

Abstract
Rationale
Elevated whole-blood serotonin (5-HT) is a robust biomarker in ~ 30% of patients with autism spectrum disorders, in which repetitive behavior is a core symptom. Furthermore, elevated whole-blood 5-HT has also been described in patients with pediatric obsessive-compulsive disorder. The 5-HT1B receptor is associated with repetitive behaviors seen in both disorders. Chronic blockade of serotonin transporter (SERT) reduces 5-HT1B receptor levels in the orbitofrontal cortex (OFC) and attenuates the sensorimotor deficits and hyperactivity seen with the 5-HT1B agonist RU24969. We hypothesized that enhanced SERT function would increase 5-HT1B receptor levels in OFC and enhance sensorimotor deficits and hyperactivity induced by RU24969.
Objectives
We examined the impact of the SERT Ala56 mutation, which leads to enhanced SERT function, on 5-HT1B receptor binding and 5-HT1B-mediated sensorimotor deficits.
Methods
Specific binding to 5-HT1B receptors was measured in OFC and striatum of naïve SERT Ala56 or wild-type mice. The impact of the 5-HT1A/1B receptor agonist RU24969 on prepulse inhibition (PPI) of startle, hyperactivity, and expression of cFos was examined.
Results
While enhanced SERT function increased 5-HT1B receptor levels in OFC of Ala56 mice, RU24969-induced PPI deficits and hyperlocomotion were not different between genotypes. Baseline levels of cFos expression were not different between groups. RU24969 increased cFos expression in OFC of wild-types and decreased cFos in the striatum.
Conclusions
While reducing 5-HT1B receptors may attenuate sensorimotor gating deficits, increased 5-HT1B levels in SERT Ala56 mice do not necessarily exacerbate these deficits, potentially due to compensations during neural circuit development in this model system.
Keywords: Serotonin, 5-HT1B, Autism spectrum disorder, ASD, Obsessive-compulsive disorder, OCD, Sensorimotor gating, Prepulse inhibition of startle, Orbitofrontal cortex, RU24969
Introduction
Elevated whole-blood serotonin (5-HT) levels are the most robust biomarker in autism spectrum disorder (ASD) (Gabriele et al. 2014; Muller et al. 2016) and have also been described in familial pediatric obsessive-compulsive disorder (OCD) (Hanna et al. 1991), suggesting that the serotonin system is perturbed in both disorders. ASD and OCD share repetitive behavior as a core symptom domain and frequently co-occur in the same individual (Bejerot 2007; Leyfer et al. 2006). Sensorimotor gating deficits have also been found in both ASD and OCD (Ahmari et al. 2012, 2016; Frankland et al. 2004; Hoenig et al. 2005; Kohl et al. 2013; McAlonan et al. 2002). Furthermore, serotonin 5-HT1B receptor agonists disrupt sensorimotor gating, and increased sensitivity to 5-HT1B agonists has been described in both ASD and OCD (Hollander et al. 2000; Novotny et al. 2004).
Prepulse inhibition (PPI) of startle has been used to study sensorimotor gating deficits in ASD and OCD (Ahmari et al. 2012, 2016; Frankland et al. 2004; Hoenig et al. 2005; Kohl et al. 2013). In rodents, 5-HT1B agonists attenuate PPI (Dulawa et al. 1997; Shanahan et al. 2009). The mixed 5-HT1A/1B agonist RU24969 diminishes the PPI response in wild-type (Dulawa et al. 1998) and 5-HT1A receptor knockout mice (Dulawa et al. 2000) but not in 5-HT1B receptor knockout mice (Dulawa et al. 1997, 1998, 2000), suggesting that RU24969 selectively modulates PPI through the 5-HT1B receptor. Furthermore, perseverative hyperlocomotion induced by RU24969 is also mediated by the 5-HT1B receptor (Shanahan et al. 2009).
While 5-HT1B receptors are most densely located on striatal neuron terminals in the globus pallidus and substantia nigra, and on Purkinjie neuron terminals in the deep cerebellar nuclei (Boschert et al. 1994; Pazos and Palacios 1985), 5-HT1B receptors in the orbitofrontal cortex (OFC) have been established as necessary and sufficient to induce PPI deficits and hyperlocomotive behavior in mice (Shanahan et al. 2011). Additionally, positive correlations between 5-HT1B receptor binding in the OFC and PPI in controls and OCD patients were observed (Pittenger et al. 2016). Functional neuroimaging studies found increased activity in the OFC and striatum in OCD patients (Saxena and Rauch 2000). The correlation between 5-HT1B receptor binding in the dorsal striatum was also implicated in PPI in rodent studies (Baldan Ramsey et al. 2011; Swerdlow et al. 2001), and PPI was significantly different from the correlation in OCD patients (Pittenger et al. 2016). Finally, 5-HT1B receptor agonism increased cFos expression, a marker of cellular activity, in the striatum of mice (Ho et al. 2016). As a result of these collective data across species, the 5-HT1B receptor has been suggested as a potential mediating factor in repetitive behavior seen in OCD (Dulawa et al. 1997; Pittenger et al. 2016).
Effective OCD treatments have been used to validate the use of 5-HT1B agonism in modeling OCD-related behavioral abnormalities. Serotonin reuptake inhibitors (SRIs) provide effective pharmacological therapy for OCD (Hollander and Pallanti 2002). In mice, treatment for 28 days, but not 7 days, with selective SRIs (SSRIs) prevented PPI attenuation by RU24969 (Shanahan et al. 2009). Knockout of the 5-HT transporter (SERT) similarly prevents PPI attenuation by RU24969 (Shanahan et al. 2009). Treatment with ketamine, a promising treatment for OCD (Rodriguez et al. 2013), for 24 h reduced the preservative hyperlocomotion deficits induced by RU24969 (Thompson et al. 2020). Chronic SRI treatment also ameliorated RU24969-induced preservative hyperlocomotion in mice, while desipramine, a norepinephrine reuptake inhibitor that is not effective at treating OCD, did not affect RU24969-induced hyperlocomotion (Ho et al. 2016; Thompson et al. 2020). Thus, the PPI and hyperlocomotor deficits seen in mice after RU24969 administration are meaningful behavioral readouts for examining OCD-related circuit abnormalities associated with 5-HT1B receptors.
Abnormal SERT function could explain elevated whole-blood 5-HT levels in autism and linkage to autism has been identified on the 17q region harboring the SLC6A4 gene that encodes SERT, particularly in families with affected males, inherited from their unaffected mothers (International Molecular Genetic Study of Autism Consortium 1998; Sutcliffe et al. 2005). Multiple rare variants of SLC6A4 have been identified in families with ASD and linkage to chromosome 17q, with the most common amino acid variant being a glycine to alanine change at position 56 in the SERT protein (Gly56Ala, referred to as Ala56) (Sutcliffe et al. 2005); however, de novo variants in SERT have not been identified at increased rates in autism (Cappi et al. 2016; Feliciano et al. 2019). A rare SERT variant, Ile425Val, has also been identified in an extended family with OCD (Wendland et al. 2008). In cellular models and lymphoblastoid cell lines, the Ala56 variant leads to increased SERT function, as do multiple other identified variants, including Val425 (Prasad et al. 2009; Prasad et al. 2005; Quinlan et al. 2019).
We previously created SERT Ala56 knock-in mice (Veenstra-Vanderweele et al. 2009; Veenstra-VanderWeele et al. 2012) that display enhanced and dysregulated SERT function as well as elevated whole-blood 5-HT levels, mimicking elevated whole-blood levels of 5-HT seen in approximately 30% of ASD patients (Gabriele et al. 2014; Piven et al. 1991). These animals also have altered firing properties of dorsal raphe neurons, including decreased basal firing and enhanced inhibition by 5-HT, as well as enhanced responses to 5-HT1A and 5-HT2 receptor agonists (Veenstra-VanderWeele et al. 2012). In addition, relative to wild-type littermates, Ala56 knock-in mice exhibit reduced ultrasonic vocalizations, decreased social dominance, and increased repetitive hanging/climbing behaviors. Here we sought to understand the impact of chronic enhanced SERT function on 5-HT1B binding and 5-HT1B-mediated PPI, hyperlocomotion, and immediate early gene activation. We hypothesized that the enhanced SERT function in Ala56 mice would lead to altered 5-HT1B receptor binding and enhanced sensitivity to RU24969, paralleling what we observed at 5-HT1A and 5-HT2 receptors in these animals.
Methods
Animals
Male homozygous SERT Ala56/Ala56 (hereafter referred to as SERT Ala56 or Ala56) (Veenstra-Vanderweele et al. 2009) and wild-type littermate control mice (Gly56/Gly56) were generated from heterozygous breeding on a pure 129S6/SvEvTac background. Only male mice were used because the SERT Ala56 variant only affects males in the human population (Sutcliffe et al. 2005). Mice were housed with same-sex littermates, two to five per cage with a 12-h light/dark cycle and food and water available ad libitum. Mice were between 8 and 14 weeks of age for experiments. All procedures were approved by the Institutional Animal Care and Use Committee at Vanderbilt University, where the experiments took place, and designed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
5-HT1B binding assay
Adult, naïve wild-type and Ala56 mice were sacrificed via decapitation; the brains were rapidly removed and frozen by immersion in isopentane refrigerated with liquid nitrogen. Eight series of coronal sections (10 μm) were obtained with a cryostat at − 20 °C and thaw-mounted onto poly-l-lysine-coated slides. Right and left hemispheres were tracked. Each slide contained one brain from each genotype.
Half of the brains were processed on 1 day and the other half were processed on a different day, balanced for genotype. The sections were thawed at room temperature for 60 min and pre-incubated for 60 min in binding buffer (65 mM Tris-HCL, 0.93 mM MgCl2 in water) at room temperature. The edges of the slides were then dried and marked with a PAP-pen (Abcam, Cambridge, MA). Total binding (TB) and nonspecific binding (NSB) solutions were prepared in binding buffer. TB: [125I]-iodocyanopindolol (1 μL/mL), pargyline (10−2 M), isoproterenol (10−2 M), 8-OH-DPAT (10−4 M), ascorbic acid (1%), and bovine serum albumen (3%). NSB: [125I]-iodocyanopindolol (1 μL/mL), pargyline (10−2 M), isoproterenol (10−2 M), 8-OH-DPAT (10−4 M), GR127935 (200 μM), ascorbic acid (1%), and bovine serum albumen (3%). Slides were allowed to incubate for 120 min in 1 mL of TB or NSB solution in a covered tray at room temperature, after which point the reactions were terminated by submerging the slides in ice-cold binding buffer followed by two washes (7.5 min each) in ice-cold binding buffer. The slides were then dipped in ice-cold water and allowed to air-dry for 4 h at room temperature. Once dry, the slides were exposed to film in the presence of radioactive standards (0.063–2.15 μCi/g) for 24 h. The films were developed using a Mini-Medical Series developer. Once dry, the films were scanned on an Epson Perfection V550 Photo Scanner at 1200 dpi.
Optical densities were measured from acquired images using ImageJ, as described in O’Reilly et al. (2016), and converted to radioactivity (μCi/g) from the standards. Readings were taken from the OFC (medial, ventral, and lateral orbitofrontal cortices) and the dorsal striatum (dSTR) (Supplemental File 1). Readings were taken from the left and right hemispheres from ~ 5 sections per hemisphere for the OFC and from ~ 3 sections per hemisphere for the dSTR. The location of each section along the anterior-posterior axis was tracked in order to determine if there was an effect of the anterior-posterior location on the binding levels. The nonspecific binding radioactivity was subtracted from the total binding radioactivity to estimate the specific binding.
Behavior
Wild-type (n = 12) and Ala56 (n = 14) mice were 8–12 weeks at the time of testing and habituated to the open-field chamber for 90 min 2 days prior to behavioral testing. Mice received an intraperitoneal injection of 0 or 10 mg/kg body weight of the 5-HT1B agonist, RU24969 dissolved in 0.9% saline, and were immediately placed in the open-field chamber for 20 min. A 10-mg/kg dose of RU24969 was chosen because it has been shown to reliably affect PPI (Dulawa et al. 1997). Locomotor activities were assessed using activity monitors measuring 27.9 × 27.9 cm (MED Associates). Each apparatus contained 16 photocells in each horizontal direction, as well as 16 photocells elevated 4 cm to measure rearing, as described previously (Veenstra-VanderWeele et al. 2012).
Following the open-field test, the mice were immediately tested for prepulse inhibition of acoustic startle using Acoustic Startle Reflex Test Compartments (MED Associates). By placing the animals in the open field immediately following injections, the start of the prepulse inhibition paradigm is similar to the timing used previously, which was 30 min post-injection (Dulawa et al. 1997). Mice were acclimated to background white noise of 65 dB for 5 min before being presented with seven trial types in six discrete, randomized blocks of trials (for a total of 42 trials) with an intertrial interval of 10 to 20 s. One trial measured baseline movement and one trial measured response to the 120 dB, 50-ms startle stimulus alone. The other five trials used an acoustic prepulse of 70, 76, 82, or 88 dB preceding the acoustic startle stimulus by 100 ms. Startle amplitude was measured every millisecond over a 65-ms period beginning at the onset of the startle stimulus. Prepulse inhibition was calculated by dividing the difference between baseline startle and startle following prepulse by baseline startle as in Veenstra-VanderWeele et al. (2012). Two days later, the same mice received the opposite drug treatment and underwent the same behavioral analysis.
cFos immunostaining
Two hours after the second injection (RU24969 or saline), Ala56 mice were anesthetized with sodium pentobarbital and transcardially perfused with 1× phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA) in PBS. The brains were extracted, stored overnight in 4% PFA at 4 °C, and transferred to PBS containing 30% sucrose until they were cut on a microtome (40-μm sections) in the coronal plane and stored in a cryoprotectant (PBS, ethylene glycol, and glycerol) at − 20 °C.
cFos immunostaining was performed on sections containing OFC and striatal regions. The sections were washed (4 × 5 min) in PBS containing 0.3% Triton X-100 (PBST), followed by incubation in 0.3% H2O2 for 30 min at room temperature and were then washed (3 × 5 min) again with PBST. The sections were blocked in PBST containing 5% Normal Donkey Serum and then incubated with rabbit polyclonal anti-cFos primary antibody (1:1000 dilution in PBST, 5% NDS, Abcam, Cambridge, MA) for 3 days at 4 °C with constant agitation. The sections were washed (PBST, 4 × 5 min), followed by incubation in donkey anti-rabbit biotinylated secondary antibody (1:250 dilution in PBST, 5% NDS, Jackson Immuno Secondary, catalog number) for 1.5 h at 4 °C. The sections were then washed (PBST, 4 ×5 min); incubated in PBST containing VECTASTAIN® Elite® avidin/biotin complex (ABC) reagent; washed (PBST, 4 × 5 min); and reacted with 3,3′-diaminobenzidine (DAB) substrate (DAB substrate kit, Vector Labs, Burlingame, CA) according to manufacturer’s instructions. The sections were washed three times in PBS and mounted on Superfrost slides.
Sections were visualized under a brightfield light microscope and images were obtained using AxioVision software. Activated cells in the ventral OFC and dSTR were counted using the ImageJ software (US National Institutes of Health, Bethesda, MD). The area of activated cells as a percentage of the total area was used for quantification.
Statistics
5-HT1B binding
Univariate analysis of variance (ANOVA) was used to determine if there was an effect of section location along the anterior-posterior axis on 5-HT1B binding levels within each hemisphere for the wild-type and Ala56 genotypes combined. Once it was determined that there was no effect of section, the activity was averaged for each brain region within each hemisphere and a univariate ANOVA was used to determine if there was an effect of hemisphere. Once it was determined that there was no effect of hemisphere, the radioactivity for each region was averaged over both hemispheres. The primary hypothesis was that 5-HT1B binding would be altered in the OFC, particularly in the ventral OFC (VO), where activation of 5-HT1B receptors was sufficient to induce PPI deficits (Shanahan et al. 2011). We hypothesized that binding could also be altered in the medial and lateral OFC (MO and LO, respectively) where drugs infused into the VO could potentially diffuse to and where a few cannula were placed in the Shanahan et al.’s (2011) study. Our secondary analysis focused on binding in the dSTR where cFos expression has been shown to be increased after RU24696 exposure (Ho et al. 2016). To analyze receptor binding activity in the OFC, a multivariate analysis of variance (MANOVA) was performed with MO, VO, and LO as dependent variables and genotype as the independent variable. A univariate ANOVA was used to determine if there was an effect of genotype in the dSTR. Differences were considered significant at p < 0.05.
Behavior
For the open field, the total distance traveled was broken into 5-min time bins over the 20-min trial. A three-way ANOVA was used to examine the effect of genotype, time bin, and RU24969 exposure. For the prepulse inhibition paradigm, a three-way ANOVA was used to examine effects of prepulse intensity, genotype, and RU24969 exposure. Post hoc analysis was performed to examine the effect of drug exposure on startle response by performing a t test at each prepulse intensity for the two genotypes combined. Bonferroni’s correction was done to account for multiple comparisons. Differences were considered significant at p < 0.05.
cFos staining
Two-way ANOVA evaluating genotype (wild-type or Ala56) and drug treatment (Saline or RU24969) was used to establish group differences or significant interactions. Differences were further investigated using Tukey’s HSD post hoc analysis. Differences were considered significant at p < 0.05.
Results
5-HT1B receptor binding is increased in the orbitofrontal cortex
There was no effect of section location along the anterior-posterior axis from which 5-HT1B receptor binding levels were measured within either hemisphere for any brain region on specific 5-HT1B binding, nor was there an effect of hemisphere (Supplemental File 2). There was a trend for genotype to affect 5-HT1B binding in the OFC (F3,7 = 4.19, p = 0.05). Compared to wild-type, Ala56 mice had increased specific 5-HT1B binding in the VO and a trend for increased binding in the LO (Fig. 1a; VO—F1,9 = 6.14, p value: 0.04; LO—F1,9 = 5.03, p = 0.052). Differences in 5-HT1B-specific binding were not detected between the wild-type and Ala56 mice in the MO (Fig. 1a; F1,9 = 1.49, p value: 0.25) or in the dSTR (Fig. 1b; F1,9 = 1.87, p = 0.21).
Fig. 1.
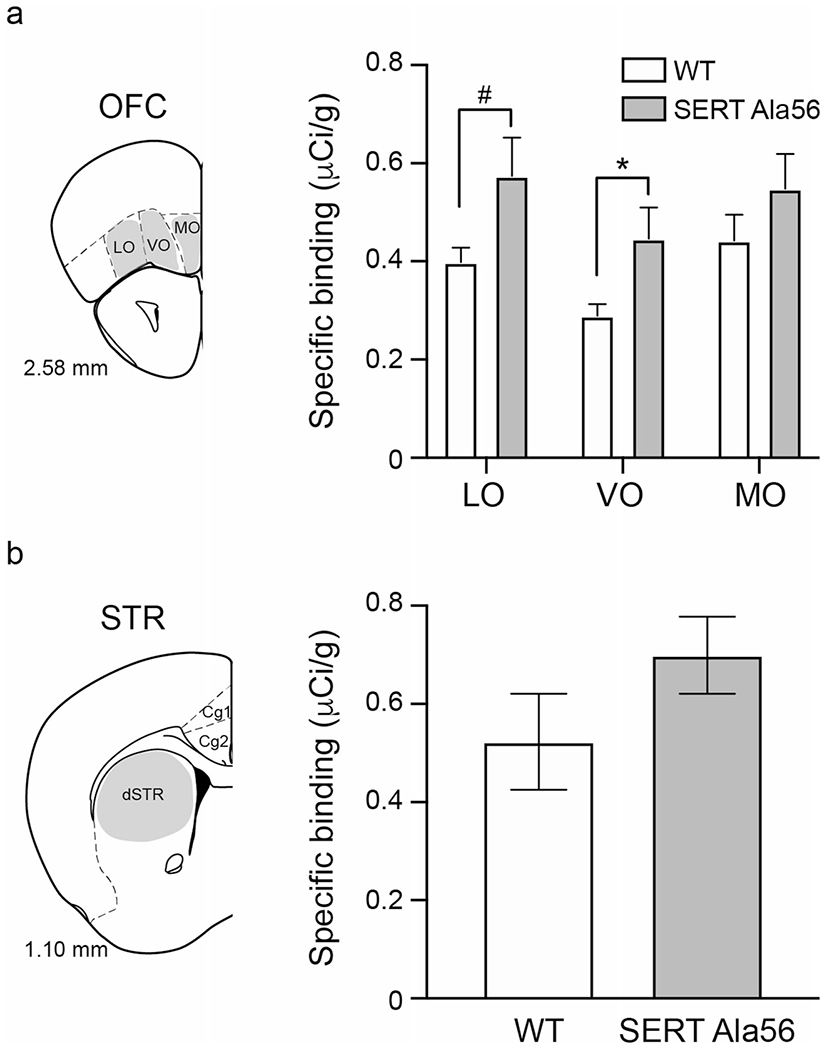
5-HT1B receptor binding is increased in the ventral OFC. a Specific binding to 5-HT1B receptors was measured in the OFC. There was more receptor binding in the ventral OFC of Ala56 than wild-type mice and a tendency for receptor binding to be increased on the lateral OFC of Ala56 mice. b There were no differences in the dorsal striatum in 5-HT1B binding between the two genotypes. LO = lateral OFC; VO = ventral OFC; MO = medial OFC. dSTR = dorsal striatum. Wild type: n = 6; Ala56: n = 5. Data are mean ± SEM. *p < 0.05; #p < 0.1
Ala56 mice show no difference in RU24969-induced hyperlocomotion or PPI deficits
Locomotor activity, measured as distance traveled, was assessed in the SERT Ala56 and wild-type mice in response to saline or RU24969 administration in the open field (Fig. 2a). There was an effect of time (F1.12,28.24 = 19.90, p < 0.0001) and RU24969 administration (F1,24 = 180.70, p < 0.0001) on locomotor activity, and there was an interaction between these two factors (time × drug F2.12,50.79 = 83.50, p < 0.0001). Differences in locomotor activity were not detected due to genotype (genotype F1,24 = 0.01, p = 0.93), nor was there an interaction between time and genotype (F3,72 = 0.70, p = 0.56), drug and genotype (F1,24 = 0.36, p = 0.56) or among the three factors (time × drug × genotype F3,72 = 1.30, p = 0.28). Although there was an effect of prepulse intensity (F3,72 = 80.60, p < 0.0001), RU24969 exposure (F1,24 = 15.48, p < 0.001), and an interaction between intensity and drug exposure (F3,72 = 9.94, p < 0.0001), there was no effect of genotype (F1,24 = 1.31, p = 0.26) on PPI (Fig. 2b), nor were there interaction effects between genotype and prepulse intensity (F3,72 = 1.84, p = 0.15); genotype and RU24969 exposure (F1,24 = 2.72, p = 0.11); or genotype, prepulse intensity, and RU24969 exposure (F3,64 = 1.25, p = 0.30). Because we found no genotype differences in response to RU24969, we combined both genotypes to evaluate the interaction between prepulse intensity and drug exposure. We found that RU24969 enhanced the startle effect at prepulse intensities of 70, 76, and 80 dB (70 dB: t192 = 4.594, p < 0.0001; 76 dB: t192 = 3.846, p < 0.001; PPI 82: t192 = 2.965, p < 0.01) but the effect was no longer present at 88 dB (t192 = 0.4328; p > 0.9999).
Fig. 2.
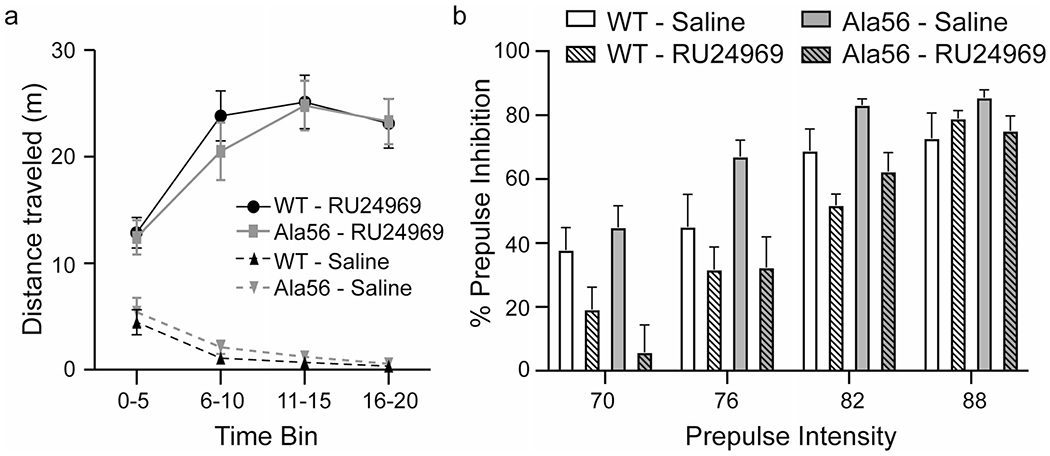
RU24969 enhances hyperlocomotive behavior and PPI similarly in Ala56 and wild-type mice. Wild-type (n = 12) and Ala56 (n = 14) mice were administered 10-mg/kg body weight RU24969 or an equivalent volume of saline and immediately tested in the open field. a Under saline conditions, both wild-type and Ala56 mice habituate to the open field, decreasing the distance they walk with time. Administration of RU24696 increases locomotor activity similarly in wild-type and Ala56 mice. b Immediately following testing in the open field, mice were tested for PPI. There were no differences in Ala56 and wild-type in percent of prepulse inhibition at any prepulse intensity. RU24969 decreased the percent of prepulse inhibition similary for both genotypes. Data are mean ± SEM
RU24969 administration alters immediate early gene expression in the orbitofrontal cortex and striatum
To examine the effect of RU24969 administration on cellular activity, cFos expression was examined in the VO of the OFC and the dSTR (Supplemental File 3) using the region delineations used to assess 5-HT1B binding (Supplemental File 1). A two-way ANOVA revealed there was no effect of drug (F1,19 = 2.58, p = 0.12) or genotype (F1,19 = 0.05, p = 83) on cFos expression in the VO (Fig. 3a), but there was an interaction (F1,19 = 8.27, p = 0.01). Post hoc analysis revealed that exposure to RU24969 significantly increased cFos expression in the OFC of the wild-type mice (Tukey’s HSD p = 0.03, Supplemental File 2), with no significant difference in the SERT Ala56 mice (p = 0.78).
Fig. 3.
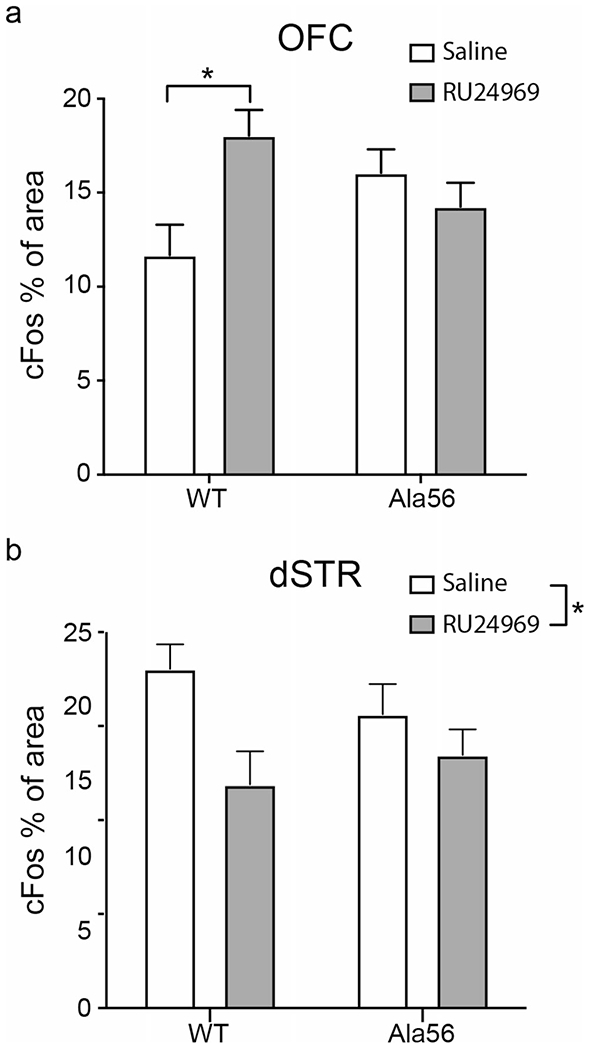
RU24969 has no effect on cFos activity in SERT Ala56 mice. Two hours following saline or RU24969 injection, brains were collected for cFos immunostaining. There were no differences between wild-type and Ala56 cFos staining in any brain region after saline injection. a cFos staining was increased in the ventral OFC (VO) of wild-type mice after RU24969 administration. b RU24969 decreased cFos staining in the dorsal striatum (dSTR). There was no effect of genotype nor was there an interaction. VO: wild-type, saline: n = 5; wild-type, RU24969: n = 5. Ala56, saline: n = 5; Ala56, RU24969: n = 5; dS: wild-type, saline: n = 5: wild-type, RU24969: n = 6; Ala56, saline: n = 5; Ala56, RU24969: n = 9; vS: wild-type, saline: n = 4; wild-type, RU24969: n = 6; Ala56, saline: n = 5; Ala56, RU24969: n = 9. Data are mean ± SEM. *p < 0.05
A two-way ANOVA on cFos expression in the dSTR (Fig. 3b, Supplemental File 3) revealed an effect of drug (F1,21 = 6.75, p = 0.02) on cFos expression, but no effect of genotype (F1,21 = 0.07, p = 0.79), and no interaction effect (F1,21 = 3.59, p = 0.22).
Discussion
Across species, serotonin 5-HT1B receptor agonism has been suggested to mediate prepulse inhibition deficits and repetitive behavior observed in OCD and ASD (Dulawa et al. 1997; Pittenger et al. 2016). Chronic administration of SRIs and genetic ablation of SERT result in decreased levels of 5-HT1B receptors in the OFC of mice, which have been shown to be necessary and sufficient for RU24969-induced hyperlocomotion and PPI deficits (Shanahan et al. 2011). Consistent with decreased SERT function yielding decreased 5-HT1B receptor levels in the OFC, Ala56 mice, whose SERT function is enhanced and dysregulated, have increased baseline levels of 5-HT1B binding in the OFC. However, the relationship between 5-HT1B receptor levels in the OFC and behavioral response to RU24969 of Ala56 mice do not neatly parallel-blunted RU24969 response to decreased 5-HT1B receptor levels. We saw no increase in either baseline or RU24969-induced hyperlocomotion or PPI deficits in Ala56 mice compared to wild-type mice. Our PPI results are, however, consistent with prior work showing that RU24969 no longer has an effect on startle response at the 88-dB prepulse intensity in this background strain (Dulawa and Geyer 2000).
The 10-mg/kg dose of RU24969 is considered a high dose and was used because it reliably alters PPI (Dulawa et al. 1997), but PPI deficits induced by 2.5-, 5-, and 10-mg/kg doses of RU24969 are similarly different from saline administration (Dulawa and Geyer 2000). While administration of 1-, 3-, 5-, or 10-mg/kg RU24969 significantly increased the distance traveled in the open field compared to saline administration in the wild-type mice (Ho et al. 2016), the doses did not linearly increase locomotor activity: locomotor activity induced by 1- and 10-mg/kg doses differed from that induced by 3-mg/kg activity, whereas the 5-mg/kg dose did not elicit a locomotor response that differed from 1, 3, or 10 mg/kg (Ho et al. 2016). These data imply that, although 10 mg/kg is a high dose, lower doses would not necessarily yield different results.
Although we did not measure the path complexity of the observed hyperlocomotion in the open field, previous data suggest that locomotor path complexity is less robustly associated with RU24969 response than is hyperlocomotion itself (Shanahan et al. 2011). Based on these data, it is possible that having fewer 5-HT1B receptors is protective against sensorimotor gating deficits, while having more receptors does not necessarily exacerbate these deficits.
It is also possible that developmental alterations in non-serotonergic circuits within the SERT Ala56 animals could offset changes in 5-HT1B receptor expression. Chronic SERT blockade with fluoxetine has been shown to reduce striatal dopamine and dopamine metabolite levels; this effect was also seen in SERT knockout mice (Morelli et al. 2011). Natural variations in SERT function have also been suggested to mediate dopamine-related behavioral phenotypes (Carneiro et al. 2009), and Ala56 mice have altered dopamine-related molecular pathways in the medial and lateral amygdala compared to the wild-type mice (O’Reilly et al. 2020). Dopamine receptor agonists, such as apomorphine, induce PPI deficits in mice (Dulawa and Geyer 1996). Thus, the discordant results between 5-HT1B binding and RU24969-induced behavior could be explained by complex effects of the SERT Ala56 variant on multiple neurotransmitter systems.
5-HT1B receptors are expressed on terminals of serotonergic neurons, where they inhibit release of 5-HT (Boschert et al. 1994; Hen 1992). Here, we find that exposure to RU24969 increased cFos activity in the OFC, perhaps due to serotonergic neurons sending an excitatory projection to inhibitory neurons in the OFC, as is seen in the PFC and hippocampus (Sun et al. 2019; Szonyi et al. 2016). RU24969 may therefore reduce the excitatory effect of serotonergic projections on inhibitory neurons, allowing primary neurons to have enhanced firing. However, 5-HT1B receptors are also present on terminals of glutamate, GABA, dopamine, and acetylcholine projecting neurons and also inhibit neurotransmitter release from these neurons (reviewed in (Sari 2004)). Thus, the role of the 5-HT1B receptor in modulating release of these other neurotransmitters may play a part in increased cFos expression in the OFC of the wild-type mice. The maintained cFos activity in Ala56 mice after RU24969 seems likely to be due to a compensatory mechanism, such as a shift in the amount of inhibition or neurotransmission in the OFC. For example, an increase in the number of 5-HT1B receptors in the OFC may lead to a baseline reduction in inhibitory signaling in Ala56 mice, resulting in slightly increased levels of excitatory activity. Further reducing the 5-HT signaling by RU24969 agonism may thus not have much of an impact on excitation in the OFC of Ala56 mice. Alternatively, having an increased number of 5-HT1B receptors in the OFC of Ala56 mice may lead to altered neurotransmission of multiple neurotransmitters, resulting in no net change in cFos activity. Neither of these possibilities was directly assessed.
Prior studies in rodents found no change in cFos mRNA in the OFC following RU24969 exposure (Ho et al. 2016), which contrasts our results showing that cFos protein expression is enhanced in the OFC of the wild-type mice. 5-HT1B binding in the OFC was significantly higher in the VO and tended to be higher in the LO of Ala56 mice compared to wild-type, which may indicate subregional differences in receptor expression. It is therefore possible that cFos mRNA expression was not different in previous studies due to the inclusion of MO (Ho et al. 2016).
In the dSTR, RU24969 exposure reduced cFos expression. This directionality is opposite to what is expected based on previous reports in which cFos mRNA or protein levels are increased by exposure to RU24969 (Ho et al. 2016; Wirtshafter and Cook 1998). Although both male and female mice respond similarly to RU24969 (Thompson and Dulawa 2019), sex differences or strain differences (C57BL/6J vs. 129S6/SvEvTac) could account for the discrepancies between the current cFos results from male mice and those reported previously in female mice (Ho et al. 2016). Additionally, although previous reports indicate that cFos levels normalize by 24 h post-injection with RU24969 (Wirtshafter and Cook 1998), we cannot discount the impact of experience on cellular function: the saline group received an injection of RU24969 48 h prior to the saline injection given for the cFos expression studies along with exposure to the open field and PPI paradigm. We note that because we did not see an interaction effect of genotype × drug exposure, we did not perform post hoc analyses to determine if the cFos expression was significantly different specifically in the wild-type mice.
The SERT Ala56 variant, as do other identified SERT variants found in the ASD population, leads to increased and dysregulated SERT function (Prasad et al. 2009; Prasad et al. 2005; Quinlan et al. 2019) and, presumably, decreased 5-HT signaling, as supported by increased 5-HT clearance from the extracellular space and increased sensitivity to 5-HT1A and 5-HT2A receptor agonists (Veenstra-VanderWeele et al. 2012). Notably, the SERT Ala56 variant leads to increased repetitive climbing/hanging behavior in mice (Veenstra-VanderWeele et al. 2012). Other gain-of-function SERT variants have been described in human genetic studies but not in mouse models; although a transgenic SERT overexpressing mouse has been created but not evaluated for response to RU24969 or baseline PPI (Dawson et al. 2011; Jennings et al. 2006, 2008). Here, we did not detect genotype differences in PPI or locomotor activity between SERT Ala56 and control mice even with relatively large sample sizes. Similarly, SERT knockout mice do not differ from control mice in PPI, although SERT knockout mice display lower levels of locomotor activity in the open field (Shanahan et al. 2009). Thus, alterations in SERT function minimally alter behavior on their own, but the downstream effects of altering SERT function may be substantial, as is seen in the SERT knockout response to RU24969 (Shanahan et al. 2009).
Although Ala56 mice display increased sensitivity to 5-HT1A and 5-HT2A/2C receptor agonists (Veenstra-VanderWeele et al. 2012), the current results suggest that not all 5-HT receptors are similarly affected. Of note, it is also possible that the increased 5-HT1A sensitivity that we previously observed counteracted any differential effect of RU24969 at 5-HT1B receptors. Because SERT inhibitors attenuate RU24969-induced PPI deficits and hyperactivity, we expected the SERT Ala56 mice to have enhanced PPI deficits and hyperactivity in response to RU24969. While we did not find altered behavioral effects in response to RU24969 administration to SERT Ala56 mice compared to the wild-type mice, our 5-HT1B binding and cFos staining results suggest that there is a difference in neural response to 5-HT1B agonists. The lack of behavioral difference in the context of altered neural response may indicate complex developmental effects of the SERT Ala56 variant or compensatory mechanisms that offset the impact of increased 5-HT uptake on 5-HT1B receptor signaling or on downstream circuit function. Thus, while SRI treatments may be effective at treating OCD, in part because they reduce 5-HT1B levels, increased 5-HT1B receptor levels due to enhanced SERT function do not fully account for the sensorimotor gating deficits seen in ASD and OCD.
Supplementary Material
Funding
This work was supported by National Institutes of Health grants MH094604 (JV), MH081066 (JV), MH114296 (SA, JV), and T32MH016434 (JV/LCS).
Conflict of interest
JV has consulted or served on advisory boards for Novartis, Roche Pharmaceuticals, and SynapDx; has received research funding from Novartis, Roche Pharmaceuticals, Forest, Seaside Therapeutics, Janssen, and SynapDx; and has received an editorial stipend from Springer and Wiley. All other authors declare that they have no competing interests.
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00213-020-05758-8.
References
- Ahmari SE, Risbrough VB, Geyer MA, Simpson HB (2012) Impaired sensorimotor gating in unmedicated adults with obsessive-compulsive disorder. Neuropsychopharmacology 37:1216–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmari SE, Risbrough VB, Geyer MA, Simpson HB (2016) Prepulse inhibition deficits in obsessive-compulsive disorder are more pronounced in females. Neuropsychopharmacology 41:2963–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldan Ramsey LC, Xu M, Wood N, Pittenger C (2011) Lesions of the dorsomedial striatum disrupt prepulse inhibition. Neuroscience 180:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerot S (2007) An autistic dimension: a proposed subtype of obsessive-compulsive disorder. Autism 11:101–110 [DOI] [PubMed] [Google Scholar]
- Boschert U, Amara DA, Segu L, Hen R (1994) The mouse 5-hydroxytryptamine1B receptor is localized predominantly on axon terminals. Neuroscience 58:167–182 [DOI] [PubMed] [Google Scholar]
- Cappi C, Brentani H, Lima L, Sanders SJ, Zai G, Diniz BJ, Reis VN, Hounie AG, Conceicao do Rosario M, Mariani D, Requena GL, Puga R, Souza-Duran FL, Shavitt RG, Pauls DL, Miguel EC, Fernandez TV (2016) Whole-exome sequencing in obsessive-compulsive disorder identifies rare mutations in immunological and neurodevelopmental pathways. Transl Psychiatry 6:e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro AM, Airey DC, Thompson B, Zhu CB, Lu L, Chesler EJ, Erikson KM, Blakely RD (2009) Functional coding variation in recombinant inbred mouse lines reveals multiple serotonin transporter-associated phenotypes. Proc Natl Acad Sci U S A 106:2047–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson N, Ferrington L, Lesch KP, Kelly PA (2011) Cerebral metabolic responses to 5-HT2A/C receptor activation in mice with genetically modified serotonin transporter (SERT) expression. Eur Neuropsychopharmacol 21:117–128 [DOI] [PubMed] [Google Scholar]
- Dulawa SC, Geyer MA (1996) Psychopharmacology of prepulse inhibition in mice. Chin J Phys 39:139–146 [PubMed] [Google Scholar]
- Dulawa SC, Geyer MA (2000) Effects of strain and serotonergic agents on prepulse inhibition and habituation in mice. Neuropharmacology 39:2170–2179 [DOI] [PubMed] [Google Scholar]
- Dulawa SC, Hen R, Scearce-Levie K, Geyer MA (1997) Serotonin1B receptor modulation of startle reactivity, habituation, and prepulse inhibition in wild-type and serotonin1B knockout mice. Psychopharmacology 132:125–134 [DOI] [PubMed] [Google Scholar]
- Dulawa SC, Hen R, Scearce-Levie K, Geyer MA (1998) 5-HT1B receptor modulation of prepulse inhibition: recent findings in wild-type and 5-HT1B knockout mice. Ann N Y Acad Sci 861:79–84 [DOI] [PubMed] [Google Scholar]
- Dulawa SC, Gross C, Stark KL, Hen R, Geyer MA (2000) Knockout mice reveal opposite roles for serotonin 1A and 1B receptors in prepulse inhibition. Neuropsychopharmacology 22:650–659 [DOI] [PubMed] [Google Scholar]
- Feliciano P, Zhou X, Astrovskaya I, Turner TN, Wang T, Brueggeman L, Barnard R, Hsieh A, Snyder LG, Muzny DM, Sabo A, Consortium S, Gibbs RA, Eichler EE, O’Roak BJ, Michaelson JJ, Volfovsky N, Shen Y, Chung WK (2019) Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med 4:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, Silva AJ (2004) Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry 9:417–425 [DOI] [PubMed] [Google Scholar]
- Gabriele S, Sacco R, Persico AM (2014) Blood serotonin levels in autism spectrum disorder: a systematic review and meta-analysis. Eur Neuropsychopharmacol 24:919–929 [DOI] [PubMed] [Google Scholar]
- Hanna GL, Yuwiler A, Cantwell DP (1991) Whole blood serotonin in juvenile obsessive-compulsive disorder. Biol Psychiatry 29:738–744 [DOI] [PubMed] [Google Scholar]
- Hen R (1992) Of mice and flies: commonalities among 5-HT receptors. Trends Pharmacol Sci 13:160–165 [DOI] [PubMed] [Google Scholar]
- Ho EV, Thompson SL, Katzka WR, Sharifi MF, Knowles JA, Dulawa SC (2016) Clinically effective OCD treatment prevents 5-HT1B receptor-induced repetitive behavior and striatal activation. Psychopharmacology 233:57–70 [DOI] [PubMed] [Google Scholar]
- Hoenig K, Hochrein A, Quednow BB, Maier W, Wagner M (2005) Impaired prepulse inhibition of acoustic startle in obsessive-compulsive disorder. Biol Psychiatry 57:1153–1158 [DOI] [PubMed] [Google Scholar]
- Hollander E, Pallanti S (2002) Current and experimental therapeutics of OCD. In: Davis K, Charney D, Coyle J, Nemeroff C (eds) Neuropsychopharmacology: the fifth generation of progress. Lippincott Williams & Wilkins, Philadelphia, pp 1648–1655 [Google Scholar]
- Hollander E, Novotny S, Allen A, Aronowitz B, Cartwright C, DeCaria C (2000) The relationship between repetitive behaviors and growth hormone response to sumatriptan challenge in adult autistic disorder. Neuropsychopharmacology 22:163–167 [DOI] [PubMed] [Google Scholar]
- International Molecular Genetic Study of Autism Consortium (1998) A full genome screen for autism with evidence for linkage to a region on chromosome 7q. Hum Mol Genet 7:571–578 [DOI] [PubMed] [Google Scholar]
- Jennings KA, Loder MK, Sheward WJ, Pei Q, Deacon RM, Benson MA, Olverman HJ, Hastie ND, Harmar AJ, Shen S, Sharp T (2006) Increased expression of the 5-HT transporter confers a low-anxiety phenotype linked to decreased 5-HT transmission. J Neurosci 26:8955–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings KA, Sheward WJ, Harmar AJ, Sharp T (2008) Evidence that genetic variation in 5-HT transporter expression is linked to changes in 5-HT2A receptor function. Neuropharmacology 54:776–783 [DOI] [PubMed] [Google Scholar]
- Kohl S, Heekeren K, Klosterkotter J, Kuhn J (2013) Prepulse inhibition in psychiatric disorders–apart from schizophrenia. J Psychiatr Res 47:445–452 [DOI] [PubMed] [Google Scholar]
- Leyfer OT, Folstein SE, Bacalman S, Davis NO, Dinh E, Morgan J, Tager-Flusberg H, Lainhart JE (2006) Comorbid psychiatric disorders in children with autism: interview development and rates of disorders. J Autism Dev Disord 36:849–861 [DOI] [PubMed] [Google Scholar]
- McAlonan GM, Daly E, Kumari V, Critchley HD, van Amelsvoort T, Suckling J, Simmons A, Sigmundsson T, Greenwood K, Russell A, Schmitz N, Happe F, Howlin P, Murphy DG (2002) Brain anatomy and sensorimotor gating in Asperger’s syndrome. Brain 125:1594–1606 [DOI] [PubMed] [Google Scholar]
- Morelli E, Moore H, Rebello TJ, Gray N, Steele K, Esposito E, Gingrich JA, Ansorge MS (2011) Chronic 5-HT transporter blockade reduces DA signaling to elicit basal ganglia dysfunction. J Neurosci 31:15742–15750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CL, Anacker AMJ, Veenstra-VanderWeele J (2016) The serotonin system in autism spectrum disorder: From biomarker to animal models. Neuroscience 321:24–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny S, Hollander E, Phillips A, Allen A, Wasserman S, Iyengar R (2004) Increased repetitive behaviours and prolactin responsivity to oral m-chlorophenylpiperazine in adults with autism spectrum disorders. Int J Neuropsychopharmacol 7:249–254 [DOI] [PubMed] [Google Scholar]
- O’Reilly KC, Perica MI, Fenton AA (2016) Memory deficits with intact cognitive control in the methylazoxymethanol acetate (MAM) exposure model of neurodevelopmental insult. Neurobiol Learn Mem 134 Pt B:294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KC, Anacker AMJ, Rogers TD, Forsberg CG, Wang J, Zhang B, Blakely RD, Veenstra-VanderWeele J (2020) A social encounter drives gene expression changes linked to neuronal function, brain development and related disorders in mice expressing the serotonin transporter Ala56 variant. Neurosci Lett 730:135027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos A, Palacios JM (1985) Quantitative autoradiographic mapping of serotonin receptors in the rat brain. I. Serotonin-1 receptors. Brain Res 346:205–230 [DOI] [PubMed] [Google Scholar]
- Pittenger C, Adams TG Jr, Gallezot JD, Crowley MJ, Nabulsi N, James R, Gao H, Kichuk SA, Simpson R, Billingslea E, Hannestad J, Bloch M, Mayes L, Bhagwagar Z, Carson RE (2016) OCD is associated with an altered association between sensorimotor gating and cortical and subcortical 5-HT1b receptor binding. J Affect Disord 196:87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piven J, Tsai GC, Nehme E, Coyle JT, Chase GA, Folstein SE (1991) Platelet serotonin, a possible marker for familial autism. J Autism Dev Disord 21:51–59 [DOI] [PubMed] [Google Scholar]
- Prasad HC, Zhu CB, McCauley JL, Samuvel DJ, Ramamoorthy S, Shelton RC, Hewlett WA, Sutcliffe JS, Blakely RD (2005) Human serotonin transporter variants display altered sensitivity to protein kinase G and p38 mitogen-activated protein kinase. Proc Natl Acad Sci U S A 102:11545–11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad HC, Steiner JA, Sutcliffe JS, Blakely RD (2009) Enhanced activity of human serotonin transporter variants associated with autism. Philos Trans R Soc Lond Ser B Biol Sci 364:163–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan MA, Krout D, Katamish RM, Robson MJ, Nettesheim C, Gresch PJ, Mash DC, Henry LK, Blakely RD (2019) Human serotonin transporter coding variation establishes conformational bias with functional consequences. ACS Chem Neurosci 10:3249–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CI, Kegeles LS, Levinson A, Feng T, Marcus SM, Vermes D, Flood P, Simpson HB (2013) Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology 38:2475–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari Y (2004) Serotonin1B receptors: from protein to physiological function and behavior. Neurosci Biobehav Rev 28:565–582 [DOI] [PubMed] [Google Scholar]
- Saxena S, Rauch SL (2000) Functional neuroimaging and the neuroanatomy of obsessive-compulsive disorder. Psychiatr Clin N Am 23:563–586 [DOI] [PubMed] [Google Scholar]
- Shanahan NA, Holick Pierz KA, Masten VL, Waeber C, Ansorge M, Gingrich JA, Geyer MA, Hen R, Dulawa SC (2009) Chronic reductions in serotonin transporter function prevent 5-HT1B-induced behavioral effects in mice. Biol Psychiatry 65:401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanahan NA, Velez LP, Masten VL, Dulawa SC (2011) Essential role for orbitofrontal serotonin 1B receptors in obsessive-compulsive disorder-like behavior and serotonin reuptake inhibitor response in mice. Biol Psychiatry 70:1039–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Li X, Ren M, Zhao M, Zhong Q, Ren Y, Luo P, Ni H, Zhang X, Zhang C, Yuan J, Li A, Luo M, Gong H, Luo Q (2019) A whole-brain map of long-range inputs to GABAergic interneurons in the mouse medial prefrontal cortex. Nat Neurosci 22:1357–1370 [DOI] [PubMed] [Google Scholar]
- Sutcliffe JS, Delahanty RJ, Prasad HC, McCauley JL, Han Q, Jiang L, Li C, Folstein SE, Blakely RD (2005) Allelic heterogeneity at the serotonin transporter locus (SLC6A4) confers susceptibility to autism and rigid-compulsive behaviors. Am J Hum Genet 77:265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL (2001) Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology 156:194–215 [DOI] [PubMed] [Google Scholar]
- Szonyi A, Mayer MI, Cserep C, Takacs VT, Watanabe M, Freund TF, Nyiri G (2016) The ascending median raphe projections are mainly glutamatergic in the mouse forebrain. Brain Struct Funct 221:735–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Dulawa SC (2019) Dissecting the roles of beta-arrestin2 and GSK-3 signaling in 5-HT1BR-mediated perseverative behavior and prepulse inhibition deficits in mice. PLoS One 14:e0211239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Welch AC, Iourinets J, Dulawa SC (2020) Ketamine induces immediate and delayed alterations of OCD-like behavior. Psychopharmacology 237:627–638 [DOI] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Jessen TN, Thompson BJ, Carter M, Prasad HC, Steiner JA, Sutcliffe JS, Blakely RD (2009) Modeling rare gene variation to gain insight into the oldest biomarker in autism: construction of the serotonin transporter Gly56Ala knock-in mouse. J Neurodev Disord 1:158–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, Cohen J, Mannangatti P, Jessen T, Thompson BJ, Ye R, Kerr TM, Carneiro AM, Crawley JN, Sanders-Bush E, McMahon DG, Ramamoorthy S, Daws LC, Sutcliffe JS, Blakely RD (2012) Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci U S A 109:5469–5474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland JR, DeGuzman TB, McMahon F, Rudnick G, Detera-Wadleigh SD, Murphy DL (2008) SERT Ileu425Val in autism, Asperger syndrome and obsessive-compulsive disorder. Psychiatr Genet 18:31–39 [DOI] [PubMed] [Google Scholar]
- Wirtshafter D, Cook DF (1998) Serotonin-1B agonists induce compartmentally organized striatal Fos expression in rats. Neuroreport 9:1217–1221 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.