

INTRODUCTION
Migraine is the third most disabling illness in the world among people aged 15 to 49 and affects ~12% of the population worldwide [73; 74]. Chronic migraineurs suffer from unilateral, throbbing headache, disruptions to sensory and motor systems, and up to one third of migraine patients also experience aura [35; 57]. This neurological disorder causes functional impairment during and between migraine attacks, and it is common for comorbidities such as depression, anxiety, and sleep disturbances to manifest and further decrease quality of life [9]. Despite the severe negative effects of migraine on patients, their families, and the economy, this public health issue remains understudied and migraine specific therapeutics remain limited.
We previously identified the delta opioid receptor (DOP) as a promising target for headache disorders [15; 40; 42; 53]. In comparison to mu opioid receptor agonists, activation of DOP produces fewer adverse effects related to gastrointestinal transit, sedation, and respiration [25; 26]; additionally DOP agonists have low abuse liability [8; 17; 43]. We, and others, have shown that DOP agonists are effective in models of migraine-associated pain, negative affect, and aura [19; 53]. We have also observed that chronic treatment with SNC80, a highly selective DOP agonist, can prevent the development of acute and chronic allodynia in a model of post-traumatic headache [40; 42]. Additionally, DOP agonist treatment can alleviate established cephalic allodynia in models of chronic migraine, post-traumatic headache, and medication overuse headache induced by opioids and triptans [42]. Considering the behavioral evidence that DOP activation could be beneficial for the treatment of migraine, the aim of this study was to investigate the mechanisms by which DOP agonists produce these effects.
Calcitonin gene-related peptide (CGRP) is a key regulator of migraine pathophysiology and is considered to be an endogenous migraine trigger [18; 20; 64]. Pioneering clinical studies showed increased CGRP in the jugular outflow during a migraine attack [29; 30]. Further, systemic injection of CGRP to migraine patients causes a delayed headache which can present with a migraine-like phenotype [33; 37]. Small molecule CGRP receptor (CGRPR) antagonists also effectively treat migraine [44; 64], and antibodies targeting CGRP or CGRPR were recently approved as migraine therapies [64]. A possible way in which DOP agonists produce anti-migraine effects is through decreased CGRP release or CGRPR signaling. DOPs have primarily been investigated for the treatment of peripheral pain conditions [28; 50], and its expression in dorsal root ganglia (DRG) and lumbar spinal cord has been extensively studied [4; 32; 58; 71; 72]. Comparatively, there have been very few studies to examine the expression of DOPs in trigeminal ganglia (TG) and trigeminal nucleus caudalis (TNC), which are the peripheral and central nervous system regions that transmit cephalic pain information respectively. The aim of this study was to investigate how CGRP expression was affected by DOP agonist treatment in a model of chronic migraine-associated pain. A further goal was to characterize the expression of DOP, CGRP, and CGRPR within the trigeminovascular complex, and determine if this expression was altered during a chronic migraine state.
MATERIALS AND METHODS
Animals
Experiments were performed on male and female C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME. USA) and DOPeGFP knockin mice weighing 20-30g, and no sex differences were observed. Mice were group housed in a 12h-12h light-dark cycle, where the lights were turned on at 07:00 and turned off at 19:00. Food and water were available ad libitum. All responses were conducted in a blinded fashion by 1-3 experimenters. Weight was recorded on each test day for all experiments. Neither treatments nor drugs significantly affected weight gain or mortality. All experimental procedures were approved by the University of Illinois at Chicago Office of Animal Care and Institutional Biosafety Committee, in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) guidelines and the Animal Care Policies of the University of Illinois at Chicago. All results are reported according to Animal Research: reporting of In Vivo Experiments (ARRIVE) guidelines.
Sensory sensitivity testing
Separate groups of animals were used for all experiments. For all behavioral experiments, mice were counterbalanced into groups following the first basal test for mechanical sensitivity. The experimenter was blinded to the drug condition being tested. No adverse effects were observed in any of the experiments. All mice were tested in a separate behavior room with low-light (~35-50 lux) and low-noise conditions, between 09:00 and 16:00. Mice were tested on a raised wire mesh rack with a plexiglass enclosure with space to test 12 mice, as described previously [41]. For all behavioral tests, mice were habituated to the testing rack for 2 days prior to the first test day, and on each test day for 20 minutes prior to the first measurement. For peripheral measurements, the plantar surface of the left hind paw was tested. For cephalic testing, mice were tested in 4 oz paper cups, to which they had been previously habituated for 1 hour over 2 days. The periorbital region caudal to the eyes and near the midline was tested. To assess mechanical sensitivity, the threshold for responses to punctate mechanical stimuli (mechanical allodynia) was tested according to the up-and-down method [14]. The region of interest was stimulated with a series of eight von Frey hair filaments (bending force ranging from 0.008g, 0.04, 0.07, 0.16, 0.4, 0.6, 1.0, 2.0). A hind paw response was defined as a lifting, shaking, or licking of the stimulated paw. A cephalic response was defined as a shaking, ducking, or vigorous grooming of the head following stimulation. The first filament tested was 0.4g. In the absence of a response, a heavier filament (up) was tried, and in the presence of a response, a lighter filament (down) was tested. This pattern was followed for a maximum of four filaments following the first response.
Nitroglycerin model of chronic migraine
Nitroglycerin (NTG) was purchased at a concentration of 5.0 mg/mL (American Reagent, NY, USA), and was freshly diluted on each test day in 0.9% saline to a concentration of 1mg/mL for a dose of 10 mg/kg. Mice were treated every other day for 9 days with vehicle or NTG (10 mg/kg, IP, Figure 1A). For hind paw experiments, basal thresholds were assessed on days 1, 3, 5, 7, and 9. For cephalic experiments, basal thresholds were assessed on days 1, 5, and 9. On test days, mechanical thresholds were measured prior to vehicle/NTG injection (basal responses) and again 2h post-NTG/VEH administration (post-treatment responses)(Figure 1B).
Figure 1:
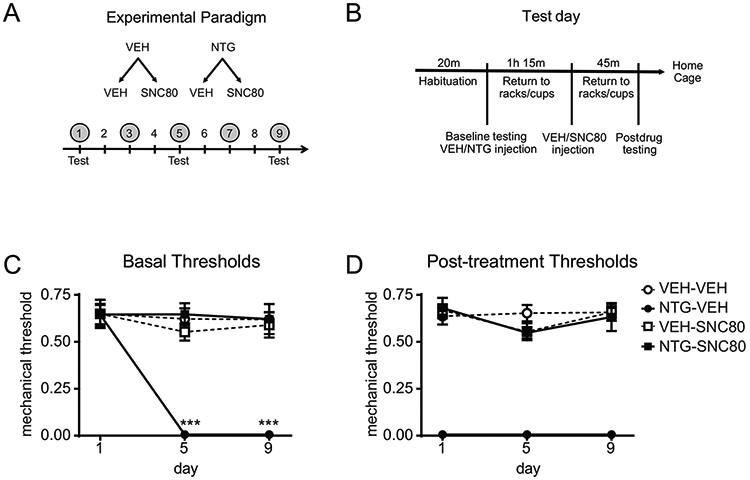
Repeated SNC80 prevents the development of acute and chronic NTG-induced cephalic allodynia. (A) Experimental paradigm. Male and female C57BL6/J mice were treated with vehicle (VEH) or NTG (10 mg/kg IP), followed by VEH or SNC80 (10 mg/kg IP), every other day for 9 days (shaded circles). Cephalic allodynia was only measured on days 1, 5 and 9 (Test). (B) Test day schematic. Baselines were measured prior to VEH/NTG administration. Post-drug thresholds were measured 45min after VEH/SNC80 administration (2h post-NTG). (C) Over 9 days, NTG-VEH mice developed a chronic allodynia as measured before treatment on that day, an effect not observed in NTG-SNC80 treated mice. p<0.001 effect of treatment, time, and interaction, mixed effects model, Holm-Sidak post hoc analysis, ***p<0.001 relative to VEH-VEH on that day. (D) On each test day, NTG evoked acute allodynia, 2 hours post-injection, an effect that was not observed in the NTG-SNC80 treated groups. p<0.001 effect of treatment only. n=6/group. DOP activation prevents the development of acute and chronic migraine-associated pain.
Chronic treatment with SNC80
To determine whether chronic DOP activation prevented the development of NTG-induced hypersensitivity, mice were treated with SNC80 (10 mg/kg, IP) 1h15 minutes after NTG administration on days 1, 3, 5, 7 and 9. For hind paw experiments, mice were tested on days 1, 3, 5, 7, and 9. For cephalic experiments, mice were tested on days 1, 5, and 9 only. On test days, mice were tested 45 minutes after the SNC80/vehicle injection (2h post-NTG).
Immunohistochemistry
Trigeminal ganglia (TG) and brains containing the trigeminal nucleus caudalis (TNC) region were collected on day 10 of mice tested in the NTG paradigm. Mice were anesthetized with Somnasol (100 μL/mouse; 390 mg/mL pentobarbital sodium; Henry Schein, SKU#024352), and perfused intracardially with 15 mL of ice-cold phosphate buffer (0.1M PB, pH 7.2) and subsequently 50 mL of ice-cold 4% paraformaldehyde (PFA)/0.1M PB (pH 7.4) over 5 minutes. TG and brains were harvested from the mice and post-fixed for 2-4h in 4% PFA/0.1M PB at 4°C. Tissue was cryoprotected in 30% sucrose/0.1M PB for 24-36 hours, or until it sank. Sections of the TG and TNC were sliced at 14 μm. Upon slicing, sections were immediately mounted onto slides. Slides were blocked with 5% normal donkey serum in 0.1M phosphate-buffered saline with 0.3% Triton X-100 (NDST) for 1 hour at room temperature. Slides were incubated overnight at room temperature with primary sheep anti-CGRP antibody (RRID AB_725809; ab22560; Abcam; 1:1000 dilution), primary chicken anti-eGFP antibody (RRID AB_300798, ab13970; Abcam; 1:1000) primary rabbit anti-eGFP antibody (RRID AB_221569; A11122; Invitrogen; 1:1000), and primary rabbit anti-RAMP1 antibodies (RRID AB_2736495; PA5-77720, Invitrogen; 1:1000; RRID AB_2801502; CRB034D95, Cambridge Research Biochemicals; 1:500), made in 1% NDST. Slides were washed with 1%NDST before incubating with a secondary antibody solution (Alexa Fluor 555 Donkey anti-Sheep (RRID AB_2535857; A21436;Life Technologies; 1:1000), Alexa Fluor 350 Donkey anti-Sheep (RRID AB_2535751; ThermoFisher, A21097, 1:200); Alexa Fluor 488 Donkey anti-Chicken (RRID AB_2340375; Jackson; 703-545-155;1:1000); Alexa Fluor 488 Donkey anti-Rabbit (RRID AB_2535792; Life technologies; A21206; 1:1000) Alexa Fluor 555 Donkey anti-Rabbit (RRID AB_162543; Life technologies; A31572; 1:1000) made in 1% NDST for 2 hours at room temperature. Slides were washed with 0.1M phosphate buffer, and cover slipped with Fluoromount G mounting solution. Images for quantification were taken by 2 observers in a blinded manner using an EVOS FL Auto Cell Imaging System, using a 20X objective. All images collected were used for analysis. For TGs between 100-500 neurons were counted/animal across several slices. For TNCs, ~10 slices were analyzed/animal and both hemispheres were analyzed. For measurement of fluorescent intensity, in the TG and for DOP+ and RAMP1+ cells in the TNC the intensity of individual cells was measured. In the TNC fluorescent intensity of CGRP as well as DOP without receptor internalization was quantified across the region of interest. Expression of CGRP, DOPeGFP, and RAMP1 was quantified by observers blinded to treatment groups. All CGRP-positive, DOPeGFP-positive, and RAMP1-positive cells from all sections were analyzed. Confocal images were taken by a Zeiss Laser Scanning Microscope (LSM) 710 using a 25X objective.
RNAScope Fluorescent In Situ Hybridization
RNAscope kit was purchased from Advanced Cell Diagnostics RNAScope Technology (ACD Bioscience). C57Bl6/J mice were anesthetized, brain and TG were collected and immediately frozen. Frozen tissue was cut on a cryostat at 14 μm, collected on slides, and processed per the manufacturer’s protocol. Every 4th section was quantified at 3 different dorso-ventral regions along the TNC. The probes used were targeted against the mouse genes for DOP and CRLR.
Experimental Design and Statistical Analyses
The specific statistical design and analyis for each experiment can be found in the figure legends and Supplementary Table 1. Data are expressed as mean + SEM. All mice tested were included in the analysis. All statistical analyses were performed by SigmaStat software, and graphs were generated using GraphPad Prism. For all behavioral experiments, a three-way repeated-measures analysis of variance (ANOVA) was performed, with treatment (VEH/NTG), drug (VEH/SNC80) and time (days) as factors. For cephalic testing a mixed measures analysis was performed as we lost one mouse on the final day of testing, and 3-way RM ANOVA does not allow for missing values. For immunohistochemical or in situ hybridization experiments comparing two groups, a two-tailed Student’s t-test was performed. For immunohistochemical experiments comparing more than two groups, a two-way ANOVA was performed. When a significant interaction occurred, subsequent Holm-Sidak post-hoc analysis was performed. A significance level of p<0.05 was used.
RESULTS
Chronic treatment with SNC80 blocks the development of acute and chronic NTG-induced pain.
To determine the effect of DOP activation on NTG-induced pain, mice were tested with repeated intermittent SNC80 in the chronic NTG model. On days 1, 3, 5, 7, and 9, mice were treated with either vehicle or NTG (10 mg/kg IP), injected with vehicle or SNC80 (10 mg/kg IP) 1h15min later, and tested 45min after than (2h post-NTG) (Figure 1A). Cephalic/periorbital allodynia was only determined on days 1, 5 and 9. On each test day (Figure 1B), both basal (prior to injections that day) and post-treatment thresholds were determined. Mice chronically treated with NTG-VEH developed a significant chronic allodynia during NTG treatment; and this effect was not observed in mice treated with NTG and SNC80 (Figure 1C). In addition, NTG also evoked an acute allodynia 2h post-administration, which was also blocked by SNC80 treatment (Figure 1D). Similar results were observed in peripheral (hind paw) testing in a separate group of mice (Supplementary Figure 1). These results demonstrate that DOP activation following NTG treatment can prevent the development of acute and chronic NTG-induced allodynia.
NTG induces an increase in CGRP expression that is blocked by SNC80.
We next determined if chronic NTG treatment increased expression of the pro-migraine peptide CGRP, and if SNC80 treatment affected this expression. Both the TG and the TNC were examined. Outlined arrowheads show representative CGRP+ cells in the TG (Figure 2A). Mice chronically treated with NTG-VEH had significantly more CGRP+ cells in the TG as compared to the VEH-VEH group (Figure 2B). In contrast, NTG-SNC80 mice had CGRP+ cell counts comparable to VEH-VEH controls (Figure 2B). We used fluorescence intensity as a proxy for how much CGRP was expressed within each CGRP+ cell. NTG-VEH mice showed increased CGRP expression/cell relative to VEH-VEH controls, an effect not observed in the NTG-SNC80 treatment group (Figure 2C). Immunohistochemical staining showed CGRP afferent endings in the superficial laminae of the TNC (Figure 2D; dashed lines outline the lateral edge of the superficial laminae of the TNC). Quantification of fluorescence intensity of this region showed that NTG-VEH mice had significantly increased CGRP expression as compared to VEH-VEH mice, an effect not observed in NTG-SNC80 mice (Figure 2E). Our findings indicated that chronic intermittent NTG increases CGRP expression in the trigeminovascular system, and chronic DOP activation can prevent this increase.
Figure 2:

SNC80 blocks chronic NTG-induced increase in CGRP. Tissue is from mice treated and tested chronically in Figure 1. (A) Representative images of CGRP expression in the trigeminal ganglia of treatment groups. Outlined arrowheads point to CGRP+ cells in the trigeminal ganglia. (B) Chronic NTG administration increases the number of CGRP+ cells in the trigeminal ganglia, an effect not observed in the NTG-SNC80 group. p<0.05 treatment, drug, and interaction, two-way ANOVA, Holm-Sidak post hoc analysis, **p<0.01 relative to VEH-VEH. ##p<0.01 relative to NTG-VEH. (C) Chronic NTG administration increased the amount of CGRP expressed within each CGRP+ cell in the trigeminal ganglia, as detected by fluorescence intensity. Mice treated with NTG-SNC80 did not show this increased expression, p<0.01 treatment, drug, and interaction, two-way ANOVA, Holm-Sidak post hoc analysis, ***p<0.001 relative to VEH-VEH, ###p<0.001 relative to NTG-VEH (D) Representative images of CGRP expression in the trigeminal nucleus caudalis of test groups. Dashed lines outline the lateral edge of the superficial laminae of the trigeminal nucleus caudalis. (E) Chronic NTG administration increased CGRP expression in the superficial layers of the trigeminal nucleus caudalis, an effect that was not observed in NTG-SNC80 mice. Two-way ANOVA, p<0.05 effect of NTG, and p<0.001 effect of SNC80. Chronic, intermittent NTG increases CGRP expression in the trigeminal ganglia and trigeminal nucleus caudalis, an effect that is prevented by DOP activation.
NTG increases CGRP and DOP expression in the trigeminal ganglia
In order to examine the expression of DOP within the trigeminal complex we used DOPeGFP knockin mice [49; 59]. We first confirmed that DOPeGFP mice showed a comparable response to chronic intermittent NTG administration. Similar to C57BL6 mice, DOPeGFP mice also developed acute and chronic cephalic allodynia to NTG (Supplementary Figure 2). We next examined the effect of chronic NTG treatment on DOP and CGRP expression and distribution. Representative images of TG taken from vehicle (VEH; Figure 3A) and NTG (Figure 3B) mice show DOP+ cells with filled arrowheads, and CGRP+ cells with outlined arrowheads. As in C57BL6 mice, chronic NTG resulted in an increase in the number and expression of CGRP in the TG of DOPeGFP mice (Figure 3C and D). Furthermore, chronic NTG also resulted in increased number and expression of DOP within the TG (Figure 3E and F). An overall analysis of co-expression of CGRP+ and DOP+ cells revealed ~40% of DOP+ cells were CGRP+DOP+ cells (Figure 3G) and ~13% of CGRP+ were CGRP+DOP+ (Figure 3H)and this percentage did not change with chronic NTG treatment. Analysis of the total cell population showed that 3.7% of the vehicle and 7.4% of the NTG groups were CGRP+DOP+ (Figure 3I), a difference that did not reach significance. Further analysis of TGs by cell size revealed that CGRP was predominantly expressed in smaller diameter cells, while DOP was mainly expressed in medium to larger diameter cells (Figure 3J). A more detailed analysis of DOP and CGRP expression across cell size revealed that larger diameter neurons showed greater co-expression, and ~40% of cells 25-30 μm in diameter co-expressed the two proteins (Figure 3K). We also examined the expression of DOP with isolectin B4 (IB4) a marker of small diameter C fibers, and neurofilament 200 (NF200), a marker of myelinated A fibers, and observed that DOP was highly expressed in myelinated cells (Supplementary Figure 3). Our results indicate that chronic migraine associated pain results in increased CGRP and DOP expression in the TG, and that co-expression of these two proteins is primarily observed in larger diameter neurons.
Figure 3:
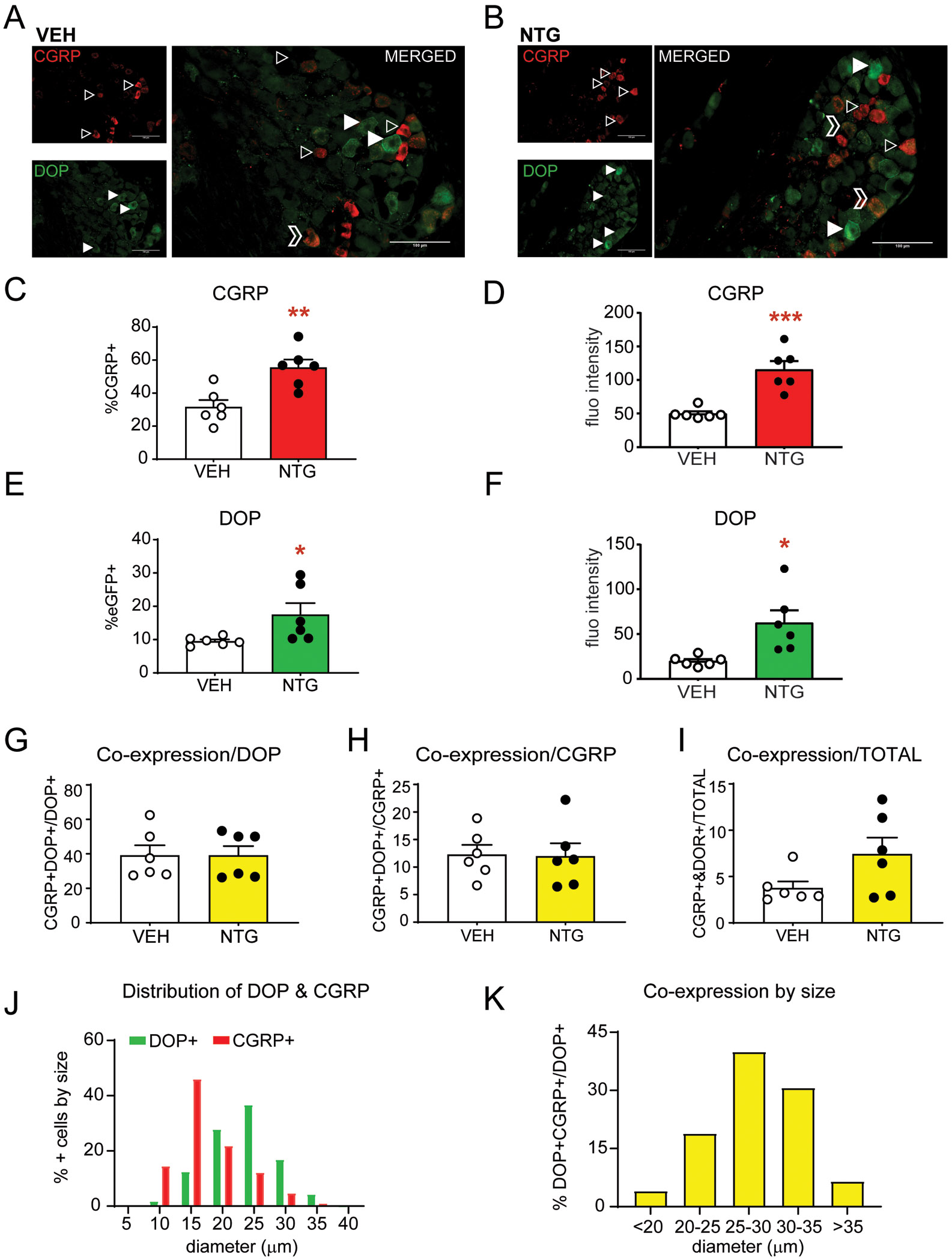
Chronic NTG increases CGRP and DOP expression in the trigeminal ganglia. DOPeGFP mice were treated chronically with VEH/NTG as described above and tissue was collected on day 10. Representative images of DOPeGFP+ and CGRP+ cells in the trigeminal ganglia after chronic (A) VEH and (B) NTG. Outlined arrowheads point to CGRP+ cells and filled in arrowheads point to DOP+ cells. Outlined chevrons point to cells co-expressing CGRP and DOP. Between 200-500 cells were quantified across multiple sections for each mouse. Chronic NTG increased (C) the percentage and (D) expression of CGRP+ cells in the TG of DOPeGFP knockin mice. Chronic NTG also increased (E) the percentage and (F) expression of DOP+ cells in the TG. *p<0.05, **p<0.01, ***p<0.001 t-test. Of the DOP+ cells ~40% were also CGRP+ (G), of the CGRP+ cells ~12% were DOP+ (H), and of the total cell population ~5% co-expressed CGRP and DOP (I). This co-expression was not altered by chronic NTG treatment. (J) Distribution of DOP+ and CGRP+ afferents by cell diameter (μm) shows that these two proteins are largely expressed in different cell populations. (K) DOP+CGRP+ co-expression analyzed by size shows that larger diameter cells show increased co-expression relative to the total DOP+ cell population in each size range. Chronic migraine-associated pain increases both CGRP and DOP and there is some co-expression of this peptide and receptor.
NTG increases CGRP and DOP expression, but not co-expression, in the trigeminal nucleus caudalis
We next determined the expression of CGRP and DOP in the TNC following chronic intermittent treatment with vehicle or NTG. Dashed lines in representative images indicate the lateral edge of the superficial laminae of the TNC (Figure 4A). CGRP expression was robustly expressed in the superficial laminae of the TNC (Figure 4A; top panel), however, DOP expression appeared to be diffusely spread across the TNC (Figure 4A; middle panel). A plot profile of the fluorescence across the TNC shows that CGRP expression is greatest in the transition from the trigeminal spinal tract through lamina I of the TNC, and that DOP expression begins to increase in the transition from lamina 1 through the deeper laminae of the TNC (Figure 4B). We measured the fluorescence intensity of DOP and CGRP in the superficial laminae of the TNC and found that NTG reproducibly increased CGRP expression in this region (Figure 4C) but did not alter DOP fluorescent intensity (Figure 4D). In order to better visualize the cellular resolution of DOP in the TNC, we administered an acute dose of SNC80 (10 mg/kg IP) to mice 2h prior to tissue collection. SNC80 produces robust internalization of DOP [49; 54; 70], therefore making it easier to identify DOP+ cells within this region (Figure 4E). Filled arrowheads in representative images show DOP+ cells in the TNC in both VEH and NTG groups. Chronic NTG caused a significant increase in the percentage of DOP+ cells (Figure 4F), and DOP expression within these cells in the TNC (Figure 4G). These results indicate that like the TG, chronic NTG results in the increased expression of both CGRP and DOP within the TNC. CGRP and DOP were not expressed in the same region therefore co-expression could not specifically be studied here.
Figure 4:

Chronic NTG increases CGRP and DOP expression in the trigeminal nucleus caudalis. (A) Representative images of DOP+ and CGRP+ expression in DOPeGFP knockin mice after chronic VEH or NTG. Dashed lines outline the lateral edge of the superficial laminae of the trigeminal nucleus caudalis. (B) Plot profile detailing fluorescence intensity from the trigeminal tract through the deeper laminae of the trigeminal nucleus caudalis. CGRP and DOP fluorescence peak in different laminae of the trigeminal nucleus caudalis. (C) NTG increases fluorescence intensity associated with CGRP staining in superficial laminae of the trigeminal nucleus caudalis but does not increase (D) DOP expression when measured this way. *p<0.05, t-test. (E) Representative images of internalized DOP+ cells in DOPeGFP knockin mice after chronic VEH or NTG. Internalization was induced by treatment with SNC80 (10 mg/kg IP) 2h prior to tissue collection. Filled arrowheads point to DOP+ cells in the trigeminal nucleus caudalis. The bottom panel is a magnified image of the inset panel. Between 100-500 cells were quantified across multiple sections for each mouse. NTG increases the (F) percentage and (G) fluorescence intensity of DOP+ cells in the superficial laminae of the trigeminal nucleus caudalis. *p<0.05, ***p<0.001, t-test. Chronic migraine-associated pain increases CGRP and DOP in central pain processing regions.
CGRP Receptor is co-expressed with DOP in the trigeminal ganglia and trigeminal nucleus caudalis
The CGRP receptor (CGRPR) is composed of multiple components, including RAMP1, which is a rate-limiting molecule for this receptor. We next examined the co-expression of DOP with RAMP1 in the TG and TNC. Representative images from the TG of DOP, CGRP, and RAMP1 staining are depicted in Figure 5A. Filled arrowheads show DOP+ cells (top panel), outlined arrowheads show CGRP+ cells (middle panel), and filled in arrows are RAMP1+ cells (bottom panel). The merged image suggests that there is high co-expression between DOP and RAMP1. Quantification across multiple animals indicate that chronic NTG does not significantly alter the percentage (Figure 5B) or intensity (Figure 5C) of RAMP1 expression in the TG. However, in both vehicle and NTG treated groups there is a high co-expression (~60%) between RAMP1+ and DOP+ cells in this region (Figure 5D). Analysis of the total cell population showed that 6.7% of the vehicle and 9.3% of the NTG groups were RAMP1+DOP+ (Figure 5E), a difference that did not reach significance. To examine this expression pattern in the TNC, we internalized DOP as described above, and immunostained for DOP, CGRP, and RAMP1. Once again, we observed high expression of DOP throughout the TNC (Figure 5F, top panel), and an enriched CGRP expression in the superficial lamina (Figure 5F, middle panel). Like DOP, RAMP1 was expressed throughout the TNC (Figure 5F, bottom panel), and was highly co-expressed with DOP (Figure 5F, merged). Similar expression patterns were observed with 2 different RAMP1 antibodies. We chose not to quantify total RAMP1+ neurons in the TNC as without DOPeGFP to mark the cell body, it was difficult to separate RAMP1+ processes from cell body staining. Of DOP+ cells in the TNC, 56.4% ± 4.5 also expressed RAMP1.
Figure 5:

DOP is co-expressed with RAMP1, a major component of the CGRP receptor. Representative images of DOP, CGRP, and RAMP1 in (A) trigeminal ganglia and (F) trigeminal nucleus caudalis from DOPeGFP knockin mice. Filled in arrowheads point to DOP+ cells, outlined arrowheads point to CGRP+ cells, filled arrows point to RAMP1+ cells, and outlined chevrons point to DOP+/RAMP1+ cells. Between 100-500 cells were quantified across multiple sections for each mouse. Chronic NTG treatment does not significantly affect (B) percentage of RAMP1+ cells or (C) RAMP1 fluorescent intensity. (D) Within DOP+ cells there is a high co-expression between RAMP1 and DOP (60-70%) ; and of the total cells counted (E) 6-9% co-express RAMP1 and DOP. (F) In the trigeminal nucleus caudalis 56.4 ± 4.5% of DOP+ cells are RAMP1+. There is high co-expression of DOP+ and RAMP1+ cells in the trigeminal complex.
The CGRP receptor is composed of multiple components, which include RAMP1, calcitonin receptor like receptor (CRLR) and receptor component protein (RCP)[21]. RAMP1 is a rate-limiting component of CGRPR, however, this protein is also a component of other receptors. In order to confirm the specificity of DOP co-expression with CGRP receptor, we next examined the co-expression of DOP with CRLR. Although there are commercially available antibodies targeting CRLR, in our hands we did not observe convincing immunohistochemical staining in our tissue. We therefore used RNAScope fluorescent in situ hybridization to examine the distribution of DOP and CRLR in the TNC of C57BL6 mice, and representative images are shown in Figure 6A. We examined the effect of chronic NTG treatment on gene expression of DOP (Figure 6B) and CRLR (Figure 6C) but did not observe a significant upregulation. Regardless of treatment group, we observed a high co-expression of DOP+/CRLR+ cells relative to total DOP+ (Figure 6D) and CRLR+ (Figure 6E) cell populations and ~40% of DOP+ cells co-expressed CRLR and ~60% of CRLR+ cells expressed DOP. These results confirm that DOP is highly co-expressed with specific components of the CGRPR. In addition, this finding validates the results observed in DOPeGFP mice in C57BL6 mice.
Figure 6:

DOP is co-expressed with the CGRP receptor component CRLR. (A) Representative images of DOP and CRLR transcripts as measured by RNAScope in situ hybridization in the trigeminal nucleus caudalis. Filled in arrowheads point to DOP+ cells, outlined arrowheads to CRLR+ cells, and outlined chevrons to DOP+/CRLR+ cells. Between 400-600 cells were quantified across multiple sections for each mouse. Chronic NTG treatment does not significantly affect (B) percentage of DOP+ cells or (C) CRLR+ cells. There is co-expression between DOP and CRLR transcripts relative to DOP+ cells (D) and CRLR+ cells (E) that is maintained after chronic NTG treatment. DOP mRNA is co-expressed with the CGRP-specific component of the CGRP receptor.
DISCUSSION
The aim of this study was to investigate the mechanisms by which DOP activation reduces migraine-associated pain. We found that repeated treatment with a DOP agonist prevented the development of acute and chronic pain in an NTG model of chronic migraine. We also observed that chronic NTG increased expression of CGRP in the trigeminovascular system, and that SNC80 blocked this upregulation. In addition, chronic migraine-associated pain also increased DOP expression. Approximately 40% of DOP+ cells and ~4-7% of total TG neurons co-expressed DOP and CGRP, and this co-expression was predominantly in larger diameter neurons. We also observed high co-expression of DOP with RAMP1 in the TG. In contrast, in the TNC there was very little co-expression between DOP and CGRP, whereas there was high co-expression between DOP and the major components of the CGRP receptor, RAMP1 and CRLR. DOPs are Gi/o G protein coupled receptors, and activation of DOP results in hyperpolarization and inhibition of cell firing as well as decreased neurotransmitter release in axon terminals [16; 26]. Our findings suggest that DOP agonists can inhibit migraine-associated pain in two ways: by inhibiting CGRP release from a proportion of TG neurons, and by potentially inhibiting pro-nociceptive signaling by the CGRPR. We propose that chronic NTG treatment results in elevated CGRP levels, and that DOP agonists counteract this effect by inhibiting CGRP release and reducing cellular excitability evoked by CRLR activation, thus inhibiting acute and chronic migraine pain (Supplementary Figure 4).
There is a need for new and mechanistically diverse migraine treatments, and our behavioral experiment indicates that DOP is such a target. We found that co-administration of the DOP agonist, SNC80, with NTG inhibited both acute allodynia, determined 2h post-NTG, as well as the development of chronic allodynia. We have previously shown similar results in this model of chronic migraine with the known migraine preventives, topiramate [52] and propranolol [65] thus strengthening the validity of this model as a screen for novel migraine prophylactics. Our previous work also shows that SNC80 can inhibit cortical spreading depression [53], which is a physiological correlate of migraine aura, and is also used to screen preventive therapies [2]. One concern with the use of DOP agonists as preventives is that chronic treatment could result in medication overuse headache/opioid induced hyperalgesia. An estimated 15% of migraineurs go on to develop medication overuse headache, which further exacerbates and increases the frequency of migraine [35; 60]. We recently showed that unlike sumatriptan or morphine, SNC80 produces limited medication overuse headache/hyperalgesia [42], thus further supporting the development of DOP agonists for chronic migraine treatment.
CGRP is a key regulator of migraine. CGRP is upregulated during a migraine attack [29; 30] and increased CGRP levels are also observed in chronic migraine patients interictally [13]. Antibodies targeting CGRP and its receptor and CGRP receptor antagonists are highly effective in treating both episodic and chronic migraine [64], and are now being prescribed regularly. In line with the clinical data, we also found that CGRP expression was upregulated in our NTG model of chronic migraine-associated pain. We observed increases in both the amount of CGRP expressed/cell as well as in the number of CGRP+ cells in TG. The TNC is the terminating region for trigeminal ganglia, and here we also observed a significant increase in CGRP expression following chronic NTG treatment. Correspondingly, other groups have also observed increased CGRP gene expression in the TG [24; 31], medulla pons, and cervical spinal cord [31] of rats treated with chronic intermittent NTG. These results agree with the notion that upregulated CRGP is a significant feature of chronic migraine; and provides mechanistic validation for the use of chronic NTG as a model for this disorder.
Along with an increase in CGRP, chronic NTG also increased DOP expression in both TG and TNC. We did not see a corresponding increase in DOP mRNA. NTG may increase DOP expression by augmenting trafficking of receptor from intracellular stores, increasing stability of existing DOP mRNA, or increasing translation of DOP mRNA to protein. Delta agonists are not highly effective in models of acute pain [25], but have demonstrated efficacy in chronic pain conditions [11; 27; 36; 49; 51; 55; 69]. This discrepancy is thought to be due to increased functionality and/or cell surface expression of DOP during chronic pain states [10; 26]. Upregulation or increased DOP function has been observed in dorsal root ganglia and lumbar spinal cord in peripheral models of inflammatory [27; 48] and neuropathic [36] pain. In addition, in a rat model of acute pulpitis DOP protein and mRNA expression was also found to be upregulated in TG [34]. Furthermore, chronic morphine [12; 39] or alcohol [68] treatment are also found to increase DOP activity in peripheral regions. Upregulation of DOP function has also been observed in response to inflammatory mediators such as arachidonic acid [63], bradykinin [45; 47], and protease activated receptor 2 [46]; and modulation of phosphatases, kinases, and cellular scaffolds [6; 7; 62] may serve as a common mechanism to upregulate DOP function in these diverse pain and drug states. DOP expression is thus dynamically regulated, and may act as a defense mechanism in response to chronic pain, headache, and/or stress [5]. Considering that headache disorders are closely linked to stress and chronic pain, increased DOP functionality could be a common thread between all these conditions.
DOPs are Gi/o G protein coupled receptors, and a predominant characteristics of DOP activation is to decrease neurotransmission, an effect that occurs predominantly through altered ion channel conductance mediated by Gβγ subunits [26]. We initially hypothesized that DOP and CGRP were co-expressed, and activation of DOP would therefore lead to decreased CRGP release. We found that overall in TG ~40% of DOP+ cells were also CGRP+, and that of the CGRP expressing cells ~12% were DOP+. We observed that CGRP expression was primarily found in smaller unmyelinated cells, similar to reports across multiple species including human [1; 3; 22; 23; 66]. In contrast, DOP was predominantly expressed in medium to large myelinated cells. A more detailed analysis of these larger diameter neurons revealed a higher co-expression of DOP and CGRP relative to the overall population. DOP expression has been well characterized in DRG using DOPeGFP mice [4]. These studies established the low co-expression between DOP and CGRP in small NF200-negative DRG neurons, and the greater co-expression in NF200+ larger diameter neurons. [4; 58]. These findings have since been confirmed by multiple RNA sequencing studies [61; 67; 75]. A previous study examining the co-expression of DOP and CGRP in rat dura and TG also found co-expression between the two molecules [56]. Cells that do co-express DOP and CGRP may be particularly important in regulating cephalic responses, and future studies will examine their projection profiles and potential role in head pain processing.
We found that there was little co-expression between DOP and CGRP in the TNC. CGRP expression was concentrated in fibers innervating superficial layers of the TNC, along with limited staining in cell bodies in deeper lamina. This pattern of expression is similar to that observed in human and rat TNC [21]. In contrast, DOP was expressed throughout the TNC, and DOP+ cells were found at the border of lamina 2 and in deeper lamina, consistent with pervious observations in the spinal cord dorsal horn [71]. Furthermore, the expression of DOP anatomically overlapped with expression of components of the CGRP receptor, CRLR and RAMP1. The observation that CRLR and RAMP1 was in deeper lamina of the spinal trigeminal tract is comparable to the expression pattern seen in rat and human TNC [21]. We propose that in the TNC DOP activation inhibits CGRP receptor expressing cells, and thus inhibits the pro-nociceptive signaling of CGRP.
This study provides further support for the development of DOP agonists for the treatment of migraine; and suggests that DOP could relieve migraine by inhibiting CGRP release from TG and inhibiting CGRP receptor signaling in both TG an TNC. Post-mortem studies also demonstrate that DOP is expressed broadly in human TG and TNC [38], thus placing DOP in an appropriate location to mediate human cephalic pain. DOP agonists may therefore help to expand and diversify the migraine therapy toolbox.
Supplementary Material
ACKNOWLEDGEMENTS
LM is a member of the Graduate Program in Neuroscience at UIC. Research was supported by NIDA R01 grant DA040688 (AP), Diversity Supplement DA040688-02S1 (LM), and W81XWH-15-1-0076 Neurosensory Research Award from the Department of Defense (G.S). G.S. is a New York Stem Cell Foundation – Robertson Investigator. The authors declare no conflicts of interest
REFERENCES
- [1].Alvarez FJ, Morris HR, Priestley JV. Sub-populations of smaller diameter trigeminal primary afferent neurons defined by expression of calcitonin gene-related peptide and the cell surface oligosaccharide recognized by monoclonal antibody LA4. J Neurocytol 1991;20(9):716–731. [DOI] [PubMed] [Google Scholar]
- [2].Ayata C, Jin H, Kudo C, Dalkara T, Moskowitz MA. Suppression of cortical spreading depression in migraine prophylaxis. AnnNeurol 2006;59(4):652–661. [DOI] [PubMed] [Google Scholar]
- [3].Bae JY, Kim JH, Cho YS, Mah W, Bae YC. Quantitative analysis of afferents expressing substance P, calcitonin gene-related peptide, isolectin B4, neurofilament 200, and Peripherin in the sensory root of the rat trigeminal ganglion. J Comp Neurol 2015;523(1):126–138. [DOI] [PubMed] [Google Scholar]
- [4].Bardoni R, Tawfik VL, Wang D, Francois A, Solorzano C, Shuster SA, Choudhury P, Betelli C, Cassidy C, Smith K, de Nooij JC, Mennicken F, O'Donnell D, Kieffer BL, Woodbury CJ, Basbaum AI, MacDermott AB, Scherrer G. Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron 2014;81(6):1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bertels Z, Pradhan AAA. Emerging Treatment Targets for Migraine and Other Headaches. Headache 2019;59 Suppl 2:50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brackley AD, Gomez R, Akopian AN, Henry MA, Jeske NA. GRK2 Constitutively Governs Peripheral Delta Opioid Receptor Activity. Cell Rep 2016;16(10):2686–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brackley AD, Sarrami S, Gomez R, Guerrero KA, Jeske NA. Identification of a signaling cascade that maintains constitutive delta-opioid receptor incompetence in peripheral sensory neurons. J Biol Chem 2017;292(21):8762–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brandt MR, Furness MS, Rice KC, Fischer BD, Negus SS. Studies of tolerance and dependence with the delta-opioid agonist SNC80 in rhesus monkeys responding under a schedule of food presentation. JPharmacolExpTher 2001;299(2):629–637. [PubMed] [Google Scholar]
- [9].Buse DC, Rupnow MF, Lipton RB. Assessing and managing all aspects of migraine: migraine attacks, migraine-related functional impairment, common comorbidities, and quality of life. Mayo Clinic proceedings 2009;84(5):422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends PharmacolSci 2007;28(1):23–31. [DOI] [PubMed] [Google Scholar]
- [11].Cahill CM, Morinville A, Hoffert C, O'Donnell D, Beaudet A. Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain 2003;101(1-2):199–208. [DOI] [PubMed] [Google Scholar]
- [12].Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. JNeurosci 2001;21(19):7598–7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cernuda-Morollon E, Larrosa D, Ramon C, Vega J, Martinez-Camblor P, Pascual J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 2013;81(14):1191–1196. [DOI] [PubMed] [Google Scholar]
- [14].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. JNeurosciMethods 1994;53(1):55–63. [DOI] [PubMed] [Google Scholar]
- [15].Charles A, Pradhan AA. Delta-opioid receptors as targets for migraine therapy. Curr Opin Neurol 2016;29(3):314–319. [DOI] [PubMed] [Google Scholar]
- [16].Corder G, Castro DC, Bruchas MR, Scherrer G. Endogenous and Exogenous Opioids in Pain. Annu Rev Neurosci 2018;41:453–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Do Carmo GP, Folk JE, Rice KC, Chartoff E, Carlezon WA Jr., Negus SS. The selective non-peptidic delta opioid agonist SNC80 does not facilitate intracranial self-stimulation in rats. EurJPharmacol 2009;604(1-3):58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dodick DW. CGRP ligand and receptor monoclonal antibodies for migraine prevention: Evidence review and clinical implications. Cephalalgia 2019;39(3):445–458. [DOI] [PubMed] [Google Scholar]
- [19].Dripps IJ, Boyer BT, Neubig RR, Rice KC, Traynor JR, Jutkiewicz EM. Role of signalling molecules in behaviours mediated by the delta opioid receptor agonist SNC80. Br J Pharmacol 2018;175(6):891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Edvinsson L, Goadsby PJ. Discovery of CGRP in relation to migraine. Cephalalgia 2019;39(3):331–332. [DOI] [PubMed] [Google Scholar]
- [21].Eftekhari S, Edvinsson L. Calcitonin gene-related peptide (CGRP) and its receptor components in human and rat spinal trigeminal nucleus and spinal cord at C1-level. BMC Neurosci 2011;12:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eftekhari S, Salvatore CA, Calamari A, Kane SA, Tajti J, Edvinsson L. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 2010;169(2):683–696. [DOI] [PubMed] [Google Scholar]
- [23].Eftekhari S, Salvatore CA, Johansson S, Chen TB, Zeng Z, Edvinsson L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res 2015;1600:93–109. [DOI] [PubMed] [Google Scholar]
- [24].Farajdokht F, Mohaddes G, Shanehbandi D, Karimi P, Babri S. Ghrelin attenuated hyperalgesia induced by chronic nitroglycerin: CGRP and TRPV1 as targets for migraine management. Cephalalgia 2017:333102417748563. [DOI] [PubMed] [Google Scholar]
- [25].Gallantine EL, Meert TF. A comparison of the antinociceptive and adverse effects of the mu-opioid agonist morphine and the delta-opioid agonist SNC80. Basic ClinPharmacolToxicol 2005;97(1):39–51. [DOI] [PubMed] [Google Scholar]
- [26].Gendron L, Cahill CM, von Zastrow M, Schiller PW, Pineyro G. Molecular Pharmacology of delta-Opioid Receptors. Pharmacol Rev 2016;68(3):631–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gendron L, Lucido AL, Mennicken F, O'Donnell D, Vincent JP, Stroh T, Beaudet A. Morphine and pain-related stimuli enhance cell surface availability of somatic delta-opioid receptors in rat dorsal root ganglia. JNeurosci 2006;26(3):953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gendron L, Mittal N, Beaudry H, Walwyn W. Recent advances on the delta opioid receptor: From trafficking to function. Br J Pharmacol 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 1993;33(1):48–56. [DOI] [PubMed] [Google Scholar]
- [30].Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 1990;28(2):183–187. [DOI] [PubMed] [Google Scholar]
- [31].Greco R, Demartini C, Zanaboni AM, Tassorelli C. Chronic and intermittent administration of systemic nitroglycerin in the rat induces an increase in the gene expression of CGRP in central areas: potential contribution to pain processing. J Headache Pain 2018;19(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Guan JS, Xu ZZ, Gao H, He SQ, Ma GQ, Sun T, Wang LH, Zhang ZN, Lena I, Kitchen I, Elde R, Zimmer A, He C, Pei G, Bao L, Zhang X. Interaction with vesicle luminal protachykinin regulates surface expression of delta-opioid receptors and opioid analgesia. Cell 2005;122(4):619–631. [DOI] [PubMed] [Google Scholar]
- [33].Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia 2010;30(10):1179–1186. [DOI] [PubMed] [Google Scholar]
- [34].Huang J, Lv Y, Fu Y, Ren L, Wang P, Liu B, Huang K, Bi J. Dynamic Regulation of Delta-Opioid Receptor in Rat Trigeminal Ganglion Neurons by Lipopolysaccharide-induced Acute Pulpitis. J Endod 2015. [DOI] [PubMed] [Google Scholar]
- [35].ICHD3b. The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia : an international journal of headache 2013;33(9):629–808. [DOI] [PubMed] [Google Scholar]
- [36].Kabli N, Cahill CM. Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain 2007;127(1-2):84–93. [DOI] [PubMed] [Google Scholar]
- [37].Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia 2002;22(1):54–61. [DOI] [PubMed] [Google Scholar]
- [38].Mennicken F, Zhang J, Hoffert C, Ahmad S, Beaudet A, O'Donnell D. Phylogenetic changes in the expression of delta opioid receptors in spinal cord and dorsal root ganglia. JComp Neurol 2003;465(3):349–360. [DOI] [PubMed] [Google Scholar]
- [39].Morinville A, Cahill CM, Aibak H, Rymar VV, Pradhan A, Hoffert C, Mennicken F, Stroh T, Sadikot AF, O'Donnell D, Clarke PB, Collier B, Henry JL, Vincent JP, Beaudet A. Morphine-induced changes in delta opioid receptor trafficking are linked to somatosensory processing in the rat spinal cord. JNeurosci 2004;24(24):5549–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moye LS, Novack ML, Tipton AF, Krishnan H, Pandey SC, Pradhan AA. The development of a mouse model of mTBI-induced post-traumatic migraine, and identification of the delta opioid receptor as a novel therapeutic target. Cephalalgia 2018:333102418777507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Moye LS, Pradhan AAA. Animal Model of Chronic Migraine-Associated Pain. Curr Protoc Neurosci 2017;80:9 60 61–69 60 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moye LS, Tipton AF, Dripps I, Sheets Z, Crombie A, Violin JD, Pradhan AA. Delta opioid receptor agonists are effective for multiple types of headache disorders. Neuropharmacology 2018;148:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Negus SS, Rosenberg MB, Altarifi AA, O'Connell RH, Folk JE, Rice KC. Effects of the delta opioid receptor agonist SNC80 on pain-related depression of intracranial self-stimulation (ICSS) in rats. J Pain 2012;13(4):317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. NEnglJMed 2004;350(11):1104–1110. [DOI] [PubMed] [Google Scholar]
- [45].Patwardhan AM, Berg KA, Akopain AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM. Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci 2005;25(39):8825–8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Patwardhan AM, Diogenes A, Berg KA, Fehrenbacher JC, Clarke WP, Akopian AN, Hargreaves KM. PAR-2 agonists activate trigeminal nociceptors and induce functional competence in the delta opioid receptor. Pain 2006;125(1-2):114–124. [DOI] [PubMed] [Google Scholar]
- [47].Pettinger L, Gigout S, Linley JE, Gamper N. Bradykinin controls pool size of sensory neurons expressing functional delta-opioid receptors. J Neurosci 2013;33(26):10762–10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pradhan A, Smith M, McGuire B, Evans C, Walwyn W. Chronic inflammatory injury results in increased coupling of delta opioid receptors to voltage-gated Ca2+ channels. Mol Pain 2013;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pradhan AA, Becker JA, Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Massotte D, Gaveriaux-Ruff C, Kieffer BL. In vivo delta opioid receptor internalization controls behavioral effects of agonists. PLoSOne 2009;4(5):e5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends PharmacolSci 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pradhan AA, Perroy J, Walwyn WM, Smith ML, Vicente-Sanchez A, Segura L, Bana A, Kieffer BL, Evans CJ. Agonist-Specific Recruitment of Arrestin Isoforms Differentially Modify Delta Opioid Receptor Function. J Neurosci 2016;36(12):3541–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pradhan AA, Smith ML, McGuire B, Tarash I, Evans CJ, Charles A. Characterization of a novel model of chronic migraine. Pain 2014;155(2):269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pradhan AA, Smith ML, Zyuzin J, Charles A. delta-Opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. Br J Pharmacol 2014;171(9):2375–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pradhan AA, Tawfik VL, Tipton AF, Scherrer G. In vivo techniques to investigate the internalization profile of opioid receptors. Methods Mol Biol 2015;1230:87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pradhan AA, Walwyn W, Nozaki C, Filliol D, Erbs E, Matifas A, Evans C, Kieffer BL. Ligand-directed trafficking of the delta-opioid receptor in vivo: two paths toward analgesic tolerance. JNeurosci 2010;30(49):16459–16468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rice FL, Xie JY, Albrecht PJ, Acker E, Bourgeois J, Navratilova E, Dodick DW, Porreca F. Anatomy and immunochemical characterization of the non-arterial peptidergic diffuse dural innervation of the rat and Rhesus monkey: Implications for functional regulation and treatment in migraine. Cephalalgia 2017;37(14):1350–1372. [DOI] [PubMed] [Google Scholar]
- [57].Russell MB, Rasmussen BK, Thorvaldsen P, Olesen J. Prevalence and sex-ratio of the subtypes of migraine. International journal of epidemiology 1995;24(3):612–618. [DOI] [PubMed] [Google Scholar]
- [58].Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 2009;137(6):1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Laustriat D, Cao YQ, Basbaum AI, Dierich A, Vonesh JL, Gaveriaux-Ruff C, Kieffer BL. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. ProcNatlAcadSciUSA 2006;103(25):9691–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schwedt TJ, Alam A, Reed ML, Fanning KM, Munjal S, Buse DC, Dodick DW, Lipton RB. Factors associated with acute medication overuse in people with migraine: results from the 2017 migraine in America symptoms and treatment (MAST) study. J Headache Pain 2018;19(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sharma N, Flaherty K, Lezgiyeva K, Wagner DE, Klein AM, Ginty DD. The emergence of transcriptional identity in somatosensory neurons. Nature 2020;577(7790):392–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shiwarski DJ, Tipton A, Giraldo MD, Schmidt BF, Gold MS, Pradhan AA, Puthenveedu MA. A PTEN-Regulated Checkpoint Controls Surface Delivery of delta Opioid Receptors. J Neurosci 2017;37(14):3741–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sullivan LC, Berg KA, Clarke WP. Dual regulation of delta-opioid receptor function by arachidonic acid metabolites in rat peripheral sensory neurons. J Pharmacol Exp Ther 2015;353(1):44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tepper SJ. History and Review of anti-Calcitonin Gene-Related Peptide (CGRP) Therapies: From Translational Research to Treatment. Headache 2018;58 Suppl 3:238–275. [DOI] [PubMed] [Google Scholar]
- [65].Tipton AF, Tarash I, McGuire B, Charles A, Pradhan AA. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia 2016;36(11):1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tsai SH, Tew JM, McLean JH, Shipley MT. Cerebral arterial innervation by nerve fibers containing calcitonin gene-related peptide (CGRP): I. Distribution and origin of CGRP perivascular innervation in the rat. J Comp Neurol 1988;271(3):435–444. [DOI] [PubMed] [Google Scholar]
- [67].Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015;18(1):145–153. [DOI] [PubMed] [Google Scholar]
- [68].van Rijn RM, Brissett DI, Whistler JL. Emergence of functional spinal delta opioid receptors after chronic ethanol exposure. Biol Psychiatry 2012;71(3):232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vicente-Sanchez A, Dripps IJ, Tipton AF, Akbari H, Akbari A, Jutkiewicz EM, Pradhan AA. Tolerance to high-internalizing delta opioid receptor agonist is critically mediated by arrestin 2. Br J Pharmacol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vicente-Sanchez A, Segura L, Pradhan AA. The delta opioid receptor tool box. Neuroscience 2016;338:145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang D, Tawfik VL, Corder G, Low SA, Francois A, Basbaum AI, Scherrer G. Functional Divergence of Delta and Mu Opioid Receptor Organization in CNS Pain Circuits. Neuron 2018;98(1):90–108.e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, Lu YJ, Zhang ZN, He SQ, Zheng HC, Wu SX, Hokfelt TG, Bao L, Zhang X. Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. ProcNatlAcadSciUSA 2010;107(29):13117–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].WHO LtB. Atlas of headache disorders and resources in the world 2011, Vol. 72, 2011. [Google Scholar]
- [74].Woldeamanuel YW, Cowan RP. Migraine affects 1 in 10 people worldwide featuring recent rise: A systematic review and meta-analysis of community-based studies involving 6 million participants. Journal of the neurological sciences 2017;372:307–315. [DOI] [PubMed] [Google Scholar]
- [75].Zheng Y, Liu P, Bai L, Trimmer JS, Bean BP, Ginty DD. Deep Sequencing of Somatosensory Neurons Reveals Molecular Determinants of Intrinsic Physiological Properties. Neuron 2019;103(4):598–616.e597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.