

Abstract
Androgen receptor (AR) signaling continues to play a dominant role in all stages of prostate cancer (PC), including castration-resistant prostate cancers (CRPC) that have developed resistance to second-generation AR antagonists such as enzalutamide. In this study, we identified a long non-coding RNA (lncRNA), NXTAR (LOC105373241), that is located convergent with the AR gene and is repressed in human prostate tumors and cell lines. NXTAR bound upstream of the AR promoter and promoted EZH2 recruitment, causing significant loss of AR (and AR-V7) expression. Paradoxically, AR bound the NXTAR promoter, and inhibition of AR by the ACK1/TNK2 small molecule inhibitor (R)-9b excluded AR from the NXTAR promoter. The histone acetyltransferase GCN5 bound and deposited H3K14 acetylation marks, enhancing NXTAR expression. Application of an oligonucleotide derived from NXTAR exon 5 (NXTAR-N5) suppressed AR/AR-V7 expression and prostate cancer cell proliferation, indicating the translational relevance of the negative regulation of AR. In addition, pharmacological restoration of NXTAR using (R)-9b abrogated enzalutamide-resistant prostate xenograft tumor growth. Overall, this study uncovers a positive feedback loop, wherein NXTAR acts as a novel prostate tumor-suppressing lncRNA by inhibiting AR/AR-V7 expression, which in turn upregulates NXTAR levels, compromising enzalutamide-resistant prostate cancer. The restoration of NXTAR could serve as a new therapeutic modality for patients who have acquired resistance to second-generation AR antagonists.
Significance
This study identifies NXTAR as a tumor suppressive lncRNA that can epigenetically downregulate AR/AR-V7 expression and provides a therapeutic strategy to reinstate NXTAR expression for treating recurrent CRPC.
Keywords: LncRNA, AR, ACK1, small molecule inhibitor, Prostate Cancer, CRPC, (R)-9b, NXTAR-N5
Graphical Abstract
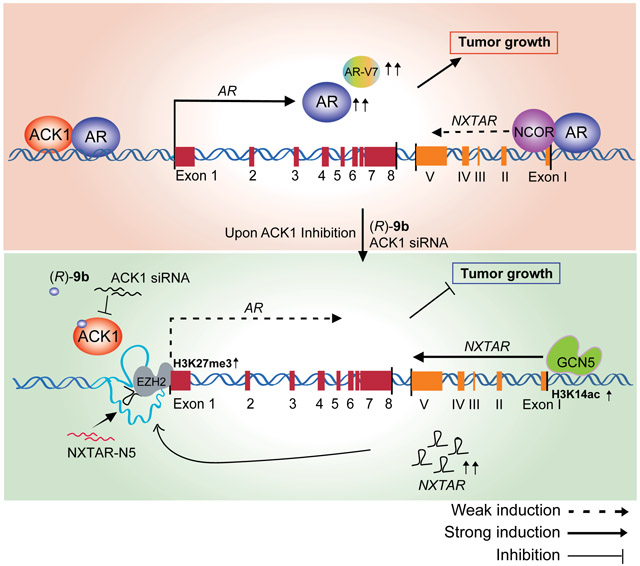
INTRODUCTION
Prostate cancer (PC) is the second most reported cancer in American men (1). The limited efficacy of androgen deprivation therapies often results in progression of the disease to lethal castration-resistant prostate cancer (CRPC) in 1 to 2 years, with reversion of androgen receptor (AR) activity (2-4). To combat CRPCs, abiraterone, an androgen biosynthesis inhibitor, and enzalutamide, a drug that inhibits AR nuclear translocation, have been widely used; however, the overall disease-free survival for these two AR antagonists as single agents has been modest (5,6), and their combination has provided little advantage (7). Robust amplification of the AR enhancer region in almost 81% of enzalutamide-treated patients reinforces the notion that AR signaling is the epicenter of drug resistance (8-10). The bypassing of AR signaling by increased expression of glucocorticoid receptor (GR) is thought to be one of the early mechanisms of enzalutamide resistance (11). A pluripotency transcription factor, SOX2, was later shown to be required for the lineage plasticity and enzalutamide resistance through the loss of TP53 and RB1 (12). AR regulating its own expression by epigenetic modification (phospho-Tyr88-H4) of its enhancer in an enzalutamide-rich environment has emerged as another mechanism to maintain high AR levels and thus confer resistance (13). In addition, many CRPC patients develop resistance by expressing an AR splice variant called AR-V7 (14) that lacks the ligand-binding domain, nullifying the effects of these two AR antagonists. Collectively, these data have firmly established that AR is not only important in the pre-CRPC scenario, but also retains a dominant role in the post-CRPC stage, including in the drug-resistant state.
The identification of a large number of noncoding RNAs (ncRNAs) has firmly established the much-awaited paradigm that the human genome is pervasively transcribed, regardless of the protein coding abilities of the resultant RNAs (15), with ncRNAs comprising almost 90% of the transcribed genome (16). Long noncoding RNAs (lncRNAs), typically >200 nt long, with mostly no evident open reading frames (ORFs) but sometimes with a polyadenylated tail, form a large portion of these ncRNAs (17,18). The surge in studying these lncRNAs has revealed their important contribution in myriad biological processes, including cell proliferation, drug resistance, metabolism, apoptosis, and senescence (19-23). LncRNAs often participate in these processes by orchestrating enzymatic protein regulation or degradation or by fine-tuning different chromatin states, indicating that deregulation of certain lncRNAs could lead to the development of a disease.
Because AR activity is paramount to all stages of PC, the importance of PC-specific or PC-regulating lncRNAs has long been speculated (24). Studies have revealed that successive binding of AR to two overexpressed lncRNAs in PC, PRNCR1 and PCGEM1, enhances its binding to androgen response elements (AREs) so as to induce AR-mediated gene expression in androgen-dependent or CRPC tumors (25). ARLNC1 (AR-regulated long noncoding RNA 1) was not only induced by the AR protein, but it also stabilized the AR transcript via RNA–RNA interaction (26). Overall, several studies have revealed the significance of lncRNA in AR signaling (27); however, direct negative regulation of AR and AR-V7 transcription by lncRNA has not yet been reported. Serendipitously, we identified a novel tumor-suppressor lncRNA, NXTAR, that not only suppressed AR and AR-V7 transcription, but also when NXTAR expression was reinstated, it inhibited the proliferation of enzalutamide-resistant PC cells and tumor development in prostate models. Here, we present key data to demonstrate the importance of the NXTAR lncRNA and its potential therapeutic utility using a therapeutic oligonucleotide derived from this lncRNA.
MATERIALS AND METHODS
Cell lines, plasmids, retroviral constructs, and siRNAs
RWPE-1 (normal human prostate cell line; CRL-11609; RRID:CVCL_3791), 22Rv1 (CRL-2505; RRID:CVCL_1045), LNCaP (CRL-1740; RRID:CVCL_1379), VCaP (CRL-2876; RRID:CVCL_2235), and PC3 (CRL-1435; RRID:CVCL_0035) cells were obtained from the American Type Culture Collection. LAPC4 and C4-2B cells were grown as described earlier (13). Cells were grown in 5% charcoal-stripped FBS (Gibco, Cat#12676029) containing medium (CSM), mimicking androgen deprivation, or serum-free medium (SFM) for experimental purposes. All the cell lines were routinely tested for Mycoplasma (ATCC, Cat#30-1012K). Small interfering (si)RNAs for AR and ACK1 are as described previously (13). NCOR1 siRNA was purchased from Horizon. pBABE-puro retroviral expression plasmids (kindly provided by Dr. Mani Sendurai) were used for NXTAR expression.
Human subjects
Patients provided written informed consent and the study was conducted under Washington University in St. Louis institutional review board approved genitourinary banking protocol (HRPO#:201411135). Human prostate tumor and adjacent normal samples were obtained from the written consented prostate cancer patients following radical prostatectomy at the Washington University School of Medicine in St. Louis, Urologic surgery. The specimens were de-identified before processing in the laboratory. The normal (far from the tumor site) and tumor (center core of cancerous lesion) tissues were dissected by a board-certified genitourinary pathologist based on MRI imaging and pathology report of the biopsies from each patient. About 2 mm of the collected tissue was H&E stained and reviewed again by pathologist, who assigned a Gleason score. All the tumor samples had Gleason score of 7-9. The specimen were washed in PBS and RNA was prepared (Qiagen).
RNA pull-down assay
A set of 5 DNA oligonucleotide probes complementary to NXTAR RNA was synthesized, with biotin labels at their 3′ end (IDT). Nonspecific oligonucleotide recognizing lacZ RNA was synthesized as controls. The sequences are shown in Supplementary Table S1. The biotinylated oligonucleotides were incubated with lysates (2h), the lysates were incubated with streptavidin magnetic beads overnight. Beads attached to RNAs were divided equally and processed for either RNA isolation or western blotting. For RNA isolation, beads were washed 3 times and then separated using proteinase K treatment. RNA was then isolated and purified, and DNA was digested in the process. The real time quantitative RT-PCR was performed on isolated RNA to check the efficiency of pull-down of NXTAR RNA. The remaining beads were heated at 80°C for 5 min in loading buffer, followed by western blotting. The blots were probed with EZH2 (CST, 5246S; RRID:AB_10694683) antibody and Actin (SCBT, sc-47778; RRID:AB_2714189) was used as the loading control.
Chromatin Isolation by RNA Purification (ChIRP) assay and NXTAR-EZH2 binding to AR promoter
Biotin labelled NXTAR-N5 (complementary to the AR promoter) and globin oligos were synthesized (IDT) (Supplementary Table S1). ChIRP were performed using lysate from formaldehyde fixed VCaP cells and oligos that were immobilized to streptavidin beads, as described earlier (28). The amount of the chromatin DNA was determined by real-time PCR. For detection of NXTAR-N5/EZH2 binding, biotin labelled NXTAR-N5 oligo and control globin oligo were incubated with VCaP cell lysates and were subjected to chromatin pull-down, followed by immunoblotting with EZH2 antibodies.
NXTAR-N5 transfection
NXTAR-N5 oligonucleotide was synthesized by IDT, and the oligonucleotide sequence is provided in Supplementary Table S1 and Fig. 7C (* represents phosphorothioate bond modifications to avoid degradation by exonucleases). Equal numbers of VCaP and 22Rv1 cells were transfected with NXTAR-N5 or globin-derived oligonucleotide using X-tremeGENE360 (Sigma-Aldrich, Cat#08724121001), and the number of live cells were counted 96 h post-transfection using trypan blue exclusion method (Sigma, Cat #T8154). RNA was prepared from these cells, and qRT-PCR [SYBR PremixEx Taq II (Tli RNase H Plus)ROX, Clontech Takara, Cat#RR82LR] for AR and AR-V7 was performed as described earlier (13).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP), quantitative RT-PCR, and ChIP-qPCR were performed as described earlier (13,29,30). Briefly, cells were either transfected, infected or treated with various inhibitors (R)-9b; CPTH2 (Sigma, Cat#C9873)], fixed in formaldehyde, lysed and ChIP was performed with antibodies recognizing AR (SCBT, sc-7305; RRID:AB_626671), GCN5 (SCBT, SC-365321; RRID:AB_10846182), H3K14ac (CST, 7627S; RRID:AB_10839410), H3K27me3 (CST, 9733S; RRID:AB_2616029), NCOR1 (Bethyl Laboratories, Cat#A301-145A; RRID:AB_873085), EZH2 (CST, 5246S; RRID:AB_10694683), or IgG (Abcam, ab2410; RRID:AB_303052), immobilized on Protein A magnetic beads (Biorad, Cat# 161-4013). The complexes were washed with ChIP buffers, eluted with elution buffer (Active Motif, Cat#53008), and subjected to reverse crosslinking, followed by RNase and proteinase K treatments. A part of soluble chromatin was processed separately at the same time and designated as input DNA. Treated ChIP DNA and input DNAs were purified using PCR-DNA purification columns (Qiagen, Cat#28106). The amount of immunoprecipitated DNA was determined by real-time PCR.
Quantitative RT-PCR
Cells were transfected, infected or treated [(R)-9b; EPZ6438, TargetMol, Cat#T1788] accordingly. RNA was used for cDNA preparation using High-Capacity cDNA Reverse Transcription Kit (Thermo Scientific Cat#4368814). Resulting cDNA was then analyzed by qPCR using an Applied Biosystems 7900 Real Time PCR System (Thermo Scientific) and SYBR Green PCR Master Mix [SYBR PremixEx Taq II (Tli RNase H Plus) ROX, Clontech Takara, Cat#RR82LR] according to the manufacturers’ instructions. Dissociation curves were generated for each plate to verify the integrity of the primers. The relative expression of RNAs was calculated using the comparative Ct or standard curve method. 18S rRNA or actin were used as internal controls. The primers used are shown in Supplementary Table S1.
Xenograft studies
All animal experiments were performed using the standards for humane care in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice breeding and colony maintenance were performed according to IACUC protocols approved by Washington University in St. Louis. 1.5x106 VCaP cells were suspended in 200 μL of PBS with 50% Matrigel (Corning, Cat#354428) and implanted subcutaneously into the dorsal flank of castrated 6-week-old male SCID mice (Charles River, Strain code:236). Once the tumors reached approximately 100 mm3, mice were randomized and injected subcutaneously with (R)-9b mesylate salt suspended in 6% Captisol (Cydex Pharmaceuticals). Tumor volumes were measured twice weekly using calipers. At the end of the study, all mice were humanely euthanized and tumors were extracted and weighed. To assess the role of NXTAR in tumor growth, 1.5x106 VCaP cells that were either infected with pBABE vector or NXTAR and selected with puromycin (Gibco, Cat#A1113803), implanted and growth of tumors was recorded. The personnel taking tumor volume measurement were blinded for the treatment groups.
Proliferation assay
The 22Rv1, VCaP, or LNCaP cells were retrovirally infected with control or NXTAR vectors and selected in puromycin medium containing 5% charcoal-stripped serum. Equal numbers of selected cells were seeded in 6-well plates followed by treatment with either (R)-9b, enzalutamide (Selleck Chemicals, Cat#S1250), or abiraterone (Selleck Chemicals, Cat#S2246), and the number of viable cells was counted by trypan blue exclusion assay.
Antibodies and western blot analysis
Cells were harvested, lysed by sonication in receptor lysis buffer (31) and 20-50 μg of protein lysates were fractionated by SDS-PAGE, and transferred onto a PVDF membrane (Immobilon, Cat# IPVH00010). After blocking in 5% nonfat dry milk (RPI, Cat#M17200) (or 3% BSA, Goldbio, Cat# A-420-500), membranes were incubated with the following monoclonal antibodies: AR (SCBT, Cat# sc-7305; RRID:AB_626671), pY276-AR (29), ACK1 (SCBT, sc-28336; RRID:AB_626629), pACK1 (Upstate, 09-142; RRID:AB_612088), EZH2, and Actin. The signals visualized by using the enhanced chemiluminescence (ECL; Pierce, 32106) system.
6XHis tagged-EZH2 purification
1mg of lysate prepared from HEK293 cells transfected with 6XHis-tagged-EZH2 plasmid was incubated with Ni-NTA agarose beads (Qiagene) overnight at 4°C. Beads were washed three times using wash buffer (20mM Tris/HCl, 200mM NaCl, 5mM imidazole; pH 7.5) and purified EZH2 was eluted using elution buffer (20mM Tris/HCl, 200mM NaCl, 200mM imidazole; pH 7.5).
EZH2 pull-down Assay
LncRNA-Protein binding study was performed as described earlier (32). Briefly, 10 pmol Biotinylated NXTAR-N5 oligo or Biotinylated Globin oligos were diluted in 40ul RNA structure buffer, heated at 90°C for 2min, immediately transferred onto ice for 2 min and then kept at RT for further 20mins. 10 uM of purified EZH2 protein was added to these biotinylated oligos with 500ul of NETN buffer and the tubes were placed at 4°C for 2hrs. Post washing with cold NETN buffer, streptavidin magnetic beads (60ul) were added into prepared oligo-protein mix and incubated at 4°C overnight. Beads were washed 3 times with NETN buffer, , followed by western blot analysis. For NXTAR-EZH2 binding studies, 10 pmol biotinylated oligos complimentary to NXTAR or lacZ oligos were incubated with 1μM RNA prepared from NXTAR overexpressing VCaP cells in hybridization buffer (50mM Tris-HCl pH 7.0, 750 mM NaCl, 1mM EDTA, 1% SDS, 15% Formamide), along with pre-washed streptavidin magnetic beads at RT for 4h. 10 uM of purified EZH2 protein was added and after incubation for 2hrs magnetic beads were washed 3 times with NETN buffer, followed by western blot analysis.
Statistical analysis
Data were summarized by descriptive statistics. Data between two groups were analyzed with unpaired Student’s t-tests using Graphpad Prism (Graphpad Software Inc., RRID:SCR_002798). The qRT-PCR gene expression in tumor normalized to matched samples of a gene was tested against one (indicating equality to matched normal samples) by one sample Wilcoxon test. All tests were 2-sided and a p-value of ≤0.05 was considered significant.
The data generated in this study are available upon request from the corresponding author.
RESULTS
AR negatively regulates the tumor-suppressor lncRNA NXTAR
We identified a convergent lncRNA (LOC105373241) positioned next to the AR gene, which we named NXTAR (Fig. 1A; Supplementary Fig. S1A). The gene is 40,180 nt long (67,784,890 to 67,744,711 nt position), encoding a 1476-nt transcript with 5 exons of 81, 94, 39, 103, and 1159 nt, respectively (Fig. 1A). NXTAR exhibited overall homology with rhesus monkey, mouse, dog, and elephant sequences, but not with chicken, Xenopus frog, zebrafish, or lamprey (Supplementary Fig. S1A). Although exon 5 showed a small stretch of homologous sequence with Xenopus, NXTAR appears to be a mammalian lncRNA, and its regions of conservation with other mammals is shown in the Supplementary Table S2.
Figure 1. NXTAR is a novel tumor suppressor lncRNA in human prostate cancer.

A, Location of NXTAR gene with respect to AR gene on chromosome X, shown in a graphical format. The exons 1-5 in NXTAR are shown in Roman numerals (I to V), to distinguish from AR exons. B-L, Total RNA isolated from paired normal human prostates and tumor samples and subjected to qRT-PCR and relative expression of NXTAR (B and F) and KLK3 (D and H) is shown. Data representing mean of relative NXTAR (C, G and J) and KLK3 (E, I and K) expression between Normal and Tumor samples is shown. L, A relative NXTAR expression in various Gleason grades of prostate tumor samples is shown. M, Total RNA isolated from various cell lines and subjected to qRT-PCR with NXTAR and 18S primers. Data are represented as mean ± SEM as in (B-L). *** p<0.001, **p < 0.01, *p ≤0.05, two-tailed Student’s t-test. NS, not significant.
Given its genomic position and convergent location (Fig. 1A), we reasoned that NXTAR could influence AR expression or might be affected by it (or both). PC cell lines were deprived of androgen, the ligand for AR, by growing them in CSM, followed by assessment of NXTAR RNA expression by qRT-PCR. LNCaP and 22Rv1cells exhibited NXTAR upregulation upon androgen deprivation (Supplementary Fig. S1B, C). This increase in NXTAR was further enhanced upon removal of growth factors by growing cells in serum-free conditions (Supplementary Fig. S1B, C), suggesting that loss of AR activity could augment NXTAR levels.
Methylation of the fourth amino acid residue from the N-terminus of histone H3, H3K4me3, is a histone modification associated with the promoters and transcription start sites (TSS) of actively transcribed genes (33). Although the AR promoter is enriched for H3K4me3 in most cancer samples, including CRPCs (34) and AR-positive LNCaP cells (Supplementary Fig. S2A), H3K4me3 deposition on the NXTAR promoter was found to be lacking in LNCaP cells (GEO:GSM945240; Supplementary Fig. S2B). These data open the possibility of an inverse correlation between AR and NXTAR expression in PC.
To test this hypothesis, we designed a primer pair that amplified the 177-bp region spanning exons IV and V of NXTAR and performed qRT-PCR of 35 human prostate cancer and paired normal prostate derived RNAs, which revealed 2 distinct scenarios. Type I: 24 out of 35 prostate cancer patients (68%) exhibited a significant decrease in NXTAR expression in tumors, compared to normal tissues (Fig. 1B, C). Among these samples, the PSA/KLK3 expression was significantly up-regulated relative to normal (Fig. 1D, E). Three out of these 10 patients (#376, 422 and 460) were treated with androgen deprivation therapy in past and were considered as CRPCs. Type II: In 11 out of 35 prostate cancer patients, NXTAR levels were either higher than normal or not significantly altered (Fig. 1F and G). The KLK3 levels in these patients are also shown (Fig. 1H and I). Data representing mean of relative NXTAR and KLK3 expression between normal and tumor samples is shown (Fig. 1J and K). In addition, we also assessed NXTAR levels in various grades of prostate cancer and observed that in low grade cancer (Gleason grade 6) there was no statistically significant change, however, NXTAR levels were significantly downregulated in Gleason grade 7 or medium-grade cancers (Fig. 1L).
Furthermore, NXTAR expression was under-expressed in various AR-positive PC cell lines, including 22Rv1, LNCaP, and LAPC4. In contrast, “normal” prostate cells (RWPE) exhibited detectable NXTAR transcript levels (Fig. 1M). The highest relative expression of NXTAR was observed in PC3 cells that have no detectable AR expression (Fig. 1M). A semi-quantitative PCR using the same set of primers showed a pattern of NXTAR expression similar to that seen in qRT-PCR of different cell lines, with the highest expression in PC3 cells (Supplementary Fig. S1D).
Inhibition of ACK1/AR signaling restores NXTAR expression, suppressing prostate tumor growth
To validate the role of AR, NXTAR expression levels were measured after depletion of AR levels in LNCaP, C4-2B, 22Rv1, and VCaP cell lines using AR siRNA (Supplementary Fig. S3A). A significant increase in NXTAR levels were seen in all PC cell lines after AR depletion (Fig. 2A-D). Because withdrawal of growth factors also enhanced NXTAR upregulation, we reasoned that growth factor-regulated tyrosine kinase signaling could suppress NXTAR expression. To determine the role of a growth factor-regulated tyrosine kinase in suppressing NXTAR expression, we focused on ACK1 because of its regulation by multiple receptor tyrosine kinases (RTKs) such as insulin receptor, HER2, PDGFR, MERTK, and EGFR (13,29,35-37). ACK1, also known as TNK2, is a non-receptor tyrosine kinase whose protein levels (by SIAH-mediated ubiquitinylation) and activation are precisely regulated; a significant increase in its activation occurs as hormone-sensitive prostate cancer progresses to CRPC (31,38-41). Kinase screen revealed that Prostate cancer stem-like cells (PCSCs) depends on ACK1 for their survival (42). Recently, we demonstrated that ACK1 regulates AR expression by epigenetically modifying the AR enhancer with a novel epigenetic mark: histone H4 Tyr88-phosphorylation (13). Recognizing the significance of ACK1/AR signaling, we developed the ACK1 small molecule inhibitor (R)-9b, which not only inhibited ACK1 kinase activity but also suppressed AR expression (13,43). LNCaP and 22Rv1 cells treated with (R)-9b exhibited robust increases in NXTAR levels in a dose-dependent manner (Fig. 2E and F). Efficient knockdown of ACK1 was obtained using two distinct sets of siRNAs (Fig. 2G). Knocking down ACK1 also led to significant induction of NXTAR levels in a time-dependent manner in LNCaP (Fig. 2H) and 22Rv1 (Figure 2I) cells.
Figure 2. AR and ACK1 negatively regulates NXTAR expression in prostate cancer cell lines.

A-D, Androgen-deprived LNCaP, C4-2B, 22Rv1 and VCaP cells were transfected with scrambled (Sc) or AR siRNA. Total RNA was isolated, followed by qRT-PCR with AR, NXTAR and 18S rRNA primers. E and F, Androgen-deprived (E) LNCaP and (F) 22Rv1 cells were treated with vehicle or (R)-9b (3.5 and 5 μM) for 48 hrs. Total RNA was isolated, followed by qRT-PCR with NXTAR and 18S rRNA primers. G, qRT-PCR was performed to assess suppression of ACK1 expression in 22Rv1 cells upon transfection with two different sets of (set I and II) ACK1 siRNA. H and I, Total RNA isolated from (H) LNCaP or (I) 22Rv1 cells in which ACK1 expression was downregulated using two different sets of siRNAs at the indicated time points and subjected to qRT-PCR. Data are represented as mean ± SEM. *** p<0.001, **p < 0.01, *p ≤0.05, two-tailed Student’s t-test. NS, not significant.
To assess whether restoration of NXTAR levels affected prostate tumor growth, enzalutamide-resistant VCaP cells were implanted subcutaneously in male severe combined immunodeficiency (SCID) mice. Once tumors became palpable, the mice were injected with either the vehicle (6% Captisol in PBS) or (R)-9b (12 or 20 mg/kg of body weight), five times a week. (R)-9b treatment significantly decreased tumor growth (Fig. 3A-C). Further, no significant decrease in body weight was observed in (R)-9b-treated mice (Fig. 3D) and no significant decreases in AR levels were noted in mice prostates or brain (Fig. 3E, F). These data suggest that (R)-9b treatment does not have off-target effects in normal tissues and may not pass the blood–brain barrier. However, the xenograft tumors exhibited a significant reduction in AR and in mRNA levels of its target genes, KLK3 and TMPRSS2 (Fig. 3G-I). In contrast, we observed a significant increase in NXTAR levels in (R)-9b-treated tumors (Fig. 3J). The immunoblotting of tumor lysates further confirmed loss of ACK1, phospho-ACK1, AR, and phospho-AR protein levels upon (R)-9b treatment (Fig. 3K). Together, these data suggest that one possible mechanism underlying the significant loss of tumor growth upon overcoming ACK1/AR signaling is the restoration of the tumor-suppressor lncRNA NXTAR.
Figure 3: (R)-9b inhibits Enzalutamide-resistant CRPC xenograft tumor growth in vivo and induces NXTAR expression.

A, Enzalutamide-resistant VCaP cells were injected subcutaneously in castrated male SCID mice. Once the tumors were palpable the mice were treated with either Vehicle (Captisol; n=7) or (R)-9b at 12mg/Kg (n=7) or 20mg/Kg (n=7) subcutaneously, five times a week. B and C, Tumor weights were recorded (B) and a photograph was taken (C). D, Graph represents weights of the vehicle and (R)-9b treated mice. E and F, Prostates (E) and brains (F) of the mice were harvested, RNA prepared, followed by qRT-PCR to determine the levels of the AR mRNA (n=4 each). G-J, Tumors were harvested, RNA prepared, followed by qRT-PCR to determine the levels of the (G) AR, (H) KLK3, (I) TMPRSS2 mRNAs and (J) NXTAR (n=4 each). For G-J, (n=4 each, 3 replicates). K, Tumor lysates (n=3 tumors in each arm) were immunoblotted by AR, pAR, pACK1, ACK1 and Actin antibodies, as shown. Data (A, B and D-J) are represented as mean ± SEM. *p ≤0.05; ** p<0.01; ***p<0.001, two-tailed Student’s t-test. NS, not significant.
Increased H3K14 acetylation on NXTAR promoter augments its expression upon ACK1 inhibition
H3K14ac is a hallmark of gene activation and it is also present on bivalent promoters that are usually inactive but primed for activation if stimulus is presented. Levels of H3K14ac correlate with the CpG content of the promoters (44). Several families of histone acetyltransferase (HAT) have been reported to mediate acetylation of Lys14 of H3, including GCN5 (45). To determine whether an increase in NXTAR upon ACK1 inhibition was due to an increase of H3K14ac deposition at its putative promoter, 22Rv1 and LNCaP cells were treated with (R)-9b alone or in combination with CPTH2, a selective GCN5 inhibitor. The significant increase in NXTAR levels observed following (R)-9b treatment was severely compromised by GCN5 inhibition in 22Rv1 and LNCaP cells (Fig. 4A-C). We checked whether this increase in NXTAR coincided with an increase in H3K14ac status in the NXTAR promoter region by designing primers for this region (designated Ppr3 and Ppr4) (Fig. 4A). ChIP revealed a significant increase in H3K14ac mark deposition at these sites in (R)-9b-treated 22Rv1 and LNCaP cells, an increase that was abrogated when (R)-9b was combined with CPTH2 (Fig. 4D, E, Supplementary Fig. S3B, C; also see Supplementary Fig. S9A-D). To further validate the role of GCN5 in deposition of H3K14ac marks, ChIP was performed using GCN5 antibodies, which revealed significant binding of GCN5 to the putative NXTAR promoter in (R)-9b-treated samples (Fig. 4F, G, and Supplementary Fig. S3D, E).
Figure 4: ACK1 inhibition induces NXTAR expression by increasing acetylation of H3K14 at NXTAR promoter.

A, Graphical representation of primers (Ppr3 and Ppr4) upstream of the NXTAR transcription start site. The numbers indicate nucleotide position. B and C, 22Rv1 (B) and LNCaP (C) cells treated with either vehicle or (R)-9b alone or in combination with a GCN5 inhibitor, CPTH2 were processed for total RNA extraction and qRT-PCR was performed with primers for NXTAR and 18S rRNA. D-E, 22Rv1 cells were treated with either vehicle or (R)-9b alone or in combination with CPTH2 and ChIP was performed using H3K14ac antibody or IgG (Supplementary Fig. S9A and B), followed by qPCR using (D) PPr4 and (E) PPr3 primers. F-G, LNCaP cells treated with (R)-9b alone or in combination with CPTH2 were subjected to ChIP using GCN5 antibody or IgG (Supplementary Fig. S9C and D), followed by qPCR using (F) Ppr4 and (G) Ppr3 primers. H-I, LNCaP cells were treated with either (R)-9b alone or in combination with CPTH2 were harvested and processed for ChIP using antibody against AR or IgG (Supplementary Fig. S9E and F), followed by qPCR using (H) PPr4 and (I) PPr3 primers. J, 22Rv1 cells were treated with either vehicle or (R)-9b, subjected to ChIP using GCN5 antibody, followed by qPCR using IRF8 binding site primers. K, VCaP cells were treated with either vehicle or (R)-9b, subjected to ChIP using NCOR1 antibody, followed by qPCR using NXTAR enhancer primers. L, VCaP cells were transfected with control siRNA and three sets of NCOR1 specific siRNAs (1, 2, 3 and pool) and the cell lysate was subjected to immunoblotting. M, RNA isolated processed from these cells was subjected to qRT-PCR using NXTAR and 18S rRNA specific primers. Data are represented as mean ± SEM. *p ≤0.05; ** p<0.01; ***p<0.001, two-tailed Student’s t-test. NS, not significant.
Androgen deprivation or suppression of AR by siRNA or (R)-9b resulted in robust transcriptional activation of NXTAR (Fig. 1 and 2). Together with the significant GCN5 binding in (R)-9b-treated samples, we reasoned that AR and GCN5 could be competing for binding the same location in the NXTAR promoter. The binding of AR at Ppr3 and Ppr4 was compromised in cells that were deprived of the growth factors or subjected to (R)-9b treatment (Fig. 4H, I, Supplementary Fig. S3F, G; also see Supplementary Fig. S9E, F). These data indicate that a decrease in AR levels or activity could promote GCN5 occupation of the NXTAR promoter, upregulating its expression by deposition of H3K14ac marks.
Nuclear receptor corepressor 1 (NCOR1) recruitment upstream of NXTAR suppressing its expression
A distal intragenic enhancer was identified within the 1st intron of the NXTAR gene (chrX: 67779247-67781971), using the ENCODE (46) (Fig. 4A, blue box). Multiple transcription factor binding sites were noticed in this region, including Nuclear receptor corepressor1 (NCOR1) which function as corepressor for AR. NCOR contains a pair of SW13/ADA2/NCoR/TFIIB (SANT) domains (47). The N-terminal SANT is a critical component of a deacetylase activation domain (DAD) that binds and activates HDAC3, promoting histone deacetylation and transcriptional repression (48). We reasoned that since inhibition of ACK1 leading to loss of AR levels reinstates NXTAR expression (Fig. 2D-I), AR might recruit corepressor such as NCOR1 to accomplish NXTAR suppression. To test this possibility, 22Rv1 cells were treated with (R)-9b, followed by ChIP with NCOR1 and GCN5 antibodies. qPCR with IRF8 binding site primers revealed increased GCN5 binding upon (R)-9b-treatment (Fig. 4J). In contrast, qPCR with NXTAR enhancer region specific primers revealed NCOR1 binding to this region, which was significantly decreased upon (R)-9b-treatment (Fig. 4K). Further, transfection with NCOR1 siRNA2, 3 and pooled siRNAs exhibited significant downregulation of NCOR1 expression (Fig. 4L), which was reflected in increased NXTAR expression (Fig. 4M). Taken together with Figure 4F-I and Supplementary Figure 3A-D, these findings uncover the molecular dynamics at NXTAR locus wherein NCOR1/AR keep NXTAR levels in check by assembling at the promoter/enhancer region, however, upon suppression of AR allows GCN5 binding to re-initiate transcriptional activity.
Epigenetic silencing of AR gene by NXTAR
BLASTN analysis revealed a sequence in exon 5 of NXTAR (the N5 region) with significant homology (in the complementary strand) to two regions of 30 and 43 nt separated by about 1607 nt in the AR promoter (Supplementary Fig. S4; see also Fig. 7A and B). These data open up the possibility that NXTAR could bind to the AR promoter, regulating its expression reciprocally. To test this hypothesis, we generated a retroviral construct expressing NXTAR and 22Rv1 and VCaP cells were infected. Upon restoration of NXTAR, a significant decrease in AR mRNA level was seen, which was also reflected in significant loss of expression of the AR target genes KLK3 and TMPRSS2 (Supplementary Fig. S5A and B). 22Rv1 cells expressed AR-V7, a variant derived from the AR locus which too was significantly compromised upon reintroduction of NXTAR (Supplementary Fig. S5A). AR protein was decreased upon NXTAR expression in a time-dependent manner in LNCaP and 22Rv1 cells (Supplementary Fig. S5C and D, respectively). This regulation of AR protein by NXTAR was further confirmed by its dose-dependent downregulation of AR in 22Rv1 cells (Supplementary Fig. S5F), in contrast, vector-transfected cells had no effect on AR levels (Supplementary Fig. S5E). Further to confirm functionality of LNCaP and 22Rv1 cells, qRT-PCR was performed; an increased AR and KLK3/PSA levels post-DHT-treatment was significantly compromised upon NXTAR restoration (Supplementary Fig. S6A-D).
To examine whether effect of ACK1 inhibitor on AR is mediated through ACK1, a ‘rescue’ experiment was performed, wherein NXTAR overexpression caused significant loss of AR levels. However, when NXTAR expression was accompanied by activated ACK1 (caACK) (49,50) expression, AR levels were restored (Supplementary Fig. S6E). As an additional evidence for interdependence of AR and NXTAR expression in imparting Enzalutamide-resistance, AR and NXTAR levels were assessed in Enzalutamide treated VCaP cells. These cells are Enzalutamide-resistant and thus exhibited marginal decrease in AR levels, which is also reflected in no change in NXTAR levels (Supplementary Fig. S6F).
To understand precisely how AR transcript levels are suppressed by NXTAR, we took a closer look at the epigenetic landscape of the AR promoter. The histone-methyltransferase enhancer of zeste homolog 2 (EZH2), a subunit of PRC2, catalyzes H3K27 trimethylation (H3K27me3), leading to chromatin compaction and subsequent silencing of genes (51). ChIP with H3K27me3 antibodies followed by qPCR using the primers spanning the region (−0.7 to +3.4 kb) around the TSS of the AR gene (Fig. 5A) revealed a significant increase in deposition of H3K27me3 marks in the AR promoter upon NXTAR restoration in LNCaP, 22Rv1 cells (Fig. 5B, Supplementary Fig. S7A, and Supplementary Fig. S9G-K and L-O) and VCaP cells (Supplementary Fig. S7C). To validate that the cause of H3K27me3 deposition is the binding of EZH2 at the AR promoter, ChIP was performed, revealing its significant binding in LNCaP cells overexpressing NXTAR (Figure 5C and Supplementary Fig. S9P-R). Similar observations were made in VCaP (Supplementary Fig. S7B) and 22Rv1 cells (Supplementary Fig. S7F-H). Further, (R)-9b treatment too caused a significant binding of EZH2 to the AR promoter in VCaP and 22Rv1 cells (Supplementary Fig. S7D, F-H), reflecting in increased H3K27me3 levels (Supplementary Fig. S7E).
Figure 5: NXTAR over-expression causes an increase in repressive methylation on AR promoter.

A, Location of primers with respect to AR gene shown in a graphical format. B, LNCaP cells were infected with either pBabe control vector or NXTAR expressing constructs and grown in androgen-free medium for 48 hrs. ChIP was performed using H3K27me3 antibody or IgG (Supplementary Fig. S9G-K and L-O), followed by qPCR with using primers spanning regions upstream and downstream of AR TSS (−0.7 kb to +3.4 kb) as shown in (A). C, LNCaP cells infected with pBabe or NXTAR expressing constructs and grown in androgen free medium for 48 hrs and ChIP was performed using EZH2 and IgG antibodies (Supplementary Fig. S9P-R), followed by qPCR. D, Biotinylated oligos complementary to NXTAR (or lacZ as control) were used to pull down NXTAR from cell lysates prepared from VCaP cells retrovirally-infected to over-express NXTAR, followed by immunoblotting for EZH2. E, VCaP cells were infected with NXTAR expressing construct and treated with EZH2 inhibitor, EPZ6438 (1μM) overnight, followed by qRT-PCR. F, NXTAR (or lacZ as control) immobilized onto streptavidin beads were incubated with purified EZH2. Pull-down were washed, followed by immunoblotting with EZH2 antibodies. Data (B, C and E) are represented as mean ± SEM. *p ≤0.05; ** p<0.01; ***p<0.001, two-tailed Student’s t-test.
To explore the potential NXTAR binding to EZH2, RNA pull-down was performed wherein DNA oligonucleotides complementary to NXTAR lncRNA (oligonucleotides complementary to lacZ as a control) were used to perform the pull-down of NXTAR RNA from VCaP cells (which was subjected to retroviral expression of NXTAR). Immunoblotting revealed that EZH2 could bind to NXTAR RNA, but not to the lacZ (Fig. 5D). NXTAR pull-down was confirmed by qPCR (Supplementary Fig. S7I). Further, treatment with EZH2 inhibitor (EPZ6438) restored the AR expression (Fig. 5E). To further assess NXTAR interaction with EZH2, we generated 6XHis-tagged EZH2 construct, which was transfected in 293T cells. EZH2 was purified using nickel affinity chromatography (binding to Ni-NTA beads), followed by elution with imidazole. NXTAR immobilized on streptavidin beads when incubated with purified EZH2, exhibited considerable binding, which was not seen with the LacZ oligo (Fig. 5F). Since, the in vitro RNA/protein binding assay was performed using tagged EZH2 purified from 293T cells, but not from bacteria, we cannot completely rule out the possibility that NXTAR could bind to proteins associated with EZH2 in PRC2 complex, e.g. EED, SUZ12, JARID2, AEBP2, RbAp46/48, and PCL (51). Taken together, these data indicate that NXTAR could bind to the AR promoter, recruit PRC2 complex, leading to silencing of AR due to the H3K27me3 marking.
Restoration of NXTAR mitigates enzalutamide-resistant prostate cancer proliferation and tumor growth
To test whether NXTAR had any effect on prostate cancer cell proliferation, 22Rv1 and VCaP cells were infected with a pBABE-puro vector or NXTAR-expressing constructs and selected using puromycin. In contrast to vector, severe loss of proliferation was seen upon NXTAR expression in VCaP (Fig. 6A) and 22Rv1 (Fig. 6B) cells. The loss of AR expression upon NXTAR expression was confirmed by immunoblotting (Supplementary Fig. S8A and B). We reasoned that a lncRNA that inhibits AR (and AR-V7) could limit the proliferation of AR antagonist-resistant prostate cancer cells. To test this hypothesis, VCaP and 22Rv1 cells expressing vector or NXTAR constructs were treated with enzalutamide, which did not result in a significant decrease in proliferation of either cell line, however, NXTAR expression significantly compromised proliferation upon enzalutamide treatment (Fig. 6C and D). Interestingly, this reduction was comparable to that observed with NXTAR overexpression alone, suggesting that NXTAR expression causing loss of AR makes enzalutamide-mediated AR inactivation irrelevant. In addition to enzalutamide, we assessed another AR antagonist that has a different mode of action: abiraterone, which suppresses androgen synthesis. Although 22Rv1 cells are sensitive to abiraterone when it is used alone, NXTAR expression further sensitized them (Fig. 6E). As a positive control, cells were treated with (R)-9b, which caused cell death in both cell lines (Supplementary Fig. S8C, D), suggesting that (R)-9b-mediated NXTAR upregulation is robust and could provide superior benefits compared with retroviral overexpression of NXTAR.
Figure 6: NXTAR restoration inhibits prostate cancer proliferation.

A and B, VCaP (A) and 22Rv1 (B) cells were infected with either pBabe vector or NXTAR expressing constructs and were seeded in androgen-deprived condition. Graph represents number of viable cells determined by trypan blue exclusion assay. C and D, VCaP (C) and 22Rv1 (D) cells infected with pBabe vector or NXTAR expressing constructs and were treated with either vehicle (10% DMSO) or enzalutamide for 9 or 5 days respectively, and viable cells were counted. E, 22Rv1 cells infected with pBabe vector or NXTAR expressing constructs, treated with either vehicle (10% DMSO) or abiraterone for 5 days and the viable cells were counted. F, 1.5 million VCaP cells that were infected either with pBabe or NXTAR vectors were implanted subcutaneously per SCID mice, and allowed to grow till they reached an average volume of ~1200mm3 for pBabe mice. G-I, The tumors were excised, photographed (G) and the tumor weights (H) and volume (I) were recorded. (J) Tumor lysates (n=3 per group) were immunoblotted. Densitometry measurement is provided below the AR blot. Data are represented as mean ± SEM. *p ≤0.05; ** p<0.01; ***p<0.001, two-tailed Student’s t-test. NS, not significant.
To examine the direct effect of NXTAR restoration on prostate tumor growth, equal numbers of VCaP infected with either pBABE vector or NXTAR constructs were injected in the SCID mice. Palpable tumors were formed around week 5 that were measured twice a week thereafter. Compared with mice injected with vector-expressing cells, mice injected with NXTAR overexpressing cells showed a significant reduction in tumor growth (Fig. 6F). The tumor burden in mice injected with NXTAR expressing cells was significantly reduced compared with that of vector-transfected cells (Fig. 6G-I). Further, the NXTAR expressing tumors exhibited significant reduction in AR protein levels (Fig. 6J).
NXTAR-N5 oligonucleotide derived from NXTAR mitigates AR expression and prostate cancer cell proliferation
As described earlier, we uncovered two regions of 30 and 43 nt upstream of the AR promoter that have complementary sequences to the N5 region of exon V of NXTAR (Supplementary Fig. S4 and Fig. 7A and B). We reasoned that binding of NXTAR upstream of the AR promoter could hinder transcription (see Fig. 7A and Graphical Abstract). To test this hypothesis, we designed an oligonucleotide corresponding to the N5 region of exon V, designated NXTAR-N5 (Fig. 7C). VCaP and 22Rv1 cells transfected with NXTAR-N5 exhibited a significant decrease in AR and AR-V7 transcription, which was also reflected in the compromised proliferation of these cells (Fig. 7D, E). The bindings of NXTAR-N5 to the upstream of AR was validated; briefly, biotinylated-NXTAR-N5 oligo was immobilized onto streptavidin beads, incubated with VCaP lysate, followed by qPCR with the primer AR Up1.1 (Fig. 7A). NXTAR-N5 bound upstream of AR, however, globin binding was significantly reduced (Fig. 7F). In addition, NXTAR-N5 interaction with EZH2 was confirmed by performing pulldown as described above (Fig. 7G). Moreover, NXTAR-N5 interaction with purified EZH2 (as described above) was confirmed by performing pulldown (Fig. 7H). Furthermore, NXTAR-N5 introduction induced histone H3K27 methylation in the AR promoter region (−0.7kb), which was confirmed by ChIP (Fig. 7I).
Figure 7: NXTAR derived oligonucleotide NXTAR-N5 suppresses prostate cancer proliferation.

A, Location of NXTAR-N5 binding region upstream of AR gene shown in a graphical format. B, BLASTN analysis showing regions of significant homology (complementarity to other strand) in the upstream regions of AR and exon 5 of NXTAR. C, The NXTAR-N5 oligonucleotide sequence is shown. *Represents a Phosphorothioate bonds modifications to avoid degradation by exonucleases. D, VCaP and 22Rv1 cells were transfected with NXTAR-N5 oligonucleotide and cell proliferation was measured using trypan blue exclusion method. E, VCaP and 22Rv1 cells were transfected with NXTAR-N5, and RNA was prepared, followed by qRT-PCR for AR and AR-V7 mRNA. F, Biotin conjugated NXTAR-N5 and Globin oligos were incubated with lysate from fixed VCaP cells and qPCR was performed for AR Up1.1 primers (see Fig. 7A). G, VCaP cell lysate was subjected to chromatin pull down using NXTAR-N5 biotin conjugated oligos, followed by immunoblotting for EZH2. Actin was used as loading control. H, NXTAR-N5 and Globin oligos were incubated with purified EZH2. Pull-down were washed, followed by immunoblotting with EZH2 antibodies. I, VCaP cells were transfected with Globin and NXTAR-N5 oligos and subjected to ChIP with H3K27me3 antibody followed by qPCR for site upstream of AR TSS (−0.7kb). Data are represented as mean ± SEM. **p<0.01. *** p<0.001.
DISCUSSION
Overall, our results demonstrate that a novel tumor-suppressor lncRNA NXTAR, located close to the AR gene, suppresses AR expression by recruiting EZH2 methyltransferase and marking the AR promoter with H3K27me3 repressive epigenetic marks. The loss of AR, in turn, allowed GCN5 acetyltransferase to bind to the NXTAR upstream region, leading to deposition of H3K14ac epigenetic marks that enhanced NXTAR expression (Graphical Abstract). To address how GCN5 is recruited, we used MotifMap, a web-based tool for genome-wide maps of regulatory elements to scan the region between enhancer and promoter. It revealed a few distinct transcription factor sites (Supplementary Table S3); the first one was the IRF-8. Interferon regulatory factors (IRFs) upon binding to a specific DNA element, also bind to coactivators such as HATs GCN5 and PCAF and become modified (52,53). ChIP with GCN5 clearly shows its binding to promoter (Ppr3/Ppr4 region) (Fig. 4F, G and Supplementary Fig. S3D-G) and adjoining IRF-8 binding region (Figure 4J). Taken together these findings reveal that NCOR1/AR assemble at enhancer/promoter region of NXTAR to maintain its low levels in prostate cancer, however, suppression of AR allows IRF-8 to recruit GCN5 to this newly vacated region, re-initiating NXTAR transcription. Significantly, this negative feed forward NXTAR-AR circuitry seems to explain low levels of NXTAR in AR-positive prostate tumors or cancer-derived cell lines. Further, interruption of this circuitry by inhibition of AR expression using the ACK1 inhibitor, (R)-9b (13) restored NXTAR levels, establishing the tumor-suppressor function of this lncRNA. Moreover, a significant loss of enzalutamide-resistant xenograft tumor growth was observed upon restoring NXTAR expression (Fig. 6). Overall, these data provide new insight into the mechanism by which prostate cancer cells maintain high AR levels continuously, i.e. suppression of NXTAR.
We did not find the downregulation of NXTAR in advanced prostate cancer in publically available databases (Supplementary Fig. S1E). Similar to other studies on lncRNAs e.g. HOTAIR in breast cancer (54), the correlation of expression with the lncRNAs and their target genes is not observed. There are several potential explanations. First, low expression of NXTAR made it hard to differentiate signal from noise in RNA-Seq data. While NXTAR was not differentially expressed in a patient cohort, a small subset of patients show a negative correlation between AR and NXTAR. Nonetheless, this data is inconclusive due to small sample size and therefore we decided not to include. Second, the AR locus is one of the most complex loci in prostate cancer. AR expression is regulated by numerous genetic and epigenetic mechanisms. In prostate cancer (particularly in advanced patients), the AR locus is constantly altered. For example, the well-known genetic alteration in prostate cancer is amplification of the AR region that results in overexpression of AR and its nearby genes including NXTAR. Although, database analysis of PC patients shows a positive correlation of expression between AR and NXTAR, this observation, however, does not negate the negative regulation of AR by NXTAR as this may be one of the many ways AR is regulated. Importantly, we show this negative regulation in our tightly controlled experimental data where the only perturbation was NXTAR expression.
What is the region within NXTAR binding to EZH2 or its interacting proteins in PRC2 complex? The initial analysis suggests that there could be 2-3 contact points between NXTAR and EZH2. The ‘RNAstructure’ MaxExpect results generated a high fidelity NXTAR RNA secondary structure composed of highly probable base pairs, shown in Supplementary Fig. 10. It shows that (a) 490-1180 nt, (b) 1270-1450 nt and (c) 290-440 nt regions form distinct secondary structures with high probability of base pairing. Interestingly, these regions are also highly conserved in rhesus monkeys (Supplementary Table S2). We believe, by virtue of high probability of base pairing and evolutionary conservation, these region/s might be involved in binding to PRC2 complex proteins.
Although at least 12 tumor-suppressor lncRNAs have been reported in androgen-dependent PC, only 3 have been shown to be involved in CRPCs: LINC00844, DRAIC, and PCAT29 (27). LINC00844 was identified to be an AR-regulated lncRNA that is downregulated in metastatic prostate cancer (55). A tumor-suppressive locus on human chromosome 15q23 has recently been reported that contains two lncRNAs, DRAIC and PCAT29, both of which are suppressed by androgen-bound AR (56,57). In contrast, NXTAR is a distinct lncRNA, both functionally and spatially; unlike the others, (i) NXTAR is convergently paired tail-to-tail with the AR gene; (ii) it negatively regulates AR and AR-V7 expression; (iii) it is, itself, negatively regulated by AR; and (iv) it’s restoration overcomes AR and AR-V7, suppressing enzalutamide-resistant xenograft tumor growth. These unique qualities of NXTAR make it a highly desirable therapeutic target for recurrent prostate cancer, a stage that has become a major cause of PC-related mortality. Future studies involving restoration of NXTAR by using the small molecule inhibitor (R)-9b or NXTAR-N5 therapeutic oligonucleotide could provide long-term beneficial response in patients with CRPC.
Supplementary Material
Acknowledgements
N.P.M. is a recipient of NIH/NCI grants (1R01CA208258 and 5R01CA227025), Prostate Cancer Foundation (PCF) grant (17CHAL06) and Department of Defense grant (W81XWH-21-1-0202). F.Y.F is supported by NIH/NCI grants (5R01CA227025).
Footnotes
Disclosure of Potential Conflicts of Interest
Washington University in St. Louis has filed a patent application ‘NXTAR-derived oligonucleotides and uses thereof’ (63/126,916). N.P.M. is named as an inventor. The Moffitt Cancer Center has filed patent application “Inhibitors of ACK1/TNK2 Tyrosine Kinase” (patent no. 9,850,216 and 10,017,478). K.M. and N.P.M. are named as inventors. ACK1 inhibitor patents have been licensed by TechnoGenesys, Inc. K.M. and N.P.M. are co-founders of TechnoGenesys, Inc., own stock, and serve as consultants.
References
- 1.Siegel RL, Miller KD and Jemal A (2020) Cancer statistics, 2020. CA: A Cancer Journal for Clinicians, 70, 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Watson PA, Arora VK and Sawyers CL (2015) Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nature Reviews Cancer, 15, 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sweeney CJ, Chen YH, Carducci M, Liu G, Jarrard DF, Eisenberger M, Wong YN, Hahn N, Kohli M, Cooney MM et al. (2015) Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N Engl J Med, 373, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lonergan PE and Tindall DJ (2011) Androgen receptor signaling in prostate cancer development and progression. J Carcinog, 10, 20–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hussain M, Fizazi K, Saad F, Rathenborg P, Shore N, Ferreira U, Ivashchenko P, Demirhan E, Modelska K, Phung et al. (2018) Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N Engl J Med, 378, 2465–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, Ozguroglu M, Ye D, Feyerabend S, Protheroe A et al. (2017) Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N Engl J Med, 377, 352–360. [DOI] [PubMed] [Google Scholar]
- 7.Attard G, Borre M, Gurney H, Loriot Y, Andresen-Daniil C, Kalleda R, Pham T, Taplin ME and collaborators P (2018) Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment. J Clin Oncol, 36, 2639–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, Foye A, Kothari V, Perry MD, Bailey AM et al. (2018) Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell, 174, 758–769 e759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, Haradhvala NJ, Freeman SS, Reed SC, Rhoades J et al. (2018) Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell, 174, 433–447 e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeda DY, Spisak S, Seo JH, Bell C, O'Connor E, Korthauer K, Ribli D, Csabai I, Solymosi N, Szallasi Z et al. (2018) A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell, 174, 422–432 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C et al. (2013) Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell, 155, 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z et al. (2017) SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science, 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahajan K, Malla P, Lawrence HR, Chen Z, Kumar-Sinha C, Malik R, Shukla S, Kim J, Coppola D, Lawrence NJ et al. (2017) ACK1/TNK2 Regulates Histone H4 Tyr88-phosphorylation and AR Gene Expression in Castration-Resistant Prostate Cancer. Cancer Cell, 31, 790–803 e798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL et al. (2014) AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med, 371, 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermüller J, Hofacker IL et al. (2007) RNA Maps Reveal New RNA Classes and a Possible Function for Pervasive Transcription. Science, 316, 1484. [DOI] [PubMed] [Google Scholar]
- 16.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S et al. (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet, 47, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quinn JJ and Chang HY (2016) Unique features of long non-coding RNA biogenesis and function. Nature Reviews Genetics, 17, 47–62. [DOI] [PubMed] [Google Scholar]
- 19.Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P et al. (2011) Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet, 43, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao X-Y and Lin JD (2015) Long Noncoding RNAs: A New Regulatory Code in Metabolic Control. Trends Biochem Sci, 40, 586–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossi MN and Antonangeli F (2014) LncRNAs: New Players in Apoptosis Control. Int J Cell Biol, 2014, 473857–473857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdelmohsen K, Panda A, Kang M-J, Xu J, Selimyan R, Yoon J-H, Martindale JL, De S, Wood WH 3rd, Becker KG et al. (2013) Senescence-associated lncRNAs: senescence-associated long noncoding RNAs. Aging Cell, 12, 890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yousefi H, Maheronnaghsh M, Molaei F, Mashouri L, Reza Aref A, Momeny M and Alahari SK (2020) Long noncoding RNAs and exosomal lncRNAs: classification, and mechanisms in breast cancer metastasis and drug resistance. Oncogene, 39, 953–974. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J et al. (2013) lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature, 500, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parolia A, Crea F, Xue H, Wang Y, Mo F, Ramnarine VR, Liu HH, Lin D, Saidy NRN, Clermont P-L et al. (2015) The long non-coding RNA PCGEM1 is regulated by androgen receptor activity in vivo. Molecular Cancer, 14, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Pitchiaya S, Cieślik M, Niknafs YS, Tien JCY, Hosono Y, Iyer MK, Yazdani S, Subramaniam S, Shukla SK et al. (2018) Analysis of the androgen receptor-regulated lncRNA landscape identifies a role for ARLNC1 in prostate cancer progression. Nat Genet, 50, 814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramnarine VR, Kobelev M, Gibb EA, Nouri M, Lin D, Wang Y, Buttyan R, Davicioni E, Zoubeidi A and Collins CC (2019) The evolution of long noncoding RNA acceptance in prostate cancer initiation, progression, and its clinical utility in disease management. Eur Urol, 76, 546–559. [DOI] [PubMed] [Google Scholar]
- 28.Chu C and Chang HY (2016) Understanding RNA-Chromatin Interactions Using Chromatin Isolation by RNA Purification (ChIRP). Methods Mol Biol, 1480, 115–123. [DOI] [PubMed] [Google Scholar]
- 29.Mahajan K, Coppola D, Rawal B, Chen YA, Lawrence HR, Engelman RW, Lawrence NJ and Mahajan NP (2012) Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. J Biol Chem, 287, 22112–22122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahajan K, Fang B, Koomen JM and Mahajan NP (2012) H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat Struct Mol Biol, 19, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C et al. (2010) Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One, 5, e9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao K, Wang X and Hu Y (2021) Identification of lncRNA-Protein Interactions by CLIP and RNA Pull-Down Assays. Methods Mol Biol, 2348, 231–242. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M et al. (2010) An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer cell, 17, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma Naomi L., Massie Charlie E., Ramos-Montoya A, Zecchini V, Scott Helen E., Lamb Alastair D., MacArthur S, Stark R, Warren Anne Y., Mills Ian G. et al. (2013) The Androgen Receptor Induces a Distinct Transcriptional Program in Castration-Resistant Prostate Cancer in Man. Cancer cell, 23, 35–47. [DOI] [PubMed] [Google Scholar]
- 35.Mahajan K and Mahajan NP (2015) ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene, 34, 4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahajan K and Mahajan NP (2010) Shepherding AKT and androgen receptor by Ack1 tyrosine kinase. J Cell Physiol, 224, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahajan K and Mahajan NP (2012) PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol, 227, 3178–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchwald M, Pietschmann K, Brand P, Gunther A, Mahajan NP, Heinzel T and Kramer OH (2013) SIAH ubiquitin ligases target the nonreceptor tyrosine kinase ACK1 for ubiquitinylation and proteasomal degradation. Oncogene, 32, 4913–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahendrarajah N, Paulus R and Kramer OH (2016) Histone deacetylase inhibitors induce proteolysis of activated CDC42-associated kinase-1 in leukemic cells. J Cancer Res Clin Oncol, 142, 2263–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahajan K, Challa S, Coppola D, Lawrence H, Luo Y, Gevariya H, Zhu W, Chen YA, Lawrence NJ and Mahajan NP (2010) Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate, 70, 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahendrarajah N, Borisova ME, Reichardt S, Godmann M, Sellmer A, Mahboobi S, Haitel A, Schmid K, Kenner L, Heinzel T et al. (2017) HSP90 is necessary for the ACK1-dependent phosphorylation of STAT1 and STAT3. Cell Signal, 39, 9–17. [DOI] [PubMed] [Google Scholar]
- 42.Mahajan NP, Coppola D, Kim J, Lawrence HR, Lawrence NJ and Mahajan K (2018) Blockade of ACK1/TNK2 To Squelch the Survival of Prostate Cancer Stem-like Cells. Sci Rep, 8, 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawrence HR, Mahajan K, Luo Y, Zhang D, Tindall N, Huseyin M, Gevariya H, Kazi S, Ozcan S, Mahajan NP et al. (2015) Development of novel ACK1/TNK2 inhibitors using a fragment-based approach. J Med Chem, 58, 2746–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karmodiya K, Krebs AR, Oulad-Abdelghani M, Kimura H and Tora L (2012) H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics, 13, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagy Z and Tora L (2007) Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene, 26, 5341–5357. [DOI] [PubMed] [Google Scholar]
- 46.Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyer LA, Latek RR and Peterson CL (2004) The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Bio, 5, 158–163. [DOI] [PubMed] [Google Scholar]
- 48.Yu JJ, Li Y, Ishizuka T, Guenther MG and Lazar MA (2003) A SANT motif in the SMRT corepressor interprets the histone code and promotes histone deacetylation. Embo J, 22, 3403–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahajan NP, Whang YE, Mohler JL and Earp HS (2005) Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res, 65, 10514–10523. [DOI] [PubMed] [Google Scholar]
- 50.Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, Earp HS and Whang YE (2007) Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc Natl Acad Sci U S A, 104, 8438–8443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Margueron R and Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature, 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masumi A, Wang IM, Lefebvre B, Yang XJ, Nakatani Y and Ozato K (1999) The histone acetylase PCAF is a phorbol-ester-inducible coactivator of the IRF family that confers enhanced interferon responsiveness. Mol Cell Biol, 19, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masumi A (2011) Histone acetyltransferases as regulators of nonhistone proteins: the role of interferon regulatory factor acetylation on gene transcription. J Biomed Biotechnol, 2011, 640610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL et al. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature, 464, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lingadahalli S, Jadhao S, Sung YY, Chen M, Hu L, Chen X and Cheung E (2018) Novel lncRNA LINC00844 Regulates Prostate Cancer Cell Migration and Invasion through AR Signaling. Mol Cancer Res, 16, 1865–1878. [DOI] [PubMed] [Google Scholar]
- 56.Malik R, Patel L, Prensner JR, Shi Y, Iyer MK, Subramaniyan S, Carley A, Niknafs YS, Sahu A, Han S et al. (2014) The lncRNA PCAT29 inhibits oncogenic phenotypes in prostate cancer. Mol Cancer Res, 12, 1081–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakurai K, Reon BJ, Anaya J and Dutta A (2015) The lncRNA DRAIC/PCAT29 Locus Constitutes a Tumor-Suppressive Nexus. Mol Cancer Res, 13, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.