability
The code used for the primary analysis is available on GitHub at https://github.com/RajLabMSSM/MyND-Analysis. Any additional code used for analysis is available upon request from the corresponding author.
Extended Data
Extended Data Fig. 1. Experimental flow outline and demographic/clinical information for subjects for monocytes isolation.
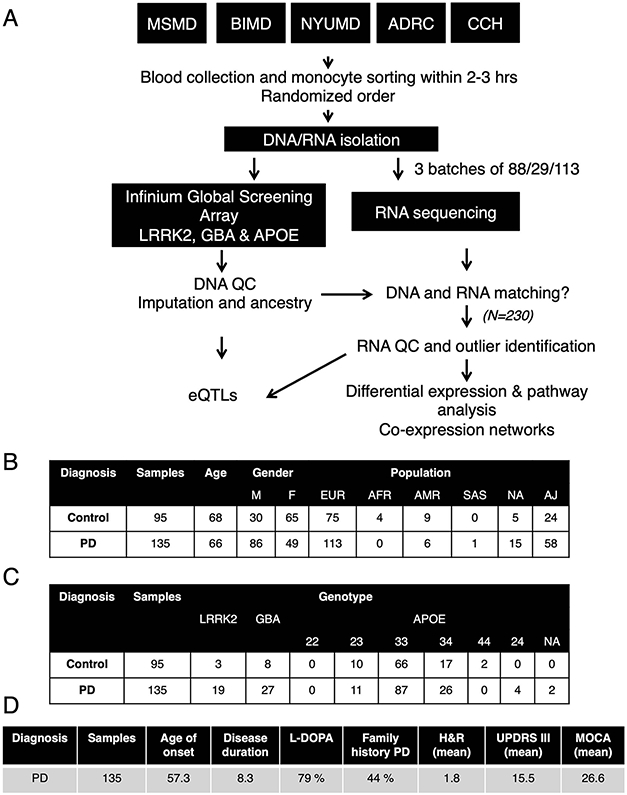
(A) Blood was collected from five independent clinics across New York City (ADRC, CCH, MSMD, BIMD, and NYUMD; details described in Methods) and transferred to the Icahn School of Medicine at Mount Sinai for monocyte sorting and RNA/DNA isolation. Samples were genotyped for common SNPs using Global Screening Array (GSA) and LRRK2, GBA and APOE were independently genotyped. RNA-seq was performed at Genewiz Inc. in three independent and randomized batches. DNA and RNA data was subjected to stringent QC, DNA data was imputed and ancestry was calculated. DNA and RNA were compared to the identification of miss-matches prior outlier identification. After QC, a total of 230 samples were used for subsequent analysis. (B) Demographic, (C) genotype and (D) clinical variables describing the 230 samples included in the study.
Extended Data Fig. 2. Differential expression analysis at the transcript level in PD and controls derived monocytes.

(A) MA plot showing the fold-change (log2 scale) at the transcript level in the y-axis and the mean of log2counts (x-axis), highlighting the DETs at FDR < 0.05 in red. (B) Volcano plot showing the fold-change (log2 scale) of transcripts between PD-monocytes (n = 135) and controls (n = 95) (x-axis) and their significance in the y-axis −log10 P-value scale). DETs at FDR < 0.05 are highlighted in red (upregulated) and blue (downregulated). Moderated t-statistic (two sided) is used for statistical test (see R package limma). (C) Pathway enrichment analysis for the upregulated (n=230 independent samples) and (D) downregulated DETs using Biological processes from GSEA. Significance is represented in the x-axis (−log10 P-value scale of the q-value). Only the 20 most significant pathways (q-value < 0.05) with a minimum overlap of 5 genes are shown. Pathways are grouped and colored by biological related processes. n=230 independent samples
Extended Data Fig. 3. Differential expression analysis at the splicing level in PD and controls derived monocytes.

(A) Histogram reflecting the counts (y-axis) and the % of missingness (x-axis). (B) Volcano plot showing the delta PSI of genes with splicing events in PD-monocytes (n = 135) and controls (n = 95) (x-axis) and their significance in the y-axis (−log10 P-value scale). DSs at FDR < 0.05 are highlighted in red (delta PSI > 0) and blue (delta PSI < 0). Positive delta-PSI indicates that the long isoform is favored whereas negative delta-PSI indicates preference for the short isoform. Chi-squared is used for statistical test (C) Pathway enrichment analysis for the DSs at FDR < 0.0.5 (left panel) DSs + DEGs at FDR < 0.05 (right panel) using Biological processes from GSEA. Significance is represented in the x-axis (−log10 P-value scale of the q-value). Only the 20 most significant pathways (q-value < 0.05) with a minimum overlap of 5 genes are shown. Pathways are grouped and colored by biological related processes. n=230 independent samples. (D) Examples of genes showing significant splicing events.
Extended Data Fig. 4. Module enrichment for biological pathways.

(A) Enrichment of modules (x-axis) containing co-expressed genes for specific biological pathways and curated gene sets (y-axis). Modules are represented by color names and are ordered by size. Enrichment for selected gene sets and GO biological processes (top panel). The size and color of the circles indicate the significance level (−log10 P-value). Enrichments for PD heritability, using stratified LD score regression (bottom panel). The size and color of circles indicate the enrichment value (from LD score) and significance level (−log10 P-value) of enrichment, respectively. Only modules that were significant at a nominal P-value < 0.05 are shown here. (B) Barchart showing Pearson correlation coefficient (r) (x-axis) of three modules (y-axis) significantly associated with PD (FDR < 0.05) determined by the module eigengene analysis. Numbers on the plot represent adjusted P-values, Two-sided Wilcoxon rank-signed test. n=230 independent samples.
Extended Data Fig. 5. Monocyte subcluster characterization by single-cell analysis.

(A) Proportions of the 3 main monocyte sub-clusters using FACS (n = 11 controls and 11 PD). No statistical differences were obtained between groups. (B) Cell proportions of the 6 sub-clusters obtained by unsupervised clustering with monocle3 in scRNA-seq (n = 3 controls and 7 PD). No statistical differences were obtained between cases and controls in cell proportions. Cluster 1 corresponds to classical monocytes and cluster 2 to intermediate monocytes. (C) Top: UMAP colored by diagnosis (green = controls, yellow = PD). Bottom: UMAP colored by CD14 and FCGR3A (CD16) marker genes expression. (D) Histogram showing the variance (y-axis) explained by the 20 first PCA components (x-axis). (E) Histogram showing the frequency of the genes colored by diagnosis (green = control, yellow = PD). (F) Expression of mitochondrial genes by each cluster and divided by diagnosis. Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range. n=10 independent donors.
Extended Data Fig. 6. Fresh microglia transcriptome analysis.

Microglia transcriptomic profiling was performed from 22 samples from 13 PD donors and 106 samples from 42 control donors. (A) Experimental workflow for the generation of human microglial transcriptomic profiles (B) Tables describing the samples included in the study (top: demographic information, middle: clinical information, bottom: brain regions). CC: Corpus Callosum; MFG: medial frontal gyrus; STG: Superior temporal gyrus; THA: thalamus; SVZ: subventricular zone; SN: substantia nigra (C) Heatmap for the expression of marker genes of different brain cell types (red: microglia, dark blue: astrocytes, green: neurons, light blue: oligodendrocytes). (D) Microglial isolation purity assessed by qPCR comparing the brain homogenate, and the positive and negative fractions after CD11b magnetic beads comparing microglial markers (P2RY12, CXCR1, TREM2) and astrocytic markers (GFAP, FGFR3). (E) Violin plot showing the % of variance (y-axis) explained by known covariates (x-axis) by variancePartition. Each dot represents a gene. (F) PCA after regressing out covariates colored by diagnosis (left panel), brain region (middle panel), postmortem interval (right panel). n=128 samples from 55 independent donors. (G) Volcano plot showing the fold-change of genes (log2 scale) between PD-microglia (22 samples from 13 donors) and controls (106 samples from 42 donors) (x-axis) and their significance (y-axis, −log10 scale). Moderated t-statistic (two sided) is used for statistical test (see R package DREAM).(H) Expression of selected mitochondria-specific genes in microglia. Adjusted gene expression levels after normalization are shown. Boxplots: the line represents the median. The boxes extend from the 25th - 75th percentile and the lines extend 1.5 times the interquartile range. n=128 samples from 55 independent donors.
Supplementary Material
Acknowledgements
We thank the study participants for providing blood samples and for their generous gifts of brain donation to the MyND study. We thank the Netherlands Brain Bank and the Neuropathology Brain Bank & Research Core at Mount Sinai for assistance in collecting human brain samples. We thank the Flow Cytometry Core and the Human Immune Monitoring Center at Icahn School of Medicine at Mount Sinai (ISMMS) for optimization of cell isolations; Genewiz Inc. for RNA-sequencing; Christos Proukakis for feedback on the manuscript; Seunghee Kim-Schulze for help optimizing PBMC and monocyte isolation; Ying-Chih Wang for help with processing RNA-seq data; Javier Fernandez-Lopez for help with data analysis; members of the Ronald Loeb Center for Alzheimer’s disease for helpful discussion; research participants and employees of 23andMe who contributed to the PD GWAS. Data used in the preparation of this article were obtained from the AMP-PD Knowledge Platform. AMP-PD – a public-private partnership – is managed by the NIH and funded by Celgene, GSK, the Michael J. Fox Foundation for Parkinson’s Research, the National Institute of Neurological Disorders and Stroke, Pfizer, Sanofi, and Verily. PPMI – a public-private partnership – is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners (names of all of the PPMI funding partners found at www.ppmi-info.org/fundingpartners). The PPMI Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit www.ppmi-info.org. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the ISMMS. T.R. supported by grants from the Michael J. Fox Foundation (Grant #14899 and #16743), US National Institutes of Health NIH NINDS R01-NS116006, NINDS U01-NS120256, NIA R01-AG054005, NIA R21-AG063130, and NIA U01 P50-AG005138. R.S-P. is supported by grants from the NIH NINDS U01-NS107016, NINDS U01-NS094148-01 and Bigglesworth Family Foundation. K.F. is supported by NIH F32 AG056098. E.N. is supported by a fellowship from the Ramon Areces Foundation (Spain). The research reported in this paper was additionally supported by the Office of Research Infrastructure of the NIH under award numbers S10OD018522 and S10OD026880.
Footnotes
Competing interests
A.M.G. served on the scientific advisory board for Denali Therapeutics from 2015-2018 and has served as a consultant to AbbVie, Biogen, Eisai, Illumina, and GSK. R.S-P. and S.B. have served as consultants to Denali Therapeutics. R.H.W. has served as consultant to Neurocine Biosciences, Inc. Other authors declare no competing financial interests.
REFERENCES
- 1.Poewe W et al. Parkinson disease. Nat Rev Dis Primers 3, 17013 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Ohnmacht J, May P, Sinkkonen L & Krüger R Missing heritability in Parkinson’s disease: the emerging role of non-coding genetic variation. Journal of Neural Transmission vol. 127 729–748 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nalls MA et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li YI, Wong G, Humphrey J & Raj T Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun 10, 994 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gagliano SA et al. Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson's diseases. Annals of Clinical and Translational Neurology vol. 3 924–933 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj T et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reynolds RH et al. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson’s disease heritability. npj Parkinson’s Disease vol. 5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bae E-J et al. Antibody-Aided Clearance of Extracellular -Synuclein Prevents Cell-to-Cell Aggregate Transmission. Journal of Neuroscience vol. 32 13454–13469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi I et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun 11, 1386 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duffy MF et al. Correction to: Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. Journal of Neuroinflammation vol. 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fellner L et al. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia vol. 61 349–360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doorn KJ et al. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathologica Communications vol. 2 90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olanow CW et al. Temporal evolution of microglia and α-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain vol. 142 1690–1700 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Grozdanov V et al. Increased Immune Activation by Pathologic α- Synuclein in Parkinson’s Disease. Ann. Neurol 86, 593–606 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Herz J, Filiano AJ, Smith A, Yogev N & Kipnis J Myeloid Cells in the Central Nervous System. Immunity 46, 943–956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harms AS et al. α-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathol Commun 5, 85 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harms AS et al. Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp. Neurol 300, 179–187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bliederhaeuser C et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 131, 379–391 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Wijeyekoon RS et al. Monocyte Function in Parkinson’s Disease and the Impact of Autologous Serum on Phagocytosis. Front. Neurol 9, 870 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braak H et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Scheperjans F, Derkinderen P & Borghammer P The Gut and Parkinson’s Disease: Hype or Hope? Journal of Parkinson’s Disease vol. 8 S31–S39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapelet G, Leclair-Visonneau L, Clairembault T, Neunlist M & Derkinderen P Can the gut be the missing piece in uncovering PD pathogenesis? Parkinsonism Relat. Disord 59, 26–31 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Klingelhoefer L & Reichmann H Pathogenesis of Parkinson disease—the gut–brain axis and environmental factors. Nat. Rev. Neurol 11, 625–636 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Schlachetzki JCM et al. A monocyte gene expression signature in the early clinical course of Parkinson’s disease. Sci. Rep 8, 10757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donega V et al. Transcriptome and proteome profiling of neural stem cells from the human subventricular zone in Parkinson’s disease. Acta Neuropathol Commun 7, 84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan E-K et al. Parkinson disease and the immune system — associations, mechanisms and therapeutics. Nature Reviews Neurology vol. 16 303–318 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Hoehn MM & Yahr MD Parkinsonism: onset, progression and mortality. Neurology 17, 427–442 (1967). [DOI] [PubMed] [Google Scholar]
- 28.Cuperfain AB, Zhang ZL, Kennedy JL & Gonçalves VF The Complex Interaction of Mitochondrial Genetics and Mitochondrial Pathways in Psychiatric Disease. Mol Neuropsychiatry 4, 52–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth J, Vogl T, Sorg C & Sunderkötter C Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 24, 155–158 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Xia C, Braunstein Z, Toomey AC, Zhong J & Rao X S100 Proteins As an Important Regulator of Macrophage Inflammation. Front. Immunol 8, 1908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sathe K et al. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-α pathway. Brain 135, 3336–3347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jinn S et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A 114, 2389–2394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maiese K, Chong ZZ, Shang YC & Wang S mTOR: on target for novel therapeutic strategies in the nervous system. Trends Mol. Med 19, 51–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao J et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordon S & Taylor PR Monocyte and macrophage heterogeneity. Nat. Rev. Immunol 5, 953–964 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Zawada AM et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 118, e50–61 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Mukherjee R et al. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci. Rep 5, 13886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grozdanov V et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol. 128, 651–663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith AM et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells. Mov. Disord 33, 1580–1590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoffman GE & Roussos P dream: Powerful differential expression analysis for repeated measures designs. doi: 10.1101/432567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smajić S et al. Single-cell sequencing of the human midbrain reveals glial activation and a neuronal state specific to Parkinson’s disease. bioRxiv (2020) doi: 10.1101/2020.09.28.20202812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q et al. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun 10, 5234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao DW, O’Connor LJ, Price AL & Gusev A Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet (2020) doi: 10.1038/s41588-020-0625-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young A, Kumasaka N, Calvert F & Hammond TR A map of transcriptional heterogeneity and regulatory variation in human microglia. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng B et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat. Neurosci 20, 1418–1426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nott A et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chattaragada MS et al. FAM49B, a novel regulator of mitochondrial function and integrity that suppresses tumor metastasis. Oncogene 37, 697–709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schapira AH et al. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 54, 823–827 (1990). [DOI] [PubMed] [Google Scholar]
- 51.Mizuno Y et al. Deficiencies in Complex I subunits of the respiratory chain in Parkinson’s disease. Biochemical and Biophysical Research Communications vol. 163 1450–1455 (1989). [DOI] [PubMed] [Google Scholar]
- 52.Teves JMY et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci 11, 737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryan BJ, Hoek S, Fon EA & Wade-Martins R Mitochondrial dysfunction and mitophagy in Parkinson’s: from familial to sporadic disease. Trends Biochem. Sci 40, 200–210 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Seibler P et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J. Neurosci 31, 5970–5976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mortiboys H et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society 64, 555–565 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mortiboys H, Johansen KK, Aasly JO & Bandmann O Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 75, 2017–2020 (2010). [DOI] [PubMed] [Google Scholar]
- 57.Annesley SJ et al. Immortalized Parkinson’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech 9, 1295–1305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parnetti L et al. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 18, 573–586 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Parnetti L et al. Cerebrospinal fluid biomarkers in Parkinson disease. Nature Reviews Neurology vol. 9 131–140 (2013). [DOI] [PubMed] [Google Scholar]
- 60.Trapnell C et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kunkle BW et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wood AR et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet 46, 1173–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weissbrod O, Hormozdiari F, Benner C & Cui R Functionally-informed fine-mapping and polygenic localization of complex trait heritability. BioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nextflow - A DSL for parallel and scalable computational pipelines. https://www.nextflow.io/ [Google Scholar]
- 66.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.GENCODE - Human Release 30. https://www.gencodegenes.org/human/release_30.html. [Google Scholar]
- 69.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feng Y-Y et al. RegTools: Integrated analysis of genomic and transcriptomic data for discovery of splicing variants in cancer. bioRxiv 436634 (2018) doi: 10.1101/436634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- 72.Picard Tools - By Broad Institute. https://broadinstitute.github.io/picard/. [Google Scholar]
- 73.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoffman GE & Schadt EE variancePartition: Interpreting drivers of variation in complex gene expression studies. doi: 10.1101/040170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gomes AV Genetics of proteasome diseases. Scientifica 2013, 637629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buja A & Eyuboglu N Remarks on Parallel Analysis. Multivariate Behav. Res 27, 509–540 (1992). [DOI] [PubMed] [Google Scholar]
- 79.Parsana P et al. Addressing confounding artifacts in reconstruction of gene co-expression networks. Genome Biol. 20, 94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langfelder P & Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics vol. 9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pedersen TL ggraph. GitHub https://github.com/thomasp85/ggraph (2017). [Google Scholar]
- 82.Stoeckius M et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Durinck S et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol (2018) doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 87.McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv [stat.ML] (2018). [Google Scholar]
- 88.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Series B Stat. Methodol 57, 289–300 (1995). [Google Scholar]
- 89.Prakash KG et al. Neuroanatomical changes in Parkinson’s disease in relation to cognition: An update. J. Adv. Pharm. Technol. Res 7, 123–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goldman JG et al. Corpus callosal atrophy and associations with cognitive impairment in Parkinson disease. Neurology 88, 1265–1272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kendi ATK, Lehericy S & Luciana M Altered diffusion in the frontal lobe in Parkinson disease. American Journal of (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martin WRW, Wayne Martin WR, Wieler M, Gee M & Camicioli R Temporal lobe changes in early, untreated Parkinson’s disease. Movement Disorders vol. 24 1949–1954 (2009). [DOI] [PubMed] [Google Scholar]
- 93.Melief J et al. Characterizing primary human microglia: A comparative study with myeloid subsets and culture models. Glia 64, 1857–1868 (2016). [DOI] [PubMed] [Google Scholar]
- 94.gtex-pipeline. (Github; ). [Google Scholar]
- 95.Stegle O, Parts L, Piipari M, Winn J & Durbin R Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc 7, 500–507 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Delaneau O et al. A complete tool set for molecular QTL discovery and analysis. bioRxiv 068635 (2016) doi: 10.1101/068635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fairfax BP et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aguet F et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. bioRxiv 787903 (2019) doi: 10.1101/787903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.summary-gwas-imputation. (Github; ). [Google Scholar]
- 100.HaploReg v4.1. https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php. [Google Scholar]
- 101.Schilder BM, Humphrey J & Raj T echolocatoR: an automated end-to-end statistical and functional genomic fine-mapping pipeline. bioRxiv 2020.10.22.351221 (2020) doi: 10.1101/2020.10.22.351221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weissbrod O et al. Functionally-informed fine-mapping and polygenic localization of complex trait heritability. BioRxiv (2019) doi: 10.1101/807792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang G, Sarkar A, Carbonetto P & Stephens M A simple new approach to variable selection in regression, with application to genetic fine-mapping. (2018) doi: 10.1101/501114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gazal S et al. Linkage disequilibrium–dependent architecture of human complex traits shows action of negative selection. Nature Genetics vol. 49 1421–1427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature Genetics vol. 47 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gazal S et al. Functional architecture of low-frequency variants highlights strength of negative selection across coding and non-coding annotations. Nature Genetics vol. 50 1600–1607 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed read counts and full eQTL summary statistics are available from Zenodo online data sharing portal at https://zenodo.org/record/4715907. Raw RNA-seq data and genotypes from the MyND cohort are made available via dbGAP (study accession ID: phs002400.v1.p1) at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002400.v1.p1. RNA-seq data and genotypes of Parkinson's Progression Markers Initiative (PPMI) cohort were obtained from the Accelerating Medicines Partnership program for Parkinson's disease (AMP-PD) Knowledge Platform. For up-to-date information on the study, https://www.amp-pd.org.