

Abstract
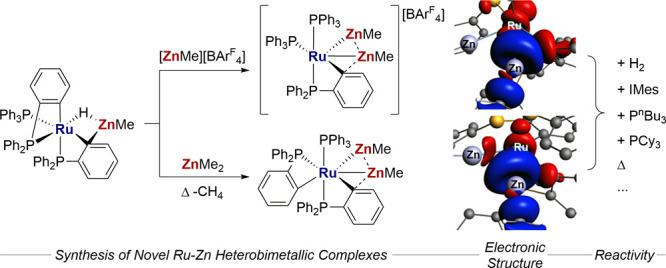
A combined experimental and computational study of the structure and reactivity of two [RuZn2Me2] complexes, neutral [Ru(PPh3)(Ph2PC6H4)2(ZnMe)2] (2) and cationic [Ru(PPh3)2(Ph2PC6H4)(ZnMe)2][BArF4] ([BArF4] = [B{3,5-(CF3)2C6H3}4]) (3), is presented. Structural and computational analyses indicate these complexes are best formulated as containing discrete ZnMe ligands in which direct Ru–Zn bonding is complemented by weaker Zn···Zn interactions. The latter are stronger in 2, and both complexes exhibit an additional Zn···Caryl interaction with a cyclometalated phosphine ligand, this being stronger in 3. Both 2 and 3 show diverse reactivity under thermolysis and with Lewis bases (PnBu3, PCy3, and IMes). With 3, all three Lewis bases result in the loss of [ZnMe]+. In contrast, 2 undergoes PPh3 substitution with PnBu3, but with IMes, loss of ZnMe2 occurs to form [Ru(PPh3)(C6H4PPh2)(C6H4PPhC6H4Zn(IMes))H] (7). The reaction of 3 with H2 affords the cationic trihydride complex [Ru(PPh3)2(ZnMe)2(H)3][BArF4] (12). Computational analyses indicate that both 12 and 7 feature bridging hydrides that are biased toward Ru over Zn.
Short abstract
Experimental and computational studies of two [Ru-Zn2R2] heterobimetallic complexes, neutral [Ru(PPh3)(Ph2PC6H4)2(ZnMe)2] and cationic [Ru(PPh3)2(Ph2PC6H4)(ZnMe)2]+, reveal Ru−ZnR and Zn···Zn interactions suggesting that the complexes are best formulated as [Ru(ZnR)2] species. The Ru−Zn bonding is supplemented by peripheral Zn···Caryl interactions to a cyclometalated phosphine ligand. Studies of the reactivity of the two complexes with respect to thermolysis, a range of Lewis bases (PR3 and NHC), and H2 reveal diverse behavior, with both Ru and Zn acting as the reaction center.
Introduction
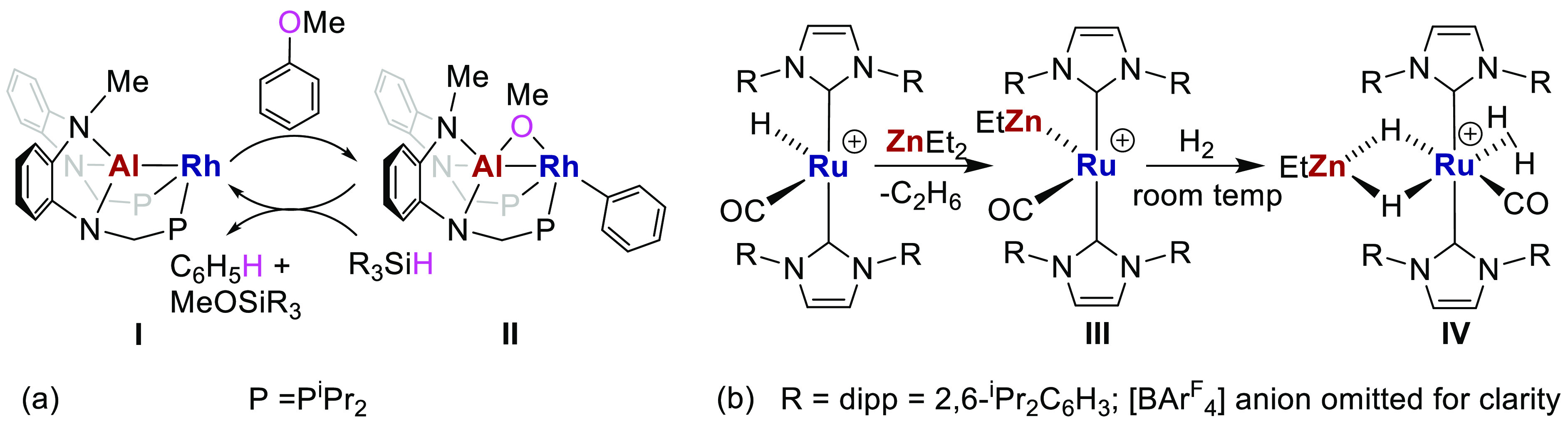
Heterobimetallic complexes comprised of a transition metal (TM) and a main group metal (MGM) are the focus of considerable interest1−3 because of the possibility that the disparate chemistry of the two partners could combine cooperatively to bring about the novel stoichiometric and/or catalytic activation of small molecules.4−9 In one recent example, shown in Scheme 1a, the challenging C–O activation of an anisole takes place across the Rh–Al bond of complex I to afford II, which upon addition of a silane, mediates catalytic C–O bond reduction.10 Complex I represents one class of heterobimetallic complexes in which the MGM forms part of a multidentate ligand on the TM center.11 Another class of complex is represented by III in Scheme 1b, in which the MGM is unsupported and unconstrained. In this particular case, both Ru and Zn centers are coordinatively unsaturated, and this “dual unsaturation” allows them to act cooperatively in the stoichiometric activation of H2 to give IV.12 We have interpreted Ru–Zn bonding within complex III and other related Ru–Zn complexes13,14 in terms of a donor–acceptor interaction between a Ru(0) metal center and Z-type Zn-based acceptor ligands.
Scheme 1. Examples of Cooperativity in Small Molecule Activation by TM–MGM Heterobimetallic Complexes.
Complex III is formed upon elimination of an alkane,15−18 an approach we have used to prepare other Ru and mono-Zn-containing products, including complex 1 in Scheme 2 that features bridging hydride and aryl ligands.12−14,19−21 Accordingly, the reaction of complex 1 with ZnMe2 resulted in further alkane elimination and formation of neutral [RuZn2Me2] complex 2.14 Alternatively, reaction with a source of [ZnMe]+ induced C–H reductive coupling in 1 and formation of cationic [RuZn2Me2] complex 3.13
Scheme 2. Syntheses of Neutral and Cationic [RuZn2Me2] Complexes 2 and 3.
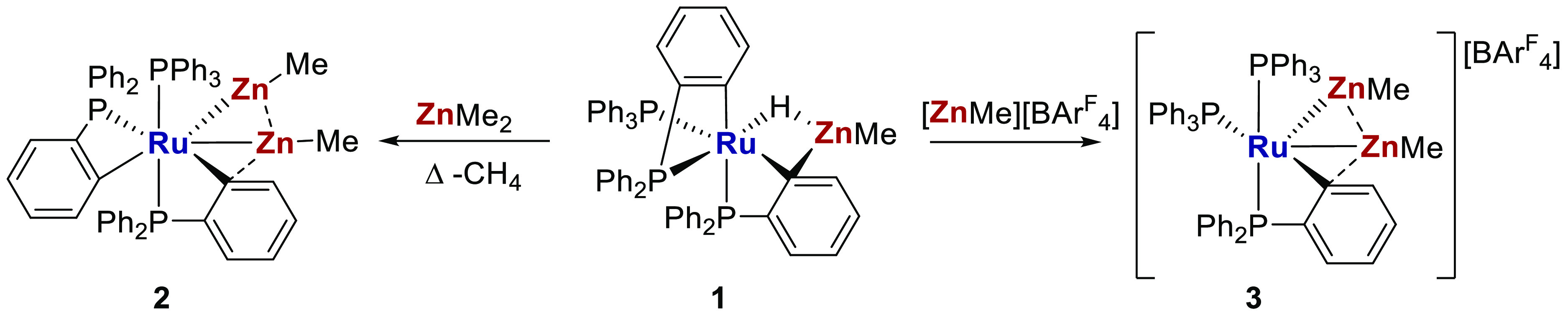
[BArF4] = [B(3,5-(CF3)2C6H3)4]. The nature of the bonding between centers connected by dashed lines is investigated herein.22
Another strategy for the preparation of [TM-Zn2R2] species23−25 involves addition of Carmona’s Cp*Zn–ZnCp* dimer to low-valent precursors.26,27 On the basis of the isolobal nature of Cp*Zn and a hydrogen atom, the coordination of the TM center to an intact Zn–Zn bond can be considered to form an all-metal analogue of a TM(η2-H2) complex. Likewise, weakening of the Zn–Zn interaction to the point where it gives two ZnCp* ligands has been compared to the oxidative cleavage of the η2-H2 ligand to form two M–H bonds, although such Zn–Zn bond cleavage is proposed to proceed without any change in the formal oxidation state.23,26 In such cases, ZnCp* and related ZnR (R = alkyl or aryl) ligands have been formulated as monovalent one-electron donors.26 Computational studies have suggested that the extent to which the Zn–Zn interaction in Cp*Zn–ZnCp* is retained upon approach to a TM is dependent on the nature of the metal itself, the surrounding ancillary ligands, and the ZnR substituents.26−28
In this context, the availability of the closely related neutral and cationic [RuZn2R2] complexes, 2 and 3, respectively, provides an opportunity to explore the analogy between {RZn-ZnR} and H2. Herein, we report computational and experimental studies to this end.
Results and Discussion
Structure and Bonding in 2 and 3
Figure 1 shows the geometries and labeling system used in the discussion of 2 and 3. In general, good agreement was seen between the experimental and fully optimized structures; however, the Zn1···C1 distances were overestimated by 0.06–0.20 Å depending on the functional used (Supporting Information). Therefore, to analyze the observed geometries, we have taken the heavy atom (i.e., non-H) positions from the crystallographic studies and optimized the H atom positions with the BP86 functional. This approach also allows for a consistent treatment of the new hydride-containing structures that we describe below, where the H atom location is intrinsically less precise.
Figure 1.

Geometries and labeling scheme used in the analyses of 2 and 3, focusing on the central RuZn2 core. Selected distances are given in angstroms.
Both 2 and 3 feature triangular RuZn2 moieties with Ru–Zn distances that are shorter than Zn–Zn distances. The Ru–Zn distances are more symmetrical in 2 (2.50/2.51 Å) than in 3 (2.41/2.55 Å) and are all within the sum of the covalent radii (Ru, 1.46 Å; Zn, 1.22 Å),29 suggesting direct Ru–Zn bonds in all cases. Zn–Zn bonding in related systems has been discussed within the limits of the Zn–Zn distance in Zn2Cp*2 (2.31 Å), and the metallic radius of Zn (1.339 Å) and the Zn–Zn distances in 2 and 3 (2.59 and 2.68 Å, respectively) are at the upper end of this range.26 A [TMZn2R2] unit with an intact η2-RZn–ZnR moiety would also be characterized by near-linear Zn–Zn–R angles and M–Zn–R angles approaching 150° (the limit for an equilateral triangle).26 In 2, the average Zn–Zn–R and Ru–Zn–Me angles are 133.9° and 165.8°, respectively, while in 3, the average Zn–Zn–R angle is 113.2° and the Ru–Zn2–Me angle is 179.4°. These data indicate that 2 and 3 are best formulated as Ru(ZnMe)2 complexes, but that 2 is slightly displaced along the continuum toward a Ru(η2-RZn–ZnR) species.26 Note that in 3, the Ru–Zn1–Me angle is smaller than expected at 161.3°, but in this case, the Me group is bent away from C1, suggesting that it is the short Zn1···C1 contact of 2.15 Å that drives this distortion. This is explored further in the electronic structure analyses below.
Figure 2a provides details of quantum theory of atoms in molecules (QTAIM) analyses of 2 and 3 with electron density contours plotted in the {RuZn1Zn2} plane. These are complemented by noncovalent interaction (NCI) plots shown in Figure 2b. For 2, bond paths between Ru and both Zn centers are consistent with the presence of Ru–Zn bonds. The associated bond critical points (BCPs) show similar electron densities, ρ(r), of ∼0.06 au, and this relatively low value, coupled with positive values of the Laplacian and small negative total energy densities (Figures S39 and S40), is consistent with a donor–acceptor (i.e., Ru → Zn) interaction between two heavy atoms.30,31Figure 2a also shows the computed delocalization indices (DI) in parentheses. These reflect the degree of shared electron density between two atomic centers32,33 and proved more discriminating than ρ(r) for the Ru–Zn interactions. Thus, a larger DI of 0.68 is associated with the shorter Ru–Zn2 bond compared to a DI of 0.52 for the Ru–Zn1 bond. DIs can also be measured between atoms not linked by a bond path and can be useful for identifying interactions in areas of flat electron density.34 A Zn1···Zn2 DI of 0.35 suggests a weak Zn···Zn interaction is present. For Zn1, this is supplemented by interaction with C1 to which a curved bond path [2.35 Å; ρ(r) = 0.049; DI = 0.26] indicates a degree of bridging character for the cyclometalated aryl group, albeit biased toward Ru [2.17 Å; ρ(r) = 0.098 au; DI = 0.76]. These stabilizing interactions are confirmed by the NCI plot of 2 that displays turquoise and blue regions along the Zn1···Zn2 and Zn1···C1 vectors, respectively.
Figure 2.
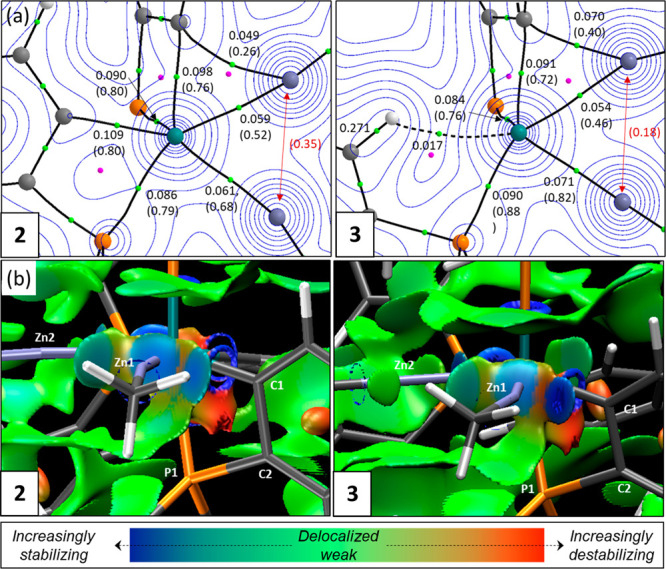
Electronic structure analysis of (left) 2 and (right) 3 focusing on key interactions around the {RuZn1Zn2} plane. (a) QTAIM molecular graphs with bond critical points (BCPs) in green and ring critical points (RCPs) in pink. Electron density, ρ(r), contour plots are shown in the {RuZn1Zn2} plane along with selected BCP (au) and delocalization indices in parentheses; delocalization indices between atoms not linked by a bond path are colored red. (b) Detail of the NCI plots viewed from above the {RuZn1Zn2} plane and looking down the Ru–Zn1 vector. Isosurfaces are generated for σ = 0.3 au and −0.07 < ρ < 0.07 au; a key showing the color scheme employed is also provided.
For 3, variations in these different interactions are seen relative to 2 that reflect changes in the interatomic distances. Thus, the Ru–Zn2 interaction strengthens [2.41 Å; ρ(r) = 0.071; DI = 0.82] while Ru–Zn1 interaction weakens [2.55 Å, ρ(r) = 0.054; DI = 0.46]. The Zn1···Zn2 interaction also weakens significantly (2.68 Å; DI = 0.18); for Zn2, this is compensated by the stronger interaction with Ru, whereas for Zn1, the interaction with C1 strengthens [2.15 Å; ρ(r) = 0.070; DI = 0.40]. The stronger Zn1···C1 interaction is also apparent in the NCI plot where a sharper blue disk along the Zn1···C1 vector is seen, and this is also consistent with the bending of the Ru–Zn1–Me angle away from C1 as noted above. A similar Zn···Caryl interaction has been noted before in a related Ru–Zn bimetallic complex [Zn···C, 2.282(2) Å13], while the longer Zn–Caryl distances in the asymmetrically bridged [ZnPh]2 dimer average 2.40 Å.35
A natural orbitals for chemical valence (NOCV) analysis confirmed the differences in the additional stabilizing interactions at the {Zn1Me}+ fragments in 2 and 3 (Figure 3). In each case, the key deformation density channel is dominated by donation from the dsp hybrid HOMO of the Ru-based fragment into the σ* LUMO of {ZnMe}+. For 2, this also shows contributions from both Zn2 and C1, whereas for 3, a larger component from C1 is apparent but no contribution from Zn2 is seen. Equivalent plots for the {Zn2Me}+ fragments are provided in Figures S46 and S47.
Figure 3.
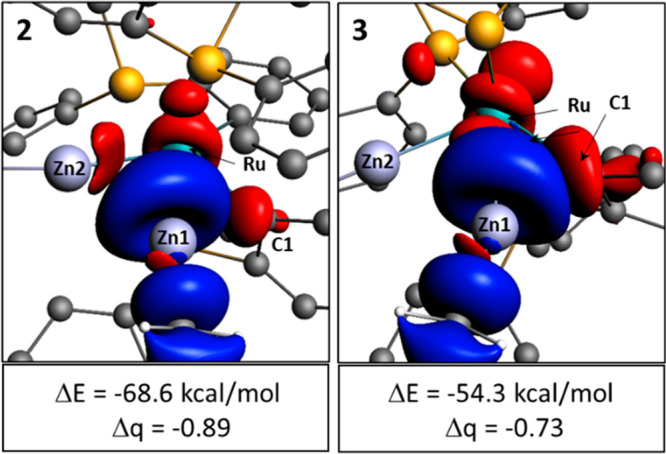
NOCV contour plots (isovalue of 0.0025 au) of the major deformation density channels in the interaction of the {Zn1Me}+ fragment with [Ru(PPh3)(PPh2C6H4)2(ZnMe)]− in 2 and with [Ru(PPh3)2(PPh2C6H4)(ZnMe)] in 3. Electron flow is shown from red to blue, and H atoms have been omitted for the sake of clarity.
Reactivity Studies
Given that 2 and 3 result from the formal introduction of ZnMe and [ZnMe]+, respectively, into 1, a series of reactivity studies were undertaken to probe the potential to reverse this process, through either thermolysis or reactions with Lewis bases. Such processes probe further the isolobality of ZnR and H, for example, the deprotonation of TM-hydrides. The reactions of 2 and 3 with H2 were also attempted, and the results are summarized in panels a and b of Scheme 3.
Scheme 3. Reactions of (a) [Ru(PPh3)(C6H4PPh2)2(ZnMe)2] 2 and (b) [Ru(PPh3)2(C6H4PPh2)(ZnMe)2][BArF4] 3.
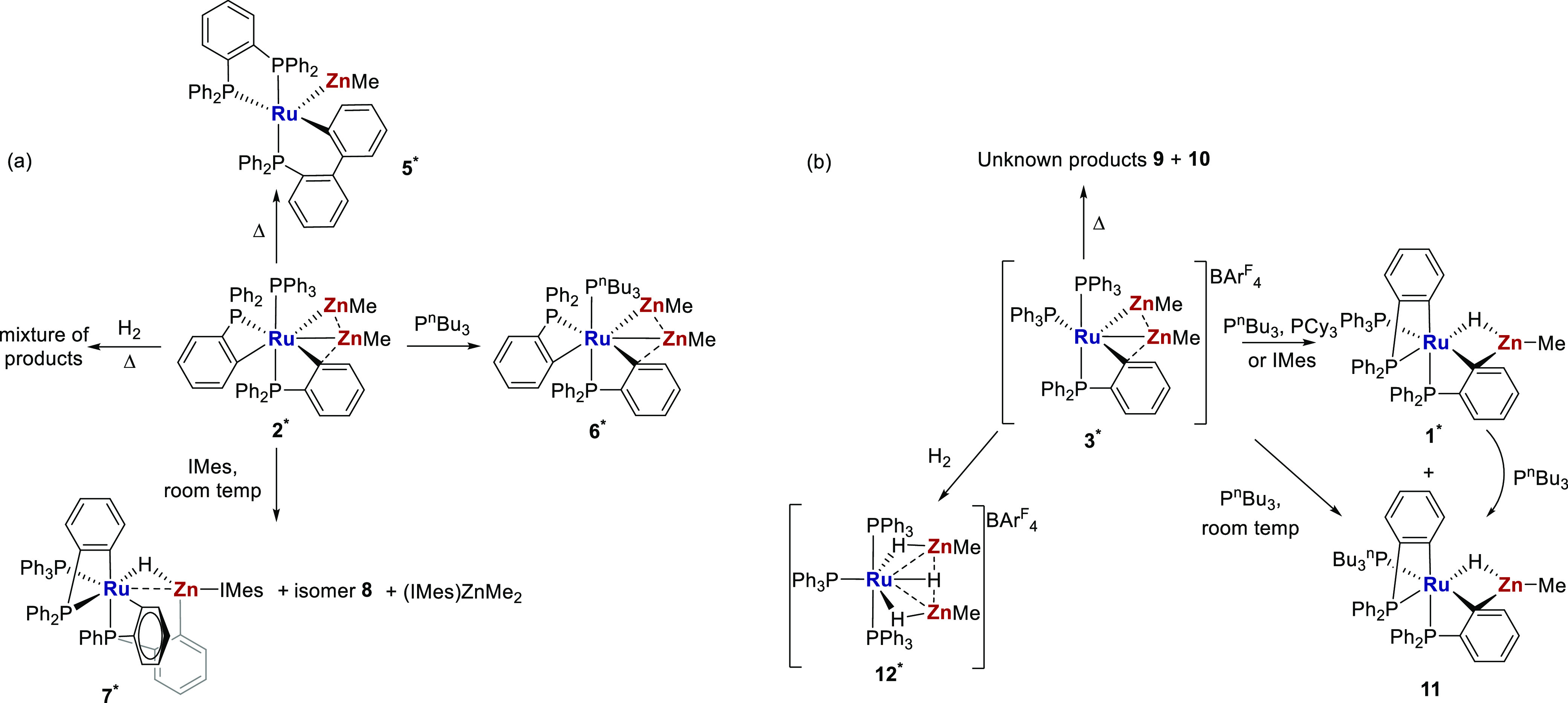
Structurally characterized complexes (reported here or previously in refs (13), (14), and (20)) are marked with asterisks.
Thermal Stabilities of 2 and 3
Heating 2 in toluene (80 °C, 2 days) resulted in the loss of ZnMe to give the previously reported [RuZnMe] complex, [Ru(dppbz)(PPh2(biphenyl′))(ZnMe)] [5; dppbz = 1,2-bis(diphenylphosphino)benzene; PPh2(biphenyl)′ = cyclometalated PPh2(biphenyl)], as the major product.20 Alongside formal elimination of ZnMe,36 the formation of 5 also requires C–H/P–C activation and C–C coupling steps to generate the dppbz and metalated Ph2P(biphenyl) ligand, although the exact sequence in which these steps take place remains unknown.37 For 3, the result of heating proved to be much less clear. 31P NMR monitoring of a reaction mixture refluxed in benzene (2 days) or refluxed in toluene (2 h) revealed formation of an initial product 9 (characterized by two coupled doublet resonances at δ 78 and 48) that, upon further heating (2 days) in toluene, converted to a second product, 10, which showed a similar pair of coupled signals at δ 53 and 46. Both compounds gave oils in all tested combinations of solvents,38 which, together with an absence of any diagnostic (i.e., non-aromatic) 1H NMR signals, makes their identities hard to determine.
Reactivity of 2 and 3 with Lewis Bases
Rather than removing either of the ZnMe ligands, the reaction of 2 with PnBu3 at room temperature led to substitution of the PPh3 ligand and formation of [Ru(PnBu3)(C6H4PPh2)2(ZnMe)2] (6). X-ray characterization (Figure 4) revealed a structure that was broadly similar to that of 2 in terms of metrics (Table S2). The ease of phosphine substitution in 2 contrasts with the difficulties reported by Fischer in attempting to exchange phosphine ligands in multi-Zn-containing Ni species.39 At the same time, the lack of reaction between 2 and PCy3 indicates how sensitive these systems are to the choice of Lewis base. In contrast, 3 reacted with both PnBu3 and PCy3, and in this case, this did incur the loss of [ZnMe]+ to give 1, together with 11 in the case of PnBu3.40,41 The findings fit with the previously observed complete conversion of 3 into 1 that is seen in THF.13 The fate of the eliminated [ZnMe]+ could not be established, but when the Lewis base is changed to the N-heterocyclic carbene IMes,42 trapping as the NHC adduct [(IMes)2ZnMe]+ was found,43 alongside formation of 1.
Figure 4.
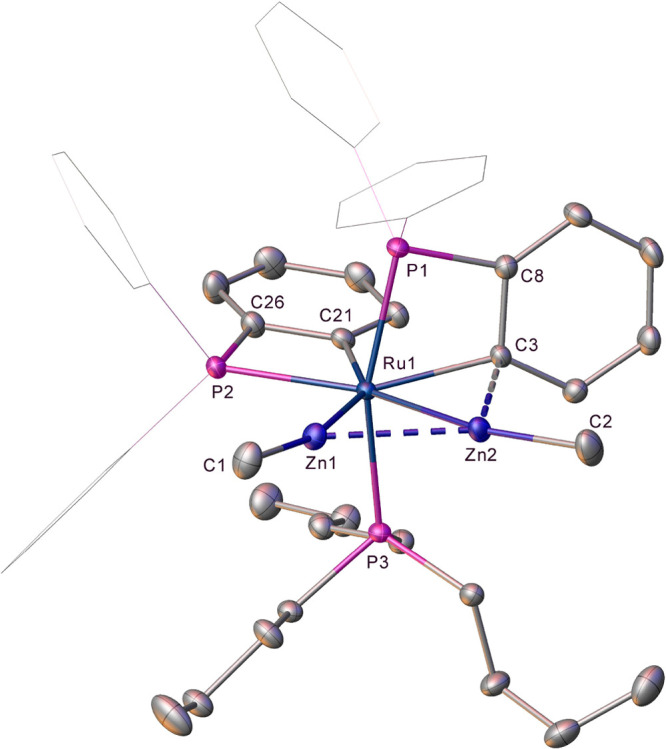
Molecular structure of 6. Ellipsoids are represented at 30% probability. Hydrogen atoms and a minor disordered component have been omitted for the sake of clarity.
A very different outcome was found when IMes was reacted with 2. This afforded [Ru(PPh3)(C6H4PPh2)(C6H4PPhC6H4Zn(IMes))H] (7) through substitution of a Me group by IMes on Zn. The formally eliminated ZnMe2 was trapped as (IMes)ZnMe2 by the second equivalent of carbene necessary to bring about the full consumption of 2.44,45
The X-ray structure of 7 (Figure 5) showed a fac arrangement of an intact PPh3 (based on P3), one phosphine that was cyclometalated onto ruthenium (based on P1), and a third phosphine ligand (based on P2) unusually metalated through two phenyl rings, one onto Ru [Ru1–C19, 2.1438(16) Å] and the second onto Zn [Zn1–C25, 2.0206(18) Å]. The sixth transition metal coordination site was occupied by a hydride ligand bridging the Ru and Zn centers [Ru1···Zn1, 2.6541(3) Å]. Computational analysis of the bonding in 7 is reserved until after discussion of the product of the reaction of 3 with H2.
Figure 5.
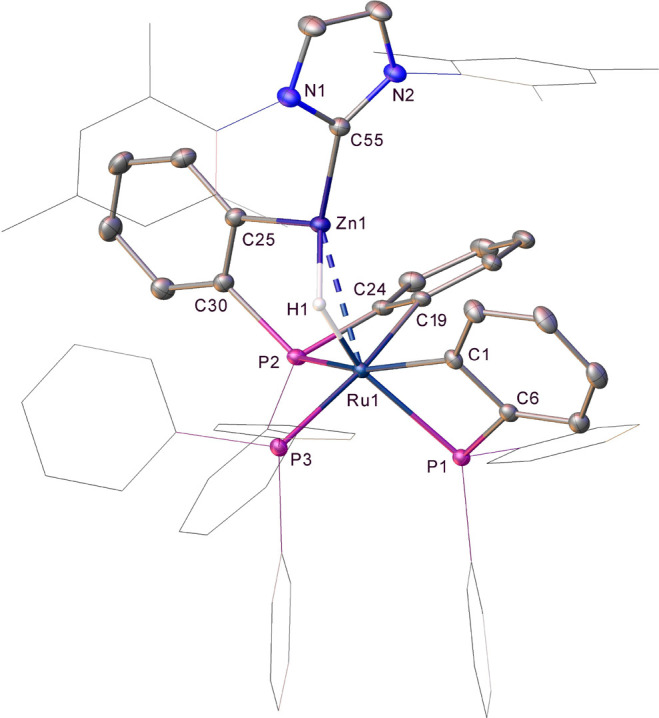
Molecular structure of 7. Ellipsoids are represented at 30% probability. Hydrogen atoms (except for H1) and the solvent have been omitted for the sake of clarity.
Redissolving a crystalline sample of the compound gave NMR signals for 7 together with a second, minor species, 8.46 The signals for 7 were consistent with the solid state structure, a doublet of doublet of doublets Ru–H–Zn resonance in the 1H NMR spectrum, with one large (pseudotrans) and two smaller 2JHP splittings, and one high-frequency 31P triplet (δ 52) for the PPh3 group, together with two lower-frequency (δ −12 and −24) doublet of doublet signals for the two cyclometalated phosphines.47,48 The presence of similar 31P chemical shifts for the minor species 8 supports it being an isomer. While 1H{31P} NMR measurements showed that both isomers feature hydride trans to a metalated phosphine, the presence of two surprisingly small 2JHP couplings (9 and 6 Hz), in addition to a large, pseudotrans splitting (46 Hz) in the 31P-coupled 1H NMR spectrum, leaves it unclear as to exactly what the structure of 8 is. Closer inspection of NMR spectra recorded shortly after combining IMes and 2 indicates that 8 is formed in the initial stages (mixing for <15 min) and is thus a kinetic product of the reaction formed prior to subsequent growth of the thermodynamic product, 7.49 The signals of 8 seen in the NMR spectra of 7 may therefore arise due to co-crystallization.
Reactivity of 2 and 3 toward H2
During our previous studies of Ru mono-Zn complexes,12,13,19,20 H2 was typically found to add across the Ru–Zn bond, as shown in Scheme 1b. Very different, contrasting behavior was seen with 2 and 3. Thus, the former did not react with H2 at room temperature and, upon being heated to 60 °C, gave only a complex mixture of products. In contrast, 3 reacted rapidly with two molecules of H2 at room temperature to reverse the phosphine cyclometalation and form the cationic dizinc trihydride complex, [Ru(PPh3)3(ZnMe)2H3][BArF4] (12). Remarkably, this transformation could also be carried out in the solid state simply by stirring a powdered sample of solid 3 under 1 atm of H2.
Complex 12 displayed high-frequency doublet and triplet 31P NMR resonances, consistent with the mer-RuP3 geometry apparent in the X-ray crystal structure (Figure 6). The 1H NMR spectrum showed two hydride signals at δ −7.3 (dtd) and −11.1 (dtt) in a 2:1 ratio.50 Upon being heated to 60 °C, 12 decomposed as evidenced by the precipitation of an insoluble red oil at the bottom of reaction solutions.
Figure 6.
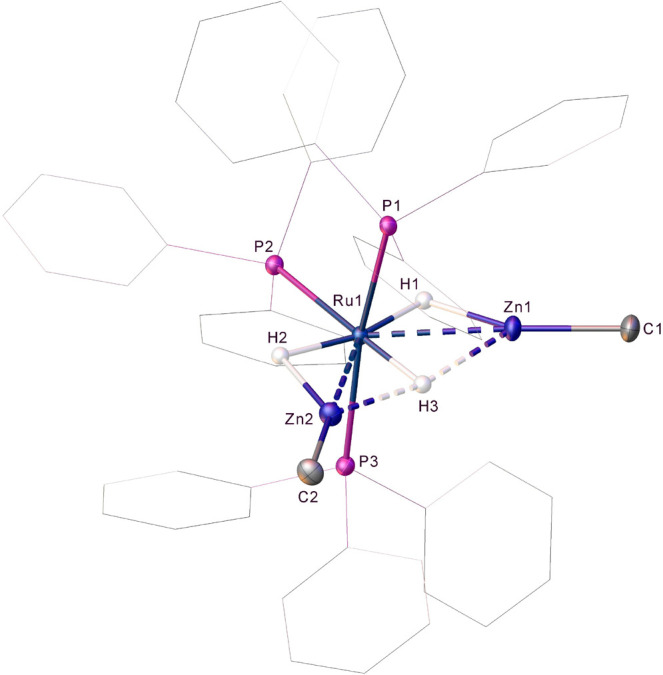
Molecular structure of the cation of 12. Ellipsoids are represented at 30% probability. Phosphine and zinc methyl hydrogen atoms have been omitted for the sake of clarity.
The molecular structure of the cation in 12 is shown in Figure 6. The equatorial positions comprised two ZnMe ligands, one PPh3 ligand, and three hydrides (which were located and refined without restraints). The coordination sphere was completed by two phosphines in a distorted trans-axial arrangement [P1–Ru–P3, 164.804(17)°]. Two of the hydrides were disposed approximately trans to one another [H1–Ru1–H2, 163.3(16)°], while the third was located trans to the equatorial PPh3 ligand [H3–Ru1–P2, 175.7(12)°]. The two Ru–Zn distances [2.5188(3) and 2.5397(3) Å] are considered further in the computational analysis of 12 below.
Structure and Bonding in 12 and 7
The computed structure of 12 and details of the QTAIM analyses are shown in panels a and b, respectively, of Figure 7, with the related NCI and NOCV analyses presented in the Supporting Information. In this case, QTAIM reveals an absence of Ru–Zn bond paths, despite Ru–Zn distances that are similar to those in 2 and 3. However, the computed DIs (Ru···Zn1, 0.38; Ru···Zn2, 0.41) indicate significant Ru···Zn interactions are still present, and this is supported by the NCI plot that shows blue stabilizing features between Ru and both Zn centers. For the outer hydrides, H1 and H2, the computed Ru–H distances of ∼1.69 Å are typical for a trans H–Ru–H arrangement and bond paths are characterized by a ρ(r) of ∼0.106 au and DIs of ∼0.69. These Ru–H bonds appear stronger than the Zn1–H1 and Zn2–H2 bonds [∼1.82 Å; ρ(r) ∼ 0.065 au; DI ∼ 0.31], and these are in turn significantly weaker than the unperturbed Zn–H bond in MeZnH [1.52 Å; ρ(r) = 0.11 au; DI = 0.89; see Supporting Information]. H1 and H2 are therefore bridging the respective Ru–Zn vectors but are strongly biased toward Ru over Zn. This is also reflected in the very low ellipticities of the Ru–H BCPs (average of 0.028) that are indicative of terminal hydride character, compared with the higher ellipticities of the Zn–H BCPs [average of 0.48 (Figure S42)].
Figure 7.

Electronic structure analyses of 12 highlighting key interactions in the {RuZn1Zn2} plane. (a) Computed structure of 12 (based on the experimental heavy atom positions with H atoms optimized with the BP86 functional). (b) QTAIM molecular graph with electron density contours shown in the {RuZn1Zn2} plane, with BCPs colored green with associated ρ(r) (au) and delocalization indices in parentheses. Delocalization indices between selected atoms not linked by a bond path are denoted in red.
The properties of the Ru–H3 bond [1.69 Å; ρ(r) = 0.107 au, DI = 0.70] are similar to those of the Ru–H1 and Ru–H2 bonds. In this case, no bond path to either Zn center is seen but DIs of 0.19 indicate some residual interactions are still present, and these are confirmed in the NCI plot that shows turquoise regions along the Zn1···H3 and Zn2···H3 vectors. The significant ellipticity of the Ru–H3 BCP (0.145) also suggests a distortion of the electron density away from a terminal Ru–H σ-bond due to the presence of the two {ZnMe}+ moieties.51 An NOCV analysis of {ZnMe}+ bonding in 12 indicates the major deformation density channels exhibit donation from Ru and both adjacent hydrides (see the Supporting Information).
A similar analysis of the bonding in compound 7 shows the hydride present, H1, to have characteristics similar to those of H1 and H2 in 12 [Ru–H1, 1.68 Å, ρ(r) = 0.108, DI = 0.68; Zn–H1, 1.79 Å, ρ(r) = 0.067, DI = 0.33]. The Ru–Zn distance of 2.65 Å is the longest of the species studied here, and no Ru–Zn bond path is computed; however, a Ru···Zn DI of 0.29 suggests some interaction between the two metal centers (Figure S41).
Overall, the {Ru(H1)Zn1(H3)} and {Ru(H2)Zn2(H3)} moieties in 12 and the {RuH1Zn} moiety in 7 can be considered as featuring asymmetrically bound bridging hydride ligands that interact more strongly with the Ru centers. A similar situation is seen in [Ru(IPr)2(CO)(ZnR)(η2-H2)(H)2]+ and [Ru(IPr)2(CO)(ZnR)(H)2]+ species (R = Et or Me),12 where, depending on the nature of the trans ligand, the hydride within a {RuHZn} moiety shows different degrees of Ru–H or Zn–H bonding character. These add to the continuum of structures that can be accessed in TM-MG heterobimetallic complexes featuring hydride ligands, the precise nature of which will depend on the coordination environment of the TM partner.52,53
Conclusions
A combined computational and experimental study has been undertaken on two [RuZn2Me2] species, neutral 2 and cationic 3. Geometrical considerations supported by computational analyses confirm the presence of direct Ru–Zn bonds in both species and suggest these are best formulated as Ru(ZnMe)2 complexes featuring discrete ZnMe ligands. Some additional stabilization may be achieved via Zn···Zn interactions, and 2 and 3 both exhibit Zn···Caryl interactions, with these being more significant in 3.
Experimentally, the two complexes exhibit diverse reactivities with thermolysis and the addition of a range of Lewis bases bringing about different outcomes with no apparent correlation to either the overall charges of the complexes or the different strengths of the Ru–Zn interactions present. 2 reacted with H2 to give a mixture of products, while in contrast, reaction of H2 with 3 led cleanly to [Ru(PPh3)3(ZnMe)2H3][BArF4] (12). Computational analyses of this complex suggest the presence of three hydride ligands that bridge the Ru–Zn vectors asymmetrically toward Ru.
12 adds to the range of transition metal complexes that feature multiple main group metals and multiple hydride ligands that have recently attracted a great deal of attention due to the unusual bonding interactions and unusual geometries they can possess.54,55 Studies of their reactivity, however, remain rare.56 In the study presented here, we have shown that both the TM and the MGM can be centers of reactivity in these heterobimetallic complexes and the factors that govern the site of reactivity will be the subject of future reports from our groups.
Experimental Section
General Comments
All manipulations were carried out under argon using standard Schlenk, high-vacuum, and glovebox techniques using dry and degassed solvents. C6D6 and THF-d8 were vacuum transferred from potassium. NMR spectra were recorded at 298 K (unless otherwise stated) on Bruker Avance 400 and 500 MHz NMR spectrometers and referenced as follows: C6D6 (1H, δ 7.16; 13C, δ 128.0), THF-d8 (1H, δ 1.72; 13C, δ 25.3), and toluene-d8 (1H, δ 2.09). 31P spectra were referenced externally to 85% H3PO4 (δ 0.0). Elemental analyses were performed by Elemental Microanalysis Ltd. (Okehampton, Devon, U.K.). Compounds 1,132,143,13 and IMes57 were prepared according to literature methods.
[Ru(PnBu3)(C6H4PPh2)2(ZnMe)2] (6)
C6D6 (0.5 mL) was added to a mixture of 2 (40 mg, 0.038 mmol) and PnBu3 (9.5 μL, 0.038 mM) in a J. Young’s resealable NMR tube. After 15 h at room temperature, the volatiles were removed and the resulting solid was recrystallized from benzene/hexane. The microcrystalline solid was washed with hexane and dried under vacuum to give 6 as a yellow solid (18 mg, 48%). 1H NMR (500 MHz, C6D6): δ 8.21–8.16 (m, 2H, Ar), 7.97–7.92 (m, 2H, Ar), 7.91–7.86 (m, 2H, Ar), 7.76 (m 1H, Ar), 7.26 (tt, J = 7.6 Hz, J = 1.5 Hz, 1H, Ar), 7.18 (dt, J = 7.6 Hz, J = 1.5 Hz, 2H, Ar), 7.11–7.04 (m, 2H, Ar), 7.03–6.85 (br, 8H, Ar), 6.84–6.72 (m, 4H, Ar), 6.54–6.49 (m, 2H, Ar), 6.40–6.35 (m, 2H, Ar), 1.71–1.52 (br m, 6H, PCH2), 1.35–1.02 (br m, 12 H, PCH2CH2CH2), 0.84 (t, 3JHH = 7.5 Hz, 9H, PCH2CH2CH2CH3), 0.47 (s, 3H, ZnMe), −0.64 (s, 3H, ZnMe). 31P{1H} NMR (202 MHz, C6D6): δ 21.3 (dd, 2JPP = 246 Hz, 2JPP = 17 Hz), −23.2 (dd, JPP = 25 Hz, JPP = 17 Hz), −26.3 (dd, 2JPP = 246 Hz, 2JPP = 25 Hz). 13C{1H} NMR (126 MHz, C6D6): δ 173.9 (dt, JCP = 13 Hz, JCP = 10 Hz, Cquaternary), 158.4 (dd, JCP = 47 Hz, JCP = 4 Hz, Cquaternary), 158.2 (dd, JCP = 48 Hz, JCP = 9 Hz, Cquaternary), 152.8 (dd, JCP = 47 Hz, JCP = 2 Hz, Cquaternary), 140.7 (dt, JCP = 20 Hz, JCP = 3 Hz, aryl CH), 140.5–140.0 (m, Cquaternary), 138.1 (d, JCP = 21 Hz, aryl CH), 133.6 (br d, JCP = 12 Hz, aryl CH), 132.7 (d, JCP = 9 Hz, aryl CH), 132.3 (d, JCP = 11 Hz, aryl CH), 131.9 (d, JCP = 10 Hz, CH), 130.8 (br d, J = 4 Hz, aryl CH), 130.6 (app t, JCP = 3 Hz, aryl CH), 130.0 (br s, aryl CH), 128.8 (br d, JCP = 16 Hz, aryl CH), 128.6 (d, JCP = 9 Hz, aryl CH), 128.3 (d, JCP = 9 Hz, aryl CH), 127.3 (d, JCP = 9 Hz, aryl CH), 127.1 (br s), 127.0 (br s), 124.2 (d, JCP = 7 Hz, aryl CH), 122.0 (d, JCP = 8 Hz, aryl CH), 31.1 (br m, JCP = 22 Hz, CH2), 26.8 (d, JCP = 3 Hz, CH2), 24.8 (d, JCP = 11 Hz, CH2), 14.0 (s, CH3), 11.5 (br m, ZnCH3), −3.6 (s, ZnCH3). Anal. Found (%): C, 60.51; H, 6.22. Calcd for C50H61P3RuZn2: C, 60.86; H, 6.23.
[Ru(PPh3)(C6H4PPh2)(PPh(C6H4)2Zn(IMes))H] (7)
A mixture of 2 (40 mg, 0.037 mmol) and IMes (23 mg, 0.074 mmol) was added to a J. Young’s resealable NMR tube. Addition of C6D6 (0.5 mL) led to an instantaneous change in color from red to orange-yellow. NMR spectroscopy revealed that consumption of the starting material took place over 3 h to give 7 as the main product. Removal of the volatiles under reduced pressure and recrystallization of the residue from benzene/hexane gave yellow crystals of 7, which were washed with hexane and dried under vacuum (33 mg, 70% yield). 1H NMR (500 MHz, THF-d8): δ 7.41 (s, 2H, Ar), 7.38 (br d, J = 6.8 Hz, 1H, Ar), 7.27 (t, J = 7.1 Hz, 1H, Ar), 7.22–7.14 (m, 4H, Ar), 7.11–7.07 (m, 1H, Ar), 7.02 (t, J = 8.0 Hz, 2H, Ar), 6.95–6.62 (m, 22H, Ar), 6.60–6.39 (m, 11H, Ar + NCH=CHN), 6.12 (t, J = 8.0 Hz, 1H, Ar), 5.87 (t, J = 7.5 Hz, 1H, Ar), 5.64 (m, 1H, Ar), 5.42 (t, J = 7.5 Hz, 1H, Ar), 2.16 (s, 6H, C6Me3H3), 2.11 (s, 6H, C6Me3H3), 1.92 (s, 6H, C6Me3H3), −10.50 (ddd, 2JHP = 53.5 Hz, 2JHP = 23.0 Hz, 2JHP = 3.5 Hz, 1H, RuH). 31P{1H} NMR (162 MHz, C6D6): δ 52.4 (t, 2JPP = 25 Hz), −11.6 (dd, 2JPP = 27 Hz, 2JPP = 15 Hz), −24.0 (dd, 2JPP = 25 Hz, 2JPP = 15 Hz). Selected 13C{1H} NMR (126 MHz, C6D6): δ 182.9 (br d, JCP = 3 Hz, CNHC), 176.9 (br dm, JCP = 65 Hz, Cipso), 168.8 (dt, JCP = 40 Hz, JCP = 4 Hz, Cipso), 166.5 (d, 1JCP = 59 Hz, Cipso), 160.6 (dd, 1JCP = 56 Hz, 3JCP = 12 Hz, Cipso), 154.4 (dd, JCP = 48 Hz, JCP = 4 Hz, Cipso), 148.3 (d, 1JCP = 36 Hz, Cipso), 21.0 (s, C6Me3H3), 19.0 (s, C6Me3H3), 18.5 (s, C6Me3H3). Selected NMR data for 8. 1H NMR (500 MHz, C6D6): δ −9.03 (ddd, 2JHP = 45.9 Hz, 2JHP = 8.8 Hz, 2JHP = 6.1 Hz, 1H, RuH). 31P{1H} NMR (162 MHz, C6D6): δ 57.4 (t, 2JPP = 18 Hz), −26.9 (t, 2JPP = 20 Hz), −30.9 (app t, 2JPP = 18 Hz). Anal. Found (%): C, 72.97; H, 5.46; N, 1.99. Calcd for C75H67N2P3RuZn·0.4C6H6·0.1C6H14: C, 72.31; H, 5.51; N, 2.16 [NMR spectroscopy confirmed the presence of benzene and hexane (Figure S31)].
Thermal Decomposition of 2
A C6D5CD3 solution (0.5 mL) of 2 (10 mg, 0.01 mmol) was heated at 80 °C for 50 h, affording a dark red-brown colored solution. 31P{1H} NMR spectroscopy showed complete consumption of the starting material together with the formation of 5(20) and a new species 4. Selected NMR data for 4. 31P{1H} NMR (162 MHz, C6D5CD3): δ 87.0 (t, 2JPP = 25 Hz), 49.3 (dd, 2JPP = 249 Hz, 2JPP = 24 Hz), 44.4 (dd, 2JPP = 249 Hz, 2JPP = 26 Hz).
Thermal Decomposition of 3
A C6D6 solution (0.5 mL) of 3 (40 mg, 0.02 mmol) was heated at 80 °C for 2 days. 31P{1H} NMR spectroscopy showed no remaining starting material and the formation of two doublet resonances for a new product 9 at δ 77.5 (JPP = 38 Hz) and δ 47.7 (JPP = 38 Hz). When a second sample was prepared and heated at 110 °C for 3 days, 31P{1H} NMR spectroscopy revealed total consumption of 3 and the appearance of two doublets at δ 52.8 (JPP = 31 Hz) and δ 46.5 (JPP = 31 Hz), which we assign to a second product, 10.
Reaction of 3 with Lewis Bases
(i) PBu3 (2.6 μL, 0.01 mmol) was added to a C6D6 (0.5 mL) solution of 3 (20 mg, 0.01 mmol) in a J. Young’s resealable NMR tube to give a homogeneous red solution. After ∼15 min, deposition of an unknown, insoluble red oil started to occur. A 1H NMR spectrum of the sample at this time showed the presence of a doublet of doublet of doublets hydride signal at δ −8.25 corresponding to 1 and a second doublet of doublet of doublets hydride signal at δ −9.07 (2JHP = 50.9, 21.8, and 12.3 Hz), for [Ru(PnBu3)(C6H4PPh2)2(ZnMe)H] (11). (ii) PCy3 (3 mg, 0.02 mmol) and 3 (20 mg, 0.01 mmol) were dissolved in C6D6 (0.5 mL) in a J. Young’s resealable NMR tube to give a homogeneous red solution. After ∼15 min, deposition of an unknown, insoluble red oil started to occur. A 1H NMR spectrum of the sample at this time showed the presence of a doublet of doublet of doublets hydride signal at δ −8.25 corresponding to 1. (iii) IMes (3 mg, 0.01 mmol) and 3 (20 mg, 0.01 mmol) were dissolved in C6D6 (0.5 mL) in a J. Young’s resealable NMR tube to give a homogeneous red solution. After ∼30 min, deposition of an unknown, insoluble red oil started to occur. A 1H NMR spectrum of the solution displayed resonances for 3 alongside the diagnostic hydride of 1. Addition of a second equivalent of IMes (3 mg, 0.01 mmol) afforded full conversion of 3 to 1. Isolation of a small number of crystals confirmed the same unit cell parameters reported for 1.
[Ru(PPh3)3(ZnMe)2H3][BArF4] (12)
A J. Young’s resealable ampule was charged with a C6H6 (5 mL) suspension of 3 (96 mg, 0.05 mmol). After being gently heated to fully dissolve the solid, the resulting red solution was degassed (three freeze–pump–thaw cycles) and H2 (1 atm) added with vigorous stirring. After 5 min, this gave a pale-yellow solution, which upon treatment with hexane (5 mL) afforded a pale-yellow crystalline sample of 12. This was collected and dried under vacuum. Yield: 76 mg (79%). An alternative route to 12 involved stirring a solid sample of 3 under H2 (1 atm) for ∼2 h, by which time the sample had changed color from red-orange to off-white. A 31P NMR spectrum in C6D6 revealed complete conversion to 12. 1H NMR (500 MHz, C6D6): δ 8.47 (s, 8H, BArF4), 7.71 (s, 4H, BArF4), 7.07 (br t, 1H, J = 9.1 Hz, 6H, Ar), 7.04–6.99 (m, 12H, Ar), 6.85 (t, J = 7.3 Hz, 9H, Ar), 6.78 (t, J = 7.4 Hz, 12H, Ar), 6.71 (td, J = 7.8 Hz, J = 1.4 Hz, 6H, Ar), −1.02 (s, 6H, ZnMe), −7.34 (dtd, 2JHP = 16.1 Hz, 2JHP = 12.5 Hz, 2JHH = 3.3 Hz, 2H, RuH), −11.06 (dtt, 2JHP = 39.5 Hz, 2JHP = 18.1 Hz, 2JHH = 3.3 Hz, 1H, RuH). 31P{1H} NMR (202 MHz, C6D6): δ 47.4 (t, 2JPP = 26 Hz), 41.8 (d, 2JPP = 26 Hz). 13C{1H} NMR (126 MHz, C6D6): δ 162.8 (1:1:1:1 q, 1JCB = 50 Hz, BArF4), 135.6–135.1 (m), 133.9 (d, JCP = 11 Hz, Cortho-PPh3), 133.7–133.2 (m, 133.6, Cipso-PPh3 overlapped with 133.4, vt, JCP = 6 Hz, Cortho-PPh3), 131.3 (s, Cpara-PPh3), 130.4–129.5 (m, 130.3, s, Cpara-PPh3 overlapped with 130.0, qq, JCF = 32 Hz, JCF = 3 Hz, BArF4), 129.2 (vt, JCP = 5 Hz, Cmeta-PPh3), 128.3 (d, JCP ∼ 5 Hz, Cmeta-PPh3, overlapped with C6D6), 125.3 (q, 1JCF = 273 Hz, BArF4), 118.1 (s, BArF4). −5.7 (s, ZnMe). Anal. Found (%): C, 55.19; H, 3.48. Calcd for C88H66BF24Zn2P3Ru: C, 55.19; H, 3.47.
X-ray Crystallography
Data for 6, 7, and 12 were obtained using an Agilent SuperNova instrument and a Cu Kα radiation source. All experiments were conducted at 150 K, and models refined using SHELXL58 via the Olex259 interface. Refinements were largely straightforward, and only points of note will be detailed herein. First, the phenyl rings based on C9 and C15 were treated for 80:20 disorder in the structure of 6. In 7, the asymmetric unit was seen to contain one molecule of the bimetallic complex and two molecules of benzene. The hydride in the former was located and refined without restraints as were the hydride ligands in 12. Unsurprisingly, the anion in the latter structure required some disorder modeling. In particular, fluorine atoms F4–F6 were treated for three-way disorder in a 0.425:0.425:0.15 ratio, while F22–F24 were modeled to take into account 50:50 disorder. Distances and ADP restraints were employed in disordered regions, to assist convergence.
Computational Details
Density functional theory calculations were performed with Gaussian 16 (revision C.01).60 Ru, Zn, and P centers were described with the Stuttgart RECPs and associated basis sets,61 and 6-31G** basis sets were used for all other atoms.62,63 A set of d orbital polarization functions was also added to P (ζd = 0.387).64 Electronic structure analyses were performed on geometries using the heavy atom positions derived from the crystallographic studies with H atom positions optimized with the BP86 functional.65,66 Details of functional testing on the fully optimized structure of 2 are provided in the Supporting Information. Quantum theory of atoms in molecules (QTAIM)67 used the AIMALL program.68 NCI calculations were based on the promolecular densities and used NCIPLOT69 with visualization via VMD.70 Natural orbitals for chemical valence (NOCV) analyses71 were performed using the Amsterdam Modeling Suite (AMS) package.72 Computed geometries are displayed with ChemCraft,73 and all geometries are supplied as a separate XYZ file (Supporting Information).
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie Grant Agreement 792674 (F.M.M.) and EPSRC (Grant EP/T019743/1 for A.-F.P., Grant EP/T019876/1 for L.S., and Grant EP/R020752/1 for N.A.R.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c02072.
Accession Codes
CCDC 2083542–2083544 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Dedication
The authors dedicate this paper to Professor Christian Bruneau in recognition of his outstanding contributions to ruthenium chemistry.
Supplementary Material
References
- Liddle S. T., Ed. Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Bouhadir G.; Bourissou D. Complexes of ambiphilic ligands: reactivity and catalytic applications. Chem. Soc. Rev. 2016, 45, 1065–1079. 10.1039/C5CS00697J. [DOI] [PubMed] [Google Scholar]
- Campos J. Bimetallic cooperation across the periodic table. Nat. Rev. Chem. 2020, 4, 696–702. 10.1038/s41570-020-00226-5. [DOI] [PubMed] [Google Scholar]
- Cammarota R. C.; Vollmer M. V.; Xie J.; Ye J. Y.; Linehan J. C.; Burgess S. K.; Appel A. M.; Gagliardi L.; Lu C. C. A bimetallic nickel-gallium complex catalyzes CO2 hydrogenation via the intermediacy of an anionic d10 nickel hydride. J. Am. Chem. Soc. 2017, 139, 14244–14250. 10.1021/jacs.7b07911. [DOI] [PubMed] [Google Scholar]
- Takaya J.; Iwasawa N. Synthesis, structure, and catalysis of palladium complexes bearing a group 13 metalloligand: Remarkable effect of an aluminum-metalloligand in hydrosilylation of CO2. J. Am. Chem. Soc. 2017, 139, 6074–6077. 10.1021/jacs.7b02553. [DOI] [PubMed] [Google Scholar]
- Yamada R.; Iwasawa N.; Takaya J. Rhodium-catalyzed C-H activation enabled by an indium metalloligand. Angew. Chem., Int. Ed. 2019, 58, 17251–17254. 10.1002/anie.201910197. [DOI] [PubMed] [Google Scholar]
- Morisako S.; Watanabe S.; Ikemoto S.; Muratsugu S.; Tada M.; Yamashita M. Synthesis of a pincer-IrV complex with a base-free alumanyl ligand and its application toward the dehydrogenation of alkanes. Angew. Chem., Int. Ed. 2019, 58, 15031–15035. 10.1002/anie.201909009. [DOI] [PubMed] [Google Scholar]
- Fujii I.; Semba K.; Li Q.-Z.; Sakaki S.; Nakao Y. Magnesiation of aryl fluorides catalyzed by a rhodium-aluminum complex. J. Am. Chem. Soc. 2020, 142, 11647–11652. 10.1021/jacs.0c04905. [DOI] [PubMed] [Google Scholar]
- Hara N.; Uemura N.; Nakao Y. C2-Selective silylation of pyridines by a rhodium-aluminum complex. Chem. Commun. 2021, 57, 5957–5960. 10.1039/D1CC00278C. [DOI] [PubMed] [Google Scholar]
- Seki R.; Hara N.; Saito T.; Nakao Y. Selective C-O bond reduction and borylation of aryl ethers catalyzed by a rhodium-aluminium heterobimetallic complex. J. Am. Chem. Soc. 2021, 143, 6388–6394. 10.1021/jacs.1c03038. [DOI] [PubMed] [Google Scholar]
- Cammarota R. C.; Clouston L. J.; Lu C. C. Leveraging molecular metal-support interaction for H2 and N2 activation. Coord. Chem. Rev. 2017, 334, 100–111. 10.1016/j.ccr.2016.06.014. [DOI] [Google Scholar]
- Riddlestone I. M.; Rajabi N. A.; Lowe J. P.; Mahon M. F.; Macgregor S. A.; Whittlesey M. K. Activation of H2 over the Ru-Zn bond in the transition metal-Lewis acid heterobimetallic species [Ru(IPr)2(CO)ZnEt]+. J. Am. Chem. Soc. 2016, 138, 11081–11084. 10.1021/jacs.6b05243. [DOI] [PubMed] [Google Scholar]
- Miloserdov F. M.; Rajabi N. A.; Lowe J. P.; Mahon M. F.; Macgregor S. A.; Whittlesey M. K. Zn-Promoted C-H reductive elimination and H2 activation via a dual unsaturated Ru-Zn intermediate. J. Am. Chem. Soc. 2020, 142, 6340–6349. 10.1021/jacs.0c01062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miloserdov F. M.; Isaac C. J.; Beck M. L.; Burnage A. L.; Farmer J. C. B.; Macgregor S. A.; Mahon M. F.; Whittlesey M. K. Impact of the novel Z-acceptor ligand bis{(ortho-diphenylphosphino)phenyl}zinc (ZnPhos) on the formation and reactivity of low-coordinate Ru(0) centers. Inorg. Chem. 2020, 59, 15606–15619. 10.1021/acs.inorgchem.0c01683. [DOI] [PubMed] [Google Scholar]
- Tebbe F. N. Lewis acidic metal alkyl-transition metal complex interactions. 1. Niobium and tantalum hydrides. J. Am. Chem. Soc. 1973, 95, 5412–5414. 10.1021/ja00797a052. [DOI] [Google Scholar]
- St Denis J. N.; Butler W.; Glick M. D.; Oliver J. P. Studies on main group metal-transition metal bonded compounds: II. The crystal and molecular structure of π-C5H5(CO)3WGa(CH3)2 and evidence for mixed organozinc and organogallium transition metal derivatives. J. Organomet. Chem. 1977, 129, 1–16. 10.1016/S0022-328X(00)93222-4. [DOI] [Google Scholar]
- Skupiński W. A.; Huffman J. C.; Bruno J. W.; Caulton K. G. Dinuclear elimination from rhenium hydrides and AlMe3 - rhenium aluminium polyhydrides. J. Am. Chem. Soc. 1984, 106, 8128–8136. 10.1021/ja00338a021. [DOI] [Google Scholar]
- Thorn D. L.; Harlow R. L. Transition metal-organoindium chemistry. Addition of alkyl-indium bonds to alkyliridium compounds and the structure of IrH(Et)(Et2In)(PMe3)3. J. Am. Chem. Soc. 1989, 111, 2575–2580. 10.1021/ja00189a033. [DOI] [Google Scholar]
- Espinal-Viguri M.; Varela-Izquierdo V.; Miloserdov F. M.; Riddlestone I. M.; Mahon M. F.; Whittlesey M. K. Heterobimetallic ruthenium–zinc complexes with bulky N-heterocyclic carbenes: Syntheses, structures and reactivity. Dalton Trans. 2019, 48, 4176–4189. 10.1039/C8DT05023F. [DOI] [PubMed] [Google Scholar]
- O’Leary N.; Miloserdov F. M.; Mahon M. F.; Whittlesey M. K. Transforming PPh3 into bidentate phosphine ligands at Ru-Zn heterobimetallic complexes. Dalton Trans. 2019, 48, 14000–14009. 10.1039/C9DT03106E. [DOI] [PubMed] [Google Scholar]
- For Ru–Ga and Ru–In complexes prepared using the same approach, see:; Riddlestone I. M.; Rajabi N. A.; Macgregor S. A.; Mahon M. F.; Whittlesey M. K. Well-defined heterobimetallic reactivity at unsupported ruthenium-indium bonds. Chem. - Eur. J. 2018, 24, 1732–1738. 10.1002/chem.201705796. [DOI] [PubMed] [Google Scholar]
- The depiction of species 3 in Scheme 2 differs from that in ref (13) and reflects the outcomes of our study that demonstrate direct Ru–Zn bonding and weak Zn···Zn and Zn···Caryl interactions.
- Bollermann T.; Gemel C.; Fischer R. A. Organozinc ligands in transition metal chemistry. Coord. Chem. Rev. 2012, 256, 537–555. 10.1016/j.ccr.2011.11.008. [DOI] [Google Scholar]
- Molon M.; Gemel C.; Fischer R. A. From AlCp*- and GaCp*-ligated ruthenium hydrides to zinc-rich heterometallic complexes. Eur. J. Inorg. Chem. 2013, 2013, 3616–3622. 10.1002/ejic.201300317. [DOI] [Google Scholar]
- Chen M.; Jiang S. J.; Maron L.; Xu X. Transition metal-induced dehydrogenative coupling of zinc hydrides. Dalton Trans 2019, 48, 1931–1935. 10.1039/C8DT04651D. [DOI] [PubMed] [Google Scholar]
- Freitag K.; Molon M.; Jerabek P.; Dilchert K.; Rosler C.; Seidel R. W.; Gemel C.; Frenking G.; Fischer R. A. Zn···Zn interactions at nickel and palladium centers. Chem. Sci. 2016, 7, 6413–6421. 10.1039/C6SC02106A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R.; Carmona E.; Galindo A. The dizinc bond as a ligand: A computational study of elongated dizinc bonds. Inorg. Chim. Acta 2018, 470, 197–205. 10.1016/j.ica.2017.06.008. [DOI] [Google Scholar]
- Ayala R.; Galindo A. A QTAIM and DFT study of the dizinc bond in non-symmetric [CpZn2Ln] complexes. J. Organomet. Chem. 2019, 898, 120878. 10.1016/j.jorganchem.2019.120878. [DOI] [Google Scholar]
- Cordero B.; Gómez V.; Platero-Prats A. E.; Revés M.; Echeverría J.; Cremades E.; Barragán F.; Alvarez S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. 10.1039/b801115j. [DOI] [PubMed] [Google Scholar]
- Lepetit C.; Fau P.; Fajerwerg K.; Kahn M. L.; Silvi B. Topological analysis of the metal-metal bond: A tutorial review. Coord. Chem. Rev. 2017, 345, 150–181. 10.1016/j.ccr.2017.04.009. [DOI] [Google Scholar]
- Macchi P.; Proserpio D. M.; Sironi A. Experimental electron density in a transition metal dimer: Metal–metal and metal–ligand bonds. J. Am. Chem. Soc. 1998, 120, 13429–13435. 10.1021/ja982903m. [DOI] [Google Scholar]
- Merino G.; Vela A.; Heine T. Description of electron delocalization via the analysis of molecular fields. Chem. Rev. 2005, 105, 3812–3841. 10.1021/cr030086p. [DOI] [PubMed] [Google Scholar]
- Outeiral C.; Vincent M. A.; Martín Pendás Á.; Popelier P. L. A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. 10.1039/C8SC01338A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J.; Evans C.; Lentz D.; Roemer M. The QTAIM approach to chemical bonding between transition metals and carbocyclic rings: A combined experimental and theoretical study of (η5-C5H5)Mn(CO)3, (η6-C6H6)Cr(CO)3, and (E)-{(η5-C5H4)CF=CF(η5-C5H4)}(η5-C5H5)2Fe2. J. Am. Chem. Soc. 2009, 131, 1251–1268. 10.1021/ja808303j. [DOI] [PubMed] [Google Scholar]
- Markies P. R.; Schat G.; Akkerman O. S.; Bickelhaupt F.; Smeets W. J. J.; Spek A. L. Coordinational behavior of solvent-free diorganylzinc compounds: The remarkable X-ray structure of dimeric diphenylzinc. Organometallics 1990, 9, 2243–2247. 10.1021/om00158a022. [DOI] [Google Scholar]
- The speciation of the eliminated ZnMe moiety was not established.
- NMR monitoring of the reaction showed that after heating for 1 day, 2 and 5 were both present, alongside a major intermediate that was tentatively assigned as the BIPHEP [BIPHEP = 2,2′-bis(diphenylphosphino)biphenyl] complex [Ru(BIPHEP)(PPh3)(ZnMe)2] (4) based on the similarity of the 31P NMR data [δ 87 (t, JPP = 25 Hz), 49 (dd, JPP = 248 Hz, JPP = 24 Hz), 44 (dd, JPP = 248 Hz, JPP = 26 Hz)] to those of Ru(BIPHEP)(PPh3)HCl [δ 87 (dd, JPP = 38 Hz, JPP = 21 Hz), 41 (dd, JPP = 305 Hz, JPP = 38 Hz), 36 (dd, JPP = 305 Hz, JPP = 21 Hz)].20 The presence of the BIPHEP ligand and two ZnMe ligands (two X-type ligands are necessary in combination with the three L-type ligands to give a neutral Ru species) implies that the C–C coupling needed for BIPHEP formation, ; Chaudret B.; Cole-Hamilton D. J.; Wilkinson G. Bis(styrene)bis(triphenylphosphine)ruthenium(0) and its reactions with triphenylphosphine and with alkenes. J. Chem. Soc., Dalton Trans. 1978, 1739–1745. 10.1039/dt9780001739. [DOI] [Google Scholar]; precedes Ru–Zn cleavage and elimination of a ZnMe group.
- These involved attempted crystallisation from C6D6, C6D6/hexane, C6D6/pentane, toluene (both room temperature and −35 °C), toluene/hexane, toluene/pentane, THF (room temperature and −35 °C), THF/hexane, THF/pentane, C6H5F (room temperature and −35 °C), and C6H5F/hexane (room temperature and −35 °C).
- Heiß P.; Hornung J.; Zhou X.; Jandl C.; Pöthig A.; Gemel C.; Fischer R. A. Combined experimental and theoretical study on hampered phosphine dissociation in heteroleptic Ni/Zn complexes. Inorg. Chem. 2020, 59, 514–522. 10.1021/acs.inorgchem.9b02798. [DOI] [PubMed] [Google Scholar]
- This was independently prepared from treatment of 1 with PnBu3.
- The mixture of 1 and 11 was found to be unstable in a C6D6 solution, and over ∼24 h, the hydride signals for both species merged into the baseline (Supporting Information). In part, this may result from the deposition of an insoluble red oil, resulting in the loss of material dissolved in solution. However, the formation of new signals in the 31P NMR spectrum, two at high frequencies and one at a low frequency, not that disimilar to those for 3 may also suggest there is further reaction of 1 or 11 with [(PnBu3)xZnMe]+, resulting in the formation of a PnBu3 analogue of 3. However, the formation of a “red oil” prevented us from isolating any such product; attempts to drive the reaction through use of >1 equiv of PnBu3 led to only increased amounts of red oil.
- IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene.
- Specklin D.; Fliedel C.; Gourlaouen C.; Bruyere J. C.; Avilés T.; Boudon C.; Ruhlmann L.; Dagorne S. N-Heterocyclic carbene based tri-organyl-Zn-alkyl cations: Synthesis, structures, and use in CO2 functionalization. Chem. - Eur. J. 2017, 23, 5509–5519. 10.1002/chem.201605907. [DOI] [PubMed] [Google Scholar]
- Schnee G. F.; Fliedel C.; Avilés T.; Dagorne S. Neutral and cationic N-heterocyclic carbene zinc adducts and the BnOH/Zn(C6F5)2 binary mixture - characterization and use in the ring-opening polymerization of β-butyrolactone, lactide, and trimethylene carbonate. Eur. J. Inorg. Chem. 2013, 2013, 3699–3709. 10.1002/ejic.201300292. [DOI] [Google Scholar]
- Rit A.; Spaniol T. P.; Maron L.; Okuda J. Mixed alkyl hydrido complexes of zinc: Synthesis, structure, and reactivity. Organometallics 2014, 33, 2039–2047. 10.1021/om500190c. [DOI] [Google Scholar]
- The 7:8 ratio was ∼1:0.15. Variable-temperature 1H NMR measurements showed this did not change between −70 and 70 °C.
- Garrou P. E. ΔR ring contributions to 31P NMR parameters of transition-metal-phosphorus chelate complexes. Chem. Rev. 1981, 81, 229–266. 10.1021/cr00043a002. [DOI] [Google Scholar]
- Mohr F.; Privér S. H.; Bhargava S. K.; Bennett M. A. Ortho-metalated transition metal complexes derived from tertiary phosphine and arsine ligands. Coord. Chem. Rev. 2006, 250, 1851–1888. 10.1016/j.ccr.2005.10.003. [DOI] [Google Scholar]
- An additional species was also formed at this early stage of the reaction but is an intermediate that was mostly consumed within ∼3 h; its identity remains unknown.
- The two hydride signals at δ −7.3 and −11.1 showed similar T1 values of 400 and 540 ms, respectively (400 MHz, 300 K).
- The computed structure of [Ru(PPh3)3H3]− (i.e., 12 with both [ZnMe]+ groups removed) shows a shortening of the Ru–H3 distance from 1.69 to 1.64 Å, consistent with the {ZnMe} groups influencing the Ru–H3 interaction in 12. In contrast, the Ru–H1 and −H2 distances are not affected.
- Ekkert O.; White A. J. P.; Crimmin M. R. Trajectory of approach of a zinc-hydrogen bond to transition metals. Angew. Chem., Int. Ed. 2016, 55, 16031–16034. 10.1002/anie.201608599. [DOI] [PubMed] [Google Scholar]
- Abdalla J. A. B.; Caise A.; Sindlinger C. P.; Tirfoin R.; Thompson A. L.; Edwards A. J.; Aldridge S. Structural snapshots of concerted double E-H bond activation at a transition metal centre. Nat. Chem. 2017, 9, 1256–1262. 10.1038/nchem.2792. [DOI] [PubMed] [Google Scholar]
- Garçon M.; Bakewell C.; Sackman G. A.; White A. J. P.; Cooper R. I.; Edwards A. J.; Crimmin M. R. A hexagonal planar transition-metal complex. Nature 2019, 574, 390–393. 10.1038/s41586-019-1616-2. [DOI] [PubMed] [Google Scholar]
- Tauchert M. E.; Okuda J. A hexagonal planar metal complex. Angew. Chem., Int. Ed. 2020, 59, 4214–4215. 10.1002/anie.201915432. [DOI] [PubMed] [Google Scholar]
- Butler M. J.; Crimmin M. R. Magnesium, zinc, aluminium and gallium hydride complexes of the transition metals. Chem. Commun. 2017, 53, 1348–1365. 10.1039/C6CC05702K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantreil X.; Nolan S. P. Synthesis of N-heterocyclic carbene ligands and derived ruthenium olefin metathesis catalysts. Nat. Protoc. 2011, 6, 69–77. 10.1038/nprot.2010.177. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian16, rev. C.01; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- Andrae D.; Häußermann U.; Dolg M.; Stoll H.; Preuß H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta. 1990, 77, 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. Self-consistent molecular orbital methods XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta. 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Hollwarth A.; Bohme M.; Dapprich S.; Ehlers A. W.; Gobbi A.; Jonas V.; Kohler K. F.; Stegmann R.; Veldkamp A.; Frenking G. A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al-Bi and f-type polarization functions for Zn Cd, Hg. Chem. Phys. Lett. 1993, 208, 237–240. 10.1016/0009-2614(93)89068-S. [DOI] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Perdew J. P. Density-functional approximation for the correlation-energy of the inhomogeneous electron-gas. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Bader R. F. W.Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, U.K., 1994. [Google Scholar]
- Keith T. A.AIMAll, ver. 17.11.14; TK Gristmill Software: Overland Park, KS, 2017. [Google Scholar]
- Contreras-García J.; Johnson E. R.; Keinan S.; Chaudret R.; Piquemal J.-P.; Beratan D. N.; Yang W. NCIPLOT: A program for plotting noncovalent interaction reagions. J. Chem. Theory Comput. 2011, 7, 625–632. 10.1021/ct100641a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Mitoraj M. P.; Parafiniuk M.; Srebro M.; Handzlik M.; Buczek A.; Michalak A. Applications of the ETS-NOCV method in descriptions of chemical reactions. J. Mol. Model. 2011, 17, 2337–2352. 10.1007/s00894-011-1023-6. [DOI] [PubMed] [Google Scholar]
- te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- Chemcraft version 1.7 (see http://www.chemcraftprog.com).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.