

Abstract
Objectives
The use of broad-spectrum cephalosporins is associated with induction of Clostridioides difficile infection (CDI). Recent knowledge on the importance of the healthy microbiota in preventing pathogen colonization/outgrowth highlights the caution needed when prescribing broad-spectrum antibiotics. The use of historical narrow-spectrum antibiotics, such as first-generation cephalosporins, is gaining increased attention once more as they have a reduced impact on the microbiota whilst treating infections. Here, the effects of two first-generation cephalosporins, compared with a third-generation cephalosporin, on the human microbiota were investigated and their propensity to induce simulated CDI.
Methods
Three in vitro chemostat models, which simulate the physiochemical conditions of the human colon, were seeded with a human faecal slurry and instilled with either narrow-spectrum cephalosporins, cefalexin and cefradine, or a broad-spectrum cephalosporin, ceftriaxone, at concentrations reflective of colonic levels.
Results
Instillation of cefalexin was associated with reduced recoveries of Bifidobacterium and Enterobacteriaceae; however, Clostridium spp. recoveries remained unaffected. Cefradine exposure was associated with decreased recoveries of Bifidobacterium spp., Bacteroides spp. and Enterobacteriaceae. These changes were not associated with induction of CDI, as we observed a lack of C. difficile spore germination/proliferation, thus no toxin was detected. This is in contrast to a model exposed to ceftriaxone, where CDI was observed.
Conclusions
These model data suggest that the minimal impact of first-generation cephalosporins, namely cefalexin and cefradine, on the intestinal microbiota results in a low propensity to induce CDI.
Introduction
The intestinal microbiota exists in a symbiotic relationship with humans, where the microbiota aids digestion of foodstuffs, immune system regulation and protection from pathogenic organisms—a term called colonization resistance. There exists a balance in clinical practices between the need to treat patients with potentially unknown microbial infections and the need to reduce the impact of those antibiotics on the resident microbiota. Antibiotics are life-saving drugs; however, their use is associated with risks, such as consequences of healthcare-associated infections and antibiotic resistance. Whilst broad-spectrum antibiotics target a wide range of pathogenic bacteria, their use impacts upon the commensal gut microbiota.1–3 The reduction in microbial diversity, even if temporary, can increase the carriage of pathogenic microorganisms or those harbouring antibiotic-resistance determinants.4,5
Clostridioides difficile infection (CDI) is the leading cause of antibiotic-associated diarrhoea and causes significant morbidity and mortality worldwide. CDI is highly associated with the use of antibiotics, where the antimicrobial action depletes the intestinal microbiota, which allows germination and outgrowth of ingested C. difficile spores. The financial burden of CDI cases on healthcare systems is estimated to be €3 billion in Europe and $4.8 billion in the USA.6–8 Broad-spectrum antibiotics, such as amoxicillin, are the most highly prescribed antibiotics and are highly associated with induction of CDI.3,9 Indeed, the use of second-, third- and fourth-generation cephalosporins is associated with a significantly increased risk of developing CDI; however, there is a low risk reported with first-generation cephalosporin prescriptions.10 The use of narrow-spectrum antibiotics has gained attention in recent years, where they have reduced impact on the microbiota, already have approval for human medical use and are inexpensive.
Cefalexin and cefradine are first-generation cephalosporins used to treat respiratory or urinary tract infections, with activity mostly against Gram-positive cocci and some Gram-negative organisms, including Streptococcus spp., Escherichia coli and Proteus spp.11 Like all cephalosporins, cefalexin and cefradine bind and inactivate the bacterial penicillin-binding proteins (PBPs). Inhibition of PBPs prevents the cross-linking of the peptidoglycan layer, which results in weakened bacterial cell walls and, ultimately, cell lysis. However, these antibiotics are susceptible to different β-lactamases. We have previously shown, using an in vitro human gut model, that ceftriaxone affects the indigenous microbiota, specifically the recoveries of Lactobacillus spp., Clostridium spp. and Enterococcus spp., which provides a niche for the onset of simulated CDI.3 Here, using this in vitro human gut model, we assessed the impact of cefalexin and cefradine on a healthy microbiota and the propensity to induce CDI, alongside a comparator antibiotic, ceftriaxone.3 This model consists of three chemostat vessels arranged in a weir cascade fashion, where each vessel mimics the physiological conditions of the proximal to distal colon.12
Materials and methods
Gut model
Setup
Three triple-staged gut models were assembled to simulate CDI induction, as previously described.12,13 Briefly, each model was composed of three chemostat vessels and maintained at physiological conditions; vessel 1 (pH 5.5 ± 0.1, 280 mL; proximal colon), vessel 2 (pH 6.2 ± 0.1, 300 mL; medial colon) and vessel 3 (pH 6.7 ± 0.1, 300 mL; distal colon). An anaerobic environment was maintained by sparging each vessel with nitrogen, and a complex growth medium connected to vessel 1 at a preestablished rate of 0.015 L/h.13
Faecal samples from five healthy donors (aged ≥60 years old with no history of antimicrobial usage in the previous 6 months) were individually screened for the presence of glutamine dehydrogenase (GDH), a constitutively expressed C. difficile-specific protein, as determined by the EIA C. DIFF CHEKTM - 60 test (TECHLAB Inc., USA). The ages of participants who provided faecal donations were chosen to represent a common risk factor for CDI.14 Each donor faecal sample was screened as negative for C. difficile by EIA C. DIFF CHEKTM. Following this, faecal samples were diluted 1:10 with pre-reduced PBS. This slurry was filtered to remove large particulate matter. Each vessel, of each model, was seeded with ∼160 mL of this slurry to start the experiment. A small aliquot of faecal slurry was kept anaerobically at 37°C and the bacterial populations were enumerated (as described below).
Ethics
The collection and use of human faeces in our gut model has been approved by the School of Medicine Research Ethics Committee, University of Leeds (MREC 15-070; Investigation of the Interplay between Commensal Intestinal Organisms and Pathogenic Bacteria). Participants were provided with a ‘Participant Information Sheet’ (PIS) detailing a lay summary of the in vitro gut model and the scientific work they are contributing to by providing a faecal donation. Within this PIS, it is explained that by providing the sample the participant is giving informed consent for that sample to be used in the gut model.
Experimental design
The experimental timeline for these models is depicted in Figure 1a. After addition of the faecal slurry, microbial populations were monitored for 14 days, without further intervention, to ensure the populations reached steady state. A 1 mL aliquot of C. difficile spores (107 spores/mL) of strain 210 [BI/NAP1/PCR ribotype (RT) 027/toxinotype III]15 was added to vessel 1 of each model. This was done to establish that the microbiota had formed colonization resistance against C. difficile germination. One week later, another dose of C. difficile spores was added to the model and each model was instilled with either cefalexin (15 mg/L, four times daily for 7 days), cefradine (15 mg/L, four times daily for 7 days) or ceftriaxone (150 mg/L, once daily for 7 days) and the microbial populations monitored for 2 weeks after the last dose. These antibiotic concentrations are reflective of the concentration found in the human colon.3,11,16 Bacterial populations, including C. difficile, were monitored daily throughout the experiment using selective and non-selective agars, as outlined previously.12 Only those counts from vessel 3 are shown.
Figure 1.

Schematic timeline of the in vitro triple-stage chemostat gut model and experimental design for each model (a). C. difficile (CD) spores were added to each model (black arrows) before addition of antibiotics (blue arrow). Microbial populations were monitored after exposure to either cefalexin (b), cefradine (c) or ceftriaxone (d). The total obligate anaerobic bacteria (black lines), Bacteroides spp. (red lines), Bifidobacterium spp. (blue lines), Clostridium spp. (green lines) and lactose-fermenting (LF) Enterobacteriaceae (orange lines) are shown as mean log10 cfu/mL from three technical replicates. Limit of detection for this assay is 1.2 log10 cfu/mL.
Preparation of C. difficile RT027 strain 210 spores
C. difficile spores for gut model inoculation were prepared as previously described.17 Briefly, C. difficile RT027 was grown in brain heart infusion (BHI) broth anaerobically at 37°C for 6 days and removed from the incubator and incubated aerobically at room temperature overnight to further induce sporulation. Growth was harvested by centrifugation and incubated with PBS supplemented with 10 mg/mL lysozyme at 37°C overnight. Samples were separated using a sucrose gradient and spores were treated with PBS supplemented with 20 ng/mL protease K and 200 nM EDTA. Spores were then separated using a sucrose gradient and washed with PBS twice before a final resuspension in 30 mL. These were enumerated and diluted to approximately 1 × 107 spores/mL for use in the models.18
Enumeration of endogenous bacteria and quantification of C. difficile toxin
Gut microbiota populations were monitored using viable enumeration on selective and non-selective agars, as described previously.12 Microbial colonies were enumerated and identified, based on colony morphology and MALDI-TOF identification. Each bacterial population was measured in triplicate (three technical replicates of a single biological replicate) in vessels 2 and 3. C. difficile total viable counts (TVCs) and spore counts were measured from all vessels; spore counts were obtained through plating serial dilutions of model fluid after alcohol shock. The limits of detection for TVCs or spore counts were 1.2 or l.5 log10 cfu/mL, respectively.
C. difficile cytotoxin was monitored using a semi-quantitative Vero cell cytotoxicity assay as previously described.12 Cytotoxin titre was expressed as log10 relative units (RU) at the highest dilution with >70% cell rounding, i.e. 10°, 1 RU; 101, 2 RU etc.
Antibiotic bioassays
The concentration of ceftriaxone in each vessel was determined by antibiotic bioassay, as previously described; E. coli ATCC 25922 was inoculated onto Mueller–Hinton agar (Oxoid, UK).3 Cefalexin and cefradine concentrations were determined by antibiotic bioassay, as before except the E. coli ATCC 25922 indicator strain was incorporated into Iso-Sensitest agar (Oxoid, UK) and either cefalexin or cefradine calibrator curve concentrations (range between 128 and 0.5 mg/L) were added randomly to each plate. The limits of detection were 0.05 mg/L for cefalexin and cefradine, and 1 mg/L for ceftriaxone.
Results
Formation of colonization resistance against C. difficile spores
Upon inoculation, the microbial populations were allowed to equilibrate for 2 weeks prior to further intervention; during this time, microbial growth patterns settled. To determine whether the established microbiota within each model conferred colonization resistance against C. difficile spore germination, we exposed each model to 107 spores/mL and monitored for germination and outgrowth. Once added to the model, C. difficile cells remained in spore form, where no evidence of germination was seen. Formation of colonization resistance against C. difficile spore germination in the models ensures that any post-antibiotic induction of CDI would be directly related to the effects of the different antibiotics rather than microbiota stability within the model.
Antibiotic-induced changes to gut microbiota populations
Cefalexin
Cefalexin instillation caused declines to the Bifidobacterium spp. (2.4 log10 cfu/mL) and lactose-fermenting Enterobacteriaceae (1.0 log10 cfu/mL) recoveries, where Bifidobacterium spp. failed to recover to pre-antibiotic levels, whilst lactose-fermenting Enterobacteriaceae recovered to levels approximately 1 log10 cfu/mL higher than pre-antibiotic levels (Figure 1b). Cefalexin promoted an initial decline in Bacteroides spp.; however, this population soon recovered to approximately 1 log10 cfu/mL higher than pre-antibiotic levels. Lactobacillus spp. also declined by 1.2 log10 cfu/mL after exposure to cefalexin but recovered after withdrawal of the antibiotic. There was no change in Enterococcus spp., and Clostridium spp. remained unchanged throughout the experiment.
Cefradine
Cefradine instillation caused similar declines to the Bifidobacterium spp. (2.0 log10 cfu/mL) and lactose-fermenting Enterobacteriaceae (1.1 log10 cfu/mL) populations compared with pre-antibiotic levels (Figure 1c). These bacterial populations recovered to pre-antibiotic levels by Day 46. Interestingly, Bacteroides spp. and Clostridium spp. remained unaffected by the antibiotic levels. Enterococcus spp. recoveries increased by 1.0 log10 cfu/mL after exposure to cefradine but decreased to pre-antibiotic levels by Day 46.
Ceftriaxone
Instillation of ceftriaxone caused widespread changes to the microbiota. Lactose-fermenting Enterobacteriaceae decreased by 3.2 log10 cfu/mL during ceftriaxone instillation; however, post-antibiotic Enterobacteriaceae increased by 1.4 log10 cfu/mL compared with pre-antibiotic levels (Figure 1d). Bacteroides spp. and Clostridium spp. showed decreases of 1.2 and 1.4 log10 cfu/mL, respectively, during ceftriaxone exposure; however, these populations recovered to pre-antibiotic levels 2 weeks after withdrawal. Enterococcus spp. were unaffected by ceftriaxone instillation; however, there was a 3.9 log10 cfu/mL increase during the dysbiosis created after antibiotic exposure. Interestingly, Bifidobacterium spp. remained unaffected by ceftriaxone (Figure 1d).
Comparing the effects of each antibiotic on the microbiota showed that cefalexin and cefradine caused the least disruption to the microbiota compared with ceftriaxone. The disruption caused by cefalexin and cefradine was characterized by reductions of −3.5-fold and −3.8-fold, respectively, to the Enterobacteriaceae population compared with pre-antibiotic levels (Figure 2a and b). Lactobacillus was also affected during cefalexin and cefradine instillation, showing reductions of −3.2-fold and −1.6-fold, respectively. However, all microbial populations had recovered to pre-antibiotic levels by the end of the experiment. Ceftriaxone caused the most disruption to the microbiota, with Lactobacillus, Enterobacteriaceae and Bacteroides populations showing −6.8-fold, −8.9-fold and −4.5-fold decreases compared with pre-antibiotic levels. The microbial dysbiosis observed was maintained for at least 2 weeks after withdrawal, with Enterobacteriaceae and Enterococcus spp. 4.6-fold and 8.1-fold higher than pre-ceftriaxone levels (Figure 2c).
Figure 2.
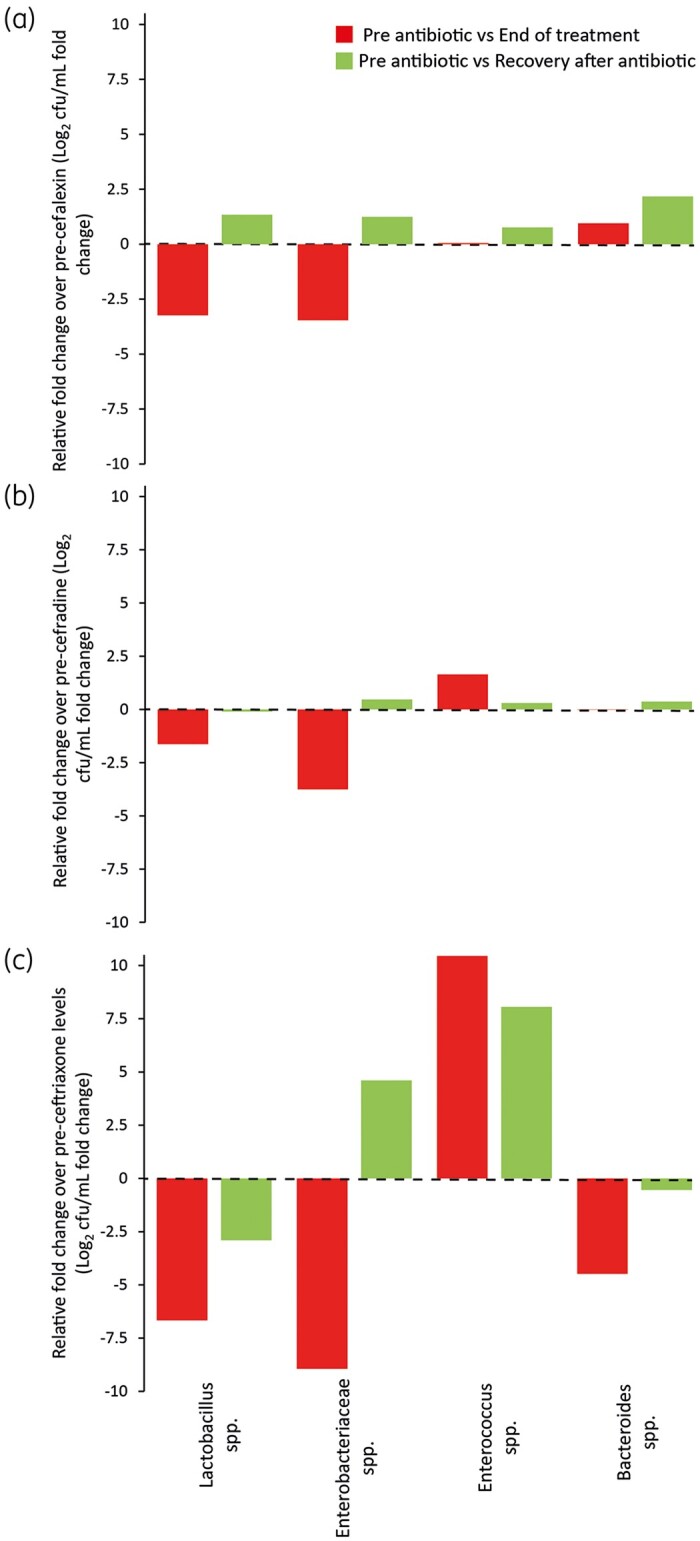
Comparative effects of cefalexin (a), cefradine (b) and ceftriaxone (c) on the human gut microbiota. Key bacterial members of the microbiota were enumerated just before antibiotics were added, when the antibiotic treatment ended and 2 weeks after antibiotic withdrawal. Results shown are the fold change in bacterial recovery of the end of antibiotic treatment (red bars) and 2 weeks after antibiotic (green bars) compared with pre-antibiotic levels.
Antibiotic disruption of the gut microbiota does not induce CDI
Instillation of cefalexin, cefradine or ceftriaxone reached peak concentrations of 4.8, 5.3 and 39.0 mg/L, respectively, in vessel 1 of each model; antibiotic concentrations were lower in vessels 2 and 3 in each model. These concentrations of cefalexin and cefradine achieved in our gut model did not cause simulated CDI. There was no divergence between C. difficile TVCs and spore recoveries after cefalexin exposure and, whilst there was limited germination after cefradine exposure, there was no toxin detected in these models (Figure 3a and b). The level of ceftriaxone-mediated microbiota disruption induced simulated CDI where the TVCs diverged from the spore counts, with peak C. difficile recoveries occurring 5 days after antibiotic instillation (Day 33; 6.4 log10 cfu/mL), which was preceded by toxin detection. Peak toxin was detected on Day 35 at 4.5 RU toxin activity (Figure 3c).
Figure 3.

Recovery of C. difficile populations in vessel 3 after instillation of either cefalexin (a), cefradine (b) or ceftriaxone (c). The C. difficile TVCs (black lines) and spore populations (grey lines) are shown as mean log10 cfu/mL from three technical replicates. Toxin production (arrows) was measured by cell toxicity assay—toxin activity was only detected after exposure to ceftriaxone (c); thus, no arrow is visible in (a) and (b). Limits of detection for this assay are 1.2 and 2.5 log10 cfu/mL for total viable and spores counts, respectively.
Discussion
The toxicity and tolerability profiles of cephalosporins make them ideal treatment options for a wide variety of infections; however, cephalosporin-resistant pathogens can exploit this and cause serious complications. C. difficile is one such pathogen, where the use of second- to fourth-generation cephalosporins is associated with a high OR of inducing CDI (OR between 3.2 and 2.14), whilst the use of first-generation cephalosporins has a lower OR of 1.36.10 The lower risk of developing subsequent CDI after first-generation cephalosporin use is likely due to the narrow spectrum of activity against bacterial populations.11 In our study, we showed that cefalexin and cefradine temporarily reduced the Enterobacteriaceae and Lactobacillus populations, which recovered to pre-antibiotic levels soon after withdrawal. In contrast, ceftriaxone caused more disruption to the microbiota, where, 2 weeks after antibiotic withdrawal, overgrowth of opportunistic pathogens Enterobacteriaceae and Enterococcus spp. was seen, and lower levels of Lactobacillus spp., compared with pre-antibiotic levels. The observed effects of ceftriaxone on the microbiota were similar to those previously reported by our group and in clinical trials.3,19 This level of ceftriaxone-induced dysbiosis created a niche that allowed C. difficile spore germination and the onset of simulated CDI, whereas cefalexin and cefradine did not induce CDI.
Environmental metabolites, such as primary bile acids and some amino acids, act as cues for disease initiation during CDI, whereas other metabolites, such as ethanolamine and succinate, are utilized during vegetative growth.20–22 Antibiotic depletion of some Clostridium spp., such as Clostridium scindens, can result in accumulation of primary bile acids, which are a known germination signal for C. difficile spores, thus initiating disease.23 However, Clostridium spp. recoveries before, during and after first-generation cephalosporin exposure remained unchanged, suggesting that the capacity to metabolize primary bile acids may have been preserved after exposure to either first-generation cephalosporin tested. In contrast, Clostridium spp. were reduced by ceftriaxone, which could have caused increased concentrations of spore germinants in our system. A reduction in Clostridium spp. and Bifidobacterium spp. was seen with other cephalosporins (ceftaroline/cefotaxime), which induced simulated CDI in our model system.3,13 Upon germination, C. difficile still must compete with the microbiota for nutrients to grow. Interestingly, we did observe limited germination and outgrowth after cefradine instillation; however, there appeared to be sufficient microbial competition that prevented sufficient vegetative growth, with no toxin being detected. It has been shown that some Clostridium spp., such as Clostridium bifermentans, can actively compete with C. difficile by utilizing the bioavailable amino acid pool, thus preventing CDI.24 Thus, the differential effect of cefalexin/cefradine versus ceftriaxone on the Clostridium spp. could impact C. difficile growth and the different propensities to induce simulated CDI. The number of different classes of antimicrobials that are high risk for inducing CDI suggests that C. difficile can metabolically adapt to different niches created during antibiotic-mediated disruption of the microbiota.25
Whilst treating the primary infection, some classes of antibiotics can subsequently lead to life-threatening complications, such as CDI. There exists a need to determine the effect of different antibiotics upon the intestinal microbiota, as preservation of a patient’s microbiota should be incorporated in the multifaceted decision-making process when choosing a treatment option. Here, we show that two narrow-spectrum cephalosporins, cefalexin and cefradine, have a limited effect on the human microbiota and this disruption represents a low risk for subsequently developing simulated CDI. Our in vitro data support the clinical findings previously reported by Slimings and Riley.10
Acknowledgements
We thank staff at the Leeds Teaching Hospital NHS Trust pharmacy for sourcing cefalexin and cefradine powders for use in the gut model.
Funding
This study was supported by University of Leeds internal funding.
Transparency declarations
M.H.W. has received honoraria for consultancy work, financial support to attend meetings and research funding from Astellas, AstraZeneca, Abbott, Actelion, Alere, Bayer, bioMérieux, Cerexa, Cubist, Da Volterra, Durata, Merck, Nabriva Therapeutics plc, Pfizer, QIAGEN, Roche, Seres Therapeutics Inc., Synthetic Biologics, Summit and The Medicines Company. I.B.M. has received support to attend meetings from TECHLAB Inc. A.M.B. has received financial support to attend meetings and research funding from Seres Therapeutics Inc., Motif Biosciences plc, Nabriva Therapeutics plc, Tetraphase Pharmaceuticals and Hayashibara Co. Ltd. All other authors: none to declare.
References
- 1. Nord CE, Brismar B, Kasholm-Tengve B et al. Effect of piperacillin/tazobactam therapy on intestinal microflora. Scand J Infect Dis 1992; 24: 209–13. [DOI] [PubMed] [Google Scholar]
- 2. Zimmermann P, Curtis N. The effect of antibiotics on the composition of the intestinal microbiota - a systematic review. J Infect 2019; 79: 471–89. [DOI] [PubMed] [Google Scholar]
- 3. Baines SD, Chilton CH, Crowther GS et al. Evaluation of antimicrobial activity of ceftaroline against Clostridium difficile and propensity to induce C. difficile infection in an in vitro human gut model. J Antimicrob Chemother 2013; 68: 1842–9. [DOI] [PubMed] [Google Scholar]
- 4. Rooney CM, Sheppard AE, Clark E et al. Dissemination of multiple carbapenem resistance genes in an in vitro gut model simulating the human colon. J Antimicrob Chemother 2019; 74: 1876–83. [DOI] [PubMed] [Google Scholar]
- 5. Buckley AM, Altringham J, Clark E et al. Eravacycline, a novel tetracycline derivative, does not induce Clostridioides difficile infection in an in vitro human gut model. J Antimicrob Chemother 2021; 76: 171–8. [DOI] [PubMed] [Google Scholar]
- 6. Aguado JM, Anttila VJ, Galperine T et al. Highlighting clinical needs in Clostridium difficile infection: the views of European healthcare professionals at the front line. J Hosp Infect 2015; 90: 117–25. [DOI] [PubMed] [Google Scholar]
- 7. Lessa FC, Mu Y, Bamberg WM et al. Burden of Clostridium difficile infection in the United States. N Engl J Med 2015; 372: 825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sheitoyan-Pesant C, Abou Chakra CN, Pépin J et al. Clinical and healthcare burden of multiple recurrences of Clostridium difficile infection. Clin Infect Dis 2016; 62: 574–80. [DOI] [PubMed] [Google Scholar]
- 9. Wilcox MH, Chalmers JD, Nord CE et al. Role of cephalosporins in the era of Clostridium difficile infection. J Antimicrob Chemother 2017; 72: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Slimings C, Riley TV. Antibiotics and hospital-acquired Clostridium difficile infection: update of systematic review and meta-analysis. J Antimicrob Chemother 2014; 69: 881–91. [DOI] [PubMed] [Google Scholar]
- 11. Hartley CL, Clements HM, Linton KB. Effects of cephalexin, erythromycin and clindamycin on the aerobic Gram-negative faecal flora in man. J Med Microbiol 1978; 11: 125–35. [DOI] [PubMed] [Google Scholar]
- 12. Moura IB, Buckley AM, Ewin D et al. Omadacycline gut microbiome exposure does not induce Clostridium difficile proliferation or toxin production in a model that simulates the proximal, medial, and distal human colon. Antimicrob Agents Chemother 2019; 63: e01581-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Freeman J, Neill FJO, Wilcox MH. Effects of cefotaxime and desacetylcefotaxime upon Clostridium difficile proliferation and toxin production in a triple-stage chemostat model of the human gut. J Antimicrob Chemother 2003; 52: 96–102. [DOI] [PubMed] [Google Scholar]
- 14. Bartlett JG, Gerding DN. Clinical recognition and diagnosis of Clostridium difficile infection. Clin Infect Dis 2008; 46: S12–18. [DOI] [PubMed] [Google Scholar]
- 15. Chilton CH, Crowther GS, Baines SD et al. In vitro activity of cadazolid against clinically relevant Clostridium difficile isolates and in an in vitro gut model of C. difficile infection. J Antimicrob Chemother 2014; 69: 697–705. [DOI] [PubMed] [Google Scholar]
- 16. Bullen BR, Ramsden CH, Kester RC. Peroperative cephradine concentrations in the gall bladder wall and bile. Curr Med Res Opin 1982; 8: 5–8. [DOI] [PubMed] [Google Scholar]
- 17. Lawley TD, Croucher NJ, Yu L et al. Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J Bacteriol 2009; 191: 5377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buckley AM, Spencer J, Candlish D et al. Infection of hamsters with the UK Clostridium difficile ribotype 027 outbreak strain R20291. J Med Microbiol 2011; 60: 1174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pletz MWR, Rau M, Bulitta J et al. Ertapenem pharmacokinetics and impact on intestinal microflora, in comparison to those of ceftriaxone, after multiple dosing in male and female volunteers. Antimicrob Agents Chemother 2004; 48: 3765–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Francis MB, Allen CA, Shrestha R et al. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog 2013; 9: e1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferreyra JA, Wu KJ, Hryckowian AJ et al. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 2014; 16: 770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nawrocki KL, Wetzel D, Jones JB et al. Ethanolamine is a valuable nutrient source that impacts Clostridium difficile pathogenesis. Environ Microbiol 2018; 20: 1419–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buffie CG, Bucci V, Stein RR et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015; 517: 205–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Girinathan B, DiBenedetto N, Worley J et al. The mechanisms of in vivo commensal control of Clostridioides difficile virulence. bioRxiv 2020; https://doi.org/10.1101/2020.01.04.894915. [Google Scholar]
- 25. Hofmann JD, Otto A, Berges M et al. Metabolic reprogramming of Clostridioides difficile during the stationary phase with the induction of toxin production. Front Microbiol 2018; 9: 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]