

Abstract
Poly (ADP-Ribose) polymerase-1 (PARP-1) is involved in the DNA repairing system by sensing and signaling the presence of DNA damage. Inhibition of PARP-1 is tested in combination with DNA damaging agents such as topoisomerase I inhibitors or ionizing radiations (RT) for the treatment of glioblastoma (GBM). Disruption of p53, widely prevalent in GBMs, plays a major role in DNA repairing system. The current study investigates whether p53 activity has an effect on the sensitivity of human GBM cells to PARP-1 inhibitors in combination with topoisomerase I inhibitor topotecan (TPT) and/or RT. Human GBM cell lines carrying a different functional status of p53 were treated with PARP-1 inhibitor NU1025, in combination with TPT and/or RT. Cytotoxic effects were examined by analyzing the antiproliferative activity, the cell cycle perturbations, and the DNA damage induced by combined treatments. PARP inhibition enhanced the antiproliferative activity, the cell cycle perturbations and the DNA damage induced by both TPT or RT in GBM cells. These effects were influenced by the p53 activity: cells carrying an active p53 were more sensitive to the combination of PARP inhibitor and RT, while cells carrying an inactive p53 displayed a higher sensitivity to the combination of PARP inhibitor and TPT. Our study suggests that p53 activity influences the differential sensitivity of GBM cells to combined treatments of TPT, RT, and PARP inhibitors.
Keywords: p53, glioblastoma, PARP inhibitor, radiotherapy, topotecan, combinatorial strategy
Introduction
Chemotherapy (temozolomide) given concurrently or after radiation therapy (RT) is the standard of care for patients with newly diagnosed glioblastoma (GBM) (1). This combination significantly increases the overall survival of GBM patients as compared to RT alone. However, treatment with chemo-RT is effective for a limited time and GBMs continue to carry a poor survival rate (2).
GBMs widely present a disruption of p53. The latter plays a major role in the DNA repairing system. It has been shown that an inactive p53 sensitizes cancer cells to DNA damaging agents such as topoisomerase I (Topo I) inhibitors (3). Topotecan (TPT), an inhibitor of Topo I, is approved for GBM treatment in the refractory and relapsed setting (4). In vitro and in vivo (5-9) evidence has demonstrated that TPT enhances the radiosensitivity of human GBM cells. However, phase 2 trials failed to demonstrate a clear benefit from concurrent administration of TPT and RT resulting in a high toxicity (10,11). On the other hand, administration of Poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors has shown great promise in the treatment of several solid tumors, including GBMs (12). PARP-1 as well as p53 is involved in the DNA repair system by sensing and signaling the presence of DNA damage. Activation of PARP-1 leads the recruitment of proteins such as p53 (13) and Topo I/II (14). In vitro and in vivo studies have shown that PARP-1 inhibitors enhance the antitumor activity of both RT and chemotherapy, including Topo I inhibitors, in GBMs (15-19). Nevertheless, there is the need to identify potential bio-markers, which might determine the sensitivity to combined treatments of PARP-1 inhibitors, TPT, and RT. 8-Hydroxy-2-methylquinazoline-4-one (NU1025) is a PARP-1 inhibitor that inhibits GBM cell growth without cytotoxicity (19,20).
In this study, we tested whether the differential sensitivity of GBM cells to combined treatments of TPT, RT, and NU1025 is influenced by the functional status of p53.
Material and Methods
Cell Culture
Human GBM cell lines D54, with an active p53 (21), and U251, with an inactive p53 (22), were obtained from American Type Culture Collection. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s nutrient mixtures F-12 (Ham’s) 1:1 (GIBCO/Invitrogen, Carlsbad, CA), supplemented with 10% foetal calf serum (FCS, Lonza, CA), 20 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, and 5 mM l-glutamine (complete medium). All cells were cultured at 37°C in a 5% CO2 atmosphere.
Chemical Reagents and Antibodies
TPT was purchased from Glaxo Smith-Kline (Brentford, UK). NU1025 was purchased from Alexis Biochemicals (San Diego, CA). 3-(4,5-dimethylthiazole-2-yl)–2,5-diphenyl-2H-tetrazolium bromide (MTT), propidium iodide (PI), and RNAse were purchased from Sigma Chemicals Co (St Louis, MO). 5-bromo-2′-deoxyuridine (BrdU) and Alexa Fluor 488 goat antimouse IgG antibody were purchased from Invitrogen (Portland, OR). Anti-BrdU Pure mouse monoclonal antibody (mAb) was purchased from BD Biosciences (San Giose, CA). Antiphospho-histone H2AX (γH2AX) (Ser139) Fluorescein isothiocyanate (FITC)-conjugated and anti-Poly (ADP-Ribose) mouse mAbs were purchased from Millipore (Billerica, MA). FITC-conjugated goat anti-mouse IgG antibody was purchased from Dako (Milan, Italy). p53-, p21-, and β-actin-specific mAbs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-conjugated IgG antibody was purchased from Amersham-Pharmacia (Buckinghamshire, UK).
Cell Proliferation and MTT Assay
Cells were seeded in triplicate in 96-well plates at a density of 2.5 × 103 cells/well and treated with the indicated doses of TPT, NU1025, and RT. Adherent cells were irradiated in medium with 250 kVp X-rays (dose rate 0.5 Gy/min). Untreated cells were used as a control. In combinatorial treatments cells were treated with NU1025 at the final concentration of 10 μM. Previous results showed that this dose effectively inhibits PARP-1 activity in two GBM cell lines tested (19). Following an up to 5 day incubation, cell proliferation was assessed by MTT assay. MTT assay was carried out as reported elsewhere (23). Cells in randomly selected fields per well were photographed at different time points using a Zeiss Inverted Fluorescence Microscope (AxioVision Software, Carl Zeiss Micro-Imaging GmbH, Germany).
Immunofluorescent Staining
Cells were seeded at a density of 1 × 105 cells, grown as monolayer on cover slips placed in cell culture dishes and treated with the indicated doses of TPT, NU1025, and RT. Untreated cells were used as a control. Following a 4 h incubation PARP-1 activity was determined by immunofluorescent detection of poly-ADP-ribosylation (PAR) of DNA as reported elsewhere (24). Cells were then stained with PI (5 μg/ml) for 30 min as an indicator of nuclear compartment and cover slips were mounted on slides. Slides were examined by Leica DMRXA fluorescent microscope. Images were captured by a QImaging CCD camera.
Cell Cycle Analysis and BrdU Incorporation
Cells were seeded in triplicate in 6-well plates at a density of 3 × 105 cells/well and treated with the indicated doses of TPT, NU1025, and RT. Untreated cells were used as a control. Following an up to 96 h incubation, cell-cycle analysis was determined by PI staining. PI staining was carried out as reported elsewhere (19). Potential blocks in cell cycle phases were determined by BrdU incorporation and PI staining. Untreated and treated cells were incubated with BrdU (20 μM) for 30 min in fresh complete medium. Then the medium was removed and fresh medium was added. Following an up to 12 h incubation cells were trypsinized and washed with PBS. Preparation of samples for FACS analysis was performed as reported elsewhere (25). Cells were analyzed by flow cytometry (FACScan, Becton Dickinson, San Josè, CA) using the CyCLOPS Summit (Cytomation, Fort Collins, CO). Distribution of the cells in cell cycle phases was evaluated by Mod Fit 2.0 (Verity Software HOUSe, Ranger, ME). For the analysis of BrdU-DNA bivariate graphs, cells were split into four categories: (i) BrdU-positive (BrdU+); (ii) G1 phase/BrdU-positive (G1+); (iii) S phase/BrdU positive (S+); and (iv) G2/M phase/BrdU-positive (G2/M+).
DNA Damage
Cells were seeded in triplicate in 6-well plates at a density of 3 × 105 cells/well and treated with the indicated doses of TPT, NU1025, and RT. Untreated cells were used as a control. DNA damage was assessed following an up to 4 h incubation by flow cytometry characterization of DNA Strand Breaks (DSB) using γH2AX expression and DNA content detection by PI staining. Preparation of samples for FACS analysis was performed as reported elsewhere (26). Cell cycle distribution and γH2AX expression were analyzed by FACScan. For the analysis of γH2AX-DNA bivariate graphs, cells were split into two categories: (i) γH2AX-positive cells (γH2AX+); and (ii) γH2AX-negative cells (γH2AX−). Mathematic difference (Δ) between % of treated (tr) and untreated (untr) γH2AX+ cells was calculated to compare the DNA damaging effect (Δ% = γH2AX+tr – γH2AX+untr).
Cell Transfection
D54 cells were seeded in 6-well plates at a density of 6 × 104 cells/well and transfected with pcRNAi vector. Two synthetic oliginucleotides were designed using the following sequences: 5′-CCGGAATTCCCGACTCCAGTGGTAATCTAC TTCAAGAGAGTAGATTACCA-CTGGAGTCTTTTTGGAACT CGAGCGG-3′ and 5′-CCGCTCGAGTTCCAAAAAGACTCC-AGTGGTAATCTACTCTCTTGAAGTAGATTACCACTGGAGT CGGGAATTCCGG-3′ (27). The resulting double stranded oligonucleotides were then cloned into the pcRNAi vector encoding resistance to geneticin (G-418), and derived from the pcDNA3.1 vector (Invitrogen) by replacing the viral promoter-cassette with the H1 promoter. The latter is specifically recognized by RNA polymerase III. Two days after transfection, geneticin G-418 (GIBCO) (200 μg/ml) was added to cell culture medium in order to enrich resistant-transfected clones (D54 p53-) (clones named P1 and P2).
Western Blot Analysis
D54 and D54 p53- cells were seeded in 6-well plates at a density of 3 × 105 cells/well and treated with doxorubicin (2.5 μg/ml). Untreated cells were used as a control. Following a 12 h incubation cells were harvested and lysed. Cell lysates were analyzed by western blot with the indicated mAbs. Western blot analysis was carried out as reported elsewhere (28).
Statistical Methods
Averages, standard deviations, and unpaired t-test were calculated using MS-Excel. Data are shown as mean ± SD of the results obtained in at least three independent experiments. Differences between groups were considered significant when the p-value was <0.05. The asterisk (***) indicates p < 0.05.
Results
Enhancement by PARP-1 Inhibition of the Antiproliferative Activity of TPT or RT in GBM Cell Lines
A titration experiment showed that the IC50 doses of TPT and RT were 10 nM and 2 Gy, respectively, for both D54 and U251 cell lines (Figs. 1A and 1B). U251 cells were more radioresistant than D54 cells (Fig. 1B) but both cell lines were highly sensitive to the antiproliferative activity of TPT (Fig. 1A). The doses of 2Gy for RT and those of 5 and 10 nM for TPT were chosen to be combined with NU1025 in order to test the antiproliferative activity of combined treatments. As shown in Figure 1C, NU1025 had no detectable effect on the proliferation of D54 and U251 cell lines. However, when combined with TPT or RT, NU1025 significantly (p < 0.05) enhanced their antiproliferative activity (Fig. 2). It is noteworthy that the two cell lines displayed a different sensitivity to the antiproliferative activity of combined treatments of TPT, RT, and NU1025. In D54 cell line RT and NU1025 combination displayed a similar antiproliferative activity to that of TPT and RT. However, both combinations inhibited the proliferation of the cells to a significantly (p < 0.05) greater extent than the combination of TPT and NU1025. In U251 cell line TPT (10 nM) and NU1025 combination inhibited the proliferation of tumor cells to a significantly (p < 0.05) greater extent than all the other combinations. In both cell lines the triple combination of TPT, RT, and NU1025 did not increase the anti-proliferative activity of the most effective double agent combinations.
Figure 1.
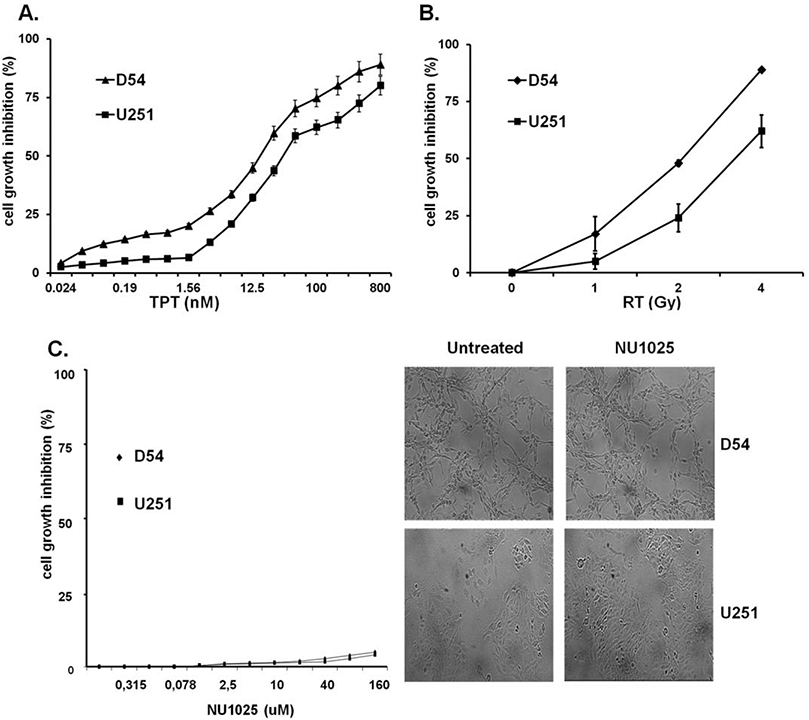
Dose dependent effects of TPT, RT, and NU1025 on the in vitro growth of human GBM cell lines. D54 and U251 cells were treated with the indicated doses of topotecan (TPT) (A), ionizing radiations (RT) (B) and NU1025 (C). Cell growth inhibition was determined by MTT assay following a five day incubation. Percentage of cell growth inhibition was calculated as the ratio of treated to untreated cells at each concentration point. Data are expressed as mean ± SD of the results obtained in three independent experiments. Representative results of D54 and U251 cells treated with NU1025 (10 μM) are shown (C, right panel).
Figure 2.
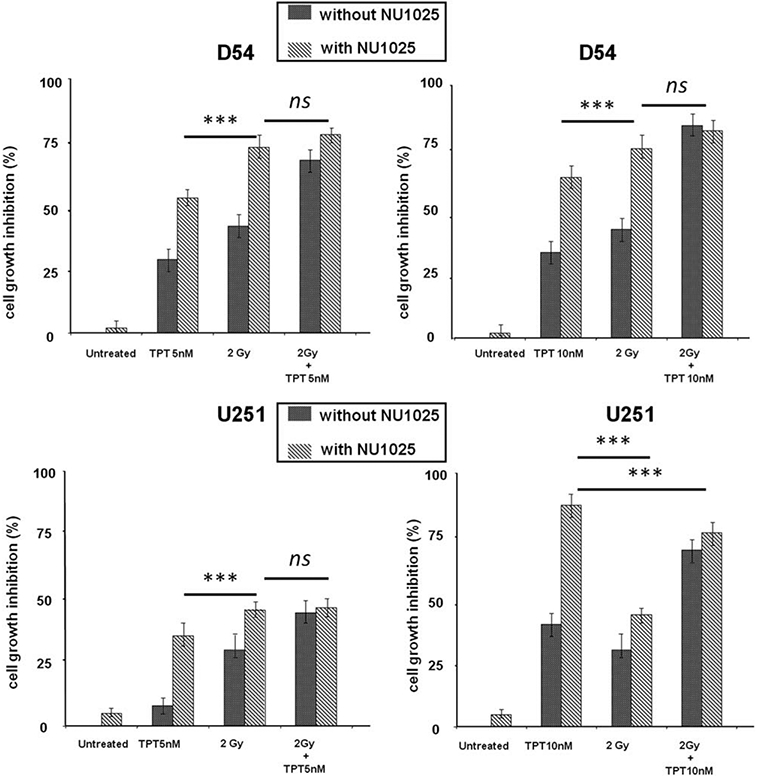
Enhancement by NU1025 of the in vitro antiproliferative activity of TPT or RT in human GBM cell lines. D54 and U251 cells were treated with the indicated doses of topotecan (TPT), ionizing radiations (RT), and NU1025 (10 μM). Cell growth inhibition was determined by MTT assay following a five day incubation. Percentage of cell growth inhibition was calculated as ratio of treated to untreated cells at each treatment. Data are expressed as mean ± SD of the results obtained in three independent experiments. *** indicates p < 0.05. ns: indicates no significant difference.
PARP-1 Inhibition Following Treatment with TPT, RT, and NU1025 in GBM Cell Lines
To demonstrate an increased activation of PARP-1 induced by TPT or RT treatment and to validate its inhibition by NU1025, cells were treated with TPT, RT, and NU1025 and the nuclear PAR-DNA levels were assessed. Following a 4 h incubation an increased nuclear PARP-1 activity was detected in untreated U251 cells as compared to that of D54 cells (Fig. 3). Treatment with TPT and RT, used as single agents, markedly increased the nuclear activation of PARP-1 in both D54 and U251 cell lines. However, treatment with NU1025 markedly inhibited the enhanced activation of PARP-1 induced by TPT and RT treatment.
Figure 3.

Inhibition by NU1025 of PARP-1 activation induced by TPT or RT in human GBM cell lines. D54 and U251 cells were treated with the indicated doses of topotecan (TPT), ionizing radiations (RT) and NU1025 (10 μM). PARP-1 activity was determined by immunofluorescent staining of poly-ADP-ribosylation (PAR) of the DNA following a 4 h incubation. PI staining was used as an indicator of nuclear compartment. Representative staining of PAR and PI staining of D54 and U251 cells treated with TPT, RT, and NU1025 are shown.
Enhancement by PARP-1 Inhibition of the Cell Cycle Perturbations Induced by TPT or RT in GBM Cell Lines
To determine the mechanisms underlying the differential anti-proliferative activity of single and combined treatments in the two cell lines, D54 and U251 cells were treated with TPT, RT, and NU1025, stained with PI and analyzed for cell cycle perturbations. As shown in Figure 4, NU1025 did not alter the cell cycle distribution as compared to untreated cells in both cell lines. In D54 cell line RT and NU1025 combination displayed a similar activity to that of TPT and RT. However, both combinations significantly (p < 0.05) increase the percentage of cells in S-phase as compared to TPT and NU1025 combination (Fig. 4A). In U251 cell line (Fig. 4B) TPT (10 nM) and NU1025 combination increased the cell death and the accumulation of cells in S- and G2/M-phase to a significantly (p < 0.05) greater extent than all the other combinations. In both cell lines the triple combination of TPT, RT, and NU1025 did not increase the cell cycle perturbations of the most effective double agent combinations.
Figure 4.
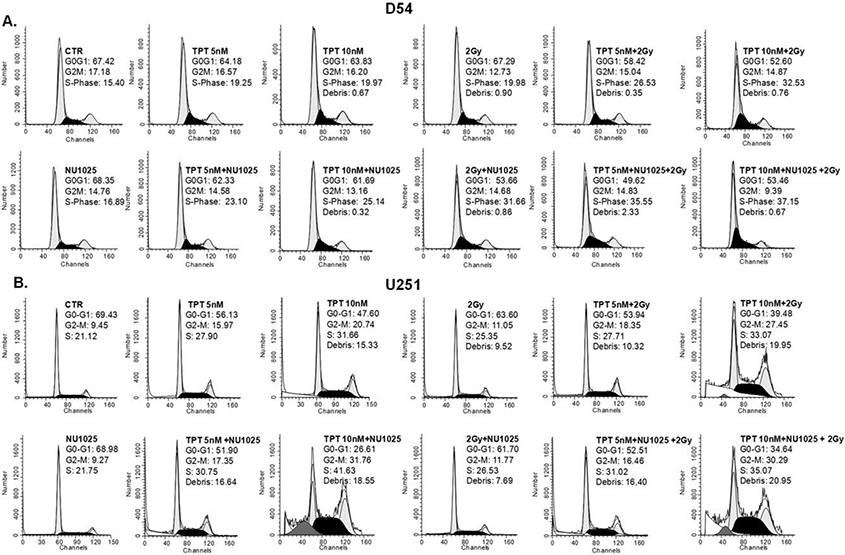
Enhancement by NU1025 of the cell cycle perturbations induced by TPT or RT in human GBM cell lines. D54 (A) and U251 (B) cells were treated with the indicated doses of topotecan (TPT), ionizing radiations (RT), and NU1025 (10 μM). DNA content was detected by PI staining following 96 h incubation. The percentage of D54 and U251 cells in the different phases of cell cycle is shown. Data refer to one of two experiments giving similar results.
To validate the differential cell cycle alterations caused by combined treatments, cells were treated with TPT, RT, and NU1025, incubated with BrdU and stained with PI. In D54 cell line RT and NU1025 combination dramatically decreased the % of BrdU+ cells as compared to untreated cells (Supporting Information Fig. 1A). Treated cells reached and were arrested in G1-phase. Similar results were obtained with TPT and RT combination (data not shown). In U251 cell line, BrdU/PI staining confirmed the strong accumulation of cells in S-phase and the block in G2/M-phase caused by the most effective combination of TPT and NU1025 (Supporting Information Fig. 1B). This effect was more evident after the gate of BrdU+ cells: at time zero cells were in G1-phase and entered in S-phase, but at 6 and 12 h all cells reached and were arrested in G2/M-phase.
Enhancement by PARP-1 Inhibition of the DNA Damage Induced by TPT or RT in GBM Cell Lines
We next investigated whether the differential antiproliferative activity and cell cycle alterations caused by combined treatments were mediated by a diverse induction of DNA damage. We determined DSB presence by γH2AX detection and PI staining in the two GBM cell lines after treatment with TPT, RT, and NU1025. As shown in Figure 5A and Supporting Information Figure 2, no significant changes were detected in D54 cell line following a 1 h incubation with TPT, RT, and NU1025 used as single agents or in combinations. An increase of γH2AX expression was found following a 4 h incubation with both TPT and RT. The latter effects were markedly enhanced (p < 0.05) by NU1025. However, RT and NU1025 combination as well as TPT and RT combination increased the DNA damage to a significantly greater extent (p < 0.05) than TPT and NU1025 combination. In U251 cell line an increase of γH2AX expression was already detected following a 1 h incubation with TPT and RT when they were used as single agents (Fig. 5B and Supporting Information Fig. 2). This effect was significantly enhanced by PARP-1 inhibition (p < 0.05). Nonetheless, TPT (10 nM) and NU1025 combination increased γH2AX expression to a significantly greater extent (p < 0.05) than all the other combinations.
Figure 5.
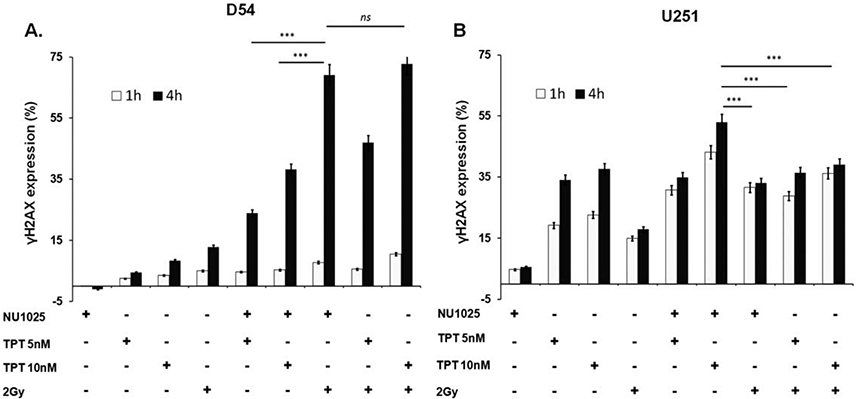
Enhancement by NU1025 of the DNA damage induced by TPT or RT in human GBM cell lines. D54 (A) and U251 (B) cells were treated with the indicated doses of topotecan (TPT), ionizing radiations (RT) and NU1025 (10 μM). DNA damage was determined by flow cytometry analysis of phosphorylated histone H2AX (γH2AX) expression plotted versus the DNA content of cells as determined by PI staining. Following an up to 4 h incubation, relative γH2AX expression was calculated as the mathematic difference (Δ) between % of treated (tr) and untreated (untr) γH2AX+ cells (Δ % = γH2AX+tr – γH2AX+untr). Data are expressed as mean ± SD of the results obtained in three independent experiments. ***Indicates p < 0.05. ns indicates no significant difference.
p53 Activity as a Biomarker of the Sensitivity of GBM Cells to PARP-1 Inhibitor in Combination with TPT and RT
Our studies indicated that NU1025 enhanced the cytotoxic effects of TPT and RT in both D54 and U251 cell lines. However, the two cell lines displayed a differential sensitivity to the antitumor activity of combined treatments. To test whether the p53 activity played a role in the differential sensitivity of GBM cells to TPT, RT, and NU1025 combinations we knocked-down p53 in D54 cells using two specific p53-RNAi and evaluated the antitumor activity of TPT, RT, and NU1025 in both D54 and D54 p53-cells. p53 as well as its downstream component p21 were strongly down-regulated in D54 p53-cells both under basal conditions and after treatment with doxorubicin (Supporting Information Fig. 3). In line with data obtained in U251 cell line, D54 p53-cells displayed an increased radio resistance as compared to D54 cells (p < 0.05) (Fig. 6A). Furthermore, although NU1025 enhanced the antiproliferative activity of both TPT and RT, TPT and NU1025 combination inhibited cell proliferation to a significantly greater extent than all the other combinations (p < 0.05) (Fig. 6B). Lastly, treatment with TPT alone or in combination with NU1025 or RT induced a significant accumulation of D54 p53-cells in S- and G2/M-phase of cell cycle. However, this effect was more marked after treatment with TPT and NU1025 as compared to all the other combinations (Fig. 6C).
Figure 6.

Antiproliferative activity of TPT, RT, and NU1025 in GBM cells carrying a differential functional status of p53. D54 and D54 p53- P1 cells were irradiated with the indicated doses of ionizing radiations (RT). Cell growth inhibition was determined by MTT assay following five day incubation. (B) D54 and D54 p53-P1 cells were treated with the indicated doses of topotecan (TPT), ionizing radiations (RT), and NU1025 (10 μM). Cell growth inhibition was determined by MTT assay following three day incubation. Percentage of cell growth inhibition was calculated as the ratio of treated to untreated cells at each concentration point. Data are expressed as mean ± SD of the results obtained in three independent experiments. ***Indicates p < 0.05. (C) D54 and D54 p53- P1 cells were treated with the indicated doses of TPT, RT, and NU1025 (10 μM). Following a three day incubation DNA content of cells was detected by PI staining. The percentage of D54 and D54 p53-cells in different phases of cell cycle is shown. Data refer to one of two experiments giving similar results.
Discussion
In the Era of targeted therapy current treatment for GBMs still involves the combination of chemotherapy and RT. So far, the combination of temozolomide and RT still represents the standard of care for GBM. This combination increases the overall survival of GBM patients as compared to RT alone. However, temozolomide and RT combination is effective for a limited time and GBM patients continue to carry a poor overall survival. There is evidence that the combination of Topo I inhibitors and RT might be an effective strategy for the treatment of GBM. However, its use is limited by an increased toxicity (9,10,29). Radio- and chemo-resistance are the most significant causes of treatment failure and toxicity. PARP inhibitors sensitize tumor cells including GBM cells to RT and to chemotherapeutic agents such as platinum derivates and Topo I inhibitors. Furthermore treatment with PARP inhibitors does not affect the toxicities and improves both the therapeutic index and the success rate of chemotherapy and RT (30-34). Nevertheless, GBM cells displayed a different sensitivity to the treatment of PARP inhibitors in combination with TPT or RT.
In this study, we show in vitro that NU1025, a PARP-1 inhibitor, selectively inhibits PARP activation, which is induced by TPT or RT in GBM cell lines. It is noteworthy that U251 GBM cells, carrying an inactive p53, displayed a high level of PARP-1 activity. This “stand alone” phenomenon has been already reported in other malignancies and is caused by the lack of a functionally active p53 (35). Treatment with NU1025 does not affect the growth of GBM cells but enhances the antiproliferative activity of both TPT and RT in GBM cells. This effect is caused by an increase of cell cycle perturbations and of induction of DNA damage of GBM cells. It is noteworthy that use of triple agent combination does not increase the sensitivity of GBM cells as compared to the most effective double combinations.
It is known that a critical event in determining sensitivity to RT or TPT is represented by the repair of DSB. Our data showed that the PARP inhibition leads to an increased sensitivity to RT or TPT by enhancing the DSB. γH2AX expression, a marker of DSB (36-38), is increased after treatment with NU1025 in combination with TPT or RT as compared to those of untreated cells and of cells treated with single agents. PARP-1 has been shown to play a major role in the DNA-damage repairing system (13,14). Therefore, the enhanced TPT and RT anti-proliferative activity is likely to reflect an inhibition of DNA-damage repairing system.
p53 is defined as the “guardian” of the genome and most of the GBMs carry an inactive p53 (39). The enhanced cell growth inhibition and increased DSB which are induced by PARP-1 inhibition in combination with TPT or RT have been shown to be independent from the activity of p53. NU1025 enhances the antiproliferative activity of TPT and RT in GBM cell lines carrying both a p53 wild-type and a mutated or down-regulated p53. However, our data showed that the functional status of p53 influences the sensitivity of GBM cells to combined treatments of TPT, RT, and PARP-1 inhibitor. Specifically, cells carrying an active p53 are more sensitive to the combination of RT and NU1025 while cells carrying a mutated or down-regulated p53 are more sensitive to TPT and NU1025 combination. These conclusions are corroborated by the data obtained analyzing the growth inhibitory effects, the cell cycle perturbations and the induction of DNA damage. The differential effects of TPT, RT, and NU1025 combinations are likely to reflect the differences in the cell cycle phase targeting of TPT and RT (40) as well as the different radiosensitivity of GBM cells, which is partially overcome by PARP-1 inhibition. Cells carrying an inactive p53 display a higher radioresistance as compared to cells carrying an active p53. Several lines of evidence indicate that the functional status of p53 plays a major role in the chemo-radiosensitivity and in the clinical outcome of GMBs (3,41-44). It is noteworthy that NU1025 in combination with RT or TPT displayed a similar or higher activity than TPT and RT combination. The latter as compared to NU1025 and TPT or RT combinations is expected to have a higher toxicity and a lower therapeutic index (10,11). These in vitro results can have clinical relevance since they expand the potential application of PARP inhibitors in combination with TPT or RT to the treatment of GBM based on the functional status of p53. They suggest differential treatments for GBM patients, which are expected to have a more favorable therapeutic index as compared to the toxic chemotherapy and RT combination. Radio- and chemo-sensitizing agents that do not carry side effects such as PARP inhibitors have the great potential to achieve selective modulation of cytotoxic drug action in tumors by increasing their efficacy and dose tolerance.
Supplementary Material
Acknowledgments
The authors wish to thank Mr. G. Borriello for his excellent technical assistance.
Grant sponsor: F.A.R.O. (Finanziamenti per l’avvio di ricerche originali)
Grant sponsor: Polo delle Scienze e delle Tecnologie per la Vita, Grant number: 0086257
Grant sponsor: National Cancer Institute PHS grants, Grant number: RO1CA138188
Grant sponsor: Fondazione Umberto Veronesi Post Doctoral Fellowship
Footnotes
Additional Supporting Information may be found in the online version of this article.
Literature Cited
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–996. [DOI] [PubMed] [Google Scholar]
- 2.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res 2004;64:6892–6899. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Zhu S, Cloughesy TF, Liau LM, Mischel PS. p53 disruption profoundly alters the response of human glioblastoma cells to DNA topoisomerase I inhibition. Oncogene 2004;23:1283–1290. [DOI] [PubMed] [Google Scholar]
- 4.Sasine JP, Feun LG. Topoisomerase I inhibitors in the treatment of primary CNS malignancies: An update on recent trends. Anticancer Agents Med Chem 2011;10:683–696. [DOI] [PubMed] [Google Scholar]
- 5.Pinel S, Chastagner P, Merlin JL, Marchal C, Taghian A, Barberi-Heyob M. Topotecan can compensate for protracted radiation treatment time effects in high grade glioma xenografts. J Neurooncol 2006;76:31–38. [DOI] [PubMed] [Google Scholar]
- 6.Fisher B, Won M, Macdonald D, Johnson DW, Roa W. Phase II study of topotecan plus cranial radiation for glioblastoma multiforme: Results of Radiation Therapy Oncology Group 9513. Int J Radiat Oncol Biol Phys 2002;53:980–986. [DOI] [PubMed] [Google Scholar]
- 7.Klautke G, Schutze M, Bombor I, Benecke R, Piek J, Fietkau R. Concurrent chemoradiotherapy and adjuvant chemotherapy with Topotecan for patients with glioblastoma multiforme. J Neurooncol 2006;77:199–205. [DOI] [PubMed] [Google Scholar]
- 8.Bernier-Chastagner V, Grill J, Doz F, Bracard S, Gentet JC, Marie-Cardine A, Luporsi E, Margueritte G, Lejars O, Laithier V, et al. Topotecan as a radiosensitizer in the treatment of children with malignant diffuse brainstem gliomas: results of a French Society of Paediatric Oncology Phase II Study. Cancer 2005;104:2792–2797. [DOI] [PubMed] [Google Scholar]
- 9.Gross MW, Altscher R, Brandtner M, Haeusser-Mischlich H, Chiricuta IC, Siegmann AD, Engenhart-Cabillic R. Open-label simultaneous radio-chemotherapy of glioblastoma multiforme with topotecan in adults. Clin Neurol Neurosurg 2005;107:207–213. [DOI] [PubMed] [Google Scholar]
- 10.Grabenbauer GG, Gerber KD, Ganslandt O, Richter A, Klautke G, Birkmann J, Meyer M. Effects of concurrent topotecan and radiation on 6-month progression-free survival in the primary treatment of glioblastoma multiforme. Int J Radiat Oncol Biol Phys 2009;75:164–169. [DOI] [PubMed] [Google Scholar]
- 11.Lesimple T, Riffaud L, Frappaz D, Ben Hassel M, Gedouin D, Bay JO, Linassier C, Hamlat A, Piot G, Fabbro M, et al. Topotecan in combination with radiotherapy in unresectable glioblastoma: A phase 2 study. J Neurooncol 2009;93:253–260. [DOI] [PubMed] [Google Scholar]
- 12.Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res 2007;13:1383–1388. [DOI] [PubMed] [Google Scholar]
- 13.Wesierska-Gadek J, Schmid G, Cerni C. ADP-ribosylation of wild-type p53 in vitro: Binding of p53 protein to specific p53 consensus sequence prevents its modification. Biochem Biophys Res Commun 1996;224:96–102. [DOI] [PubMed] [Google Scholar]
- 14.Simbulan-Rosenthal CM, Rosenthal DS, Iyer S, Boulares AH, Smulson ME. Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J Biol Chem 1998;273:13703–13712. [DOI] [PubMed] [Google Scholar]
- 15.Griffin RJ, Curtin NJ, Newell DR, Golding BT, Durkacz BW, Calvert AH. The role of inhibitors of poly(ADP-ribose) polymerase as resistance-modifying agents in cancer therapy. Biochimie 1995;77:408–422. [DOI] [PubMed] [Google Scholar]
- 16.Bowman KJ, Newell DR, Calvert AH, Curtin NJ. Differential effects of the poly (ADP-ribose) polymerase (PARP) inhibitor NU1025 on topoisomerase I and II inhibitor cytotoxicity in L1210 cells in vitro. Br J Cancer 2001;84:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tentori L, Portarena I, Graziani G. Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol Res 2002;45:73–85. [DOI] [PubMed] [Google Scholar]
- 18.Tentori L, Graziani G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol Res 2005;52:25–33. [DOI] [PubMed] [Google Scholar]
- 19.Cimmino G, Pepe S, Laus G, Chianese M, Prece D, Penitente R, Quesada P. Poly(-ADPR)polymerase-1 signalling of the DNA damage induced by DNA topoisomerase I poison in D54(p53wt) and U251(p53mut) glioblastoma cell lines. Pharmacol Res 2007;55:49–56. [DOI] [PubMed] [Google Scholar]
- 20.Sabisz M, Wesierska-Gadek J, Skladanowski A. Increased cytotoxicity of an unusual DNA topoisomerase II inhibitor compound C-1305 toward HeLa cells with downregulated PARP-1 activity results from re-activation of the p53 pathway and modulation of mitotic checkpoints. Biochem Pharmacol 2010;79:1387–1397. [DOI] [PubMed] [Google Scholar]
- 21.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–760. [DOI] [PubMed] [Google Scholar]
- 22.Fuxe J, Akusjarvi G, Goike HM, Roos G, Collins VP, Pettersson RF. Adenovirus-mediated overexpression of p15INK4B inhibits human glioma cell growth, induces replicative senescence, and inhibits telomerase activity similarly to p16INK4A. Cell Growth Differ 2000;11:373–384. [PubMed] [Google Scholar]
- 23.Mosmann T Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63. [DOI] [PubMed] [Google Scholar]
- 24.Burkle A, Chen G, Kupper JH, Grube K, Zeller WJ. Increased poly(ADP-ribosyl)ation in intact cells by cisplatin treatment. Carcinogenesis 1993;14:559–561. [DOI] [PubMed] [Google Scholar]
- 25.Jacobberger JW, Sramkoski RM, Stefan T. Multiparameter cell cycle analysis. Methods Mol Biol 2011;699:229–249. [DOI] [PubMed] [Google Scholar]
- 26.Huang X, Halicka HD, Darzynkiewicz Z. Detection of histone H2AX phosphorylation on Ser-139 as an indicator of DNA damage (DNA double-strand breaks). Curr Protoc Cytom 2004;Chapter 7(Unit 7):27. [DOI] [PubMed] [Google Scholar]
- 27.Hao DL, Liu CM, Dong WJ, Gong H, Wu XS, Liu DP, Liang CC. Knockdown of human p53 gene expression in 293-T cells by retroviral vector-mediated short hair-pin RNA. Acta Biochim Biophys Sin (Shanghai) 2005;37:779–783. [DOI] [PubMed] [Google Scholar]
- 28.Sabbatino F, Wang Y, Wang X, Flaherty KT, Yu L, Pepin D, Scognamiglio G, Pepe S, Kirkwood JM, Cooper ZA, et al. PDGFRalpha up-regulation mediated by sonic hedgehog pathway activation leads to BRAF inhibitor resistance in melanoma cells with BRAF mutation. Oncotarget 2014;5:1926–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piroth MD, Gagel B, Pinkawa M, Stanzel S, Asadpour B, Eble MJ. Postoperative radiotherapy of glioblastoma multiforme: analysis and critical assessment of different treatment strategies and predictive factors. Strahlenther Onkol 2007;183:695–702. [DOI] [PubMed] [Google Scholar]
- 30.Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature 1980;283:593–596. [DOI] [PubMed] [Google Scholar]
- 31.Ueno AM, Tanaka O, Matsudaira H. Inhibition of gamma-ray dose-rate effects by D2O and inhibitors of poly(ADP-ribose) synthetase in cultured mammalian L5178Y cells. Radiat Res 1984;98:574–582. [PubMed] [Google Scholar]
- 32.Thraves P, Mossman KL, Brennan T, Dritschilo A. Radiosensitization of human fibroblasts by 3-aminobenzamide: an inhibitor of poly(ADP-ribosylation). Radiat Res 1985;104:119–127. [PubMed] [Google Scholar]
- 33.Kelland LR, Tonkin KS. The effect of 3-aminobenzamide in the radiation response of three human cervix carcinoma xenografts. Radiother Oncol 1989;15:363–369. [DOI] [PubMed] [Google Scholar]
- 34.Papeo G, Forte B, Orsini P, Perrera C, Posteri H, Scolaro A, Montagnoli A. Poly(-ADP-ribose) polymerase inhibition in cancer therapy: are we close to maturity? Expert Opin Ther Pat 2009;19:1377–1400. [DOI] [PubMed] [Google Scholar]
- 35.Nguewa PA, Fuertes MA, Alonso C, Perez JM. Pharmacological modulation of Poly(-ADP-ribose) polymerase-mediated cell death: exploitation in cancer chemotherapy. Mol Pharmacol 2003;64:1007–1014. [DOI] [PubMed] [Google Scholar]
- 36.Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol 2003;81:123–129. [DOI] [PubMed] [Google Scholar]
- 37.Banath JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res 2003;63:4347–4350. [PubMed] [Google Scholar]
- 38.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol 2003;79:351–358. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi R, Giannini C, Sarkaria JN, Schroeder M, Rogers J, Mastroeni D, Scrable H. p53 isoform profiling in glioblastoma and injured brain. Oncogene 2012;32:3165–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorczyca W, Gong J, Ardelt B, Traganos F, Darzynkiewicz Z. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor agents. Cancer Res 1993;53:3186–3192. [PubMed] [Google Scholar]
- 41.Martin S, Janouskova H, Dontenwill M. Integrins and p53 pathways in glioblastoma resistance to temozolomide. Front Oncol 2012;2:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pena-Rico MA, Calvo-Vidal MN, Villalonga-Planells R, Martinez-Soler F, Gimenez-Bonafe P, Navarro-Sabate A, Tortosa A, Bartrons R, Manzano A. TP53 induced glycolysis and apoptosis regulator (TIGAR) knockdown results in radiosensitization of glioma cells. Radiother Oncol 2011;101:132–139. [DOI] [PubMed] [Google Scholar]
- 43.Puca R, Nardinocchi L, Porru M, Simon AJ, Rechavi G, Leonetti C, Givol D, D’Orazi G. Restoring p53 active conformation by zinc increases the response of mutant p53 tumor cells to anticancer drugs. Cell Cycle 2011;10:1679–1689. [DOI] [PubMed] [Google Scholar]
- 44.Ruano Y, Ribalta T, de Lope AR, Campos-Martin Y, Fiano C, Perez-Magan E, Hernandez-Moneo JL, Mollejo M, Melendez B. Worse outcome in primary glioblastoma multiforme with concurrent epidermal growth factor receptor and p53 alteration. Am J Clin Pathol 2009;131:257–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.