

Abstract
Acute respiratory distress syndrome (ARDS) due to coronavirus disease 2019 and other etiologies results from injury to the alveolar epithelial cell (AEC) barrier resulting in noncardiogenic pulmonary edema, which causes acute respiratory failure; recovery requires epithelial regeneration. During physiological regeneration in mice, type 2 AECs (AEC2s) proliferate, exit the cell cycle, transiently assume a transitional state, then differentiate into type 1 AECs (AEC1s); in humans, persistence of the transitional state is associated with pulmonary fibrosis. It is unknown whether transitional cells emerge and differentiate into AEC1s without fibrosis in human ARDS and why transitional cells differentiate into AEC1s during physiological regeneration but persist in fibrosis. We hypothesized that incomplete but ongoing AEC1 differentiation from transitional cells without fibrosis may underlie persistent barrier permeability and acute respiratory failure in ARDS. Immunostaining of postmortem ARDS lungs revealed abundant transitional cells without fibrosis. They were typically cuboidal or partially spread, sometimes flat, and occasionally expressed AEC1 markers. Immunostaining and/or single-cell RNA sequencing revealed that transitional cells in mouse models of physiological regeneration, ARDS, and fibrosis express markers of cell cycle exit but only in fibrosis express a specific senescence marker. Thus, in severe, fatal early ARDS, AEC1 differentiation from transitional cells is incomplete, underlying persistent barrier permeability and respiratory failure but ongoing without fibrosis; senescence of transitional cells may be associated with pulmonary fibrosis.
Coronavirus disease 2019 (COVID-19) remains a global scourge, claiming thousands of lives daily and straining health care systems worldwide. In its most severe form, COVID-19 manifests as the acute respiratory distress syndrome (ARDS). COVID-19 ARDS causes acute hypoxemic respiratory failure with high mortality rates.1, 2, 3, 4, 5 However, the underlying mechanism by which COVID-19 ARDS results in refractory hypoxemia and high mortality is unknown.
Critical to the pathogenesis of ARDS is injury to the alveolar epithelium, which forms a tight barrier that maintains dry airspaces and permits gas exchange.6 The alveolar epithelium comprises type 1 AECs (AEC1s) and type 2 AECs (AEC2s). AEC1s cover >95% of the alveolar surface, thus playing a critical role in barrier integrity, and are exquisitely thin, facilitating efficient gas exchange.7,8 AEC2s are cuboidal, produce surfactant, and serve as progenitors.9 During ARDS, AEC death results in barrier permeability, leading to flooding of the airspaces with proteinaceous edema fluid, which in turn causes severe hypoxemia, often necessitating mechanical ventilation, and death.6 Conversely, regeneration of the alveolar epithelium is critical for the restoration of barrier integrity, resolution of edema, liberation from the ventilator, and survival.10,11 The state of epithelial injury and regeneration in COVID-19 ARDS has not been well characterized histologically. We speculated that incomplete alveolar regeneration may result in ongoing barrier permeability, pulmonary edema, and death from acute hypoxemic respiratory failure in COVID-19 ARDS.
The principal progenitor responsible for alveolar regeneration is the AEC2,9,12, 13, 14 although other progenitors may contribute.15, 16, 17 AEC2s proliferate and then spread and differentiate into AEC1s to restore normal alveolar architecture and barrier integrity. A mouse model showed that AEC2-to-AEC1 differentiation occurs in a nongradual manner.18 After proliferation, AEC2s exit the cell cycle and assume a transitional state before differentiating into AEC1s. The transitional state is characterized by markers of cell cycle exit, down-regulation of AEC2 markers, a cuboidal or partially spread morphology, and expression of unique signature genes, including Krt8. This state was subsequently identified in diverse mouse models of lung injury, suggesting that it is a conserved stage of regeneration regardless of the cause of injury.19, 20, 21, 22, 23 During physiological regeneration in mice, the transitional state is transient; lineage tracing studies confirmed that transitional cells differentiate into AEC1s, restoring normal alveolar structure and function.8,18,19,24
It is unknown which stage of alveolar regeneration (AEC2 proliferation, acquisition of the transitional state, or AEC1 differentiation) may be incomplete in COVID-19. Autopsy studies of mechanically ventilated patients who died of acute respiratory failure have shown that the histology of COVID-19 ARDS is classic diffuse alveolar damage (DAD) and includes AEC2 hyperplasia.25, 26, 27, 28, 29 This finding suggests that proliferation may be preserved and that the prolonged barrier permeability and poor clinical outcomes in COVID-19 ARDS may be associated with incomplete conversion of AEC2s into the transitional state or differentiation of transitional cells into AEC1s.
Idiopathic pulmonary fibrosis (IPF) is caused by irreversible scarring of the lungs widely believed to arise from impaired alveolar regeneration after chronic or repetitive injury. IPF is characterized by persistence of transitional cells with a paucity of mature AEC1s, suggesting that ineffectual differentiation of transitional cells into AEC1s may be the specific regenerative defect underlying the pathogenesis of fibrosis.19, 20, 21, 22,24,30,31 Because the transitional state has a transcriptomic signature of cell cycle arrest,18,19,30,31 the persistence of transitional cells in IPF is consistent with emerging literature suggesting that epithelial cells in IPF are senescent.32, 33, 34, 35, 36, 37, 38, 39 Recent case reports identified transitional AECs in the fibrosis that develop after months of nonresolving COVID-19 ARDS, so-called “fibroproliferative ARDS.”40,41 In these cases, the transitional cells exist in a milieu of extensive matrix deposition and architectural distortion that was deemed irreversible, necessitating lung transplantation. In fact, the presence of transitional cells on biopsy sample was proposed as a potential biomarker of irreversible fibrosis.40 However, it is unknown whether transitional cells are inevitably associated with fibrosis in humans. Moreover, why transitional cells differentiate into AEC1s to restore normal alveolar architecture and function in mouse models of physiological regeneration, whereas they persist and may beget fibrosis in human IPF and fibroproliferative ARDS, is a fundamental unanswered question in the field.
We hypothesized that in early (ie, within the first 14 days of presentation) fatal COVID-19 ARDS, after severe epithelial injury, AEC2s proliferate and assume the transitional state, but that AEC1 differentiation from the transitional state is incomplete, resulting in ongoing barrier permeability, noncardiogenic pulmonary edema, and ventilator dependence, and mortality. It was further speculated that in early human ARDS, as in mouse models of physiological regeneration, proliferating AEC2s exit the cell cycle and transiently adopt the transitional state but retain the capacity to differentiate into AEC1s, restoring normal alveolar architecture without fibrosis. However, in IPF and fibroproliferative ARDS after months of chronic or repetitive injury, AECs evolve into a state of permanent cell cycle arrest, or senescence, losing capacity for an AEC1 fate, and fibrosis ensues.19,30, 31, 32, 33, 34, 35 To explore these hypotheses, postmortem lung tissue of patients with COVID-19 ARDS who died of acute respiratory failure within 14 days of presentation was studied. For comparison, postmortem lung tissue from patients with early fatal ARDS of other etiologies and explanted lung tissue from patients with IPF undergoing lung transplantation was studied. Tissue was examined for evidence of AEC2 proliferation, transitional cells, AEC1 differentiation, senescence, and fibrosis. Using existing single-cell RNA sequencing (scRNAseq) data sets and immunostaining, the gene expression profiles of transitional cells were compared in two mouse models of physiological regeneration without fibrosis [lipopolysaccharide (LPS) and pneumonectomy], early human COVID-19 ARDS, and human IPF, focusing on markers of cell cycle exit and senescence. The findings advance our basic understanding of physiological and pathologic alveolar regeneration and have implications for clinical prognosis and management. Ultimately, investigation of the cellular and molecular mechanisms underlying ineffectual alveolar regeneration in ARDS and fibrosis may lead to novel therapies to promote physiological regeneration, thus accelerating restoration of barrier integrity, resolution of edema, liberation from the ventilator and survival in ARDS, and preventing fibrosis in fibroproliferative ARDS and IPF.
Materials and Methods
Clinical Data and Tissue Acquisition
This study was approved by the University of Michigan Institutional Review Board. Patients were diagnosed with COVID-19 by using real-time quantitative PCR for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) on a nasopharyngeal, sputum, or bronchoalveolar lavage sample. Medical records were reviewed. Formalin-fixed, paraffin-embedded lung tissue was obtained from the autopsies of patients with COVID-19 who died at the University of Michigan Hospital. Normal lung from deceased individuals rejected for lung transplantation was obtained from Gift of Life Michigan. IPF lung was explanted tissue from patients who underwent lung transplantation at the University of Michigan Hospital.
Histology and Immunostaining
Sections were stained for hematoxylin and eosin (H&E) or trichrome and interpreted by a board-certified pathologist. For immunostaining, deparaffinized sections were boiled in Dako Target Retrieval Solution (S1699, Agilent Technologies, Santa Clara, CA). The samples were blocked with 5% donkey serum in Tris-buffered saline with 0.05% Tween and incubated with antibodies against KRT8 (TROMA-1, 1:20, University of Iowa Developmental Studies Hybridoma Bank, Iowa City, IA), HTI-56 (TB-29AHT1-56, 1:10, Terrace Biotech, San Francisco, CA), tomato lectin (FL-1171, 1:250, Vector Laboratories, Burlingame, CA), α-smooth muscle actin–Cy3 (C6198, 1:200, Sigma-Aldrich, St. Louis, MO), pro–surfactant protein C (proSPC) (AB3786, 1:500, MilliporeSigma, Burlington, MA), TP53 (ab131442, 1:100, Abcam, Cambridge, United Kingdom), CDKN2A/p16INK4a (ab108349, 1:250, Abcam), club cell secretory protein (sc-365992, 1:100, Santa Cruz Biotechnology, Dallas, TX), and/or SARS nucleocapsid protein (NB100-56576, 1:50-1:500, Novus Biologicals, Littleton, CO) overnight at 4°C. Secondary antibodies, including fluorescein isothiocyanate–conjugated anti-rat IgG (712-095-153, 1:250, Jackson ImmunoResearch Laboratories, West Grove, PA), Cy3-conjugated anti-rat IgG (711-167-003, 1:250, Jackson ImmunoResearch Laboratories), Cy5-conjugated anti-rat IgG (712-175-153, 1:250, Jackson ImmunoResearch Laboratories), Alexa-Fluor 555–conjugated anti-mouse IgG (A-31570, 1:250, Invitrogen, Carlsbad, CA), Cy3-conjugated anti-rabbit IgG (711-165-152, 1:250, Jackson ImmunoResearch Laboratories), and Cy5-conjugated anti-rabbit IgG (711-175-152, 1:250, Jackson ImmunoResearch Laboratories), were applied for 1 hour at room temperature. For fluorescence in situ hybridization, sections were stained with SARS-CoV-2 probe (#848561, ACDBio, Newark, CA) as previously described.18 The slides were counterstained with DAPI 1:10,000 in phosphate-buffered saline and mounted on standard glass slides with Prolong Gold (P36930, Invitrogen) antifade reagent. Images were acquired on a BX53 microscope (Olympus, Tokyo, Japan) or Aperio scanner (Leica Microsystems, Buffalo Grove, IL). Exposure settings were not always equal due to red blood cell autofluorescence but were set by using isotype control antibodies. In all images, blue pseudocolor is DAPI.
Analysis of scRNAseq Data Sets
scRNAseq data sets were interrogated for gene expression levels. Data sets were obtained from the NCBI Gene Expression Omnibus (https://www.nci.nlm.nih.gov/geo; accession numbers GSE113049,18 GSE138585,21 GSE136831,30 GSE106960,42 and GSE135893).31 Unique molecular identifier count matrices were reprocessed to obtain average log-normalized expression values for each cell type annotated in the respective study. In the mouse LPS scRNAseq study, mice had been treated with 45 μg LPS intratracheally and euthanized 7 days later.18 In the pneumonectomy scRNAseq study, mice had been subjected to pneumonectomy of the right lobe and euthanized 7 or 21 days later.21 In both studies, lineage-labeled AEC2s were sequenced. In the human IPF studies, the lungs of patients with IPF had been subjected to scRNAseq.30,31 The AEC2 state was identified as “naive AEC2s” from the LPS data set,18 subpopulation II from the pneumonectomy data set,21 and AEC2s from the human IPF data sets.30,31 The transitional state was identified as the “cell cycle arrest” cluster in the LPS data set,18 “subpopulation I” from the pneumonectomy day 7 wild-type mice,21 and the “aberrant basaloid”30 and “KRT5–/KRT17+”31 clusters from the human IPF data sets. The AEC1 state was identified as “naive AEC1s” from the LPS data set,18 AEC1s from a data set used as a control for the pneumonectomy data set,42 and AEC1s from the human IPF data sets.30,31
Results
Clinical Presentation
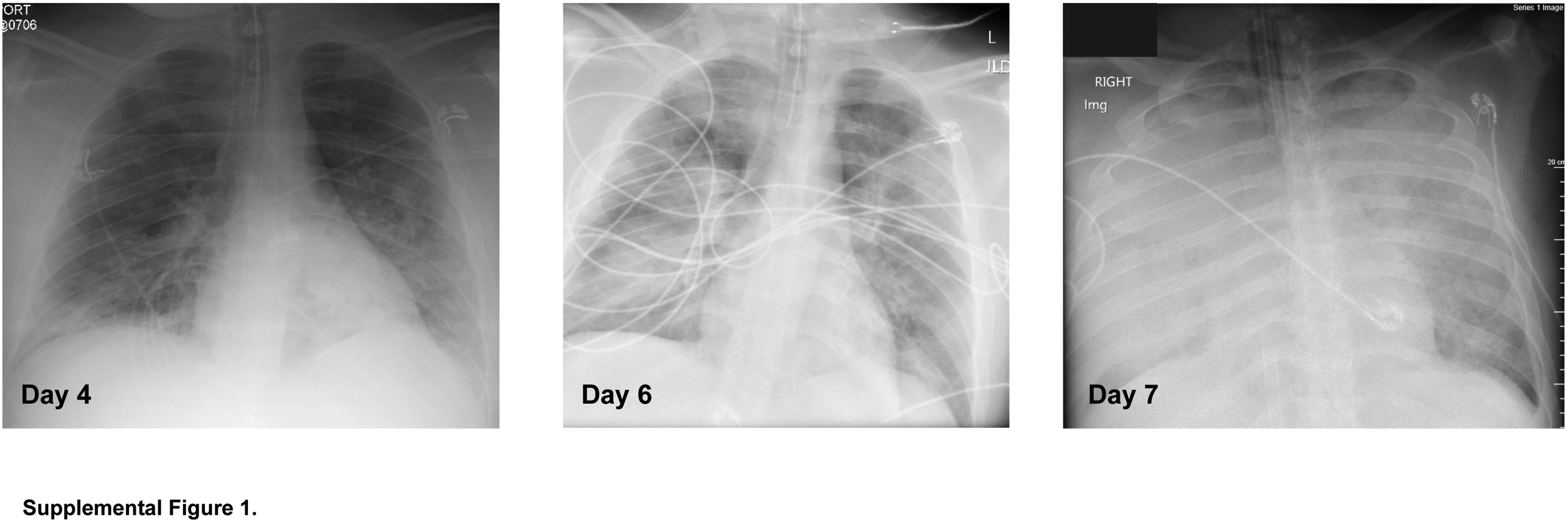
All patients with ARDS presented with acute hypoxemic respiratory failure and radiographic evidence of pulmonary edema (Supplemental Figure S1 and Supplemental Tables S1 and S2). All patients died within 2 weeks of hospitalization. Patients #1 to #3 were diagnosed with COVID-19 by SARS-CoV-2 PCR. Patients #4 to #6 were hospitalized in 2018. Clinical autopsy reports revealed DAD.
Severe Epithelial Damage in COVID-19 ARDS
To characterize epithelial injury in COVID-19 ARDS, histologic lung sections stained with H&E were examined. Consistent with prior reports,25, 26, 27, 28 the histology of COVID-19 ARDS was acute DAD. There was diffuse airspace filling with edema, fibrin, and hyaline membranes (Figure 1, A-D), indicative of increased epithelial permeability. Interstitial and alveolar inflammation, predominantly macrophages and lymphocytes, was present. Desquamated epithelial cells with elongated morphology were observed (Figure 1D). Such cells are commonly designated AEC1s43 but are often, and in this case were, thicker and cover less surface area than AEC1s,44 raising new doubt as to their identity. Regardless, the radiographic and histologic findings observed in COVID-19 ARDS indicated epithelial injury.
Figure 1.

Epithelial injury and proliferation in coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome (ARDS). Hematoxylin and eosin staining of lungs from patients with COVID-19 reveals acute diffuse alveolar damage (DAD) with edema (asterisks) (A), fibrin (star) (B), hyaline membranes (open arrowheads) (C), and desquamated epithelial cells (arrows) (D). E: Immunostaining for the type 1 alveolar epithelial cell (AEC1) markers HTI-56 and tomato lectin reveals a diffusely speckled pattern with frequent small defects in staining (arrowhead) and septal thickening consistent with interstitial edema and/or inflammation (asterisk). F: Immunostaining for the AEC1 markers HTI-56 and tomato lectin reveals occasional areas in which staining was completely absent from one or both sides of the alveolar septa (indicated by red dashed tracing), consistent with denudation. Septal thickening indicated by asterisk. G: Immunostaining for the type 2 AEC (AEC2s) marker pro–surfactant protein C (proSPC) reveals vast areas devoid of mature AEC2s with rare AEC2s remaining. H: Hematoxylin and eosin reveals hyperplastic cuboidal epithelial cells (arrow). n = 3. Scale bars = 50 μm. Original magnification: ×200 (A–D); ×40 (H).
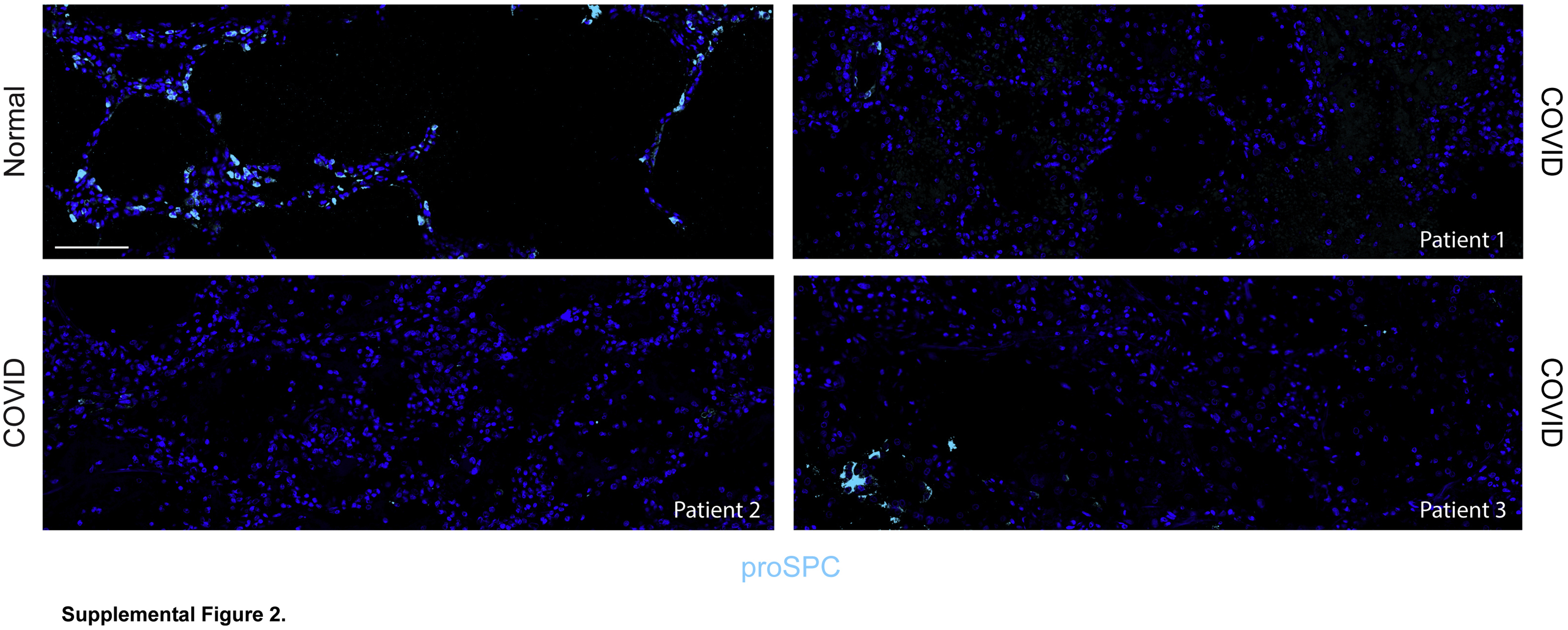
To specifically assess the extent of structural damage to AEC1s and AEC2s, sections were stained for AEC1 and AEC2 markers. AEC1 markers appeared diffuse and speckled throughout the lungs, consistent with extensive AEC1 injury (Figure 1, E and F). The septa were thickened, likely due to the presence of edema and inflammatory cells in the interstitial space (Figure 1, E and F). Small defects in staining were frequent (Figure 1E). Complete absence of staining from one or both sides of the alveolar septa was present but infrequent (Figure 1F) and was interpreted as AEC1 loss resulting in denuded septa. Staining for the AEC2 marker proSPC revealed vast areas devoid of mature (proSPC+) AEC2s with only rare proSPC+ AECs remaining (Figure 1G and Supplemental Figure S2). The near-complete absence of proSPC+ cells may indicate AEC2 death or SPC down-regulation. Taken together, the clinical, radiographic, and histologic data indicated severe, extensive epithelial damage, resulting in noncardiogenic pulmonary edema, in turn leading to acute hypoxemic respiratory failure.
AEC2s Proliferate and Assume the Transitional State in COVID-19 ARDS
To assess whether surviving AEC2s are mobilized to regenerate the injured epithelium, histologic sections stained with H&E were again examined. Hyperplastic cuboidal epithelial cells (Figure 1H), a common histologic feature of DAD that has historically been termed “AEC2 hyperplasia,” were observed.43 Indeed, proSPC staining revealed rare hyperplastic AEC2s (Supplemental Figure S3). The hyperplastic AEC2s were hypertrophic, consistent with previous observations in mouse models of lung injury.45 However, hyperplasia of mature (proSPC+) AEC2s was exceedingly rare amid vast areas devoid of mature AEC2s (Figures 1G and Supplemental Figure S2). This finding suggests that the majority of the hyperplastic epithelial cells observed by H&E staining do not retain a mature AEC2 phenotype. Regardless, the presence of hyperplastic cuboidal epithelial cells on H&E suggested that AEC2s (or other progenitors) had successfully proliferated in an attempt to replace damaged AECs.
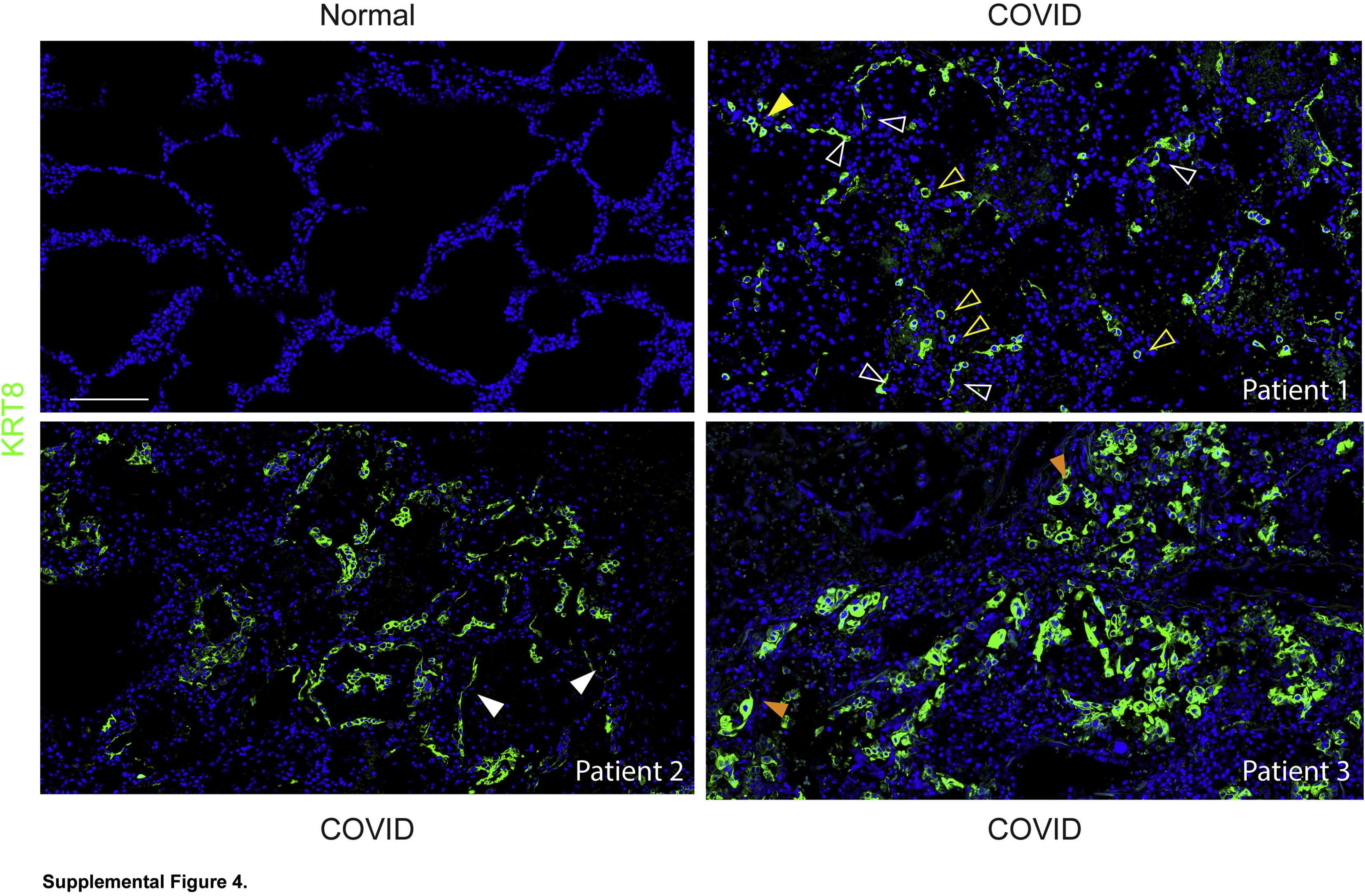
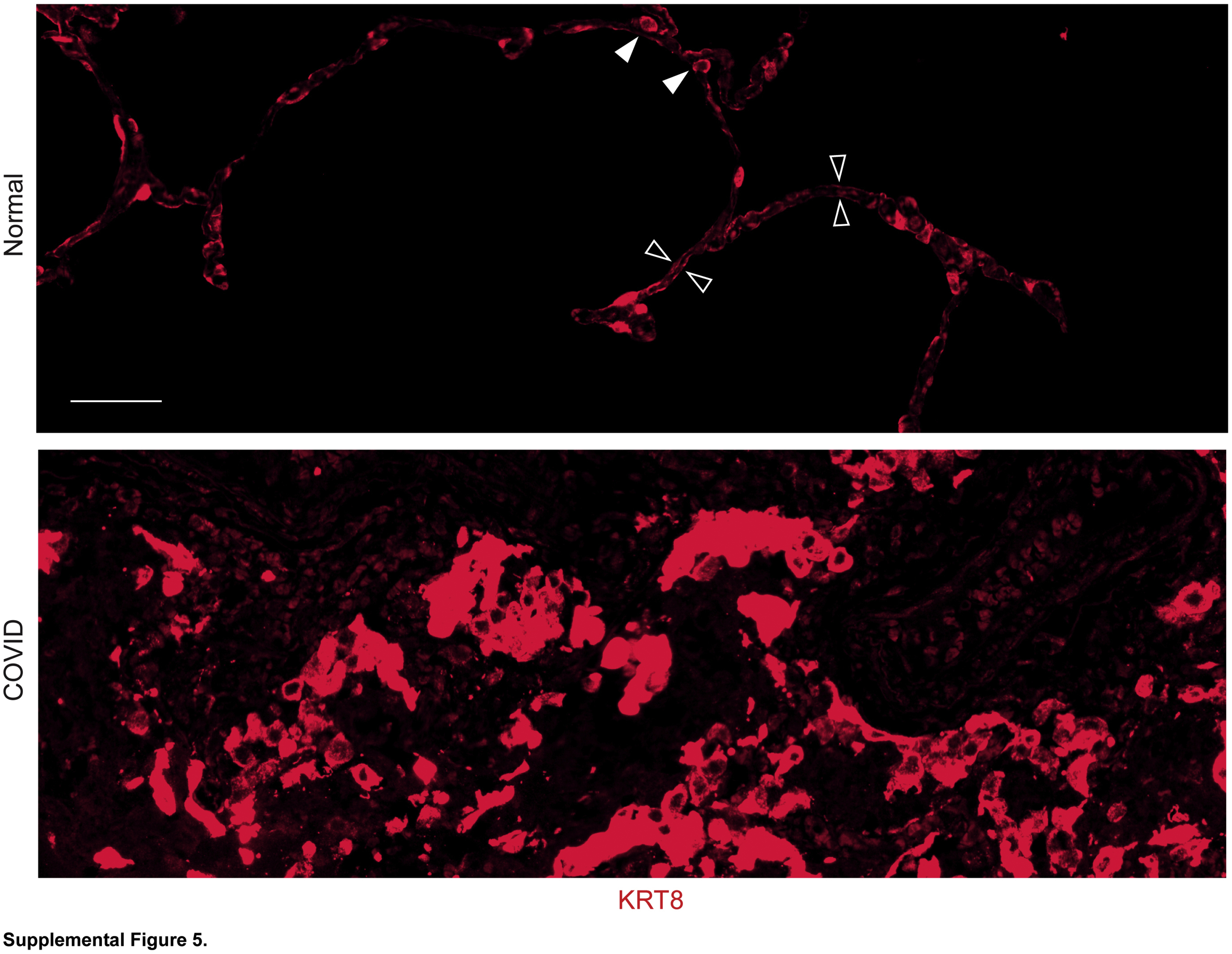
To determine whether regenerating AEC2s were able to assume the transitional state, sections were stained for the transitional state marker KRT8. KRT8hi transitional cells were abundant throughout the COVID-19 lungs but absent in normal lungs (Figure 2, A and B and Supplemental Figure S4). [AEC1s and AEC2s in normal lungs express KRT8 at much lower levels than transitional cells in injured lungs (Supplemental Figure S5).18, 19, 20, 21, 22,24,30,31] Some transitional cells were cuboidal; some existed as single cells, and others were doublets or hyperplastic, suggesting recent cell division (Figure 2A and Supplemental Figure S4). However, the morphology of transitional cells was often partially spread and occasionally flat, approaching AEC1 morphology (Figure 2A and Supplemental Figure S4). The cuboidal hyperplastic, partially spread, and flat transitional cells typically existed as monolayers along structurally normal septa (Figure 2A and Supplemental Figure S4), consistent with organized proliferation and ongoing differentiation into AEC1s. Occasional transitional cells were hypertrophic, bizarrely shaped, and disorganized (Figure 2A and Supplemental Figure S4). KRT8hi transitional cells did not express SPC (Figure 2B), consistent with known down-regulation of AEC2 markers as cells assume the transitional state.18, 19, 20, 21, 22,30,31 [Although transitional cells may arise from club-like progenitors,15,20,30,31 bronchiolization was not observed, and transitional cells did not express club cell secretory protein, a club cell marker (Supplemental Figure S6).] Taken together, these data show that transitional cells arise within the first 2 weeks of onset of human ARDS. The abundance of transitional cells (Figure 2A and Supplemental Figure 4) suggests that the paucity of proSPC+ cells may be attributable to both AEC2 death due to cytopathic viral infection and successful acquisition of the transitional state.
Figure 2.

Epithelial proliferation yields abundant transitional cells with incomplete type 1 alveolar epithelial cell (AEC1) differentiation in coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. A: Immunostaining reveals KRT8hi transitional cells that are sometimes cuboidal and isolated (open yellow arrowhead) or existing in pairs (closed yellow arrowhead) or hyperplastic (orange arrowhead), suggestive of recent cell division. Transitional cells are often partially spread (open white arrowhead) and occasionally flat (closed white arrowheads). B: Transitional cells do not express pro–surfactant protein C (proSPC) and are abundant in areas devoid of proSPC+ cells. C: Sections were stained for KRT8 and the AEC1 marker HTI-56. KRT8hi transitional cells fill gaps denuded of AEC1s on alveolar septa. Some are cuboidal (open yellow arrowheads); most are partially spread (open white arrowheads). Occasional transitional cells approach a flat AEC1 morphology but do not express AEC1 markers (closed white arrowhead), with rare exception (closed yellow arrowheads). Flat cells that express AEC1 markers but not KRT8 (closed green arrowhead) were interpreted as native AEC1s that were not damaged during lung injury. Findings suggest ongoing organized, albeit incomplete, AEC1 differentiation. Rare cells display bizarre morphologies and/or have sloughed into the airspaces (orange arrowheads), consistent with haphazard regeneration. n = 3. Scale bars = 50 μm.
Incomplete Differentiation of Transitional Cells into Mature AEC1s in COVID-19 ARDS
Because progenitors successfully proliferated and assumed the transitional state, we hypothesized that incomplete differentiation of transitional cells into mature AEC1s may underlie the persistent barrier permeability, ongoing edema, and poor clinical outcomes in COVID-19. Supporting this hypothesis, although the transitional cells appeared to be in the process of differentiating into AEC1s, they were typically cuboidal or partially spread, only occasionally assuming the flat morphology of AEC1s (Figure 2A). To further elucidate the extent to which the transitional cells had differentiated into AEC1s, lung sections were co-stained for KRT8 and AEC1 markers (Figure 2C and Supplemental Figure S7). In most cases, transitional cells expanded along alveolar septa, filling gaps denuded of AEC1s (Figure 2C). Again, some cells were cuboidal (Figure 2C), but most cells were partially spread (Figure 2C). However, some had a flat morphology (Figure 2C), and occasional transitional cells expressed AEC1 markers (Figure 2C). Flat cells expressing AEC1 markers but not KRT8 were interpreted as native AEC1s that withstood injury (Figure 2C), although nascent AEC1s that down-regulated KRT8 after differentiation cannot be excluded. Rarely, transitional cells assumed bizarre morphologies and/or had sloughed off the septa into the airspaces (Figure 2C).
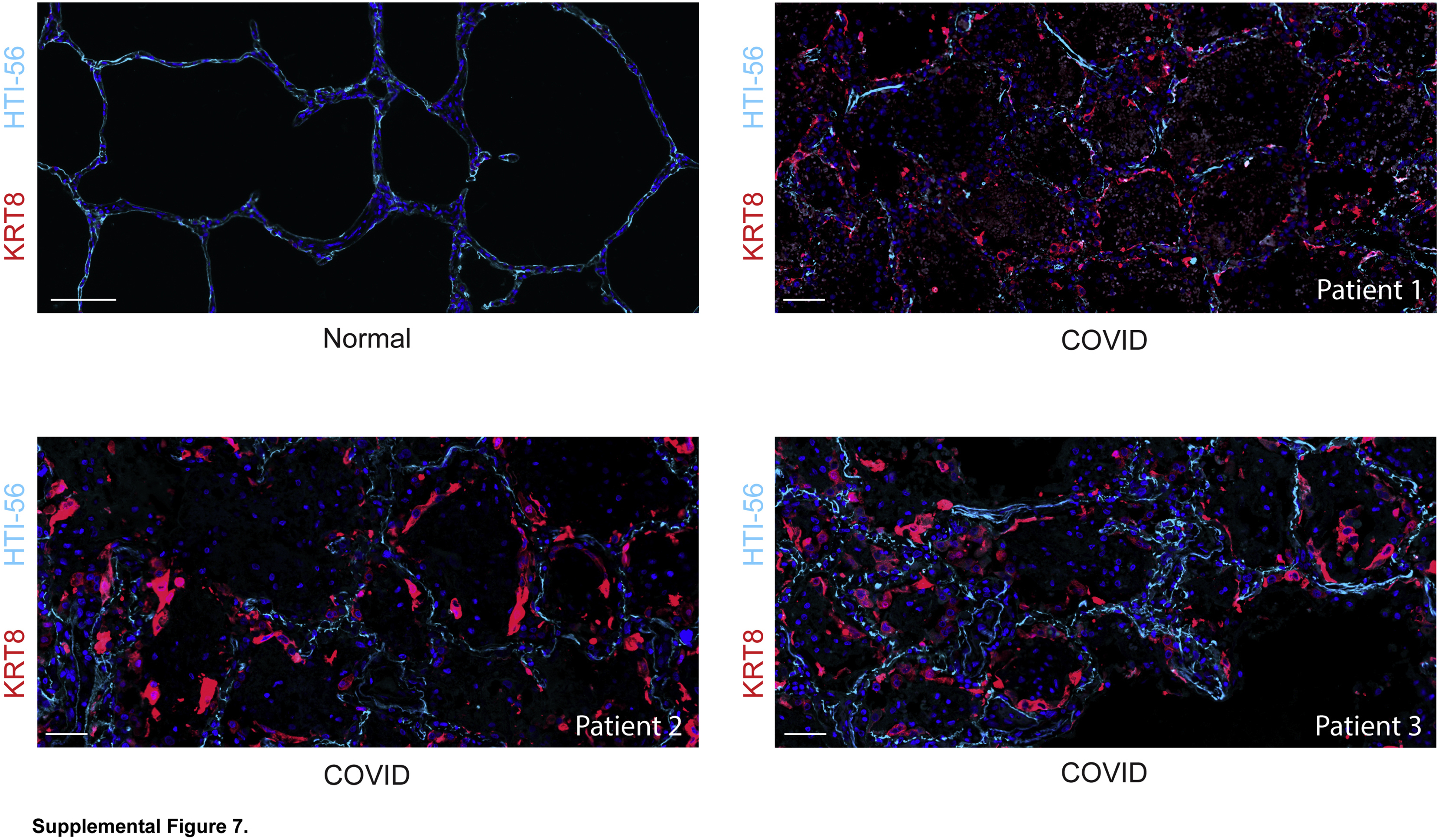
Although there was some variability between patients and across the tissue of each patient, the general histology, AEC1 damage, paucity of mature AEC2s, and abundance of transitional cells were present in all patients (Supplemental Figures S2, S6, S7, and S8). The predominant organization of transitional cells in a monolayer on alveolar septa filling gaps denuded of AEC1s and displaying increasingly spread morphologies without AEC1 marker expression (Figure 2 and Supplemental Figure S7) suggested that differentiation along a linear trajectory into mature AEC1s was ongoing but incomplete. Thus, the progressive edema, ongoing ventilator dependence, and high mortality observed in COVID-19 ARDS may be due to incomplete differentiation of transitional cells into AEC1s.
SARS-CoV-2 Was Not Detected in COVID-19 ARDS Autopsy Specimens
To assess for ongoing viral infection, which may contribute to the persistence of the transitional state, the tissue was examined for the presence of SARS-CoV-2. SARS-CoV-2 was not detected by immunostaining with an antibody against nucleocapsid protein (data not shown). Fluorescence in situ hybridization using a probe against SARS-CoV-2 failed to detect viral RNA, although staining with control probes suggested RNA degradation in the fixed tissue (data not shown). Viral inclusions were not observed, consistent with prior studies.46
Because SARS-CoV-2 infects AEC2s due to high expression of the viral receptor angiotensin-converting enzyme 2, and mature AEC2s were rare (Figure 1G and Supplemental Figure S2), we hypothesized that the apparent lack of ongoing viral infection may be due to a lack of angiotensin-converting enzyme 2 expression by transitional cells and AEC1s. Interrogation of publicly available scRNAseq data sets revealed that AEC2s down-regulate angiotensin-converting enzyme 2 as they assume the transitional state and differentiate into AEC1s (Supplemental Figure S9). Although it is difficult to make definitive conclusions based on negative data, the inability to detect SARS-CoV-2, absence of viral inclusions, and paucity of AECs that express the viral receptor argue against ongoing viral infection.
Absence of Fibrosis in COVID-19 ARDS
In humans, persistence of the transitional state has previously been observed only in the setting of fibrosis, whether due to IPF or fibroproliferative ARDS, leading to speculation that this finding may be pathognomonic of fibrosis.19,20,22,24,30,31,40,41 However, it is unknown whether transitional cells are inevitably associated with fibrosis in humans.40,41 In mouse models of physiological regeneration such as the LPS model and the pneumonectomy model, transitional cells appear transiently and then differentiate into AEC1s to restore normal alveolar architecture without fibrosis.18,21 We therefore hypothesized that in early COVID-19 ARDS, transitional cells, which appear to be in the process of physiological AEC1 differentiation (Figure 2C), exist without fibrosis. Indeed, no excessive collagen deposition, myofibroblast accumulation, or architectural distortion was observed in early COVID-19 ARDS, in contrast to IPF (Figure 3, A and B). Moreover, whereas in IPF the transitional cells overlie fibroblastic foci (Figure 3C) and line honeycomb cysts,22 the transitional cells in COVID-19 ARDS overlie structurally normal alveolar septa (Figure 3A and Supplemental Figure S7). Finally, in early COVID-19 ARDS, nascent AEC1s appear to be emerging from transitional cells (Figure 2, A and C and Supplemental Figure S7), whereas IPF is characterized by a paucity of AEC1s (Figure 3B).
Figure 3.

Absence of fibrosis in early coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. A: Trichrome highlighted (in blue) basement membranes and vascular adventitia in normal and COVID-19 lungs and collagen deposition with marked fibrosis in idiopathic pulmonary fibrosis (IPF). B: Immunostaining revealed abundant myofibroblasts in IPF but not in COVID-19. Open arrowheads indicate smooth muscle cells; closed arrowheads indicate myofibroblasts. C: Serial sections were stained for hematoxylin and eosin (H&E) (left panel) or KRT8 (right panel). Transitional cells (black and white arrowheads) overlie fibroblastic focus (black and white asterisks). n = 3. Scale bars = 50 μm. α-SMA, α-smooth muscle actin.
Non–COVID-19 ARDS Characterized by Accumulation of Transitional Cells without Fibrosis
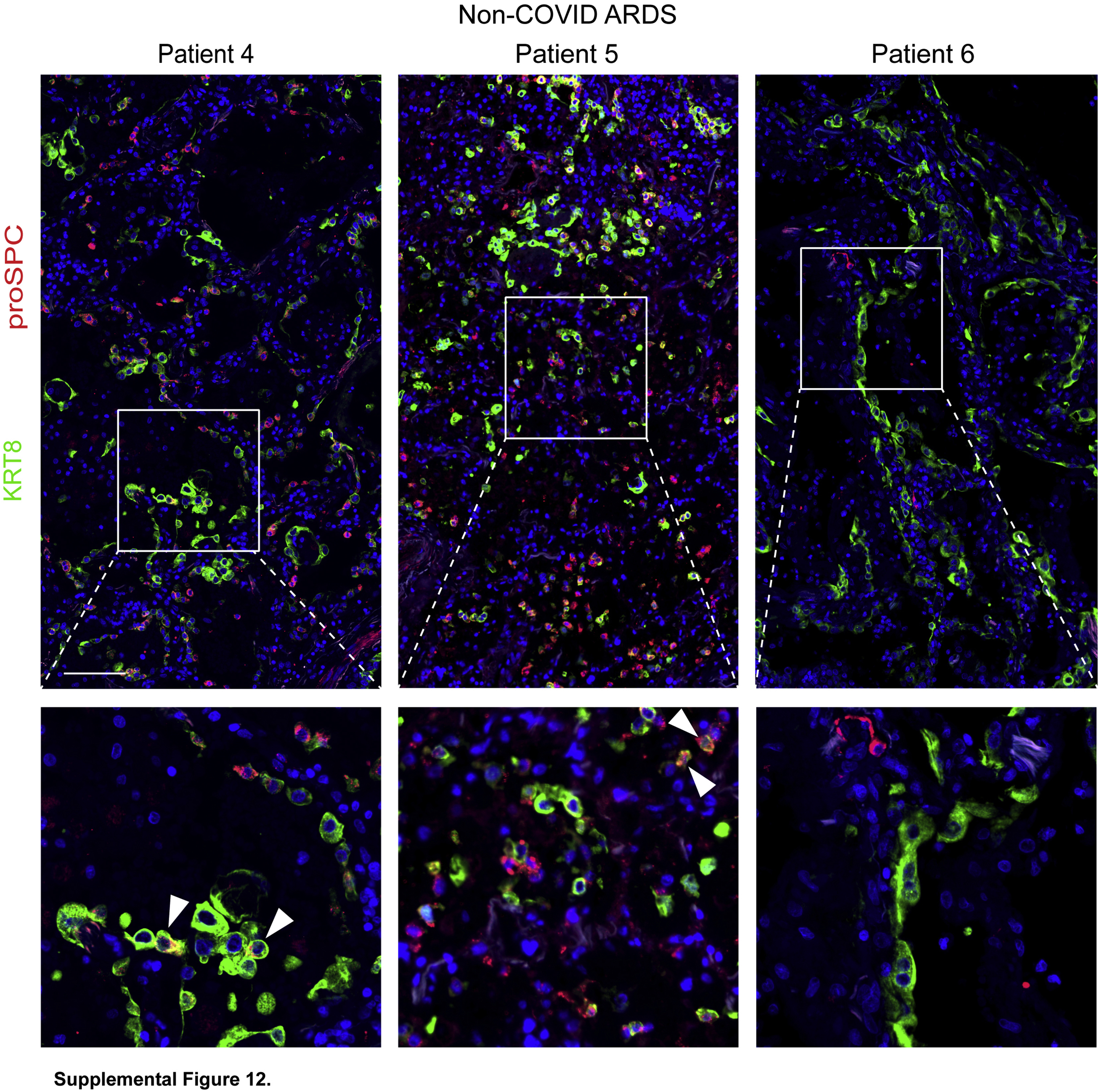
Although initial studies suggested high mortality rates for COVID-19 ARDS,1 recent evidence indicates that clinical outcomes of COVID-19 ARDS may be similar to those of ARDS from other etiologies.47 Because the histology of COVID-19 ARDS is identical to that of ARDS of other etiologies [classic DAD (Figure 1, A-D and Supplemental Figure S8)26, 27, 28,48], we hypothesized that the state of regeneration observed here is common to fatal early ARDS regardless of etiology. To address this hypothesis, postmortem lung tissue from patients who died within 14 days of ARDS of etiologies other than COVID-19 were immunostained. These patients had acute DAD (Supplemental Figure S10) with AEC1 damage (Supplemental Figure S11A), AEC2 proliferation (Supplemental Figures S11B and S12), abundant transitional cells (Supplemental Figure S11C), and incomplete AEC1 differentiation (Supplemental Figure S11D). However, in contrast to COVID-19 ARDS lungs, which were largely devoid of mature AEC2s, mature AEC2s were typically present throughout the non–COVID-19 ARDS lungs (Supplemental Figures S11B and S12). In addition, occasional cells were proSPC+ KRT8hi (Supplemental Figure S12). The non–COVID-19 ARDS lungs were without fibrosis (Supplemental Figure S13).
Transitional Cells Are Senescent in IPF but Not in ARDS
It is unknown why in mouse models of physiological alveolar regeneration and early human ARDS, transitional cells appear capable of differentiating into AEC1s with restoration of normal alveolar architecture, whereas in IPF, transitional cells persist with a paucity of AEC1s, and fibrosis ensues. To confirm that the transcriptome of transitional cells is highly conserved across diverse mouse models of physiological alveolar regeneration, human ARDS, and human IPF, expression of transitional state markers was assessed by immunostaining and interrogation of existing scRNAseq data sets. Transitional cells in the LPS and pneumonectomy mouse models of physiological alveolar regeneration and human IPF shared the unique gene expression signature of the transitional state (Figure 4A). The fact that the transcriptome of the transitional state was highly conserved across two mouse models of physiological alveolar regeneration and IPF underscores the enigma of the vastly divergent pathologic outcomes. During physiological regeneration, and likely in early human ARDS (Figure 2C and Supplemental Figure S7), transitional cells emerge as proliferating AEC2s exit the cell cycle and express markers of cell cycle arrest but ultimately differentiate into AEC1s,18,21 whereas AECs in IPF have previously been identified as senescent.30, 31, 32, 33, 34, 35,40 We therefore speculated that the critical difference underlying the divergent outcomes between physiological regeneration, early human ARDS, and fibrosis may be that while in physiological regeneration and early human ARDS, transitional cells assume a transient state of cell cycle arrest as proliferating AEC2s exit the cell cycle in anticipation of terminal differentiation, over time in the context of ongoing or repetitive injury, they evolve into a permanent state of cell cycle arrest, or senescence, and fibrosis ensues.
Figure 4.

Transitional cells in mouse models of physiological regeneration, coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome, and idiopathic pulmonary fibrosis (IPF) exist in a state of cell cycle exit but are senescent only in IPF. A, B, and D: Single-cell RNA sequencing data sets from two mouse models of physiological regeneration, lipopolysaccharide (LPS)18 and pneumonectomy (PNX),21 and human IPF30,31 were interrogated. C and E: Lung sections were immunostained. A: As type 2 alveolar epithelial cells (AEC2s) assume the transitional state, they down-regulate AEC2 markers and up-regulate transitional markers that are conserved in mouse models of physiological regeneration and human IPF. Type 1 AECs (AEC1s) express low levels of transitional markers and high levels of AEC1 markers. In mouse models of physiological regeneration, COVID-19 acute respiratory distress syndrome, and IPF, transitional cells express general markers of cell cycle exit (B and C), but only in IPF do they express CDKN2A/p16, a highly specific marker of senescence (D and E). C and E: Asterisks indicate fibroblastic foci. n = 3. Scale bars = 50 μm. CCND1, cyclin D1; CDKN1A, cyclin-dependent kinase inhibitor 1A; CDKN2A, cyclin-dependent kinase inhibitor 2A; CDKN2B, cyclin-dependent kinase inhibitor 2B; HOPX, HOP homeobox; KRT8, keratin 8; LAMP3, lysosome-associated membrane glycoprotein 3; PDPN, podoplanin; SFN, stratifin; TGFB1, transforming growth factor-beta 1; TP53, tumor protein P53.
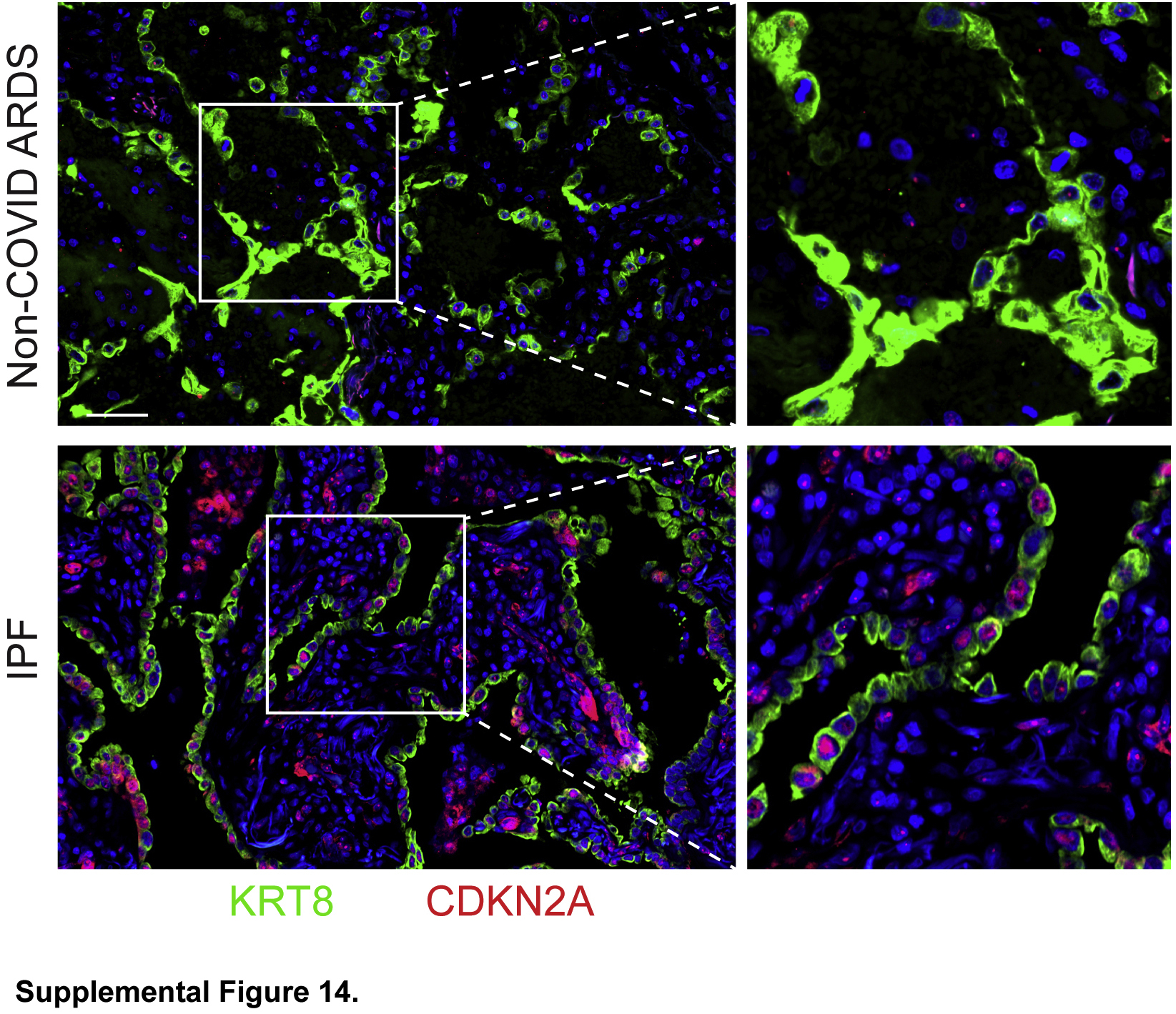
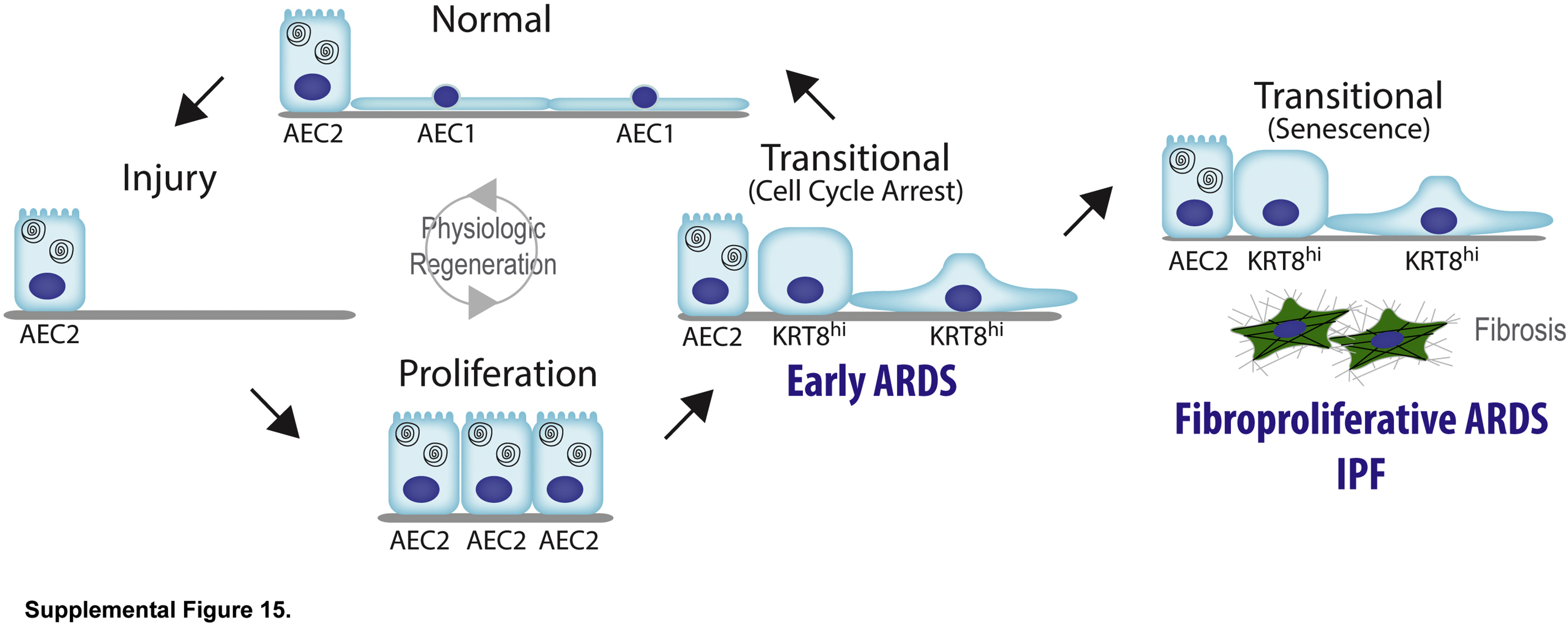
To confirm that transitional cells in fibrosis but not mouse models or human ARDS are senescent, the expression of general markers of cell cycle arrest, p21 (CDKN1A), p15 (CDKN2B), p53 (TP53), and cyclin D1 (CCND1), and a more specific marker of senescence, p16 (CDKN2A),49 were confirmed in mouse models, early human ARDS, and human IPF. General markers of cell cycle arrest are expressed in all three conditions (Figure 4, B and C), but p16 is expressed only in IPF (Figure 4, D and E and Supplemental Figure S14). These findings were conserved across the patients studied. Notably, both general markers of cell cycle arrest and p16 are expressed in transitional cells in the fibrosis associated with fibroproliferative ARDS.40 Taken together, these data suggest that in physiological regeneration in mice and early human ARDS, proliferating AEC2s undergo cell cycle exit and adopt the transitional state transiently before differentiating into AEC1s to restore normal alveolar architecture. However, in fibrosis due to IPF or fibroproliferative ARDS, transitional AECs eventually become permanently arrested, or senescent, losing capacity for an AEC1 fate and begetting fibrosis (Supplemental Figure S15).
Discussion
Herein, we performed a comprehensive examination of epithelial injury and regeneration in early, fatal ARDS due to COVID-19 and other etiologies, with comparison versus IPF. The early ARDS lungs were characterized by extensive epithelial damage and a regenerative response in which AEC2s (or other progenitors) proliferated and assumed the transitional state. Residual mature AEC2s were rare in the COVID-19 lungs but not in the non–COVID-19 lungs. Some transitional cells were hyperplastic and cuboidal. However, most displayed a partially spread morphology, filling gaps on alveolar septa denuded of AEC1s, suggesting they were in the process of AEC1 differentiation. The transitional cells occasionally assumed a flat AEC1 morphology and rarely expressed AEC1 markers, suggesting that AEC1 differentiation was ongoing but incomplete. Rare transitional cells appeared to have sloughed off into the airspaces. Similar to mouse models of physiological regeneration and in contrast to IPF and fibroproliferative ARDS, transitional cells existed on structurally normal alveolar septa without fibrosis and expressed markers of cell cycle exit but not senescence. Taken together, these data suggest that transitional cells arise in early human ARDS without fibrosis and that AEC1 differentiation from the transitional state with restoration of normal alveolar architecture and barrier integrity is ongoing but incomplete in early fatal ARDS, whereas impaired AEC1 differentiation and ensuing fibrosis are irreversible in fibroproliferative ARDS and IPF.
In ARDS, injury to the alveolar epithelial barrier results in noncardiogenic pulmonary edema, which causes severe hypoxemia frequently necessitating mechanical ventilation and leading to death.6 Conversely, epithelial repair is associated with clinical recovery and survival.10,11 However, whether poor clinical outcomes in ARDS may be linked to ineffectual epithelial regeneration is unknown. This study revealed that progenitors proliferate and assume the transitional state but that incomplete AEC1 differentiation from the transitional state likely explains the persistent barrier permeability, ongoing pulmonary edema, ventilator dependence, and death in these patients.
Future research should explore whether specific factors actively delay AEC1 differentiation from the transitional state. An intriguing possibility would be that infection of transitional cells by SARS-CoV-2 maintains cells in the transitional state. Viral nucleocapsid protein or viral inclusions were not detected. Although negative results are difficult to interpret, the absence of active viral infection would be consistent with prior work showing that patients with COVID-19 rarely have live virus after day 8 of infection despite amplification of viral RNA by PCR.29,40,50,51 Moreover, it is unlikely that transitional cells become infected by SARS-CoV-2 because they do not express its receptor, angiotensin-converting enzyme 2 (Supplemental Figure S9).20,22 In fact, given the dramatic paucity of mature AEC2s in COVID-19 ARDS (Figure 1G and Supplemental Figure S2), lack of persistent SARS-CoV-2 infection may be due to depletion of the AEC2 reservoir. Persistence of the transitional state, also observed in non–COVID-19 ARDS, is likely due to signals from within the transitional cells or from the injured lung milieu. Previous work suggested that transforming growth factor-β maintains the transitional state18; other transcriptional pathways differentially activated in the transitional and AEC1 states such as Sox418, 19, 20, 21 merit further investigation. In addition, unchecked inflammation, which is known to maintain the transitional state24 and contribute to poor clinical outcomes in ARDS,52 may delay AEC1 differentiation. The nongradual nature of differentiation suggests that activation of some process is necessary to induce transitional cells to differentiate into AEC1s. Elucidation of the cellular and molecular mechanisms regulating AEC1 differentiation may ultimately lead to novel therapies to accelerate AEC1 differentiation, barrier restitution, liberation from the ventilator, and survival in ARDS.
Transitional cells had previously been identified in humans only in the setting of fibrosis, whether in the context of IPF or fibroproliferative ARDS.19,20,22,24,31,40,41 In fact, it has been proposed that persistence of the transitional state with impaired AEC1 differentiation is the specific regenerative defect driving the pathogenesis of fibrosis and that the presence of transitional cells on lung biopsy specimens may serve as a biomarker of fibrosis in prolonged ARDS, indicating to the clinician that lung transplant may be the only viable therapeutic option.19, 20, 21, 22,40 However, it was unknown whether transitional cells are inevitably associated with fibrosis in humans. During physiological regeneration in mice, proliferating AEC2s exit the cell cycle and transiently assume the transitional state before differentiating into AEC1s to restore normal alveolar architecture and function without fibrosis.18,21 Here, we show for the first time that transitional cells arise in early human ARDS without fibrosis. Although AEC1 differentiation from the transitional state was incomplete in early ARDS, the appearance of cuboidal, partially spread, and flat transitional cells organized in a monolayer on structurally normal alveolar septa without fibrosis suggests that these cells may retain the capacity for physiological AEC1 differentiation with restoration of normal alveolar architecture and barrier integrity, as occurs in mouse models of physiological regeneration.18,21 This finding provides a pathophysiological rationale that clinical recovery may be possible and justifies ongoing aggressive supportive care, including extracorporeal membrane oxygenation, to allow time for complete regeneration. Of course, had the patients studied here survived and experienced additional insults to the epithelium from prolonged mechanical ventilation and hospital-acquired pneumonias, the transitional cells may have eventually lost the capacity for AEC1 differentiation and become senescent with ensuing fibrosis. However, the novel finding of the current study is that transitional cells arise early during regeneration in ARDS in the absence of fibrosis, at which time they appear to retain capacity for AEC1 differentiation and restoration of normal lung structure and function.
By comparing and contrasting the transitional state in mouse models of physiological regeneration, early human ARDS, and human IPF, our study provides insight into the fundamental question of why injury sometimes resolves with physiological regeneration and other times leads to fibrosis. We confirmed that the transcriptomes of the transitional state in two mouse models of physiological regeneration and human IPF are highly conserved. Taken together, these findings raise the pivotal question of why transitional cells may maintain the capacity to differentiate into AEC1s and restore normal alveolar structure in mouse models of physiological regeneration and early human ARDS but persist in the transitional state, leading to fibrosis, in IPF and fibroproliferative ARDS. We discovered that although transitional cells in physiological regeneration in mice, early human ARDS, and IPF all express markers of cell cycle exit, only in IPF do they express a specific marker of senescence. We therefore propose the novel paradigm that in mouse models of physiological regeneration and early human ARDS, proliferating AEC2s exit the cell cycle and transiently adopt the transitional state but retain the capacity to differentiate into AEC1s, restoring normal alveolar architecture without fibrosis; in IPF and fibroproliferative ARDS, however, transitional AECs evolve into a permanent state of cell cycle arrest, or senescence, losing capacity for an AEC1 fate and promoting fibrosis (Supplemental Figure S15).
Additional investigation will be necessary to confirm this hypothesis and to determine whether senescent transitional cells actually cause fibrosis and the underlying mechanisms. Because transitional cells lie in close spatial proximity to fibroblasts in the fibroblastic foci (Figure 4C)22,30 and express profibrotic genes, including transforming growth factor-β (Figure 4A),18,30,31 we further speculate that transitional cells may promote fibrosis by directly stimulating fibroblasts to become myofibroblasts and deposit matrix. If evolution from a transient state of cell cycle exit into a permanent state of senescence proves to be the critical switch that irreversibly diverts physiological regeneration toward fibrosis, elucidation of the mechanisms by which transitional cells become senescent and by which senescent transitional cells activate fibroblasts may lead to the development of novel therapies to promote physiological regeneration and prevent fibrosis in IPF and fibroproliferative ARDS.
This comprehensive immunostaining of DAD lungs for AEC2, transitional cell, and AEC1 markers yielded some unanticipated discoveries that are clinically relevant and may shift paradigms in our basic understanding of the pathology of DAD. We found that both sloughed epithelial cells long believed to be AEC1s and some hyperplastic epithelial cells long believed to be AEC2s are actually transitional cells. These discoveries not only suggest a longstanding misidentification of epithelial cell states in DAD but provide additional insight into clinical outcomes. The dramatic paucity of mature, surfactant-producing AEC2s in COVID-19, whether due to cytopathic viral infection or acquisition of the SPC– transitional state, likely contributes to surfactant deficiency. Surfactant deficiency causes atelectasis, which exacerbates hypoxemia and poor lung compliance and leads to compensatory alveolar overdistension and ventilator-induced lung injury.6 The dramatic down-regulation of surfactant shown here may be one reason why corticosteroids, which stimulate surfactant production,53 have clinical efficacy in COVID-19.54 The fact that many desquamated and hyperplastic epithelial cells long believed to be AEC1s and AEC2s, respectively, are actually transitional cells suggests that the histologic entity known as “acute DAD” represents a significantly more advanced state of regeneration than previously recognized. The acute injury phase of DAD probably occurs within the first few hours to days of clinically manifest ARDS and is unlikely to be captured on autopsy specimens.
Our study has several limitations. First, only six patients with ARDS were studied. However, the findings were highly uniform across patients with ARDS of various etiologies, suggesting generalizability. Because ARDS will remain a prevalent and devastating cause of morbidity and mortality long beyond the COVID-19 pandemic,6 the findings presented here will have enduring impact. Findings were also highly conserved across the patients with IPF studied. Second, all patients with COVID-19 ARDS died after mechanical ventilation; however, the histology of the patients with non–COVID-19 ARDS who were not ventilated was identical. Third, although a failure of barrier restitution due to incomplete AEC1 differentiation likely contributes to poor clinical outcomes, causality has not been established. The patients studied here died of refractory acute respiratory failure due to noncardiogenic pulmonary edema, and the histologic appearance of transitional cells (Figure 2, Supplemental Figures S4 and S7) certainly does not seem compatible with barrier integrity. Fourth, as mentioned, we have not excluded the possibility that some transitional cells arise from progenitors other than AEC2s,15, 16, 17 particularly in COVID-19 ARDS given the paucity of AEC2s. Fifth, although we speculate that transitional cells retain the ability to differentiate into AEC1s in early human ARDS and lose the capacity for AEC1 differentiation in fibroproliferative ARDS and IPF, the potential fates of these cells are unknown. Without the ability to acquire serial tissue samples, intervene, and perform lineage tracing, it is difficult to definitively establish causality and cell fate in observational human studies. Interpretation of cell fate dynamics remains speculative. Moreover, although p16 is considered specific for senescence, distinguishing senescence from transient cell cycle arrest is not straightforward.49 Finally, the notion that evolution from a transient state of cell cycle arrest to a truly senescent state is the critical defect underlying the irreversible switch from physiological regeneration toward fibrosis has not been proven.
In conclusion, the findings presented here suggest that in early fatal human ARDS, physiological AEC1 differentiation from transitional cells is incomplete, thus underlying prolonged barrier permeability and respiratory failure but, as in physiological regeneration in mice, is ongoing without fibrosis. These findings establish a foundation for future mechanistic studies to dissect the molecular mechanisms by which transitional cells can differentiate into AEC1s during physiological regeneration and early ARDS and may lose capacity for an AEC1 fate and/or promote fibrogenesis in fibroproliferative ARDS and fibrosis. Such mechanistic studies may ultimately lead to novel therapies to promote AEC1 differentiation, thus accelerating restoration of barrier integrity, clearance of edema fluid, liberation from the ventilator, and survival in severe ARDS and prevent fibrosis in fibroproliferative ARDS and IPF.
Acknowledgments
We thank Rachel Dyal for acquisition of tissue and Bethany Moore, Marc Peters-Golden, Elizabeth Redente, Michael Matthay, Michael Beers, and Guy Zimmerman for thoughtful discussions.
Footnotes
See related Commentary on page 406
Supported by NIHR01HL131608 and R01HL147920 (R.L.Z.), R01HL1127203 (S.K.H.), and T32HL007749 (F.W.).
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2021.11.014.
Author Contributions
R.L.Z. conceived and designed the study; C.T., M.A., N.V., S.K.H., K.A.R., F.W., and C.F. acquired and analyzed data; C.T., C.F., and R.L.Z. interpreted data; C.T. and R.L.Z. drafted or revised the manuscript; all authors approved the final manuscript. R.L.Z. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Supplemental Data
Progression of bilateral infiltrates consistent with pulmonary edema in a patient with coronavirus disease 2019. Chest radiographs from a patient (patient #1) with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome taken on the indicated day of hospitalization. The patient died on hospital day 9.
{kind=link}
Massive loss of mature type 2 alveolar epithelial cells (AEC2s) observed in all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Immunostaining for the AEC2 marker pro–surfactant protein C (proSPC) revealed vast areas devoid of proSPC+ AEC2s with rare areas of proSPC+ AEC2s remaining. n = 3. Scale bar = 100 μm.
{kind=link}
Type 2 alveolar epithelial cell (AEC2) hyperplasia is rare in coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Immunostaining for the AEC2 marker pro–surfactant protein C (proSPC) revealed rare areas of hyperplastic mature AEC2s. n = 3. Scale bar = 50 μm.
{kind=link}
KRT8hi transitional cells were abundant throughout lungs of all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Immunostaining for KRT8 revealed KRT8hi transitional cells that were cuboidal and isolated (open yellow arrowheads), occurring in pairs (closed yellow arrowhead) or hyperplastic, suggesting recent cell division, partially spread (open white arrowheads), or, in rare cases, flat (closed white arrowheads). Occasional KRT8hi transitional cells were hypertrophic and bizarrely shaped (closed orange arrowheads). Some images also shown in Figure 2. n = 3. Scale bar = 200 μm.
{kind=link}
Normal type 1 alveolar epithelial cells (AECs) and type 2 AECs express much lower levels of KRT8 than transitional cells. Lung sections were stained for KRT8. Expression of KRT8 in normal type 1 AECs (open arrowheads) and type 2 AECs (closed arrowheads) was observed with very high exposure times. Equal exposure times were used for both panels, which allowed low levels of K8 expression in normal lungs to be detected but resulted in oversaturation in transitional cells in coronavirus disease 2019 (COVID-19) lungs due to high levels of K8 expression. n = 3. Scale bar = 50 μm.
{kind=link}
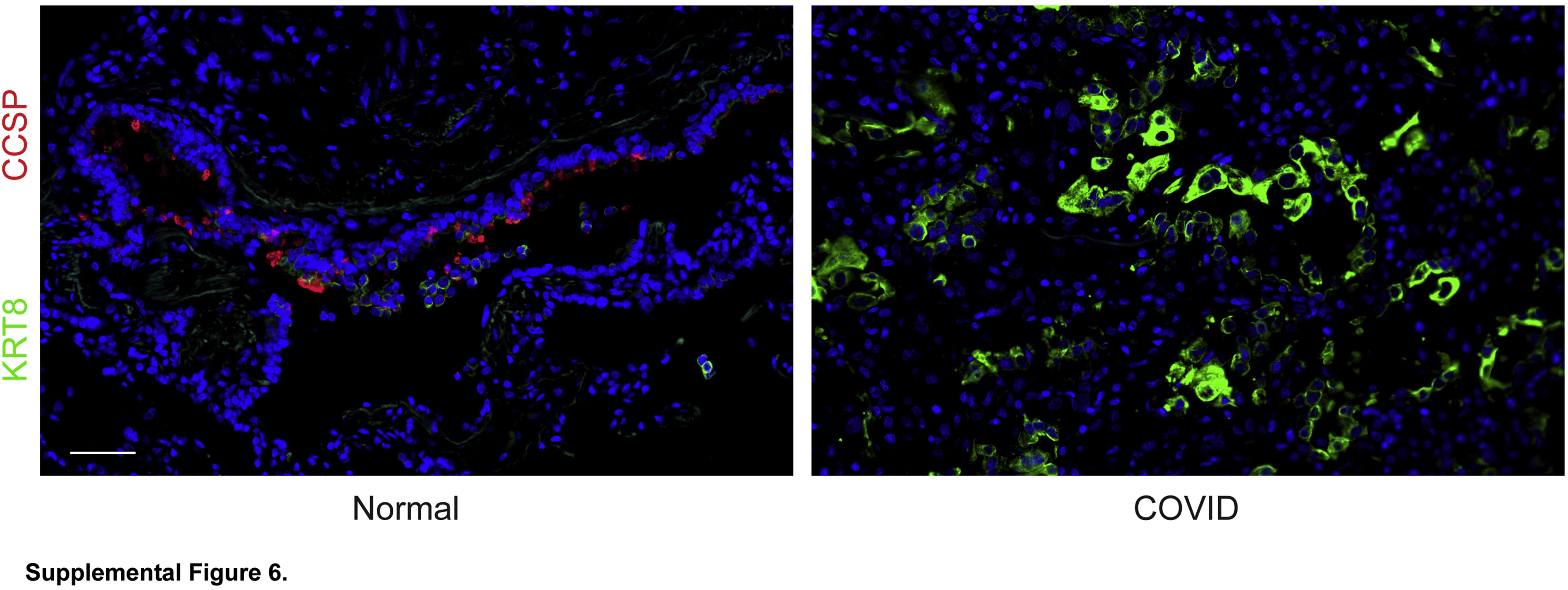
KRT8hi transitional cells do not express club cell secretory protein (CCSP), a club cell marker. Club cells in normal airways express CCSP. Transitional cells in coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome do not express CCSP. n = 3. Scale bar = 50 μm.
{kind=link}
Abundance of KRT8hi transitional cells with incomplete differentiation into mature type 1 alveolar epithelial cells (AEC1s) observed in all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. KRT8hi transitional cells filled gaps denuded of AEC1s, were cuboidal, partially spread, or approaching a flat AEC1 morphology but typically did not express AEC1 markers. These findings were believed to represent ongoing organized, physiological, albeit incomplete, AEC1 regeneration. Although all morphologies were present in all patients, Patient #3 tended to have more bizarrely shaped cells. Some images also shown in Figure 2. n = 3. Scale bars = 100 μm.
{kind=link}
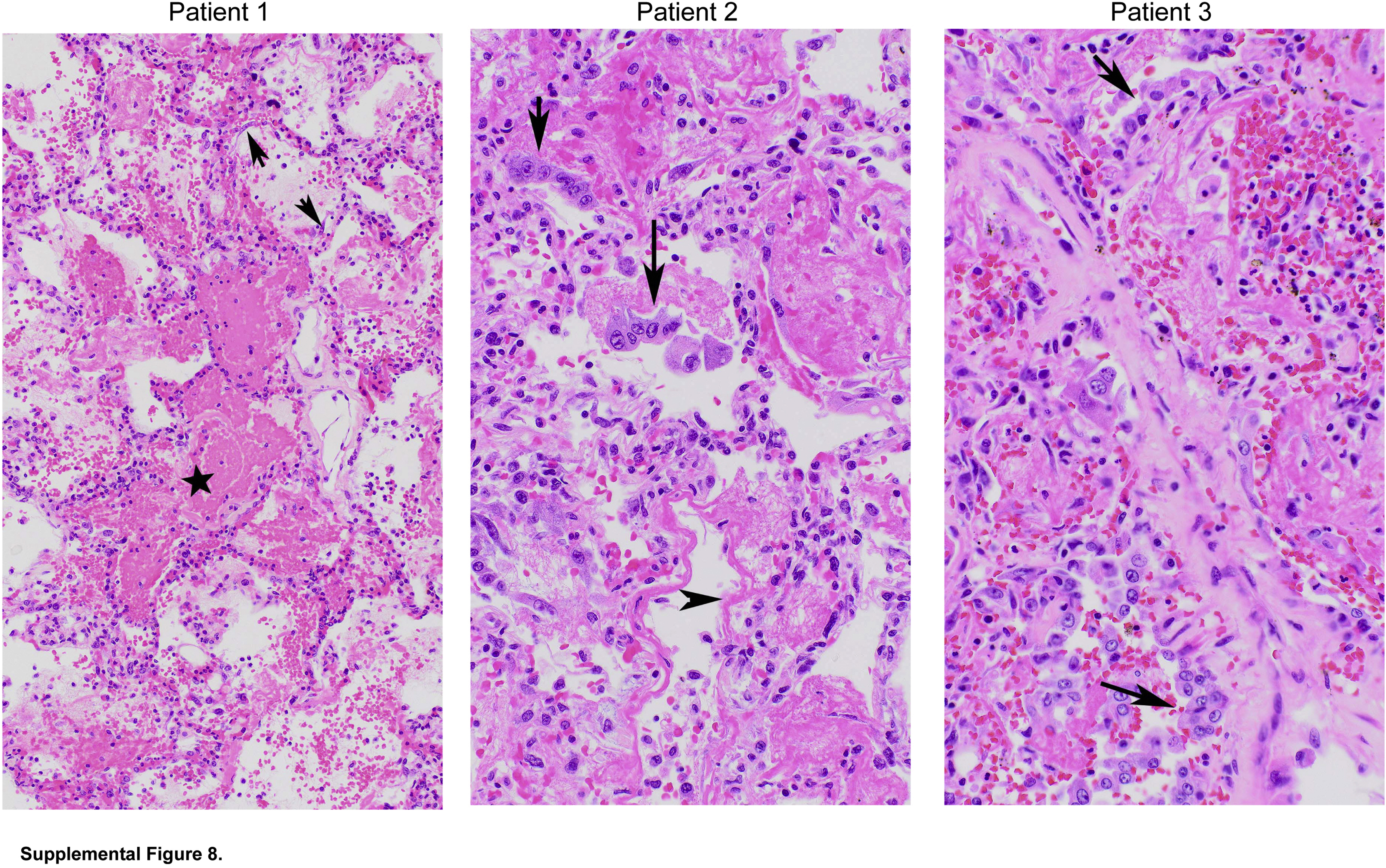
Histology of all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome is acute diffuse alveolar damage. Lung sections were stained with hematoxylin and eosin. Patient 1: Early acute diffuse alveolar damage with intra-alveolar fibrin (star) and degenerating alveolar epithelial cells (AECs) (arrows) within alveolar space (magnification ×100). Patient 2: Acute diffuse alveolar damage with AEC hyperplasia (arrows) and hyaline membranes (arrowhead) (magnification ×200). Patient 3: Acute diffuse alveolar damage with AEC hyperplasia (arrows) (magnification ×200). Some images also shown in Figure 1.
{kind=link}
Transitional cells and type 1 alveolar epithelial cells (AECs) express lower levels of the severe acute respiratory syndrome coronavirus 2 receptor angiotensin-converting enzyme 2 (ACE2). Single-cell RNA sequencing data sets from two mouse models of physiological regeneration, lipopolysaccharide (LPS)20 and pneumonectomy (PNX),23 and normal and idiopathic pulmonary fibrosis (IPF) human lung31,32 were interrogated for expression of the severe acute respiratory syndrome coronavirus 2 receptor ACE2. Transitional cells and type 1 AECs express much lower levels of ACE2 than type 2 AECs.
{kind=link}
Histology of all patients with non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome is acute diffuse alveolar damage. Lung sections were stained with hematoxylin and eosin. Scale bar = 100 μm.
{kind=link}
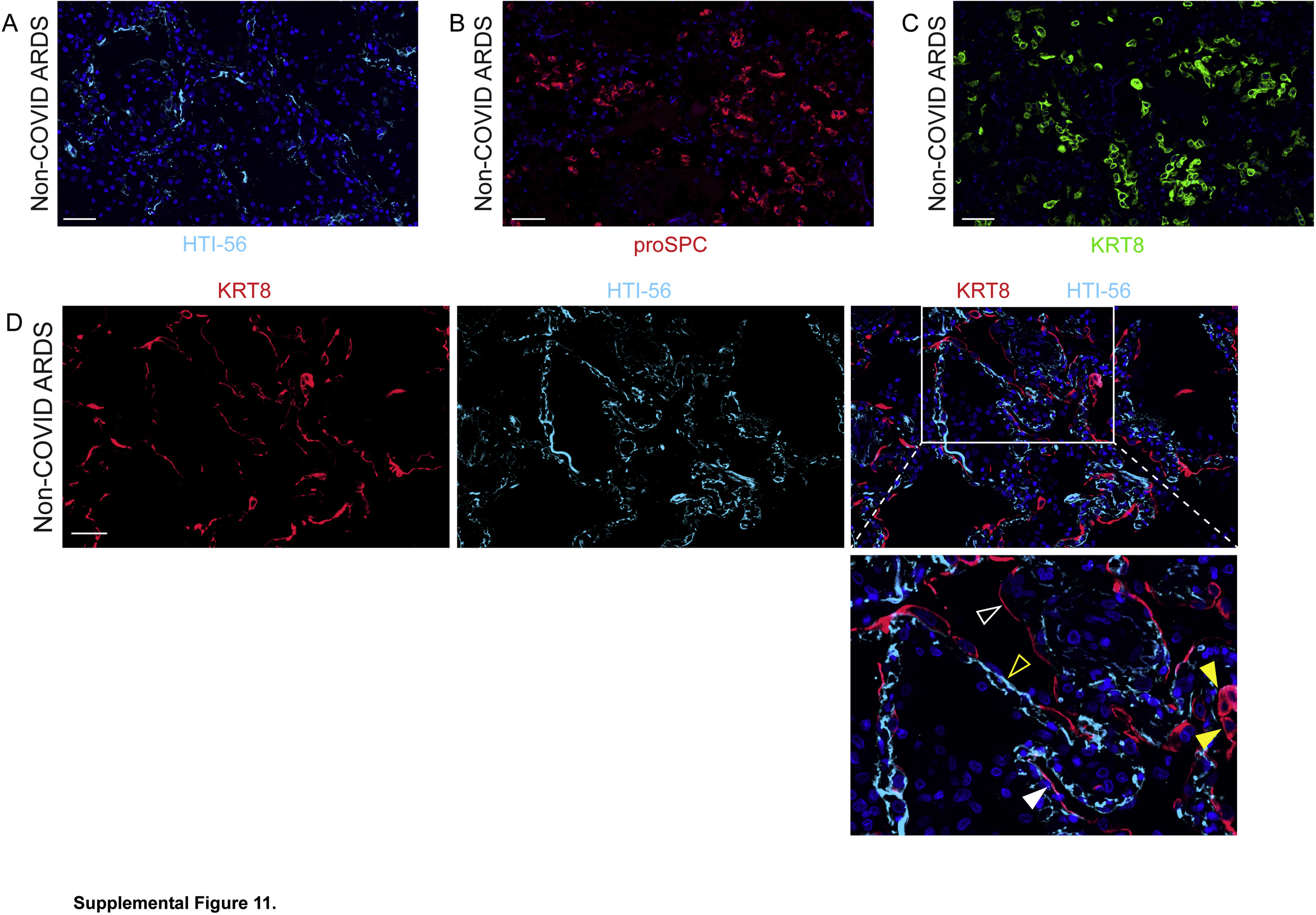
Type 1 alveolar epithelial cell (AEC1) injury, type 2 AEC proliferation, abundant transitional cells, and ongoing but incomplete AEC1 differentiation in non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Lung sections were stained for HTI-56, KRT8, and pro–surfactant protein C. A: AEC1 loss. B: Type 2 AEC hyperplasia. C: Abundance of transitional cells. D: KRT8hi transitional cells fill gaps denuded of AEC1s on alveolar septa. Some are cuboidal (closed yellow arrowheads); some are partially spread, some approach a flat AEC1 morphology but do not express AEC1 markers (open white arrowhead), and rare cells express AEC1 markers (closed white arrowhead). Flat cells that express AEC1 markers but not KRT8 (open yellow arrowhead) were interpreted as native AEC1s that were not damaged during lung injury. Findings suggest ongoing organized, albeit incomplete, AEC1 differentiation. n = 3. Scale bars = 50 μm.
{kind=link}
Type 2 alveolar epithelial cells and transitional cells in all patients with non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Lung sections were stained for KRT8 and pro–surfactant protein C (proSPC). All patients exhibited abundant transitional cells and type 2 alveolar epithelial cells. Occasional pro–surfactant protein C+ KRT8hi cells were observed in non–COVID-19 acute respiratory distress syndrome lungs (arrowheads). n = 3. Scale bar = 100 μm.
{kind=link}
Absence of fibrosis in early non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome (ARDS). A: Trichrome highlighted (in blue) basement membranes and vascular adventitia in normal and non–COVID-19 ARDS lungs and collagen deposition with marked fibrosis in idiopathic pulmonary fibrosis (IPF). B: Immunostaining demonstrated abundant myofibroblasts in idiopathic pulmonary fibrosis (asterisk) but not non–COVID-19 ARDS. n = 3. Scale bars: 200 μm (A); 50 μm (B).
{kind=link}
Transitional cells in idiopathic pulmonary fibrosis (IPF) but not non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome are senescent. Lung sections were immunostained. KRT8hi transitional cells in IPF but not non–COVID-19 acute respiratory distress syndrome express CDKN2A/p16, a highly specific marker of senescence. n = 3. Scale bar = 50 μm.
{kind=link}
Proposed lineage trajectories in physiological alveolar regeneration, early acute respiratory distress syndrome (ARDS), and fibrosis. During physiological regeneration after lung injury resulting in alveolar epithelial cell (AEC) death, surviving type 2 AECs (AEC2s) proliferate, then exit the cell cycle and transiently adopt the transitional state but ultimately differentiate into mature type 1 AECs. In early ARDS, after AEC death, surviving AEC2s proliferate, then exit the cell cycle and adopt the transitional state, and are in the process of organized type 1 AEC differentiation; however, incomplete differentiation is associated with ongoing barrier permeability, pulmonary edema, ventilator dependence, and mortality. In fibroproliferative ARDS and idiopathic pulmonary fibrosis (IPF), after AEC death, surviving AEC2s proliferate, then exit the cell cycle and adopt the transitional state but evolve into a permanent state of cell cycle arrest, or senescence, and fibrosis ensues. Adapted with permission of the American Thoracic Society. Copyright © 2021 American Thoracic Society. All rights reserved. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, Zemans RL: Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med 2020, 201:1443–1447. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society. Readers are encouraged to read the entire article for the correct context at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7258651. The authors, editors, and The American Thoracic Society are not responsible for errors or omissions in adaptations.
{kind=link}
References
- 1.Richardson S., Hirsch J.S., Narasimhan M., Crawford J.M., McGinn T., Davidson K.W., the Northwell COVID-19 Research Consortium. Barnaby D.P., Becker L.B., Chelico J.D., Cohen S.L., Cookingham J., Coppa K., Diefenbach M.A., Dominello A.J., Duer-Hefele J., Falzon L., Gitlin J., Hajizadeh N., Harvin T.G., Hirschwerk D.A., Kim E.J., Kozel Z.M., Marrast L.M., Mogavero J.N., Osorio G.A., Qiu M., Zanos T.P. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area [Erratum appeared in JAMA 2020, 323:2098] JAMA. 2020;323:2052–2059. doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karagiannidis C., Mostert C., Hentschker C., Voshaar T., Malzahn J., Schillinger G., Klauber J., Janssens U., Marx G., Weber-Carstens S., Kluge S., Pfeifer M., Grabenhenrich L., Welte T., Busse R. Case characteristics, resource use, and outcomes of 10 021 patients with COVID-19 admitted to 920 German hospitals: an observational study. Lancet Respir Med. 2020;8:853–862. doi: 10.1016/S2213-2600(20)30316-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gamberini L., Tonetti T., Spadaro S., Zani G., Mazzoli C.A., Capozzi C., Giampalma E., Bacchi Reggiani M.L., Bertellini E., Castelli A., Cavalli I., Colombo D., Crimaldi F., Damiani F., Fogagnolo A., Fusari M., Gamberini E., Gordini G., Laici C., Lanza M.C., Leo M., Marudi A., Nardi G., Ottaviani I., Papa R., Potalivo A., Russo E., Taddei S., Volta C.A., Ranieri V.M., ICU-RER COVID-19 Collaboration Factors influencing liberation from mechanical ventilation in coronavirus disease 2019: multicenter observational study in fifteen Italian ICUs. J Intensive Care. 2020;8:80. doi: 10.1186/s40560-020-00499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grasselli G., Zangrillo A., Zanella A., Antonelli M., Cabrini L., Castelli A., Cereda D., Coluccello A., Foti G., Fumagalli R., Iotti G., Latronico N., Lorini L., Merler S., Natalini G., Piatti A., Ranieri M.V., Scandroglio A.M., Storti E., Cecconi M., Pesenti A., COVID-19 Lombardy ICU Network Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. JAMA. 2020;323:1574–1581. doi: 10.1001/jama.2020.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auld S.C., Caridi-Scheible M., Blum J.M., Robichaux C., Kraft C., Jacob J.T., Jabaley C.S., Carpenter D., Kaplow R., Hernandez-Romieu A.C., Adelman M.W., Martin G.S., Coopersmith C.M., Murphy D.J., the Emory COVID-19 Quality and Clinical Research Collaborative ICU and ventilator mortality among critically ill adults with coronavirus disease 2019. Crit Care Med. 2020;48:e799–e804. doi: 10.1097/CCM.0000000000004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matthay M.A., Zemans R.L., Zimmerman G.A., Arabi Y.M., Beitler J.R., Mercat A., Herridge M., Randolph A.G., Calfee C.S. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18. doi: 10.1038/s41572-019-0069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobbs L.G., Johnson M.D., Vanderbilt J., Allen L., Gonzalez R. The great big alveolar TI cell: evolving concepts and paradigms. Cell Physiol Biochem. 2010;25:55–62. doi: 10.1159/000272063. [DOI] [PubMed] [Google Scholar]
- 8.Jansing N.L., McClendon J., Henson P.M., Tuder R.M., Hyde D.M., Zemans R.L. Unbiased quantitation of alveolar type II to alveolar type I cell transdifferentiation during repair after lung injury in mice. Am J Respir Cell Mol Biol. 2017;57:519–526. doi: 10.1165/rcmb.2017-0037MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barkauskas C.E., Cronce M.J., Rackley C.R., Bowie E.J., Keene D.R., Stripp B.R., Randell S.H., Noble P.W., Hogan B.L.M. Type 2 alveolar cells are stem cells in adult lung. J Clin Invest. 2013;123:3025–3036. doi: 10.1172/JCI68782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthay M.A., Wiener-Kronish J.P. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis. 1990;142(Pt 2):1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- 11.Ware L.B., Matthay M.A. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 12.Evans M.J., Cabral L.J., Stephens R.J., Freeman G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp Mol Pathol. 1975;22:142–150. doi: 10.1016/0014-4800(75)90059-3. [DOI] [PubMed] [Google Scholar]
- 13.Nabhan A.N., Brownfield D.G., Harbury P.B., Krasnow M.A., Desai T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018;359:1118–1123. doi: 10.1126/science.aam6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zacharias W.J., Frank D.B., Zepp J.A., Morley M.P., Alkhaleel F.A., Kong J., Zhou S., Cantu E., Morrisey E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature. 2018;555:251–255. doi: 10.1038/nature25786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kathiriya J.J., Brumwell A.N., Jackson J.R., Tang X., Chapman H.A. Distinct airway epithelial stem cells hide among club cells but mobilize to promote alveolar regeneration. Cell Stem Cell. 2020;26:346–358.e4. doi: 10.1016/j.stem.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H., Matsumoto K., Brockway B.L., Rackley C.R., Liang J., Lee J.-H., Jiang D., Noble P.W., Randell S.H., Kim C.F., Stripp B.R. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells. 2012;30:1948–1960. doi: 10.1002/stem.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaughan A.E., Brumwell A.N., Xi Y., Gotts J.E., Brownfield D.G., Treutlein B., Tan K., Tan V., Liu F.C., Looney M.R., Matthay M.A., Rock J.R., Chapman H.A. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature. 2015;517:621–625. doi: 10.1038/nature14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riemondy K.A., Jansing N.L., Jiang P., Redente E.F., Gillen A.E., Fu R., Miller A.J., Spence J.R., Gerber A.N., Hesselberth J.R., Zemans R.L. Single cell RNA sequencing identifies TGF[beta] as a key regenerative cue following LPS-induced lung injury. JCI Insight. 2019;5:e123637. doi: 10.1172/jci.insight.123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi Y., Tata A., Konkimalla A., Katsura H., Lee R.F., Ou J., Banovich N.E., Kropski J.A., Tata P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol. 2020;22:934–946. doi: 10.1038/s41556-020-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strunz M., Simon L.M., Ansari M., Kathiriya J.J., Angelidis I., Mayr C.H., Tsidiridis G., Lange M., Mattner L.F., Yee M., Ogar P., Sengupta A., Kukhtevich I., Schneider R., Zhao Z., Voss C., Stoeger T., Neumann J.H.L., Hilgendorff A., Behr J., O'Reilly M., Lehmann M., Burgstaller G., Königshoff M., Chapman H.A., Theis F.J., Schiller H.B. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat Commun. 2020;11:3559. doi: 10.1038/s41467-020-17358-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu H., Yu Y., Huang H., Hu Y., Fu S., Wang Z., Shi M., Zhao X., Yuan J., Li J., Yang X., Bin E., Wei D., Zhang H., Zhang J., Yang C., Cai T., Dai H., Chen J., Tang N. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells. Cell. 2020;180:107–121.e17. doi: 10.1016/j.cell.2019.11.027. [DOI] [PubMed] [Google Scholar]
- 22.Jiang P., Gil de Rubio R., Hrycaj S.M., Gurczynski S.J., Riemondy K.A., Moore B.B., Omary M.B., Ridge K.M., Zemans R.L. Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med. 2020;201:1443–1447. doi: 10.1164/rccm.201909-1726LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joshi N., Watanabe S., Verma R., Jablonski R.P., Chen C.-I., Cheresh P., Markov N.S., Reyfman P.A., McQuattie-Pimentel A.C., Sichizya L., Lu Z., Piseaux-Aillon R., Kirchenbuechler D., Flozak A.S., Gottardi C.J., Cuda C.M., Perlman H., Jain M., Kamp D.W., Budinger G.R.S., Misharin A.V. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur Respir J. 2020;55:1900646. doi: 10.1183/13993003.00646-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi J., Park J.-E., Tsagkogeorga G., Yanagita M., Koo B.-K., Han N., Lee J.-H. Inflammatory signals induce AT2 cell-derived damage-associated transient progenitors that mediate alveolar regeneration. Cell Stem Cell. 2020;27:366–382.e7. doi: 10.1016/j.stem.2020.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carsana L., Sonzogni A., Nasr A., Rossi R.S., Pellegrinelli A., Zerbi P., Rech R., Colombo R., Antinori S., Corbellino M., Galli M., Catena E., Tosoni A., Gianatti A., Nebuloni M. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020;20:1135–1140. doi: 10.1016/S1473-3099(20)30434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian S., Xiong Y., Liu H., Niu L., Guo J., Liao M., Xiao S.-Y. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. 2020;33:1007–1014. doi: 10.1038/s41379-020-0536-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fox S.E., Akmatbekov A., Harbert J.L., Li G., Brown J.Q., Vander Heide R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med. 2020;8:681–686. doi: 10.1016/S2213-2600(20)30243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konopka K.E., Nguyen T., Jentzen J.M., Rayes O., Schmidt C.J., Wilson A.M., Farver C.F., Myers J.L. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 infection is morphologically indistinguishable from other causes of DAD. Histopathology. 2020;77:570–578. doi: 10.1111/his.14180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaefer I.-M., Padera R.F., Solomon I.H., Kanjilal S., Hammer M.M., Hornick J.L., Sholl L.M. In situ detection of SARS-CoV-2 in lungs and airways of patients with COVID-19. Mod Pathol. 2020;33:2104–2114. doi: 10.1038/s41379-020-0595-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams T.S., Schupp J.C., Poli S., Ayaub E.A., Neumark N., Ahangari F., Chu S.G., Raby B.A., DeIuliis G., Januszyk M., Duan Q., Arnett H.A., Siddiqui A., Washko G.R., Homer R., Yan X., Rosas I.O., Kaminski N. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6:eaba1983. doi: 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Habermann A.C., Gutierrez A.J., Bui L.T., Yahn S.L., Winters N.I., Calvi C.L., Peter L., Chung M.-I., Taylor C.J., Jetter C., Raju L., Roberson J., Ding G., Wood L., Sucre J.M.S., Richmond B.W., Serezani A.P., McDonnell W.J., Mallal S.B., Bacchetta M.J., Loyd J.E., Shaver C.M., Ware L.B., Bremner R., Walia R., Blackwell T.S., Banovich N.E., Kropski J.A. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6:eaba1972. doi: 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schafer M.J., White T.A., Iijima K., Haak A.J., Ligresti G., Atkinson E.J., Oberg A.L., Birch J., Salmonowicz H., Zhu Y., Mazula D.L., Brooks R.W., Fuhrmann-Stroissnigg H., Pirtskhalava T., Prakash Y.S., Tchkonia T., Robbins P.D., Aubry M.C., Passos J.F., Kirkland J.L., Tschumperlin D.J., Kita H., LeBrasseur N.K. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. 2017;8:14532. doi: 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuwano K., Kunitake R., Kawasaki M., Nomoto Y., Hagimoto N., Nakanishi Y., Hara N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996;154(Pt 2):477–483. doi: 10.1164/ajrccm.154.2.8756825. [DOI] [PubMed] [Google Scholar]
- 34.Lomas N.J., Watts K.L., Akram K.M., Forsyth N.R., Spiteri M.A. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol. 2012;5:58–71. [PMC free article] [PubMed] [Google Scholar]
- 35.Chilosi M., Poletti V., Murer B., Lestani M., Cancellieri A., Montagna L., Piccoli P., Cangi G., Semenzato G., Doglioni C. Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of deltaN-p63. Lab Invest. 2002;82:1335–1345. doi: 10.1097/01.lab.0000032380.82232.67. [DOI] [PubMed] [Google Scholar]
- 36.Yao C., Guan X., Carraro G., Parimon T., Liu X., Huang G., Mulay A., Soukiasian H.J., David G., Weigt S.S., Belperio J.A., Chen P., Jiang D., Noble P.W., Stripp B.R. Senescence of alveolar type 2 cells drives progressive pulmonary fibrosis. Am J Respir Crit Care Med. 2021;203:707–717. doi: 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alder J.K., Barkauskas C.E., Limjunyawong N., Stanley S.E., Kembou F., Tuder R.M., Hogan B.L.M., Mitzner W., Armanios M. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112:5099–5104. doi: 10.1073/pnas.1504780112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minagawa S., Araya J., Numata T., Nojiri S., Hara H., Yumino Y., Kawaishi M., Odaka M., Morikawa T., Nishimura S.L., Nakayama K., Kuwano K. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-[beta]-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–L401. doi: 10.1152/ajplung.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auyeung V.C., Sheppard D. Stuck in a moment: does abnormal persistence of epithelial progenitors drive pulmonary fibrosis? Am J Respir Crit Care Med. 2021;203:667–669. doi: 10.1164/rccm.202010-3898ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bharat A., Querrey M., Markov N.S., Kim S., Kurihara C., Garza-Castillon R., Manerikar A., Shilatifard A., Tomic R., Politanska Y., Abdala-Valencia H., Yeldandi A.V., Lomasney J.W., Misharin A.V., Budinger G.R.S. Lung transplantation for patients with severe COVID-19. Sci Transl Med. 2020;12:eabe4282. doi: 10.1126/scitranslmed.abe4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J., Wu H., Yu Y., Tang N. Pulmonary alveolar regeneration in adult COVID-19 patients. Cell Res. 2020;30:708–710. doi: 10.1038/s41422-020-0369-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y., Tang Z., Huang H., Li J., Wang Z., Yu Y., Zhang C., Li J., Dai H., Wang F., Cai T., Tang N. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc Natl Acad Sci U S A. 2018;115:2407–2412. doi: 10.1073/pnas.1719474115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cardinal-Fernández P., Lorente J.A., Ballén-Barragán A., Matute-Bello G. Acute respiratory distress syndrome and diffuse alveolar damage. New insights on a complex relationship. Ann Am Thorac Soc. 2017;14:844–850. doi: 10.1513/AnnalsATS.201609-728PS. [DOI] [PubMed] [Google Scholar]
- 44.Stone K.C., Mercer R.R., Gehr P., Stockstill B., Crapo J.D. Allometric relationships of cell numbers and size in the mammalian lung. Am J Respir Cell Mol Biol. 1992;6:235–243. doi: 10.1165/ajrcmb/6.2.235. [DOI] [PubMed] [Google Scholar]
- 45.Jansing N.L., Patel N., McClendon J., Redente E.F., Henson P.M., Tuder R.M., Hyde D.M., Nyengaard J.R., Zemans R.L. Flow cytometry underestimates and planimetry overestimates alveolar epithelial type 2 cell expansion after lung injury. Am J Respir Crit Care Med. 2018;198:390–392. doi: 10.1164/rccm.201709-1838LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z., Shi L., Wang Y., Zhang J., Huang L., Zhang C., Liu S., Zhao P., Liu H., Zhu L., Tai Y., Bai C., Gao T., Song J., Xia P., Dong J., Zhao J., Wang F.-S. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sjoding M.W., Admon A.J., Saha A.K., Kay S.G., Brown C.A., Co I., Claar D., McSparron J.I., Dickson R.P. Comparing clinical features and outcomes in mechanically ventilated patients with COVID-19 and the acute respiratory distress syndrome. Ann Am Thorac Soc. 2021;18:1876–1885. doi: 10.1513/AnnalsATS.202008-1076OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borczuk A.C., Salvatore S.P., Seshan S.V., Patel S.S., Bussel J.B., Mostyka M., Elsoukkary S., He B., Del Vecchio C., Fortarezza F., Pezzuto F., Navalesi P., Crisanti A., Fowkes M.E., Bryce C.H., Calabrese F., Beasley M.B. COVID-19 pulmonary pathology: a multi-institutional autopsy cohort from Italy and New York City. Mod Pathol. 2020;33:2156–2168. doi: 10.1038/s41379-020-00661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharpless N.E., Sherr C.J. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- 50.Wölfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Müller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C., Hoelscher M., Bleicker T., Brünink S., Schneider J., Ehmann R., Zwirglmaier K., Drosten C., Wendtner C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- 51.Bullard J., Dust K., Funk D., Strong J.E., Alexander D., Garnett L., Boodman C., Bello A., Hedley A., Schiffman Z., Doan K., Bastien N., Li Y., Van Caeseele P.G., Poliquin G. Predicting infectious severe acute respiratory syndrome coronavirus 2 from diagnostic samples. Clin Infect Dis. 2020;71:2663–2666. doi: 10.1093/cid/ciaa638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grant R.A., Morales-Nebreda L., Markov N.S., Swaminathan S., Querrey M., Guzman E.R., et al. NU Script Study Investigators Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature. 2021;590:635–641. doi: 10.1038/s41586-020-03148-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robertson B. Corticosteroids and surfactant for prevention of neonatal RDS. Ann Med. 1993;25:285–288. doi: 10.3109/07853899309147876. [DOI] [PubMed] [Google Scholar]
- 54.RECOVERY Collaborative Group. Horby P., Lim W.S., Emberson J.R., Mafham M., Bell J.L., Linsell L., Staplin N., Brightling C., Ustianowski A., Elmahi E., Prudon B., Green C., Felton T., Chadwick D., Rege K., Fegan C., Chappell L.C., Faust S.N., Jaki T., Jeffery K., Montgomery A., Rowan K., Juszczak E., Baillie J.K., Haynes R., Landray M.J. Dexamethasone in hospitalized patients with Covid-19. N Engl J Med. 2020;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Progression of bilateral infiltrates consistent with pulmonary edema in a patient with coronavirus disease 2019. Chest radiographs from a patient (patient #1) with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome taken on the indicated day of hospitalization. The patient died on hospital day 9.
Massive loss of mature type 2 alveolar epithelial cells (AEC2s) observed in all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Immunostaining for the AEC2 marker pro–surfactant protein C (proSPC) revealed vast areas devoid of proSPC+ AEC2s with rare areas of proSPC+ AEC2s remaining. n = 3. Scale bar = 100 μm.
Type 2 alveolar epithelial cell (AEC2) hyperplasia is rare in coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Immunostaining for the AEC2 marker pro–surfactant protein C (proSPC) revealed rare areas of hyperplastic mature AEC2s. n = 3. Scale bar = 50 μm.
KRT8hi transitional cells were abundant throughout lungs of all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Immunostaining for KRT8 revealed KRT8hi transitional cells that were cuboidal and isolated (open yellow arrowheads), occurring in pairs (closed yellow arrowhead) or hyperplastic, suggesting recent cell division, partially spread (open white arrowheads), or, in rare cases, flat (closed white arrowheads). Occasional KRT8hi transitional cells were hypertrophic and bizarrely shaped (closed orange arrowheads). Some images also shown in Figure 2. n = 3. Scale bar = 200 μm.
Normal type 1 alveolar epithelial cells (AECs) and type 2 AECs express much lower levels of KRT8 than transitional cells. Lung sections were stained for KRT8. Expression of KRT8 in normal type 1 AECs (open arrowheads) and type 2 AECs (closed arrowheads) was observed with very high exposure times. Equal exposure times were used for both panels, which allowed low levels of K8 expression in normal lungs to be detected but resulted in oversaturation in transitional cells in coronavirus disease 2019 (COVID-19) lungs due to high levels of K8 expression. n = 3. Scale bar = 50 μm.
KRT8hi transitional cells do not express club cell secretory protein (CCSP), a club cell marker. Club cells in normal airways express CCSP. Transitional cells in coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome do not express CCSP. n = 3. Scale bar = 50 μm.
Abundance of KRT8hi transitional cells with incomplete differentiation into mature type 1 alveolar epithelial cells (AEC1s) observed in all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. KRT8hi transitional cells filled gaps denuded of AEC1s, were cuboidal, partially spread, or approaching a flat AEC1 morphology but typically did not express AEC1 markers. These findings were believed to represent ongoing organized, physiological, albeit incomplete, AEC1 regeneration. Although all morphologies were present in all patients, Patient #3 tended to have more bizarrely shaped cells. Some images also shown in Figure 2. n = 3. Scale bars = 100 μm.
Histology of all patients with coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome is acute diffuse alveolar damage. Lung sections were stained with hematoxylin and eosin. Patient 1: Early acute diffuse alveolar damage with intra-alveolar fibrin (star) and degenerating alveolar epithelial cells (AECs) (arrows) within alveolar space (magnification ×100). Patient 2: Acute diffuse alveolar damage with AEC hyperplasia (arrows) and hyaline membranes (arrowhead) (magnification ×200). Patient 3: Acute diffuse alveolar damage with AEC hyperplasia (arrows) (magnification ×200). Some images also shown in Figure 1.
Transitional cells and type 1 alveolar epithelial cells (AECs) express lower levels of the severe acute respiratory syndrome coronavirus 2 receptor angiotensin-converting enzyme 2 (ACE2). Single-cell RNA sequencing data sets from two mouse models of physiological regeneration, lipopolysaccharide (LPS)20 and pneumonectomy (PNX),23 and normal and idiopathic pulmonary fibrosis (IPF) human lung31,32 were interrogated for expression of the severe acute respiratory syndrome coronavirus 2 receptor ACE2. Transitional cells and type 1 AECs express much lower levels of ACE2 than type 2 AECs.
Histology of all patients with non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome is acute diffuse alveolar damage. Lung sections were stained with hematoxylin and eosin. Scale bar = 100 μm.
Type 1 alveolar epithelial cell (AEC1) injury, type 2 AEC proliferation, abundant transitional cells, and ongoing but incomplete AEC1 differentiation in non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Lung sections were stained for HTI-56, KRT8, and pro–surfactant protein C. A: AEC1 loss. B: Type 2 AEC hyperplasia. C: Abundance of transitional cells. D: KRT8hi transitional cells fill gaps denuded of AEC1s on alveolar septa. Some are cuboidal (closed yellow arrowheads); some are partially spread, some approach a flat AEC1 morphology but do not express AEC1 markers (open white arrowhead), and rare cells express AEC1 markers (closed white arrowhead). Flat cells that express AEC1 markers but not KRT8 (open yellow arrowhead) were interpreted as native AEC1s that were not damaged during lung injury. Findings suggest ongoing organized, albeit incomplete, AEC1 differentiation. n = 3. Scale bars = 50 μm.
Type 2 alveolar epithelial cells and transitional cells in all patients with non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome. Lung sections were stained for KRT8 and pro–surfactant protein C (proSPC). All patients exhibited abundant transitional cells and type 2 alveolar epithelial cells. Occasional pro–surfactant protein C+ KRT8hi cells were observed in non–COVID-19 acute respiratory distress syndrome lungs (arrowheads). n = 3. Scale bar = 100 μm.
Absence of fibrosis in early non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome (ARDS). A: Trichrome highlighted (in blue) basement membranes and vascular adventitia in normal and non–COVID-19 ARDS lungs and collagen deposition with marked fibrosis in idiopathic pulmonary fibrosis (IPF). B: Immunostaining demonstrated abundant myofibroblasts in idiopathic pulmonary fibrosis (asterisk) but not non–COVID-19 ARDS. n = 3. Scale bars: 200 μm (A); 50 μm (B).
Transitional cells in idiopathic pulmonary fibrosis (IPF) but not non–coronavirus disease 2019 (COVID-19) acute respiratory distress syndrome are senescent. Lung sections were immunostained. KRT8hi transitional cells in IPF but not non–COVID-19 acute respiratory distress syndrome express CDKN2A/p16, a highly specific marker of senescence. n = 3. Scale bar = 50 μm.
Proposed lineage trajectories in physiological alveolar regeneration, early acute respiratory distress syndrome (ARDS), and fibrosis. During physiological regeneration after lung injury resulting in alveolar epithelial cell (AEC) death, surviving type 2 AECs (AEC2s) proliferate, then exit the cell cycle and transiently adopt the transitional state but ultimately differentiate into mature type 1 AECs. In early ARDS, after AEC death, surviving AEC2s proliferate, then exit the cell cycle and adopt the transitional state, and are in the process of organized type 1 AEC differentiation; however, incomplete differentiation is associated with ongoing barrier permeability, pulmonary edema, ventilator dependence, and mortality. In fibroproliferative ARDS and idiopathic pulmonary fibrosis (IPF), after AEC death, surviving AEC2s proliferate, then exit the cell cycle and adopt the transitional state but evolve into a permanent state of cell cycle arrest, or senescence, and fibrosis ensues. Adapted with permission of the American Thoracic Society. Copyright © 2021 American Thoracic Society. All rights reserved. Jiang P, Gil de Rubio R, Hrycaj SM, Gurczynski SJ, Riemondy KA, Moore BB, Omary MB, Ridge KM, Zemans RL: Ineffectual type 2-to-type 1 alveolar epithelial cell differentiation in idiopathic pulmonary fibrosis: persistence of the KRT8hi transitional state. Am J Respir Crit Care Med 2020, 201:1443–1447. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society. Readers are encouraged to read the entire article for the correct context at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7258651. The authors, editors, and The American Thoracic Society are not responsible for errors or omissions in adaptations.