

Abstract
Cardiac injury triggers an acute immune response that drives tissue healing and remodeling via the activation of compensatory mechanisms. Over time, remodeling and inflammation become chronic and have adverse effects that lead to a depression of cardiac function and eventual heart failure. Cardiac inflammation is characterized by dynamic spatial and temporal crosstalk between the resident cells of the heart and recruitment of circulating leukocytes. Until recently, the cardiomyocyte has not been accepted as a direct contributor to cardiac inflammation. It has now emerged as a key initiator of the acute immune response via its ability to produce cytokines and may also synchronize leukocyte recruitment post-injury. This review will focus on the role of the cardiomyocyte in the acute immune response to ischemic and non-ischemic injury and the mechanisms by which it may influence the course of cardiac remodeling and failure.
Introduction
Interest in the role of cardiac inflammation in heart disease was ignited in the 1950’s by a seminal study in which Elster et al. observed elevated C-reactive protein, a marker of systemic inflammation, in the circulation of patients with chronic heart failure [1]. Since that time, extensive clinical observations and preclinical studies have highlighted a positive correlation between inflammatory cytokines and cardiac pathology across a wide range of etiologically distinct models of disease, thus implicating the innate immune response in the development of heart failure [2]. Despite substantial evidence supporting the beneficial effects of modulating the immune response in animal models of heart failure, most phase III clinical trials targeting inflammatory mediators failed to improve or even worsened cardiovascular outcome [2–4]. Fortuitously, the field rebounded with the positive outcomes of the of the 2019 CANTOS trial which was shown to decrease adverse cardiac events and re-hospitalization in patients with heart failure [5,6]. Advances in the field of innate immunity and cardiac inflammation have revealed that most nucleated cells are capable of some immune function, and notably this includes the cardiomyocytes themselves [7–9,10•]. Although the extent to which the cardiomyocyte contributes to inflammation is not well appreciated, several studies have shown improvements in cardiac remodeling and function when cardiomyocyte-specific processes are targeted. In this review, we will discuss our current understanding of the contribution of cardiomyocytes to inflammation in the diseased heart and define potential new therapeutic targets for treating cardiomyopathies.
Inflammation, cardiac remodeling, and heart failure
In response to hemodynamic insults or stresses, an acute inflammatory response is initiated to help the heart compensate and adapt to noxious stimuli and return to homeostasis. Cardiac compensatory mechanisms consist of complex signaling cascades that generate numerous cellular and tissue changes that alter the geometry, size, and function of the heart. Collectively, this alteration in cardiac tissue and chamber properties is referred to as cardiac remodeling and can be quantified by cardiomyocyte hypertrophy and tissue fibrosis. Cardiac remodeling is a coping mechanism that helps the heart maintain and normalize its pump function despite hemodynamic stressors such as pressure overload, volume overload, and ischemia. If these compensatory mechanisms are successful in eliminating cardiac stress and tissue injury, the inflammatory responses elicited by the initial stress are also resolved [2]. Conversely, persistence of pathological stimuli can lead to a dysregulated activation of the immune response and a sustained, low-grade inflammatory state termed para-inflammation [2]. In this scenario, the continual process of cardiac remodeling becomes maladaptive over time, and compensatory mechanisms are eventually exhausted, wherein the heart transitions into failure. It is currently well recognized that the immune system and its inflammatory effector molecules are crucial drivers of adverse remodeling and the progression to heart failure, that is, the cytokine hypothesis [2].
Innate immunity and the cardiomyocyte as an amateur immune cell
The purpose of the immune system is to protect the host against pathogenic infection and promote tissue repair after injury [2]. Subdivided into the innate and adaptive systems, the innate response is rapidly activated as a nonspecific inflammatory reaction to tissue injury, whereas the adaptive system mounts a highly specific response mediated by B-cell and T-cell lymphocytes; both systems are activated in response to chronic cardiac insult [2,11,12]. Initiation of the innate immune response is well recognized to occur through signal transduction by activated Pattern Recognition Receptors (PRRs), and there are two classes of PRRs which are categorized by their cellular location. These receptors recognize a wide assortment of endogenous molecules released by injured or dying cells, collectively known as damage/danger associated molecular patterns (DAMPs). Toll-like receptors (TLRs) belong to the first class of membrane-bound PRRs and represent the most widely studied PRR in heart failure; indeed, TLRs are upregulated in both the failing human and murine hearts [13,14]. The second class of PRRs are cytosolic sensors of DAMPs and include the NOD-like receptor (NLR) [15–17]. Although receptor activation of the various PRRs recruit different signal transduction pathways, they all converge on canonical pro-inflammatory transcription factors such as nuclear factor kappa B (NFκβ), interferon regulatory factor 3 (IRF3), and activator protein 1 (AP-1) [4,18]. The subsequent induction of gene expression is required for expression of a range of cytokine, chemokine, and inflammasome associated proteins. These inflammatory mediators produced by these genes typically act locally to potentiate inflammation through the recruitment and activation of leukocytes at the site of injury. However, in the setting of massive injury or chronic inflammatory conditions such as heart failure, cytokines may be produced at high enough concentrations that they also flood the systemic circulation.
While it is recognized that cardiomyocytes can generate cytokines in response to both mechanical stretch and DAMP-PRR activation, their contribution to cardiac inflammation has been considered limited in comparison to that of professional immune and stromal cells [19–22]. Cardiomyocyte-generated cytokines can act locally in autocrine, paracrine, and juxtacrine manners, and a growing number of recent reports demonstrate the role of cardiomyocytes as initiators of an inflammatory domino effect via crosstalk with their more immunoreactive cardiac-localized neighbors, that is, fibroblasts, endothelial cells, and resident immune cells. Thus, the persistent, low-grade inflammation characteristic of heart failure may be mediated by chronically stressed cardiomyocytes. Targeting inflammatory processes in cardiomyocytes could represent a more direct approach to dampening the cardiac immune response and one that may be applicable across differing models of heart failure.
Cardiomyocytes in ischemic injury
In ischemic injury such as that which occurs in acute myocardial infarction (MI), the rapid onset of massive myocardial cell death and consequent release of intracellular mediators is the most likely mechanism initiating cardiac inflammation and leukocyte recruitment (reviewed in detail by Adamo et al. [2]). The acute inflammatory phase is characterized by increased expression of proinflammatory cytokines and the rapid arrival of circulating neutrophils, which then enhance the subsequent recruitment of monocytes [23,24]. This initial proinflammatory phase is followed by a reparative phase marked by macrophage-mediated clearance of necrotic tissue, production of profibrotic cytokines (i.e. TGF-β and IL-10), collagen scar formation, and angiogenesis; this stage is also marked by the arrival of the adaptive immune lymphocytes, the T cells and B cells [25,26].
Induction of the acute inflammatory stage in MI is known to occur through PRR recognition of multiple DAMPs including extracellular matrix fragments, lectins, nucleotides, calcium binding proteins, and more. However, beyond extensive studies utilizing genetic deletion of toll-like receptors, the exact DAMPs and signal transduction pathways are not yet fully determined. A compelling study by King et al. provided novel insights into the mechanism by which dying cardiomyocytes contribute to the initiation of inflammation in the infarcted myocardium. Self-DNA released from the nucleus and/or mitochondria of dying cardiomyocytes is recognized by the cytosolic PRR, cyclic GMP-AMP synthase (cGAS), and elicits an inflammatory response via signal transduction through the cGAS-STING-IRF3 pathway [27•]. The acute inflammatory response is critical in determining the success of cardiac repair through its influence on the extent and type of infiltrating leukocytes, survival of border zone cardiomyocytes, the size of the infarct, and hence overall adverse remodeling [28].
In addition to death and DAMP release, the cardiomyocyte is highly sensitive to the pleiotropic cytokine TGF-β and may thereby contribute to leukocyte recruitment [29]. Intriguingly, blockade of TGF-β-Smad3 signaling has different effects in cardiac fibroblasts versus myocytes [30]. Whereas blocking the TGF-β signaling pathway in the fibroblasts perturbed post-infarct wound healing, the cardiomyocyte-specific response to the blockade enhanced adverse remodeling, perhaps by interfering with appropriate monocyte and neutrophil recruitment [30]. Neutrophils govern the transition from acute inflammation to the reparative phase by influencing both the recruitment and reparative functions of monocytes [29–32]. Indeed, Horckmans et al. demonstrated that neutrophil depletion adversely affected post-infarct repair and cardiac dysfunction in mice [33]. When tissue injury is appropriately resolved, the reparative cells undergo apoptosis, and the myocardium adapts to maintain normal contraction of a scarred ventricle. Conversely, failure to reach resolution results in para-inflammation that continually exacerbates processes involved in adverse remodeling. Directly targeting cardiomyocyte-specific processes involving inflammation, myocyte death, and cell damage may all represent approaches to fine tuning and DAMPening the pro-inflammatory response in both the acute and reparative phases of MI [34,35].
Cardiomyocytes in non-ischemic injury
As described above, ischemic stress produces extensive cardiomyocyte necrosis, and the consequent release of danger signals leads to recruitment of bone marrow derived myeloid cells, producing an intense, feed-forward inflammatory response. Marked cardiac inflammation is also observed, however, in the non-ischemic, pressure-overloaded myocardium despite the absence of detectable cardiomyocyte necrosis. As such, research models of non-ischemic stress have been used to identify the connection between hemodynamic stress and leukocyte mobilization. These interventions include both chemical and surgical induction of left ventricular (LV) pressure overload, primarily represented by infusion of adrenergic agonists or angiotensin and surgical constriction of the transverse aorta (TAC). The distinct temporal stages of inflammation, cardiac remodeling, and heart failure in TAC make it the most frequently employed model in studies examining the effects of inflammation in disease progression of non-ischemic injury. In mice, the acute response to TAC is characterized by an initial decline in ejection fraction (EF) observed up to three days post-surgery, before development of compensatory hypertrophy [36,37]. This is followed by the compensatory stage at approximately one week, a stage represented by concentric hypertrophy and recovery of cardiac function. At approximately 3–4 weeks post-surgery, the heart enters the decompensated stage, and adverse remodeling produces progressive fibrosis, LV chamber dilation, and decline in ejection fraction.
Multiple studies investigating the progression of cardiac remodeling in pressure overload have observed an early decline in EF during the hyperacute phase cited above [36,37]. This is classically thought to result from a sudden increase in afterload that precedes the ability of the heart to compensate via concentric hypertrophy. Cardiac physiology dictates, however, that the myocyte should be able to increase cytosolic calcium concentrations and thereby generate enough contractile force to maintain normal function despite the initial lack of remodeling. Indeed, Baier et al. recently demonstrated that calcium/calmodulin-dependent protein kinase II (CaMKII) plays a role in the instant adaptation to TAC-induced mechanical stress by hyperphosphorylating sarcoplasmic proteins and enhancing calcium transients [37]. Our early and more recent studies showing rapid CaMKII activation in pressure overload and further demonstrate that this kinase mediates NFKβ activation and transcription of proinflammatory genes exclusively in the cardiomyocyte by three days post TAC [10•,38]. Moreover, by using mice with cardiomyocyte-specific deletion of CaMKII, as well as by isolating myocytes from these mice, we showed that the cardiomyocyte accounts for up to 80% of proinflammatory gene transcripts in the whole heart at these early time-points. Thus, before cardiac compensation, the myocyte is generating cytokines that may be meant to contribute to the adaptation to stress but when sustained, become maladaptive mediators contributing to decompensation to heart failure.
Our previous studies presented several lines of evidence indicating that cardiac inflammation is directly initiated by the cardiomyocyte and that this contributes to leukocyte recruitment by a process that can be dissociated from overt myocyte death (Figure 1) [10•,39,40]. In this case, activated CaM kinase II (CaMKII) is the mediator of NFκβ activation and its proinflammatory sequalae. If there is myocyte damage, cytosolic DNA, which is sensed by the cGAS/STING pathway, can result in transcriptional activation of interferon regulatory factors and other chemoattractants important in the recruitment of leukocytes [16]. The cardiomyocyte is a potent source of inflammatory cytokines, and gene deletion of one single cytokine such as the major monocyte chemoattractant protein 1 (MCP1) decreases overall cardiac inflammation and dysfunction in response to TAC [39]. Cardiomyocyte-generated cytokines observed at early times after TAC include not only MCP1 but also CXCL1, IL-18, IL-6, and IL-33, all of which may contribute to the local inflammatory milieu of the heart. Other investigators using the TAC model have shown cytokine immunoreactivity localized to the lining of the vasculature, which also implicates the endothelial cells as a significant source of cytokines preceding immune cell infiltration [19,21,41]. As such, cardiomyocyte-generated cytokines may contribute primarily to initiating an inflammatory domino effect by activating their more immunoreactive neighbors (i.e. resident immune cells, endothelial cells, and fibroblasts). Indeed, the contribution of CaMKII-mediated inflammatory cardiomyocyte signaling that occurs at early times after TAC appears to be most critical for to subsequent leukocyte recruitment and adverse remodeling [10•]. In line with this finding, the Walsh and Prabhu laboratories demonstrated that the progression to heart failure requires the recruitment of neutrophils and monocytes during the early phase of TAC [42 ,43••,44•]. Conversely, antagonizing cardiomyocyte CaMKII signaling at two weeks after TAC or preventing late-stage leukocyte recruitment do not significantly impact in the progression of heart failure [39,42,45]. At this point, once a complement of leukocytes have been recruited to the heart, the role of the cardiomyocyte may shift to that of regulating local inflammatory processes and sustaining inflammation by the juxtacrine/autocrine actions of cardiokines such as MCP1 and IL-1β. Targeting cardiomyocyte-specific inflammation may nonetheless work to prevent the development of the para-inflammatory state and cell death during the later stages of TAC. These studies shed insight on designing therapeutic interventions, the efficacy of which may be dependent on timing and phase of cardiac remodeling.
Figure 1.
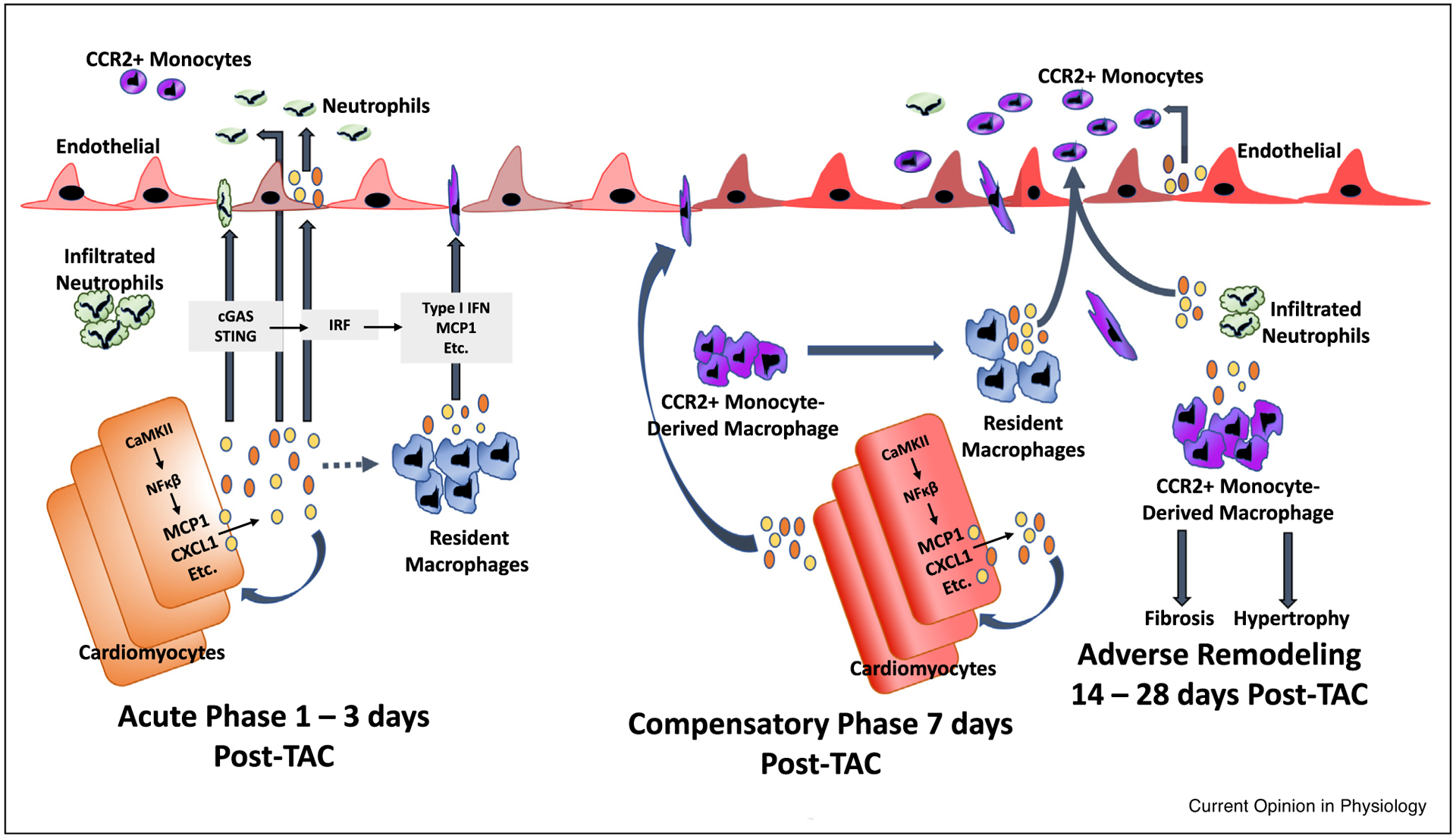
Inflammatory cascade initiated by cardiomyocytes in pressure overload (TAC) induced injury - the role of the cardiomyocyte as a major initiator of inflammation may be of paramount importance in the development of heart failure. During the acute phase (1–3 days post-TAC), cardiomyocytes express cytokines such as MCP1 and CXCL1 in response to CaMKII-mediated NFkB activation. Likewise, the presence of cytosolic DNA may activate the cGAS/STING pathway to induce type I interferon expression [16]. Cardiomyocyte-generated chemokines and other cytokines may be released into the circulation to mediate, feedback in a juxtacrine or autocrine manner, recruitment of monocytes and neutrophils, and locally activate tissue resident macrophages and endothelial cells [52]. Bajpai et al. demonstrated that distinct subsets of tissue-resident cardiac macrophages are crucial in the extravasation of bone-marrow derived neutrophils and monocytes after injury [46••,53••]. At approximately seven days post-TAC, cardiomyocyte-specific inflammatory processes mediate a significant influx of CCR2+ monocyte-derived macrophages into the myocardium [9]. The progression to adverse remodeling and heart failure, characterized by excess fibrosis and hypertrophy, requires infiltration of CCR2+ monocyte-derived macrophages, which replace the resident macrophage population; these results validate the widespread appreciation for the role of CCR2+ monocytes in TAC induced heart failure [43••]. TAC-induced adverse remodeling and heart failure has also been shown by Wang et al. to require neutrophil infiltration, and neutrophil depletion reduces the absolute number of recruited monocytes [44•]. Since infiltrated neutrophils appear earlier than recruited CCR2+ monocytes, it is possible that resident macrophages and infiltrated neutrophils synergistically recruit circulating CCR2+ monocyte-derived macrophages; disruption of any of these processes prevents downstream responses that lead to adverse remodeling.
Conclusion
Cardiac insults invoke an innate host response to protect against invaders and resolve tissue injury. Advances in molecular techniques such as single-cell RNA sequencing and lineage tracing capabilities, complemented by extensive recent research in this area, reveal intricacies of cell types and cell heterogeneity involved in cardiac remodeling that are far more complex than initially envisioned [11,25,27•,28,43••,46••,47]. There is compelling evidence that the cardiomyocyte is an initiator of cardiac inflammation in non-ischemic injury and may set the course of adverse remodeling and the progression to heart failure, in large part by coordinating leukocyte recruitment. Because of the limits of this review, the role of lymphocytes in cardiac repair was only briefly mentioned, but investigators have identified diverse subsets of resident dendritic and mast cells, T cells and B cells that significantly coordinate cardiac healing and fibrosis [3,12,26,48]. The convergence of complex cellular crosstalk synchronizes leukocyte recruitment and function in a spatiotemporal manner. The multifaceted coordination of leukocyte heterogeneity combined with the pleiotropic nature of most cytokines may explain the difficult nature of targeting inflammation after the onset of heart failure. The cardiomyocyte is arguably a more direct target for treating acute and sustained cardiac inflammation and breaking the vicious cycle between para-inflammation and tissue injury. A better understanding of the processes that occur in the myocyte and its milieu could enable specific targeting of the inflammatory process in cardiomyocytes and represents a viable option to reduce cytokine production and inflammation in alleviating chronic heart failure [49–51].
Acknowledgements
This work was supported by NIH grants 5R01HL145459 to JHB and SM. VKN was supported by NIH T32 training grant to RSR and JC.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as
• of special interest
•• of outstanding interest
- 1.Elster SK, Braunwald E, Wood HF: A study of C-reactive protein in the serum of patients with congestive heart failure. Am Heart J 1956, 51:533–541. [DOI] [PubMed] [Google Scholar]
- 2.Adamo L, Rocha-Resende C, Prabhu SD, Mann DL: Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 2020, 17:269–285. [DOI] [PubMed] [Google Scholar]
- 3.Swirski FK, Nahrendorf M: Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol 2018, 18:733–744. [DOI] [PubMed] [Google Scholar]
- 4.Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M et al. : The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail 2018, 20:445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T et al. : Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019, 139:1289–1299. [DOI] [PubMed] [Google Scholar]
- 6.Abbate A, Trankle CR, Buckley LF, Lipinski MJ, Appleton D, Kadariya D et al. : Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J Am Heart Assoc 2020, 9:e014941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodriguez-Nuevo A, Zorzano A: The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Lavine KJ, Epelman S, Evans SA, Weinheimer CJ, Barger PM et al. : Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J Am Heart Assoc 2015, 4:e001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toth AD, Schell R, Levay M, Vettel C, Theis P, Haslinger C et al. : Inflammation leads through PGE/EP3 signaling to HDAC5/MEF2-dependent transcription in cardiac myocytes.. EMBO Mol Med 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.•.Suetomi T, Miyamoto S, Brown JH: Inflammation in nonischemic heart disease: initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am J Physiol Heart Circul Physiol 2019, 317: H877–H890. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a model of surgically induced pressure overload (TAC) in cardiomyocyte-specific CaMKII knockout mice, the authors demonstrate the role of the myocyte in initiating cardiac inflammation. This is the first study that implicates the cardiomyocyte as a key player in generating inflammatory mediators and in activation of the NLRP3 inflammasome, steps involved in leukocyte recruitment and adverse remodeling.
- 11.Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T et al. : Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 2019, 139:206–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carrillo-Salinas FJ, Ngwenyama N, Anastasiou M, Kaur K, Alcaide P: Heart inflammation: immune cell roles and roads to the heart. Am J Pathol 2019, 189:1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu FY, Fan D, Yang Z, Tang N, Guo Z, Ma SQ et al. : TLR9 is essential for HMGB1-mediated post-myocardial infarction tissue repair through affecting apoptosis, cardiac healing, and angiogenesis.. Cell Death Dis 2019, 10:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rotter Sopasakis V, Sandstedt J, Johansson M, Lundqvist A, Bergstrom G, Jeppsson A et al. : Toll-like receptor-mediated inflammation markers are strongly induced in heart tissue in patients with cardiac disease under both ischemic and non-ischemic conditions. Int J Cardiol 2019, 293:238–247. [DOI] [PubMed] [Google Scholar]
- 15.Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X et al. : NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol 2020:101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu D, Cui YX, Wu MY, Li L, Su LN, Lian Z et al. : Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Heart Circul Phhysiol 2020, 318:H1525–H1537. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi T, Shishido T, Kinoshita D, Watanabe K, Toshima T, Sugai T et al. : Cardiac nuclear high-mobility group Box 1 ameliorates pathological cardiac hypertrophy by inhibiting DNA damage response. JACC Basic Transl Sci 2019, 4:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palomer X, Roman-Azcona MS, Pizarro-Delgado J, Planavila A, Villarroya F, Valenzuela-Alcaraz B et al. : SIRT3-mediated inhibition of FOS through histone H3 deacetylation prevents cardiac fibrosis and inflammation. Signal Transduct Targeted Ther 2020, 5:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghigo A, Franco I, Morello F, Hirsch E: Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc Res 2014, 102:270–280. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB et al. : IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J 2018, 39:60–69. [DOI] [PubMed] [Google Scholar]
- 21.Aoyagi T, Matsui T: The cardiomyocyte as a source of cytokines in cardiac injury. J Cell Sci Ther 2011, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Yang X, Jiang G, Yu Y, Wu J, Su Y et al. : HMGB1 enhances mechanical stress-induced cardiomyocyte hypertrophy in vitro via the RAGE/ERK1/2 signaling pathway. Int J Mol Med 2019, 44:885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marinkovic G, Koenis D, de Camp L, Jablonowski R, Graber N, de Waard V et al. : S100A9 links inflammation and repair in myocardial infarction. Circul Res 2020. [DOI] [PubMed] [Google Scholar]
- 24.Prame Kumar K, Nicholls AJ, Wong CHY: Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res 2018, 371:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanton RM, Carrillo-Salinas FJ, Alcaide P: T-cell recruitment to the heart: friendly guests or unwelcome visitors? Am J Physiol Heart Circul Physiol 2019, 317:H124–H140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adamo L, Staloch LJ, Rocha-Resende C, Matkovich SJ, Jiang W, Bajpai G et al. : Modulation of subsets of cardiac B lymphocytes improves cardiac function after acute injury. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.•.King KR, Aguirre AD, Ye YX, Sun Y, Roh JD, Ng RP Jr et al. : IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 2017, 23:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]; Although it is widely accepted that monocyte-derived macrophages infiltrate the myocardium after myocardial infarction, the signals that regulate their recruitment and activation are less clear. King et al. demonstrate that self-DNA released from dying cardiomyocytes governs the responses of a Type I interferon-responsive macrophage subset found in the murine heart after MI.
- 28.Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA et al. : An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanna A, Frangogiannis NG: The role of the TGF-beta superfamily in myocardial infarction. Front Cardiovasc Med 2019, 6:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A et al. : Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation 2018, 137:707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferraro B, Leoni G, Hinkel R, Ormanns S, Paulin N, Ortega-Gomez A et al. : Pro-angiogenic macrophage phenotype to promote myocardial repair. J Am Coll Cardiol 2019, 73:2990–3002. [DOI] [PubMed] [Google Scholar]
- 32.Marwick JA, Mills R, Kay O, Michail K, Stephen J, Rossi AG et al. : Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-kappaB activation.. Cell Death Dis 2018, 9:665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M et al. : Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 2017, 38:187–197. [DOI] [PubMed] [Google Scholar]
- 34.Leoni G, Soehnlein O: (Re) solving repair after myocardial infarction. Front Pharmacol 2018, 9:1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA et al. : Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 2018, 186:73–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glasenapp A, Derlin K, Wang Y, Bankstahl M, Meier M, Wollert KC et al. : Multimodality imaging of inflammation and ventricular remodeling in pressure-overload heart failure. J Nucl Med 2020, 61:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baier MJ, Klatt S, Hammer KP, Maier LS, Rokita AG: Ca(2+)/calmodulin-dependent protein kinase II is essential in hyperacute pressure overload. J Mol Cell Cardiol 2020, 138:212–221. [DOI] [PubMed] [Google Scholar]
- 38.Zhang T, Miyamoto S, Brown JH: Cardiomyocyte calcium and calcium/calmodulin-dependent protein kinase II: friends or foes? Recent Progr Hormone Res 2004, 59:141–168. [DOI] [PubMed] [Google Scholar]
- 39.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S et al. : Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca(2+)/calmodulin-dependent protein kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018, 138:2530–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J: CaMKIIdelta-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen WY, Hong J, Gannon J, Kakkar R, Lee RT: Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc Natl Acad Sci U S A 2015, 112:7249–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel B, Ismahil MA, Hamid T, Bansal SS, Prabhu SD: Mononuclear phagocytes are dispensable for cardiac remodeling in established pressure-overload heart failure. PLoS One 2017, 12:e0170781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.••.Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M et al. : CCR2(+) monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci 2018, 3:230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]; The Prabhu lab previously demonstrated that monocytes were unnecessary for the progression to heart failure once adverse remodeling was established in response to TAC. In this follow up study, the authors show that TAC-induced heart failure actually does require the initial infiltration of CCR2+ monocyte-derived macrophages which occurs at early times.
- 44.•.Wang Y, Sano S, Oshima K, Sano M, Watanabe Y, Katanasaka Y et al. : Wnt5a-mediated neutrophil recruitment has an obligatory role in pressure overload-induced cardiac dysfunction. Circulation 2019, 140:487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]; The key role of monocytes in TAC-induced heart failure have been widely accepted and appreciated. In this study Wang et al. were amongst the first to demonstrate a key role for neutrophils and Wnt5 signaling in adverse remodeling of the pressure overloaded mouse myocardium.
- 45.Mohamed BA, Elkenani M, Jakubiczka-Smorag J, Buchholz E, Koszewa S, Lbik D et al. : Genetic deletion of calcium/ calmodulin-dependent protein kinase type II delta does not mitigate adverse myocardial remodeling in volume-overloaded hearts. Sci Rep 2019, 9:9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.••.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I et al. : Tissue Resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circul Res 2019, 124:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]; These studies by Bajpai et al. reveal that both mice and human hearts contain resident CCR2− and CCR2+ macrophage subsets that are both ontologically and functionally distinct. After cardiac injury, CCR2+ resident macrophages participate in the recruitment of CCR2+ monocytes from the periphery.
- 47.Martini E, Kunderfranco P, Peano C, Carullo P, Cremonesi M, Schorn T et al. : Single-cell sequencing of mouse heart immune infiltrate in pressure overload-driven heart failure reveals extent of immune activation. Circulation 2019, 140:2089–2107. [DOI] [PubMed] [Google Scholar]
- 48.Ngwenyama N, Salvador AM, Velazquez F, Nevers T, Levy A, Aronovitz M et al. : CXCR3 regulates CD4+ T cell cardiotropism in pressure overload-induced cardiac dysfunction.. JCI Insight 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Omiya S, Omori Y, Taneike M, Murakawa T, Ito J, Tanada Y et al. : Cytokine mRNA degradation in cardiomyocytes restrains sterile inflammation in pressure-overloaded hearts. Circulation 2020, 141:667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mentkowski KI, Lang JK: Exosomes engineered to express a cardiomyocyte binding peptide demonstrate improved cardiac retention in vivo. Sci Rep 2019, 9:10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Almeida Paiva R, Martins-Marques T, Jesus K, Ribeiro-Rodrigues T, Zuzarte M, Silva A et al. : Ischaemia alters the effects of cardiomyocyte-derived extracellular vesicles on macrophage activation. J Cell Mol Med 2019, 23:1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation Immunity, 40 (2014), pp. 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.••.Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M et al. : The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 2018, 24:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]; These studies by Bajpai et al. reveal that both mice and human hearts contain resident CCR2− and CCR2+ macrophage subsets that are both ontologically and functionally distinct. After cardiac injury, CCR2+ resident macrophages participate in the recruitment of CCR2+ monocytes from the periphery.