

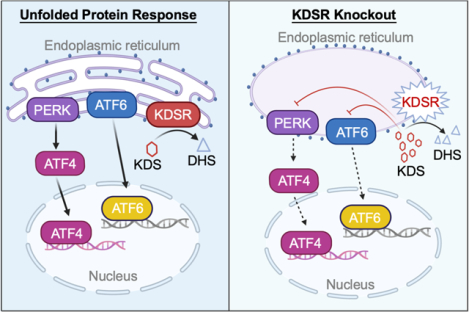
Abstract
Sphingolipids and their metabolic pathways have been implicated in disease development and therapeutic response; however, the detailed mechanisms remain unclear. Using a sphingolipid network focused CRISPR/Cas9 library screen, we identified an endoplasmic reticulum (ER) enzyme, 3-Ketodihydrosphingosine reductase (KDSR), to be essential for leukemia cell maintenance. Loss of KDSR led to apoptosis, cell cycle arrest, and aberrant ER structure. Transcriptomic analysis revealed the indispensable role of KDSR in maintaining the unfolded protein response (UPR) in ER. High-density CRISPR tiling scan and sphingolipid mass spectrometry pinpointed the critical role of KDSR’s catalytic function in leukemia. Mechanistically, depletion of KDSR resulted in accumulated 3-ketodihydrosphingosine (KDS) and dysregulated UPR checkpoint proteins PERK, ATF6, and ATF4. Finally, our study revealed the synergism between KDSR suppression and pharmacologically induced ER-stress, underscoring a therapeutic potential of combinatorial targeting sphingolipid metabolism and ER homeostasis in leukemia treatment.
Keywords: Sphingolipid, 3-Ketodihydrosphingosine, Metabolism, KDS, KDSR, ER, Unfolder protein response, CRISPR gene body scan, leukemia, ER-stress
Graphical Abstract
INTRODUCTION
Leukemia is a class of aggressive hematological diseases characterized by aberrant proliferation, defected differentiation, and occluded apoptosis of hematopoietic stem/progenitor cells (1, 2). Although advanced therapies such as allogeneic hematopoietic stem cell transplantation and molecularly targeted therapies have been developed (3), the overall survival of the more malignant subtypes such as acute myeloid leukemia (AML) or acute lymphoid leukemia (ALL) remains slender (4). Identifying novel effectors with selective dependency in leukemia is much needed to expand more effective regimens for leukemia treatment.
Sphingolipids are lipid components involved in regulating the lipid bilayer fluidity and membrane protein trafficking (5). The sphingolipid-rich microdomains, i.e., the “lipid rafts,” are associated with membrane receptor signaling and mediate cellular processes such as proliferation, apoptosis, differentiation, and stress-response (6). Sphingolipids in mammalian cells are synthesized de novo or recycled through the salvage pathways (7). The de novo pathway converts palmitoyl-CoA and L-serine to 3-ketodihydrosphingosine (KDS) by serine palmitoyltransferase (SPTLC1). KDS is then reduced by KDS reductase (KDSR) to dihydrosphingosine (DHS), which was later converted to ceramides (Cer) by ceramide synthases (CERS1/2, etc.). The salvage pathway of sphingolipids begins with the degradation of sphingomyelin (SM) or glucosylceramides (GlcCer) to become Cer, which is further converted to sphingosine (SP) (7). The homeostasis of sphingolipid species plays a critical role in cellular physiological processes, and dysregulation of sphingolipids and their metabolic enzymes has been implicated in leukemia (8–10). For example, accumulation of dihydroceramide (DHCer) and Cer promotes apoptosis in leukemia cells (11, 12), while sphingosine-1-phosphate (S1P), a downstream product of Cer, is anti-apoptotic (13). Moreover, sphingolipid enzymes such as ceramidase and sphingosine kinase are upregulated in leukemia and associated disease progression (14, 15). It appears that sphingolipid metabolism is involved in leukemia development and therapeutic responses; however, the detailed pathological roles of the sphingolipid enzymes and their metabolites in diverse cellular membrane compartments in leukemic cells remain unclear.
Endoplasmic reticulum (ER) is a cellular organelle essential for lipid biosynthesis and protein folding (16). Accumulation of misfolded proteins in ER leads to ER stress and triggers apoptosis (17). Elevated ER stress also induces unfolded protein response (UPR), a cellular process that restores the ER homeostasis through refolding or promoting the degradation of misfolded proteins (18). UPR consists of three major pathways mediated by protein kinase R-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring protein-1a (IRE1α)(18). Persistent UPR has been observed in leukemic cells to drive prosurvival signaling and chemotherapy resistance (19–21). Moreover, pharmacological inhibitions of the UPR that aggravate ER stress have been proposed for leukemia treatment (22–24). Targeting ER homeostasis/UPR alone or in combination with other interventions may open up novel therapeutic opportunities for diseases prone to ER stress.
In this study, we conducted an unbiased CRISPR/Cas9 library screen by targeting all major enzymes in the sphingolipid metabolism pathways and identified an essential requirement of KDSR, an ER membrane protein that converts KDS to DHS (25), in leukemia. Using the genetic (CRISPR tiling scan), transcriptomic (RNA-seq), and metabolomic (mass spectrometry) profiling, we revealed that KDSR is critical for leukemia survival through its involvement in the lipid metabolism and UPR pathway. KDSR thus represents a novel target for clinical applications in conjunction with UPR pharmacological modulators.
METHODS
Cell lines and culture conditions
MV4-11, MOLM13, KOPN-8, RS4;11, CCRF-CEM, Jurkat, SET2, Jeko-1, Ramos, 293T, U251, SW620, and A673 cells were obtained from ATCC. Hut78 cells were received from Steven Rosen (City of Hope Cancer Center). MV4-11, MOLM13, KOPN-8, RS4;11, CCRF-CEM, Jurkat, SET2, Jeko-1, and Ramos cells were cultured in RPMI 1640 (Thermo Fisher Scientific) medium supplemented with 10% fetal bovine serum (FBS) (Omega Scientific). Hut78 was cultured in RPMI 1640 medium with 15% FBS. 293T, U251, SW620, and A673 cells were cultured in DMEM (Gibco) with 10% FBS. Penicillin/streptomycin (Gibco), GlutaMax (Gibco) and plasmocin (0.5 μg/ml; Invitrogen) were added to all media. All cells were cultured in 37°C incubator with 5% CO2. Cells stably expressing the Cas9 endonuclease were established via transduction of LentiCas9-blast (Addgene) lentivirus and select by blasticidin (20 μg/ml; Gibco).
Molecular cloning and lentiviral transduction
Guide RNA sequences of sphingolipid metabolism focused CRISPR library (targeting 29 sphingolipid pathway enzymes at 25 sgRNAs per gene), KDSR high-density CRISPR tiling library (122 sgRNAs targeting the coding exons of KDSR), together with 22 sgRNAs targeting commonly essential genes (e.g., MYC, BRD4, RPA3, etc.) and 40 sgRNAs targeting non-essential sequences were designed using the Genetic Perturbation Platform (Broad Institute)(26). Briefly, sgRNA oligonucleotides were synthesized via microarray (CustomArray) and cloned into the ipUSEPR lentiviral sgRNA vector (hU6-driven sgRNA co-expressed with EF-1α-driven red fluorescent protein [RFP] and puromycin-resistance gene) using the BsmBI (NEB) restriction sites. A full-length KDSR cDNA contains synonymous mutations (bypass the targeting by sgKDSR) was designed using the CLC Main Workbench (Qiagen), synthesized by gBlock Gene Fragments (IDT), and cloned into the ECEG lentiviral vector (EF-1α-driven transgene co-expressed with green fluorescent protein [GFP]) using Gibson Assembly kit (NEB). The final plasmids were validated via Sanger sequencing (Eton Bioscience). Lentivirus was produced in HEK293 cells (ATCC) by co-transfecting ipUSEPR or ECEG vectors with the packaging plasmids pPAX2 (Addgene) and pMD2.G (Addgene). For lentiviral infection, target cells were mixed with the viral solution and polybrene (10 μg/ml; MilliporeSigma) and centrifuged at 1,000 rpm, 37°C for 60 min.
CRISPR library screens
The lentivirus of sphingolipid metabolism focused, and KDSR high-density tiling CRISPR libraries were pre-titrated to obtain ~15 % infection (monitored by flow cytometry for RFP expression) in the Cas9-expressing MV4-11 and MOLM13 cells. Briefly, cells were infected with the CRISPR library (3 replicates each screen) and selected by puromycin (1 μg/ml; Gibco). The library-transduced cells were subcultured every four days for a total of 28 days. Genomic DNA from each screen culture was collected at the start (day 0) and end (day 28) timepoints. The integrated sgRNA in each sample was PCR-amplified (NEBNext Ultra II Q5; NEB) using primers DCF01 5’-CTTGTGGAAAGGACGAAACACCG-3’ and DCR03 5’-CCTAGGAACAGCGGTTTAAAAAAGC-3’ for high-throughput sequencing (NextSeq550, Illumina). To quantify sgRNA reads, 20-nucleotide sequences that matched the sgRNA backbone structure (5’ prime CACCG and 3’ prime GTTT) were extracted and mapped to the library sgRNA sequences using Bowtie2 (27). The frequency for individual sgRNAs was calculated as the read counts of each sgRNA divided by the total read counts matched to the library. For the sphingolipid gene panel screen, the CRISPR score was defined as a fold-change in the frequency of individual sgRNAs between the start (day 0) and end (day 28) of the screen samples. For the CRISPR tiling scan, the CRISPR tiling score was defined as a log10-fold change in the frequency of individual sgRNAs between the start (day 0) and end (day 28) of the screened samples and normalized by the median score of the negative control sgRNA (defined as 0.00; sgRNA targeting non-essential sequences) and the median score of the positive control sgRNA (defined as −1.00; sgRNA targeting MYC, BRD4, RPA3, etc.) within the screen data. The under-represented sgRNAs (5 out of 122; less than 5% of the average frequency) in the library were excluded from the analysis. The CRISPR tiling score was further interpolated by Gaussian kernel smoothing in R (28).
Flow cytometric assays
For competition cell culture assays, Cas9-expressing cells were transduced with the ipUSEPR sgRNA (RFP-positive) or ECEG cDNA (GFP-positive) constructs in 96-well plates at ~50% infection. The cell cycle was measured by Click-iT™ Plus EdU Alexa Fluor 647 Assay Kits (Invitrogen). The cellular apoptosis was detected using Annexin V-FITC Apoptosis Detection Kit (Invitrogen) and Active Caspase3 Staining Kit (Invitrogen). The cell viability was detected by 4’,6-diamidino-2-phenylindole (DAPI; Invitrogen) dye exclusion. The δ score of drug combination was defined as:
where V%(0), V%(T), V%(K), and V%(T+K) are the observed cell viability under no drug, tunicamycin alone, KDS alone, and tunicamycin plus KDS combination, respectively. Data were obtained by high-throughput flow cytometry using an Attune NxT flow cytometer with an autosampler (ThermoFisher).
Western blotting
Cells were harvested and lysed in LDS sample buffer (Invitrogen) at 5×106 cells/mL, separated electrophoretically using Bolt 4–12% Bis-Tris plus gels (Invitrogen), and transferred onto PVDF membranes (0.2 μm pore size) using PVDF Mini Stacks and iBlot 2 (Invitrogen). Membranes were probed with rabbit antibodies against KDSR (ab153960, Abcam; 1:1000), PERK (5683T, Cell Signaling Technology; 1:1000), ATF4 (ab184909, Abcam; 1:1000), ATF6 (65880T, Cell Signaling Technology; 1:1000), IRE1α (3294T, Cell Signaling Technology; 1:1000), and histone H3 (ab1791, Abcam; 1:1000) at 4°C overnight. After washing, the membranes were incubated with HRP-linked goat anti-rabbit IgG antibody (31460, Invitrogen; 1: 200,000) at room temperature for 1 hour. Chemiluminescent signals were developed using the SuperSignal West Femto Substrate (ThermoFisher) and detected using a ChemiDoc imaging system (Bio-Rad).
RNA-seq and data analysis
Total RNA was extracted using the RNeasy kit (QIAGEN) and submitted for mRNA library prep and sequencing (Novogene). Raw sequence reads were mapped to the human genome (GRCh37) using STAR v2.5.3 (29), and the frequency of genes was calculated using featureCounts v1.5.1 (30). The raw counts were then normalized using the trimmed mean of M values (TMM) method and compared using Bioconductor package “edgeR”(31). Differentially expressed genes were identified if reads per kilobase per million (RPKM) ≥ 1 in at least one sample, fold change ≥ 2, and P ≤ 0.05. Gene set enrichment analysis (GSEA) was performed using the GSEA v4.1.0 (UC San Diego and BROAD Institute) (32).
Mass spectrometry of sphingolipids
One million cells from each condition were extracted with hexane, methyl acetate, acetonitrile, water (4:4:3:4, v/v/v/v). The middle polar lipid layer was collected, evaporated to dryness, and dissolved in 100 ul buffer containing 50 nM heavy-isotype labeled internal standards for analysis. An Agilent 6490 triple quadrupole coupled with a 1290 UPLC system was used to separate the sphingolipids and quantify by multiple reaction monitoring (MRM) in positive ion mode and the sulfatides in negative ion mode at City of Hope Mass Spectrometry and Proteomics Core Facility.
Transmission Electron Microscopy (TEM)
Cells were fixed with 2.5% glutaraldehyde, 0.1 M sodium cacodylate buffer (Na(CH3)2AsO2 ·3H2O), pH7.2, at 4°C. Samples were then prepared through post-fixation with osmium tetroxide, serial dehydration with ethanol, and embedment in Eponate (33). Ultra-thin sections (~70 nm thickness) were acquired by ultramicrotomy, post-stained, and examined on an FEI Tecnai 12 transmission electron microscope equipped with a Gatan OneView CMOS camera at City of Hope Electron Microscopy Core Facility.
Quantification And Statistical Analysis
All experiments were performed in biological triplicate unless otherwise indicated. The number of replicates for each experiment can be found in the figure legends. Graphs present as mean ± SD of triplicated experiments. Figures were produced using Biorender (https://app.biorender.com/biorender-templates), Prism (GraphPad), Excel and PowerPoint (Microsoft). Flow cytometry was analysis using Attune NxT (ThermoFisher) and Flow Jo (BD). Statistics was performed using Student’s t-test in Prism (GraphPad). Triplicated experiments were performed to have 80% power in detecting a minimal 30% difference at a statistical significance of P < 0.05 using two-sided t-test analysis with an assumed standard deviation of 10% per group.
RESULTS
Sphingolipid metabolism focused CRISPR/Cas9 library screens identify essential roles of de novo sphingolipid biosynthesis in leukemia cells
To identify critical sphingolipid modulators in leukemia, we developed a lentiviral CRISPR library of total 787 sgRNAs targeting the coding regions of 29 sphingolipid metabolism enzymes (7) at a density of 25 sgRNAs per gene, together with 22 sgRNAs targeting commonly essential genes (e.g., MYC, BRD4, RPA3) and 40 sgRNAs targeting non-essential sequences. Using this library, we conducted screens in Cas9-expressing MV4-11 (biphenotypic leukemia) and MOLM13 (AML) cells, and the frequencies of each integrated sgRNA sequence at day 0 and day 28 post-transduction were measured by high-throughput sequencing (Figure 1A; library sequences and read counts shown in Table S1). The top hit from these parallel screens revealed KDSR, an enzyme mediating KDS-to-DHS conversion, as an essential sphingolipid modulator in these leukemia cells (Figures 1B and 1C). Evaluation of the CRISPR scores over the sphingolipid network unveiled functional hotspots mediating de novo sphingolipid biosynthesis in ER, including SPTLC1, KDSR, CERS1 and CERS2, in leukemia maintenance (Figure 1D).
Figure 1. Sphingolipid metabolism focused CRISPR/Cas9 screens in leukemia.
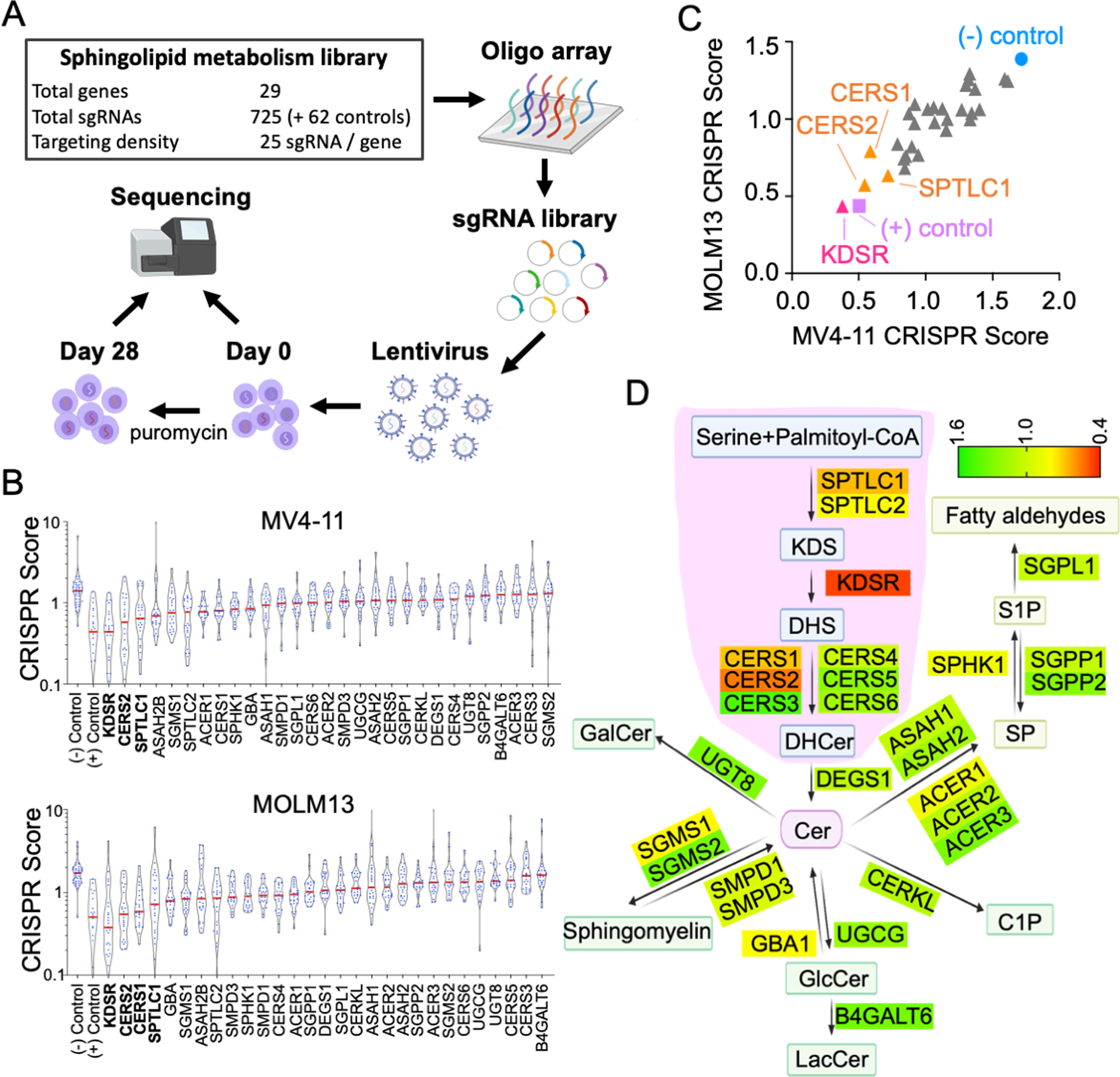
(A) Outline of the sphingolipid enzyme focused CRISPR/Cas9 screen coupled with high-throughput sequencing in leukemia cells. (B) Violin plot of the CRISPR score of 29 sphingolipid enzymes screened in Cas9-expressing MV4-11 and MOLM13 cells. The red line indicates the median value of the 25 sgRNAs (blue dots) targeting each gene. (C) Comparison of the median CRISPR score between MV4-11 and MOLM13 cells. (D) Annotation of CRISPR/Cas9 screen results over the sphingolipid metabolism network. The score of each enzyme represents the median CRISPR score averaged from MV4-11 and MOLM13 cells. The area highlighted in pink indicates the de novo biosynthesis pathway of sphingolipids in ER.
To validate the library screen results, we transduced Cas9-expressing MV4-11 and MOLM13 cells with sgRNAs targeting KDSR (sgKDSR#1 and sgKDSR#2). Using an RFP (co-expressed with sgRNA) flow cytometric growth competition assay (Figure S1), we found that cells transduced with sgKDSR were selectively outcompeted compared to cells transduced with sgRNA targeting non-essential sequences (sgLUC targeting Firefly luciferase; sgREN targeting Renilla luciferase) (Figure 2A). Furthermore, ectopic expression of a KDSR cDNA with synonymous mutations (bypass the targeting by sgKDSR) completely rescued the survival of these leukemic cells (Figure 2B), indicating the suppression of leukemia by CRISPR/Cas9 and sgKDSR can be attributed to the disruption of KDSR expression.
Figure 2. KDSR is essential for leukemia cells.
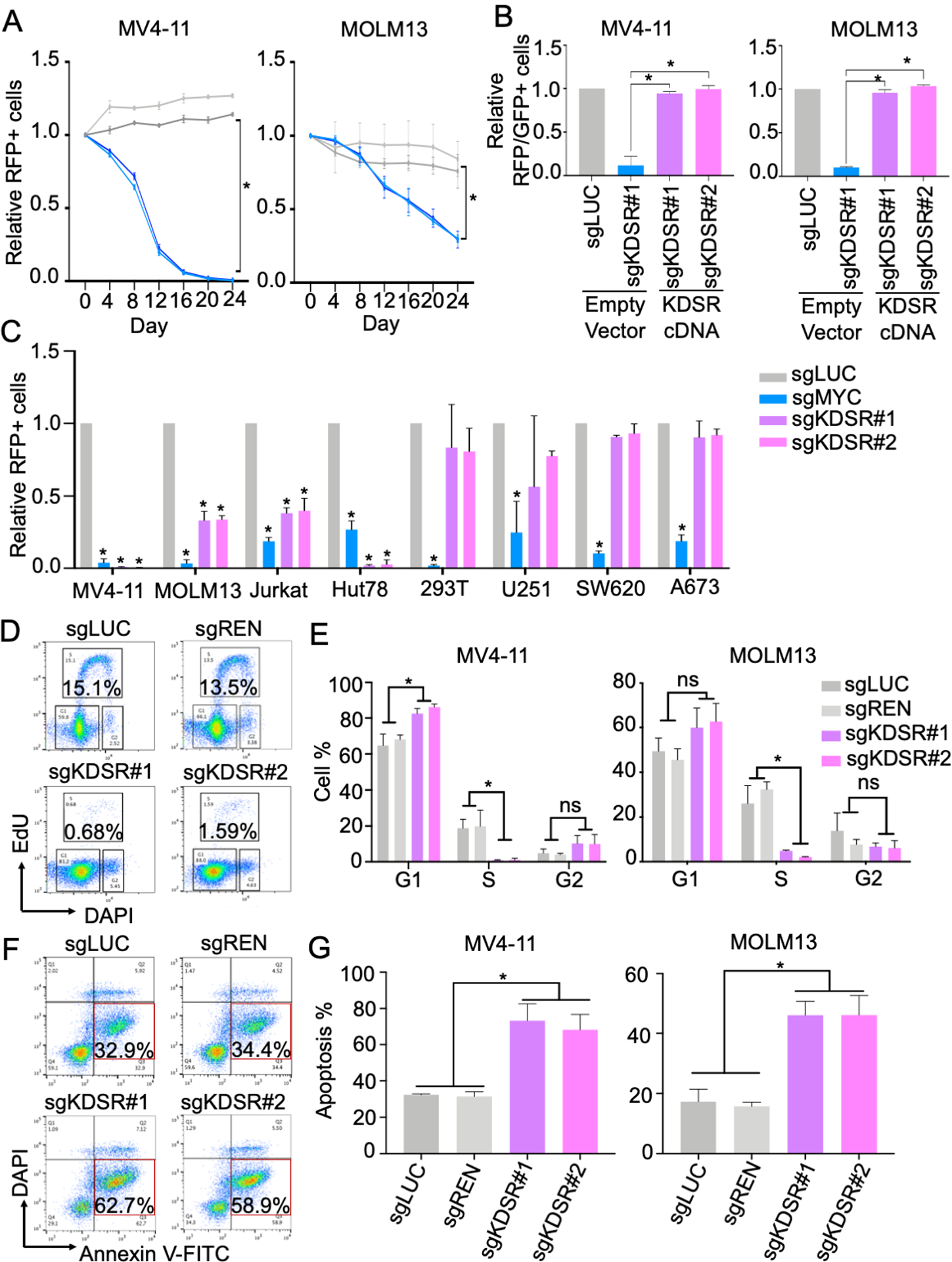
(A) Growth competition assay of MV4-11 and MOLM13 cells transduced with RFP-labeled negative control sgRNAs (sgLUC and sgREN; grey lines) and sgRNAs targeting KDSR (sgKDSR#1 and sgKDSR#2; blue lines). (B) Effect of ectopically expressed KDSR cDNA (GFP-labeled) on the cell numbers of sgKDSR (RFP-labeled) transduced MV4-11 and MOLM13 cells. (C) Growth competition assay of KDSR depletion in eight cancer cell models. (D,E) Cell cycle monitored by EdU and DAPI, and (F,G) cellular apoptosis detected by Annexin V in MV4-11 and MOLM13 cells. Data are represented as mean ± SD of triplicated experiments. *P < 0.01 using Student’s t-test.
To examine the impact of KDSR over diverse cell types, we CRISPR/Cas9 targeted KDSR in two additional blood cancer types, Jurkat (T-ALL) and Hut78 (T cell lymphoma), and compared to the non-leukemic cell types, including 293T (transformed embryonic kidney cell), U251 (glioblastoma), SW620 (colon cancer), and A673 (Ewing’s sarcoma) (Figure 2C). Efficient CRISPR-editing and cell suppression were observed in these eight cell models evidenced by significant depletion of the RFP-positive cells with sgRNA targeting a general cancer essential gene MYC (sgMYC). In contrast, depletion of the RFP-positive cells in sgKDSR transduced cultures was only observed in the hematopoietic cell types including MV4-11, MOLM13, Jurkat and Hut78 (Figure 2C). Further analysis of the genome-wide CRISPR/Cas9 screen databases (DepMap Portal; Achilles v.20Q2; BROAD Institute) revealed a significantly stronger gene effect Z-score of KDSR in leukemia compared to other cancer types (Figure S2A). Gene rank analysis indicated comparable potency between targeting KDSR and BRD4 (an essential gene in leukemia and other MYC-dependent cancers)(34) in multiple leukemia cell models (Figure S2B). Together, our pathway-focused CRISPR/Cas9 screen highlights a unique vulnerability of leukemia to the disruption of de novo sphingolipid biosynthesis pathway, particularly during the KDS-to-DHS transition via KDSR.
KDSR is required for leukemia cell maintenance and ER homeostasis
To investigate how KDSR supports leukemia maintenance, we monitored the cell cycle, apoptosis, and gene expression profiling upon KDSR disruption by CRISPR/Cas9 in both MV4-11 and MOLM13 cells. Using EdU (5-ethynyl-2’-deoxyuridine) incorporation and DAPI (4′,6-diamidino-2-phenylindole) DNA content labeling, we observed a significant reduction of the S phase in the sgKDSR transduced cells (Figure 2D, E). Annexin V staining revealed a drastic induction of Annexin V-positive/DAPI-negative population, indicating increased cellular apoptosis in the KDSR disrupted cells (Figure 2F, G). A similar apoptotic phenotype was also observed in the sgKDSR transduced cells monitored by the activation of caspase 3 (Figure S3).
Transcriptomic analysis through RNA-seq and Gene Set Enrichment Analysis (GSEA)(32) revealed the unfolded protein response (UPR) pathway (35) was amongst the top suppressed gene sets in the KDSR disrupted leukemia cells (Figure 3A–D). The UPR is activated in response to the accumulation of unfolded or misfolded proteins in the lumen of the endoplasmic reticulum (ER), with several critical checkpoint proteins including PERK, ATF6, and IRE1α (36). Whereas the protein level of IRE1α was unchanged in sgKDSR transduced MV4-11 cells, immunoblotting revealed a drastic reduction of PERK (and its downstream effector ATF4)(36) and ATF6 upon KDSR depletion (Figure 3E; left). In contrast, the expression of these UPR modulators was not affected in the KDSR-independent 293T cells (Figure 3E; right). Given that KDSR is a protein anchored to the ER-membrane (25), we sought to investigate the impact of KDSR depletion on ER structures in the leukemia cells. Wright-Giemsa stain confirmed the cellular morphology of MV4-11 cells was not impacted by the KDSR depletion (Figure 3F); however, the appearance of ER (Figure 3G; red arrows) was deformed (swollen with reduced ribosomal density) when observed by transmission electron microscopy (TEM). This is in contrast to the elongated tubular ER observed in the control leukemia cells. On the other hand, the normal ER morphology was unaffected by KDSR depletion in the 293T and SW620 cells (Figure 3H), consistent with the unchanged UPR modulators in these KDSR-independent cell types. In summary, our results suggest a selective requirement of KDSR-mediated UPR pathways and ER homeostasis in leukemia maintenance.
Figure 3. KDSR depletion dysregulates the UPR pathway and ER structure.
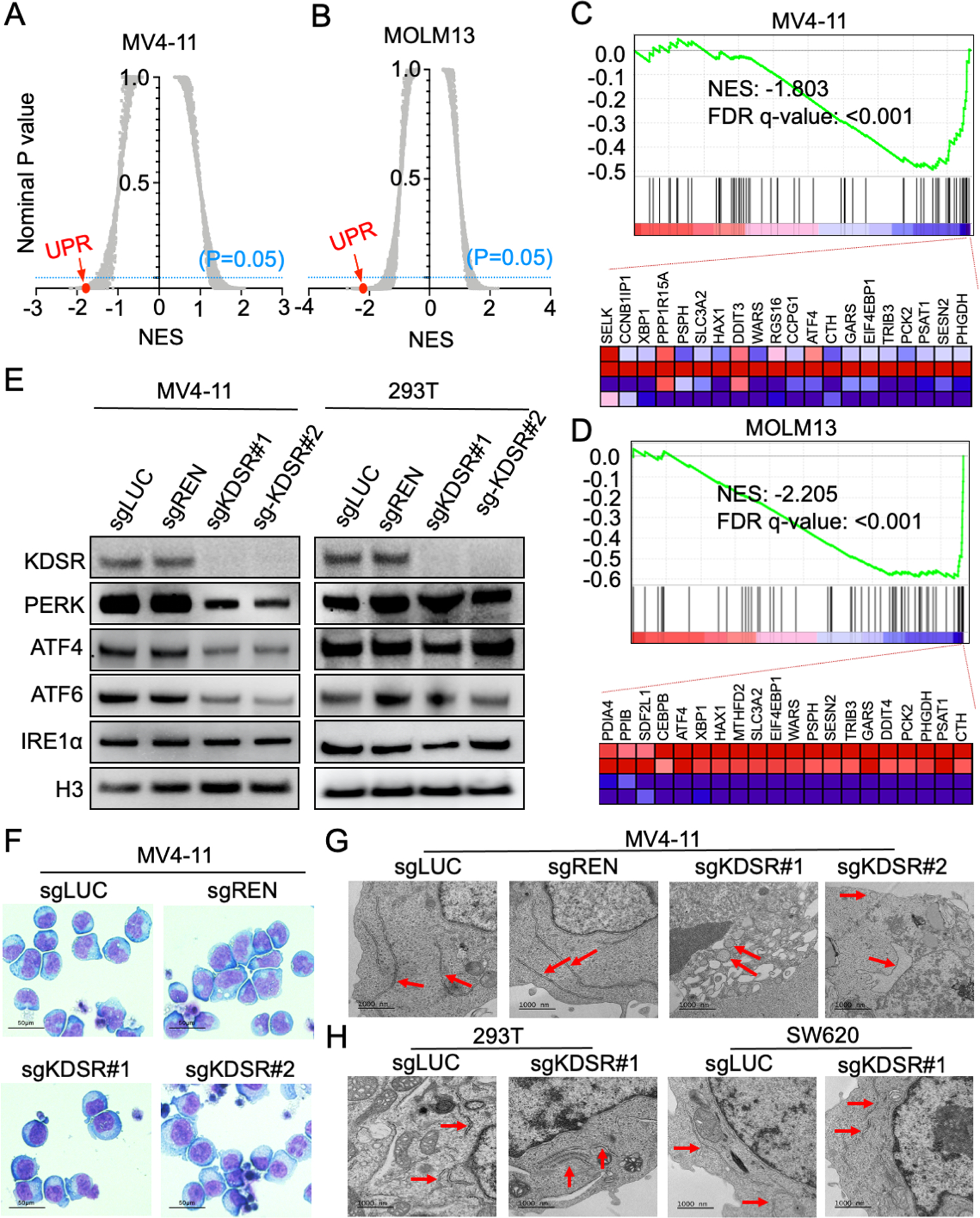
(A,B) Summary of gene sets affected by KDSR depletion using GSEA analysis. Each plot indicates one gene set from the GSEA Molecular Signature Database (MSigDB). NES: Normalized enrichment score. UPR: Unfolded protein response gene set. (C,D) GSEA analysis showing changes in expression of the UPR genes in MV4-11 and MOLM13 upon KDSR depletion. Heatmap showing the top 20 depleted genes in sgKDSR conditions. (E) Western blot of KDSR and UPR checkpoint proteins in MV4-11 and 293T cells transduced with control and KDSR-targeting sgRNAs. (F) Wright-Giemsa stain of MV4-11 cells and (G,H) TEM of MV4-11, 293T, and SW620 cells transduced with control and KDSR-targeting sgRNAs. Red arrows indicate ER.
KDSR mediates sphingolipid metabolism and ER stress response in leukemia cells
To investigate which domains of KDSR are essential to the leukemia cells, we applied an unbiased high-density CRISPR tiling scan (37–39) in MV4-11 cells (Figure 4A; CRISPR screen steps refer to Figure 1A). This approach allows a saturation mutagenesis screen of the KDSR coding exons with an average density of 8.1 base pairs (or 2.7 amino acids) per sgRNA (Table S2). Using a local-smoothing strategy that accounts for the overall behaviour of neighbouring sgRNAs, we observed a depletion of clusters of sgRNAs and smoothened CRISPR tiling scores targeting the N-terminal signal peptide (residues M1 – P25) and C-terminal ER-transmembrane domain (residues G271 – I314), suggesting the requirement of ER membrane targeting for KDSR. The CRISPR tiling scan also revealed the critical roles of KDSR’s NADP binding motif (residues V36 – T60) and catalytic core (residues E139 – M202), indicating the involvement of KDSR’s enzymatic activity (Figure 4B)(25) in leukemia maintenance.
Figure 4. Accumulation of KDS after KDSR depletion dysregulates the UPR pathway and ER structure.
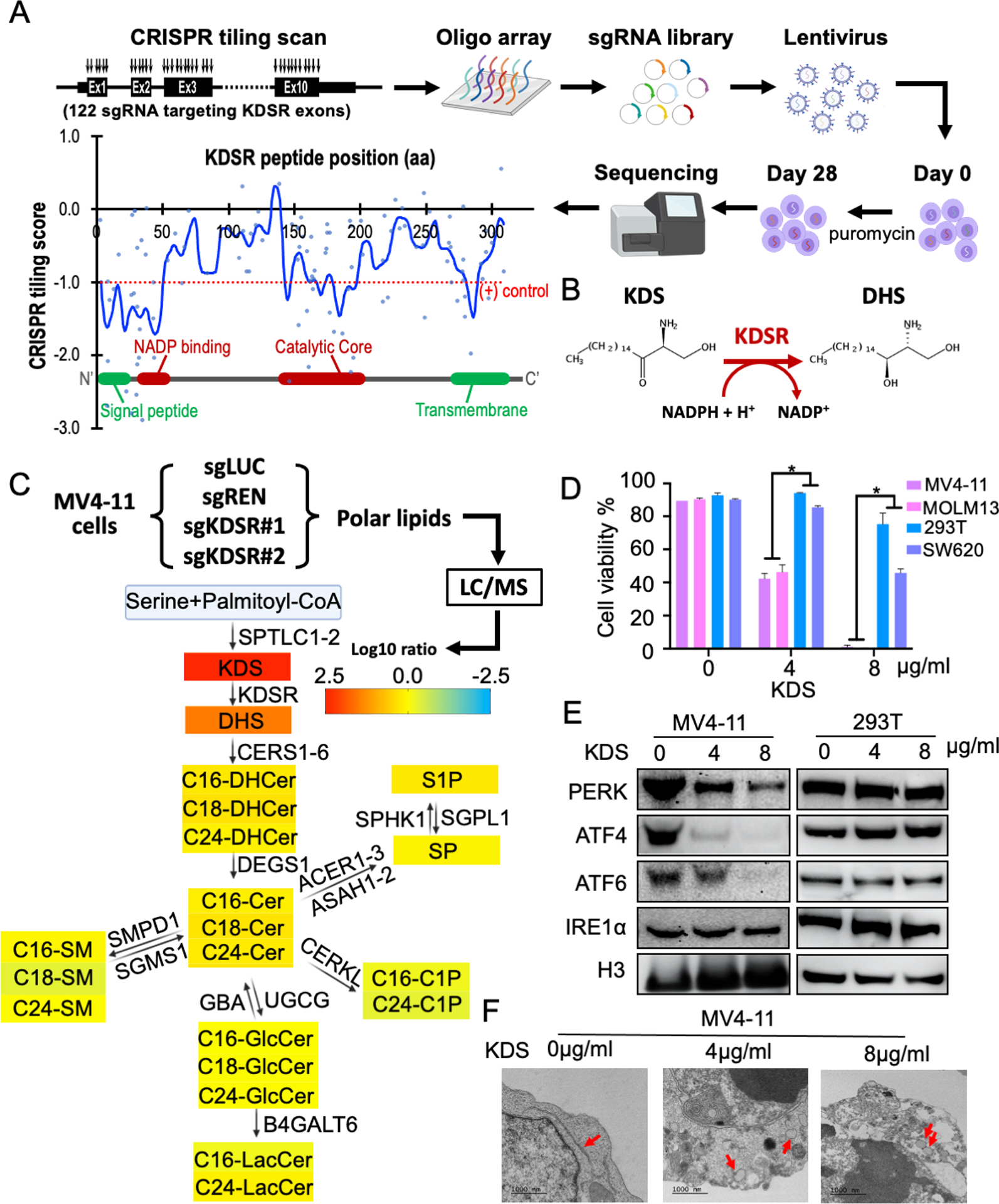
(A) High-density CRISPR tiling scan of KDSR in MV4-11 cells. Blue line indicates the smoothened model of CRISPR tiling score derived from 122 sgRNAs (blue dots) targeting the coding exons of KDSR. Red dotted line indicates the median value of the positive control sgRNAs. (B) Catalytic reaction of KDSR. (C) Annotation of sphingolipid alterations over the sphingolipid metabolism network. The value represents the average log10-fold change of each sphingolipid upon KDSR-depletion in MV4-11 cells observed by mass spectrometry. (D) Summary of cell viability in MV4-11, MOLM13, 293T, and SW620 cells with KDS treatment. Data are represented as mean ± SD of triplicated experiments. *P < 0.01 using Student’s t-test. (E) Western blot of KDSR and UPR checkpoint proteins in MV4-11 and 293T cells treated with KDS. (F) TEM of MV4-11 cells treated with KDS. Red arrows indicate ER.
The requirement of domains involved in the enzymatic function prompted us to investigate the impact of KDSR depletion on sphingolipid homeostasis. We evaluated 33 sphingolipid components by mass spectrometry and revealed a pronounced accumulation of KDS (more than 200-fold increase) in KDSR depleted MV4-11 cells (Figure 4C and S4), whereas the amount of other sphingolipid species was less affected. We then sought to investigate whether accretion of KDS can suppress leukemia cells. Our results revealed that direct addition of KDS in culture medium for 48 hours led to significant cytotoxicity (Figure 4D), loss of UPR proteins (Figure 4E), and aberrant ER structures (Figure 4F) in MV4-11 leukemia cells. The unaffected cell survival and ER stress response in the KDSR-independent 293T and SW620 cells argues for a unique sensitivity of leukemic cells to KDSR depletion via KDS accumulation and ER malfunction.
We then sought to investigate the relationship between KDSR and ER stress response. Consistent with previous reports(40, 41), leukemia cells (MV4-11 and MOLM13) exhibited pronounced sensitivities to multiple ER stress inducers including tunicamycin, tanespimycin, thapsigargin, and bortezomib (Figure 5A and S5). Interestingly, the KDSR-independent cell types (293T and SW620) are remarkably more tolerant of the chemically-induced ER stress. We further examined tunicamycin, a compound that blocks N-linked glycosylation and protein folding in the ER lumen (42, 43). Our results revealed a dose-dependent disruption of UPR proteins and ER morphology in MV4-11 cells (Figure 5B and C), resembling the effects triggered by KDSR-depletion (Figure 3E, G) and KDS overload (Figure 4E, F). Based on these observations, we hypothesized that treatments leading to ER stress might synergize with KDSR/KDS perturbation to suppress leukemia maintenance. Indeed, CRISPR/Cas9 depletion of KDSR significantly increased the sensitivity of MV4-11 and MOLM13 leukemia cells to tunicamycin treatment (Figure 5D). Similarly, KDS synergized with tunicamycin to suppress cell viability specifically in MV4-11 (δ score = 5.75) and MOLM13 (δ score = 2.46) leukemia cells, but not in the KDSR-independent 293T (δ score = 1.03) and SW620 (δ score = 1.31) cells (Figure 6). Furthermore, we observed similar synergistic interactions between KDS and tunicamycin in other hematopoietic disease models including KOPN-8 (B-ALL; δ score = 1.96), RS4;11 (B-ALL; δ score = 2.06), CCRF-CEM (T-ALL; δ score = 3.78), Jurkat (T-ALL; δ score = 7.03), SET2 (acute megakaryoblastic leukemia; δ score = 6.58), Jeko-1 (Mantle cell lymphoma; δ score = 3.47), Ramos (Burkitt lymphoma; δ score = 4.94), and Hut78 (T cell lymphoma; δ score = 5.81) cells (Figure 6). In summary, our study revealed the critical roles of KDSR in sphingolipid metabolism and ER homeostasis, representing a novel therapeutic candidate in hematopoietic malignancies.
Figure 5. Targeting KDSR sensitizes the leukemia cells to ER stress inducer tunicamycin.
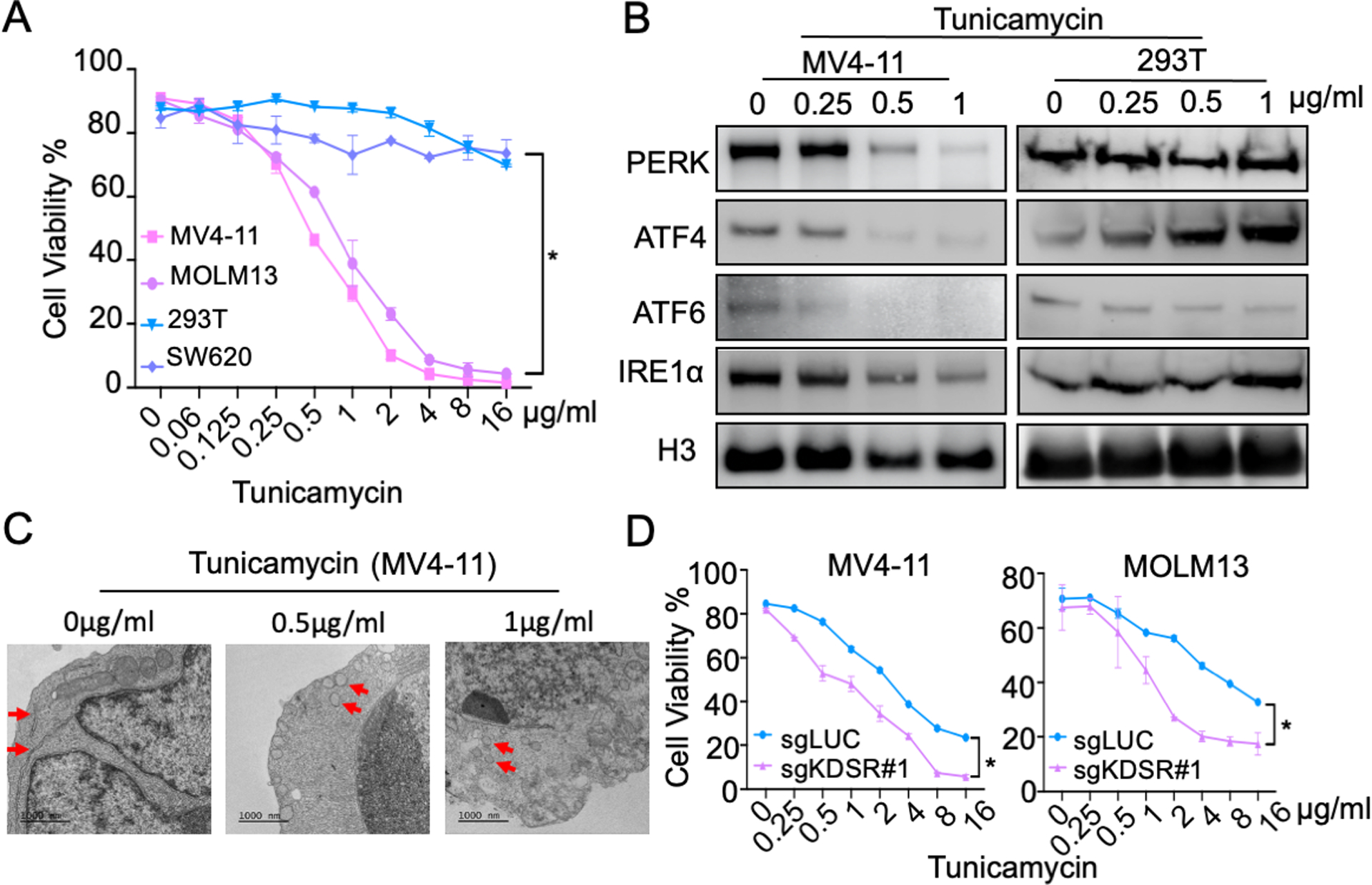
(A) Effect of tunicamycin on cell viability of KDSR-dependent (MV4-11, MOLM13) and -independent (293T, SW620) cells. (B) Western blot of KDSR and UPR checkpoint proteins in MV4-11 and 293T cells treated with tunicamycin. (C) TEM of MV4-11 cells treated with tunicamycin. Red arrows indicate ER. (D) Effect of tunicamycin on cell viability of MV4-11 and MOLM13 cells transduced with sgKDSR#1. Data are represented as mean ± SD of triplicated experiments. *P < 0.01 using Student’s t-test.
Figure 6. KDS synergizes with tunicamycin in leukemia and lymphoma.
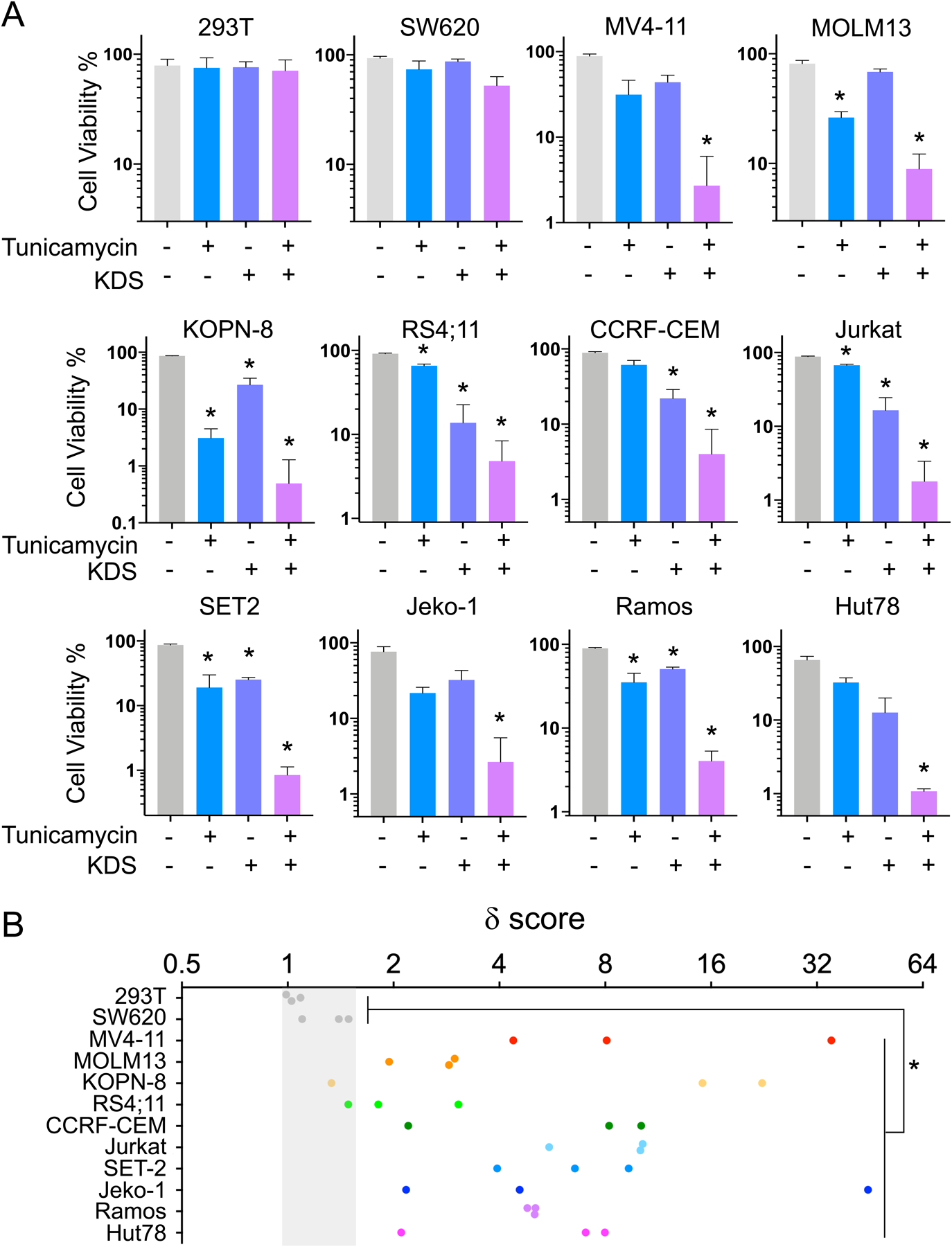
(A) Viability of cells treated with vehicle (grey), tunicamycin alone (blue), KDS alone (purple), or the combination of tunicamycin and KDS (pink). Data are represented as mean ± SD of triplicated experiments. *P < 0.001 compare to vehicle using Student’s t-test. (B) δ score of leukemia and lymphoma cells (colored dots) compared to 293T and SW620 cells (grey dots). *P < 0.001 using Student’s t-test.
DISCUSSION
In this study, we uncovered that KDSR plays a crucial role in leukemia cell survival by maintaining the UPR, a cellular response triggered by ER stress typically caused by the accumulation of misfolded proteins (18). Leukemia is characterized by the accumulation of gene mutations and chromosomal translocations that the encoding proteins are frequently prone to misfolding (23, 44, 45). It has been reported that sustained UPR exists in multiple leukemia types and plays a vital role in disease progression and therapeutic resistance (19, 20, 46–48). Thus, targeting ER stress and UPR mechanisms represents attractive therapeutic options to the leukemia treatment (49–53).
Using the sphingolipid metabolism focused CRISPR/Cas9 genetic screens (Figure 1), we identified KDSR to be critical for leukemia cell maintenance. While targeting KDSR exerted slower cytotoxicity as compared to targeting other known leukemia essential genes (e.g., MYC, RPS20, PCNA, and BRD4), disruption of these fundamental mechanisms (e.g., RPS20 for protein translation and PCNA for DNA replication) triggered cell death in almost all the cell types screened in the DepMap database. Conversely, we observed a stronger KDSR-dependency in leukemia than non-leukemia cell types (Figure 2C and S2A), supporting the selective role of KDSR in leukemia maintenance. It is also worth mentioning that compare to the DepMap database (using 3 – 4 sgRNAs targeting each gene; screen coverage typically around 100 – 200 cells per sgRNA), our focused library screen utilized 25 sgRNAs targeting each gene with a coverage of approximately 1,000 cells per sgRNA. This strategy provides higher resolution and statistical confidence to determine the candidate gene function, thereby offering orthogonal and non-redundant evaluations to the publicly available genome-wide CRISPR library screens.
KDSR, also known as FVT-1 (25, 54),is an ER transmembrane protein that catalyzes the transition of KDS to DHS during the de novo sphingolipid biosynthesis. Prior studies implicated the involvement of KDSR in thrombocytopenia (55, 56), keratoderma (57, 58), and hepatic injury (59); however, the linkage of KDSR to leukemia development and therapeutics has not been described. Our sphingolipid profiling revealed that disruption of KDSR led to a drastic accumulation of its upstream substrate KDS, whereas the majority of sphingolipid species were not significantly impacted. It was reported that DHS, the direct downstream product of KDSR, can be synthesized from hexadecenal and ethanolamine phosphate through an alternative pathway in yeast (60). Although the pathways that bypass KDSR to generate DHS have not been identified in mammalian cells, our results implicate the blockade of de novo sphingolipid biosynthesis in KDSR-disrupted cells is compensated by alternative mechanisms similar to what was observed in the primitive organisms.
On the other hand, our CRISPR tiling scan identified the essential roles of KDSR’s catalytic core and the NADP binding motif (Figure 4A). Overloading of KDS also resembled the selective killing effect on leukemia cells triggered by KDSR-depletion. Of note, direct addition of KDS to the leukemia cell cultures exerted a faster cytotoxic effect (within 48 hours of treatment) compared to the genetic suppression of the KDSR gene (takes a longer period to accumulate an excessive KDS), indicating the aberrant KDS level as an effector leading toward impaired ER structure and UPR function upon KDSR knockout. These findings suggest the reductase function of KDSR and the metabolism of KDS is likely involved in leukemia maintenance, arguing the potential of targeting KDSR’s NADP binding site or the critical catalytic residues in its enzymatic core for therapeutics development. Additionally, the amphiphilic nature of KDS (contains a hydrophilic head and a hydrophobic tail) may provide unique opportunities for future combinational therapy. For example, KDS can be formulated as “liposomes” to increase its stability in the aqueous phase, and the inner space of the engineered liposomes is eligible for carrying tunicamycin or other ER stress inducers. We envision an adequate evaluation of the KDSR/ER combinational targeting in preclinical models will provide novel pharmacotherapeutic opportunities for leukemia treatment.
The relationship between KDSR/KDS and ER-stress/UPR has not been elucidated previously. Here we showed that both KDSR depletion and KDS overload disrupt the ER morphology. Swollen ER was also observed in other ER protein dysregulations (61–63) including Atlastin, a GTPase required for homotypic fusion of ER membrane (64). Interestingly, the ER density (i.e., ER count per view; Figure S6) in the KDSR-dependent cell models (MV4-11 and MOLM13) was significantly lower than the insensitive cell types (293T and SW620). Given the high nuclear/cytoplasmic ratio typically observed in leukemia cells, the net ER number (and capacity) could be constrained by the limited cytoplasmic space in leukemia, which may contribute to a pronounced sensitivity to KDSR/KDS disruption and ER stress. We also speculate that the obstruction of ER membrane dynamics may perturb the stability of proteins embedded in it, including the key UPR components (65). Interestingly, while the level of PERK and ATF6 was eliminated upon KDSR inhibition or KDS treatment, the expression of another UPR effector IRE1α was not affected. The selective exhaustion of UPR components raises a hypothesis that the sphingolipids such as KDS may play signaling roles in controlling the diverse UPR machinery. Indeed, DHS and DHCer were recently reported to activate ATF6 directly on its luminal domain (66), supporting the possibility that KDS (a precursor of DHS/DHCer) may inhibit the UPR activation through competing against DHS/DHCer.
In conclusion, our study highlighted that KDSR-mediated KDS metabolism is required for leukemia maintenance. Disruption of KDSR (or KDS homeostasis) synergizes with pharmacologically induced ER stress, providing critical rationales toward a more effective combinatorial therapy against leukemia. Furthermore, the insights into the roles of sphingolipid metabolic enzymes in malignancies and therapeutic responses may prompt future efforts aiming to discover novel classes of molecules targeting their catalytic cores. Although the ER stress/UPR pathways are recognized to play important roles in multiple diseases, studies on ER-targeted therapy have been focused primarily on inhibiting a single mechanism to off-balance the ER. The dynamic interplays between sphingolipid metabolism and the therapeutic outcome are just beginning to gain recognition. This study thus represents one of the emerging research fields that explores how the specific lipid metabolites coordinate in a broad spectrum of biological processes such as membrane dynamics, organelle homeostasis, leukemogenesis and therapeutic responses.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the American Society of Hematology (ASH Scholar Award to C.-W.C.), Alex’s Lemonade Stand Foundation (ALSF Innovation Award to C.-W.C.), National Institutes of Health Grants CA197498, CA233691, CA236626 (to C.-W.C.), Graduate Academic Exchange Scholarship of Fujian Medical University (to Q.L.), and Riggs-Union International Exchange Scholarship of Fujian Medical University Union Hospital (to Q.L.). Research reported in this publication included work performed in the City of Hope’s Mass Spectrometry and Proteomics Core Facility (Dr. Gabriel Gugiu) and Electron Microscopy Core Facility (Drs. Zhuo Li and Ricardo Zerda) supported by the National Cancer Institute of the National Institutes of Health P30 Grant CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATION
- KDS
3-ketodihydrosphingosine
- DHS
dihydrosphingosine
- DHCer
dihydroceramide
- Cer
ceramide
- GlcCer
glucosylceramide
- GalCer
galactosylceramide
- LacCer
lactosylceramide
- SM
sphingomyelin
- C1P
ceramide-1-phosphorate
- SP
sphingosine
- S1P
sphingosine-1-phosphorate
Footnotes
CONFLICT OF INTEREST
The authors declare no competing interests.
AVAILABILITY OF BIOLOGICAL MATERIALS
Cas9-expressing MV4-11 and MOLM13 leukemia cells will be available upon request. CRISPR gene-tiling library for KDSR will be available upon request. All other biological materials are commercially available.
CODE AVAILABILITY STATEMENT
The computational codes/tool packages used in this study are available through developers and venders, including Genetic Perturbation Platform, CLC Main Workbench, Bowtie2, and Gaussian kernel smoothing in R.
DATA AVAILABILITY STATEMENT
Datasets can be found in Tables S1 and S2. RNA-Seq data generated in this study is available in the Gene Expression Omnibus (GEO: GSE167688). Other data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Warner JK, Wang JC, Hope KJ, Jin L, Dick JE. Concepts of human leukemic development. Oncogene. 2004;23(43):7164–77. [DOI] [PubMed] [Google Scholar]
- 2.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15(9):494–501. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Jiang PYZ, Sun H, Zhang X, Jiang Z, Li Y, et al. Advances in targeted therapy for acute myeloid leukemia. Biomark Res. 2020;8:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zjablovskaja P, Florian MC. Acute Myeloid Leukemia: Aging and Epigenetics. Cancers (Basel). 2019;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogretmen B Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18(1):33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327(5961):46–50. [DOI] [PubMed] [Google Scholar]
- 7.Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis AC, Wallington-Beddoe CT, Powell JA, Pitson SM. Targeting sphingolipid metabolism as an approach for combination therapies in haematological malignancies. Cell Death Discov. 2018;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezombes C, de Thonel A, Apostolou A, Louat T, Jaffrezou JP, Laurent G, et al. Overexpression of protein kinase Czeta confers protection against antileukemic drugs by inhibiting the redox-dependent sphingomyelinase activation. Mol Pharmacol. 2002;62(6):1446–55. [DOI] [PubMed] [Google Scholar]
- 10.El-Assaad W, Kozhaya L, Araysi S, Panjarian S, Bitar FF, Baz E, et al. Ceramide and glutathione define two independently regulated pathways of cell death initiated by p53 in Molt-4 leukaemia cells. Biochem J. 2003;376(Pt 3):725–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarvis WD, Fornari FA Jr., Browning JL, Gewirtz DA, Kolesnick RN, Grant S. Attenuation of ceramide-induced apoptosis by diglyceride in human myeloid leukemia cells. J Biol Chem. 1994;269(50):31685–92. [PubMed] [Google Scholar]
- 12.Wang H, Maurer BJ, Liu YY, Wang E, Allegood JC, Kelly S, et al. N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Mol Cancer Ther. 2008;7(9):2967–76. [DOI] [PubMed] [Google Scholar]
- 13.Baran Y, Salas A, Senkal CE, Gunduz U, Bielawski J, Obeid LM, et al. Alterations of ceramide/sphingosine 1-phosphate rheostat involved in the regulation of resistance to imatinib-induced apoptosis in K562 human chronic myeloid leukemia cells. J Biol Chem. 2007;282(15):10922–34. [DOI] [PubMed] [Google Scholar]
- 14.Tan SF, Liu X, Fox TE, Barth BM, Sharma A, Turner SD, et al. Acid ceramidase is upregulated in AML and represents a novel therapeutic target. Oncotarget. 2016;7(50):83208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wallington-Beddoe CT, Powell JA, Tong D, Pitson SM, Bradstock KF, Bendall LJ. Sphingosine kinase 2 promotes acute lymphoblastic leukemia by enhancing MYC expression. Cancer Res. 2014;74(10):2803–15. [DOI] [PubMed] [Google Scholar]
- 16.Schwarz DS, Blower MD. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell Mol Life Sci. 2016;73(1):79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattarai KR, Chaudhary M, Kim HR, Chae HJ. Endoplasmic Reticulum (ER) Stress Response Failure in Diseases. Trends Cell Biol. 2020;30(9):672–5. [DOI] [PubMed] [Google Scholar]
- 18.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–29. [DOI] [PubMed] [Google Scholar]
- 19.Tanimura A, Yujiri T, Tanaka Y, Hatanaka M, Mitani N, Nakamura Y, et al. The anti-apoptotic role of the unfolded protein response in Bcr-Abl-positive leukemia cells. Leuk Res. 2009;33(7):924–8. [DOI] [PubMed] [Google Scholar]
- 20.Sudsaward S, Khunchai S, Thepmalee C, Othman A, Limjindaporn T, Yenchitsomanus PT, et al. Endoplasmic reticulum stress, unfolded protein response and autophagy contribute to resistance to glucocorticoid treatment in human acute lymphoblastic leukaemia cells. Int J Oncol. 2020;57(3):835–44. [DOI] [PubMed] [Google Scholar]
- 21.Zhou C, Martinez E, Di Marcantonio D, Solanki-Patel N, Aghayev T, Peri S, et al. JUN is a key transcriptional regulator of the unfolded protein response in acute myeloid leukemia. Leukemia. 2017;31(5):1196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martelli AM, Paganelli F, Chiarini F, Evangelisti C, McCubrey JA. The Unfolded Protein Response: A Novel Therapeutic Target in Acute Leukemias. Cancers (Basel). 2020;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masciarelli S, Capuano E, Ottone T, Divona M, Lavorgna S, Liccardo F, et al. Retinoic acid synergizes with the unfolded protein response and oxidative stress to induce cell death in FLT3-ITD+ AML. Blood Adv. 2019;3(24):4155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goda AE, Erikson RL, Sakai T, Ahn JS, Kim BY. Preclinical evaluation of bortezomib/dipyridamole novel combination as a potential therapeutic modality for hematologic malignancies. Mol Oncol. 2015;9(1):309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kihara A, Igarashi Y. FVT-1 is a mammalian 3-ketodihydrosphingosine reductase with an active site that faces the cytosolic side of the endoplasmic reticulum membrane. J Biol Chem. 2004;279(47):49243–50. [DOI] [PubMed] [Google Scholar]
- 26.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34(2):184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527(7577):192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 31.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mascorro JA, Bozzola JJ. Processing biological tissues for ultrastructural study. Methods Mol Biol. 2007;369:19–34. [DOI] [PubMed] [Google Scholar]
- 34.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adamson B, Norman TM, Jost M, Cho MY, Nunez JK, Chen Y, et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell. 2016;167(7):1867–82 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89. [DOI] [PubMed] [Google Scholar]
- 37.Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016;6(8):900–13. [DOI] [PubMed] [Google Scholar]
- 38.Schoonenberg VAC, Cole MA, Yao Q, Macias-Trevino C, Sher F, Schupp PG, et al. CRISPRO: identification of functional protein coding sequences based on genome editing dense mutagenesis. Genome Biol. 2018;19(1):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33(6):661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosati E, Sabatini R, Rampino G, De Falco F, Di Ianni M, Falzetti F, et al. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood. 2010;116(15):2713–23. [DOI] [PubMed] [Google Scholar]
- 41.Morin MJ, Porter CW, McKernan P, Bernacki RJ. The biochemical and ultrastructural effects of tunicamycin and D-glucosamine in L1210 leukemic cells. J Cell Physiol. 1983;114(2):162–72. [DOI] [PubMed] [Google Scholar]
- 42.Lehle L, Tanner W. The specific site of tunicamycin inhibition in the formation of dolichol-bound N-acetylglucosamine derivatives. FEBS Lett. 1976;72(1):167–70. [DOI] [PubMed] [Google Scholar]
- 43.Yoo J, Mashalidis EH, Kuk ACY, Yamamoto K, Kaeser B, Ichikawa S, et al. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol. 2018;25(3):217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan AKN, Chen CW. Rewiring the Epigenetic Networks in MLL-Rearranged Leukemias: Epigenetic Dysregulation and Pharmacological Interventions. Front Cell Dev Biol. 2019;7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Federici L, Falini B. Nucleophosmin mutations in acute myeloid leukemia: a tale of protein unfolding and mislocalization. Protein Sci. 2013;22(5):545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schardt JA, Mueller BU, Pabst T. Activation of the unfolded protein response in human acute myeloid leukemia. Methods Enzymol. 2011;489:227–43. [DOI] [PubMed] [Google Scholar]
- 47.Haefliger S, Klebig C, Schaubitzer K, Schardt J, Timchenko N, Mueller BU, et al. Protein disulfide isomerase blocks CEBPA translation and is up-regulated during the unfolded protein response in AML. Blood. 2011;117(22):5931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giuli MV, Diluvio G, Giuliani E, Franciosa G, Di Magno L, Pignataro MG, et al. Notch3 contributes to T-cell leukemia growth via regulation of the unfolded protein response. Oncogenesis. 2020;9(10):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kharabi Masouleh B, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2014;111(21):E2219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun H, Lin DC, Guo X, Kharabi Masouleh B, Gery S, Cao Q, et al. Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget. 2016;7(14):18736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pellegrini P, Selvaraju K, Faustini E, Mofers A, Zhang X, Ternerot J, et al. Induction of ER Stress in Acute Lymphoblastic Leukemia Cells by the Deubiquitinase Inhibitor VLX1570. Int J Mol Sci. 2020;21(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kharabi Masouleh B, Chevet E, Panse J, Jost E, O’Dwyer M, Bruemmendorf TH, et al. Drugging the unfolded protein response in acute leukemias. J Hematol Oncol. 2015;8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zi J, Han Q, Gu S, McGrath M, Kane S, Song C, et al. Targeting NAT10 Induces Apoptosis Associated With Enhancing Endoplasmic Reticulum Stress in Acute Myeloid Leukemia Cells. Front Oncol. 2020;10:598107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krebs S, Medugorac I, Rother S, Strasser K, Forster M. A missense mutation in the 3-ketodihydrosphingosine reductase FVT1 as candidate causal mutation for bovine spinal muscular atrophy. Proc Natl Acad Sci U S A. 2007;104(16):6746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takeichi T, Torrelo A, Lee JYW, Ohno Y, Lozano ML, Kihara A, et al. Biallelic Mutations in KDSR Disrupt Ceramide Synthesis and Result in a Spectrum of Keratinization Disorders Associated with Thrombocytopenia. J Invest Dermatol. 2017;137(11):2344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bariana TK, Labarque V, Heremans J, Thys C, De Reys M, Greene D, et al. Sphingolipid dysregulation due to lack of functional KDSR impairs proplatelet formation causing thrombocytopenia. Haematologica. 2019;104(5):1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boyden LM, Vincent NG, Zhou J, Hu R, Craiglow BG, Bayliss SJ, et al. Mutations in KDSR Cause Recessive Progressive Symmetric Erythrokeratoderma. Am J Hum Genet. 2017;100(6):978–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huber M, Chiticariu E, Bachmann D, Flatz L, Hohl D. Palmoplantar Keratoderma with Leukokeratosis Anogenitalis Caused by KDSR Mutations. J Invest Dermatol. 2020;140(8):1662–5 e1. [DOI] [PubMed] [Google Scholar]
- 59.Park KH, Ye ZW, Zhang J, Hammad SM, Townsend DM, Rockey DC, et al. 3-ketodihydrosphingosine reductase mutation induces steatosis and hepatic injury in zebrafish. Sci Rep. 2019;9(1):1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skrzypek MS, Nagiec MM, Lester RL, Dickson RC. Analysis of phosphorylated sphingolipid long-chain bases reveals potential roles in heat stress and growth control in Saccharomyces. J Bacteriol. 1999;181(4):1134–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Akiyama M, Hatanaka M, Ohta Y, Ueda K, Yanai A, Uehara Y, et al. Increased insulin demand promotes while pioglitazone prevents pancreatic beta cell apoptosis in Wfs1 knockout mice. Diabetologia. 2009;52(4):653–63. [DOI] [PubMed] [Google Scholar]
- 62.Esk C, Lindenhofer D, Haendeler S, Wester RA, Pflug F, Schroeder B, et al. A human tissue screen identifies a regulator of ER secretion as a brain-size determinant. Science. 2020;370(6519):935–41. [DOI] [PubMed] [Google Scholar]
- 63.Orso G, Pendin D, Liu S, Tosetto J, Moss TJ, Faust JE, et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460(7258):978–83. [DOI] [PubMed] [Google Scholar]
- 64.Moss TJ, Andreazza C, Verma A, Daga A, McNew JA. Membrane fusion by the GTPase atlastin requires a conserved C-terminal cytoplasmic tail and dimerization through the middle domain. Proc Natl Acad Sci U S A. 2011;108(27):11133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hetz C The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. [DOI] [PubMed] [Google Scholar]
- 66.Tam AB, Roberts LS, Chandra V, Rivera IG, Nomura DK, Forbes DJ, et al. The UPR Activator ATF6 Responds to Proteotoxic and Lipotoxic Stress by Distinct Mechanisms. Dev Cell. 2018;46(3):327–43 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets can be found in Tables S1 and S2. RNA-Seq data generated in this study is available in the Gene Expression Omnibus (GEO: GSE167688). Other data that support the findings of this study are available from the corresponding author upon reasonable request.