

Abstract
Human artificial chromosomes (HACs) are gene delivery vectors that have been used for decades for gene functional studies. HACs have several advantages over viral‐based gene transfer systems, including stable episomal maintenance in a single copy in the cell and the ability to carry up to megabase‐sized genomic DNA segments. We have previously developed the alphoidtetO‐HAC, which has a single gene acceptor loxP site that allows insertion of an individual gene of interest using Chinese hamster ovary (CHO) hybrid cells. The HAC, along with a DNA segment of interest, can then be transferred from donor CHO cells to various recipient cells of interest via microcell‐mediated chromosome transfer (MMCT). Here, we detail a protocol for loading multiple genomic DNA segments or genes into the alphoidtetO‐HAC vector using an iterative integration system (IIS) that utilizes recombinases Cre, ΦC31, and ΦBT. This IIS‐alphoidtetO‐HAC can be used for either serially assembling genomic loci or fragments of a large gene, or for inserting multiple genes into the same artificial chromosome. The insertions are executed iteratively, whereby each round results in the insertion of a new DNA segment of interest. This is accompanied by changes of expression of marker fluorescent proteins, which simplifies screening of correct clones, and changes of selection and counterselection markers, which constitutes an error‐proofing mechanism that removes mis‐incorporated DNA segments. In addition, the IIS‐alphoidtetO‐HAC carrying the genes can be eliminated from the cells, offering the possibility to compare the phenotypes of human cells with and without functional copies of the genes of interest. The resulting HAC molecules may be used to investigate biomedically relevant pathways or the regulation of multiple genes, and to potentially engineer synthetic chromosomes with a specific set of genes of interest. The IIS‐alphoidtetO‐HAC system is expected to be beneficial in creating multiple‐gene humanized models with the purpose of understanding complex multi‐gene genetic disorders. Published 2021. This article is a U.S. Government work and is in the public domain in the USA. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: Integration of the first DNA segment of interest into the IIS‐alphoidteto‐HAC
Basic Protocol 2: Integration of a second DNA segment of interest into the IIS‐alphoidteto‐HAC
Basic Protocol 3: Integration of a third DNA segment of interest into the IIS‐alphoidteto‐HAC
Support Protocol: Fluorescence in situ hybridization analysis for the circular IIS‐alphoidtetO‐HAC
Keywords: DNA assembly, gene delivery, HAC, human artificial chromosome, IIS, iterative integration system
INTRODUCTION
Human artificial chromosomes (HACs) are vectors for gene delivery and expression in target cells. HACs are highly stable and behave as an extra chromosome in host cells, and are maintained independently from the host cell genome. Because HACs can carry megabase (Mb)‐sized DNA segments, they have been successfully used for gene expression studies, the development of animal models of human diseases and cell re‐programming, and have potential for use in gene therapy (Hiratsuka et al., 2011; Ikeno, & Hasegawa, 2020; Katona, 2015; Kouprina, Earnshaw, Masumoto, & Larionov, 2013; Kouprina, Tomilin, Masumoto, Earnshaw, & Larionov, 2014; Moralli, & Monaco, 2020; Moriwaki, Abe, Oshimura, & Kazuki, 2020; Oshimura, Uno, Kazuki, Katoh, & Inoue, 2015; Sinenko, Ponomartsev, & Tomilin, 2021).
HACs are constructed either by a “top down” or “bottom up” approach. The “top down” HACs are engineered from natural chromosomes by telomere‐directed truncation of the p‐ and q‐arms using telomere‐containing vectors, which leads to the replacement of natural telomeres by synthetic telomere repeats (Farr et al., 1995; Heller, Brown, Burgtorf, & Brown, 1996). The “bottom up” HACs, on the other hand, are de novo artificial chromosomes generated from bacterial artificial chromosomes (BACs) carrying high‐order centromeric DNA repeats (HORs; Harrington, Van Bokkelen, Mays, Gustashaw, & Willard, 1997; Ikeno et al., 1998). After transfection into human cells, BAC DNA undergoes multimerization (20 to 30 fold) and a functional kinetochore is assembled, leading to HAC formation. Such HACs have a circular structure.
A decade ago, we devised the “bottom up” alphoidtetO‐HAC from a synthetic alphoid DNA array (Nakano et al., 2008). The array consists of a 343‐bp synthetic dimer unit, amplified by rolling circle amplification (RCA) and then by transformation‐associated recombination (TAR) in yeast (Ebersole et al., 2005) to up to 50 kb in size. The array was then multimerized up to 1.1 Mb after transfection into human HT1080 cells, leading to alphoidtetO‐HAC formation (Fig. 1A and 1B; Kouprina et al., 2012; Nakano et al., 2008). Into each dimer ∼3,000 copies of the 42‐bp tetracycline operator (tetO) sequence, the binding site for Escherichia coli tetracycline repressor (tetR), are incorporated in place of the CENP‐B box, which can then be targeted specifically with tetR‐fusion proteins. Targeting of the alphoidtetO array with specific tetR fusion proteins disturbs kinetochore function, leading to HAC loss (Fig. 1C). Such a unique feature of the alphoidtetO‐HAC, i.e., to be eliminated from the cells along with the loaded gene, gives researchers the possibility to compare the phenotypes of human cells with and without a gene of interest (Kim et al., 2011; Kononenko et al., 2014; Kouprina et al., 2018). This provides a proper interpretation of gene complementation analysis and a control for phenotypic changes attributed to expression of HAC‐encoded genes. The original alphoidtetO‐HAC‐based gene delivery vector contains a single gene loading loxP site for the site‐specific integration of one segment of exogenous DNA (Iida et al., 2010). The HAC is fitted with a Cre‐loxP hypoxanthine phosphoribosyl transferase (HPRT) reconstitution system. While this system was quite successful at creating transgenic cells for gene functional studies (Kim et al., 2011; Kononenko et al., 2014; Ponomartsev et al., 2020), it has some limitations. For instance, this system can be used for integrating only a single genomic DNA segment into the HAC molecule at time (Current Protocols article: Liskovykh, Larionov, & Kouprina, 2021).
Figure 1.
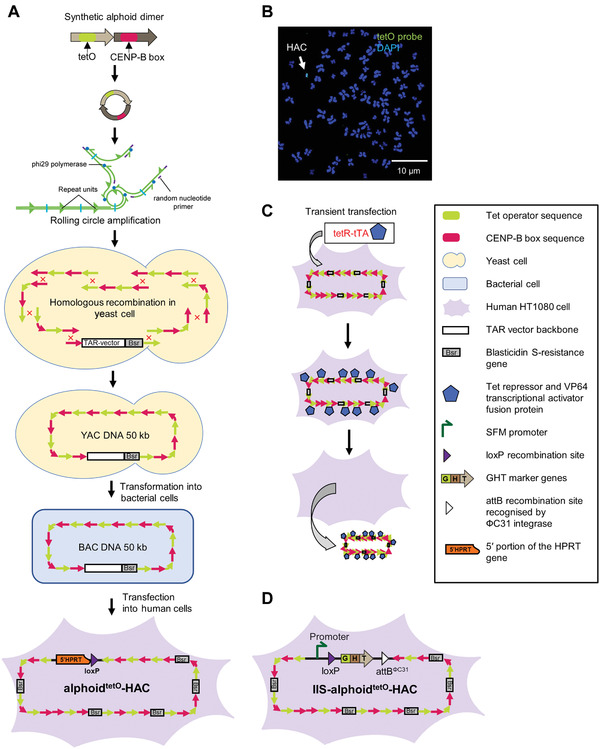
Generation of the alphoidtetO‐HAC using a synthetic alphoid DNA array. (A) A 343‐bp synthetic alphoid dimer consists of two monomers. One monomer is derived from a chromosome 17 alphoid type I 16‐mer unit and contains a CENP‐B box, a nucleotide motif involved in centromere formation. The second monomer is a wholly synthetic sequence derived from alphoid DNA consensus with the sequence corresponding to the CENP‐B box replaced by a 42‐bp tetO motif. A dimer is amplified up to ∼3‐5 kb in size by rolling circle amplification (RCA) in vitro using phi29 DNA polymerase. Then the RCA‐amplified fragments are assembled by transformation‐associated recombination (TAR) cloning in yeast, leading to formation of an ∼50 kb synthetic array cloned into a YAC/BAC vector. A hybrid circular YAC/BAC vector contains a blasticidin resistance marker (a bsr gene). The YAC/BAC molecules are then moved from yeast to bacterial cells for further BAC DNA isolation. After transfection of 50 kb input BAC DNA into human HT1080 cells, the alphoidtetO‐HAC is formed. Formation of the alphoidtetO‐HAC is accompanied by multimerization of input 50 kb DNA up to 1.1 Mb. (B) FISH analysis of the alphoidtetO‐HAC in human HT1080 cells. FISH analysis was performed using a fluorescein peptide nucleic acid (PNA)‐labeled probe for the tetO sequence (see Support Protocol). A white arrow indicates the HAC (green), while the endogenous chromosomes are labeled blue (DAPI). (C) Loss of the alphoidtetO‐HAC from recipient cells may be induced by the transcriptional activator (tTA) fused with the tet‐repressor (tetR) targeting the tetO‐HAC kinetochore (Kim et al., 2011; Kononenko et al., 2014). (D) The IIS‐alphoidtetO‐HAC was developed after insertion of the integration platform cassette into the alphoidtetO‐HAC. The integration platform cassette consists of the SFM promoter driving the expression of the GHT marker, a loxP site present between the promoter and the marker, and the attBΦC31 site for the ΦC31 integrase. The GHT marker is a fusion of enhanced green fluorescent protein (eGFP), hygromycin‐B‐phosphotransferase (hph), and thymidine kinase (TK). Abbreviations: BAC, bacterial artificial chromosome; YAC, yeast artificial chromosome; HAC, human artificial chromosome; FISH, fluorescence in situ hybridization; SFM, SV40 enhancer plus ferritin; GHT, eGFP‐hph‐TK.
The assembly of multiple genes on the same HAC molecule, and the subsequent transfer of this vector into desired recipient cells, could have multiple applications in functional genomics. Therefore, construction of an alphoidtetO‐HAC vector containing multi‐integration sites, allowing insertion of an unlimited number of genomic DNA fragments or genes, was our next goal. For this, we built the iterative integration system (IIS), which utilizes three recombinases, Cre, ΦC31, and ΦBT1, and combined it with the alphoidtetO‐HAC (Lee et al., 2018). This IIS‐alphoidtetO‐HAC carries an integration platform cassette consisting of the SV40 enhancer plus ferritin (SFM) promoter driving the expression of the eGFP‐hph‐TK (GHT) marker, a loxP site present between the promoter and the marker, and the attBΦC31 site for the ΦC31 integrase (Fig. 1D). The GHT marker is a fusion protein composed of a mutually exclusive selection marker, a mutually exclusive counterselection marker, and a mutually exclusive fluorescent marker. The GHT marker is a fusion of enhanced green fluorescent protein (eGFP), hygromycin‐B‐phosphotransferase (hph), and thymidine kinase (TK). Such an IIS‐alphoidtetO‐HAC can be used to either serially assemble large Mb‐sized genomic loci or genes from multiple smaller manageable segments of a very large gene or to serially insert multiple genes into the same HAC molecule. Such HAC molecules may be used for investigating different biomedically relevant pathways or the regulation of multiple genes. The IIS‐alphoidtetO‐HAC system may be used to create multiple‐gene humanized models and has potential for gene therapy for polygenic diseases (Fig. 2).
Figure 2.
Multiple applications of the IIS‐alphoidtetO‐HAC system.
The iterative integration system is illustrated in Figure 3. It is a cyclic system, where two markers, GHT and PCF, substitute each other as a new genomic DNA segment is added to the integration sites. The PAC‐mCherry‐FcyFur (PCF) marker is a fusion protein composed of a mutually exclusive selection marker, a mutually exclusive counterselection marker, and a mutually exclusive fluorescent marker. The PCF marker is a fusion of puromycin‐N‐acetyltransferase (Puro), a red fluorescent protein (mCherry), and cytosine deaminase‐uracil phosphoribosyl transferase (FcyFur). The PCF marker makes the cells appear red upon fluorescence microscopy, and resistant to puromycin and sensitive to 5‐fluorocytosine. The GHT marker, on the other hand, makes the cells appear green, and resistant to hygromycin B and sensitive to ganciclovir. A 2A self‐cleaving peptide sequence is placed between each element. As a consequence, these three proteins are transcribed as a single mRNA but are produced as separate proteins. As mentioned, the IIS‐alphoidtetO‐HAC system uses three recombinase enzymes, i.e., Cre, ΦC31, and ΦBT1. Cre is a bidirectional enzyme that catalyzes the recombination between two substrate loxP sites and generates two product loxP sites. Recombinases ΦC31 and ΦBT1 are unidirectional enzymes that recombine an attachment bacteria (attB) site and an attachment phage (attP) site to produce attR and attL sites that are not substrates for further reaction.
Figure 3.
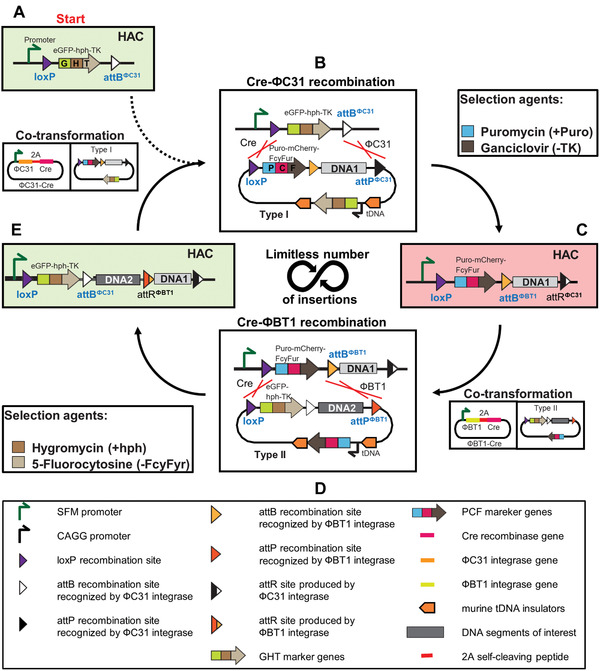
Schematic of the perpetual serial integration of DNA segments into the IIS‐alphoidtetO‐HAC by the iterative integration system (IIS). (A) The starting integration platform cassette on the human artificial chromosome (HAC). In the empty platform cassette, the SV40 enhancer plus ferritin (SFM) promoter drives the expression of the eGFP‐hph‐TK (GHT) marker. The cells express enhanced green fluorescence protein (eGFP; cells look green), and are hygromycin resistant (hph) and ganciclovir sensitive (TK). (B) Type I carrier vector bearing the first DNA segment of interest (DNA1) is integrated into the platform cassette of the HAC by Cre recombinase and ΦC31 integrase, which are themselves expressed from the plasmid A139. Recombination between a Type I carrier vector and a platform cassette by Cre recombinase and ΦC31 integrase leads to replacement of the GHT marker by the Puro‐mCherry‐FcyFur (PCF) marker and integration of the DNA of interest (DNA1) into the platform cassette. Cells with correct integration are selected for using puromycin and ganciclovir. (C) Structure of the platform cassette after the first round of DNA integration. The PCF marker is expressed. Therefore, the cells express red fluorescence (mCherry; cells now look red) and are puromycin (Puro) resistant and 5‐fluorocytosine (FcyFur) sensitive. (D) Recombination between a Type II carrier vector bearing the second DNA segment of interest (DNA2) and a platform cassette by Cre recombinase and ΦBT1 integrase, which are expressed from the plasmid A135‐JH, leads to replacement of the PCF marker by the GHT marker and DNA2 integration into the platform cassette. The integration event is selected for using hygromycin and 5‐fluorocytosine. (E) Structure of the platform cassette after the second round of DNA integration. The cells express the GHT marker and, thus, the green florescence protein eGFP (cells look green). They once again become hygromycin resistant (hph) and ganciclovir sensitive (TK). This structure is identical to the starting cassette aside from the integration of DNA segments of interest, DNA1 and DNA2.
The IIS‐alphoidtetO‐HAC system starts with the HAC carrying the integration platform cassette. The HAC is propagated in donor Chinese hamster ovary (CHO) cells (Fig. 3A). The cells express eGFP. To insert the first genomic DNA segment of interest (hereafter referred to as DNA1) into the HAC, the cells are co‐transformed with two plasmids, i.e., the A139 plasmid that expresses ΦC31 integrase and Cre recombinase (Fig. 4A) and the Type I carrier vector A167 (Fig. 4B) that contains the PCF marker and carries DNA1 (Fig. 3A). Expression of ΦC31 and Cre causes two recombination events, loxP‐loxP and attBΦC31‐attPΦC31, correspondingly, between the Type I carrier vector and the integration platform cassette of the HAC (Fig. 3B). The recombination reaction removes the GHT marker from the SFM promoter and replaces it with the PCF marker, loxP, and attBΦBT1 sites, and the DNA1 segment from the Type I carrier vector A167, while deleting all other vector components (Fig. 3C). The promoter within the platform cassette now drives the PCF marker. The cells that successfully completed both recombination reactions lose eGFP and sensitivity to ganciclovir, and gain red fluorescence (mCherry), resistance to puromycin, and sensitivity to 5‐fluorocytosine.
Figure 4.
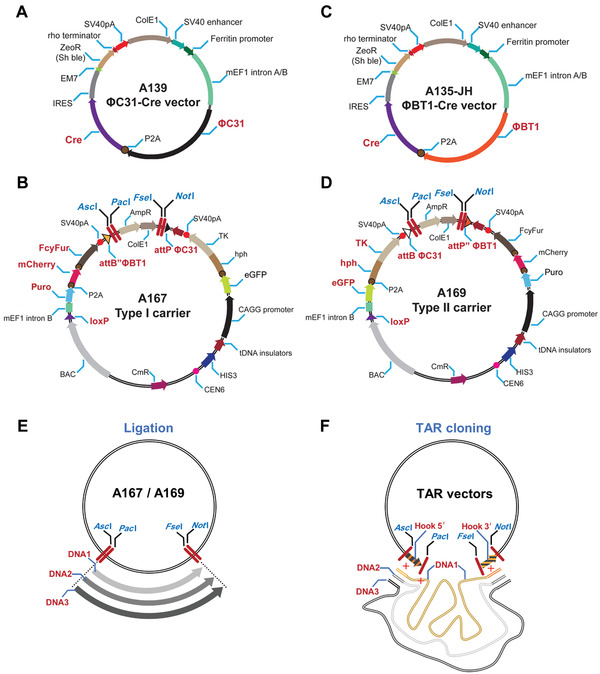
Scheme of the vectors used in the IIS‐alphoidtetO‐HAC system. (A) A139 vector expressing ΦC31 integrase and Cre recombinase and (B) Type I carrier vector A167 to deliver the first genomic DNA fragment (DNA1) and to perform every odd‐numbered round of DNA integration. A167 vector has a promoterless Puro‐mCherry‐FcyFur (PCF) marker and a constitutively active eGFP‐hph‐TK (GHT) marker under the CAGG promoter in its vector backbone. (C) A135‐JH vector expressing ΦBT1 integrase and Cre recombinase and (D) Type II carrier vector A169 to deliver the second genomic DNA fragment (DNA2) and to perform every even‐numbered round of DNA integration. A169 vector has a promoterless GHT marker and a constitutively active PCF marker under the CAGG promoter in its vector backbone. A139 and A135‐JH expression vectors carry zeomycin resistance (ZeoR). Both Type I and Type II carrier vectors are used to integrate DNA segments of interest into the platform cassette of the IIS‐alphoidtetO‐HAC. These vectors contain both a BAC cassette containing F′ origin of replication (low‐copy maintenance) and a pBR322 origin of replication (ColE1) to make the vectors multicopy. ColE1 origin is removed once a large DNA segment of interest (DNA1, DNA2, or DNAn) is added to the vector. (E) DNA fragments are inserted into Type I and Type II carrier vectors A167 and A169 via ligation into unique 8‐bp restriction sites, i.e., AscI/PacI/FseI/NotI (marked in blue). (F) DNA fragments are inserted into Type I and Type II carrier vectors A167 and A169 by TAR cloning in yeast S. cerevisiae (Kouprina et al., 2021). In this case, AscI/PacI and FseI/NotI sites are used to insert the hook sequences homologous to the 5′ and 3′ ends of DNA segments of interest. For TAR isolation of DNA segments of interest from total genomic DNA, A167 and A169 vectors contain CEN6 (a yeast centromere sequence) and HIS3 (a yeast selectable marker) for proper propagation and selection of the TAR‐cloned material in yeast. A BAC cassette allows direct transfer of the TAR‐cloned DNA material from yeast to bacterial cells for further BAC DNA isolation. Abbreviations: BAC, bacterial artificial chromosome; TAR, transformation‐associated recombination.
To insert a second DNA fragment of interest (DNA2) into the HAC, the cells are co‐transformed with the A135‐JH vector expressing ΦBT1 integrase and Cre recombinase (Fig. 4C), and the Type II carrier vector A169 (Fig. 4D), which contains DNA2 (Fig. 3C). Cre and ΦBT1 expression causes two recombination events, loxP‐loxP and attBΦBT1‐attPΦBT1, respectively, between the Type II vector and the platform cassette (Fig. 3D). This leads to the replacement of the PCF marker by the GHT marker and the attBΦBT1 site, followed by the insertion of DNA2 from the Type II carrier vector. As a result, the platform cassette in the HAC will now contain a loxP site, an expressed GHT marker (eGFP), and an attBΦBT1 site (Fig. 3E). Selection with hygromycin B and counterselection with 5‐fluorocytosine ensures that only cells that have correctly undergone the second round of assembly will survive. Untransformed parental cells and cells with incomplete recombination are killed by this double selection.
After two rounds of recombination, the integration platform cassette is once again where it started, except that two genomic DNA segments of interest (DNA1 and DNA2) have now been integrated into the HAC (Fig. 3E). The GHT marker is expressed, and the cells once again express eGFP, and are resistant to hygromycin B and sensitive to ganciclovir. Further rounds of DNA fragment insertions can be repeated indefinitely as required (DNA3, DNA4,…DNAn). The final IIS‐alphoidtetO‐HAC carrying the required number of genomic DNA fragments can then be successfully moved from hamster donor CHO cells to different recipient cells by microcell‐mediated chromosome transfer (MMCT; Current Protocols article: Liskovykh et al., 2021; Liskovykh, Lee, Larionov, & Kouprina, 2016).
In this article, we describe three basic protocols (Fig. 5). In Basic Protocol 1, the user will integrate the Type I carrier vector A167, carrying the first genomic DNA fragment of interest (DNA1), into the IIS‐alphoidtetO‐HAC. In Basic Protocol 2, the user will integrate the Type II carrier vector A169, carrying a second genomic DNA fragment (DNA2), into the IIS‐alphoidtetO‐HAC. In Basic Protocol 3, the user will integrate the Type I carrier vector A167, carrying a third genomic DNA fragment (DNA3), into the IIS‐alphoidtetO‐HAC. The recombinant assay vectors will be transfected into hamster donor CHO cells containing the IIS‐alphoidtetO‐HAC, using a combination of either A167 plus A139 (ΦC31 integrase and Cre recombinase) vectors or A169 plus A135‐JH (ΦBT1 integrase and Cre recombinase) vectors. By following these protocols, the user will be able integrate three genomic DNA fragments into the same IIS‐alphoidtetO‐HAC molecule.
Figure 5.
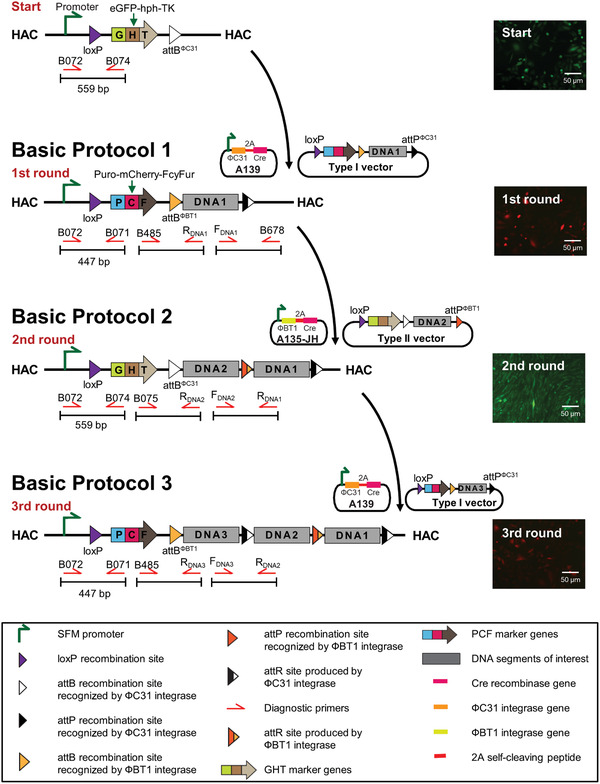
Schematic of serial integration of three DNA fragments into the IIS‐alphoidtetO‐HAC using the Type I carrier vector A167 carrying either DNA1 or DNA3 and the Type II carrier vector A169 carrying DNA2. Starting point: The empty integration platform cassette on the IIS‐alphoidtetO‐HAC includes the SV40 enhancer plus ferritin (SFM) promoter (green arrow to the right) that drives expression of the eGFP‐hph‐TK (GHT) marker containing cells that exhibit green fluorescence, and are hygromycin resistant (hph) and ganciclovir sensitive (TK). Basic Protocol 1: Structure of the platform cassette after the first round of DNA integration. The first round involves integration of the Type I carrier vector A167 carrying DNA1 into the platform cassette by ΦC31 integrase and Cre recombinase. Recombination between the Type I carrier vector A167 and the platform cassette leads to replacement of the GHT marker by the Puro‐mCherry‐FcyFur (PCF) marker and insertion of the first genomic DNA segment (DNA1). After the first round of DNA integration, the PCF marker is expressed. Therefore, the cells exhibit red fluorescence (mCherry), and are puromycin resistant (Puro) and 5‐fluorocytosine sensitive (FcyFur). Basic Protocol 2: Structure of the platform cassette after the second round of DNA integration. The second round involves recombination of the Type II carrier vector A169 carrying DNA2 and the platform cassette by ΦBT1 integrase and Cre recombinase. The PCF marker is replaced by the GHT marker and insertion of the second genomic DNA fragment (DNA2) into the platform cassette. The integration event is selected using hygromycin (hph) and 5‐fluorocytosine (FcyFur). The cells exhibit green fluorescence. Basic Protocol 3: Structure of the platform cassette after the third round of DNA integration. The GHT marker is replaced by the PCF marker and the third genomic DNA fragment (DNA3) is inserted into the platform cassette. The cells exhibit red fluorescence (mCherry) and are once again puromycin resistant (Puro) and 5‐fluorocytosine sensitive (FcyFur).
STRATEGIC PLANNING
Before experiments with the IIS‐alphoidtetO‐HAC system, the user must first determine if the alphoidtetO‐HAC containing the “empty” integration platform cassette can be transferred via the MMCT technique from the donor CHO cells into the recipient cells of interest, as HAC transfer to some cell lines may be challenging. We recommend that the user apply the improved MMCT protocol (Current Protocols article: Liskovykh et al., 2021), which has been found to be efficient for many cell cultures, human immortalized mesenchymal stem cells, pluripotent cells (ES, iPS), and mouse embryonic fibroblast primary cultures.
The iterative integration system described in this article is carried out in hamster CHO cells, but it may be performed directly in the recipient cells of interest as well. If the user desires to conduct the iterative integration of the genomic DNA fragments within the cell line of interest, the user has to confirm that after MMCT transfer of the IIS‐alphoidtetO‐HAC into the cell line of interest, the HAC is maintained in an autonomous form and as a single copy per cell. Such a control experiment can be carried out by fluorescence in situ hybridization (FISH), as previously described (Kim et al., 2011; Kononenko et al., 2014; see also Support Protocol). This experiment is important because some cell lines used in the laboratory are very karyotypically unstable, displaying a wide variation of chromosome number even among cells of the same colony. This large‐scale chromosome instability spills over to the HAC and may lead to multiple copies of the alphoidtetO‐HAC carrying the integration platform cassette, making the iterative integration system unworkable. The karyotype of the cell line can be determined by a standard FISH. If over 80% of the cells are able to maintain the HAC as a single copy, further work can be performed.
The user should remember that the described IIS‐alphoidtetO‐HAC system is a cyclic system where two markers, GHT and PCF, substitute each other as a new genomic DNA segment is added to the integration sites. At each round of DNA integration, only a single copy of a selection/counterselection marker is present in each cell. As such, the development of cell resistance may be slower when a selection marker is changed. In some cell lines, resistance to the selection agents (hygromycin B and puromycin) may be lower than expected. The counterselectable markers (TK and FcyFur) are also more vulnerable to silencing. Therefore, at each round of DNA integration, when a counterselectable marker is changed, it is advisable to apply the counterselection as soon as possible (see details in Critical Parameters, Selection and counterselection agent concentration section).
Before starting the basic protocols, the user should insert genomic DNA fragments of interest into the Type I and Type II carrier vectors A167 and A169. We recommend that the user apply the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9‐mediated TAR cloning approach to isolate the desired DNA fragments (DNA1, DNA2, DNA3,…DNAn; Current Protocols article: Kouprina, Kim, & Larionov, 2021). TAR cloning allows selective and efficient isolation of full‐size genes or chromosomal regions up to 300 kb in size from total genomic DNA as circular YAC/BAC molecules in Saccharomyces cerevisiae (Kouprina, & Larionov, 2008). For these experiments, Type I A167 and Type II A169 carrier vectors may be used to construct TAR vectors (Fig. 4F). The targeting hook sequences homologous to 5′ and 3′ ends of the target genomic regions/genes may be inserted into the AscI/PacI and FseI/NotI sites of A167 and A169 vectors, respectively. Before TAR cloning, the A167 and A169 vectors containing the hooks should be linearized by PacI/FseI to expose the hooks for recombination with the genomic sequences homologous to the hook sequences. Because the A167 and A169 vectors contain YAC [the HIS3 marker and CEN6 (centromere from chromosome 6)] and BAC (F′ origin of replication) cassettes (see Fig. 5B and 5D), TAR‐cloned molecules may be isolated in yeast cells and then moved directly to bacterial cells. In E. coli cells, the BAC molecules are then isolated to provide enough material for transfections into hamster CHO cells carrying the IIS‐alphoidtetO‐HAC gene delivery vector. Alternatively, the DNA fragments of interest may be added to A167 or A169 vectors via ligation into the unique 8‐bp restriction sites (AscI/PacI/FseI/NotI; Fig. 4E). Type I carrier vector A167 delivers the first genomic DNA fragment (DNA1) of interest and is then used for every subsequent odd‐numbered round of DNA integration into the IIS‐alphoidtetO‐HAC. Type II carrier vector A169 delivers the second genomic DNA fragment (DNA2) and is then used for every subsequent even‐numbered round of DNA integration into the IIS‐alphoidtetO‐HAC.
Basic Protocol 1. INTEGRATION OF THE FIRST DNA SEGMENT OF INTEREST INTO THE IIS‐alphoidtetO‐HAC
Here we describe insertion of the Type I carrier vector carrying the first DNA segment of interest (Type I DNA1) into the integration platform cassette of the IIS‐alphoidtetO‐HAC, propagated in hamster CHO cells (Figs. 5 and 6). At the starting point, the cells should express eGFP, and be resistant to hygromycin B and sensitive to ganciclovir. A PCR against the HAC carrying the “empty” integration platform cassette with the diagnostic primers B072/B074 should yield a fragment of 559 bp (Fig. 5).
Figure 6.
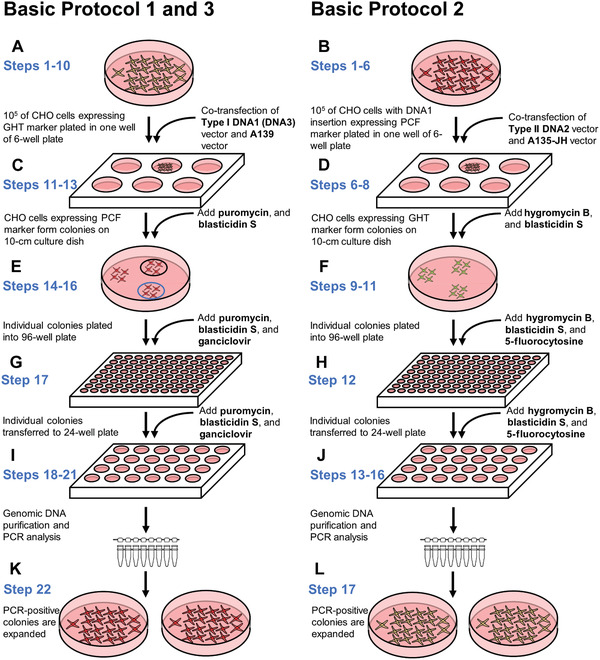
Cell culture steps in the basic protocols. Each round of integration requires similar cell culture procedures. Herein, the steps of the protocols are presented side by side to highlight the key differences between them. The first round starts with hamster CHO cells carrying the IIS‐alphoidtetO‐HAC. Afterwards, any subsequent round starts with the cells obtained during the preceding round. (A and B) The first procedure in each round is co‐transfection of a specific carrier vector carrying a genomic DNA fragment, i.e., Type I DNA1, Type II DNA2, and Type I DNA3, along with either (A) A139 or (B) A135‐JH vector. (C and D) Cells in which correct integration has occurred exhibit a change in the expression of marker genes (eGFP to mCherry or mCherry to eGFP) and can form colonies under selection with either (C) puromycin and blasticidin S or (D) hygromycin B and blasticidin S. (E and F) Individual colonies with proper fluorescence are transferred into a 96‐well plate. Additional counterselection agents, i.e, (E) ganciclovir or (F) 5‐fluorocytosine, are supplemented to remove cells with incorrect integration. (G and H) The colonies are transferred to a 24‐well plate and grown under selection. (I and J) Genomic DNA is purified from individual colonies and PCR‐analyzed to confirm proper integration. (K and L) Colonies with PCR‐confirmed integration of the DNA fragment are transferred to 10‐cm dishes for FISH analysis and for preparation of frozen stocks.
To perform Basic Protocol 1, the user will co‐transfect CHO cells carrying the IIS‐alphoidtetO‐HAC with the Type I carrier vector A167 carrying DNA1 and the A139 vector. Vector A139 expresses ΦC31 integrase and Cre recombinase. Then the user will culture the cells in puromycin/blasticidin S medium. After ∼10 days of selection, the user will pick up the colonies exhibiting red fluorescence (mCherry is expressed) and culture them in puromycin/ganciclovir/blasticidin S medium.
To verify recombination at the loxP site, the user will carry out a PCR reaction with the primer pairs B072/B071 and B074/B072 (Fig. 5). B071 primer corresponds to the PCF marker. B072 corresponds to the SFM promoter sequence. B074 primer corresponds to the GHT marker. After the first and every next odd‐numbered round of integration, a PCR reaction with B072/B071 primers should give a 447‐bp product, while PCR with B074/B072 primers should be negative. Thus the user will confirm the insertion of the PCF marker and elimination of the GHT marker.
To verify recombination between attBФC31 and attPФC31 sites and integration of DNA1 into the IIS‐alphoidtetO‐HAC, the user should choose a forward primer complementary to a 3′‐end region of DNA1 (FDNA1; Fig. 5). B678 primer corresponding to the HAC backbone sequence can be used as a reverse primer (e.g., R3/B678 primers for VHL gene insertion; see Fig. 7A and 7B).
Figure 7.
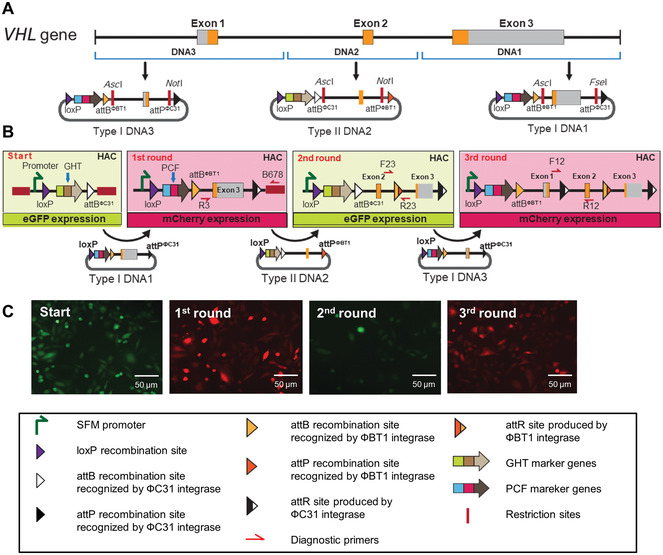
Schematic of VHL gene reconstitution by serial integration into the IIS‐alphoidtetO‐HAC. The VHL gene is located on chromosome 3 (positions 10137959‐10154492; GHCH38/hg38). The gene contains three exons. Mutations in the gene are associated with the Von Hippel‐Lindau (VHL) syndrome, a dominantly inherited hereditary cancer syndrome predisposing individuals to a variety of malignant and benign tumors of the eye, brain, spinal cord, kidney, pancreas, and adrenal glands. (A) Construction of Type I vectors carrying genomic fragments DNA1 and DNA3, encompassing exon 3 and exon 1, respectively, and Type II vector carrying a genomic DNA2 fragment encompassing exon 2 of the VHL gene. DNA1 (exon 3) and DNA3 (exon 1) were inserted into the AscI/NotI sites of the Type I carrier vector A167. DNA2 (exon 2) was inserted into the AscI/NotI sites of the Type II carrier vector A169. (B) Three rounds of insertion of the VHL fragments into the IIS‐alphoidtetO‐HAC carrying the integration platform cassette are shown. Round 1: A diagnostic PCR for DNA1 insertion was performed with primers R3/B678. Round 2: A diagnostic PCR for DNA2 insertion was performed with primers F23/R23. Round 3: A diagnostic PCR for DNA3 insertion was performed with diagnostic primers F12/R12. The position of the primer pairs is shown. (C) Representative images of how cell fluorescence changes after each round of DNA insertion are shown.
To verify the integrity of the newly inserted attBФBT1 site, the user will need to select a reverse primer complementary to a 5′‐end region of DNA1 (RDNA1; Fig. 5). B485, complementary to the PCF sequence, can be used as a forward primer.
Materials
A139 vector expressing ΦC31 integrase and Cre recombinase [available upon request from Developmental Therapeutics Branch, National Cancer Institute (NCI), National Institutes of Health (NIH)]
Type I carrier vector A167 carrying DNA1 (Type I DNA1, see Strategic Planning; concentration of vectors should be 0.1‐0.5 μg/μl; all vectors available upon request from Developmental Therapeutics Branch, NCI, NIH)
Hypoxanthine phosphoribosyl transferase (HPRT)‐deficient Chinese hamster ovary (CHO) cells (JCRB0218) carrying the IIS‐alphoidtetO‐HAC [alphoidtetO‐HAC containing the integration platform cassette, i.e., the promoter and the GHT marker components, and recombinase recognition sites, loxP and attBΦC31; GHT compound marker is composed of a fusion of eGFP, P2A self‐cleaving peptide, hygromycin phosphotransferase (hph), and viral thymidine kinase (TK); available upon request from Developmental Therapeutics Branch, NCI, NIH]
Cell culture freezing medium (CFM; see recipe)
F12 growth medium (see recipe)
F12 round I selection medium (see recipe)
Blasticidin S HCl (10 mg/ml; Thermo Fisher Scientific, cat. no. A1113903)
Ganciclovir solution, 10 mg/ml (see recipe)
Opti‐MEM medium (Thermo Fisher Scientific, cat. no. 51985034)
ViaFect transfection reagent (Promega, cat. no. E4981)
PBS (Thermo Fisher Scientific, cat. no. 10010‐023)
0.25% trypsin (Thermo Fisher Scientific, cat. no. 25200056)
GeneRuler 1 kb plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331)
Nuclease‐free water (Quality Biological, cat. no. 351‐029‐721)
DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504)
TaKaRa Ex Taq® DNA Polymerase (Takara Bio, cat. no. RR001C)
Agarose (MilliporeSigma, cat. no. A9539)
-
Diagnostic primers (to confirm insertion of DNA fragments):
B072: 5′‐CCAGTTGCGTGCGTGGAA‐3′
B071: 5′‐CGCACCGTGGGCTTGTA‐3′
B074: 5′‐GCCGGACACGCTGAACTT‐3′
B485 : 5′‐GTGCAAGAAGATTATGAAGCAG‐3′
RDNA1: Designed by the user
FDNA1: Designed by the user
B678: 5′‐GCCTCTCTCTTTTATGAAGCTTCC‐3′
6‐well culture plates (Thermo Fisher Scientific, cat. no. 140675)
96‐well culture plates (Thermo Fisher Scientific, cat. no. 167008)
24‐well culture plates (Thermo Fisher Scientific, cat. no. 142475)
Cloning cylinders (Thermo Fisher Scientific, cat. no. 09‐552‐20)
Cryovial (Thermo Fisher Scientific, cat. no. 5012‐0012)
1.7‐ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
15‐ml centrifuge tubes (Corning Falcon, cat. no. 352196)
50‐ml centrifuge tubes (Corning Falcon, cat. no. 352070)
10‐ml disposable pipets (Corning Falcon, cat. no. 356551)
10‐cm culture dishes (Thermo Fisher Scientific, cat. no. 174902)
Cell culture incubator
‐80°C freezer
Liquid nitrogen tank
Standard Hemocytometer (Weber Scientific, cat. no. 3048‐12)
PCR thermocycler
NanoDrop spectrophotometer (Thermo Fisher Scientific, cat. no. ND‐2000)
Refrigerated centrifuge
Refrigerated microcentrifuge
Inverted fluorescence microscope with filters of eGFP and mCherry (e.g., Zeiss AXIO)
Sub‐Cell GT Horizontal Electrophoresis System (BioRad, cat. no. 1704401)
Gel documentation system
-
1
Remove cryovial containing the frozen CHO cells carrying the IIS‐alphoidtetO‐HAC from a liquid nitrogen storage and immediately place it into a 37°C water bath for 1 min to thaw.
-
2
Transfer thawed cells to a 15‐ml centrifuge tube. Add 5 ml pre‐warmed to 37°C F12 growth medium. Centrifuge at 300 × g for 3 min at room temperature.
-
3
Discard supernatant. Resuspend cell pellet in 10 ml pre‐warmed to 37°C F12 growth medium supplemented with 5 μg/ml blasticidin S. Transfer cell suspension onto a 10‐cm culture dish. Incubate dish 2‐4 days in a cell culture incubator at 37°C in 5% CO2 atmosphere until the cells reach 50%‐80% confluency.
-
4
Wash cells once with 2 ml PBS, add 1.5 ml 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times. Count cells with a hemocytometer. Plate 1 × 105 CHO cells in one well of a 6‐well plate in 2 ml F12 growth medium without antibiotics. Incubate plate at 37°C in 5% CO2 atmosphere overnight so that the cells are ∼70%‐80% confluent at the time of transfection (i.e., the next day, Fig. 6A).
-
5
The next day, mix 1 μg Type I DNA1, 0.1 μg A139, and 200 μl Opti‐MEM medium without serum in a sterile 1.7‐ml tube. Mix gently by tapping.
-
6
Mix the ViaFect transfection reagent gently before use, then add 10 μl directly to the mix from step 5.
-
7
Mix gently and incubate 5 min at room temperature.
-
8
After incubation, add the mixture from the previous step directly to the well with the cells from step 4.
Do not mix by pipetting, just add drop by drop into the well.
Changing the medium before transfection is not necessary.
-
9
Shake plate vigorously backward‐forward, left‐right before placing back into the incubator.
-
10
Incubate cells overnight (typically 16‐18 hr) at 37°C in 5% CO2 atmosphere (Fig. 6B).
-
11
The next day, wash cells with 1 ml PBS once, add 300 μl 0.25% trypsin, and incubate 5 min at 37°C.
-
12
Add 4 ml F12 round I selection medium, resuspend cells by pipetting up and down five to seven times, and transfer suspension onto a 10‐cm culture dish containing 6 ml F12 round I selection medium.
-
13
Let cells grow until individual colonies become visible by the naked eye when you remove the medium from the dish. This usually takes 10‐14 days. Change F12 round I selection medium every 2‐3 days.
-
14
With a permanent marker, make circles on the bottom of the culture dish around colonies that are well isolated from other colonies (Fig. 6E).
-
15
Check the fluorescence of the cells in the circled colonies under the microscope. Mark only those colonies that have cells exhibiting red but no green fluorescence.
The number of colonies formed on the dish may vary from one to one hundred depending on recombination efficacy. It is advisable that at least ten colonies be picked up for further analysis. Scale up steps 4‐15 if you do not have enough colonies.
-
16
Pick up several individual colonies. To do this, wash cells once with 5 ml PBS, apply a cloning cylinder around a colony, and add 30 μl 0.25% trypsin into the cylinder's well. Incubate cells 5 min at 37°C. Add 150 μl F12 round I selection medium supplemented with 5 μg/ml of ganciclovir into the cylinder. Resuspend cells well by pipetting up and down five to seven times and transfer suspension into one well of a 96‐well plate. Incubate plate at 37°C in 5% CO2 atmosphere.
-
17
Continue to grow cells for an additional 3‐7 days, until the cultures reach 90%‐100% confluency, changing the F12 round I selection medium supplemented with 5 μg/ml ganciclovir every 3 days. Once confluent, wash wells once with 100 μl PBS and add 50 μl 0.25% trypsin. Incubate cells 5 min. Transfer cell suspension from each well of a 96‐well plate to a separate well of a 24‐well plate. Add 0.5 ml F12 round I selection medium with 5 μg/ml ganciclovir to each well of the 24‐well plate with cells. Grow cells until 90%‐100% confluency, changing the medium every 2‐3 days (Fig. 6G).
Addition of ganciclovir allows selection against the cells with an incorrect insertion (Fig. 8B). Approximately half of the initially picked colonies can be selected at this step.
Figure 8.
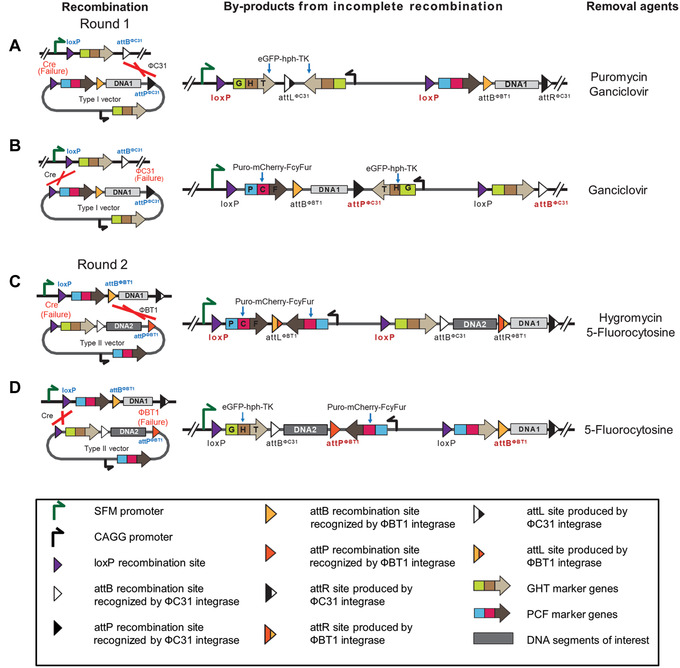
Error‐catching mechanism within the IIS‐alphoidtetO‐HAC system. Recombination reactions mediated by Cre, ΦC31, and ΦBT1 do not always go to completion. Because maintenance, storage, and screening of vertebrate cell colonies are very labor‐intensive compared to that for bacteria or yeast colonies, an error‐catching system was designed into the IIS to lighten the workload. (A and B) Products of incomplete recombination reactions involving the Type I carrier vector A167 retain an actively expressed GHT marker. (C and D) Products of incomplete recombination reactions between the Type II carrier vector A169 retain an expressed PCF marker. The selection agents to remove each misassembled product are listed. This figure was adapted from Lee et al. (2018).
-
18
Wash cells once with 200 μl PBS, add 100 μl 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times and transfer 80 μl of the suspension into new separate 1.7‐ml tubes. Add 0.5 ml F12 round I selection medium to each well to regrow the cells. Continue culturing cells in the plate at 37°C in 5% CO2 atmosphere.
It is important not to cross contaminate the culture. Use separate pipet tips for each individual colony.
-
19
While the cells are growing, check whether the fragment DNA1 from Type I carrier vector was inserted into the IIS‐alphoidtetO‐HAC. Spin down cells in the 1.7‐ml tubes (from step 18) at 300 × g for 3 min at room temperature and isolate genomic DNA using the DNeasy Blood & Tissue Kit, per the manufacturer's instructions. Elute genomic DNA in 50 μl water.
-
20Use each genomic DNA sample to run four PCR reactions:
- With diagnostic primer pairs B071/B072;
- With B074/B072 to confirm loxP recombination;
- With FDNA1 and B678 to confirm attPФC31/attBФC31 recombination;
- With forward B485 and RDNA1 to confirm integrity of attBФBT1 site.
Set up PCR reactions with TaKaRa Ex Taq DNA Polymerase, per the manufacturer's instructions, using 1 μl of the genomic DNA solution (from step 19) as a template. Set up 25‐μl total reactions and use the following PCR conditions after initial denaturation: 94°C 20 s, 60°C 30 s; 72°C 30 s; for 30 cycles (Fig. 6I).
-
21
Run samples in a 1.5% agarose gel.
PCR with B071/B072 should give a 447‐bp product, while PCR with B074/B072 primers should be negative. PCRs with FDNA1/B678 and with B485/RDNA1 should give products based on the user's design. It is advisable to sequence PCR products to ensure PCR accuracy.
-
22
Expand each colony with the PCR‐confirmed DNA1 fragment insertion (left growing in step 18) on a 10‐cm culture dish until cells reach ∼70%‐80% confluency (Fig. 6K). Wash cells once with 2 ml PBS, add 1.5 ml 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times. Count cells with a hemocytometer. Use 1 × 106 cells to start FISH (see Support Protocol).
FISH analysis on a metaphase spread is needed to confirm that the IIS‐alphoidtetO‐HAC carrying a DNA1 fragment from the A167 vector remains autonomous and has not integrated into the chromosomes.
-
23
Use 2 × 106 cells to prepare frozen stocks. Spin cells down at 300 × g for 3 min at room temperature. Discard supernatant. Resuspend cell pellet in 1 ml F12 growth medium and add 1 ml CFM (2×). Mix well and add 0.5 ml of the mixture to four cryovials. Place cryovials into the freezing box and place it at ‐80°C. For long‐term storage (up to several years), place cells in the liquid nitrogen tank the next day.
After the first round of insertion, the recombination reaction removes the GHT marker from its promoter and replaces it with the PCF marker, loxP and attBΦBT1 sites, and a DNA fragment from the Type I carrier vector A167 (DNA1), while deleting all other vector components. The promoter within the platform cassette now drives the PCF marker. The cells that successfully completed recombination reactions lose green fluorescence (eGFP), resistance to hygromycin (hph), and sensitivity to ganciclovir (TK), and gain red fluorescence (mCherry), resistance to puromycin (Puro), and sensitivity to 5‐fluorocytosine (FcyFur; Fig. 5).
Colonies that have passed PCR and FISH verification are suitable for the second round of DNA insertion (Basic Protocol 2).
Basic Protocol 2. INTEGRATION OF A SECOND DNA SEGMENT OF INTEREST INTO THE IIS‐alphoidtetO‐HAC
Here we describe insertion of the Type II carrier vector A169 carrying a second DNA segment of interest (DNA2) into the IIS‐alphoidtetO‐HAC, after the first round of DNA insertion (Basic Protocol 1; Figs. 5 and 6). The HAC is propagated in hamster CHO cells. To perform Basic Protocol 2, the user will co‐transfect the Type II carrier vector carrying DNA2 and the A135‐JH vector. The A135‐JH vector expresses ΦBT1 integrase and Cre recombinase. The user will culture the cells in hygromycin B/blasticidin S medium. After 10 days of selection, the user will pick up the colonies expressing green fluorescence (i.e., eGFP is expressed) and then expand them in medium containing hygromycin B/5‐fluorocytosine/blasticidin S.
After the second round of DNA insertion and every subsequent even‐numbered round of integration, a PCR reaction with B072/B074 primers should give a 559‐bp product, while PCR with B071/B072 primers should be negative (Fig. 5). Thus, the user will confirm the insertion of the GHT marker and elimination of the PCF marker.
To verify recombination between attBΦBT1 and attPΦBT1 sites and integration of DNA2, the user should choose primers from the sequence around a newly formed junction. A forward primer should correspond to a 3′‐end region of DNA2 (FDNA2). A reverse primer (RDNA1) should anneal to a 5′‐end region of the DNA fragment inserted in the first round (DNA1; e.g., F23/R23 primers after the second round of VHL gene integration; see Fig. 7B).
To verify the integrity of the newly inserted attBФC31 site, the user should choose a reverse primer complementary to a 5′‐end region of DNA2 (RDNA2; Fig. 5). B075, corresponding to the GHT sequence, can be used as a forward primer.
Materials
A135‐JH vector expressing ΦBT1 integrase and Cre recombinase (available upon request from Developmental Therapeutics Branch, NCI, NIH)
Type II carrier vector A169 carrying DNA2 (Type II DNA2, see Strategic Planning; concentration of vectors should be 0.1‐0.5 μg/μl; all vectors available upon request from Developmental Therapeutics Branch, NCI, NIH)
CHO cells carrying the IIS‐alphoidtetO‐HAC with DNA1 fragment inserted (from Basic Protocol 1)
Cell culture freezing medium (CFM; see recipe)
F12 growth medium (see recipe)
F12 round I selection medium (see recipe)
F12 round II selection medium (see recipe)
5‐Fluorocytosine solution, 10 mg/ml (InvivoGen, cat. no. sud‐5fc)
Opti‐MEM medium (Thermo Fisher Scientific, cat. no. 51985034)
ViaFect transfection reagent (Promega, cat. no. E4981)
PBS (Thermo Fisher Scientific, cat. no. 10010‐023)
0.25% trypsin (Thermo Fisher Scientific, cat. no. 25200056)
GeneRuler 1 kb plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331)
Nuclease‐free water (Quality Biological, cat. no. 351‐029‐721)
DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504)
TaKaRa Ex Taq® DNA Polymerase (Takara Bio, cat. no. RR001C)
Agarose (MilliporeSigma, cat. no. A9539)
-
Diagnostic primers to confirm the insertions of DNA fragments:
B072: 5′‐CCAGTTGCGTGCGTGGAA‐3′
B071: 5′‐CGCACCGTGGGCTTGTA‐3′
B074: 5′‐GCCGGACACGCTGAACTT‐3′
B075: 5′‐GGCTCCATACCGACGATAT‐3′
RDNA2: Designed by user
FDNA2: Designed by user
RDNA1: Designed by user
6‐well culture plates (Thermo Fisher Scientific, cat. no. 140675)
96‐well culture plates (Thermo Fisher Scientific, cat. no. 167008)
24‐well culture plates (Thermo Fisher Scientific, cat. no. 142475)
Cloning cylinders (Thermo Fisher Scientific, cat. no. 09‐552‐20)
1.7‐ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
15‐ml centrifuge tubes (Corning Falcon, cat. no. 352196)
50‐ml centrifuge tubes (Corning Falcon, cat. no. 352070)
10‐ml disposable pipets (Corning Falcon, cat. no. 356551)
10‐cm culture dishes (Thermo Fisher Scientific, cat. no. 174902)
Cryovial (Thermo Fisher Scientific, cat. no. 5012‐0012)
Cell culture incubator
‐80°C freezer
Liquid nitrogen tank
Standard Hemocytometer (Weber Scientific, cat. no. 3048‐12)
PCR thermocycler
NanoDrop spectrophotometer (Thermo Fisher Scientific, cat. no. ND‐2000)
Refrigerated centrifuge
Refrigerated microcentrifuge
Inverted fluorescence microscope with filters of eGFP and mCherry (e.g., Zeiss AXIO)
Sub‐Cell GT Horizontal Electrophoresis System (BioRad, cat. no. 1704401)
Gel documentation system
-
1
Remove cryovial containing the frozen CHO cells carrying the IIS‐alphoidtetO‐HAC with DNA1 inserted from a liquid nitrogen storage and immediately place it into a 37°C water bath for 1 min to thaw.
-
2
Transfer thawed cells to a 15‐ml centrifuge tube. Add 5 ml pre‐warmed to 37°C F12 growth medium. Centrifuge at 300 × g for 3 min at room temperature.
-
3
Discard supernatant. Resuspend cell pellet in 10 ml pre‐warmed to 37°C F12 round I selection medium. Transfer cell suspension onto a 10‐cm culture dish. Incubate dish in the cell culture incubator at 37°C in 5% CO2 atmosphere until the cells reach 50%‐80% confluency (Fig. 6B).
-
4
Wash cells once with 2 ml PBS, add 1.5 ml 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times. Count cells with a hemocytometer. Plate 1 × 105 CHO cells in one well of a 6‐well plate in 2 ml F12 growth medium without antibiotics. Incubate plate at 37°C in 5% CO2 atmosphere overnight so that the cells are ∼70%‐80% confluent at the time of transfection (i.e., the next day).
-
5
The next day, mix 1 μg Type II DNA2, 0.1 μg A135‐JH, and 200 μl of Opti‐MEM medium without serum in a sterile 1.7‐ml tube. Mix gently by tapping.
-
6
Repeat steps 6‐11 of Basic Protocol 1.
-
7
Add 4 ml F12 round II selection medium, resuspend cells by pipetting up and down five to seven times, and transfer suspension onto a 10‐cm culture dish containing 6 ml F12 round II selection medium (Fig. 6D).
-
8
Let cells grow until individual colonies become visible by the naked eye when you remove medium from the dish. This usually takes 10‐14 days. Change F12 round II selection medium every 2‐3 days.
-
9
With a permanent marker, make circles on the bottom of the culture dish around colonies that are well isolated from other colonies.
-
10
Check the fluorescence of the cells in the circled colonies under the microscope. Mark those colonies with the cells exhibiting green but not red fluorescence (Fig. 6F).
The number of colonies formed on the dish may vary from one to one hundred depending on recombination efficacy. It is advisable to pick up at least ten colonies for further analysis. Scale up steps 4‐10 if you do not have enough colonies.
-
11
Pick up several individual colonies. To do this, wash cells once with 5 ml PBS, apply a cloning cylinder around a colony, and add 30 μl 0.25% trypsin into the cylinder's well. Incubate cells 5 min. Add 150 μl F12 round II selection medium supplemented with 100 μg/ml of 5‐fluorocytosine into the cylinder. Resuspend cells by pipetting up and down five to seven times and transfer suspension into one well of a 96‐well plate. Incubate plate at 37°C in 5% CO2 atmosphere.
-
12
Continue to grow the cells for an additional 3‐7 days until the culture reaches 90%‐100% confluency, changing the F12 round II selection medium supplemented with 100 μg/ml 5‐fluorocytosine every 3 days. After cells reach confluency, wash wells once with 100 μl PBS and add 50 μl 0.25% trypsin. Incubate cells 5 min. Transfer the cell suspension from each well of the 96‐well plate to a separate well of a 24‐well plate. Add 0.5 ml F12 round II selection medium with 100 μg/ml 5‐fluorocytosine to each well of the 24‐well plate with the cells. Grow cells until 90%‐100% confluency, changing the medium every 2‐3 days (Fig. 6H).
Addition of 5‐fluorocytosine allows selection against the cells with an incorrect insertion (Fig. 8D). Approximately half of the initially picked colonies can be selected against at this step.
-
13
Wash cells once with 200 μl PBS, add 100 μl 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times and transfer 80 μl of each suspension into separate 1.7‐ml tubes. Add 0.5 ml F12 round II selection medium to each well to regrow the cells. Continue culturing cells in the plate at 37°C in 5% CO2 atmosphere.
-
14
While the cells are growing, check whether the DNA2 fragment from the Type II carrier vector has been inserted into the IIS‐alphoidtetO‐HAC. Spin down cells in the 1.7‐ml tubes (from step 13) at 300 × g for 3 min at room temperature and isolate genomic DNA using DNeasy Blood & Tissue Kit, per the manufacturer's instructions. Elute genomic DNA in 50 μl water (Fig. 6J).
-
15Use each genomic DNA sample to run four PCR reactions:
- With diagnostic primer pairs B071/B072;
- With B074/B072 to confirm loxP recombination;
- With FDNA2 and RDNA1 to confirm attPФBT1/attBФBT1 recombination;
- With forward B075 and RDNA2 to confirm integrity of attBФC31 site.
Set up PCR reactions with TaKaRa Ex Taq DNA Polymerase, per the manufacturer's instructions, using 1 μl of the genomic DNA solution (from step 14) as a template. Total volume of the reaction is 25 μl. Use the following PCR conditions after initial denaturation: 94°C 20 s, 60°C 30 s, 72°C 30 s; 30 cycles.
-
16
Run samples in a 1.5% agarose gel.
PCR with B074/B072 primers should give a 559‐bp product, while PCR with B071/B072 primers should be negative. PCRs with FDNA2/RDNA1 and with B075/RDNA2 should give products based on the user's design. It is advisable to sequence PCR products to ensure PCR accuracy.
-
17
Expand each colony with the PCR‐confirmed DNA2 fragment insertion (on the plate, from step 13) on a 10‐cm culture dish, until the cells reach ∼70%‐80% confluency (Fig. 6L). Wash cells once with 2 ml PBS, add 1.5 ml 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times. Count cells with a hemocytometer. Use 1 × 106 cells to start FISH (see Support Protocol).
FISH analysis on a metaphase spread is needed to confirm that the IIS‐alphoidtetO‐HAC carrying DNA1 and DNA2 fragments remains autonomous and has not integrated into the chromosomes.
-
18
Prepare frozen stocks as described in Basic Protocol 1, step 23.
After the second round of integration, the IIS‐alphoidtetO‐HAC will carry the DNA fragments from the first set of Type I and Type II carrier vectors (DNA1 and DNA2). The GHT marker will be expressed. Therefore, the cells will once again express eGFP, and will be hygromycin resistant (hph) and ganciclovir sensitive (TK; Fig 5).
Clones that have passed PCR and FISH verification are suitable for a third round of insertion.
Basic Protocol 3. INTEGRATION OF A THIRD DNA SEGMENT OF INTEREST INTO THE IIS‐alphoidtetO‐HAC
Here we describe insertion of the Type I carrier vector A167 carrying a third DNA segment of interest (DNA3) into the IIS‐alphoidtetO‐HAC after two previous rounds of DNA insertion (Basic Protocols 1 and 2; Figs. 5 and 6). The HAC is propagated in hamster donor CHO cells. After Basic Protocol 2, if users want to add an additional DNA segment, they will essentially repeat Basic Protocol 1 with some modifications (see below). Such cycles (Basic Protocols 1 to 3) can then be repeated for any number of segments. To perform Basic Protocol 3, the user will co‐transfect and select the cells as in Basic Protocol 1. As such, after the procedure, the cells will exhibit red fluorescence (mCherry is expressed).
After the third round of DNA insertion and every subsequent odd‐numbered round of integration, a PCR reaction with B071/B072 primers should give a 447‐bp product, while PCR with B072/B074 primers should be negative (Fig. 5). Thus, the user will confirm the insertion of the GHT marker and elimination of the PCF marker.
To verify recombination between attBФC31 and attPФC31 sites and integration of DNA3, the user should choose primers from the sequence around a newly formed junction. A forward primer should correspond to a 3′‐end region of DNA3 (FDNA3). A reverse primer should anneal to a 5′‐end region of DNA2 (RDNA2; Fig. 5; e.g., F12/R12 primers for VHL gene insertion; see Fig. 6B).
To verify the integrity of the newly inserted attBФBT1 site, the user should choose a reverse primer complementary to a 5′‐end region of DNA3 (RDNA3). B485, corresponding to the PCF sequence, can be used as a forward primer (Fig. 5).
As mentioned, this protocol is almost identical to Basic Protocol 1, with the following important differences: (1) the starting cell culture is the cells derived from a previous round of insertion (Basic Protocol 2), (2) Type I carrier vector contains a new DNA fragment (DNA3), and (3) a new pair of diagnostic primers (FDNA3/ RDNA2) is used.
Materials
A135‐JH vector expressing ΦBT1 integrase and Cre recombinase (available upon request from Developmental Therapeutics Branch, NCI, NIH)
Type I carrier vector A167 carrying DNA3 (Type I DNA3, see Strategic Planning; concentration of vectors should be 0.1‐0.5 μg/μl; all vectors available upon request from Developmental Therapeutics Branch, NCI, NIH)
CHO cells carrying the IIS‐alphoidtetO‐HAC with DNA1 and DNA2 fragments inserted (from Basic Protocol)
Cell culture freezing medium (CFM; see recipe)
F12 growth medium (see recipe)
F12 round I selection medium (see recipe)
F12 round II selection medium (see recipe)
Ganciclovir solution, 10 mg/ml (see recipe)
Opti‐MEM medium (Thermo Fisher Scientific, cat. no. 51985034)
ViaFect transfection reagent (Promega, cat. no. E4981)
PBS (Thermo Fisher Scientific, cat. no. 10010‐023)
0.25% trypsin (Thermo Fisher Scientific, cat. no. 25200056)
GeneRuler 1 kb plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331)
Nuclease‐free water (Quality Biological, cat. no. 351‐029‐721)
DNeasy Blood & Tissue Kit (Qiagen, cat. no. 69504)
TaKaRa Ex Taq® DNA Polymerase (Takara Bio, cat. no. RR001C)
Agarose (MilliporeSigma, cat. no. A9539)
-
Diagnostic primers to confirm the insertions of DNA fragments:
B072: 5′‐CCAGTTGCGTGCGTGGAA‐3′
B071: 5′‐CGCACCGTGGGCTTGTA‐3′
B074: 5′‐GCCGGACACGCTGAACTT‐3′
B485: 5′‐GTGCAAGAAGATTATGAAGCAG‐3′
RDNA3: Designed by user
FDNA3: Designed by user
RDNA2: Designed by user
6‐well culture plates (Thermo Fisher Scientific, cat. no. 140675)
96‐well culture plates (Thermo Fisher Scientific, cat. no. 167008)
24‐well culture plates (Thermo Fisher Scientific, cat. no. 142475)
Cloning cylinders (Thermo Fisher Scientific, cat. no. 09‐552‐20)
Cryovial (Thermo Fisher Scientific, cat. no. 5012‐0012)
1.7‐ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
15‐ml centrifuge tubes (Corning Falcon, cat. no. 352196)
50‐ml centrifuge tubes (Corning Falcon, cat. no. 352070)
10‐ml disposable pipets (Corning Falcon, cat. no. 356551)
10‐cm culture dishes (Thermo Fisher Scientific, cat. no. 174902)
Cell culture incubator
‐80°C freezer
Liquid nitrogen tank
Standard Hemocytometer (Weber Scientific, cat. no. 3048‐12)
PCR thermocycler
NanoDrop spectrophotometer (Thermo Fisher Scientific, cat. no. ND‐2000)
Refrigerated centrifuge
Refrigerated microcentrifuge
Inverted fluorescence microscope with filters of eGFP and mCherry (e.g., Zeiss AXIO)
Sub‐Cell GT Horizontal Electrophoresis System (BioRad, cat. no. 1704401)
Gel documentation system
-
1
Remove cryovial containing the frozen CHO cells carrying the IIS‐alphoidtetO‐HAC with DNA1 and DNA2 fragments from a liquid nitrogen storage and immediately place it into a 37°C water bath for 1 min to thaw.
-
2
Transfer thawed cells into a 15‐ml centrifuge tube. Add 5 ml pre‐warmed to 37°C F12 growth medium. Centrifuge at 300 × g for 3 min at room temperature.
-
3
Discard supernatant. Resuspend cell pellet in 10 ml pre‐warmed to 37°C F12 round II selection medium. Transfer cell suspension onto a 10‐cm culture dish. Incubate dish in a cell culture incubator at 37°C in 5% CO2 atmosphere until cells reach 50%‐80% confluency.
-
4
Wash cells once with 2 ml PBS, add 1.5 ml 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times. Count cells with a hemocytometer. Plate 1 × 105 CHO cells in one well of a 6‐well plate in 2 ml F12 growth medium without antibiotics. Incubate plate at 37°C in 5% CO2 atmosphere overnight so that the cells are ∼70%‐80% confluent at the time of transfection (i.e., the next day).
-
5
The next day, mix 1 μg Type I DNA3, 0.1 μg A135‐JH vector, and 200 μl Opti‐MEM medium without serum in a sterile 1.7‐ml tube. Mix gently by tapping.
-
6
Repeat steps 6‐17 of Basic Protocol 1.
-
7
Wash cells once with 200 μl PBS, add 100 μl 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times and transfer 80 μl of the suspension into new separate 1.7‐ml tubes. Add 0.5 ml F12 round I selection medium to each well to regrow cells. Continue culturing cells in the plate at 37°C in 5% CO2 atmosphere.
It is important not to cross contaminate the culture. Use separate pipet tips for each individual colony.
-
8
While the cells are growing, check whether the fragment DNA3 from the Type I carrier vector was inserted into the IIS‐alphoidtetO‐HAC. Spin cell suspension in the 1.7‐ml tube (from step 7) down at 300 × g for 3 min at room temperature and isolate genomic DNA using the DNeasy Blood & Tissue Kit, per the manufacturer's instructions. Elute genomic DNA in 50 μl water.
-
9Use each genomic DNA sample to run four PCR reactions:
- With diagnostic primer pairs B071/B072;
- With B074/B072 to confirm loxP recombination;
- With FDNA3 and RDNA2 to confirm attPФC31/attBФC31 recombination;
- With forward B485 and RDNA3 to confirm integrity of attBФBT1 site.
Set up PCR reactions with TaKaRa Ex Taq DNA Polymerase, per the manufacturer's instructions, using 1 μl of the genomic DNA solution (from step 7) as a template. Total volume of the reaction is 25 μl. Use the following PCR conditions after initial denaturation: 94°C 20 s, 60°C 30 s, 72°C 30 s; 30 cycles.
-
10
Run samples in a 1.5% agarose gel.
PCR with B071/B072 should give a 447‐bp product, while PCR with B074/B072 primers should be negative. PCRs with FDNA3/RDNA2 and with B485/RDNA3 should give the products based on the user's design. It is advisable to sequence PCR products to ensure PCR accuracy.
-
11
Expand each colony with the PCR‐confirmed DNA3 fragment insertion (from the plate in step 7) on a 10‐cm culture dish until the cells reach ∼70%‐80% confluency. Wash cells once with 2 ml PBS, add 1.5 ml 0.25% trypsin, and incubate 5 min at 37°C. Resuspend cells by pipetting up and down five to seven times. Count cells with a hemocytometer. Use 1 × 106 cells to start FISH (see Support Protocol).
FISH analysis on a metaphase spread is needed to confirm that the IIS‐alphoidtetO‐HAC carrying DNA1, DNA2, and DNA3 fragments remain autonomous and has not integrated into the host chromosomes.
-
12
Prepare frozen stocks as described in Basic Protocol 1, step 23.
After the third round of integration, the IIS‐alphoidtetO‐HAC will carry two fragments derived from the Type I carrier vector (DNA1 and DNA3) and a fragment from the Type II carrier vector (DNA2). The PCF marker will be expressed. Therefore, the cells will exhibit red fluorescence (mCherry), and will be puromycin resistant (Puro) and 5‐fluorocytosine sensitive (FcyFur; Fig. 5).
FLUORESCENCE IN SITU HYBRIDIZATION ANALYSIS FOR THE CIRCULAR IIS‐alphoidtetO‐HAC
This protocol describes the steps to confirm the presence of the circular IIS‐alphoidtetO‐HAC in an autonomous form in cells. The IIS‐alphoidtetO‐HAC contains a unique tetO sequence, allowing detection via FISH. Hybridization with a fluorophore‐labeled peptide nucleic acid (PNA) probe (Alexa488‐OO‐ACCACTCCCTATCAG) on metaphase spreads is a robust method to visualize the HAC.
Materials
HAC‐containing CHO cells (from Basic Protocol 1, step 22; from Basic Protocol 2, step 17; from Basic Protocol 3, step 11)
F12 growth medium (see recipe)
0.25% trypsin (Thermo Fisher Scientific, cat. no. 25200056)
PBS (Thermo Fisher Scientific, cat. no. 10010‐023)
Nuclease‐free water (Quality Biological, cat. no. 351‐029‐721)
Deionized (DI) water
KaryoMAX™ Colcemid™ Solution in PBS (Thermo Fisher Scientific, cat. no. 15212012)
KaryoMAX™ Potassium Chloride Solution (Thermo Fisher Scientific, cat. no. 10575090)
Methanol (MilliporeSigma, cat. no. 322415)
Absolute Ethanol, 200 proof (Thermo Fisher Scientific, cat. no. T038181000)
Acetic acid (MilliporeSigma, cat. no. 695092)
Formaldehyde fixation solution (see recipe)
Hybridization buffer (see recipe)
Wash solution (see recipe)
50 μM PNA probe (see recipe)
VECTASHIELD Vibrance Antifade Mounting Medium with DAPI (Vector Laboratories, cat. no. H‐1800)
Microscope slides (Denville, cat. no. M1021)
Micro Cover Glass #1, 22 mm × 50 mm (Electron Microscopy Sciences, cat. no. 72200‐40)
Parafilm® M (MilliporeSigma, cat. no. P7793)
1.7‐ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
15‐ml centrifuge tubes (Corning Falcon, cat. no. 352196)
10‐ml disposable pipets (Corning Falcon, cat. no. 356551)
10‐cm culture dishes (Thermo Fisher Scientific, cat. no. 174902)
Slide warmer (Thermo Fisher Scientific, cat. no. 12‐594)
Wheaton Coplin staining jars (MilliporeSigma, cat. no. S5516)
Slide holder (Jaece Industries, cat. no. L500‐C)
Pipetman L P200L, 20‐200 μl (Gilson, cat. no. FA10005M)
Cell culture incubator
‐80°C freezer
Refrigerated centrifuge
Refrigerated microcentrifuge
Inverted fluorescence microscope with filters of DAPI and Alexa Fluor 488 (e.g., Zeiss AXIO)
-
1
Incubate 1 × 106 HAC‐containing CHO cells at 37°C in 5% CO2 atmosphere in 10 ml F12 growth medium in a 10‐cm culture dish for 18‐24 hr.
-
2
Remove old medium from the culture dish and add 10 ml F12 growth medium supplemented with 100 μl KaryoMAX™ colcemid. Incubate cells at 37°C in 5% CO2 atmosphere for 4 hr.
-
3
Collect medium from the culture dish into a 15‐ml centrifuge tube.
-
4
Add 2 ml PBS to the dish, swirl, and collect PBS into the same 15‐ml centrifuge tube.
-
5
Add 1.5 ml 0.25% trypsin to the cells and incubate 5 min at 37°C. Collect cell suspension into the same 15‐ml centrifuge tube.
-
6
Spin down cells in the 15‐ml centrifuge tube at 300 × g for 3 min at room temperature.
-
7
Discard supernatant. Resuspend cells in 10 ml PBS. Spin down at 300 × g for 3 min at room temperature.
-
8
Discard supernatant. Gently resuspend cells in 10 ml KaryoMAX™ potassium chloride solution, pre‐warmed to 37°C.
-
9
Incubate cells at 37°C for 20 min.
-
10
Prepare the desired amount of fresh fixative solution by mixing three volumes of methanol with one volume of acetic acid. Put on ice.
You will need 5 ml per sample.
-
11
Transfer tube with the cell suspension into ice. Add 50 μl fixative solution. Mix by inverting the tube.
-
12
Centrifuge at 480 × g for 5 min in a pre‐cooled (4°C) centrifuge.
It is important to use centrifugation without brake to avoid cell attrition, preventing loss of material.
-
13
Carefully pipet out as much supernatant as possible, taking care not to touch the cell pellet. Slowly add 1 ml fixative solution to the tube. Do not mix. Incubate cells on ice for 30 min.
-
14
Gently re‐suspend pellet by finger tapping and centrifuge suspension at 480 × g for 5 min at 4°C.
-
15
Carefully pipet out as much supernatant as possible, taking care not to touch the cell pellet. Gently re‐suspend pellet in 1 ml fresh fixative solution. Centrifuge suspension at 480 × g for 5 min at 4°C.
-
16
Repeat step 15 two more times.
-
17
Finally, re‐suspend cells in 1 ml fixative solution; cells can be stored at ‐20°C in tightly closed tubes for up to 1 year.
This is a safe stopping point.
-
18
Treat slides with 50 ml 40% methanol in Coplin jar for 1 hr.
Slides can be stored in 40% (v/v) methanol in Coplin jar sealed with Parafilm at 4°C up to several years.
-
19
Replace methanol with 50 ml 100% ethanol and incubate 1 hr. Put slides in a slide holder and let them air dry at room temperature.
-
20
Drop 20 μl of the fixed cells suspension from step 17 onto microscope slides from an ∼10 cm height using a Gilson Pipetman. Place slides on a slide warmer (55°C) where they can be left for 1 hr or longer.
-
21
Examine slides under a phase contrast microscope to determine if the spreads are suitable for staining and counting.
Metaphase chromosome spreads should be easy to find and well separated from each other.
The slides can be stored at ‐20°C in a slide box for up to 1 month.
-
22
Put slides into a Coplin jar with 50 ml PBS for 15 min to rehydrate.
-
23
Replace PBS with 50 ml formaldehyde fixation solution for 2 min.
-
24
Wash slides in a Coplin jar with 50 ml PBS three times for 2 min.
-
25
Soak slides in a Coplin jar with 50 ml 70% (v/v) ethanol for 2 min.
-
26
Soak slides in a Coplin jar with 50 ml 85% (v/v) ethanol for 2 min.
-
27
Soak slides in a Coplin jar with 50 ml 100% ethanol for 2 min.
-
28
Put slides in a slide holder and let them air dry at room temperature.
-
29
Pre‐heat slide warmer to 85°C.
-
30
For each slide, mix 0.2 μl PNA probe in 20 μl hybridization buffer to a final concentration of 500 nM.
-
31
Pre‐warm slides on a slide warmer for 5 min.
-
32
Heat hybridization buffer containing the PNA probe at 85°C for 5 min.
-
33
Add 20 μl hybridization buffer containing the PNA probe onto each slide. Cover with a coverslip.
-
34
Heat slides in a slide warmer at 85°C for 10 min.
-
35
Place slides in a box onto wet paper towels. Incubate in the dark at room temperature for 2 hr.
-
36
Immerse slides in wash solution to remove coverslip.
-
37
Wash slides in 50 ml wash solution in a Coplin jar twice at 60°C for 10 min.
-
38
Wash slides with wash solution at room temperature for 10 min.
-
39
Wash with 50 ml PBS in a Coplin jar three times for 5 min.
-
40
Soak in a Coplin jar with 70% (v/v) ethanol for 5 min.
-
41
Soak in a Coplin jar with 85% (v/v) ethanol for 5 min.
-
42
Soak in a Coplin jar with 100% ethanol for 5 min.
-
43
Dry slides at room temperature.
-
44
Add 25 μl VECTASHIELD mounting medium with DAPI to the slides. Cover with a coverslip. Place slides in the dark at room temperature overnight to let the mounting medium solidify.
Mounted slides can be stored in the dark at ‐20°C for up to a month.
-
45
Observe slides under the fluorescence microscope with filters for DAPI and Alexa Fluor 488.
The intact HAC should be visible as a single separate dot in both channels and should be separated from the host chromosomes (Fig. 1B, white arrow).
REAGENTS AND SOLUTIONS
Cell culture freezing medium (CFM), 2×
60 ml F12 medium (Thermo Fisher Scientific, cat. no. 31765035)
20 ml FBS (Thermo Fisher Scientific, cat. no. 26140)
20 ml DMSO (MilliporeSigma, cat. no. D2650‐100ML)
Store at 4°C for up to 1 year.
F12 growth medium
500 ml F12 medium (Thermo Fisher Scientific, cat. no. 31765035)
50 ml FBS (Thermo Fisher Scientific, cat. no. 26140)
5 ml PenStrep (Thermo Fisher Scientific, cat. no. 15070063)
Store at 4°C for up to 1 month.
F12 round I selection medium
100 ml F12 growth medium (see recipe)
40 μl puromycin dihydrochloride (10 mg/ml; Thermo Fisher Scientific, cat. no. A1113803)
40 μl blasticidin S HCl (10 mg/ml; Thermo Fisher Scientific, cat. no. A1113903)
Store at 4°C for up to 1 month.
F12 round II selection medium
100 ml F12 growth medium (see recipe)
200 μl hygromycin B (50 mg/ml; Thermo Fisher Scientific, cat. no. 10687010)
40 μl blasticidin S HCl (10 mg/ml; Thermo Fisher Scientific, cat. no. A1113903)
Store at 4°C for up to 1 month.
Formaldehyde fixation solution
10 ml formaldehyde solution for molecular biology, 36.5%‐38% in H2O (MilliporeSigma, cat. no. F8775)
90 ml PBS (Thermo Fisher Scientific, cat. no. 10010‐023)
Store at room temperature for up to 1 month.
Ganciclovir solution, 10 mg/ml
1 g ganciclovir (Thermo Fisher Scientific, cat. no. 461710010)
90 ml cell culture grade water (Thermo Fisher Scientific, cat. no. MT25055CV)
10 ml HCl, 1 N (MilliporeSigma, cat. no. H9892)
Store at ‐20°C in 1‐ml aliquots for up to 1 year.
Thawed aliquots can be stored at 4°C for up to 1 week.
Hybridization buffer
200 μl 1 M Tris, pH 7.4 (KD Medical, cat. no. RGF‐3340)
3.8 ml nuclease‐free water (Quality Biological, cat. no. 351‐029‐721)
6 ml formamide DI (American Bioanalytical, cat. no. AB00600)
50 mg blocking reagent (Roche, cat. no. 11096176001)
Store at 4°C for up to 1 year.
PNA probe
Custom PNA probe corresponding to a tetO sequence (PNA BIO; as a lyophilized powder). Probe sequence is Alexa488‐OO‐ACCACTCCCTATCAG.
Resuspend a 5 nmol lyophilized PNA powder stock in 100 μl formamide DI (American Bioanalytical, cat. no. AB00600) to make a 50 μM stock.
Store in 10‐μl aliquots at ‐70°C, protected from light, for up to 1 year.
After thawing, heat at 55°C for 5 min to ensure complete dissolution.
Wash solution
10 ml 20× SSC (Quality Biological, cat. no.351‐003‐131)
90 ml DI water
100 μl Tween 20 (Bio‐Rad, cat. no. 170‐6531)
Store at room temperature for up to 1 month.
COMMENTARY
Background Information
There are several methods to produce transgenic cells for gene functional studies. One of the methods relies on transfection of BAC DNA carrying a gene(s) of interest into host cells (Arii, Kato, Kawaguchi, Tohya, & Akashi, 2009; Auriche et al., 2010; Hall et al., 2008; Head et al., 2007; Hibbitt et al., 2007; Illenye et al., 2004; Poser et al., 2008; Tsuji et al., 2006). Another popular approach is based on transduction with viruses or virus‐based delivery vectors carrying small‐sized genes (not bigger than 5 kb) or cDNA (Gimpel et al., 2021; Huang et al., 2021; Immidisetti, Nwagwu, Adamson, Patel, & Carbonell, 2021; Ma, Hill, Hoang, & Wen, 2021; Mijanović et al., 2020; Wang, Scheitler, Wenger, & Elder, 2021; Zhang, Wu, Zhang, & Xia, 2021). Both methods, however, may lead to random integrations of BACs or viruses into host chromosomes. As a result, expression of the cloned genes may be subject to position effects and the number of copies integrated. Even non‐integrated adeno‐associated recombinant viruses (rAAVs), which currently are the most attractive viral vectors, along with retro‐ and lentiviral vectors, have several serious disadvantages for use in gene therapy and even in gene functional studies, such as low cloning capacity and lack of long‐term transgene expression. Another method is based on integration of a gene into a “hot spot” of a mammalian genome using a bacteriophage P1‐derived Cre recombinase or ΦC31 integrase (Luo et al., 2013). However, efficiency of gene integration with such a system is very low. In addition, all of these approaches are typically applicable to a single gene or DNA fragment.
In this article, we describe a protocol for the IIS‐alphoidtetO‐HAC system, which addresses most of the problems associated with virus‐based gene delivery vectors and BACs because the HAC‐based vectors, including the alphoidtetO‐HAC, replicate and segregate as natural chromosomes, independently from the host genome, and have the ability to carry Mb‐size gene/fragment inserts. The IIS‐alphoidtetO‐HAC system includes the iterative integration system (IIS), which potentially allows adding an unlimited number of genomic DNA fragments. This feature makes the IIS‐alphoidtetO‐HAC a more versatile vector, an aim that was shared by several groups in the field that also developed their own multi‐integrase systems in combination with artificial chromosome‐based vectors (Honma et al., 2018; Suzuki, Kazuki, Oshimura, & Hara, 2014; Toth et al., 2014; Yamaguchi et al., 2011; Yoshimura et al., 2015). The IIS‐alphoidtetO‐HAC system has several notable advantages compared to some of these other multi‐integrase systems. First, any desired number of genomic DNA fragments can be inserted into the IIS‐alphoidtetO‐HAC. There is no inherent upper limit to the system, unlike other structurally simpler multi‐integration systems. Second, each step of gene/fragment insertion is accompanied by a visible change in cell fluorescence, which simplifies the screening of correct clones. Third, the IIS‐alphoidtetO‐HAC system can assemble large genes from intron‐exon‐intron segments because it only integrates the DNA segments of interest without extraneous plasmid DNA and leaves only a small 35‐ to 55‐bp scar site between adjacent DNA segments (e.g., between the first and second DNA segments of interest). Fourth, to ensure integrity of assembly, the IIS‐alphoidtetO‐HAC system has an error‐proofing mechanism that selects against colonies in which the recombination reaction did not proceed to completion (see below for details and Fig. 8). Finally, the alphoidtetO‐HAC may be removed from the cells, offering a unique possibility to compare the phenotypes of human cells with and without functional copies of the genes under study.
At its core, the IIS is a marker capture‐switching system. Each round of integration causes the marker in the promoter‐marker pair to be exchanged (see Fig. 3), and this change in the marker allows for the screening and selection of successful integration events. While this feature added complexity, the re‐use of markers means an unlimited number of integrations of DNA fragments could be accomplished by using only two markers and the number of recombinase proteins in the system is limited to only three, i.e., Cre, ΦC31, and ΦBT1. In the IIS‐alphoidtetO‐HAC system, each step of gene/DNA fragment loading uses the recombinase Cre and ΦC31 or Cre and ΦBT1, and is accompanied by an exchange between the eGFP‐hph‐TK (GHT) or PAC‐mCherry‐FcyFur (PCF) marker. These markers are compound markers that consist of three parts, a positive selection marker (hph) to select for cells when a new DNA fragment is added to the HAC, a counter‐selection marker (TK or FcyFur) to exclude the cells when this event does not happen, and a fluorescence marker (eGFP or mCherry) to monitor the recombination reaction (exchange between the GHT and PCF markers). The three components of the compound markers are transcribed as a single mRNA molecule but produced as separate proteins due to the presence of the 2A self‐cleaving peptide between each component.
There are several reasons to use this marker‐capture‐switching system versus other multi‐integrase systems combined with HAC‐based vectors (Honma et al., 2018; Suzuki et al., 2014; Toth et al., 2014; Yamaguchi et al., 2011; Yoshimura et al., 2015). First, there are a limited number of commercially available selection agents for use in mammalian cell cultures (e.g., blasticidin, hygromycin, puromycin). Multi‐integration systems that use a new marker for each DNA insertion would rapidly run out of useable markers and be unable to go beyond four or five insertions. Such systems also leave no marker for the user to use in other projects aside from gene integration within that cell line. Our IIS system is a cyclic system, where only two markers, GHT and PCF, substitute each other as a new genomic DNA segment is added to the integration sites. Second, at the time when the IIS was built, the number of recombinase proteins that were optimized for expression and efficient activity in vertebrate cells was extremely limited. Only Cre was widely used; ΦC31 had just begun to be used in mammalian cells, and ΦBT1 had yet to see much use or optimization in yeast, and much less in vertebrate cells. These integrases are bacteriophage proteins that in their native state become less effective at temperatures higher than room temperature, in which vertebrate cells are typically cultured at.
A disadvantage of our marker‐capture‐switching system, on the other hand, is complexity. Compared to other multi‐integration systems (Honma et al., 2018; Suzuki et al., 2014; Toth et al., 2014; Yamaguchi et al., 2011; Yoshimura et al., 2015), the IIS uses two recombinase proteins per integration cycle and has many more moving parts. However, we hope that as the IIS has a limited number of recombinases in use, optimized conditions could be found and with time, mutant recombinases with improved tolerance to elevated temperatures and lower rates of protein aggregation would eventually be developed.
The fluorescence color component was added to the compound marker to simplify the screening of correct clones that have successfully undergone the integration reaction. This is necessary, as screening of mammalian colonies is far more time consuming and labor intensive than in yeast or bacteria. In addition, the counterselectable markers were added to the compound markers to form an error‐proofing mechanism in the IIS‐alphoidtetO‐HAC system. Recombinase‐mediated reactions can fail to go to completion, and growth, maintenance, screening, and storage of candidate vertebrate colonies is far slower, more labor intensive, and space limited than with either yeast or bacteria. It was thus desirable to remove as many faulty colonies as possible by drug selection and visual inspection, keeping only the best candidates for subsequent detailed characterization. If either of the two recombination reactions fails, this failure event can be selected against and screened out by the error‐proofing design of the IIS‐alphoidtetO‐HAC system (Fig. 8). As illustrated, the backbone of each carrier vector has its own constitutively active compound marker. Hence, if recombination by Cre fails but ΦC31 occurs (Fig. 8A), the Type I carrier A167 vector will integrate into the platform cassette but the PCF marker it carries will remain promoterless. Cells carrying this error are removed by selection with puromycin and counter‐selection with ganciclovir. Alternatively, if recombination by ΦC31 fails but Cre occurs (Fig. 8B), the Type I carrier vector A167 will integrate into the construction platform, and the SFM promoter will capture the PCF marker, leaving the original GHT marker promoterless. However, the backbone of the Type I carrier vector A167 is retained and a fully expressed GHT marker under the CAGG promoter remains. Hence, cells generated by such a failure event are puromycin resistant and have both red and green fluorescence. These cells can be removed either by counter selection using ganciclovir against the thymidine kinase component (TK) of the GHT marker or by visual inspection when the colonies are formed. To counteract the loss of TK gene activity by heterochromatin silencing, the GHT marker is protected by flanking murine transfer RNA (tRNA) genes functioning as insulators (Ebersole et al., 2011; Lee et al., 2013). Similarly, the products of incomplete recombination between Type II carrier plasmid A169 with the active PCF cassette may be identified and removed. Selection with hygromycin and counter selection with 5‐fluorocytosine ensures that only cells that have correctly undergone the second round of assembly will survive (Fig. 8C and 8D). Untransformed parental cells and cells with incomplete recombination are killed by this double selection.
The IIS‐alphoidtetO‐HAC system is expected to be beneficial in various fields of study, such as multiple‐gene humanized models, polygenic disease models, reprogramming, and gene expression systems. In addition, the IIS‐alphoidtetO‐HAC system opens new opportunities for synthetic biology in multicellular eukaryotes. It provides a unique tool to engineer synthetic chromosomes carrying large segments of genomic DNA, such as segments containing long‐range genetic elements required for appropriate regulation of gene expression or multiple copies of genes (Moriwaki et al., 2020).
Critical Parameters
Always check the IIS‐alphoidtetO‐HAC system before proceeding to the next step
Due to the large time investment associated with vertebrate cell culture, the user should proceed cautiously. All recombination recognition sites (loxP, attBΦC31, attBΦBT1) should be sequence verified after each round of recombination as in the presence of recombinase proteins, these sites experience higher than normal rates of DNA sequence changes.
Maintenance of the IIS‐alphoidtetO‐HAC and its copy number
While the IIS‐alphoidtetO‐HAC is stable within a cell line over the short term, it is advised that continuous blasticidin S selection be used to ensure that the majority of cells in the culture retain the HAC over the long term. Furthermore, when restarting a cell culture from colonies, we advise that a FISH analysis be done to ascertain that only a single copy of the IIS‐alphoidtetO‐HAC is present within the cell. While rare, we have on occasion recovered colonies that have spontaneously increased the HAC copy number. These colonies should be discarded as the IIS‐alphoidtetO‐HAC system is designed to function as a single copy per cell.
Selection and counterselection agent concentration
Our iterative integration system includes two counterselectable markers (thymidine kinase and FcyFur) that can be selected against and removed by ganciclovir and 5‐fluorocytosine, respectively. However, in vertebrate cells, the counterselectable marker's expression may also be lost via heterochromatin silencing, allowing the cell to survive without physically losing the counterselectable marker gene. It is recommended that the user determines an upper tolerance limit of the cell line of interest to these counterselectable agents. Ideally, the maximum tolerated concentration of counterselectable agents should be used in the experiments. However, if the cost of the counterselectable agents is an issue, the user has to determine the minimum concentration of the counterselectable agents needed to kill the cells containing the IIS‐alphoidtetO‐HAC on a confluent 10‐cm petri dish before spontaneous silencing of the counterselectable markers occurs, which would lead to a significant background of resistant colonies.
Note that the issue described above should be taken into account when the IIS system is carried out in the cell line of interest different from hamster CHO cells. Determination of the minimum concentration of the counterselectable agents in the cell line of interest is a complicated multistep process. First, the IIS‐alphoidtetO‐HAC should be transferred from the donor hamster CHO cells to the recipient cell line of interest using MMCT (Current Protocols article: Liskovykh et al., 2021). In this cell line, the HAC will be maintained using blasticidin S, as the HAC contains the bsr gene in its backbone (Nakano et al., 2008). The HAC also carries the empty integration platform cassette that contains the GHT marker (i.e., eGFP‐hph‐TK). Once the HAC transfer is completed, the user should use this cell line to determine the minimum concentration of ganciclovir needed to kill all the cells on a 10‐cm confluent petri dish before the spontaneous emergence of resistant colonies. It is highly recommended that the experiment be extended to at least 2 to 3 weeks to determine how frequent spontaneous resistance colonies emerge at specific concentrations of ganciclovir used and when. These data are very important to determine the background of false positives the user may experience. It is also recommended that the cells be detached from the plate and allowed to reattach 3 days after selection to help remove dead cells.
The next step is to test the PCF compound marker, which is a fusion of Puro‐mCherry‐FcyFur, and to determine the minimum concentration of 5‐fluorocytosine needed to kill the cells expressing this marker. For this purpose, the user should obtain cells with the HAC carrying a single copy of the PCF marker. The most time‐efficient manner to obtain such a cell line is to use the IIS system in the HAC and integrate an empty Type I carrier vector A167 into the platform cassette. This will replace the GHT marker by the PCF marker and also will help determine the concentration of the ΦC31‐Cre expressing vector (A139) needed to trigger the recombination event. The user should determine the minimum concentration of 5‐fluorocytosine on a confluent 10‐cm petri dish before the spontaneous emergence of resistant colonies. The experiment should be extended to at least 2 to 3 weeks to determine if there is any late emergence of spontaneous resistant colonies. Removal of dead cells can be aided by detaching the cells from the plate and allowing them to reattach 3 days after selection started.
Checking the IIS‐alphoidtetO‐HAC system (optional)
Once the concentration of the Cre‐integrase vectors has been determined, we recommend that users familiarize themselves with the IIS and attempt a trial run of the system in the cell line of interest or in hamster CHO cells. Due to IIS complexity, the user should integrate an empty Type I carrier vector A167 into the platform cassette of the HAC with the ΦC31‐Cre expressing vector (A139) and then integrate the Type II carrier vector A169 into the platform cassette with the ΦBT1‐Cre vector (A135‐JH). It is easier to integrate empty carrier vectors as their smaller sizes make them less difficult to purify in larger quantities and more molecules can be transformed per unit mass DNA transfected.
Quantity of Cre‐integrase vectors
The site‐specific integrases ΦC31 and ΦBT1 function as tetramers and when overexpressed, they tend to form nonfunctional protein aggregates. Hence, overexpression of integrase proteins can result in reduced recombination activity. As a promoter's activity changes between cell lines, it is recommended that the user tests different concentrations of Cre‐integrase vectors with a set amount of the empty Type I and Type II carrier vectors (A167 and A169) to determine the optimum concentrations that will yield the highest amount of recombination activity, being mindful that “less can be more.”
Transfection with the carrier vectors A167 and A169 carrying large genomic DNA fragments
If the transfecting carrier vectors with the inserted genomic DNA fragments are ten to twenty times larger than the empty vectors, we recommend that the user re‐optimize the DNA‐transfection agent ratio. A BAC carrying the eGFP gene of similar size to the loaded carrier vectors would be a good test subject to find the optimal DNA‐transfection agent ratio.
Endotoxin‐free DNA
The negative impact of endotoxins varies by cell line. We recommend the ΦC31‐Cre (A139) and ΦBT1‐Cre (A135‐JH) vectors as well as Type I and Type II carrier vectors (A167 and A169) carrying the genomic DNA fragments be purified using commercially available endotoxin‐free kits. However, it has been observed that the buffers and solutions of some endotoxin‐free kits are not sterile. We, thus, highly recommend that the user filter‐sterilize all solutions before use to avoid downstream fungal contamination of the cell culture. In addition, the use of E. coli strains with reduced endotoxins, such as endotoxin‐free ClearColi™ BL21(DE3) Electrocompetent Cells (BIOSEARCH Technologies, cat. no. 60810‐1), could be helpful.
High DNA purity and quality
Before transfection, Type I and Type II carrier vectors (A167 and A169) carrying genomic DNA fragments >30 kb (BAC DNA) should be checked by contour‐clamped homogeneous electric field gel electrophoresis (CHEF). This step is necessary to confirm that the BAC DNA is intact and has been isolated predominantly as covalently closed circular molecules (cccDNA). All measures should be taken to ensure that only circular BAC DNAs are transfected into the cells. This includes preserving DNA integrity during preparation using cold solutions and removal of nicked and linearized DNA molecules with exonucleases. In addition, linear molecules have a higher rate of random integration into the host genome compared to covalently closed circular supercoiled DNA, which would reduce the efficiency of the IIS system and lead to a higher background.
Furthermore, the toxicity of transfection agents limits the quantity of DNA that can be transfected into a cell. Thus, BAC DNA preparation has to be pure, without any contaminating bacterial genomic DNA that will reduce the actual amount of BAC DNA that can be transfected into the cells and integrated into the IIS‐alphoidtetO‐HAC. This limitation becomes a significant factor when we transfect large BAC DNA molecules as each microgram of BAC DNA has fewer BAC molecules. Removal of bacterial genomic DNA can be accomplished by exonuclease treatment.
Troubleshooting
See Table 1 for a list of common problems with the protocols, their causes, and potential solutions.
Table 1.
Troubleshooting
| Problem | Possible cause | Solution |
|---|---|---|
|
Sectored colonies A single colony is composed of red and green cells |
Proper integration into the IIS‐alphoidtetO‐HAC occurred late during the colony's growth and did not occur in all cells Concentration of counterselection agent insufficient to kill cells with incorrect marker |
Cells with the correct marker can be rescued by dispersing the colony to single cells and sub‐cloning Concentration of counterselection agent should be increased |
| Cells are simultaneously red and green |
This can happen if ФC31 fails during the first round of integration (Fig. 8B) or if ФBT1 fails during the second round of integration (Fig. 8D). In that case, a single cell expresses both PCF and GHT markers. Those cells can be eliminated only by ganciclovir during the first round or by 5‐fluorocytosine during the second round (Figs. 8B and 8D). Concentration of counterselection agent, ganciclovir or 5‐fluorocytosine, is insufficient to kill the cells expressing the wrong marker |
Increase concentration of counterselection agent used until two‐colored cells are dead. Then, restart experiment from the beginning, using this higher concentration of counterselection agent. Increase concentration of the appropriate selection agent |
| Colorless (non‐fluorescent) colonies |
Insufficient concentration of selection agent (hygromycin or puromycin) to prevent marker silencing due to heterochromatin spread Selection agents degraded |
Slowly increase concentration of the appropriate selection agent in a stepwise manner. Check for reappearance of fluorescence as selection agent selects for cells with increased marker expression. If fluorescence does not reappear with increased concentration of selection agent AND the colony remained viable, check that the selection agent has not degraded from prolonged storage at 4°C or excessive heating from repeated freeze‐thaw cycles. Discard old selection agent and aliquot fresh stock of selection agents to vials of smaller volume before use. If selection agents are kept frozen, storage at ‐80°C is suggested. |
| Phenotype not matching expected genotype: “Fakes” | Heterochromatin silencing of one marker, giving the impression that one of the markers has been lost |
The colonies under investigation should be split with one half preserved and other half for study Screen out colonies that are “faking” their genotype by using gradually increasing concentrations of selection agents to select for gene reactivation Alternatively, apply histone deacetylase (HDAC) inhibitor to (i.e., trichostatin a) to help reverse heterochromatin silencing. Be aware that the HDAC concentration used may kill the cells and/or cause HAC destabilization. |
| Large numbers of “fake” colonies |
Insufficient concentration of counterselection agent used Too much time given for counterselection marker to degrade, allowing emergence of marker silencing by heterochromatin |
Increase concentration of counterselection agent used Decrease the time between transfection and application of counterselectable agent |
| On application of selection agent, cells look unhealthy but do not detach from plate | Lower than normal concentration of puromycin or hygromycin used |
Perform transformation in one well of a 6‐well plate, then 1 day after selection has begun, disperse cells to a 10‐cm plate. Dying cells do not reattach. Increase concentration of selection agent, if possible |
| Few colonies of the correct fluorescence obtained | If carrier vector has a large genomic insert, there may be insufficient molecules being transfected into host cell |
Make sure all plasmid and BAC used are free of bacterial genomic DNA. Use exonuclease to remove linearized DNA. Instead of co‐transfecting the integrase plasmid and carrier vector, these transfections can be done sequentially. First transfect the host cells at lower than normal confluence with the integrase plasmid. Then, allow the cell culture to recover for a day before transfecting the same cells with the carrier vector. |
| No colonies obtained |
HAC lost Selection agents Too much integrase expression vector used, leading to overexpression of integrase protein which form inactive protein aggregates. Too little carrier vectors used. Mutation in attBΦC31, attBΦBT1, or loxP sites Endotoxin contamination of DNA used in transfection Standard 37°C incubation temperature of mammalian cell culture reduces activity of ΦC31 and ΦBT1 bacteriophage integrase |
Maintain HAC selection with blasticidin Give sufficient time for building up of new selection marker and degradation of old counterselection marker. Both the timing and concentration are factors. Restart experiment with empty vector and optimize quantity of integrase expression plasmid or carrier vector used before proceeding to actual experiment DNA sequence the platform cassette to make sure the integrase attachment sites have not mutated. Pick a different colony to use if mutations have occurred. Repurify vector with endotoxin‐free kit. Remember to use wash buffer and elute buffer that are both endotoxin free and filter sterilized. E. coli strains with reduced endotoxin may be used to produce the DNA. Lower post‐transformation incubation temperature to 30°C for 2 days to promote recombinase activity before returning to normal temperature. Optimum temperature may vary with cell line. |
Understanding Results
Usually, the average number of colonies with correct fluorescence (+mCherry/‐eGFP or +eGFP/‐mCherry), after co‐transfection of 1 μg of Type I carrier vector A167 and 0.1 μg of A139 vector or 1 μg Type II carrier vector A169 and 0.1 μg of A135‐JH vector into ∼1 × 105 CHO cells carrying the IIS‐alphoidtetO‐HAC, is equal to 20 to 40. After PCR analysis with the corresponding diagnostic primers, the number of colonies with the correct insertion is approximately ten to twenty. Typically, the yield of colonies for the basic protocols depends on the quality of BAC DNA and the length of the desired DNA fragments to be inserted. Usually, the average number of colonies with correct fluorescence after co‐transfection of 1 μg of BAC DNA of 30 to 50 kb in size (A167 or A169 vectors carrying genomic DNA fragments of interest) and 0.1 μg of A139 or A135‐JH vectors and ∼1 × 105 CHO cells is ten to fifteen. The user should expect fewer colonies if transfection is performed using a BAC >50 kb. After negative selection and PCR with the corresponding diagnostic primers, the number of colonies with the correct insertion will typically be about two to five. Usually, FISH analysis reveals that 80% to 90% of the colonies contain the intact HAC that stably propagates in the cells and is maintained as one copy per cell. About 10% to 20% of the colonies may possess HAC aberrations such as integration of the HAC into the host chromosomes or HAC instability or multiplication.
Time Considerations
The full procedure depends on the number of genomic DNA fragments of interest to be inserted into the IIS‐alphoidtetO‐HAC. Each round of DNA insertion described in the basic protocols can be completed within 4 weeks. The user should start with exponentially growing hamster CHO cells. For each round of insertion of the genomic DNA fragments of interest into the IIS‐alphoidtetO‐HAC, the transfection itself takes 1 day to perform. The rest of the time is required for selection and growth of the colonies. The speed of colony formation and growth depends on the characteristics of the cells used, either hamster CHO cells or another recipient cells. Generally, rapidly proliferating cells form colonies faster. The user should be aware that the selective agents (puromycin, ganciclovir or hygromycin B, and 5‐fluorocytosine) that affect cell growth and colony formation may be significantly slower under these conditions. The user should also consider that the preparation steps for the protocols may take 2 to 3 weeks. These steps include either TAR cloning of the genomic DNA fragments of interest or ligation of them into Type I and Type II carrier vectors A167 and A169 (see Strategic Planning).
Author Contributions
Nicholas C. O. Lee: Conceptualization, methodology, writing original draft; Nikolai S. Petrov: Methodology, writing original draft; Vladimir Larionov: Conceptualization, funding acquisition; Natalay Kouprina: Conceptualization, writing original draft, supervision.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the NIH Intramural Research Program, National Cancer Institute, Center for Cancer Research, U. S. (N.K., N.C.P, V.L.; ZIA BC010413).
Lee, N. C. O. , Petrov, N. S. , Larionov, V. , & Kouprina, N. (2021). Assembly of multiple full‐size genes or genomic DNA fragments on human artificial chromosomes using the iterative integration system. Current Protocols, 1, e316. doi: 10.1002/cpz1.316
Contributor Information
Nicholas C. O. Lee, Email: nicholasleechengooi@gmail.com.
Natalay Kouprina, Email: kouprinn@mail.nih.gov.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Literature Cited
- Arii, J. , Kato, K. , Kawaguchi, Y. , Tohya, Y. , & Akashi, H. (2009). Analysis of herpesvirus host specificity determinants using herpesvirus genomes as bacterial artificial chromosomes. Microbiology and Immunology, 53(8), 433–441. doi: 10.1111/j.1348-0421.2009.00147.x. [DOI] [PubMed] [Google Scholar]
- Auriche, C. , Di Domenico, E. G. , Pierandrei, S. , Lucarelli, M. , Castellani, S. , Conese, M. , … Ascenzioni, F. (2010). CFTR expression and activity from the human CFTR locus in BAC vectors, with regulatory regions, isolated by a single‐step procedure. Gene Therapy, 17(11), 1341–1354. doi: 10.1038/gt.2010.89. [DOI] [PubMed] [Google Scholar]
- Ebersole, T. , Okamoto, Y. , Noskov, V. N. , Kouprina, N. , Kim, J. H. , Leem, S. H. , … Larionov, V. (2005). Rapid generation of long synthetic tandem repeats and its application for analysis in human artificial chromosome formation. Nucleic Acids Research, 33(15), e130. doi: 10.1093/nar/gni129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole, T. , Kim, J. H. , Samoshkin, A. , Kouprina, A. , Pavlicek, A. , White, R. , … Larionov, V. (2011). tRNA genes protect a reporter gene from epigenetic silencing in mouse cells. Cell Cycle, 10, 2779–2791. doi: 10.4161/cc.10.16.17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr, C. J. , Bayne, R. A. L. , Kipling, D. , Mills, W. , Critcher, R. , & Cooke, H. J. (1995). Generation of a human X‐derived minichromosome using telomere‐associated chromosome fragmentation. EMBO Journal, 14, 5444–5454. doi: 10.1002/j.1460-2075.1995.tb00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimpel, A. L. , Katsikis, G. , Sha, S. , Maloney, A. J. , Hong, M. S. , Nguyen, T. N. T. , … Braatz, R. D. (2021). Analytical methods for process and product characterization of recombinant adeno‐associated virus‐based gene therapies. Molecular Therapy—Methods & Clinical Development, 20, 740–754. doi: 10.1016/j.omtm.2021.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, D. C. , Johnson‐Pais, T. L. , Grubbs, B. , Bernal, R. , Leach, R. J. , & Padalecki, S. S. (2008). Maspin reduces prostate cancer metastasis to bone. Urologic Oncology, 26(6), 652–658. doi: 10.1016/j.urolonc.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Harrington, J. J. , Van Bokkelen, G. , Mays, R. W. , Gustashaw, K. , & Willard, H. F. (1997). Formation of de novo centromeres and construction of first‐generation human artificial microchromosomes. Nature Genetics, 15, 345–355. doi: 10.1038/ng0497-345. [DOI] [PubMed] [Google Scholar]
- Head, K. , Gong, S. , Joseph, S. , Wang, C. , Burkhardt, T. , Rossi, M. R. , … Cowell, J. K. (2007). Defining the expression pattern of the LGI1 gene in BAC transgenic mice. Mammalian Genome, 18(5), 328–337. doi: 10.1007/s00335-007-9024-6. [DOI] [PubMed] [Google Scholar]
- Heller, R. , Brown, K.E. , . Burgtorf, C. , & Brown, W.R. A. (1996). Mini‐chromosomes derived from the human Y chromosome by telomere directed chromosome breakage. Proceedings of the National Academy of Sciences of the USA, 93, 7125–7130. doi: 10.1073/pnas.93.14.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbitt, O. C. , Harbottle, R. P. , Waddington, S. N. , Bursill, C.A. , Coutelle, C. , Channon, K.M. , & Wade‐Martins, R. (2007). Delivery and long‐term expression of a 135 kb LDLR genomic DNA locus in vivo by hydrodynamic tail vein injection. The Journal of Gene Medicine, 9(6), 488–497. doi: 10.1002/jgm.1041. [DOI] [PubMed] [Google Scholar]
- Hiratsuka, M. , Uno, N. , Ueda, K. , Kurosaki, H. , Imaoka, N. , Kazuki, K. , … Oshimura, M. (2011). Integration‐free iPS cells engineered using human artificial chromosome vectors. PLoS One, 6(10), e25961. doi: 10.1371/journal.pone.0025961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma, K. , Abe, S. , Endo, T. , Uno, N. , Oshimura, M. , Ohbayashi, T. , … Kazuki, Y. (2018). Development of a multiple‐gene‐loading method by combining multi‐integration system‐equipped mouse artificial chromosome vector and CRISPR‐Cas9. PLoS One, 13(3), e0193642. doi: 10.1371/journal.pone.0193642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, L. , Wan, J. , Wu, Y. , Tian, Y. , Yao, Y. , Yao, S. , … Xu, H. (2021). Challenges in adeno‐associated virus‐based treatment of central nervous system diseases through systemic injection. Life Sciences, 270, 119142. doi: 10.1016/j.lfs.2021.119142. [DOI] [PubMed] [Google Scholar]
- Iida, Y. , Kim, J. H. , Kazuki, Y. , Hoshiya, H. , Takiguchi, M. , Hayashi, M. , … Oshimura, M. (2010). Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Research, 17(5), 293–301. doi: 10.1093/dnares/dsq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illenye, S. , & Heintz, N. H. (2004). Functional analysis of bacterial artificial chromosomes in mammalian cells: Mouse Cdc6 is associated with the mitotic spindle apparatus. Genomics, 83(1), 66–75. doi: 10.1016/S0888-7543(03)00205-2. [DOI] [PubMed] [Google Scholar]
- Ikeno, M. , Grimes, B. , Okazaki, T. , Nakano, M. , Saitoh, K. , Hoshino, H. , … Masumoto, H. (1998). Construction of YAC‐based mammalian artificial chromosomes. Nature Biotechnology, 16(5), 431–439. doi: 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- Ikeno, M. , & Hasegawa, Y. (2020). Applications of bottom‐up human artificial chromosomes in cell research and cell engineering. Experimental Cell Research, 390(1), 111793. doi: 10.1016/j.yexcr.2019.111793. [DOI] [PubMed] [Google Scholar]
- Immidisetti, A. V. , Nwagwu, C. D. , Adamson, D. C. , Patel, N. V. , & Carbonell, A. M. (2021). Clinically explored virus‐based therapies for the treatment of recurrent high‐grade glioma in adults. Biomedicines, 9(2), 138. doi: 10.3390/biomedicines9020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona, R. L. (2015). De novo formed satellite DNA‐based mammalian artificial chromosomes and their possible applications. Chromosome Research, 23, 143–157. doi: 10.1007/s10577-014-9458-0. [DOI] [PubMed] [Google Scholar]
- Kazuki, Y. , & Oshimura, M. (2011). Human artificial chromosomes for gene delivery and the development of animal models. Molecular Therapy, 19, 1591–1601. doi: 10.1038/mt.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki, Y. , Uno, N. , Abe, S. , Kajitani, N. , Kazuki, K. , Yakura, Y. , … Tomizuka, K. (2020). Engineering of human induced pluripotent stem cells via human artificial chromosome vectors for cell therapy and disease modeling. Molecular Therapy‐Nucleic Acids, 23, 629–639. doi: 10.1016/j.omtn.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Kononenko, A. , Erliandri, I. , Kim, T. A. , Nakano, M. , Iida, Y. , … Kouprina, N. (2011). Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proceedings of the National Academy of Sciences of the USA, 108, 20048–20053. doi: 10.1073/pnas.1114483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko, A. V. , Bansal, R. , Lee, N. C. , Grimes, B. R. , Masumoto, H. , Earnshaw, W. C. , … Kouprina, N. (2014). A portable BRCA1‐HAC (human artificial chromosome) module for analysis of BRCA1 tumor suppressor function. Nucleic Acids Research, 42(21), e164. doi: 10.1093/nar/gku870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , & Larionov, V. (2008). Selective isolation of genomic loci from complex genomes by transformation‐associated recombination (TAR cloning) in yeast Saccharomyces cerevisiae. Nature Protocols, 3(3), 371–377. doi: 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- Kouprina, N. , Samoshkin, A. , Erliandri, I. , Nakano, M. , Lee, H. S. , Fu, H. , … Larionov, V. (2012). Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synthetic Biology, 1(12), 590–601. doi: 10.1021/sb3000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Earnshaw, W. C. , Masumoto, H. , & Larionov, V. (2013). A new generation of human artificial chromosomes for functional genomics and gene therapy. Cellular and Molecular Life Sciences, 70, 1135–1148. doi: 10.1007/s00018-012-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Tomilin, A. N. , Masumoto, H. , Earnshaw, W. C. , & Larionov, V. (2014). Human artificial chromosome‐based gene delivery vectors for biomedicine and biotechnology. Expert Opinion on Drug Delivery, 11, 517–535. doi: 10.1517/17425247.2014.882314. [DOI] [PubMed] [Google Scholar]
- Kouprina, N. , Petrov, N. , Molina, O. , Liskovykh, M. , Pesenti, E. , Ohzeki, J. I. , … Larionov, V. (2018). Human artificial chromosome with regulated centromere: A tool for genome and cancer studies. ACS Synthetic Biology, 7(9), 1974–1989. doi: 10.1021/acssynbio.8b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina, N. , Kim, J. H. , & Larionov, V. (2021). Highly selective, CRISPR/Cas9‐mediated isolation of genes and genomic loci from complex genomes by TAR cloning in yeast. Current Protocols, 1(8), e207. doi: 10.1002/cpz1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, N. C. O. , Kononenko, A. V. , Lee, H. S. , Tolkunova, E. N. , Liskovykh, M. A. , Masumoto, H. , … Kouprina, N. (2013). Protecting a transgene expression from the HAC‐based vector by different chromatin insulators. Cellular and Molecular Life Sciences, 70(19), 3723–3737. doi: 10.1007/s00018-013-1362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, N. C. O. , Larionov, V. , & Kouprina, N. (2015). Highly efficient CRISPR/Cas9‐mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Research, 43(8), e55. doi: 10.1093/nar/gkv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, N. C. O. , Kim, J. H. , Petrov, N. S. , Lee, H. S. , Masumoto, H. , Earnshaw, W. C. , … Kouprina, N. (2018). Method to assemble an unlimited number of genomic DNA fragments on a human artificial chromosome with regulated kinetochore. ACS Synthetic Biology, 7(1), 63–67. doi: 10.1021/acssynbio.7b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liskovykh, M. , Lee, N. C. O. , Larionov, V. , & Kouprina, N. (2016). Moving towards a higher efficiency of microcell‐mediated chromosome transfer (MMCT). Molecular Therapy—Methods & Clinical Development, 3, 16043. doi: 10.1038/mtm.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liskovykh, M. , Larionov, V. , & Kouprina, N. (2021). Highly efficient microcell‐mediated chromosome transfer of HACs bearing a specific gene into mammalian cells. Current Protocols, 1(9), e236. doi: 10.1002/cpz1.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. , Liu, J. , Wang, Y. , Su, J. , Wu, Y. , Hu, G. , … Zhang, Y. (2013). PhiC31 integrase‐mediated genomic integration and stable gene expression in the mouse mammary gland after gene electrotransfer. The Journal of Gene Medicine, 15(10), 356–365. doi: 10.1002/jgm.2723. [DOI] [PubMed] [Google Scholar]
- Ma, X. Y. , Hill, B. D. , Hoang, T. , & Wen, F. (in press). (2021). Virus‐inspired strategies for cancer therapy. Seminars in Cancer Biology, doi: 10.1016/j.semcancer.2021.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijanović, O. , Branković, A. , Borovjagin, A. , Butnaru, D. V. , Bezrukov, E. A. , Sukhanov, R. B. , … Ulasov, I. (2020). Battling neurodegenerative diseases with adeno‐associated virus‐based approaches. Viruses, 12(4), 460. doi: 10.3390/v12040460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moralli, D. , & Monaco, Z. L. (2020). Gene expressing human artificial chromosome vectors: Advantages and challenges for gene therapy. Experimental Cell Research, 390(1), 111931. doi: 10.1016/j.yexcr.2020.111931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriwaki, T. , Abe, S. , Oshimura, M. , & Kazuki, Y. (2020). Transchromosomic technology for genomically humanized animals. Experimental Cell Research, 390(2), 111914. doi: 10.1016/j.yexcr.2020.111914. [DOI] [PubMed] [Google Scholar]
- Nakano, M. , Cardinale, S. , Noskov, V. N. , Gassmann, R. , Vagnarelli, P. , Kandels‐Lewis, S. , … Masumoto, H. (2008). Inactivation of a human kinetochore by a specific targeting of chromatin modifiers. Developmental Cell, 14, 507–522. doi: 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimura, M. , Uno, N. , Kazuki, Y. , Katoh, M. , & Inoue, T. (2015). A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio‐medical challenges. Chromosome Research, 23(1), 111–133. doi: 10.1007/s10577-014-9459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomartsev, S. V. , Sinenko, S. A. , Skvortsova, E. V. , Liskovykh, M. A. , Voropaev, I. N. , … Tomilin, A. N. (2020). Human AlphoidtetO artificial chromosome as a gene therapy vector for the developing hemophilia a model in mice. Cells, 9(4), 879. doi: 10.3390/cells9040879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser, I. , Sarov, M. , Hutchins, J. R. , Hériché, J. K. , Toyoda, Y. , . Pozniakovsky, A. , … Hyman, A. A. (2008). BAC TransgeneOmics: A high‐throughput method for exploration of protein function in mammals. Nature Methods, 5(5), 409–415. doi: 10.1038/nmeth.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinenko, S. A. , Ponomartsev, S. V. , & Tomilin, A. N. (2021). Pluripotent stem cell‐based gene therapy approach: Human de novo synthesized chromosomes. Cellular and Molecular Life Sciences, 78(4), 1207–1220. doi: 10.1007/s00018-020-03653-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T. , Kazuki, Y. , Oshimura, M. , & Hara, T. (2014). A novel system for simultaneous or sequential integration of multiple gene‐loading vectors into a defined site of a human artificial chromosome. PLoS One, 9(10), e110404. doi: 10.1371/journal.pone.0110404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth, A. , Fodor, K. , Praznovszky, T. , Tubak, V. , Udvardy, A. , Hadlaczky, G. , … Katona, R. L. (2014). Novel method to load multiple genes onto a mammalian artificial chromosome. PLoS One, 9, e85565. doi: 10.1371/journal.pone.0085565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, A. B. , Sugyo, A. , Sudo, H. , Sagara, M. , Ishikawa, A. , Ohtsuki, M. , … Harada, Y. N. (2006). Defective repair of radiation‐induced DNA damage is complemented by a CHORI‐230‐65K18 BAC clone on rat chromosome 4. Genomics, 87(2), 236–242. doi: 10.1016/j.ygeno.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Wang, J. L. , Scheitler, K. M. , Wenger, N. M. , & Elder, J. B. (2021). Viral therapies for glioblastoma and high‐grade gliomas in adults: A systematic review. Neurosurgical Focus , 50(2), E2. doi: 10.3171/2020.11.FOCUS20854. [DOI] [PubMed] [Google Scholar]
- Yamaguchi, S. , Kazuki, Y. , Nakayama, Y. , Nanba, E. , Oshimura, M. , & Ohbayashi, T. (2011). A Method for producing transgenic cells using a multi‐integrase system on a human artificial chromosome vector. PLoS One, 6, e17267. doi: 10.1371/journal.pone.0017267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura, Y. , Nakamura, K. , Endo, T. , Kajitani, N. , Kazuki, K. , Kazuki, Y. , … Ohbayashi, T. (2015). Mouse embryonic stem cells with a multi‐integrase mouse artificial chromosome for transchromosomic mouse generation. Transgenic Research, 24, 717–727. doi: 10.1007/s11248-015-9884-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Wu, W. , Zhang, J. , & Xia, X. (2021). Merits of the 'good' viruses: The potential of virus‐based therapeutics. Expert Opinion on Biological Therapy, 21(6), 731–740. doi: 10.1080/14712598.2021.1865304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.