

Abstract
In vitro and ex vivo intracellular delivery methods hold the key for releasing the full potential of tissue engineering, drug development, and many other applications. In recent years, there has been significant progress in the design and implementation of intracellular delivery systems capable of delivery at the same scale as viral transfection and bulk electroporation but offering fewer adverse outcomes. This review strives to examine a variety of methods for in vitro and ex vivo intracellular delivery such as flow-through microfluidics, engineered substrates, and automated probe-based systems from the perspective of throughput and control. Special attention is paid to a particularly promising method of electroporation using micro/nanochannel based porous substrates, which expose small patches of cell membrane to permeabilizing electric field. Porous substrate electroporation parameters discussed include system design, cells and cargos used, transfection efficiency and cell viability, and the electric field and its effects on molecular transport. The review concludes with discussion of potential new innovations which can arise from specific aspects of porous substrate-based electroporation platforms and high throughput, high control methods in general.
Keywords: intracellular delivery, electroporation, porous substrate, localized cell electroporation
Graphical Abstract
This review strives to examine a variety of methods for in vitro and ex vivo intracellular delivery such as flow-through microfluidics, engineered substrates, and automated probe-based systems from the perspective of throughput and control. Special attention is paid to a particularly promising method of electroporation using micro/nanochannel based porous substrates, which expose small patches of cell membrane to permeabilizing electric field.
1. Introduction
In vitro intracellular delivery is an extremely important field of research containing untapped potential due to the inability, until recently, to reap the benefits of high-throughput and highly controllable delivery within the same method. Simply put, intracellular delivery entails the transport of membrane impermeable molecules across the cell membrane. The most prominent form of intracellular delivery is gene delivery, but the molecules are not limited to nucleic acids and can consist of proteins, drugs, or any other impermeable molecule. Although in vivo intracellular delivery is a promising field in its own right, in vitro intracellular delivery’s potential lies in the simplicity and control afforded by culture in an artificial environment. In vitro intracellular delivery avoids the complex problems that occur in vivo such as localization within a particular tissue, avoidance of biological filtration systems like the blood brain barrier, liver, and kidneys, delivery without generation of a severe immune response, and complex interactions resulting from the presence of multiple cell types. Additionally, in vitro intracellular delivery allows environmental modification in the form of substrate stiffness and culture media to encourage stem cell differentiation[1] and somatic cell reprogramming and transdifferentiation[2–5].
In vitro intracellular delivery has medical, industrial, and analytical applications. Particularly interesting medical applications are adoptive immunotherapy and tissue engineering. Adoptive immunotherapy includes chimeric antigen receptor T (CAR-T) cell therapy, in which T cells are reprogrammed through in vitro gene delivery to target cancer cells before injection into a patient’s bloodstream[6, 7]. Tissue engineering relies on in vitro gene delivery to reprogram a patient’s somatic cells such as fibroblasts or adipocytes into induced pluripotent stem cells (iPSCs), which can differentiate into any cells present within the adult body[8, 9]. In vitro intracellular delivery has applications in industry such as increasing biomolecular production through the generation of monoclonal cell lines[10–13], or expediting analysis of the pharmacological effects a particular drug has on primary cells[14]. Furthermore, in vitro intracellular delivery of fluorophores and molecular beacons allows biointerrogation of cell types to better understand biomolecular mechanisms.
Despite the numerous opportunities provided by in vitro intracellular delivery, significant obstacles exist for a delivery system to be capable of fulfilling all these applications. The primary obstacle is delivering impermeable molecules across the cell membrane intact, a nontrivial task which requires physical disruption of the membrane or endocytosis followed by endosomal escape prior to lysosomal degradation. The process is further complicated because maintaining cell viability after delivery is crucial, so any disruption in the membrane must be temporary, and since many molecules for intracellular delivery are cytotoxic, the delivered quantity must be well controlled. Moreover, if DNA is being delivered, the delivery method must contain a mechanism for transporting the DNA through the cytoplasm and nuclear membrane to enable transcription within the nucleus. Finally, many of the applications require overcoming all of these obstacles quickly and consistently for populations of millions of cells.
One of the primary challenges of reviewing in vitro and ex vivo intracellular delivery methods is determining a classification system that encompasses the diverse spectrum of methods. Classification is nontrivial as it frames the subject and guides the conclusions drawn from it. Until now, intracellular delivery methods have been limited to classification based on the mechanism they use to bypass the cell membrane such as viral, chemical, and physical. For in vitro and ex vivo intracellular delivery methods, a new framework is needed for two reasons. First, a framework is needed to place an emphasis on system feasibility rather than the underlying vector-based or membrane disruption mechanisms themselves. In vitro and ex vivo intracellular delivery is a highly valuable field with no industry standard capable of fulfilling the numerous potential applications. Therefore, all prospective systems should be analyzed within the context of fulfilling these unmet needs. Second, a framework is needed to emphasize that the result of various delivery methods depends more on the scale and application of the mechanism than the mechanism itself. For example, bulk electroporation, probe-based electroporation, and flow-through microfluidic electroporation, despite all using electroporation as a delivery mechanism, have outcomes more in common with bulk sonoporation, single-cell injection, and microfluidic cell squeezing, respectively, than they do with each other. To satisfy these needs, this review proposes a new throughput and control classification framework for in vitro and ex vivo intracellular delivery methods. The classification system described here is not intended to replace the traditional method-based classification, but rather as a supplement to provide further insight into the field of in vitro and ex vivo intracellular delivery. Following discussion of this framework, porous substrate electroporation, one of the intracellular delivery methods with the greatest potential for industry-wide adoption, is further explored by dissecting commonalities between existing systems. Finally, an outlook of the field of in vitro and ex vivo intracellular delivery methods is provided.
2. Throughput and Control
The many opportunities and challenges of in vitro and ex vivo intracellular delivery have produced significant research interest and resulted in the proliferation of numerous methods for intracellular delivery[15, 16]. However, these methods predominantly fall into one of two categories: high throughput methods able to induce intracellular delivery into millions of cells within a short time by relying on broad, stochastic processes; and extremely precise, high control methods which are able to provide high uniformity, viability, and dosage control, yet their complexity often requires them to be performed at the single-cell scale. Throughput is a spectrum with no defined number separating low throughput from high throughput and each category spans multiple orders of magnitude. However, in general we use the term low throughput to refer to methods capable of delivering to individual cells up to hundreds of cells within a few hours; we use high throughput to refer to methods that can be scaled to deliver to hundreds of thousands or more cells within the same timeframe. For an in vitro intracellular delivery platform to fulfil the diverse and difficult applications envisioned, both high throughput and high levels of control are essential. As a result, we have classified all existing in vitro and ex vivo intracellular delivery methods into one of three categories: high throughput, low control methods; low throughput, high control methods; and high throughput, high control methods (Figure 1).
Figure 1. Throughput and Control Classification of In Vitro/Ex Vivo Intracellular Delivery.

Tree structure of the three main categories of in vitro/ex vivo intracellular delivery, their subcategories, and specific methods. High throughput, low control methods are shown in yellow; low throughput, high control methods are shown in blue; and high throughput, high control methods are shown in green.
2.1. High Throughput, Low Control Methods
High throughput, low control intracellular delivery methods were the first to be used to transfect a large number of cells but have limited mechanisms for control and thus result in highly stochastic delivery. High throughput, low control intracellular delivery methods consist of vector mediated delivery such as viral and chemical gene delivery, and bulk physical processes like bulk electroporation and bulk sonoporation. Although they can be used for in vitro intracellular delivery, vector mediated delivery methods are uniquely suited to in vivo delivery because they can be administered through ingestion, injection, or absorption through the skin[15, 17, 18]. In contrast, they are poor methods for in vitro intracellular delivery because while vector concentration can be controlled, delivery is determined by diffusion and endocytosis, meaning there is no way of precisely controlling the number of vectors administered to each cell. Likewise, bulk physical processes are ill suited for in vitro intracellular delivery because they exhibit non-uniform delivery across populations based on the distribution of cells and physical set-up of the device.
Viruses naturally contain highly efficient mechanisms that allow them to bypass the cell membrane and deliver their genetic information for replication, making modified viral vectors a natural candidate for intracellular gene delivery. Viral vectors have been engineered from many different types of viruses, especially adenoviruses, adeno-associated viruses, herpes simplex viruses, lentiviruses, and retroviruses[19–21]. Viral vectors are engineered to have their genetic code responsible for viral replication replaced with the desired genes for delivery, requiring replication to occur in genetically engineered bacteria or mammalian cells[22, 23]. Utilizing this intrinsic mechanism means the sizes of the delivered genes are limited by an upper threshold defined by the virus type, and molecules other than nucleic acids, such as proteins, cannot be delivered virally[21, 24]. More significantly, implementation of viral vectors is concerning due to evidence of viruses spontaneously regaining their ability to replicate, and oncogenesis through off-target effects[21].
Chemical vectors were created to utilize the advantages provided by vector based intracellular delivery while negating the risks associated with viral vectors. Chemical vectors are particles consisting of nucleic acids electrostatically complexed to inorganic nanoparticles, or cationic lipids (lipoplexes) and polymers (polyplexes)[25]. Lipoplexes and polyplexes allow the nucleic acids to enter the cell through endocytosis, which is otherwise prevented by the negative charge present on the nucleic acids. Following endosomal internalization, chemical vectors have specially designed coatings to induce endosomal rupture prior to lysosomal maturation, thereby preventing degradation of the nucleic acids[26]. Magnetofection is a chemical vector method that also relies on the application of bulk physical stimuli in the form of a magnetic field. During magnetofection, a polyplex with associated magnetic nanoparticles is guided to targeted cells via magnetic fields[27]. Despite these innovations, chemical vectors are still unable to match the delivery efficiency of viral vectors and bulk physical methods[25, 28]. This is particularly apparent in hard-to-transfect cells such as stem, progenitor, and primary cells[29]. Increasing concentrations of chemical vectors high enough to counteract their low efficiency often results in cytotoxicity.
In addition to vector-based methods, high throughput, low control in vitro intracellular delivery is possible using physical stimuli applied to large populations of cells simultaneously. These bulk physical methods include bulk electroporation, bulk photoporation, and bulk sonoporation. Bulk Electroporation (BEP) is the most widely used bulk physical method and refers to an electroporation method where a high voltage is applied to a dielectric chamber which can contain thousands to millions of cells[30]. The applied voltage produces a transmembrane potential (TMP) that causes a rearrangement of the membrane resulting in pore formation, allowing for intracellular cargo translocation by diffusion for small molecules or endocytosis for larger molecules[31]. When the voltage is removed, the cell membranes self-repair, closing the pores. Electroporation pulses can be adjusted to control the extent of permeabilization of the cell membrane through amplitude and pulse duration. The main drawback of BEP is the electric field is known to be nonuniform both from the large distance between the electrodes[32] and from distortion of the electric field due to the proximity of cells[33, 34]. This nonuniform electric field affects the TMP generated on the cells in suspension and thus results in some cells being irreversibly permeabilized while others have an insufficient TMP for intracellular delivery to occur. When certain combinations of pulse duration and electric field strength are applied, significant Joule heating of an electrolyte solution can occur[35]. Since bulk electroporation applies an electric field to the entire solution surrounding the cells and some regions of the solution are exposed to more intense electric fields, it is possible that Joule heating of cells is more significant in bulk electroporation than other more localized forms of electroporation. Another form of intracellular delivery, bulk photoporation uses a laser focused onto a cluster of cells to irradiate and transiently permeabilize cells. Light induced intracellular delivery has been referred to as optoporation[16], optoinjection[36], laserfection[37], and optical transfection[38], among others. In this review we refer to any form of light induced membrane permeabilization as photoporation for simplicity. The mechanism of bulk photoporation typically involves a secondary interaction such as substrate induced pressure fluctuation[39] or substrate mediated thermal effects [40]. Bulk photoporation is limited in scale when compared to BEP. Bulk photoporation has lower throughput than electroporation, coupling this with high cell lysis[39, 41] led to photoporation studies more focused on the single cell level. Lastly, sonoporation refers to the use of ultrasound acoustic waves to produce membrane poration through pressure fluctuation induced stresses[42], stable microbubble cavitation, or inertial microbubble cavitation[43–45].
2.2. Low Throughput, High Control Methods
Low throughput, high control in vitro and ex vivo intracellular delivery methods are an alternative to the lack of control, predictability and efficiency of high throughput, low control delivery methods. Low throughput, high control methods fulfill the need for precise interrogation of individual cells to discern underlying pathways and mechanisms. These methods also provide means of performing difficult transfections on highly valuable cells such as primary cells. Notably, since these methods employ an enclosed volume and can apply bidirectional forces to the cell through modulation of pressure or voltage, they require much lower quantities of expensive reagents, and can be used for intracellular extraction as well as delivery. For low throughput, high control methods, there are two main approaches of introducing cargo into the cell: membrane penetration and membrane poration. Methods of membrane penetration include micro-/nano-injection and ballistic cargo delivery. Membrane poration methods include single cell electroporation, single cell photoporation, and single cell sonoporation. These methods are highly focused techniques which allow single cell manipulation and interrogation with exceedingly sensitive equipment. As a result, they are time consuming and require a highly trained technician, which leaves a desire for higher throughput methods. In this section, methods of membrane penetration and membrane poration will be further discussed.
The main form of membrane penetration intracellular delivery is micro-/nano-injection. Microinjection was the first form of intracellular injection dating back to 1911[46]. With the evolution of technology and fabrication techniques, microinjection has evolved into nano-injection using tips on the order of ~100 nm[47, 48]. Micro-/nano-injection provides direct intracellular delivery through penetration of the cell membrane to deliver precise amounts of cargo to the cytosol or nucleus[49, 50]. Recent advancements in this area include electrophoretic injection control[47] and fluid force microscopy[51]. An additional method of low throughput, high control membrane penetration is ballistic cargo delivery. Ballistic cargo delivery entails coating cargo in heavy metal particles and propelling them towards cells with enough momentum to penetrate the membrane[16, 52]. Ballistic cargo delivery has shown the ability to introduce large cargo but lacks control and consistency when compared to other methods of high control delivery.
An alternative to membrane penetration, single cell membrane poration can be induced by highly targeted electric fields, high intensity light, or sound waves. For electric fields, single cell electroporation (SCE) uses BEP principles scaled down to the single cell level. This can be categorized as micro/nano electroporation, depending on the scale. The focused target of the electric field decreases the necessary voltage, reduces joule heating, and ultimately increases cell viability and transfection efficiency[30, 53, 54]. The narrowed focus also leads to more detailed insight into the physical and electrical characteristics of the cell[16, 55]. Through careful variation of the electric pulse parameters (amplitude, duration, frequency) and the electrophoretic solution, the electroporation parameters can be optimized based on cell type to facilitate high transfection efficiencies[16, 56]. Notable methods of singe cell electroporation are nano-fountain probe electroporation (NFP-E) and nanochannel electroporation. NFP-E uses a conductive atomic force microscopy probe which has a microfluidic channel. The probe approaches the cell and applies a targeted electric field to induce poration before delivering the cargo through the microfluidic channel[13, 57, 58]. Nanochannel electroporation contains two microchannels connected by a nanochannel. One microchannel contains the cell and the other contains the cargo to be delivered. The cell is electroporated and the cargo is driven into the cytosol by electrophoretic forces[59–61]. SCE offers a highly controlled method of intracellular delivery without the risks associated with BEP. This has made it a widely used technique and serves as the basis for many of the high throughput, high control systems. Photoporation is similar to SCE in that membrane poration is induced by targeted high intensity light focused on single cells. Photoporation has been shown to deliver a variety of cargo including plasmid DNA, mRNA, siRNA, peptide, and proteins among other molecules[16, 62]. Lastly, similarly to the methods used for bulk sonoporation, acoustics have been used for single cell sonoporation[63, 64].
2.3. High Throughput, High Control Methods
Following development of the low throughput, high control methods, researchers began looking for ways to adapt these methods to meet the high throughput requirements of many in vitro intracellular applications. The resulting high throughput, high control methods can be broadly classified into three main categories: automated probe-based systems, flow-through microfluidic systems, and complex substrates (Figure 2). Since micromanipulator based intracellular delivery methods already rely on electronics for their motion, automated probe-based systems are the natural solution to their low throughput caused by high precision and lack of parallel delivery capability. These systems have the potential to provide the highest control of any high throughput, high control methods, but unfortunately, are still unable to scale as well as other high throughput methods. The second main category, flow-through microfluidic systems, encompass both microfluidic squeezing and microfluidic electroporation methods. These methods have the capability to be extremely high throughput by increasing flow rate and the number of microfluidic channels, yet also require cellular suspension, which is undesirable for sensitive, adherent cells, and exhibit less control over their delivery mechanisms than automated probe-based methods or complex substrate methods. Complex substrate methods encompass a diverse spectrum of devices primarily grouped into nanostructure-based and electroporation-based methods, with some overlap between the two. Complex substrates may not provide the level of control of automated probe-based methods or the throughput of flow-through microfluidic systems, but they are simpler to develop than automated probe-based systems and provide a desirable adherent environment with more control than flow-through microfluidic systems. There are a few systems that fall outside of these three categories, notably nanostructure stamping which involves apically penetrating a population of cells using an array of nanostructures[65–68]. This method is not widely used and suffers from many of the same drawbacks discussed with nanostructure substrates, while incurring additional complexity by requiring the nanostructures to be applied rather than allowing spontaneous penetration.
Figure 2. High Throughput, High Control Methods.

Simplified depictions of high throughput, high control methods next to actual images of each method. The electroporation polarities shown are for delivering negative cargos. A. Automated probe-based injection. i. automated injection of zebrafish embryos [87] ii-iii. Different magnifications of an atomic force microscope tip with attached carbon nanotube needle (scale bar = 8 μm and 500 nm, respectively) [48] iv. cell held using a vacuum during injection [50] v-vi. real and simulated deformation during injection (needle diameter = 10 μm) [77] B. Automated probe-based electroporation. i-ii. image processing showing nuclear site in green and cytoplasmic site in red, followed by automated electrode positioning [90] iii. nanofountain probe electroporation (cell size ~ 10–20 μm) [58] iv. An improved version of nanofountain probe using silicon nitride for a soft touch (scale bar = 30 μm) [13]. C. Flow-through microfluidic electroporation. i. vortex microfluidic electroporation (scale bar ~ 720 μm) [97] ii. microfluidic electroporation device (scale bar = 6 mm) [96] iii. sawtooth microfluidic electroporation (scale bar = 40 μm) [95] D. Flow-through microfluidic mechanoporation, including cell squeezing and hydroporation. i. microfluidic constrictions for cell squeezing (scale bar ~ 250 μm) [92] ii-iii. microfluidic constrictions showing single and double deformation, respectively (scale bar = 10 μm) [103] iv. hydrodynamic shearing in hydroporation [105] v. spiral hydroporation [104] E. Nanostructures. i. nanoneedles (scale bar = 2 μm) [110] ii. cell adherent to nanostraws with false color added (scale bar = 10 μm) [121] iii. primary T cells on nanowires with false color added (scale bar = 10 μm) [115] iv. internalized nanowires with the cytoplasm dyed green and the cell membrane dyed red (scale bar = 10 μm) [111] v. neurons adherent to nanowires with false color added (scale bar = 10 μm) [114] vi-vii. silicon nanotubes used for biomolecular cargo delivery (scale bars = 1 um and 10 μm, respectively)[118] G. Patterned electrode electroporation. i. electrode electroporation device with multiple inputs [149] ii. clover electrodes (scale bar = 5 mm) [143] iii. interdigited electrodes [136] iv. 3D interdigited electrodes (scale bar = 800 μm) [141] H. Porous substrate electroporation. i. anodic alumina membrane (scale bar = 1 μm) [155] ii. polycarbonate membrane microfluidic device (scale bar = 12 mm) [153] iii. porous array with nanostructure trapping mechanism (scale bar = 200 μm) [161] iv. DAPI stain showing cell seating on porous array (scale bar = 100 μm) [164]. Permission is needed.
2.3.1. Automated Probe-Based Methods
Automated probe-based methods have been developed for microinjection and single-cell electroporation (SCE) systems. These automated systems use the fundamentals associated with their single-cell variant but with the addition of platform control, cell detection, and automated cargo delivery. Automated probe-based systems are the most controlled delivery process of any high throughput systems due to their requirement that they treat each cell individually. However, these methods are also perhaps the most expensive, complex, and difficult to scale of any high throughput system. Evolution of automated probes into higher throughput systems will require faster injection, faster cell recognition, and faster cell positioning[69]. These methods remain promising particularly for analytical applications where subcellular processes are studied, but their cost and poor scalability remain significant obstacles to widespread medical and industrial adoption. Targeted delivery, i.e. the delivery of cargos to subcellular regions and particularly the nucleus, has not been widely demonstrated in intracellular delivery methods other than probe-based methods. Automated microinjection and SCE both use micromanipulator platforms coupled with cell detection methods to locate and precisely transfect cells, therefore we will first discuss cell detection and platform control before discussing each cellular contact method individually.
Automated probes utilize image processing algorithms[69] to precisely control the movement of a micro-/nano- positioning platform. Despite the widespread accessibility of cell detection image processing algorithms[70, 71], image processing for an in vitro intracellular delivery system remains nontrivial due to cell transparency, clustering and overlapping in confluent populations, and varying morphologies[72, 73]. These challenges initially led to the development of automated microinjection systems focused on using zebra fish embryo due to their large size and defining features[74, 75]. Additionally, many image processing algorithms rely on fluorophores to identify cells, but these fluorophores may be undesirable in some applications due to their toxicity or potential for mutagenesis[76].
Automated microinjection (Figure 2A) delivers the cargo through injection as discussed in the previous section. To automate delivery, these systems require force measurements from the needle to determine when penetration has occurred[77]. Numerous methods of micro-force sensors were developed for the needle such as piezoelectric sensors[78], piezoresistive sensors[79], and MEMS capacitive force sensors[80, 81]. In addition to the automated zebra fish embryo microinjection devices, systems have been developed for smaller and harder to transfect cells[69, 82–87]. In contrast, automated single-cell electroporation systems (Figure 2B) do not penetrate the cell membrane but rather closely approach or slightly contact the membrane. Therefore, a method of membrane detection is necessary to automate the process. Electroporation relies on circuitry to induce poration; this same circuitry can be used to detect the cell membrane[58, 88, 89]. As the conductive probe approaches and seals against the cell membrane, a significant decrease in current can be detected. This method has been used to automate SCE devices via capillary probe electroporation[90], and nano-fountain probe electroporation[13, 57, 91].
2.3.2. Flow-Through Microfluidic Methods
Flow-through microfluidic devices offer throughput comparable to bulk methods while retaining localized cellular control. This category of devices can be divided into two categories: electroporation and mechanoporation. Flow-through microfluidic electroporation uses microfluidic channels to flow cells like a fluid and rapidly position them in close proximity to electrodes for a localized membrane permeabilization. Mechanoporation refers to mechanically induced membrane permeabilization through shear forces created by physical interaction such as passage constriction (cell squeezing) or fluid crossflows (hydroporation). Further, methods using both these techniques in series have been developed for high throughput nuclear DNA delivery[92]. Microfluidic devices can be fabricated using standard lithographic techniques and provide the highest throughput method of any of the high throughput, high control methods, and are not expensive to produce. However, microfluidic systems rely on cell to cell consistency and this can result in inconsistent delivery or clogging. Furthermore, flow-through devices rely on suspended cells making it a more stressful process for adherent cells.
The method of flow-through microfluidic electroporation (Figure 2C) uses embedded electrodes within microfluidic channels to permeabilize the cell membranes as they flow past the electrodes. The cells are suspended in a solution which contains the cargo to be delivered, and upon electroporation intracellular delivery takes place. Various methods and channel geometries have been explored for increased throughput and viability[93–97]. The microscale channels allow for narrow electrode gaps resulting in lower voltage requirements[94] and the microfluidic flow further helps to reduce negative effects such as joule heating and gas bubble evolution[98]. Notable studies include single cell impedence measurements for a flow through device[99] demonstrating single cell control. Furthermore, constant voltage flow-through electropartion systems have been designed with oil droplets[100] and varying channel width[101] which show the potential simplicity of flow-through electroporation devices. Some drawbacks of microfluidic electroporation are that it can require complex electrode geometry which are difficult to fabricate and additionally optimizing the electrical parameters for efficient transfection and high viability is a time consuming process.
The other main category of flow-through microfluidics, mechanoporation (Figure 2D), encompasses microfluidic cell squeezing and hydroporation. Cell squeezing devices are microfluidic devices that transfer molecules into cells by permeabilizing the cell membrane though deformation. In these devices, a solution of cells and cargo is flowed into microchannels which constrict the cells, typically the channel width is ~40–50% of the cell diameter. As a result, cells are squeezed as they flow into the constriction, and the cell membrane is temporarily permeabilized. After delivery, the cells are often left at room temperature for a few minutes to permit resealing. Cell speed, channel dimensions, and the number of constrictions are parameters that influence the delivery. In these devices, the transfection efficiency increases by increasing the flow rate and decreasing the gap size, while the cell viability decreases. This method has a high throughput transfection result can in some cases they can deliver into 1 million cells per second[102], and macromolecules with a variety of ranges delivered into cells[103]. Although targeted nuclear delivery has been previously limited to probe-based methods, Ding et al. developed a combined cell squeezing and electric-field-driven transport integrated in a single microfluidic device capable of accelerated nuclear delivery of plasmid DNA, and they reported higher delivery efficiency with low voltages compared to microfluidic electroporation alone[92]. One of the drawbacks of squeezing devices is that they are not able to perform for many experiments due to their small sizes, and the channels will be clogged and reduce the efficiency of the devices. Hydroporation is functionally similar to microfluidic cell squeezing, but instead of constrictions, a perpendicular fluid cross flow induces cell poration to allow for intracellular delivery. This method has the highest delivery efficiency and cell viability of all microfluidic techniques[104, 105]. Delivery efficiency in hydroporators is a balance between delivery and viability that can be controlled through the Reynolds number of the fluid flow. Notably, nanomaterials with a variety of sizes ranging up to 2000 kDa have been delivered using a platform based on spiral vortex and vortex breakdown due to the flow at the cross and T-junctions[104].
2.3.3. Engineered Substrate Methods
Engineered substrates is a term we have chosen to encompass all methods where substrates containing micro- or nanoscale features are used for intracellular delivery[106–108]. These substrates are often used to apply localized electroporation to randomly deposited cells, but some can be used with other physical stimuli such as photoporation[109] and possibly sonoporation. These methods include one dimensional nanostructures, patterned electrodes, and substrates containing micro- or nanopores.[106–108] Although these methods can require sophisticated manufacturing processes, many of these processes have already been developed within the microelectronics and microfiltration industries for efficient production at an industrial scale. As such, these substrates are readily scalable and can be widened and stacked to process numerous cells simultaneously, and are inexpensive to produce. Like probe-based methods, these systems are primarily suited for in situ intracellular delivery of adherent cells but can be utilized for suspended cell delivery using cell trapping or centrifugation. These systems can also be placed in series with other microfluidic processes such as cell sorting. Although engineered substrates do not provide as much control as automated probe-based methods or as high of throughput as flow-through microfluidic methods, they offer a compromise of many of the advantages of in situ adherent delivery while remaining highly scalable. Due to this balance of throughput and control, we consider engineered substrate methods the most promising subset of all high throughput, high control methods.
Nanostructure substrates (Figure 2E) are a group of methods utilizing one-dimensional structures such as nanoneedles[107, 110], nanowires[111–117], nanotubes[118, 119], and nanostraws[120–123] with diameters small enough that when cells are adhered to them they either induce spontaneous penetration, alter membrane permeabilization, or elicit endocytosis for molecular delivery. There is an ongoing debate as to which of these mechanisms or combination of mechanisms is primarily responsible for delivery using nanostructures[124]. Following penetration, the cell membrane seals around the base of the structure. Intracellular delivery can occur through molecules adsorbed to the surface, as is the case with solid nanoneedles, solid nanopillars, solid nanowires, and nanotubes sealed at one end; or by permanent intracellular access provided by hollow nanostructures such as hollow nanoneedles, nanotubes, and nanowires. Both approaches have disadvantages: solid and sealed nanostructures do not permit molecular extraction and only allow a single delivery without detaching and reattaching the cells, whereas hollow nanostructures cause continuous delivery of extracellular molecules and continuous leakage of intracellular molecules as long as the cell remains adhered. Nanostructures may be uniformly or stochastically spaced depending on the fabrication process. Furthermore, nanostructures are fragile and require more complex manufacturing than some other engineered substrates. Conversely, nanostructures are the only engineered substrates that do not require electroporation or other physical stimuli for delivery, and thus do not require any electronics.
To resolve adsorption and continued leakage when using nanostructures, some researchers have combined nanostructures with electroporation (Figure 2F)[125–134]. Nanostructures that are slightly too wide to cause spontaneous penetration are coated with a conductive material along the outside of the structure. Once cells are adhered to the substrate, electroporation can be used similarly to a valve to open the membrane at the end of each nanostructure. This allows for prolonged intracellular access lasting days or weeks, during which molecules can be added or extracted as desired. Electroporation increases the delivery efficiency of nanostructures and the tight seal at the cell membrane-substrate interface reduces the voltage necessary for electroporation by elevating the electric field applied to the interface. Electroporation in conjunction with nanostructures, including nanostraws and nanotubes, may form a tighter seal compared to porous substrates due to the high aspect ratio of the 1D structures[118, 133]. In addition to intracellular delivery, the conductive nanostructures can be used to record intracellular electrical measurements, although this requires nanostructures for each cell to be connected to a different electrode[132]. Nanostructure electroporation has many of the same disadvantages as nanostructures, namely fragility and manufacturing, the latter of which is even more difficult with the added conductive layers. Furthermore, electroporation requires additional electronics attached to the substrates.
Patterned electrode substrates (Figure 2G) are substrates similar to printed circuit boards, containing thin conductive paths exposed to cells. These electrodes can be made in many different patterns, including interdigited[135–141], ring-dot[142], and clover[143], among others[144–150]. These electrodes should be as closely spaced as possible to localize the electric field to individual cells and avoid the harmful effects associated with bulk electroporation. One advantage of patterned electrode substrates is they can be used to understand electroporation parameters by modifying the electrode geometry, as is the case with clover electrodes and electric field strength[143]. Moreover, the proximity of the electrodes and high electric field strength between them induces corrosion and requires substrate replacement or replating for repeated use. Since patterned electrode substrates do not have a cargo reservoir separate from the cell culture chamber like other substrate methods, higher quantities of expensive reagents are necessary.
The last category of high throughput, high control systems, porous substrate systems (Figure 2H) are substrates containing numerous micro- and nanopores on which cells are seeded and electroporated. Porous substrates can consist of commercially available membranes with random pore distribution[151–159], or uniform arrays of pores on silicon chips[160–170]. Like nanostructures, cells seal around the pores which limits the electric field exposure to discrete regions of each cell. Nanostructures may generate tighter seals at the membrane-substrate interface, but it has been shown that prior coating of substrates with extracellular matrix proteins can significantly enhance this seal in porous substrate-based methods to achieve high efficiency electroporation[171]. Although sharing many similarities to other engineered substrate methods, porous substrates benefit from increased robustness due to their lack of fragile nanostructures and corrosion-prone electrodes. Additionally, porous substrates are easier to produce because they require less material to be removed than nanostructures and do not require conductive layers like patterned electrode substrates.
3. High Throughput, Highly Controllable Porous Substrate Electroporation
Porous substrate methods are uniquely capable of achieving widespread adoption and making many of the aspirations of in vitro and ex vivo intracellular delivery a reality. Within high throughput, high control methods, it is our opinion that engineered substrates have the best balance of throughput and control due to their capacity for highly scalable in situ delivery. More specifically, porous substrates have been shown to perform similarly to more complicated alternative engineered substrates[119, 126, 141, 171]. Furthermore, evidence gathered using porous substrates is substantial enough to show promise amongst a variety of cell types using many different cargos, but there are significant opportunities for future research to better understand the fundamental processes involved, further optimize existing systems, and to use these systems in more complex biological applications. The following section serves as a review of the recent research efforts in the design and application of porous substrate electroporation systems in terms of design features, cell culture, cargo limitations, and choices of electroporation parameters (Figure 3). While porous substrates can be utilized with other methods of membrane permeabilization such as photoporation [109] and these methods are certainly worthy of further investigation, this section is devoted specifically to porous substrate electroporation systems because they have been much more widely utilized.
Figure 3. Porous Substrate Electroporation.

A. The 4 cell trapping mechanisms that have been demonstrated. From left: nanostructure, vacuum, magnetic tweezers, and dielectrophoresis. B. A cell adhered to a porous substrate and undergoing electroporation. Equivalent circuit elements are shown near their corresponding features. C. A magnified view of the cell-channel interface showing the voltage drop along the channel. D. The transition from hydrophobic pores to hydrophilic pores that occurs during electroporation. E. A further magnified view of the cell membrane showing the 3 nm radii pores that form at high voltage as predicted by Mukherjee et al. Under the current electrode configuration, electrophoresis extracts positive cargos and delivers negative cargos. From left, the cargos propidium iodide (PI), linear DNA, and bovine serum albumin (BSA) are shown to scale. F. An electroporation waveform consisting of two trains, each with three unipolar square bilevel pulses. The parameters high voltage (HV), low voltage (LV), high voltage duration (t1), low voltage duration (t2), and train interval (t3) are shown. Single level and exponential decay pulses are also shown.
3.1. Theory of Electrical Potential and Molecular Transport
It is generally accepted that the application of an electric field leads to the formation of hydrophilic pores from hydrophobic pores on the cell membrane (Figure 3D), which allows for the transport of molecules across it[172, 173]. The formation and evolution of these transient hydrophilic pores are modeled using the Smoluchowski equation:
| 1) |
where is the size density of electropores per unit membrane area, is the pore diffusion coefficient in the pore radius space is the thermal energy, and is the energy difference between cell membranes with and without hydrophilic electropores. The energy is a function of the effective membrane tension and the transmembrane potential (TMP) , which is the potential difference developed across the cell membrane on the application of the electric field. The Smoluchowski equation along with appropriate boundary conditions has been frequently used in combination with electric field and molecular transport models to estimate and optimize bulk electroporation based delivery[174–178].
More recently, this equation was incorporated in a multiphysics model to predict the molecular transport in porous substrate based localized electroporation platforms [152]. In this model it was assumed that the electric field is localized at the interface of the nanopores and the cell membrane, which was indirectly observed during delivery experiments in other studies[59, 179]. This made it possible to use an equivalent circuit approximation to estimate the localized electric field (Figure 3B). The physical components of the system such as cell membrane, nanopore resistance, buffer and contact impedance were assumed to be passive electrical circuit components and the following charge conservation equation was solved:
| 2) |
where is the potential drop across any component, is the conductivity that depends on the density of electropores in case of the cell membrane and is the permittivity. The molecular transport was assumed to be diffusive and electrophoretic and solved using the Nernst-Planck equation:
| 3) |
where, is the concentration of the molecular specie moving across the nanopore, is the diffusion coefficient, is the charge of the specie and is the elementary charge constant. The electrophoretic nature of the transport was also verified by researchers using localized electroporation based platforms[151, 180].
This multiphysics model allowed for the optimization of physical parameters involved in localized electroporation such as pore density, size, and electroporation pulse profiles. Additionally, a few key conclusions were drawn from the model predictions that were supported by the experimental data. First, it was inferred that an optimal voltage exists for efficient electroporation and delivery of large molecules. Voltages lower than optimal do not lead to the formation of sufficient pores and very high voltages lead to the formation of excessive small pores that cannot expand enough to permit the entry of large molecules. This prediction corroborated well with experimental results reported in the study and data reported in literature using similarly designed localized electroporation platforms[151, 181]. Second, the model predicted that the cell membrane tension plays a key role in efficient delivery during electroporation and higher membrane tension allows for a greater amount of cargo to be delivered with better uniformity. Osmolarity induced membrane tension was used to validate this conclusion. Moreover, this conclusion is not surprising considering mechanical perturbation methods, such as physical squeezing and hydrodynamic deformation that increase the membrane tension and eventual permeabilization, have emerged as efficient methods of intracellular delivery[15, 16]. Overall, the multiphysics model provides a useful framework that can qualitatively guide the design of localized electroporation platforms and optimization of experimental conditions.
Although the continuum scale models provide valuable insights into the mechanisms of localized electroporation mediated delivery, they use several assumptions regarding the electropore dynamics and molecular transport that are reflected in the model parameters. These parameters are difficult to quantify accurately, because of which the model estimates have uncertainties. To obtain robust quantitative predictions the uncertainties need to be quantified using systematically designed experimental validations. Additional information that complement the continuum models can be obtained from molecular dynamics simulations capturing the interactions of the delivery cargo and the lipid pores[182, 183]. Such multiscale approaches may also provide explanations for experimental observations unique to localized electroporation, for example, the uniform distribution of molecular cargo in the cytoplasm suggesting direct electrophoretic entry bypassing the endocytic pathway[59].
3.2. System Design
There are many parameters related to system design that affect the performance of porous substrate systems (Table 1) including electrode materials and whether cell trapping mechanisms are used, as well as pore diameter, length, density, and whether the pores are uniformly distributed. The two electrodes can consist of the same material or different materials. These electrodes tend to be made of noble metals such as gold, silver, or platinum to resist electrolytic corrosion, or glass with a thin conductive layer to allow viewing under a microscope while retaining fluids and providing structural support. Multiple cell trapping methods have been used with porous substrates to increase the likelihood of cell positioning over pores, and to ensure tight contact between the cell and pore opening (Figure 3A). These cell trapping methods include nanostructures around the pores[161], vacuum[156, 160, 163, 167, 168], dielectrophoresis[165], and magnetic tweezers[163] but must be carefully administered to prevent harming the cells. Nanostructure trapping entails fabricating structures such as retaining walls around each pore using the same etching techniques as used in fabricating the pores. Vacuum trapping consists of creating a pressure differential through each pore. In dielectrophoresis, a nonuniform electric field is applied to the cells which polarizes them and results in their movement. Finally, magnetic tweezers involve delivering magnetic particles into cells and applying a magnetic field to guide them. Additional cell trapping methods such as optical tweezers, acoustic tweezers, and hydrodynamic tweezers exist but to our knowledge have not yet been demonstrated for cell positioning prior to porous substrate electroporation.
Table 1.
Design Parameters of Porous Substrate Systems
| Type | Substrate Material | Electrode Material | Cell Trapping Mechanism | Pore Diameter (nm) | Pore Length (μm) | Pore Density (cm−2) | Ref. |
|---|---|---|---|---|---|---|---|
| Membrane | Track-Etched Polycarbonate | Titanium | 100 | 20 | 2E7 | [151] | |
| ITO-Coated Glass | 200 | 25 | 5E8 | [152, 158] | |||
| U: Silver/Silver Chloride, L: Gold-Coated Glass | 600 2000 | 24 23 | 4E7 3E6 | [153] | |||
| Track-Etched PET | Silver | Vacuum | U: 3000 L: 400 | [156] | |||
| U: Track-Etched PET, L: Anodic Alumina | U: 3000 L: 200 | U: 10 L: 60 | U: 8.5E5 L: 8.0E8 | [157] | |||
| Anodic Alumina | Platinum-Coated Glass | 20 | 45 | 1E11 | [154] | ||
| Gold-Coated Upper and Lower Membrane Surfaces | Vacuum | 100–200 | 60 | [155] | |||
| Array | DRIE and photo-lithographed silicon | U: Copper, L: Gold-Coated Glass | Vacuum | 650 | 20 | 4E6 | [160] |
| U: Neon Transfection System Electrode, L: Gold-Coated Glass | Nano-structures | 400 | 10 | [161] | |||
| U: ITO-Coated Glass, L: Gold-Coated Glass | * | 300 | 10 | 4E4 | [162] | ||
| U: Platinum, L: Gold-Coated Glass | Magnetic Tweezers or Vacuum | 5000 5000 | 30 30 | 1E3 4E4 | [163] | ||
| Dielectro-phoresis | 650 | [164] | |||||
| U: ITO-Coated Glass, L: Gold-Coated Glass | Dielectro-phoresis | 300 | 10 | 4E4 | [165] | ||
| 500 | 20–25 | 4E4 | [166] | ||||
| Vacuum (L) | 1000 (U) 2500–3500 (L) | 10 (U) 12–13 (L) | 1.6E6 (U) 2.5E5 (L) | [167] | |||
| Gold-Coated Glass | Vacuum | 2000 5000 | 1 | 1.8E4 | [168] |
U and L denote upper and lower, respectively, in instances where there is a difference between electrodes or when porous substrates are used both above and below.
Optical tweezers were used in this study but are only mentioned for positioning cells in the 2D channels, not for positioning on the porous substrates.
The geometry and distribution of pores are fundamental in determining how much of the cell membrane is exposed to the electric field and the electric field strength itself. Longer, narrower, and fewer pores increase the voltage drop across the pores which thereby increases the electric field strength within the pores and decreases the electric field strength at the cell membrane. The electric field strength influences the electrokinetic movement of cargos within the pores. Pore diameters range from 20–5000 nm with most located in the range of 100–650 nm. Pore lengths vary from 1–60 μm with most falling in the range of 10–30 μm. Pore densities range from 1E3–1E11 cm−2 with a common range of 4E4–8E8 cm−2. When not mentioned in the literature, values for pore dimensions and pore density were gathered from product catalogs, or in the case of pore densities, approximated from other available dimensions. Porous substrates can be divided into two main categories based on uniformity of pore distribution: membranes and arrays. These two categories share similar delivery mechanisms but have significant differences in their fabrication and implementation. Porous membrane systems have membranes with randomly distributed pores, while porous array systems are made of etched and photolithographed silicon with uniformly spaced pores.
The advantage of porous membrane systems is how simple they are to design and manufacture, which is why they were used for some of the earliest porous substrate systems[154, 156]. Porous membrane systems primarily utilize track-etched polymer membranes, which have been used commercially for filtration purposes, although anodic alumina membranes have also been used[154, 155, 157]. Moreover, track etching is a simpler process than multiple deep reactive ion etching (DRIE) and photolithographical steps, and the membranes required for these systems are already commercially available, making porous membrane systems easier to scale up for medical and industrial applications. In addition to their availability, porous membranes are much softer and more elastic than silicon, making them more physiologically relevant than silicon which has a stiffness of over 100 GPa[184], much higher than the 20 GPa stiffness of cortical bone[185]. Substrate stiffness has been shown to significantly affect cell behavior through proliferation[186], differentiation[1], and reprogramming[2].
Despite these advantages of porous membranes with respect to porous array systems, their simplicity and ease of use have drawbacks due to their stochastic fabrication process. The construction of these membranes entails ion bombardment of thin polymer sheets, producing randomly spaced and oriented channels as they pass through the material. After ion bombardment and subsequent ultraviolet light exposure, the reactivity of the tracks to an etchant is increased[187], meaning exposure time to the etchant is used to control the size of the pores, which are homogenous in size but can be tuned to be tens of nanometers to several micrometers in diameter. Channels can be created with a smooth and uniform cross section[188], although significant variation in length can exist from channel to channel, when channels are angled substantially from perpendicular to the substrate. Variations in channel length complicate delivery modeling and optimization compared to arrays, which are more uniformly fabricated. Furthermore, the random distribution of pores can cause variable delivery between cells. However, variation in the number of exposed pores per cell may be insignificant when utilizing membranes with a sufficiently high pore density, which can result in hundreds or even thousands of pores beneath each cell.
Porous array systems seek to resolve the complications resulting from nonuniform pores found in track-etched membranes[169, 170]. DRIE and photolithography are used to create silicon wafers with uniformly spaced and uniformly dimensioned micro- and nanochannels[189]. Metal-assisted chemical etching (MACE) using nanosphere lithography has been demonstrated as an alternative to DRIE for creating uniform arrays of pores[190, 191], but arrays fabricated with MACE have not yet been combined with porous substrate electroporation. Contrary to porous membrane systems, porous array systems utilize a single pore beneath each cell, and there are more options for substrate modification than porous membranes, which can only primarily adjust pore size, pore density, and surface coating.
Porous array systems have drawbacks when compared to porous membrane systems due to their increased complexity. The fabrication process for porous array systems is much more difficult, requiring the use of cleanrooms, and different substrates may need to be fabricated for different cell types. Porous arrays are also more fragile than porous membranes which limits the number of times they can be reused. Additionally, to take advantage of the uniform channels, cells must be precisely positioned over each pore, which requires employing cell trapping methods.
3.3. Cells Used
Perhaps the most important data concerning porous substrate systems are the cell types that have been used with it, including their corresponding viability, efficiency, and dosage control, because these are the metrics by which all intracellular delivery methods are evaluated. Viability is the percentage of cells that remain living after delivery, efficiency is the percentage of original cells containing cargo after delivery, and dosage control is a measure of how variable the amount of cargo is from cell to cell. These metrics are the basis of all discussion surrounding the feasibility of any intracellular delivery system, yet can be very difficult to interpret because they are confounded by many aspects of a system including cell type, cargo size, electrical parameters, substrate pore size and distribution, and whether surface coatings were used. Moreover, viability and efficiency are tradeoffs that must be balanced because the more disruptive a method is, the more cargo will be delivered (higher efficiency) and the more likely the cell will die (lower viability). Of these three metrics, viability and efficiency are widely reported across intracellular delivery methods, whereas discussion of dosage control is limited to highly controllable systems, and indeed even a minority of porous substrate systems discuss it[160, 163, 165, 168]. As a result, viability and efficiency are the two primary metrics discussed here.
Viability is a binary indicator of cell health often measured using a propidium iodide (PI) and calcein AM live-dead assay or trypan blue exclusion assay. Although viability is a simple parameter to measure, it neglects to inform whether cellular processes are functioning at or near predelivery levels in surviving cells. To better understand the harmful effects elicited by intracellular delivery, Tay and Melosh have proposed more detailed metrics such as intracellular calcium levels and RNA transcriptomics[192]. Furthermore, viability measurements are often taken at inconsistent time points after delivery, with some authors measuring hours afterwards, and others measuring days afterwards.
The second primary metric, efficiency, encompasses the broad term delivery efficiency, which is efficiency irrespective of cargo type, and transfection efficiency, which is strictly defined as the percentage of cells containing delivered nucleic acids. Transfection efficiency is the focus of this section because it is necessary for many intracellular delivery applications and is generally a higher standard than delivery efficiency, which can include smaller proteins and fluorophores. Transfection efficiency can vary significantly depending on the cargo delivered because nucleic acids such as molecular beacons, oligonucleotides, and messenger RNA (mRNA) have higher transfection efficiency since they are smaller than DNA and do not require nuclear localization for their delivery to be observed. Transfection efficiency reported as a result of genetic editing using CRISPR/Cas9 or alternatives is even lower because the cargo must not only be delivered to the nucleus but must also successfully edit the gene of interest. For reference, Cao et al. measured transfection efficiencies of porous membrane systems using 4 different cell lines and reported efficiencies of 75–80% for mRNA, 40–80% for plasmid DNA, and approximately 25% for CRISPR/Cas9 genetic editing[151].
Transfection efficiency is known to be heavily dependent on the type of cell used, with immortalized cells being the easiest to transfect, and cells such as primary and stem cells being known as “hard to transfect” cell types. Although the majority of studies with porous substrate systems have been performed on immortal cells, their effectiveness has also been demonstrated on primary and stem cells, including particularly difficult to transfect neurons [153] and cardiomyocytes[161]. Furthermore, despite their aptitude for transfecting adherent cells, porous substrate systems have demonstrated the ability to transfect multiple suspended cell lines as well as primary leukocytes using centrifugation or cell trapping.
Table 2 contains all cell types used with porous substrate electroporation and relevant data such as cell culture surface coatings, viability, and transfection efficiency. Cell culture coatings are predominantly used to increase cellular adhesion around the pores, except for polyethylene glycol (PEG) -silane which was used increase cell detachment between experiments[163]. Values for viability and transfection efficiency were taken from the same experimental conditions since the 2 parameters are tradeoffs and when multiple values were provided, the highest combined values were included. If percentages were not explicitly stated, values were taken from bar graphs and rounded down to the nearest 5%. If multiple studies used a cell line, the highest values were included and the study was cited. For some cell lines, population-wide data was not reported, but transfection was shown through fluorescent images and graphs of fluorescent intensity. BEAS-2B and HL-60 are the only cell lines listed that were not transfected. BEAS-2B was used to understand cell trapping and fluorophores were delivered to HL-60 but not nucleic acids, as such, the transfection efficiency listed for HL-60 is delivery efficiency and is noted in the table.
Table 2.
Cell Types Used in Porous Substrate Systems
| Type | Organism | Cell Line | Description | Surface Coating | Viability | Transfection Efficiency | Ref. |
|---|---|---|---|---|---|---|---|
| Immortal | Human | A375 | Melanoma | >90% | >90% | [168] | |
| BEAS-2B | Transformed Bronchial Epithelial | Gelatin, BSA, PEG | [168] | ||||
| HEK293 | Embryonic Kidney | Poly-L-Lysine or Fibronectin | >90% | 80% | [151] | ||
| HeLa | Cervical Epithelial | Poly-L-Lysine [151] or Fibronectin [151, 154] | >95% [151] | >80% [151] | [151, 153, 154, 159] | ||
| HL-60 | Promyeloblast, Suspended | >90% | 65%* | [155] | |||
| HT1080 | Connective Tissue | Fibronectin [152] | 50% [153] | [152, 153, 159] | |||
| Jurkat | T Lymphocyte, Suspended | Poly-L-Lysine or Fibronectin [151], PEG-Silane [163] | >95% [151] | 75% [151] | [151, 163] | ||
| KG1a | Promyeloblast, Suspended | PEG-Silane | 96% | [163] | |||
| K562 | Lymphoblast, Suspended | PEG-Silane [163] | 92% [163] | 83.4% [163] | [163, 164] | ||
| MDA-MB231 | Mammary Epithelial | Fibronectin | >99% [152] | 70% [152] | [152, 158] | ||
| NK-92 | Natural Killer, Suspended | 90% | 74% | [165] | |||
| Mouse | NIH3T3 | Embryonic Fibroblast | Poly-L-Lysine or Fibronectin [151] | >95% [151] | 75% [151] | [151, 156] | |
| Rat | H9C2 | Embryonic Cardio-myoblast | [160, 165, 166] | ||||
| Primary | Human | HCF-a | Myofibroblast | 95.6% | 90.5% | [168] | |
| None | Leukocyte, Suspended | PEG-Silane | [163] | ||||
| Mouse | None | Cardiomyocyte | 86% | 86% | [161] | ||
| None | Embryonic Fibroblast | >90% [161] | >90% [161] | [160, 161] | |||
| None | Neuron | Poly-D-Lysine | >90% | [153] | |||
| Stem | Human | GBM157 | Glioma Stem | >70% | [162] | ||
| GBM528 | Glioma Stem | [162] | |||||
| Mouse | CCE | Embryonic Stem | Gelatin [167] | >85% [167] | [157, 167] |
Delivery efficiency, not transfection efficiency. No nucleic acids were delivered in this study.
3.4. Cargo Properties
A wide variety of cargos have been delivered or extracted using porous substrate electroporation systems (Table 3). These cargos range from tiny ions, small molecules, and fluorophores, to massive nucleic acids. Although studies have primarily focused on delivery of these cargos, extraction of intracellular molecules using porous substrates has also been demonstrated[152]. Cargo parameters influencing successful intracellular delivery or extraction include molecular charge, size, and the composition of the delivery solution.
Table 3.
Cargo Delivered Using Porous Substrate Systems
| Type | Molecule | Mass (kDa) | Size | Charge | Concentration | Solvent | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Small | Ion | Co2+ | 0.059 | 0.125 nm rh | + | [152] | ||
| Small Molecule Drug | Dacarbazine | 0.182 | 10 μM | PBS | [168] | |||
| Temozolomide | 0.194 | 5 μM | PBS | [162] | ||||
| Fluorophore | Lucifer Yellow CH | 0.457 | 0.49 rh | - | 1000[155] −2000 μg/mL[154] | DMEM[154], PBS[155] | [154, 155] | |
| PI | 0.668 | 0.6 nm rh | + | 2[153]−20[152] μg/mL, 100 μM[167] | PBS[153], High Glucose DMEM[167] | [152, 153, 159, 160, 163, 165, 167] | ||
| Oligonucleotide | Anti-miR-363 | - | 0.005–5 μM | PBS | [162] | |||
| FAM-ODN | 5.684 | 18 nt | - | [160, 163, 165, 166] | ||||
| Micro RNA | miR-29 | ~6.9–7.5 | 21–23 nt | - | [161] | |||
| Molecular Beacon | CD44 | - | PBS | [162] | ||||
| CD133 | - | PBS | [162] | |||||
| FAM-VIM | 13.9 | 45 nt | - | [168] | ||||
| GATA2 | - | [163, 164] | ||||||
| Large | Protein | Alexa Fluor 488 BSA | 66.5 | 3 nm rh | - | 2500 μg/mL | Hypo-osmolar buffer [152], Iso-osmolar buffer [152], PBS [158] | [152, 158] |
| Cas9 RNP | Varies | Varies | - | 10 μM | [151] | |||
| mCherry STIM1 | 98 | + | [151] | |||||
| PTPs | Varies | Varies | Varies | Extracted | Extracted | [158] | ||
| tdTomato | 54.2 | - | Extracted | Extracted | [152] | |||
| Messenger RNA | mCherry mRNA | ~228 | 711 nt | - | 0.1 μg/mL | [151] | ||
| Plasmid DNA | CS1-CAR | 9 kb | - | [165] | ||||
| GFP | ~2000 | 3.3 kb | - | 0.2[151] – 1000[153] μg/mL | DMEM [153] | [151, 153, 159] | ||
| gWiz GFP | ~3500 | 5.8 kb | - | <50 μg/mL | [156] | |||
| gWiz SEAP | ~4000 | 6.6 kb | - | 5[167] −100[157] μg/mL | High Glucose DMEM[157, 167] | [156, 157, 167] | ||
| mCherry | ~2400 | 4 kb | - | 20 μg/mL | [152] | |||
| NF2 CRISPR/ Cas9 KO | >5500 | >9 kb | - | [168] | ||||
| OSKM pCAG | ~7900 | 13 kb | - | [160] | ||||
| pDsRed-C1 | ~2900 | 4.7 kb | - | 100 μg/mL | [154] | |||
| pmaxGFP | ~2100 | 3.5 kb | - | 5 μg/mL[167] | High Glucose DMEM[167] | [160, 161, 165, 167] | ||
| Linear DNA | YOYO-1-λ | ~29500 | 48.5 kb | - | 0.03 μg/mL | Tris-EDTA buffer | [156] |
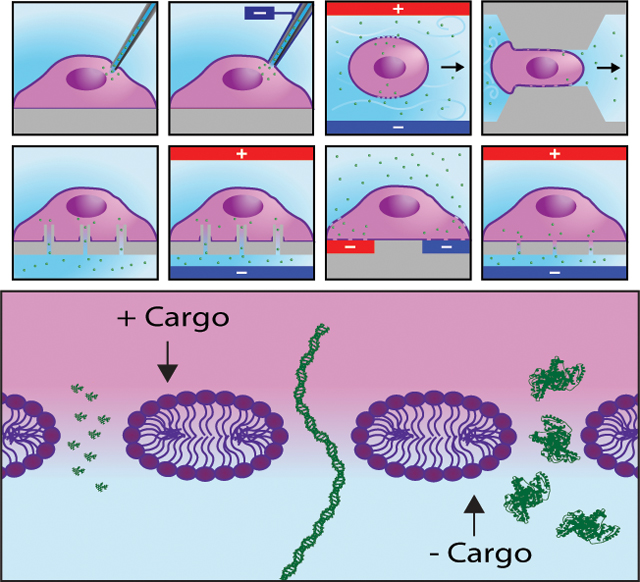
Charge plays a significant role in cargo delivery because the localized electroporation employed by porous substrates is electrophoretically dominated. Therefore, the charge of the cargo must be known to use proper electrode polarity for delivery. When delivering negatively charged molecules, the anode must be placed on the cell culture side of the porous substrate, and the cathode must be placed on the side of the porous substrate containing the cargo. Likewise, the polarity of the electrodes must be reversed when delivering positive cargo. An additional consideration is that electrophoresis occurs in both directions during electroporation. While one polarity of molecules is being delivered into the cells, molecules of the other polarity are extracted from the cells.
The role of cargo size in intracellular delivery using porous substrates is more complex. Although pore formation has not been observed in porous substrate electroporation, Mukherjee et al.[152] presents a computational model which predicts higher voltages increase pore diameter up to a critical voltage, after which the pores collapse and additional voltage generates smaller pores but in a greater number. The theoretical size of these high voltage pores is 3 nm in radius, meaning molecules smaller than these pores are delivered in greater quantities as voltage is increased due to an increase in the electrophoretic force (Figure 3E). Conversely, molecules greater than 3 nm in radius are maximally delivered at voltages just below the critical voltage. The results of this simulation were corroborated with the delivery of different sized molecules in the same paper, and are supported by data from other researchers in this field[181, 193]. For comparison, the size and mass of each cargo was listed when known. Nucleic acids are primarily measured in the number of base pairs, from which mass estimates were calculated using formulas presented by Thermo Fisher Scientific.
Additional cargo factors governing delivery are solvent formulation and cargo concentration. The results from cargo delivery are associated with the concentration of cargo within the delivered solution, yet many of these cargos are cytotoxic when delivered at too high of concentration. Furthermore, solvents used for delivery must be electrically conductive and biocompatible, with phosphate buffered saline (PBS) and Dulbecco’s modified Eagle’s medium (DMEM) commonly used. The delivery solution should also contain a pH buffering system to minimize the toxic pH change caused by electrolysis[136]. Furthermore, diluting the delivery molecules in a hypo-osmolar buffer increases membrane tension through intracellular swelling, thereby reducing the voltage required for permeabilization[194], and perhaps more importantly, increasing the radii of pores produced at high voltage[152].
3.5. Electroporation Waveforms
Researchers have been optimizing waveforms for electroporation for decades, with an emphasis placed on square and exponential decay waveforms. Bipolar square waveforms have been shown to be more efficient than unipolar exponential decay and square waveforms in bulk electroporation because the membrane is permeabilized on both sides of the cell[195]. However, unipolar square pulses are favorable for porous substrate delivery because the cargo is located on one side of the cell and generally unipolar, thus unidirectional electrophoresis is thought to result in greater delivery, but to our knowledge this has not been demonstrated. As such, most studies on porous substrate electroporation have utilized unipolar square pulses, although a few earlier studies were performed with exponentially decaying waveforms[157, 167]. To the best of our knowledge, no studies have been performed showing the effects of different waveforms specifically applied to porous substrate systems, only the effects of varying voltage, frequency, and number of pulses of unipolar square waveforms.
Comparing waveforms between papers is valuable for understanding which parameters are often used, both as a starting point for new researchers and to point out conditions where data is lacking. Unfortunately, this comparison proved difficult due to many waveforms being incompletely explained, and listed parameters often used different terminology from author to author. For this reason, we created a standardized terminology for describing unipolar waveforms (Figure 3F). Using this framework, we fit the various parameters from each paper into Table 4. Pulses were assumed to be square and single level (low voltage was assumed to be 0) unless otherwise noted. The number of trains was assumed to be 1 and there was assumed to be no train interval unless otherwise noted. Low voltage time was calculated as the difference between the inverse of the frequency and the pulse duration (high voltage time). Where applicable, the cargo size for the applied waveform was listed, reflecting the differences in waveform optimization mentioned in the cargo section for small and large cargo. In accordance with the findings by Mukherjee et al.[152], small cargos are strictly defined as molecules containing radii less than 3 nm, although due to the lack of information for many molecules, as well as the fact that nucleic acids are narrow but can be extraordinarily long, small cargos are defined in this paper as having radii less than 3 nm and a mass less than 50 kDa. This definition includes ions, small molecules, fluorophores, oligonucleotides, micro RNA, and molecular beacons, while proteins, messenger RNA, and DNA are classified as large cargos. It should be noted that this distinction has not been extensively supported and is provided for comparison purposes only.
Table 4.
Electroporation Waveforms Applied to Porous Substrate Systems
| Pulse Shape | Cargo Size | High Voltage (V) | Low Voltage (V) | High Voltage Duration (ms) | Low Voltage Duration (ms) | Pulse Frequency (Hz) | Pulses/ Train | Number of Trains | Train Interval (ms) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Single Level Square | Small | 0 | 10 | None | None | 1 | 1 | None | [164] | |
| 1–4 | 0 | 20 | 1000 | 1 | 10 | 1 | None | [155] | ||
| 10 | 0 | 10 | 5 | 1 | None | [163] | ||||
| 3–15 | 0 | 20 | None | None | 1 | 1 | None | [154] | ||
| 15 | 0 | 0.5 | 1–3 | 1 | None | [152] | ||||
| 20 | 0 | 2.5 | 2.5 | 200 | 200 | 2 | 1000 | [153] | ||
| 10–25 | 0 | 20 | None | None | 1 | 1 | None | [168] | ||
| 15–140 | 0 | 5–30 | 1000 | 1 | 2–5 | 1 | None | [160] | ||
| 15–140 | 0 | 5–30 | 1000 | 1 | 5 | 1 | None | [165] | ||
| 15–140 | 0 | 5–30 | 1 | None | [166] | |||||
| 50–200 | 0 | 5–20 | 1–10 | 1 | None | [161] | ||||
| 200–250 | 0 | 5–20 | 2–5 | 1 | None | [162] | ||||
| Large | * | 0 | 500 | 500 | 1 | 5 | 1 | None | [156] | |
| 4 | 0 | 50 | 10 | 1 | None | [163] | ||||
| 6 | 0 | 20 | None | None | 1 | 1 | None | [154] | ||
| 10–50 | 0 | 5 | 45 | 20 | 100–500 | 1 | None | [152] | ||
| 15–90 | 0 | 0.2 | 49.8 | 20 | 400–2400 | 1 | None | [151] | ||
| 100 | 0 | 20 | None | None | 1 | 1 | None | [165] | ||
| 200 | 0 | 10 | 1 | None | [161] | |||||
| Bilevel Square | Large | 80–100 | 10 | 0.25 | 3 | 200 | 50 | 8 | 500 | [153] |
| 30 | 10 | 0.5 | 2.5 | 0.5–10 | 200–400 | 1 | None | [158] | ||
| Exponential Decay | Large | * | 0 | None | None | 1 | 1 | None | [157] | |
| Both | * | 0 | None | None | 1 | 1 | None | [167] |
Values are provided for the electric field but not the voltage.
Commonalities between chosen waveforms can be seen in Table 4. Applied voltages range from 1 to 250 V, with common values being 15–140 V. Applied voltage depends on the resistance of the system and therefore varies depending on the substrates chosen. Low voltage is almost always 0 V, with the notable exception of Kang et al., who used bilevel pulses to deliver large cargo[153]. The high voltage duration ranges from 0.2–500 ms, but common values are 5–30 ms. Low voltage duration varies from 2.5–1000 ms, and low voltage duration is generally much longer than the high voltage duration, although rarely the durations are equivalent. Pulse frequencies have been reported from 1–200 Hz. The number of pulses applied per train range from 1–2400 pulses, while the number of trains is generally 1 and no more than 10. When multiple trains were used, train intervals were 500–1000 ms. Overall, there is a significant amount of data regarding waveform specifications that has not been published, making it difficult to draw conclusions. Furthermore, there are significant variations between the many applied waveforms, signifying the pressing need for further waveform optimization with respect to porous substrate systems.
4. Conclusion and Outlook
This review examined intracellular delivery methods from the perspectives of throughput and control. Currently, viral transfection, lipid transfection and bulk electroporation represent the most widely used high throughput methods for gene delivery. Flow-through microfluidic based mechanoporation and electroporation possess the highest throughput capacity of any existing methods and eliminate some of the drawbacks of viral transfection and bulk electroporation, while automated probe-based methods provide subcellular control but are lacking in throughput potential. Engineered substrate methods offer a balance by maintaining high throughput capacity while further improving control aspects such as delivery efficiency, cell viability and dosage control over the dominant methods used today. Porous substrate electroporation is a particularly promising and under investigated engineered substrate method which we reviewed in detail.
Within porous substrate electroporation specifically, there are a few potential areas where significant improvement can be made in the coming years. First, control of cell-ECM and basal membrane tension may facilitate larger pore formation with a tight seal between the substrate and the cell membrane, the translocation of large plasmids with complex shapes, and the delivery of controlled amounts of cargos. Second, the choice of porous substrate materials, the selection of micro/nanochannel dimensions, and the design of electrical waveforms all play a vital role in regulating the generation of electrical potential both at the membrane and at the micro/nanochannels, and in determining the transport of cargo molecules across the micro/nanochannels in the context of complicated electrokinetic landscapes. In addition, for porous substrate arrays, new techniques for patterning cells are prerequisite to facilitate high throughput requirements. Lastly, porous substrate electroporation has predominantly been used in proof of concept studies, therefore studies should begin investigating more complex applications such as temporal sampling of molecules of interest for real-time monitoring in live cells; batch production and extraction of industrial proteins; and transdifferentiation and iPSC reprogramming.
Beyond porous substrate-based electroporation, automated probe-based methods and flow-through microfluidic methods are well suited to other applications. Flow-through microfluidics are unparalleled for applications containing non-adherent cells such as analysis or modification of blood cells. Meanwhile, automated-probe based methods are uniquely capable for studying fundamental biological mechanisms in adherent cells across large populations. However, challenges remain with intracellular delivery in general. First, to fulfill challenging applications such as high throughput, uniform iPSC reprogramming, intracellular delivery systems must be able to both process millions of cells and be viewed in the context of current state-of-the-art single cell analysis, in which the heterogeneity of individual cells is examined. In this regard, there is a need for systems that deliver with similar control to single-cell delivery platforms while maintaining high throughput. In addition, most intracellular delivery methods outside of probe-based methods have been unable to demonstrate rapid delivery of DNA to the nucleus, often requiring many hours for translation to be detected. Lastly, innovative biological assays are needed to provide more accurate metrics for examining cell health after delivery for precise quantification of unintended side effects.
Acknowledgements
RY acknowledges the funding support from the NSF (award #1826135, #1936065), the NIH National Institutes of General Medical Sciences P20GM113126 (Nebraska Center for Integrated Biomolecular Communication) and P30GM127200 (Nebraska Center for Nanomedicine), the Nebraska Collaborative Initiative and Nebraska EPSCoR FIRST award. HDE acknowledges the support of the NIH R21 Award Number GM132709-01 and the National Cancer Institute of the National Institutes of Health (NIH) under Award Number U54CA199091.
Footnotes
Competing Interests
HDE declares majority ownership of Infinitesimal LLC, a company commercializing bio-tools for cell manipulation and analysis. The other authors declare no competing interests.
Contributor Information
Justin Brooks, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE, 68588, USA.
Grayson Minnick, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE, 68588, USA.
Prithvijit Mukherjee, Department of Mechanical Engineering, Northwestern University, Evanston, IL, 60208, USA.
Arian Jaberi, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE, 68588, USA.
Lingqian Chang, School of Biological Science and Medical Engineering, Beihang University, Beijing, 100191, China.
Horacio D. Espinosa, Department of Mechanical Engineering, Northwestern University, Evanston, IL, 60208, USA; Theoretical and Applied Mechanics Program, Northwestern University, Evanston, IL, 60208, USA.
Ruiguo Yang, Department of Mechanical and Materials Engineering, University of Nebraska-Lincoln, Lincoln, NE, 68588, USA; Nebraska Center for Integrated Biomolecular Communication, University of Nebraska-Lincoln, Lincoln, NE, 68588, USA.
References
- [1].Engler AJ; Sen S; Sweeney HL; Discher DE, Cell 2006, 126 (4), 677–689. [DOI] [PubMed] [Google Scholar]
- [2].Gerardo H; Lima A; Carvalho J; Ramos JRD; Couceiro S; Travasso RDM; Pires das Neves R; Grãos M, Scientific Reports 2019, 9 (1), 9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Guo J; Wang Y; Sachs F; Meng F, Proceedings of the National Academy of Sciences 2014, 111 (49), E5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yu J; Chau KF; Vodyanik MA; Jiang J; Jiang Y, PLOS ONE 2011, 6 (3), e17557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen G; Gulbranson DR; Hou Z; Bolin JM; Ruotti V; Probasco MD; Smuga-Otto K; Howden SE; Diol NR; Propson NE; Wagner R; Lee GO; Antosiewicz-Bourget J; Teng JMC; Thomson JA, Nature Methods 2011, 8 (5), 424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].June CH; O’Connor RS; Kawalekar OU; Ghassemi S; Milone MC, Science 2018, 359 (6382), 1361–1365. [DOI] [PubMed] [Google Scholar]
- [7].Feins S; Kong W; Williams EF; Milone MC; Fraietta JA, Am. J. Hematol 2019, 94 (S1), S3–S9. [DOI] [PubMed] [Google Scholar]
- [8].Apostolou E; Stadtfeld M, Curr. Opin. Genet. Dev 2018, 52, 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lakshmipathy U; Vemuri MC, Pluripotent Stem Cells: Methods and Protocols. Springer: 2013. [Google Scholar]
- [10].Lai T; Yang Y; Ng SK, Pharmaceuticals (Basel) 2013, 6 (5), 579–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wurm FM, Nature Biotechnology 2004, 22 (11), 1393–1398. [DOI] [PubMed] [Google Scholar]
- [12].Pilbrough W; Munro TP; Gray P, PLOS ONE 2009, 4 (12), e8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang R; Lemaître V; Huang C; Haddadi A; McNaughton R; Espinosa HD, Small 2018, 14, 1702495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li J; Wang B; Juba BM; Vazquez M; Kortum SW; Pierce BS; Pacheco M; Roberts L; Strohbach JW; Jones LH; Hett E; Thorarensen A; Telliez J-B; Sharei A; Bunnage M; Gilbert JB, ACS Chemical Biology 2017, 12 (12), 2970–2974. [DOI] [PubMed] [Google Scholar]
- [15].Stewart MP; Sharei A; Ding X; Sahay G; Langer R; Jensen KF, Nature 2016, 538 (7624), 183–192. [DOI] [PubMed] [Google Scholar]
- [16].Stewart MP; Langer R; Jensen KF, Chemical Reviews 2018, 118, 7409–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bowman K; Leong KW, International journal of nanomedicine 2006, 1 (2), 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nishikawa M; Yamauchi M; Morimoto K; Ishida E; Takakura Y; Hashida M, Gene Ther. 2000, 7 (7), 548–555. [DOI] [PubMed] [Google Scholar]
- [19].Kootstra NA; Verma IM, Annu. Rev. Pharmacol. Toxicol 2003, 43 (1), 413–439. [DOI] [PubMed] [Google Scholar]
- [20].Zhang X; Godbey W, Adv. Drug Delivery Rev 2006, 58 (4), 515–534. [DOI] [PubMed] [Google Scholar]
- [21].Lundstrom K, Diseases 2018, 6 (2), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Thomas CE; Ehrhardt A; Kay MA, Nat. Rev. Genet 2003, 4 (5), 346–358. [DOI] [PubMed] [Google Scholar]
- [23].van der Loo JC; Wright JF, Hum. Mol. Genet 2016, 25 (R1), R42–R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kay MA; Glorioso JC; Naldini L, Nat. Med 2001, 7 (1), 33–40. [DOI] [PubMed] [Google Scholar]
- [25].Yin H; Kanasty RL; Eltoukhy AA; Vegas AJ; Dorkin JR; Anderson DG, Nat. Rev. Genet 2014, 15 (8), 541–555. [DOI] [PubMed] [Google Scholar]
- [26].Hsu CYM; Uludağ H, Drug Target J. 2012, 20 (4), 301–328. [DOI] [PubMed] [Google Scholar]
- [27].Plank C; Zelphati O; Mykhaylyk O, Advanced drug delivery reviews 2011, 63 (14–15), 1300–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ramamoorth M; Narvekar A, Journal of clinical and diagnostic research: JCDR 2015, 9 (1), GE01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jones CH; Chen C-K; Ravikrishnan A; Rane S; Pfeifer BA, Mol. Pharm 2013, 10 (11), 4082–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chang L; Li L; Shi J; Sheng Y; Lu W; Gallego-Perez D; Lee LJ, Lab on a Chip 2016, 16 (21), 4047–4062. [DOI] [PubMed] [Google Scholar]
- [31].Gehl J, Acta Physiologica Scandinavica 2003, 177 (4), 437–447. [DOI] [PubMed] [Google Scholar]
- [32].Pliquett U; Gift EA; Weaver JC, Bioelectrochem. Bioenerg 1996, 39 (1), 39–53. [DOI] [PubMed] [Google Scholar]
- [33].Canatella PJ; Karr JF; Petros JA; Prausnitz MR, Biophys. J 2001, 80 (2), 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pucihar G; Kotnik T; Valič B; Miklavčič D, Ann. Biomed. Eng 2006, 34 (4), 642. [DOI] [PubMed] [Google Scholar]
- [35].Weaver JC; Smith KC; Esser AT; Son RS; Gowrishankar T, Bioelectrochemistry 2012, 87, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Krasieva TB; Chapman CF; LaMorte VJ; Venugopalan V; Berns MW; Tromberg BJ In Cell permeabilization and molecular transport by laser microirradiation, Optical Investigations of Cells In Vitro and In Vivo, International Society for Optics and Photonics: 1998; pp 38–44. [Google Scholar]
- [37].Rhodes K; Clark I; Zatcoff M; Eustaquio T; Hoyte KL; Koller MR, Methods in cell biology 2007, 82, 309–333. [DOI] [PubMed] [Google Scholar]
- [38].Stevenson DJ; Gunn-Moore FJ; Campbell P; Dholakia K, Journal of the Royal Society Interface 2010, 7 (47), 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Soughayer JS; Krasieva T; Jacobson SC; Ramsey JM; Tromberg BJ; Allbritton NL, Analytical chemistry 2000, 72 (6), 1342–1347. [DOI] [PubMed] [Google Scholar]
- [40].Zhang H; Wang J; Hu M; Li B.-c.; Li H; Chen T.-t.; Ren K-F; Ji J; Jing Q.-m.; Fu G.-s., Biomaterials Science 2019, 7 (12), 5177–5186. [DOI] [PubMed] [Google Scholar]
- [41].Rau KR; Guerra A III; Vogel A; Venugopalan V, Applied Physics Letters 2004, 84 (15), 2940–2942. [Google Scholar]
- [42].de Paula DMB; Valero-Lapchik VB; Paredes-Gamero EJ; Han SW, The journal of gene medicine 2011, 13 (7–8), 392–401. [DOI] [PubMed] [Google Scholar]
- [43].Lentacker I; De Cock I; Deckers R; De Smedt S; Moonen C, Advanced drug delivery reviews 2014, 72, 49–64. [DOI] [PubMed] [Google Scholar]
- [44].Yang Y; Li Q; Guo X; Tu J; Zhang D, Ultrasonics Sonochemistry 2020, 105096. [DOI] [PubMed] [Google Scholar]
- [45].Van Wamel A; Kooiman K; Harteveld M; Emmer M; Folkert J; Versluis M; De Jong N, Journal of controlled release 2006, 112 (2), 149–155. [DOI] [PubMed] [Google Scholar]
- [46].Barber MA, The Journal of Infectious Diseases 1911, 348–360. [Google Scholar]
- [47].Simonis M; Hübner W; Wilking A; Huser T; Hennig S, Scientific reports 2017, 7, 41277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vakarelski IU; Brown SC; Higashitani K; Moudgil BM, Langmuir 2007, 23 (22), 10893–10896. [DOI] [PubMed] [Google Scholar]
- [49].Zhang Y; Yu L-C, Current opinion in biotechnology 2008, 19 (5), 506–510. [DOI] [PubMed] [Google Scholar]
- [50].Zhang Y; Yu LC, BioEssays 2008, 30 (6), 606–10. [DOI] [PubMed] [Google Scholar]
- [51].Guillaume-Gentil O; Potthoff E; Ossola D; Franz CM; Zambelli T; Vorholt JA, Trends in biotechnology 2014, 32 (7), 381–388. [DOI] [PubMed] [Google Scholar]
- [52].Johnston SA; Tang D.-c., Gene gun transfection of animal cells and genetic immunization. In Methods in cell biology, Elsevier: 1994; Vol. 43, pp 353–365. [DOI] [PubMed] [Google Scholar]
- [53].Movahed S; Li D, Microfluidics and Nanofluidics 2011, 10 (4), 703–734. [Google Scholar]
- [54].Sun M; Duan X, Nanotechnology and Precision Engineering 2019. [Google Scholar]
- [55].Huang Y; Rubinsky B, Biomedical Microdevices 1999, 2 (2), 145–150. [Google Scholar]
- [56].Gresch O; Engel FB; Nesic D; Tran TT; England HM; Hickman ES; Körner I; Gan L; Chen S; Castro-Obregon S, Methods 2004, 33 (2), 151–163. [DOI] [PubMed] [Google Scholar]
- [57].Giraldo-Vela JP; Kang W; McNaughton RL; Zhang X; Wile BM; Tsourkas A; Bao G; Espinosa HD, Small 2015, 11 (20), 2386–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kang W; Yavari F; Minary-Jolandan M; Giraldo-Vela JP; Safi A; McNaughton RL; Parpoil V; Espinosa HD, Nano Lett. 2013, 13 (6), 2448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Boukany PE; Morss A; Liao W.-c.; Henslee B; Jung H; Zhang X; Yu B; Wang X; Wu Y; Li L, Nature nanotechnology 2011, 6 (11), 747. [DOI] [PubMed] [Google Scholar]
- [60].Gao K; Huang X; Chiang CL; Wang X; Chang L; Boukany P; Marcucci G; Lee R; Lee LJ, Mol Ther 2016, 24 (5), 956–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gao K; Li L; He L; Hinkle K; Wu Y; Ma J; Chang L; Zhao X; Perez DG; Eckardt S; McLaughlin J; Liu B; Farson DF; Lee LJ, Small 2014, 10 (5), 1015–23. [DOI] [PubMed] [Google Scholar]
- [62].Xiong R; Samal SK; Demeester J; Skirtach AG; De Smedt SC; Braeckmans K, Advances in Physics: X 2016, 1 (4), 596–620. [Google Scholar]
- [63].Fan Z; Liu H; Mayer M; Deng CX, Proceedings of the National Academy of Sciences 2012, 109 (41), 16486–16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Meng L; Cai F; Jiang P; Deng Z; Li F; Niu L; Chen Y; Wu J; Zheng H, Applied Physics Letters 2014, 104 (7), 073701. [Google Scholar]
- [65].Lindstrom ZK; Brewer SJ; Ferguson MA; Burnett SH; Jensen BD, Journal of Nanotechnology in Engineering and Medicine 2014, 5 (2). [Google Scholar]
- [66].Sessions JW; Skousen CS; Price KD; Hanks BW; Hope S; Alder JK; Jensen BD, Springerplus 2016, 5 (1), 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Teichert GH; Burnett S; Jensen BD, Journal of Micromechanics and Microengineering 2013, 23 (9). [Google Scholar]
- [68].Zhang B; Shi Y; Miyamoto D; Nakazawa K; Miyake T, Sci Rep 2019, 9 (1), 6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chi Z; Xu Q; Zhu L, IEEE Access 2020, 8, 8520–8532. [Google Scholar]
- [70].Carpenter AE; Jones TR; Lamprecht MR; Clarke C; Kang IH; Friman O; Guertin DA; Chang JH; Lindquist RA; Moffat J, Genome biology 2006, 7 (10), R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Spontón H; Cardelino J, Image Processing On Line 2015, 5, 90–123. [Google Scholar]
- [72].Long X; Cleveland WL; Yao YL, IEEE Transactions on Information Technology in Biomedicine 2005, 9 (3), 407–412. [DOI] [PubMed] [Google Scholar]
- [73].Mualla F; Schöll S; Sommerfeldt B; Maier A; Hornegger J, IEEE transactions on medical imaging 2013, 32 (12), 2274–2286. [DOI] [PubMed] [Google Scholar]
- [74].Wang W; Liu X; Gelinas D; Ciruna B; Sun Y, PloS one 2007, 2 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhao Y; Sun H; Sha X; Gu L; Zhan Z; Li WJ, Micromachines 2019, 10 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lulevich V; Shih Y-P; Lo SH; Liu G.-y., The Journal of Physical Chemistry B 2009, 113 (18), 6511–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ladjal H; Hanus JL; Ferreira A, IEEE Trans Biomed Eng 2013, 60 (9), 2461–71. [DOI] [PubMed] [Google Scholar]
- [78].Xie Y; Zhou Y; Lin Y; Wang L; Xi W, Sensors 2016, 16 (4), 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lu Z; Chen PC; Nam J; Ge R; Lin W, Journal of micromechanics and microengineering 2007, 17 (2), 314. [Google Scholar]
- [80].Sun Y; Nelson BJ, Biomedical Materials 2007, 2 (1), S16. [DOI] [PubMed] [Google Scholar]
- [81].Wei Y; Xu Q, IEEE Transactions on Automation Science and Engineering 2018, 16 (2), 931–945. [Google Scholar]
- [82].Wang W; Sun Y; Zhang M; Anderson R; Langille L; Chan W, Rev Sci Instrum 2008, 79 (10), 104302. [DOI] [PubMed] [Google Scholar]
- [83].Liu X; Fernandes R; Gertsenstein M; Perumalsamy A; Lai I; Chi M; Moley KH; Greenblatt E; Jurisica I; Casper RF, PloS one 2011, 6 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Becattini G; Mattos LS; Caldwell DG, IEEE J Biomed Health Inform 2014, 18 (1), 83–93. [DOI] [PubMed] [Google Scholar]
- [85].Chow YT; Chen S; Liu C; Liu C; Li L; Kong CWM; Cheng SH; Li RA; Sun D, IEEE/ASME Transactions on Mechatronics 2015, 21 (2), 838–850. [Google Scholar]
- [86].Permana S; Grant E; Walker GM; Yoder JA, IEEE/ASME Transactions on Mechatronics 2016, 21 (5), 2391–2404. [Google Scholar]
- [87].Xu Q, Micromachines for Biological Micromanipulation. Springer International: Cham, Switzerland, 2018. [Google Scholar]
- [88].Bae C; Butler PJ, Biotechniques 2006, 41 (4), 399–400, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sakaki K; Esmaeilsabzali H; Dechev N; Burke RD; Park EJ, IEEE Transactions on Automation Science and Engineering 2012, 9, 226–236. [Google Scholar]
- [90].Sakaki K; Esmaeilsabzali H; Massah S; Prefontaine GG; Dechev N; Burke RD; Park EJ, IEEE Trans. Biomed. Eng 2013, 60 (11), 3113–23. [DOI] [PubMed] [Google Scholar]
- [91].Kang W; McNaughton RL; Yavari F; Minary-Jolandan M; Safi A; Espinosa HD, Journal of laboratory automation 2014, 19 (1), 100–109. [DOI] [PubMed] [Google Scholar]
- [92].Ding X; Stewart MP; Sharei A; Weaver JC; Langer RS; Jensen KF, Nature Biomedical Engineering 2017, 1 (3), 0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fox M; Esveld D; Valero A; Luttge R; Mastwijk H; Bartels P; van den Berg A; Boom R, Analytical and bioanalytical chemistry 2006, 385 (3), 474. [DOI] [PubMed] [Google Scholar]
- [94].Geng T; Lu C, Lab on a Chip 2013, 13 (19), 3803–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lu H; Schmidt MA; Jensen KF, Lab Chip 2005, 5 (1), 23–9. [DOI] [PubMed] [Google Scholar]
- [96].Adamo A; Arione A; Sharei A; Jensen KF, Anal Chem 2013, 85 (3), 1637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ouyang M; Hill W; Lee JH; Hur SC, Scientific Reports 2017, 7 (1), 44757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chang A-Y; Liu X; Tian H; Hua L; Yang Z; Wang S, Scientific reports 2020, 10 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ye Y; Zhao W; Chen J; Cheng J; Wei X; Zhang L; Mao H; Li M; Zhao Y; Huang C. In Single-Cell Electroporation and Real-Time Electrical Monitoring on a Microfluidic Chip, 2020 IEEE 33rd International Conference on Micro Electro Mechanical Systems (MEMS), IEEE: 2020; pp 1040–1043. [Google Scholar]
- [100].Zhan Y; Wang J; Bao N; Lu C, Analytical Chemistry 2009, 81 (5), 2027–2031. [DOI] [PubMed] [Google Scholar]
- [101].Geng T; Zhan Y; Wang H-Y; Witting SR; Cornetta KG; Lu C, Journal of Controlled Release 2010, 144 (1), 91–100. [DOI] [PubMed] [Google Scholar]
- [102].Kollmannsperger A; Sharei A; Raulf A; Heilemann M; Langer R; Jensen KF; Wieneke R; Tampé R, Nature Communications 2016, 7 (1), 10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Modaresi S; Pacelli S; Subham S; Dathathreya K; Paul A, Advanced Therapeutics 2020, 3 (1), 1900130. [Google Scholar]
- [104].Kang G; Carlson DW; Kang TH; Lee S; Haward SJ; Choi I; Shen AQ; Chung AJ, ACS Nano 2020, 14 (3), 3048–3058. [DOI] [PubMed] [Google Scholar]
- [105].Kizer ME; Deng Y; Kang G; Mikael PE; Wang X; Chung AJ, Lab Chip 2019, 19 (10), 1747–1754. [DOI] [PubMed] [Google Scholar]
- [106].Higgins SG; Becce M; Belessiotis-Richards A; Seong H; Sero JE; Stevens MM, Advanced Materials 2020, 32 (9), 1903862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].He G; Hu N; Xu AM; Li X; Zhao Y; Xie X, Adv. Funct. Mater 2020, 30 (21), 1909890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lestrell E; Patolsky F; Voelcker NH; Elnathan R, Mater. Today 2020, 33, 87–104. [Google Scholar]
- [109].Wu YC; Wu TH; Clemens DL; Lee BY; Wen X; Horwitz MA; Teitell MA; Chiou PY, Nat Methods 2015, 12 (5), 439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Wang Y; Yang Y; Yan L; Kwok SY; Li W; Wang Z; Zhu X; Zhu G; Zhang W; Chen X; Shi P, Nat Commun 2014, 5, 4466. [DOI] [PubMed] [Google Scholar]
- [111].Berthing T; Bonde S; Rostgaard KR; Madsen MH; Sorensen CB; Nygard J; Martinez KL, Nanotechnology 2012, 23 (41), 415102. [DOI] [PubMed] [Google Scholar]
- [112].Prinz CN, J Phys Condens Matter 2015, 27 (23), 233103. [DOI] [PubMed] [Google Scholar]
- [113].Shalek AK; Gaublomme JT; Wang L; Yosef N; Chevrier N; Andersen MS; Robinson JT; Pochet N; Neuberg D; Gertner RS; Amit I; Brown JR; Hacohen N; Regev A; Wu CJ; Park H, Nano Lett 2012, 12 (12), 6498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Shalek AK; Robinson JT; Karp ES; Lee JS; Ahn DR; Yoon MH; Sutton A; Jorgolli M; Gertner RS; Gujral TS; MacBeath G; Yang EG; Park H, Proc Natl Acad Sci U S A 2010, 107 (5), 1870–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Yosef N; Shalek AK; Gaublomme JT; Jin H; Lee Y; Awasthi A; Wu C; Karwacz K; Xiao S; Jorgolli M; Gennert D; Satija R; Shakya A; Lu DY; Trombetta JJ; Pillai MR; Ratcliffe PJ; Coleman ML; Bix M; Tantin D; Park H; Kuchroo VK; Regev A, Nature 2013, 496 (7446), 461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Chen Y; Aslanoglou S; Gervinskas G; Abdelmaksoud H; Voelcker NH; Elnathan R, Small 2019, 15 (47), 1904819. [DOI] [PubMed] [Google Scholar]
- [117].Seong H; Higgins SG; Penders J; Armstrong JP; Crowder SW; Moore AC; Sero JE; Becce M; Stevens MM, ACS Nano 2020, 14 (5), 5371–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Chen Y; Aslanoglou S; Murayama T; Gervinskas G; Fitzgerald LI; Sriram S; Tian J; Johnston AP; Morikawa Y; Suu K, Advanced Materials 2020, 2000036. [DOI] [PubMed] [Google Scholar]
- [119].Golshadi M; Wright LK; Dickerson IM; Schrlau MG, Small 2016, 12 (22), 3014–3020. [DOI] [PubMed] [Google Scholar]
- [120].Higgins SG; Stevens MM, Extracting the contents of living cells. Science 2017, pp 379–380. [DOI] [PubMed] [Google Scholar]
- [121].VanDersarl JJ; Xu AM; Melosh NA, Nano Lett 2012, 12 (8), 3881–3886. [DOI] [PubMed] [Google Scholar]
- [122].Xu AM; Kim SA; Wang DS; Aalipour A; Melosh NA, Lab Chip 2016, 16 (13), 2434–2439. [DOI] [PubMed] [Google Scholar]
- [123].Xu AM; Wang DS; Shieh P; Cao Y; Melosh NA, Chembiochem 2017, 18 (7), 623–628. [DOI] [PubMed] [Google Scholar]
- [124].Chen Y; Wang J; Li X; Hu N; Voelcker NH; Xie X; Elnathan R, Advanced Materials 2020, 2001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Cao Y; Hjort M; Chen H; Birey F; Leal-Ortiz SA; Han CM; Santiago JG; Paşca SP; Wu JC; Melosh NA, Proceedings of the National Academy of Sciences of the United States of America 2017, 114, E1866–E1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Cao Y; Chen H; Qiu R; Hanna M; Ma E; Hjort M; Zhang A; Lewis RS; Wu JC; Melosh NA, Science Advances 2018, 4 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Caprettini V; Cerea A; Melle G; Lovato L; Capozza R; Huang JA; Tantussi F; Dipalo M; De Angelis F, Scientific Reports 2017, 7, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].He G; Chen H-J; Liu D; Feng Y; Yang C; Hang T; Wu J; Cao Y; Xie X, Advanced Materials Interfaces 2018, 5 (10). [Google Scholar]
- [129].He G; Yang C; Hang T; Liu D; Chen HJ; Zhang AH; Lin D; Wu J; Yang BR; Xie X, ACS Sens 2018, 3 (9), 1675–1682. [DOI] [PubMed] [Google Scholar]
- [130].Jokilaakso N; Salm E; Chen A; Millet L; Guevara CD; Dorvel B; Reddy B Jr.; Karlstrom AE; Chen Y; Ji H; Chen Y; Sooryakumar R; Bashir R, Lab Chip 2013, 13 (3), 336–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Tay A; Melosh N, Advanced Therapeutics 2019, 2 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Xie C; Lin Z; Hanson L; Cui Y; Cui B, Nat Nanotechnol 2012, 7 (3), 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Xie X; Xu AM; Leal-Ortiz S; Cao Y; Garner CC; Melosh NA, ACS Nano 2013, 7 (5), 4351–4358. [DOI] [PubMed] [Google Scholar]
- [134].Zu Y; Huang S; Lu Y; Liu X; Wang S, Sci Rep 2016, 6, 38661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Garcia-Sanchez T; Sanchez-Ortiz B; Vila I; Guitart M; Rosell J; Gomez-Foix AM; Bragos R, J Membr Biol 2012, 245 (10), 617–24. [DOI] [PubMed] [Google Scholar]
- [136].Li Y; Wu M; Zhao D; Wei Z; Zhong W; Wang X; Liang Z; Li Z, Scientific Reports 2015, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lin YC; Li M; Wu CC, Lab Chip 2004, 4 (2), 104–8. [DOI] [PubMed] [Google Scholar]
- [138].Meir A; Rubinsky B, RSC Adv. 2014, 4 (97), 54603–54613. [Google Scholar]
- [139].Novickij V; Tabasnikov A; Smith S; Grainys A; Novickij J, IETE Technical Review 2015, 32 (3), 196–203. [Google Scholar]
- [140].Olbrich M; Rebollar E; Heitz J; Frischauf I; Romanin C, Applied Physics Letters 2008, 92 (1). [Google Scholar]
- [141].Xu Y; Su S; Zhou C; Lu Y; Xing W, Bioelectrochemistry 2015, 102, 35–41. [DOI] [PubMed] [Google Scholar]
- [142].Wu M; Zhao D; Wei Z; Zhong W; Yan H; Wang X; Liang Z; Li Z, Anal Chem 2013, 85 (9), 4483–91. [DOI] [PubMed] [Google Scholar]
- [143].Wu M; Xiao Y; Zhao D; Liang Z; Li Z. In A microchip for in vitro parameter determination of cancer electrochemotherapy, Transducers and Eurosensors, Barcelona, Spain, IEEE: Barcelona, Spain, 2013. [Google Scholar]
- [144].Chen SC; Santra TS; Chang CJ; Chen TJ; Wang PC; Tseng FG, Biomedical Microdevices 2012, 14, 811–817. [DOI] [PubMed] [Google Scholar]
- [145].He H; Chang DC; Lee YK, Bioelectrochemistry 2006, 68 (1), 89–97. [DOI] [PubMed] [Google Scholar]
- [146].He H; Chang DC; Lee YK, Bioelectrochemistry 2007, 70 (2), 363–8. [DOI] [PubMed] [Google Scholar]
- [147].Homhuan S; Zhang B; Sheu FS; Bettiol AA; Watt F, Biomed Microdevices 2012, 14 (3), 533–40. [DOI] [PubMed] [Google Scholar]
- [148].S. Moon; W. Li; K. Xu, 2019.
- [149].Santra TS; Chang HY; Wang PC; Tseng FG, Analyst 2014, 139 (23), 6249–58. [DOI] [PubMed] [Google Scholar]
- [150].Xu Y; Yao H; Wang L; Xing W; Cheng J, Lab Chip 2011, 11 (14), 2417–23. [DOI] [PubMed] [Google Scholar]
- [151].Cao Y; Ma E; Cestellos-Blanco S; Zhang B; Qiu R; Su Y; Doudna JA; Yang P, Proceedings of the National Academy of Sciences 2019, 116 (16), 7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Mukherjee P; Nathamgari SSP; Kessler JA; Espinosa HD, ACS Nano 2018, 12 (12), 12118–12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Kang W; Giraldo-Vela JP; Nathamgari SSP; McGuire T; McNaughton RL; Kessler JA; Espinosa HD, Lab Chip 2014, 14 (23), 4486–4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ishibashi T; Takoh K; Kaji H; Abe T; Nishizawa M, Sensors and Actuators B: Chemical 2007, 128 (1), 5–11. [Google Scholar]
- [155].Chen Z; Akenhead MA; Sun X; Sapper H; Shin HY; Hinds BJ, Advanced Healthcare Materials 2016, 5 (16), 2105–2112. [DOI] [PubMed] [Google Scholar]
- [156].Fei Z; Wang S; Xie Y; Henslee BE; Koh CG; Lee LJ, Analytical Chemistry 2007, 79 (15), 5719–5722. [DOI] [PubMed] [Google Scholar]
- [157].Fei Z; Wu Y; Sharma S; Gallego-Perez D; Higuita-Castro N; Hansford D; Lannutti JJ; Lee LJ, Analytical Chemistry 2013, 85 (3), 1401–1407. [DOI] [PubMed] [Google Scholar]
- [158].Mukherjee P; Berns EJ; Patino CA; Hakim Moully E; Chang L; Nathamgari SSP; Kessler JA; Mrksich M; Espinosa HD, Small 2020, 16 (26), e2000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Kang W; Nathamgari SSP; Giraldo-Vela JP; McNaughton RL; Kessler J; Espinosa HD In Optimization of a microfluidic device for localized electroporation of cells, 14th IEEE International Conference on Nanotechnology, 18–21 Aug. 2014; 2014; pp 1–3. [Google Scholar]
- [160].Chang L; Bertani P; Gallego-Perez D; Yang Z; Chen F; Chiang C; Malkoc V; Kuang T; Gao K; Lee LJ; Lu W, Nanoscale 2016, 8 (1), 243–252. [DOI] [PubMed] [Google Scholar]
- [161].Chang L; Gallego-Perez D; Chiang C-L; Bertani P; Kuang T; Sheng Y; Chen F; Chen Z; Shi J; Yang H; Huang X; Malkoc V; Lu W; Lee LJ, Small 2016, 12 (43), 5971–5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Gallego-Perez D; Chang L; Shi J; Ma J; Kim S-H; Zhao X; Malkoc V; Wang X; Minata M; Kwak KJ; Wu Y; Lafyatis GP; Lu W; Hansford DJ; Nakano I; Lee LJ, Nano Letters 2016, 16 (9), 5326–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Chang L; Howdyshell M; Liao W-C; Chiang C-L; Gallego-Perez D; Yang Z; Lu W; Byrd JC; Muthusamy N; Lee LJ; Sooryakumar R, Small 2015, 11 (15), 1818–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Bertani P; Lu W; Chang L; Gallego-Perez D; James Lee L; Chiang C; Muthusamy N, Journal of Vacuum Science & Technology B 2015, 33 (6), 06F903. [Google Scholar]
- [165].Chang L; Gallego-Perez D; Zhao X; Bertani P; Yang Z; Chiang C-L; Malkoc V; Shi J; Sen CK; Odonnell L; Yu J; Lu W; Lee LJ, Lab Chip 2015, 15 (15), 3147–3153. [DOI] [PubMed] [Google Scholar]
- [166].Bertani P; Chang LQ; Gallego-Perez D; Malkoc V; Lee LJ; Lu In W. 3D Si-based nanochannel platform for robust cell electroporation, 2015 73rd Annual Device Research Conference (DRC), 21–24 June 2015; 2015; pp 83–84. [Google Scholar]
- [167].Fei Z; Hu X; Choi H.-w.; Wang S; Farson D; Lee LJ, Analytical Chemistry 2010, 82 (1), 353–358. [DOI] [PubMed] [Google Scholar]
- [168].Dong Z; Jiao Y; Xie B; Hao Y; Wang P; Liu Y; Shi J; Chitrakar C; Black S; Wang Y-C; Lee LJ; Li M; Fan Y; Chang L, Microsystems & Nanoengineering 2020, 6 (1), 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Khine M; Ionescu-Zanetti C; Blatz A; Wang L-P; Lee LP, Lab Chip 2007, 7 (4), 457–462. [DOI] [PubMed] [Google Scholar]
- [170].Suzuki T; Yamamoto H; Ohoka M; Okonogi A; Kabata H; Kanno I; Washizu M; Kotera H. In High throughput cell electroporation array fabricated by single-mask inclined UV lithography exposure and oxygen plasma etching, TRANSDUCERS 2007–2007 International Solid-State Sensors, Actuators and Microsystems Conference, IEEE: 2007; pp 687–690. [Google Scholar]
- [171].Cao Y; Ma E; Cestellos-Blanco S; Zhang B; Qiu R; Su Y; Doudna JA; Yang P, Proceedings of the National Academy of Sciences 2019, 116 (16), 7899–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Weaver JC; Chizmadzhev YA, Bioelectrochemistry and Bioenergetics 1996, 41 (2), 135–160. [Google Scholar]
- [173].Chen C; Smye SW; Robinson MP; Evans JA, Medical and Biological Engineering and Computing 2006, 44 (1), 5–14. [DOI] [PubMed] [Google Scholar]
- [174].Joshi RP; Schoenbach KH, Physical Review E 2000, 62 (1), 1025–1033. [DOI] [PubMed] [Google Scholar]
- [175].Son RS; Smith KC; Gowrishankar TR; Vernier PT; Weaver JC, The Journal of Membrane Biology 2014, 247 (12), 1209–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Son RS; Gowrishankar TR; Smith KC; Weaver JC, IEEE Transactions on Biomedical Engineering 2016, 63 (3), 571–580. [DOI] [PubMed] [Google Scholar]
- [177].Smith KC; Son RS; Gowrishankar TR; Weaver JC, Bioelectrochemistry 2014, 100, 3–10. [DOI] [PubMed] [Google Scholar]
- [178].Krassowska W; Filev PD, Biophysical Journal 2007, 92 (2), 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Yang Z; Shi J; Xie J; Wang Y; Sun J; Liu T; Zhao Y; Zhao X; Wang X; Ma Y; Malkoc V; Chiang C; Deng W; Chen Y; Fu Y; Kwak KJ; Fan Y; Kang C; Yin C; Rhee J; Bertani P; Otero J; Lu W; Yun K; Lee AS; Jiang W; Teng L; Kim BYS; Lee LJ, Nature Biomedical Engineering 2020, 4 (1), 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Cao Y; Chen H; Qiu R; Hanna M; Ma E; Hjort M; Zhang A; Lewis RS; Wu JC; Melosh NA, Science Advances 2018, 4 (10), eaat8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Nathamgari SSP; Mukherjee P; Kessler JA; Espinosa HD, Proceedings of the National Academy of Sciences 2019, 116 (46), 22909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Kotnik T; Rems L; Tarek M; Miklavčič D, Annual Review of Biophysics 2019, 48 (1), 63–91. [DOI] [PubMed] [Google Scholar]
- [183].Casciola M; Tarek M, Biochimica et Biophysica Acta (BBA) - Biomembranes 2016, 1858 (10), 2278–2289. [DOI] [PubMed] [Google Scholar]
- [184].Hopcroft MA; Nix WD; Kenny TW, Journal of Microelectromechanical Systems 2010, 19 (2), 229–238. [Google Scholar]
- [185].Guimarães CF; Gasperini L; Marques AP; Reis RL, Nature Reviews Materials 2020, 5 (5), 351–370. [Google Scholar]
- [186].Mih JD; Marinkovic A; Liu F; Sharif AS; Tschumperlin DJ, Journal of Cell Science 2012, 125 (24), 5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Cornelius TW; Apel PY; Schiedt B; Trautmann C; Toimil-Molares ME; Karim S; Neumann R, Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2007, 265 (2), 553–557. [Google Scholar]
- [188].Ferain E, Legras R, Nuclear Instruments and Methods in Physics Research Section B: Beam Interactions with Materials and Atoms 2001, 116–122. [DOI] [PubMed] [Google Scholar]
- [189].Brooks J; Jaberi A; Yang R, Microfluidic Device for Localized Electroporation. In Electroporation Protocols, Springer: 2020; pp 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Alhmoud H; Brodoceanu D; Elnathan R; Kraus T; Voelcker NH, Prog. Mater Sci 2019, 100636. [DOI] [PubMed] [Google Scholar]
- [191].Elnathan R; Isa L; Brodoceanu D; Nelson A; Harding FJ; Delalat B; Kraus T; Voelcker NH, ACS Appl. Mater. Interfaces 2015, 7 (42), 23717–23724. [DOI] [PubMed] [Google Scholar]
- [192].Tay A; Melosh N, Acc. Chem. Res 2019, 52 (9), 2462–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Cao Y; Ma E; Cestellos-Blanco S; Qiu R; Su Y; Doudna JA; Yang P, Proceedings of the National Academy of Sciences 2019, 116 (46), 22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Golzio M; Mora MP; Raynaud C; Delteil C; Teissié J; Rols MP, Biophysical journal 1998, 74 (6), 3015–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Tekle E; Astumian RD; Chock PB, Proceedings of the National Academy of Sciences 1991, 88 (10), 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]