

Abstract
In the bone marrow (BM) microenvironment, where breast cancer (BC) disseminated tumour cells (DTCs) can remain dormant for decades, NG2+/Nestin+ mesenchymal stem cells (MSCs) promote hematopoietic stem cell quiescence. Here, we reveal that periarteriolar BM-resident NG2+/Nestin+ MSCs can also instruct BC DTCs to enter dormancy. NG2+/Nestin+ MSCs produce TGFβ2 and BMP7 and activate a quiescence pathway dependent on TGFBRIII and BMPRII, which via p38-kinase result in p27 induction. Genetic depletion of MSCs or conditional knock-out of TGFβ2 in MSCs using an NG2-CreER driver led to bone metastatic outgrowth of otherwise dormant p27+/Ki67- DTCs. Also ER+ BC patients without systemic recurrence displayed higher frequency of TGFβ2 and BMP7 detection in the BM. Our results provide a direct proof that HSC dormancy niches control BC DTC dormancy and suggest that aging or extrinsic factors that affect the NG2+/Nestin+ MSC niche homeostasis may result in a break from dormancy and BC bone relapse.
Introduction
Metastases, which are derived from disseminated tumour cells (DTCs), are the major source of cancer-related deaths from solid cancers1. Post-extravasation DTCs can remain in a dormant state from prolonged periods2 and years to decades can lapse before dormant DTCs emerge as overt lesions. We postulate that targeting dormant DTCs is the shortest path to change patient outcomes by curtailing metastatic outgrowth. However, to achieve this goal we must understand the cancer cell intrinsic and micro-environmental mechanisms that control DTC dormancy and reactivation.
The bone marrow (BM) is a site where dormant DTCs are found and where metastasis can develop in various cancers after prolonged periods of clinical “remission”2–9. In trying to understand how the BM microenvironment might control DTC dormancy, we8,10 and others2,4,11,12 found that in both humans and mice, this microenvironment is a restrictive site for metastasis initiation. This is due to the presence of several cues, such as TGFβ210, BMP713,14, GAS615–17 and LIF12,18, which induce DTC dormancy. Studies in mostly 2D or 3D in vitro models, proposed that mesenchymal stem cells (MSCs)19, vascular endothelial cells11,20 and/or osteoblasts16,21,22 may be the source of dormancy cues for cancer cells. However, the function of such niche cells in vivo has not been formally tested.
There is a long-standing hypothesis that the niches that control hematopoietic stem cell (HSC) dormancy may instruct DTCs to become dormant23. A prior study in prostate cancer has drawn a connection between the HSC niche and dormancy of DTCs24. While informative, these studies did not functionally dissect in depth and in vivo the role of key cell types that regulate HSC dormancy in regulating DTC quiescence. Further, the niche influence has been inferred from the analysis of cancer cells recovered from the BM or from indirect competition assays. Thus, there is still a critical need to understand the cellular components and how BM niches enforce DTC dormancy.
Dormancy of HSCs in the BM is a robust and long-lived process that, if unperturbed, results in HSCs dividing and self-renewing only 4 to 5 times in the lifetime of a mouse25. Such powerful mechanism of quiescence and self-renewal cycles, might explain how DTCs, if responsive to niche signals, could persist for decades in the BM of breast cancer (BC) patients. The HSC microenvironment in the bone marrow is a complex multicellular network promoting HSC dormancy, self-renewal and differentiation into lineage-committed progenitors26. Previous studies have revealed that peri-arteriolar stromal cells, enriched in MSC activity, innervated by the sympathetic nervous system, and expressing the neural markers NG2 and Nestin (mesenchymal stem and progenitor cells, hereafter referred to as NG2+/Nestin+ MSCs for simplicity), are critical for the control of HSC quiescence and hematopoiesis27. Importantly, aging-induced alterations causing replicative stress damage or sympathetic neuropathy from infiltrating leukaemia cells, for example, eliminates the control of HSC dormancy by NG2+/Nestin+ MSCs and can fuel malignancy28,29. We and others also discovered that TGFβ2 and BMP7 in the BM milieu are key inducers of DTC dormancy in various epithelial cancers10,27. However, the source of these cytokines has remained elusive.
Here, we show using in vitro and in vivo assays that NG2+/Nestin+ MSCs are a source of TGFβ2 and BMP7 and that both the niche MSCs and the cytokines maintain the dormancy of different cancer cell types via TGFβRIII and BMPRII respectively, p38 and p27 signalling. Furthermore, depletion of the NG2+/Nestin+ MSCs or knockout of TGFβ2 specifically from NG2+/Nestin+ MSCs led to metastatic outgrowth in the BM. Also in 3D organoid assays “revitalized” but not “aged” NG2+/Nestin+ MSCs can reprogram malignant cells into a dormancy-like phenotype. Lastly, detectable BMP7 and TGFβ2 levels were observed at higher frequency in the BM of estrogen receptor positive (ER+) BC patients without systemic recurrence compared to patients with systemic recurrence and BMP7 presence was associated with a longer time to metastatic recurrence after therapy. Our results, pinpoint a functional link between the niches that control adult hematopoietic stem cell quiescence and DTC dormancy.
Results
Depletion of NG2+/Nestin+ MSCs from the BM niche reactivates dormant E0771 DTCs.
The BM frequently harbours dormant DTCs in both humans and mice2,4,8,10–12. As observed in humans and prior models of BM DTC detection, in both spontaneous (MMTV-HER2, 30 and Fig1a) and experimental metastatic BC mouse models (intra-cardiac injected E0771-GFP (Fig1b) and MMTV-PyMT-CFP cells) we could only find few DTCs in the BM of femurs, sternum and calvaria bones (150–400 DTCs per million BM cells). In wild type animals these DTCs were non-metastatic or developed lesions at low frequency in the bones (~5–16%, Fig1d and 5d). In contrast, these mice showed 100% incidence of metastasis to the lung (data not shown), which is frequently a permissive site for metastasis10.
Figure 1. Depletion of NG2+/Nestin+ MSCs awakens dormant DTCs in the BM.
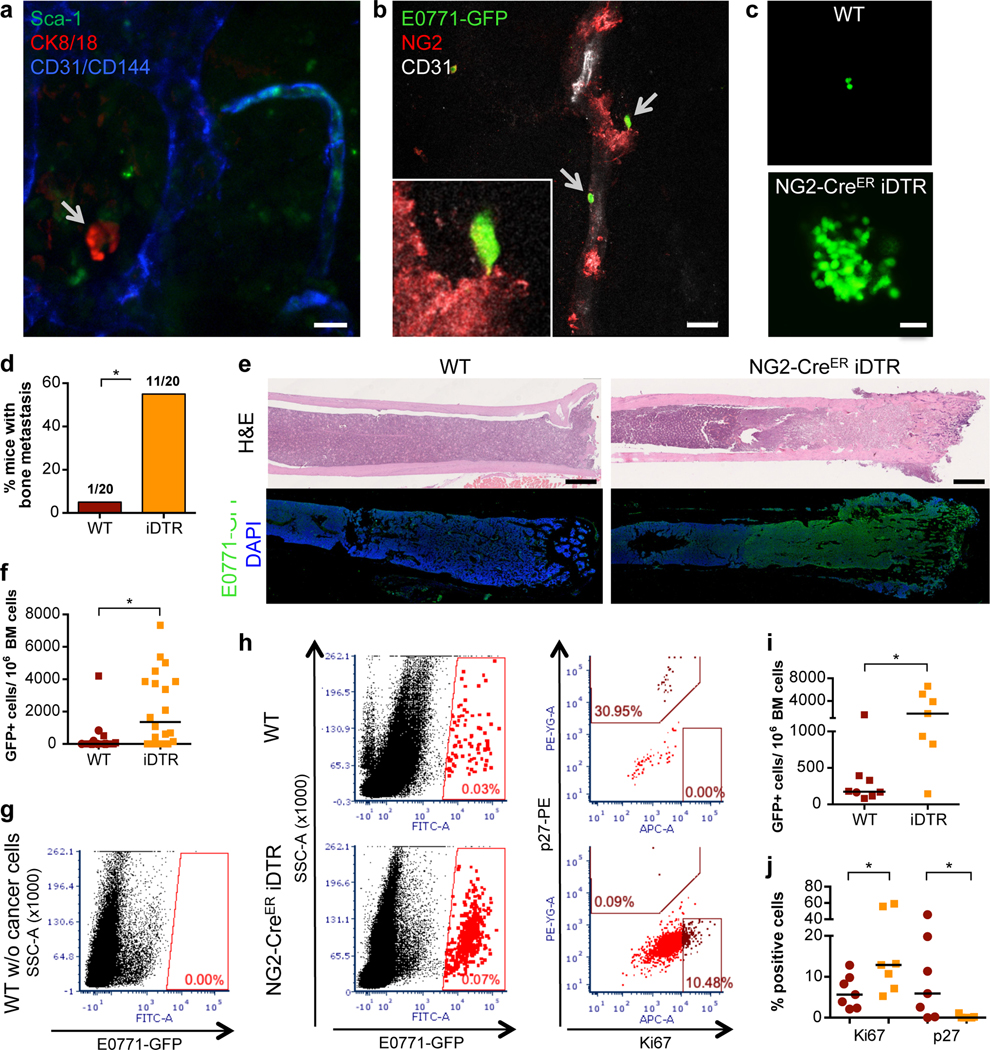
a-b. Whole bone imaging of MMTV-Neu (GEM model, CK8/18+ cancer cells, representative image of 2 independent experiments) and E0771-GFP (intra-cardiac injected) BC cells (representative image of 5 independent experiments). Scale bars: 10μm (a), 20μm (b). c-f. Effect of NG2+/Nestin+ MSC depletion prior to BM seeding by DTCs (n=20 mice per condition from 3 independent experiments. Experimental design in ED Fig1a). c. Representative images of E0771-GFP+ DTC clusters in BM flushes from wild type (WT) and NG2+/Nestin+ MSC depleted mice (NG2-CreERiDTR). Scale bar 50μm. d. Incidence of bone metastasis (>1000 GFP+ DTCs/106 BM cells) 2 weeks after cancer cell injections (2-tailed Fisher’s exact test, p=0.001). e. H&E and GFP staining of WT and NG2-CreERiDTR bones. Scale bars, 500μm. f. Number of E0771-GFP cells per million of BM cells after BM flushing, counted manually (median, 2-tailed Mann–Whitney U-test, p=0.0004). g-h. Representative plots and gates used in FACS for detection and characterization of E0771-GFP cells in BM flushes. i. Number of E0771-GFP cancer cells per million of BM cells, counted by FACS (n=15 mice, median, 2-tailed Mann–Whitney U-test, p=0.021). j. Percentage of E0771-GFP cells Ki67high and p27+ (FACS) (n=14, median, 2-tailed Mann–Whitney U-test, p=0.038 and 0.05). *p≤0.05 (p-values indicated above).
Different factors and BM stromal cells were proposed to induce and maintain DTC dormancy11,16,19–22. However, in vivo studies functionally linking specific BM cells and dormancy-inducing factors to DTC dormancy are still missing. NG2+/Nestin+ MSCs are critical inducers of HSC quiescence and hematopoiesis27. Thus, we set out to test whether, like dormancy of HSCs27, the NG2+/Nestin+ MSCs could also induce dormancy of DTCs. To this end we took advantage of the C57BL/6 NG2-CreERiDTR mouse model27,31. Treatment of mice with 2 mg of tamoxifen (TAM) for 5 days leads to Cre recombinase activation and the expression of the diphtheria toxin receptor (DTR) in periarteriolar NG2+ cells, which upon 2-day treatment with 250ng of diphtheria toxin (DT) causes a targeted depletion of NG2+ cells27,31. This strategy reproducibly led to the depletion of ~50% of NG2+/Nestin+ MSCs (measured as CD45-Ter119-CD31-PDGFRa+CD51+ MSCs (ED Fig1a–c), which overlap with NG2+/Nestin+ MSCs27,31) in the BM of long bones. Twenty-four hours after DT treatment, we performed intra-cardiac injection of 2×105 E0771-GFP cells per mouse (ED Fig1a and Suppl Movie1) and two weeks later, we measured the burden of BM DTCs. All mice had E0771-GFP+ DTCs in the BM detectable by FACS (Fig1g–h), corroborating the injection efficiency and that these cells can persist in the BM at a low burden in all injected mice. Only 5% (1 out of 20) of wild-type (WT) mice showed more than 1000 DTCs/million BM cells (manually counted after BM flush). In contrast, 55% (11 out of 20) of NG2-CreERiDTR mice with a deficiency of NG2+/Nestin+ MSCs displayed a build-up of E0771-GFP+ colonies in the BM (Fig1c,d). Consistently, histological analysis confirmed that indeed the BM of WT animals exhibited normal histology, while NG2-CreERiDTR mice revealed large areas of bone metastasis and replacement of the BM by E0771-GFP+ cancer cells (Fig1e). Further, quantification of the DTC burden (both manual counting, Fig1f, and FACS assisted, Fig1i) showed a striking increase in number of DTCs in the BM compartment of NG2-CreERiDTR compared to WT mice. FACS staining of Ki67 and p27 also showed a significant increase in the percentage of Ki67+ E0771-GFP+ cells and decrease in p27+ DTCs was detected in NG2-CreERiDTR compared with WT mice (Fig1h–j). The results were reproduced using a MMTV-PyMT-CFP cell line with only 1 out of 5 (20%) control animals showing greater than 103 cancer cells/106 BM cells, while 3 out of 5 (60%) showed reactivation. The burden of cancer cells in the iDTR group animal that reactivated (60%) increased by 10-fold and mice with high burden numbers showed a correlative increase in the percentage of Ki67+ DTCs upon depletion of NG2+/Nestin+ MSCs (ED Fig1d–g). We conclude that a 50% reduction in NG2+/Nestin+ MSCs (measured as CD45-Ter119-CD31-PDGFRa+CD51+ MSCs) can cause bone-damaging metastatic reactivation of otherwise dormant DTCs.
Further characterization revealed that in WT mice, 75% of the solitary E0771-GFP DTCs were positive for p27 (quiescence regulator10, Fig2a,b) and 54% for pATF2 (p38 activated TF32, Fig2d,e). In contrast, these same DTCs were 100% negative for Ki67 and pH3 (Fig2a,c and Fig2d,f), supporting a dormant phenotype (increased expression of pATF2 and p27 and decreased Ki67 and pH3) as reported in different cancer models10,33,34. In mice depleted of NG2+/Nestin+ MSCs, E0771-GFP+ resumed proliferation as evidenced by a reduction in the percentage of p27+ (28%) and pATF2+ (2%) cells and increase of Ki67+ (17%) and pH3+ (16%) in DTC clusters, which are not frequent in WT animals (Fig2a,c and Fig2d,f) and corroborated an awakening of DTCs and shift to a proliferative state.
Figure 2. Dormant to proliferative shift of BM E0771-GFP cancer cells upon depletion of NG2+/Nestin+ MSCs.
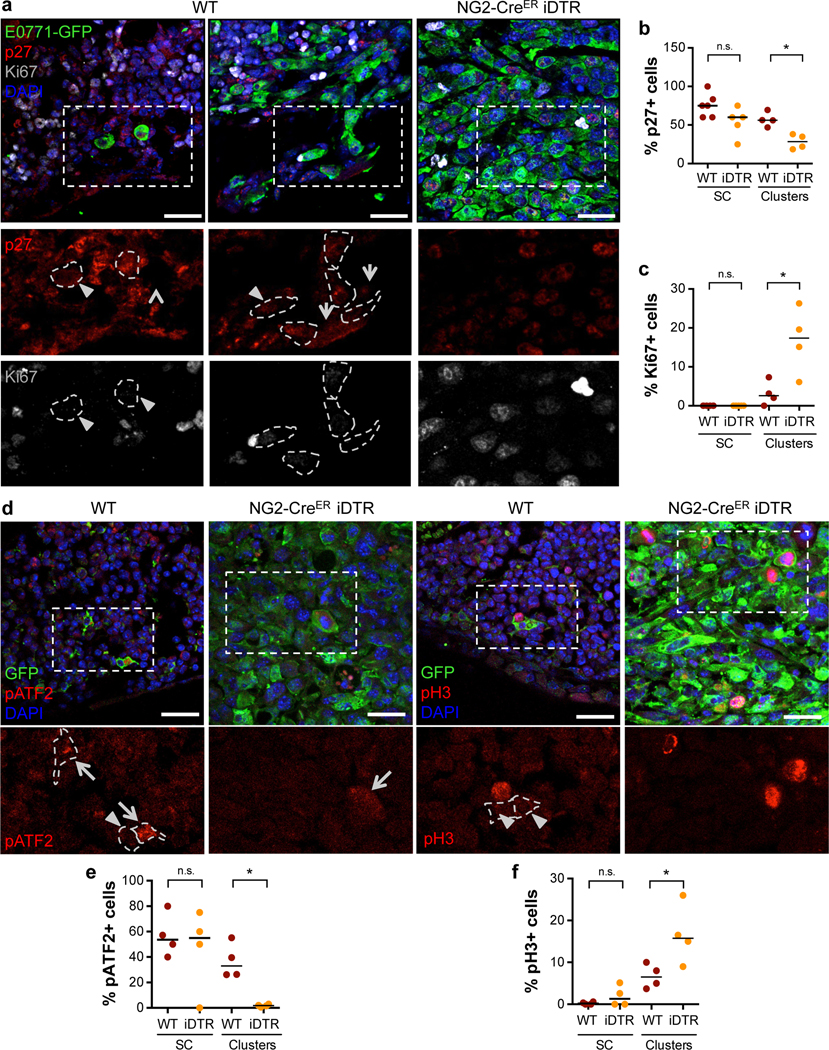
a. Representative images of p27 and Ki67 in E0771-GFP cells in WT and NG2-CreERiDTR bones. Single cells and small clusters are shown in WT mice and a metastasis in NG2-CreERiDTR. Scale bars 25μm; arrows, positive cells; arrowheads, negative cells; dotted lines, GFP+ cells border. b-c. Percentage of p27+ (b) and Ki67+ (c) E0771-GFP cells detected by immunofluorescence (n=6 WT and 5 iDTR mice, median, 2-tailed Mann–Whitney U-test, p=0.029 (b) and 0.05 (c)). d-f. Representative images (d, Scale bars 25μm; arrows, positive cells; arrowheads, negative cells) and quantification of pATF2+ (e) and pH3+ (f) E0771-GFP DTCs in WT and NG2-CreERiDTR bones (n=4 WT and 4 iDTR mice, 2-tailed Mann–Whitney U-test, p=0.03 (e) and 0.05 (f)). *p≤0.05 (p-values indicated above), n.s. not significant.
DT and DTR-mediated cell death can induce inflammation when certain cell types are targeted35–37. However, we previously reported no difference in the number of leukocytes, expression of CXCL12 and KITL (SCF) and no change in vascular volume in the NG2-CreERiDTR model27. Nevertheless, we performed a multiplex ELISA for pro-inflammatory cytokines in the BM supernatant of WT and iDTR mice. We found no differences between the control and iDTR mice in the abundance levels of IL1β, GM-CSF, IL2, IL4, IL6, IL10, IL12p70, MCP-1 or TNFα levels (IFNγ was undetectable – ED Fig2a). Thus, reactivation of dormant DTCs upon NG2+/Nestin+ MSCs depletion cannot be attributed to changes in the inflammatory cytokines we measured.
To exclude that the increased DTC burden after two weeks was not due to differences in vascular permeability or extravasation efficiency between WT and NG2-CreERiDTR mice, a group of animals was analysed 24 hours after intra-cardiac delivery of E0771-GFP cells and dextran-TexasRed injection (ED Fig2b). No differences in the amount of 70,000 MW dextran-TexasRed extravasation in the BM was observed after NG2+/Nestin+ MSC depletion as determined using image analysis (ED Fig2c), reproducing prior results27, and suggesting no obvious alteration in vessel permeability. Further, DTC burden was quantified after expansion of E0771-GFP+ cells 1 week in vitro (due to no or low detection of E0771-GFP+ in fresh BM flush). Similar numbers of E0771-GFP+ cells were found in WT and NG2-CreERiDTR mice (ED Fig2d) arguing that the differences in final metastatic burden were not due to higher vessel permeability and/or early enhanced extravasation of DTCs.
We next tested whether the depletion of NG2+/Nestin+ MSC affected DTC expansion once cancer cells had seeded and established a foothold within the BM niche. To this end we used the same NG2-CreERiDTR mouse model but the treatment with TAM for 5 days was followed by intra-cardiac injection of E0771-GFP cells, which were allowed to lodge for 72 hours in unperturbed niches and only then we did 2 daily treatments with DT (ED Fig2e). Importantly, depletion of NG2+/Nestin+ MSCs after DTCs had lodged in the BM niche also stimulated metastatic outbreak; only a third (1 out of 6) of control WT mice displayed significant expansion of E0771-GFP cells in the BM flushes, while all NG2-CreERiDTR mice (5 out of 5) had a significant increase in frequency of cancer cells per BM cells (ED Fig2f). We conclude that NG2+/Nestin+ MSCs are directly or indirectly responsible for maintaining pro-dormancy niches for DTCs in the BM.
NG2+/Nestin+ MSCs show enhanced production of the dormancy inducers TGFβ2 and BMP7.
We had shown that TGFβ2 in the BM induced DTC dormancy10, while other groups identified BMP7 as an inducer of DTC dormancy13. However, the functional source of TGFβ2 or other factors in the BM in vivo remained unidentified. We wondered whether NG2+/Nestin+ MSCs may regulate dormancy by producing cues, such as TGFβ2. To address this question, we sorted BM CD31-CD45- Nestin-GFPbright (from now on called Nestin-GFP+) and CD31-CD45- Nestin-GFP- cells (from now on called Nestin-GFP-) (Suppl Fig1a) and measured mRNA levels of TGFβ1, BMP2 (involved in dormant DTC reactivation34,38), TGFβ2 and BMP7 (dormancy inducing cues10,13,34). Nestin-GFP+ cells showed higher mRNA levels of TGFβ2 and BMP7 than Nestin-GFP- cells (Fig2a). In addition, because it has been shown that other niche cells have high expression of TGFβ2 39,40, we compared TGFβ1, TGFβ2 and BMP7 mRNA levels in MSCs, osteo-progenitor cells (OPCs) and endothelial cells (ECs). To this end, we sorted MSCs (CD45-CD31-Ter119-PDGFRa+CD51+), OPCs (CD45-Ter119-CD31-ALCAM+, OPCs) and ECs (CD45-Ter119-CD31+vEcad+, ECs) from WT mice (Suppl Fig1b) and we found that TGFβ1 is equally expressed by MSCs, OPCs and ECs, TGFβ2 was higher in MSCs and OPCs and BMP7 was uniquely produced by MSCs (Fig3b). Thus, MSCs are not an exclusive source of TGFβ2 but are the only niche cells producing both pro-dormancy TGFβ2 and BMP7 cues. Further, ELISA measurements of the BM supernatants from NG2-CreERiDTR mice showed a significant decrease in TGFβ2 and BMP7 levels but not TGFβ1 or TGFβ3 upon NG2+/Nestin+ MSC depletion compared with WT mice (Fig3c), suggesting that pro-dormancy niches established by NG2+/Nestin+ MSCs are a source of TGFβ2 and BMP7 in the BM but that other cells may compensate for the production and total levels of TGFβ1 and TGFβ3. Imaging of TGFβ2 and BMP7 in relation to the Nestin-GFP cells revealed that BMP7 was more predominant in the growth plate (Fig2d) and appeared to accumulate as a secreted factor around the Nestin-GFP+ cells (BMP7 is an extracellular matrix bound factor41,42) or co-localizing with the Nestin-GFP signal (Fig3d–e). While TGFβ2 was found homogeneously distributed across the BM. We could also find it co-localizing with the NG2 and Nestin-GFP signals supporting their production by MSCs (Fig3d–f). We conclude that pro-dormancy NG2+/Nestin+ MSCs are a source of TGFβ2 and BMP7 in the BM.
Figure 3. NG2+/Nestin+ MSCs are as a source of pro-dormancy factors TGFβ2 and BMP7 in the BM.
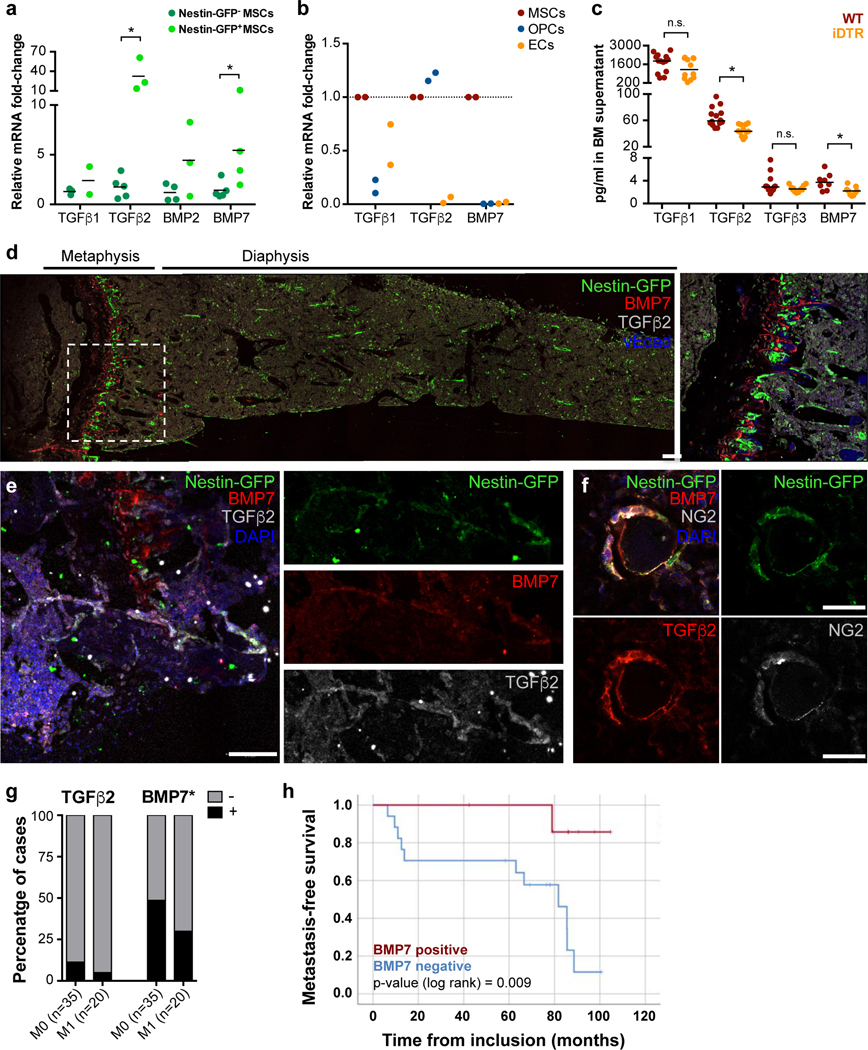
a. mRNA levels of sorted CD45-CD31-Nestin-GFP- and Nestin-GFPbright MSCs from Nestin-GFP mice (n=4 independent experiments, median, 2-tailed Mann–Whitney U-tests, p=0.027 (TGFβ2) and 0.031 (BMP7)). b. mRNA levels of sorted MSCs, OPCs and ECs (MSC population was used as reference, n=2 independent experiments, one-way ANOVA with Geissen-Greenhouse correction, p=0.034 (TGFb2) and 0.005 (BMP7)) c. TGFβ1, 2 and 3 and BMP7 levels in BM supernatant of WT and NG2-CreERiDTR mice 2 weeks after TAM and DT treatments (n=24 mice from 2 independent experiments, median, 2-tailed Mann–Whitney U-tests, p<0.0001 (TGFb2), and 0.011 (BMP7)). d-f. Imaging of Nestin-GFP+ MSCs in Nestin-GFP mice using IF. Dormancy factors TGFβ2 (white, d-e; red, f) and BMP7 (red, d-e) are expressed near MSCs (Nestin-GFP: green d-f; NG2: white, f; representative images of 3 independent experiments). Scale bars: 100μm (d-e) and 25uμm (f). Dotted rectangles (d), high-magnification insert. g-h. Analysis of n=55 ER+ BC patients with (M1) or without (M0) evidence of systemic recurrence (data from SATT clinical study9. g. TGFβ2 and BMP7 levels from BM plasma samples from (2-tailed Fisher’s exact test, p=0.009). h. Metastasis-free survival analysis of patients who did not receive any secondary chemotherapy, excluding treatment-related interpretation bias, separated by detectable BMP7 levels compared to patients with no BMP7. *p≤0.05 (p-values indicated above), n.s. not significant.
TGFβ2 and BMP7 detection in BM supernatant from ER+ BC patients.
Given that the lower levels of TGFβ2 and BMP7 upon NG2+/Nestin+ MSC depletion were associated with reactivation of dormant DTCs leading to metastatic outgrowth (Fig1), we tested the hypothesis that the abundance of these two factors in the bone marrow of BC patients might provide some information on their progression. To this end we tested a small subset of BM plasma samples from early BC patients with ER positive disease, the BC subtype most often experiencing tumor dormancy and late recurrences in the bone, among other sites. These samples were part of a clinical study monitoring DTC status after completion of standard adjuvant anthracycline-containing chemotherapy9. TGFβ2 and BMP7 levels from BM plasma were detected using multiplex ELISA and revealed that patients without systemic recurrence (M0) during follow up had 2.28- and 1.62-fold higher frequency of detectable TGFβ2 and BMP7, respectively, compared to patients with systemic recurrence (M1) (Fig3g). Importantly, metastasis-specific survival analysis of the subgroup of patients who did not receive any secondary chemotherapy, excluding treatment-related interpretation bias, pointed to a markedly improved distant disease-free survival for patients with detectable BMP7 compared to patients with no BMP7 (Fig3h) (regardless of the abundance levels). Survival analysis was not possible for TGFβ2 due to the low number of patients showing detectable TGFβ2 levels. This small pilot analysis supports expanding the analysis of the association between detectable levels of factors such as TGFβ2 and BMP7 and time to metastatic recurrence in ER+ patients after therapy.
NG2+/Nestin+ MSCs activate TGFBRIII and BMPRII signalling and a low ERK/p38 signalling ratio in cancer cells.
We next tested whether NG2+/Nestin+ MSCs could activate key dormancy pathways downstream of TGFβ2 and BMP7 signalling in cancer cells. To this end, we optimized a co-culture system of sorted NG2+/Nestin+ MSCs and various human and mouse cancer cells. In this assay, CD45-CD31-Nestin-GFP cells sorted from the BM were co-cultured (1:1 ratio) with cancer cells on top of matrigel at low density. The co-cultures were followed for up to 4 days to ensure that the MSCs retain functionality43. We monitored the frequency at which single cancer cells (to mimic solitary DTC biology) remain in a solitary state, or progressed to small and large cancer cell clusters, a measure of proliferative capacity (ED Fig3a and Fig4a). The co-cultures revealed that the majority of control (no MSCs added) and also E0771 BC cells co-cultured with Nestin-GFP- cells progressed to large clusters; compared to control cells, Nestin-GFP- cells had some growth inhibitory effect on E0771 cells as we observed ~15% increase in the accumulation of cells in single cell state. However, arrest of cancer cells in a single cell or doublet state was dramatically increased in the presence of Nestin-GFP+ MSCs (Fig4b). Similar results were obtained with human head and neck squamous carcinoma T-HEp3 cells (ED Fig3b) and mouse BC MMTV-PyMT-CFP cells (ED Fig3c). Treatment of E0771 cells in these conditions with TGFβ2, BMP7 or BM conditioned media (BM-CM) from WT TGFβ2+/+ mice also led to the accumulation of growth arrested single cancer cells (ED Fig3d,h). This effect was partially reversed when using BM-CM from mice heterozygous for TGFβ2 (+/−) (ED Fig3h). Thus, Nestin-GFP+ MSCs, which show enhanced expression of TGFβ2 and BMP7, were able to strongly suppress proliferation of different cancer cells similarly to the purified cues or BM-CM from mice with a full gene complement of TGFβ2. Mechanistic analysis revealed that the growth suppression was not due to apoptosis in the co-cultures (measured by c-Cas3 levels), but due to a reduction in p-Rb levels and increase in p-ATF2 (a p38 pathway target) and p27 only observed in the cells co-cultured with Nestin-GFP+ MSCs (Fig4c–f) or treated with TGFβ2, BMP7 or TGFβ2+/+ BM-CM (ED Fig3e–k). These changes were more evident in solitary E0771 cells, suggesting that in this state cancer cells may be more responsive to dormancy cues. Importantly, TGFβ2 and BMP7 on their own could induce these molecular changes supporting that the Nestin-GFP+ MSCs may be activating these pathways through those cues. These results were corroborated using biochemical approaches, which showed that in 2D cultures of E0771 cells TGFβ2 and BMP7 activated SMAD2 and SMAD1/5 phosphorylation respectively and that both converged on the phosphorylation of ATF2 and upregulation of p27 protein levels (ED Fig3l), required for dormancy onset10. BM-CM from WT TGFβ2 mice led to similar changes in p-SMAD2, p-ATF2 and p27 (ED Fig3l) as observed in response to TGFβ2 and BMP7; however, BM-CM from mice heterozygous for TGFβ2 (+/−) still activated these pathways suggesting that loss of one TGFβ2 allele cannot fully eliminate the immediate response on the pathway activation within a few hours, but it can reduce the proliferative response over longer periods of treatment (ED Fig3h).
Figure 4. NG2+/Nestin+ MSCs activate TGFβ2 and BMP7 growth inhibitory signalling in cancer cells.

a. Representative images of 3D co-cultures of E0771 cells with sorted Nestin-GFP- or + MSCs for 4 days. Top left: a single cell, a doublet and cluster of cancer cells. Scale bar 50μm; Bottom left: A NG2+/Nestin+ MSC (PDGFRα+, red) near a cancer cell cluster. Scale bar 50μm; Centre and right: Representative images of positive cells for cleaved caspase-3 (apoptotic cells), pRb (proliferative cells), p-ATF2 (p38-pathway activation) and p27 (quiescent cells) markers. Scale bars 10μm (representative images of 3 independent experiments). b. Percentage of E0771 cells in a single cell (p=0.01 and 0.03), doublet or cell cluster (p=0.04 and 0.02) states in the indicated co-cultures (n=3 independent experiments, mean and SEM, 2-tailed Mann–Whitney tests). c-f. Percentage of E0771 cancer cells positive for cleaved Caspase 3 (c-Cas-3), phospho-Rb, phospho-ATF2 and p27 in co-culture with Nestin-GFP- (dark green) or Nestin-GFP+ MSCs (bright green) (n=2 independent experiments, 4 wells per condition, mean, minimum and maximum). g-j. T-HEp3 cells with different bio-sensors were reversed transfected with siRNA for TGFBRIII and BMPRII followed by co-culture in trans-wells with sorted Nestin-GFP- or + cells. g. Representative images of T-HEp3 cells with ELK-Gal4::GFP (GFP+ when ERK1/2 pathway is active), p38-Clover (when p38 is active cytoplasmic signal predominates) and p27K-mVenus (mVenus signal indicates cell cycle arrest) bio-sensors (representative images of 3 independent experiments). Scale bar 25μm. h-j. Quantification of the T-HEp3 bio-sensor cell lines after transfection followed by 24-hour co-culture using trans-wells (n=3 independent experiments each, mean, minimum and maximum, 2-tailed Mann–Whitney tests, p=0.05). *p≤0.05, p-values indicated above.
Having established that E0771 cells were responsive to TGFβ2 and BMP7 and that Nestin-GFP+ MSCs produce these factors, we tested whether the MSC effect was contact dependent and if growth suppression was indeed linked to signalling downstream of these cues. To this end, we used T-HEp3 cells engineered to express an ELK-GAL4::hrGFP biosensor to monitor ERK activity44, a p38 shuttle-Clover biosensor that shows clover cytoplasmic over nuclear signal when p38 is active45 and a p27K-mVenus reporter where protein is stabilized and accumulated in the nucleus upon growth arrest46. T-HEp3-biosensor cells were transfected with siRNAs for TGFBRIII and BMPRII (ED Fig4a) and then cultured in the bottom of plates while Nestin-GFP- or Nestin-GFP+ cells were plated in upper level of the trans-wells. These experiments showed that Nestin-GFP- cells do not lead to differences in cancer cell ERK, p38 or p27 biosensors activities and that TGFBRIII and BMPRII knockdown (KD) also do not change basal biosensors activities (Fig4g–j). In contrast, Nestin-GFP+ MSCs led to significant induction of p38 and p27 activity in cancer cells, even without being in direct contact, and these activations were completely negated by KD of TGFBRIII and BMPRII. Accordingly, Nestin-GFP+ MSCs significantly reduced ERK activity in cancer cells, which was also reversed by TGFBRIII, but not significantly by BMPRII knockdown (Fig4g–j). Both TGFβ2 and BMP7 activated the p27 and p38 biosensors while inhibiting ELK activity in cancer cells (ED Fig4b–d), supporting that they are faithful reporters and that Nestin-GFP+ MSCs can directly modulate key dormancy pathways via TGFβ2 and BMP7. In all cases, TGFβ2- or BMP7-dependent effects were reversed by KD of TGFBRIII in the case of TGFβ2 and BMPRII in the case of BMP7 (ED Fig4b–d). The above results provide strong evidence supporting that NG2+/Nestin+ MSCs secrete TGFβ2 and BMP7, to inhibit the mitogenic (ERK1/2) pathway and activate the growth arrest (p38 and p27) pathway.
The ability of long-term cultured NG2+/Nestin+ MSCs niche cells to maintain HSC self-renewal ex vivo is markedly diminished due to loss of the expression of niche factors in cultured MSCs43. The loss of these factors is also observed in MSCs passaged 3–5 times in culture43 (MSCs Ct); however, revitalized MSCs (rMSCs) engineered to express Klf7, Ostf1, Xbp1, Irf3 and Irf7 restore the synthesis of HSC niche factors of BM-derived cultured MSCs43. Interestingly, passaged NG2+/Nestin+ MSCs (MSCs Ct, MSCs kept in culture for 3–5 passages that show loss of function) were not able to suppress growth of E0771 cancer cells. In contrast, rMSCs that have the ability to maintain HSC self-renewal ex vivo were able to suppress proliferation of E0771 cells (ED Fig4e), which correlated with the specific upregulation of TGFβ2 (not TGFβ1) and BMP7 in rMSCs comparted to control MSCs (ED Fig4f). These data further support that functional NG2+/Nestin+ MSCs are able to suppress cancer cell growth and that alterations of these cells may impair their dormancy-inducing function.
Conditional deletion of TGFβ2 in NG2+/Nestin+ MSCs triggers DTC escape from dormancy in the BM.
The above experiments support strong evidence that NG2+/Nestin+ MSCs are critical for cancer cell dormancy onset and maintenance in the BM niche (Fig1). Additionally, we provide evidence that these MSCs produce significant amounts of TGFβ2 and BMP7 (Fig3) and that they activate dormancy signalling pathways in cancer cells (Fig4). However, these experiments still do not prove that the dormancy cues derived from NG2+/Nestin+ MSCs in the niche in vivo are required for dormancy. To address this missing link, we focused on TGFβ2 because of our knowledge on the role of this cytokine in dormancy induction and its higher levels in NG2+/Nestin+ MSCs (Fig3a and 40).
We crossed NG2-CreER mice with TGFβ2flox mice to generate specific conditional deletion of this cytokine in the NG2 lineage. We confirmed decreased levels of TGFβ2 in NG2+/Nestin+ MSCs (sorted CD45-Ter119-CD31-PDGFRa+CD51+ MSCs, which contain NG2+/Nestin+ MSCs) from the BM of NG2-CreERTGFβ2 mice tamoxifen induction (Fig5a–c), but no differences in TGFβ1 levels supporting the specificity of the gene disruption (ED Fig5a). Although the NG2-CreER driver is mainly restricted to peri-arteriolar Nestin-GFP+ stromal cells, it can also be detected in chondrocytes, osteocytes, and rarely in osteoblasts47,48. To determine whether the conditional KO strategy we selected may impact TGFβ2 in those cells, we sorted MSCs, OPCs and ECs from WT and NG2-CreERTGFβ2 (KO) mice. Only TGFβ2 levels in MSCs decreased significantly upon tamoxifen treatment, supporting that the depletion is specific for TGFβ2 in MSCs (ED Fig5b). To test whether disruption of TGFβ2 production in the NG2+/Nestin+ MSC niche in NG2-CreERTGFβ2 mice would affect DTC behaviour we injected E0771-GFP cells into the left ventricle of the heart of animals and euthanized them 2 weeks later. All mice had detectable E0771-GFP+ DTCs in the BM by FACS (5f), corroborating the injection efficiency. However, only ~16% (4 out of 24) of wild-type (WT) mice shown more than 1000 DTCs/million BM cells (manually counted after BM flush), while 48% (14 out of 29) of NG2-CreERTGFβ2 (KO) mice displayed a build-up of E0771-GFP+ colonies in the BM (Fig5d). Consistently, quantification of the DTC burden (both manual counting, Fig5e, and by FACS, Fig5f) showed an increase in number of DTCs in the BM compartment of TGFβ2 KO mice compared with WT. Additional analysis revealed that metastatic masses in TGFβ2 KO mice showed a decrease in the percentage of p27+ cells (23%) and an increase in the number of Ki67+cells (10%) compared to solitary DTCs and small DTC clusters in WT mice (57% p27+ / 3% Ki67+) (Fig5h,i). A similar difference was found by FACS staining of Ki67, where we found an increase in the percentage of E0771-GFP+ Ki67high cells (Fig5g). We conclude that niches containing NG2+/Nestin+ MSCs and that produce TGFβ2 are required for BC cell dormancy induction in the BM compartment.
Figure 5. Conditional knock out of TGFβ2 in NG2+/Nestin+ MSCs awakens dormant DTCs in the BM.

a. 7-week old NG2-CreER- or + TGFβ2f/f mice were i.p. treated daily with tamoxifen (TAM) for 5 days followed by intra-cardiac injection of 2×105 E0771-GFP cells. Mice were euthanized and the organs collected 2 weeks after . b-c. Sorting strategy (b) and TGFβ2 mRNA levels (c) confirming the efficiency of TGFβ2 knockout in NG2+/Nestin+ MSCs (sorted using CD45-Ter119-CD31-PDGFRa+CD51+ markers) in NG2-CreERTGFβ2 mice upon TAM treatments compared with WT mice (n=4 independent experiments, median, 2-tailed Mann–Whitney U-tests, p=0.014 and 0.029). d. Incidence of bone metastasis (>1000 GFP+ DTCs/106 BM cells) 2 weeks after cancer cell injections (n=24 WT and 25 KO mice from 4 independent experiments, 2-tailed Fisher’s exact test, p=0.021). e. Number of E0771-GFP cancer cells per million of BM cells after BM flushing of WT and NG2-CreERTGFβ2 mice, counted manually (n=24 WT and 25 KO mice from 4 independent experiments, median, 2-tailed Mann–Whitney U-tests, p=0.016). f. Number of E0771-GFP cancer cells per million of BM cells after BM flushing, counted by FACS (n=7 WT and 12 KO mice, median, 2-tailed Mann–Whitney U-tests, p=0.004). g. Percentage of Ki67high E0771-GFP cancer cells (FACS, n=7 and 12 mice per condition, median, 2-tailed Mann–Whitney U-tests, p=0.05). h. Representative images of single cells and small clusters in WT and a metastasis in NG2-CreERiDTR. Scale bars 25μm; arrows, positive cells; arrowheads, negative cells; dotted lines, GFP+ cells border. i. Percentage of E0771-GFP cancer cells p27+ (p=0.029) and Ki67+ (p=0.029) detected by immunofluorescence (n=4 WT and 4 KO mice, median, 2-tailed Mann–Whitney U-tests). *p≤0.05, p-values indicated above.
Discussion
Building on previous studies11,20, we show that the BM perivascular niche plays an important role in DTC dormancy. However, here we make a leap in our understanding of this biology by revealing a specific BM cell type, namely NG2+/Nestin+ MSCs, that maintains both HSC27 and DTC dormancy in this organ. We further reveal the mechanism behind this biology, where NG2+/Nestin+ MSCs secrete TGFβ2 and BMP7 to signal through TGFBRIII and BMPRII leading to activation of SMAD, p38 and p27 pathways and cancer dormancy. Our work provides functional evidence for the long-held notion that HSC dormancy niches support DTC dormancy. Our findings are also deeply linked to the “seed and soil” theory of metastasis49. However, our findings reshape this paradigm to show that the “seed and soil” relationship in BM is not for growth of DTCs but rather for the induction and maintenance of dormancy. In fact, our data support that when the “soil” is negatively altered metastasis ensue. Thus, certain organs rather than being a proper “soil” for metastasis may indeed be homeostatic and their normal function as the “soil” is pro-dormancy; when they are altered or damaged they lose their metastasis suppressing function.
Despite our efforts, the low number of DTCs in the majority of control mice (150–400 DTCs per million BM cells) we used precluded mapping statistically their location in relation to vascular structures. Thus, we focused on a functional analysis. We now identify the active role of NG2+/Nestin+ MSCs in inducing and maintaining DTC dormancy in vivo. We went a step further and proved in vivo the crucial role of TGFβ2 produced by NG2+/Nestin+ MSCs. Future studies will test the function of BMP7 in these and other BM cells. Our data also suggests that NG2+/Nestin+ MSCs regulated niches are primarily pro-dormancy rather than only pro-survival, otherwise when the niche was disrupted in the NG2-CreERiDTR model, we would see DTC clearance instead of reactivation. However, it is possible that oncogenic signalling in the E0771 cells (K-Ras (activating), MKK4 and p53 (both inactivating mutants)50 provide absent survival signals that could be altered when we eliminate NG2+/Nestin+ MSCs. It is interesting that even carrying MKK4 (p38 and JNK upstream activator) and p53-inactivating mutations (and K-Ras active mutant) E0771 efficiently activated p38 signalling and CDK inhibitors expression. Perhaps TGFβ2 and BMP7 signalling is more reliant on MKK3/6 signalling for p38 activation. It also supports that niche cues can override potent oncogene signalling, further solidifying the notion that microenvironmental mechanisms may be dominant over genetics51,52 if cancer cells can still “read” host homeostatic signals.
We also report no major differences in vessel permeability, abundance of pro-inflammatory cytokines and DTC early seeding after NG2+/Nestin+ MSCs depletion. Thus, the enhanced DTC growth in NG2+/Nestin+ MSCs depleted before or after seeding in the BM was not due to a seeding/colonization phenotype or a widespread inflammatory response. However, we cannot rule out that circumscribed and limited inflammation in the sites where the DT kills the iDTR expressing NG2+/Nestin+ MSCs, could lead to tumor cell awakening. An important next question will be to define how cancer cells with different genetics and from different cancer types might affect the HSC niche to support or block a positive loop feeding dormancy of both HSC and DTCs in an homeostatic normal-like BM environment. Our results may also start to define what genetic alterations may allow cancer cells to escape microenvironmental control by NG2+/Nestin+ MSCs. For example, genetic alterations that affect TGFBRIII53–55 and BMPRII38,56 have been linked to increased bone metastasis in breast and prostate cancer, indicating that such alterations may allow DTCs to escape homeostatic control. These genetic changes detected in DTCs may serve as biomarkers to help monitor more closely patients at risk of relapse. To this end we provide promising data that monitoring for example BMP7 presence or absence in ER+ breast cancer patients can inform on the time to metastatic relapse. Patients that presumably had intact or functional NG2+/Nestin+ MSC and/or other niches and had detectable BMP7 were less prone to develop metastasis.
It is important to highlight that different cell types may produce the same or other dormancy cues, as we observed that OPCs also make TGFβ2 (Fig3b) and Nestin-GFP- cells slightly increase cancer cell growth arrest (Fig4b). Also, the driver we used, NG2-CreER, is mainly restricted to MSCs but it can also target chondrocytes, osteocytes and few osteoblasts (data not shown and 48). However, our data from NG2-CreERTGFβ2 mice supports that the NG2-CreER promoter is restricted to MSCs and not OPCs or ECs. Thus, the loss of TGFβ2 in NG2+/Nestin+ MSCs is the main change that allows cancer cell to avoid entering dormancy in the BM. However, our experimental design did not allow testing the direct role of TGFβ2 produced by MSCs in dormancy maintenance. It is possible as other cell types in the BM niche are also key contributors to the pro-dormancy HSC niche. To this end, it was previously described that osteoblasts are also a source of TGFβ222,40, but in our mouse models these cells do not seem to compensate for the loss of TGFβ2 in NG2+/Nestin+ MSCs or for the 50% loss of NG2+/Nestin+ MSCs after DT treatment. NG2+/Nestin+ MSCs may also produce additional factors to TGFβ2 and BMP7, which would explain why the penetrance of the phenotype seems greater when we compare DTC burden in the NG2-CreERiDTR model (Fig1) vs. the NG2-CreERTGFβ2 (Fig5).
It is known that aging affects the proper function of HSC niches by downregulation of niche factors by passaged MSCs43 and that in aged mice the BM microenvironment shows lower levels of TGFβ2 and BMP757. Our results offer a new opportunity to understand how aging, inflammation and the cancer cells themselves may alter MSCs functionality leading to a disruption of the BM pro-dormancy niche, DTC awakening and bone metastasis formation (Fig6). Finally, a remaining question, but beyond the scope of our study, is whether the function of NG2+/Nestin+ MSCs as pro-dormancy niche orchestrators is limited to the BM or whether it has similar roles in other metastasis target organs. Overall, we propose that this work shifts our understanding on how homeostatic mechanisms that control adult stem cell quiescence may govern dormancy of disseminated cancer cells and control the timing of metastasis. Our work reshapes the paradigm of metastasis by revealing how the homeostatic BM microenvironment actually serves mainly as a metastasis suppressive “soil” via dormancy induction.
Figure 6. Schematic representation of the model by which HSC niches induce dormancy of breast cancer DTCs in the BM.

We propose that niches that control HSC dormancy, such as NG2+/Nestin+ MSCs, are responsible for producing TGFβ2 and BMP7 and inducing breast cancer DTCs dormancy in the BM (left). Upon a reduction in the number of NG2+/Nestin+ MSCs (upper right) or in the production of TGFβ2 by MSCs (lower right), the homeostatic control is lost, dormancy interrupted and metastatic growth ensues. We hypothesize that perturbations of this niche caused by aging, osteoporosis or other alterations (center), may cause the niche to reduce its homeostatic control and allow for awakening of dormant DTCs in the BM. Individual elements of this original scheme were adapted from https://smart.servier.com following the terms of use and copyright license which authorize the use of their images.
Methods
Animals.
Nes–GFP58 were bred in and obtained from the Frenette’s laboratory at Albert Einstein Institute. B6.Cg-Tg(Cspg4-cre/Esr1*)BAkik/J (NG2-CreER - Jackson Laboratory, stock 008538), C57BL/6-Gt(ROSA)26 Sortm1(HBEGF) Awai/J (iDTR – Jackson Laboratory, stock 007900) and Tgfβ2flox (59, gift from Mohamad Azhar’s lab) were maintained on the C57BL/6J background and bred and crossed in our facilities. Mice were kept at 20–26°C, 30–70% humidity, 12-hour dark/light cycles, with food (LabDiet 5053 rodent diet) and water (reverse osmosis, R/O water) accessible at all times. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Icahn School of Medicine at Mount Sinai. All NG2-CreERiDTR and NG2-CreERTGFβ2 mice were genotyped using the primers listed below, number-tagged and allocated according with their genotype. 7 to 8-week-old female mice were used in all experiments. Primers: NG2-CreER F, TTAATCCATATTGGCAGAACGAAAACG; NG2-CreER R, CAGGCTAAGTGCCTTCTCTACA; DTR F, ACGGAGAATGCAAATATGTGAAGGA; DTR R, ACACCTCTCTCCATGGTAACCT; ROSA WT F, TTCCCTCGTGATCTGCAACTC; ROSA WT R, CTTTAAGCCTGCCCAGAAGACT; TGFβ2f/f F, GCTGGCGCCGGAAC; TGFβ2f/f R, GCGACTATAGAGATATCAACCACTTTGT; TGFβ2 WT F, TGGCCTAGAAAGCCAGATTACAC; TGFβ2 WT F, GGAGAGGCGAGTAAAGGAGAAG.
Induction of NG2-CreER-mediated recombination and iDTR-mediated cell depletion.
7 to 8-week old female mice were injected intraperitoneally with 2mg of tamoxifen (Sigma, T5648) dissolved in corn oil daily for 5 days to induce the CreER-mediated recombination. In case of NG2-CreERiDTR, mice were also injected intraperitoneally with 250ng of diphtheria toxin (Millipore, 322326) dissolved in serum chloride daily for 2 days for iDTR-mediated cell depletion.
Metastasis assays.
2×105 E0771-GFP or MMTV-PyMT-CFP cells were intra-cardiac injected with echo-guidance using micro ultrasound Vevo2100, Transducer MS-250, 21MHz and Vevo LAB 3.1.1 software (FUJIFILM VisualSonics Inc.) (Supplementary Movie 1). 24 hours or 2 weeks later, mice were euthanized and organs were collected and processed. In a group of mice, 70,000 MW Lysine fixable Dextran Texas Red (Invitrogen) was retro-orbital injected 15 minutes prior to euthanasia and mice were perfused with PBS. Bone marrow (BM) cells were flushed from femurs and tibias, red-blood-cell lysis buffer (Lonza) was used for 2 minutes followed by quantification of the GFP+ cells and normalization to the total number of BM cells.
Flow cytometry and cell sorting.
Bone marrow cells were flushed, incubated in red-blood cell lysis buffer (Lonza) for 2 minutes and remaining cells were permeabilized with 0.05% Triton (when using intracellular antibodies) and stained using antibodies and dilutions described in the Nature Research Reporting Summary linked to this article for 15min at 4°C in the dark. In the case of host BM cell sorting (Nestin-GFP, MSCs, OPCs and ECs), 3–4 mice cells were pooled per experiment and condition. All experiments were performed using BD FACSAria II equipped with FACS Diva software (BD Biosciences). Dead cells and debris were excluded by FCS, SSC and DAPI (4’,6-diamino-2-phenylindole) (Fisher Scientific) staining profiles. Data were analysed with FACS Diva (BD Biosciences) or FCS Express Cytometry 7 (De Novo) softwares.
Histopathology.
After dissection, bones were fixed in 4% paraformaldehyde (PFA, Thermoscientific) for 24 hours. Bones were then decalcified in 14% EDTA, renewed every other day for 7–10 days at 4°C with agitation. Bones were processed, embedded in paraffin and sections were cut. H&E and immunofluorescences were performed and slides were scanned using NanoZoomer S60 Digital slide scanner and NDP.view2 software (Hamamatsu).
Immunofluorescence.
Tissue sections were submitted to hydration in xylene and a graded alcohol series, slides were steamed in 10 mM citrate buffer (pH 6) for 40 minutes for antigen retrieval. Fixed 3D cultures were permeabilized using 0.5% Triton X-100 in PBS for 20 minutes. Both sections and co-cultures were blocked with 3% Bovine Serum Albumin (BSA, Fisher Bioreagents) and 5% normal goat serum (NGS, Gibco PCN5000) in PBS for 1 hour at room temperature. Primary antibodies described in the Nature Research Reporting Summary linked to this article were incubated overnight at 4°C, followed by washing and secondary antibodies (Invitrogen, 1:1000) incubation at room temperature for 1 hour in the dark. Slides were mounted with ProLong Gold Antifade reagent with DAPI (Invitrogen, P36931) and images were obtained using Leica Software on a Leica SPE confocal microscope. All quantifications were done using double blind method (quantification of coded samples and de-codification upon completion to interpret the results across multiple animals).
Whole-mount staining.
Calvaria bones were fixed overnight at 4°C in 4% PFA, blocked overnight with agitation in 3% BSA-5% NGS, followed by a 24 hour incubation at 4°C with agitation with primary antibodies and dilutions described in the Nature Research Reporting Summary linked to this article. 10 hours of washing was followed by secondary antibodies (Invitrogen, 1:1000) incubation at 4°C, overnight with agitation and 10-hour wash. Images were obtained using Leica Software on a Leica SPE confocal microscope.
Sternal bones were collected and transected with a surgical blade into 2–3 fragments, 10 minutes after retro-orbital injection of antibodies and dilutions described in the Nature Research Reporting Summary linked to this article. The fragments were bisected sagittally for the bone marrow cavity to be exposed, fixed in 4% PFA and immunofluorescence staining was performed as above described. Images were acquired using a ZEISS AXIO examiner D1 microscope (Zeiss) with a confocal scanner unit, CSUX1CU (Yokogawa).
Quantitative PCR.
Sorted cells were processed using Cell-to-CT 1-Step Power SYBR Green kit (Invitrogen, A25600). GAPDH was used as housekeeping control for all experiments. Primers: Mouse TGFβ1 F, CCT GAG TGG CTG TCT TTT GA; Mouse TGFβ1 R, GCT GAA TCG AAA GCC CTG TA; Mouse TGFβ2 F, TAA AAT CGA CAT GCC GTC CC; Mouse TGFβ2 R, GAG ACA TCA AAG CGG ACG AT; Mouse BMP2 F, TGG AAG TGG CCC ATT TAG AG; Mouse BMP2 R, TGA CGC TTT TCT CGT TTG TG; Mouse BMP7 F, GAA AAC AGC AGC AGT GAC CA; Mouse BMP7 R, GGT GGC GTT CAT GTA GGA GT; Mouse GAPDH F, AAC TTT GGC ATT GTG GAA GGG CTC; Mouse GAPDH R, TGG AAG AGT GGG AGT TGC TGT TGA.
Human samples.
The BM plasma samples analyzed for TGFβ2 and BMP7 were 55 female selected from early breast cancer patients included in the SATT study9, between 18 and 65 years old (median age 48). In this study the early breast cancer patients, who had completed adjuvant chemotherapy, were monitored for DTCs 2–3 months (BM1) and 8–9 months (BM2) after completion of adjuvant anthracycline-containing chemotherapy. If DTC positive at 8–9 months, the patients received secondary chemotherapy intervention with docetaxel, followed by subsequent DTC monitoring 1 (BM3) and 12 months (BM4) after docetaxel. Patients with disappearance of DTCs (80%) experienced excellent prognosis compared to the patients with DTC persistence (20%)9. The BM aspirates (from posterior iliac crest bilaterally) collected for DTC analyses were diluted 1:1 in PBS and separated by density centrifugation using Lymphoprep (Axis-Shield, Oslo, Norway) as previously described9. Later on in the inclusion period the plasma supernatant after density centrifugation was collected (if possible) and stored initially at −20 °C, and later transferred to −80 °C. From the available BM plasma samples, we initially performed a limited analysis of plasma samples from patients being DTC positive at either BM1 or BM2, or experiencing systemic relapse without DTC presence. Of these, 55 had ER positive disease, including 30 patients receiving secondary intervention with docetaxel chemotherapy, and 25 patients with no secondary treatment intervention. The original SATT study9 was approved by the Research committee at the Cancer Clinic, Oslo University Hospital and by the Regional Ethical Committee (reference number S-03032). Written informed consent was obtained from all patients. The study is registered in Clin Trials Gov (registration number NCT00248703). Use and analysis of these samples in the present study is covered by these originally obtained approvals and written informed consent.
ELISA.
Mouse BM supernatant was collected after BM flush centrifugation and stored at −80°C with phosphatase and protease inhibitors (ThermoScientific). TGFβ and pro-inflammatory ELISAs were performed by Eve Technologies Corp. (Calgary, Alberta) using the Luminex™ 100 and 200 systems (Luminex, Austin, TX, USA), Eve Technologies’ TFG-β 3-Plex Discovery Assay® (MilliporeSigma, Burlington, Massachusetts, USA) and Eve Technologies’ Mouse Focused 10-Plex Discovery Assay® (MilliporeSigma, Burlington, Massachusetts, USA). BMP7 ELISA was performed using RayBio kit (RayBio, ELM-BMP7) following manufactures instructions.
Patient BM supernatants were analyzed for TGFβ2 and BMP7 by multiplex ELISA at Human Immune Monitoring Center (HIMC) at Mount Sinai. ELISA assays were performed using human TGFβ2 (R & D System, DY302) and BMP7 (R & D System, DY354) kits following manufacture’s recommendation. Data were analyzed using a Four Parameter Logistic Fit (4PL) method.
Cell culture.
E0771 (CH3 BioSystems), E0771-GFP (gift from John Condeelis’ lab) and MMTV-PyMT-CFP (gift from Jay Debnath) breast cancer cell lines were cultured in RPMI (Gibco) supplemented with 10% foetal bovine serum (Gemini), 10mM HEPES (Corning), 100 units/ml penicillin and 100ug/ml streptomycin (Corning). The tumorigenic HEp3 (T-HEp3)60 head and neck squamous cell carcinoma (HNSCC) PDX line and Ct MSCs and rMSCs43 were generated and maintained as described previously.
T-HEp3 cells were engineered to express an ELK-GAL4::hrGFP44, p38 shuttle-Clover45 and p27K-mVenus46 biosensors. T-HEp3 with the biosensors were reversely transfected with siRNAs (siTGFBRIII, Invitrogen 32274204; siBMPRII, Oncogene SR300456B) using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions.
Trans-well co-cultures were performed using permeable trans-well assays (Corning), in which transfected cells were plated in the bottom of the well and sorted Nestin-GFP- or + cells in the permeable inserts.
Low-density 3D organoid co-cultures.
500–1000 cells (E0771 only or with MSCs) were seeded in 400μl assay medium (each cell line media with reduced FBS content (2–5%) plus 2% matrigel) in 8-well chamber slides (Falcon) on top of 50μl of growth factor-reduced matrigel (Corning). Cultures were treated every 24 hours starting at day 0 with TGFβ2 (R&D), BMP7 (R&D) or BM-conditioned media (BM-CM). Single cells, doublets and clusters were quantified after 4 days and the cultures were fixed with 4% paraformaldehyde (PFA) for 20 minutes.
Western blot.
Samples were collected in RIPA buffer and protein concentrations were calculated using Coomassie Plus protein assay (Thermo Scientific) and a standard BSA curve. Samples were then boiled for 8 minutes at 95°C in sample buffer (0.04 M Tris-HCl pH 6.8, 1% SDS, 1% β –mercaptoethanol and 10% glycerol). 8–10% SDS–PAGE gradient gels were run in running buffer (25 mM Tris, 190 mM glycine, 0.1% SDS) and transferred to PVDF membranes in transfer buffer (25 mM Tris, 190 mM glycine, 20% methanol). Membranes were then blocked in 5% milk in TBS-T (Tris-buffered saline containing Tween-20) buffer. Primary antibodies described in the Nature Research Reporting Summary linked to this article were incubated overnight at 4°C, washed with TBS-T buffer, HRP-conjugated secondary antibodies were left at room temperature for 1 hour and washed with TBS-T buffer. Western blot development was done using Amersham ECL Western Blot Detection (GE, RPN 2106) and GE ImageQuant LAS 4010.
Statistics and Reproducibility.
Sample sizes were chosen empirically, no statistical method was used to predetermine sample size and no exclusion criteria were applied. The experiments were not randomized, mice were number-tagged and quantifications were done in coded samples to reduce operator bias. Statistical analyses were done using Prism Software and differences were considered significant if p<0.05. Unless otherwise specified, 3 or more independent experiments were performed, all values were included and median, interquartile range and 2-tailed Mann–Whitney U-tests, incidence and Fisher’s exact test or mean and SEM and 2-tailed Student’s t-tests were performed.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability.
The data generated during the current study that support the reported findings are available in the manuscript, including in the provided Source Data files, and from the corresponding author on reasonable request.
Extended Data
Extended Data 1. Control of depletion of NG2+/Nestin+ MSCs and awakening of dormant PyMT-CFP DTCs in the BM.

a. 7-week old NG2-CreERiDTR mice (NG2-CreER- or +) were daily injected with tamoxifen for 5 days, followed by 1-day rest and 2 days with diphtheria toxin (DT). 24 hours later 2×105 E0771-GFP cells were intra-cardiac injected and 2 weeks later mice were euthanized. b-c. FACS plots (b) and quantifications (c) confirming the depletion of NG2+/Nestin+ MSCs (CD45-Ter119-CD31-PDGFRα+CD51+) in NG2-CreERiDTR mice upon TAM and DT treatments compared with WT mice (n=5 WT and 5 iDTR mice, median, 2-tailed Mann–Whitney U-test, p=0.008). d-g. Detection and characterization of MMTV-PyMT-CFP cells in BM flushes by FACS (n=5 WT and 5 iDTR mice). e. Incidence of bone metastasis (>1000 GFP+ DTCs/106 BM cells) 2 weeks after cancer cell injections (2-tailed Fisher’s exact test). f. Number of MMTV-PyMT-CFP cancer cells per million BM cells (median, 2-tailed Mann–Whitney U-test). g. Percentage of Ki67high E0771-GFP cancer cells (median, 2-tailed Mann–Whitney U-test). *p≤0.05 (p-values indicated above). n.s. not significant.
Extended Data 2. Pro-inflammatory cytokine status, vessel permeability and BM DTC seeding in NG2+/Nestin+ MSCs-depleted mice.
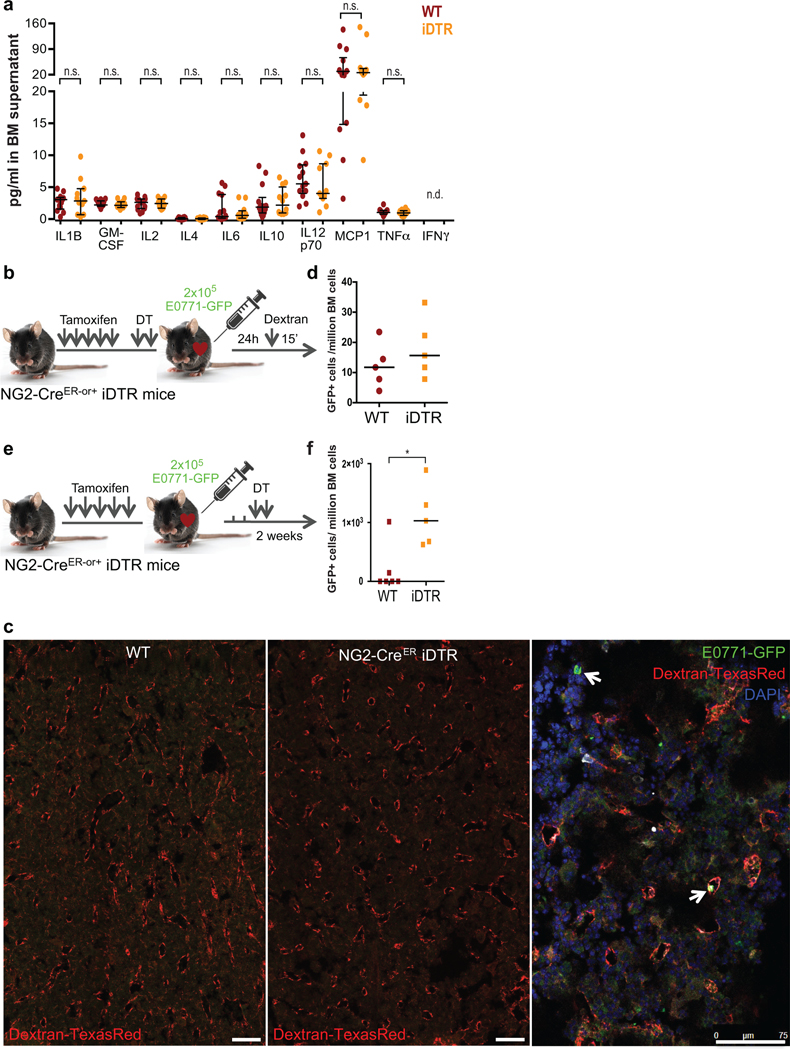
a. Levels of pro-inflammatory cytokines in BM supernatant of WT and NG2-CreERiDTR mice (n=13 WT and 11 iDTR mice, from 2 independent experiments, median and interquartile range, 2-tailed Mann–Whitney tests, *p≤0.05, n.s. not significant, n.d. not detected). b-d. NG2-CreERiDTR mice (NG2-CreER- or +) were daily i.p. injected with tamoxifen for 5 days followed by a rest day and 2 i.p. injections of DT. E0771-GFP cells were intra-cardiac injected and 24 hours after 70K Dextran-TexasRed was injected 15 minutes prior euthanasia. c. Representative images of Dextran extravasation in perfused bones. Arrows, E0771-GFP cells. d. Number of E0771-GFP cells detected after 1 week of in vitro expansion of the BM aspirates collected 24 hours after injection into mice (n=5 WT and 5 iDTR mice, median). e-f. NG2-CreERiDTR mice (NG2-CreER- or +) were daily i.p. injected with TAM for 5 days, followed by intra-cardiac injection of E0771-GFP cancer cells. Cells were allowed to disseminate and extravasate for 72 hours followed by 2 i.p. injections of DT. f. Number of E0771-GFP cells/million BM cells (n=5 WT and 5 iDTR mice, median and 2-tailed Mann–Whitney tests, p=0.015). *p≤0.05 (p-values indicated above), n.s. not significant.
Extended Data 3. Effect of NG2+/Nestin+ MSCs, TGFβ2 and BMP7 on signalling pathways and growth cancer cells.

a. 3D Matrigel assay used to track solitary cell to cluster growth. Single cells were plated on top of Matrigel in low density and the percentage of single cells, doublets and clusters was quantified 4 days after. b-c. Co-culture of human HNSCC PDX-derived T-HEp3 (b, n=4 independent experiments, 4 wells per condition, mean and SEM, 2-tailed Mann–Whitney tests p=0.04) and mouse BC MMTV-PyMT (c, n=2 independent experiments, 4 wells per condition, mean) cells with sorted Nestin-GFP- and Nestin-GFP+ MSCs for 4 days. d-k. E0771 cells were treated every day for 4 days with TGFβ2, BMP7 or bone marrow conditioned media (BM-CM) of TGFβ2+/+ or TGFβ2+/− mice. d and h. Percentage of cancer cells in a single cell, doublet or cluster state with the indicated treatments (d, n=5 independent experiments, 4 wells per condition each, mean and SEM, 2-tailed Mann–Whitney tests, p=0.03 and 0.005; h, n=4 independent experiments, 4 wells per condition each, mean and SEM, 2-tailed Mann–Whitney tests, p=0.000005, 0.0001, 0.002 (Ct vs. TGFβ2+/+: CS, doublets and clusters respectively) and 0.02 (TGFβ2+/+ vs. TGFβ2+/− clusters)). e-g and i-k. Percentage of positive cells for c-Cas-3, p-ATF2 and p27 upon the indicated treatments (n=2 independent experiments, 4 wells per condition each, mean, minimum and maximum). l. Western blots for the indicated antigens detected in E0771 cells treated for 24 hours with the TGFβ2, BMP7 and different BM-CM preparations. Molecular weight markers in kDa. *p≤0.05, p-values indicated above.
Extended Data 4. Effect of TGFβ2 and BMP7 on signalling pathways (using ELK, p38 and p27 sensors) and MSCs in E0771 cancer cell growth.

a-d. T-HEp3 cells with ERK, p38 and p27 activity biosensors were reversed transfected with control siRNA or siRNAs for TGFBRIII and BMPRII followed by 24-hour treatments with TGFβ2 and BMP7. a. TGFBRIII and BMPRII mRNA levels 48 hours after transfection with the indicated siRNAs (n=3 independent experiments, mean, minimum and maximum, 2-tailed Mann–Whitney tests, p=0.01 (TGFbR3) and 0.002 (BMPR2)). b-d. Quantification of the T-HEp3-biosensors activity (n=3 independent experiments each, mean, minimum and maximum, 2-tailed Mann–Whitney tests, p=0.05). e. Percentage of E0771 cells in a single cell (p=0.002), doublet or cluster (p=0.005) state after co-culture with Control (passaged) or revitalized (r) MSCs (n=4 independent experiments, 4 wells per condition per experiment, mean and SEM, 2-tailed Mann–Whitney tests). f. qPCR of TGFβ1, TGFβ2 and BMP7 from Control and rMSCs (n=2). *p≤0.05, p-values indicated above.
Extended Data 5. NG2-CreERTGFβ2 mouse model controls.

a. TGFβ1 mRNA levels in NG2+/Nestin+ MSCs (sorted using CD45-Ter119-CD31-PDGFRα+CD51+ markers) in NG2-CreERTGFβ2 mice upon TAM treatments compared with WT mice (n=4 independent experiments). b. TGFβ1, TGFβ2 and BMP7 mRNA levels in MSCs (CD45-CD31-Ter119-PDGFRα+CD51+), osteo-progenitor (CD45-Ter119-CD31-ALCAM+, OPCs) and endothelial (CD45-Ter119-CD31+vEcad+, ECs) cells from NG2-CreERTGFβ2 mice upon TAM treatments compared with WT mice (n=2 independent experiments). *p≤0.05, p-values indicated above.
Supplementary Material
Acknowledgments
We thank the Aguirre-Ghiso lab for helpful discussions and the assistance of the Dean’s Flow Cytometry CoRE, Microscope CoRE and Small Animal Imaging CoRE at BioMedical Engineering and Imaging Institute, Icahn School of Medicine at Mount Sinai. We are grateful to Ms. Colette Prophete (Frenette’s lab) for mouse husbandry. We thank M. Rypdal, A.V. Pladsen and O.C. Lingjærde for preparing the BC DTC material and data. The BC DTC work in Oslo has received funding from The Norwegian Cancer Association and Norwegian Health Region South-East (H.G. Russnes and B. Naume). Grant support: The National Institute of Health /National Cancer Institute CA109182, CA216248, CA218024, CA196521 and the Samuel Waxman Cancer Research Foundation Tumor Dormancy Program to J.A. Aguirre-Ghiso; HL069438, DK056638, DK116312 and DK112976 to P.S. Frenette. Portuguese Foundation for Science and Technology (SFRH/BD/100380/2014) to A.R. Nobre. MD/PhD program of the University of Lyon and the Ecole Normale Supérieure of Lyon to E. Risson. Susan G. Komen Career Catalyst Research (CCR18547848), the NCI Career Transition Award (K22CA196750), NIH (R01CA244780) and Tisch Cancer Institute NIH Cancer Center Grant (P30-CA196521) to JJ Bravo-Cordero. Instituto Serrapilheira/Serra-1708-15285 to A Birbrair. NIH (HL126705, CA218578, R01HL145064) and American Heart Association (17GRNT33650018) to M. Azhar.
Footnotes
Competing Interests
JAAG is a scientific co-founder of, scientific advisory board member and equity owner in HiberCell and receives financial compensation as a consultant for HiberCell, a Mount Sinai spin-off company focused on the research and development of therapeutics that prevent or delay the recurrence of cancer.
References
- 1.Chaffer CL & Weinberg RA A perspective on cancer cell metastasis. Science vol. 331 1559–1564 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Sherry MM, Greco FA, Johnson DH & Hainsworth JD Metastatic breast cancer confined to the skeletal system. An indolent disease. Am. J. Med. 81, 381–386 (1986). [DOI] [PubMed] [Google Scholar]
- 3.Engel J et al. The process of metastasisation for breast cancer. Eur. J. Cancer 39, 1794–1806 (2003). [DOI] [PubMed] [Google Scholar]
- 4.H??semann Y. et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell 13, 58–68 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Sanger N. et al. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int. J. Cancer (2011) doi: 10.1002/ijc.25895. [DOI] [PubMed] [Google Scholar]
- 6.Braun S. et al. Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N. Engl. J. Med. (2000) doi: 10.1056/NEJM200002243420801. [DOI] [PubMed] [Google Scholar]
- 7.Chéry L. et al. Characterization of single disseminated prostate cancer cells reveals tumor cell heterogeneity and identifies dormancy associated pathways. Oncotarget 5, 9939–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borgen E. et al. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. (2018) doi: 10.1186/s13058-018-1049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naume B. et al. Clinical outcome with correlation to disseminated tumor cell (DTC) status after DTC-guided secondary adjuvant treatment with docetaxel in early breast cancer. J. Clin. Oncol. 32, 3848–3857 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Bragado P. et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 15, 1351–61 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlson P. et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell Biol. (2019) doi: 10.1038/s41556-018-0267-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson RW et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 18, 1078–1089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi A. et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 208, 2641–55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao H. et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taichman RS et al. GAS6 Receptor Status Is Associated with Dormancy and Bone Metastatic Tumor Formation. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yumoto K. et al. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 6, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung Y. et al. Endogenous GAS6 and Mer receptor signaling regulate prostate cancer stem cells in bone marrow. Oncotarget 7, 25698–25711 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yue X. et al. Leukemia inhibitory factor promotes EMT through STAT3- dependent miR-21 induction. Oncotarget (2015) doi: 10.18632/oncotarget.6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agarwal P. et al. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 24, 769–784.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15, 807–17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawson MA et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu-Lee LY et al. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbRIII–p38MAPK–pS249/ T252RB pathway. Cancer Res. 78, 2911–2924 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cackowski FC & Taichman RS Parallels between hematopoietic stem cell and prostate cancer disseminated tumor cell regulation. Bone vol. 119 82–86 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shiozawa Y. et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Invest. 121, 1298–1312 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson A. et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 135, 1118–1129 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Pinho S. & Frenette PS Haematopoietic stem cell activity and interactions with the niche. Nature Reviews Molecular Cell Biology vol. 20 303–320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunisaki Y. et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanoun M. et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell Niche. Cell Stem Cell (2014) doi: 10.1016/j.stem.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maryanovich M. et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. (2018) doi: 10.1038/s41591-018-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harper KL et al. Mechanism of early dissemination and metastasis in Her2+mammary cancer. Nature (2016) doi: 10.1038/nature20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinho S. et al. PDGFR and CD51 mark human Nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 210, 1351–1367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranganathan AC, Adam AP, Zhang L. & Aguirre-Ghiso JA Tumor cell dormancy induced by p38SAPK and ER-stress signaling: An adaptive advantage for metastatic cells? Cancer Biology and Therapy (2006) doi: 10.4161/cbt.5.7.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fluegen G. et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat. Cell Biol. 19, 120–132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sosa MS et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 6, 1–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Männ L. et al. CD11c.DTR mice develop a fatal fulminant myocarditis after local or systemic treatment with diphtheria toxin. Eur. J. Immunol. (2016) doi: 10.1002/eji.201546245. [DOI] [PubMed] [Google Scholar]
- 36.Christiaansen AF, Boggiatto PM & Varga SM Limitations of Foxp3+ Treg depletion following viral infection in DEREG mice. J. Immunol. Methods (2014) doi: 10.1016/j.jim.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bennett CL et al. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J. Cell Biol. (2005) doi: 10.1083/jcb.200501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kobayashi A. et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med 208, 2641–2655 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu C. et al. Stem cell factor is selectively secreted by arterial endothelial cells in bone marrow. Nat. Commun. (2018) doi: 10.1038/s41467-018-04726-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tikhonova AN et al. The bone marrow microenvironment at single-cell resolution. Nature (2019) doi: 10.1038/s41586-019-1104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vukicevic S, Latin V, Chen ping, Batorsky R. & Reddi AH Localization of osteogenic protein-1 (bone morphogenetic protein-7) during human embryonic development: High affinity binding to basement membranes. Biochem. Biophys. Res. Commun. 198, 693–700 (1994). [DOI] [PubMed] [Google Scholar]
- 42.Gregory KE et al. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J. Biol. Chem. 280, 27970–27980 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Nakahara F. et al. Engineering a haematopoietic stem cell niche by revitalizing mesenchymal stromal cells. Nature Cell Biology (2019) doi: 10.1038/s41556-019-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aguirre-Ghiso JA, Ossowski L. & Rosenbaum SK Green fluorescent protein tagging of extracellular signal-regulated kinase and p38 pathways reveals novel dynamics of pathway activation during primary and metastatic growth. Cancer Res. 64, 7336–7345 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Regot S, Hughey JJ, Bajar BT, Carrasco S. & Covert MW High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oki T. et al. A novel cell-cycle-indicator, mVenus-p27K. Sci. Rep. 4, 4012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asada N. et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. (2017) doi: 10.1038/ncb3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou BO, Yue R, Murphy MM, Peyer JG & Morrison SJ Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell (2014) doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paget S. THE DISTRIBUTION OF SECONDARY GROWTHS IN CANCER OF THE BREAST. Lancet (1889) doi: 10.1016/S0140-6736(00)49915-0. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y. et al. Immunocompetent mouse allograft models for development of therapies to target breast cancer metastasis. Oncotarget 8, 30621–30643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boudreau N. & Bissell MJ Extracellular matrix signaling: Integration of form and function in normal and malignant cells. Curr. Opin. Cell Biol. 10, 640–646 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kenny PA & Bissell MJ Tumor reversion: Correction of malignant behavior by microenvironmental cues. International Journal of Cancer vol. 107 688–695 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turley RS et al. The type III transforming growth factor-β receptor as a novel tumor suppressor gene in prostate cancer. Cancer Res. 67, 1090–1098 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Ajiboye S, Sissung TM, Sharifi N. & Figg WD More than an accessory: Implications of type III transforming growth factor-β receptor loss in prostate cancer. BJU International vol. 105 913–916 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong M. et al. The type III TGF-β receptor suppresses breast cancer progression. J. Clin. Invest. 117, 206–217 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim IY et al. Loss of expression of bone morphogenetic protein receptor type II in human prostate cancer cells. Oncogene 23, 7651–7659 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Singh A. et al. Angiocrine signals regulate quiescence and therapy resistance in bone metastasis. JCI Insight (2019) doi: 10.1172/jci.insight.125679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mignone JL, Kukekov V, Chiang AS, Steindler D. & Enikolopov G. Neural Stem and Progenitor Cells in Nestin-GFP Transgenic Mice. J. Comp. Neurol. 469, 311–324 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Ishtiaq Ahmed AS, Bose GC, Huang L. & Azhar M. Generation of mice carrying a knockout-first and conditional-ready allele of transforming growth factor beta2 gene. Genesis 52, 817–826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ossowski L. & Reich E. Changes in malignant phenotype of a human carcinoma conditioned by growth environment. Cell 33, 323–333 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during the current study that support the reported findings are available in the manuscript, including in the provided Source Data files, and from the corresponding author on reasonable request.