

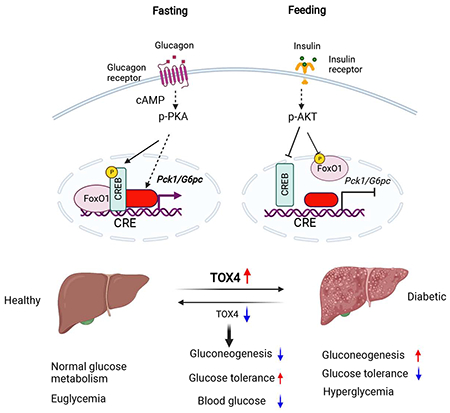
Summary
Increased hepatic glucose production (HGP) contributes to hyperglycemia in Type 2 Diabetes. Hormonal regulation on this process is primarily, but not exclusively mediated by the AKT-FoxO1 pathway. Here we show that cAMP and dexamethasone regulate the high mobility group superfamily member TOX4 to mediate HGP, independent of the insulin receptor/FoxO1 pathway. TOX4 inhibition decreased glucose production in primary hepatocytes and liver, and increased glucose tolerance. Combined genetic ablation of TOX4 and FoxO1 in liver had additive effects on glucose tolerance and gluconeogenesis. Moreover, TOX4 ablation failed to reverse the metabolic derangement brought by insulin receptor knockout. TOX4 expression is increased in livers of patients with steatosis and diabetes, and in diet-induced obese and db/db mice. In the latter two murine models, knockdown Tox4 decreased glycemia and improved glucose tolerance. We conclude that TOX4 is an insulin receptor-independent regulator of HGP and a candidate contributor to the pathophysiology of diabetes.
2. eTOC blurb:
Wang et al. have identified TOX4 as an alternative pathway to IR-FoxO1 in hormone control of hepatic glucose production. Liver TOX4 expression is increased in patients or experimental animals with diabetes, while TOX4 inhibition decreased glycemia and improved glucose tolerance, suggesting that TOX4 contributes to the pathophysiology of diabetes.
Graphical Abstract
Introduction
The liver plays a central role in glucose homeostasis by producing glucose during fasting through glycogenolysis and gluconeogenesis, and by oxidizing it or converting it into glycogen and triglycerides in response to nutrients (Han et al., 2016; Rui, 2014). As the main source of glucose during fasting, HGP is tightly controlled to maintain euglycemia. Failure to do so leads to hyperglycemia, the defining diagnostic and primary pathological contributor to T2D (Lin and Accili, 2011).
HGP is regulated by direct hepatic mechanisms as well as by nutrient flux and hormonal signals from other organs (Han et al., 2016; Lin and Accili, 2011). In hepatocytes, the balance of insulin vs. glucagon/counterregulatory hormones regulates enzymatic flux through glycolysis and oxidative phosphorylation, driving carbons into oxidative or synthetic pathways. In addition to substrate-driven flux (Samuel and Shulman, 2016), hormones also regulate transcription of genes encoding rate-limiting enzymes in these processes. Traditional examples include glucokinase (GCK), pyruvate dehydrogenase kinase-4 (PDK4), glucose-6-phosphatase (G6PC) and phosphoenolpyruvate-carboxykinase (PCK1) (Granner, 2015).
Unlike substrate fluxes, which can be assessed using isotopic and spectroscopic methods in vivo, transcription can only be assessed ex vivo (i.e., with biopsies). As a result, our knowledge of transcriptional regulation derives primarily from animal models. However, to the extent that it has been possible to match the two approaches, key elements identified by genetic and cellular biological methods have been validated. Thus, the time course of insulin inhibition of glycogenolysis in dogs matches the time course of FoxO1 inactivation (Edgerton et al., 2009). Whether this mechanism is altered in diabetes remains to be seen. Diabetic patients biopsied at the time of bariatric surgery do not have changes to mRNA levels encoding G6PC or PCK1/2 (Samuel et al., 2009), but this may well be an effect of the very low-calorie diet that precedes the surgery (Jackness et al., 2013). Fatty acids released from adipocytes promote HGP and impair the effect of insulin in mice, but only when the key transcriptional mechanism of HGP control through Akt/FoxO1 has been disabled (Lin et al., 2011; Titchenell et al., 2016). In insulin-sensitive mice, fatty acid infusions have no effect on insulin suppression of HGP (Titchenell et al., 2016). This is consistent with the modest effect of fatty acid infusions on insulin suppression of HGP in humans (Lewis et al., 1997).
Our knowledge of transcriptional regulators of hormone-dependent HGP derives primarily from transgenic and knockout mice. There are ~1,200 transcription factors (TFs) in liver (Vaquerizas et al., 2009), a sizable fraction of which is functionally regulated by hormones (Wang et al., 2019). FoxO1 plays a dominant but non-exclusive role in glycogenolysis, as demonstrated by the fact that its ablation impairs, but does not entirely abolish hormonal regulation of glucose production (Haeusler et al., 2010; Matsumoto et al., 2007). Genetic epistasis studies show that FoxO1 is the primary mediator of insulin receptor (O. Sullivan et al., 2015), IRS1/2 (Dong et al., 2008; Kubota et al., 2008), and AKT1/2 signaling (Lu et al., 2012; Titchenell et al., 2015). However, the extent to which this signaling module is required for insulin regulation of HGP remains disputed (Lu et al., 2012; O. Sullivan et al., 2015; Titchenell et al., 2015), consistent with the involvement of additional TFs. cAMP response element binding protein (CREB) and its co-activators such as CREB binding protein (CBP)/p300, CREB regulated transcription co-activator 2 (CRTC2), peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α) are also playing an essential role in mediating glucagon and other stress hormone-regulated gluconeogenesis (Erion et al., 2009; He et al., 2009; Herzig et al., 2001; Koo et al., 2005; Yoon et al., 2001).
To address this knowledge gap, we employed DNA affinity purification from the hormone-responsive Pck1 promoter and mass spectrometry in hepatocytes and liver to identify hormone-regulated TFs (Wang et al., 2019). Using this strategy, we found Thymocyte selection-associated high mobility group box factor 4 (TOX4), a member of the high mobility group box superfamily. TOX proteins play important roles in the immune system and in cancer (Scott et al., 2019), possibly as components of the PTW/PP1 phosphatase complex regulating chromatin structure and cell fate (Lee et al., 2010; Vanheer et al., 2019). Overlaying genetic, transcriptomic, and mouse metabolic phenotypes, a correlation of TOX4 expression with obesity, steatosis and hyperglycemia has emerged (Gottmann et al., 2020), further suggesting a metabolic role of TOX4.
In the present study, we employed loss-of-function approaches to explore the function of TOX4 in hepatic metabolism in dietary and genetic models of diabetes and surveyed its expression in the human diabetic liver. We show that TOX4 regulates HGP in an insulin receptor-and FoxO1-independent manner and that its expression is increased in liver biopsies of diabetic patients with nonalcoholic fatty liver disease (NAFLD). Thus, TOX4 represents a candidate alternative metabolic pathway to the main insulin-dependent transcriptional regulation.
Results
TOX4 controls hormone-regulated gluconeogenic gene expression
We used a biotin-labeled murine Pck1 promoter sequence (Pck1p, −600 to +69 bp) as a bait for DNA pulldown assays in primary hepatocytes (Wang et al., 2019). Mass spectrometry revealed that TOX4 is recruited to the Pck1 promoter following treatment with dexamethasone and cAMP (D/C), and that binding is decreased when insulin is added to the medium, similar to FoxO1 and CREB1 (Figure 1A, and S1A–C). We assessed the nuclear-cytoplasmic distribution of TOX4 but found no apparent effect of D/C on nuclear TOX4, while insulin slightly increased cytoplasmic TOX4 levels (Figure 1B). However, D/C treatment increased and insulin decreased TOX4 protein levels in primary hepatocytes (Figure S1D). Consistent with these findings, liver TOX4 levels were induced ~three-fold by fasting (Figure 1C–D), without changes to Tox4 mRNA in primary hepatocytes (Figure S1E) and liver (Figure S1F), one of the two organs expressing the highest levels of TOX4 (Figure S1G).
Figure 1. TOX4 controls hormone-regulated gluconeogenic gene expression.
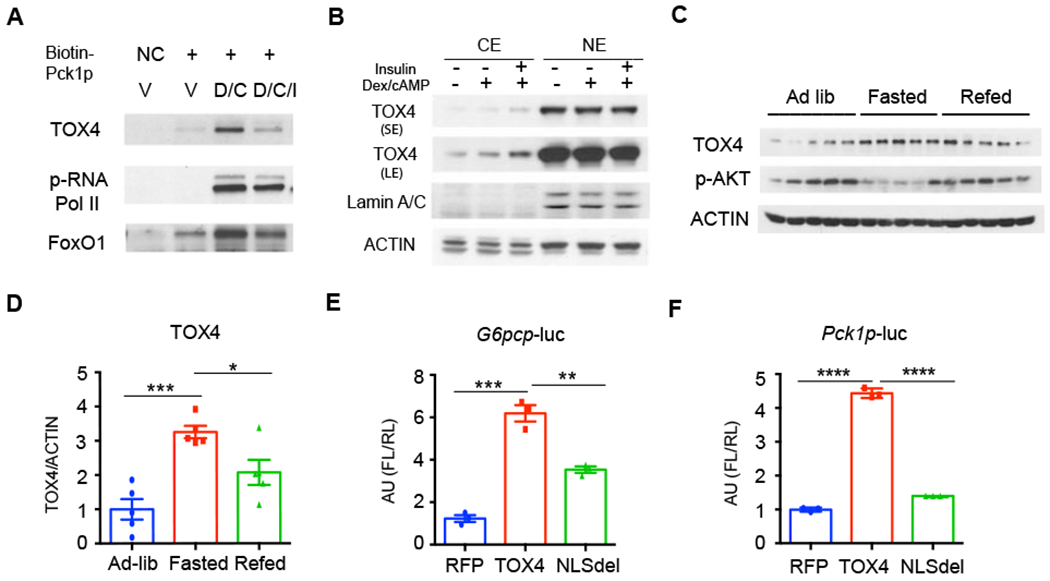
(A) Western blot (WB) of biotin-Pck1 promoter (Pck1p) pulldown of nuclear protein extracts from primary hepatocytes of 10-week-old C57BL/6J (WT) male mice treated with vehicle (V), Dexamethasone/cAMP (D/C) for 6 hrs, D/C for 6hrs then 100nM insulin for the last 30 min (D/C/I). A biotin-labeled primer oligo was used as negative control (NC).
(B) Distribution of TOX4 in cytoplasmic (CE) and nuclear extracts (NE) of primary hepatocytes treated as in A. SE: short exposure, LE: Long exposure.
(C-D) Representative gel and quantification of TOX4 in liver of ad lib, fasted and refed mice (n=5).
(E-F) G6pc promoter (G6pcp)- (E) or Pck1p-firefly luciferase (FL) (F) reporter gene regulation by wild-type or nuclear localization sequence mutant (NLSdel) TOX4 in 293 cells. pCMV-renilla luciferase and RFP were used as controls (n=3).
* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001 by one-way ANOVA. Data are presented as means ± SEM.
To explore whether TOX4 regulates the expression of gluconeogenic genes, we performed luciferase reporter assays with G6pc promoter (G6pcp) and Pck1p. TOX4 transfection activated both by ~5-fold (Figure 1E–F). This effect required an intact TOX4 nuclear localization sequence, encoded by amino acids 213-218, as a deletion mutant lacking this hexapeptide was unable to regulate either gene (Figure 1E–F). We saw similar effects when we deleted the TOX4 HMG DNA binding domain (Figure S1H–I). Thus, TOX4 can regulate the transcription of gluconeogenic genes.
Tox4 knockdown reduces gluconeogenesis and improves glucose tolerance
To determine whether this molecular function translates into a role in HGP, we performed loss-of-function studies using adenovirus encoding Tox4 shRNA (Tox4-sh). Following transduction of primary hepatocytes, we observed a ~90% decrease of TOX4 levels compared with Ctrl shRNA (Ctrl-sh) (Figure 2A). D/C induction of glucose production from pyruvate and lactate decreased by 30%, while inhibition by insulin was largely preserved in Tox4-sh-transduced primary hepatocytes (Figure 2B). Accordingly, gene expression analyses showed that the ability of D/C to induce Pck1 and G6pc was decreased by ~20% and 60%, respectively (Figure 2C). Western blots confirmed the reduction of TOX4, G6PC, PCK1, and Fructose-1,6-bisphosphatase (FBP1) in Tox4-sh-treated primary hepatocytes (Figure S2A). We obtained the same results using siRNA to lower Tox4 mRNA (Figure S2B–D). These data indicate that TOX4 is required for full induction of HGP by D/C. We also performed gain-of-function studies to explore TOX4’s function in glucose metabolism using Ad-TOX4. Overexpression TOX4 in either primary hepatocytes or mouse liver only slightly increased D/C-induced HGP but did not affect glucose metabolism in vivo (Figure S2E–I).
Figure 2. Tox4 knockdown reduces gluconeogenesis and improves glucose tolerance.
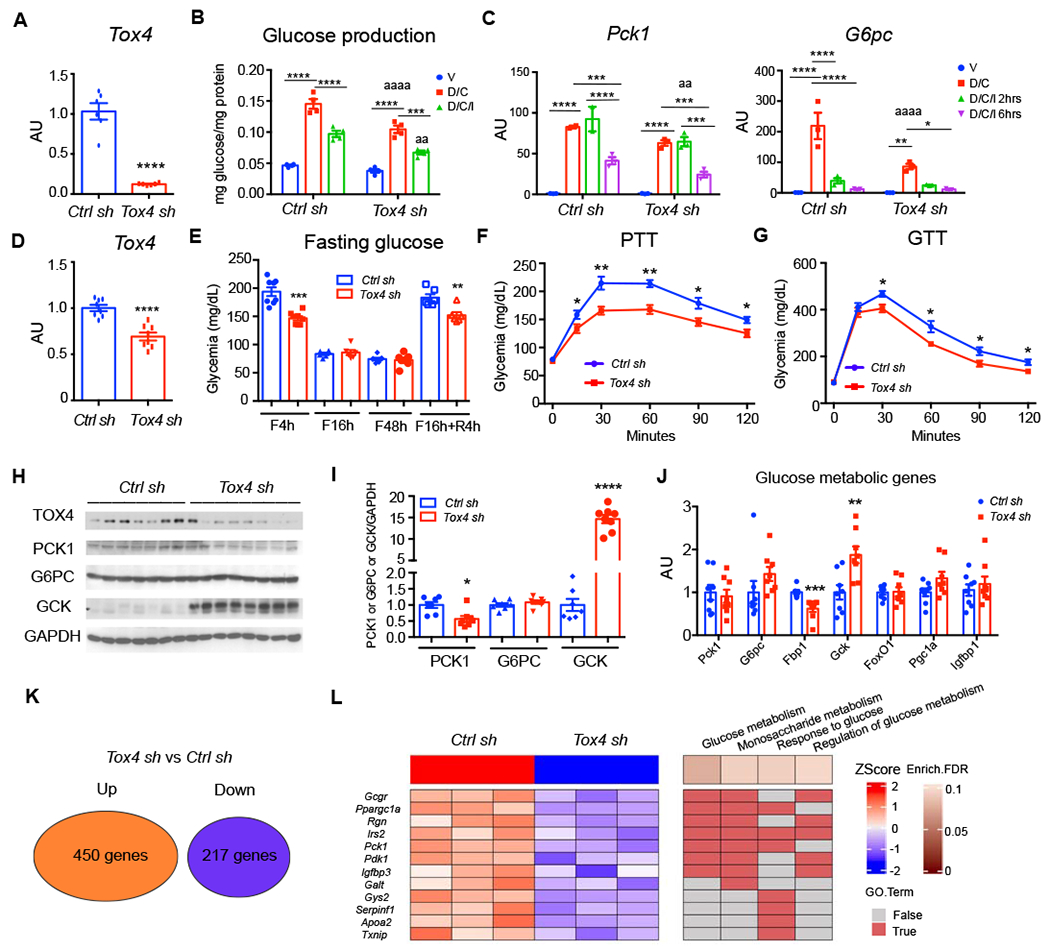
(A) Tox4 mRNA in primary hepatocytes following transduction with Ad-Tox4 shRNA (Tox4 sh, KD) or Ad-Ctrl shRNA (Ctrl sh) (n=6).
(B-C) Glucose production from pyruvate (B) (n=4), and Pck1 and G6pc mRNA (C) (n=3) in Ctrl or Tox4 sh-treated primary hepatocytes.
(D) Liver Tox4 mRNA. 10-week-old WT male mice received Ctrl sh (n=7) or Tox4 sh (n=8) and were killed after 2 weeks.
(E) Glucose levels in Ctrl sh- and Tox4 sh-treated mice at the indicated time points of fasting and refeeding.
(F) Pyruvate tolerance test (PTT) on day 6 post virus injection.
(G) Glucose tolerance tests (GTT) on day 12 post virus injection.
(H-I) WB (H) and quantification (I) of liver lysates from Ctrl sh- or Tox4 sh-treated mice after a 4-hr fast.
(J) Liver mRNA expression of glucose metabolic genes in Ctrl sh- or Tox4 sh-treated mice after 4-hr fasting;
(K) Summary of differentially expressed genes identified from RNA-seq of Ctrl sh- and Tox4 sh-treated livers (n=3 for each group). Log2FC>0.5 and FDR<0.1 were used to define the differentially expressed genes.
(L) Heatmap of down-regulated genes associated with glucose metabolism and their enriched biological processes (BPs) from gene ontology analysis.
* p<0.05, **/aa p<0.01, ***p<0.001, ****/aaaa p<0.0001. Asterisks indicate within group or Ctrl sh-treated mice comparisons, “a” indicate comparison with Ctrl sh group, by 2-tailed student’s t-test in A, D-K and 2-way ANOVA in B-C. Data are presented as means ± SEM.
Next, to assess the function of TOX4 in liver, we administered the Tox4-sh adenovirus to mice. Compared with Ctrl-sh, we achieved a 40% reduction of mRNA levels two weeks after adenovirus injection (Figure 2D). Expression of other Tox isoforms did not change in Tox4 KD liver (Figure S3A), suggesting that there is no redundancy with other Tox genes. This level of TOX4 inhibition did not affect body weight (Figure S3B), but increased liver weight by 20% (Figure S3C). It significantly reduced glycemia after a 4-hr fast or 4-hr refeeding (Figure 2E), without affecting plasma insulin (Figure S3D), lipid levels (Figure S3E–G) or alanine aminotransferase (ALT), a marker of liver function (Figure S3H). Further gene expression analysis of adipose tissue revealed that mRNAs encoding lipogenic genes were increased while Tox4 as well as adipogenic and lipolysis genes were unaffected in Tox4 sh-treated mice (Figure S3I). It suggests a modest shift in lipogenesis vs lipolysis, likely secondary to the liver effect. Metabolic tests revealed a 20-25% reduction of glucose levels following pyruvate administration (Figure 2F), and ~50mg/dl reduction of glucose levels in GTT (Figure 2G). These data were associated with a 40% decrease of PCK1 and a 13-fold increase in glucokinase (GCK), measured after a 4-hr fast (Figure 2H–I). Based on luciferase reporter assay, TOX4 itself did not affect Gck promoter activity (Figure S3J), but showed an additive effect with HNF4α-induced Gck transcription, suggesting that TOX4 regulates Gck indirectly. Periodic acid-Schiff (PAS) staining and measurements of liver glycogen content did not show differences between Ctrl sh and Tox4 sh-treated mice after a 16-hr fast (Figure S3K–L).
To interrogate the genome-wide target genes of TOX4, we performed RNA-seq from livers of 16-hr fasted animals following TOX4 KD. Differential gene expression analysis showed that levels of 450 mRNAs increased, and 217 decreased compared to controls (Figure 2K, S4A). Gene ontology analyses found that up-regulated genes were associated with immune response and viral infection (Figure S4B), while down-regulated genes were enriched in carboxylic acid and lipid metabolism (Figure S4C). Importantly, glucose metabolism genes, such as Pck1, Ppargc1a, Igfbp3, Gcgr, Irs2 were substantially decreased in TOX4 KD liver, consistent with reduced gluconeogenesis (Figure 2L), while Gck, Hk1, Hk3 increased (Figure S4D), further confirming the role of TOX4 in glycolysis. Thus, reduced levels of liver TOX4 are associated with decreased glucose production and potentially increased glycolysis. Of special note is the substantial decrease of Gcgr mRNA (Figure S4E), which is expected to lower the response to glucagon and predispose to hypoglycemia (Longuet et al., 2013).
TOX4 ablation lowers hepatocyte glucose production and improves glucose tolerance
To evaluate the chronic effects of TOX4 ablation in hepatocytes, and to rule out a contribution from other liver cell types to the process, we somatically ablated TOX4 using albumin-Cre to excise floxed Tox4 alleles (TLKO) (Figure S5A–B). Gene expression studies showed decreased Tox4, but not Tox2 or Tox3 mRNA (Figure S5C), and protein measurements confirmed that the ablation was liver-specific (Figure 3A). In primary hepatocytes from TLKO mice, D/C-induced glucose production decreased by nearly half, and residual gluconeogenesis was refractory to inhibition by insulin (Figure 3B). This was accompanied by a 25% decrease of PCK1 levels, while G6PC was unaffected (Figure 3C, Figure S5D). Thus, TOX4 ablation decreases glucose production ex vivo.
Figure 3. TOX4 ablation lowers hepatocyte glucose production and improves glucose tolerance.
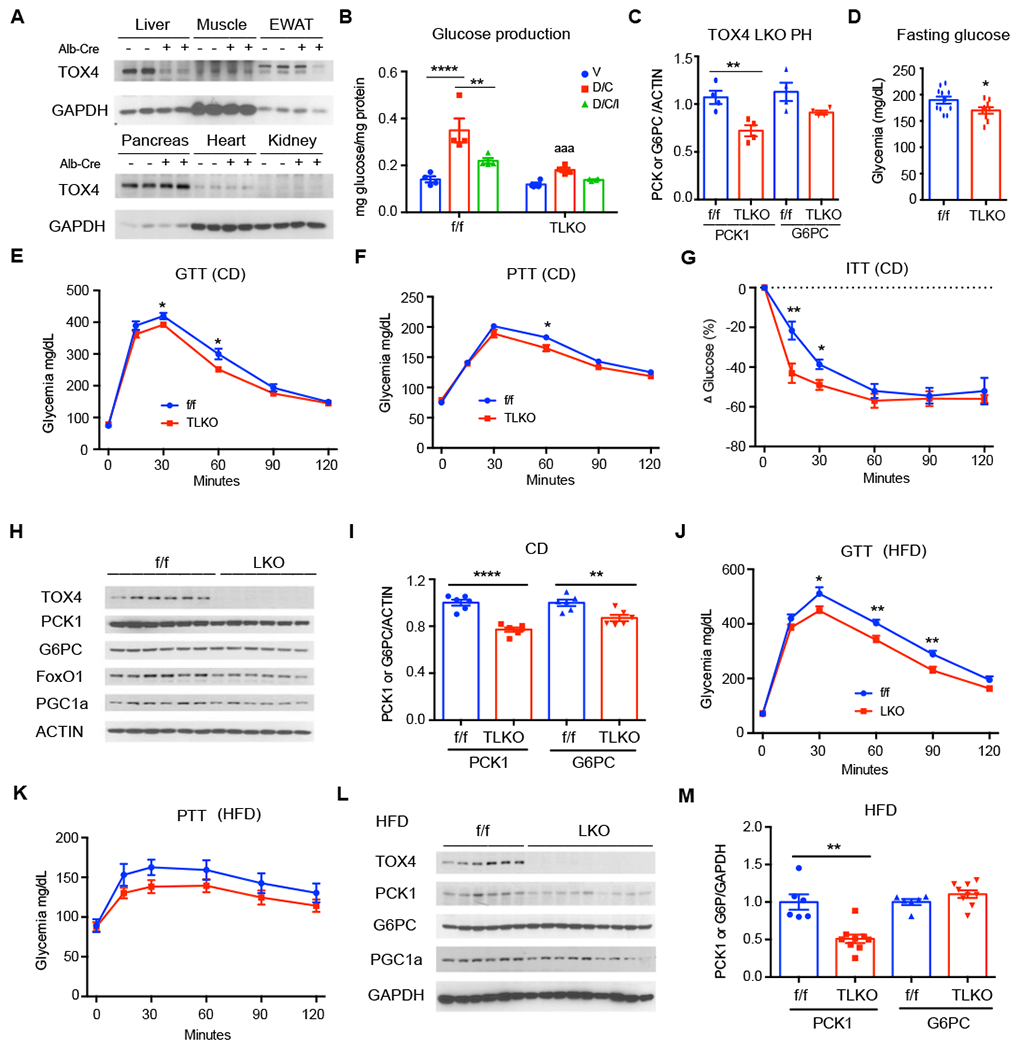
(A) TOX4 WB in tissues of TOX4 f/f and LKO mice.
(B) Glucose production in TOX4 f/f and LKO primary hepatocytes (n=4).
(C) Quantification of PCK1 and G6PC from WB of TOX4 f/f and LKO primary hepatocytes treated with or without D/C (n=4) for 6-hr (Fig.S5D).
(D) Fasting glucose in chow-fed (CD) TOX4 f/f (n=11) and LKO mice (n=9) after 4-hr fast.
(E-G) GTT in 9-week-old (E), PTT in 12-week-old (F) and insulin tolerance test (ITT, G) in 10-week old TOX4 f/f (n=11) and TOX4 LKO (n=9) mice on CD.
(H-I) WB (H) and quantification (I) of liver extracts from 16-hr-fasted, CD-fed TOX4 f/f and LKO mice (n=6,6).
(J-K) GTT after 4 weeks (I) and PTT after 7 weeks of HFD in TOX4 f/f (n=8) and LKO (n=8) mice.
(L-M) WB of liver extracts from 7-week HFD-fed TOX4 f/f and LKO mice.
*p<0.05, ** p<0.01, aaa p<0.001, **** p<0.0001, 2-way ANOVA in B and 2-tailed student’s t-test in C-M. “a” indicates comparison with f/f group. Data are presented as means ± SEM.
We investigated the metabolic effects in chow- and high fat diet (HFD)-fed mice. Neither group showed differences in body weight compared to control littermates (Figure S5E). Chowfed TLKO mice showed a 10% reduction of 4-hr fasting glucose levels (Figure 3D), with modest improvement of glucose tolerance (Figure 3E) and virtually unchanged pyruvate tolerance (Figure 3F) and increased insulin sensitivity (Figure 3G) compared to controls. Accordingly, PCK1 and G6PC were only reduced by ~25% and 10%, respectively (Figure 3H–I). We obtained similar results in mice fed HFD for one month, with normal glucose and insulin levels (Figure S5F–G), and a modest improvement of glucose, but not pyruvate tolerance or insulin sensitivity (Figure 3J–K, Figure S5H). Levels of PCK1 were further decreased to ~50% of control, while those of G6PC were unchanged (Figure 3L–M). We also saw a significant increase of Gck mRNA in chow- but not in HFD-fed mice, and no changes to other gluconeogenic genes (Figure S5J–K). The milder phenotype of constitutive vs. acute ablation suggests that chronic compensation offsets the phenotype seen following induced ablation. We have observed a similar effect in studies of FoxO1, and have identified mechanisms of transcriptional resiliency in the regulation of glucose metabolism genes that may account for this difference (Kitamoto et al., 2021).
TOX4 and FoxO1 work parallelly in regulating HGP
The gluconeogenic function of TOX4 phenocopies FoxO1 function (Matsumoto et al., 2007). To investigate their relationship, we generated TOX4/FoxO1 double knockouts (DKO). Body weight of single and double KO were similar to WT (Figure S6A), as were fasting glucose and insulin levels (Figure S6B–C). DKO mice displayed additive effects on glucose tolerance (Figure 4A–B) and pyruvate tolerance compared to single KO mice (Figure 4C–D). In contrast, liver weight increased equally slightly in single and double knockouts (Figure S6D). Liver glycogen measurements revealed no differences among these mice, despite seeming differences in the intensity of PAS staining (Figure S6E–F). PCK1 mRNA and protein levels showed a striking 90% decrease in DKO mice compared to WT, and a nearly 70% decrease compared to O1LKO (Figure 4E–G). G6pc and Ppargc1a showed a similar trend, while the decrease of Fbp1 was less pronounced (Figure 4G). These data indicate that TOX4 and FoxO1 have additive effects on glucose production and glucose tolerance, possibly through their transcriptional control of Pck1, G6pc, and Ppargc1a, while other genes, such as Fbp1, show redundant regulation.
Figure 4. TOX4 and FoxO1 work parallelly in regulating HGP.
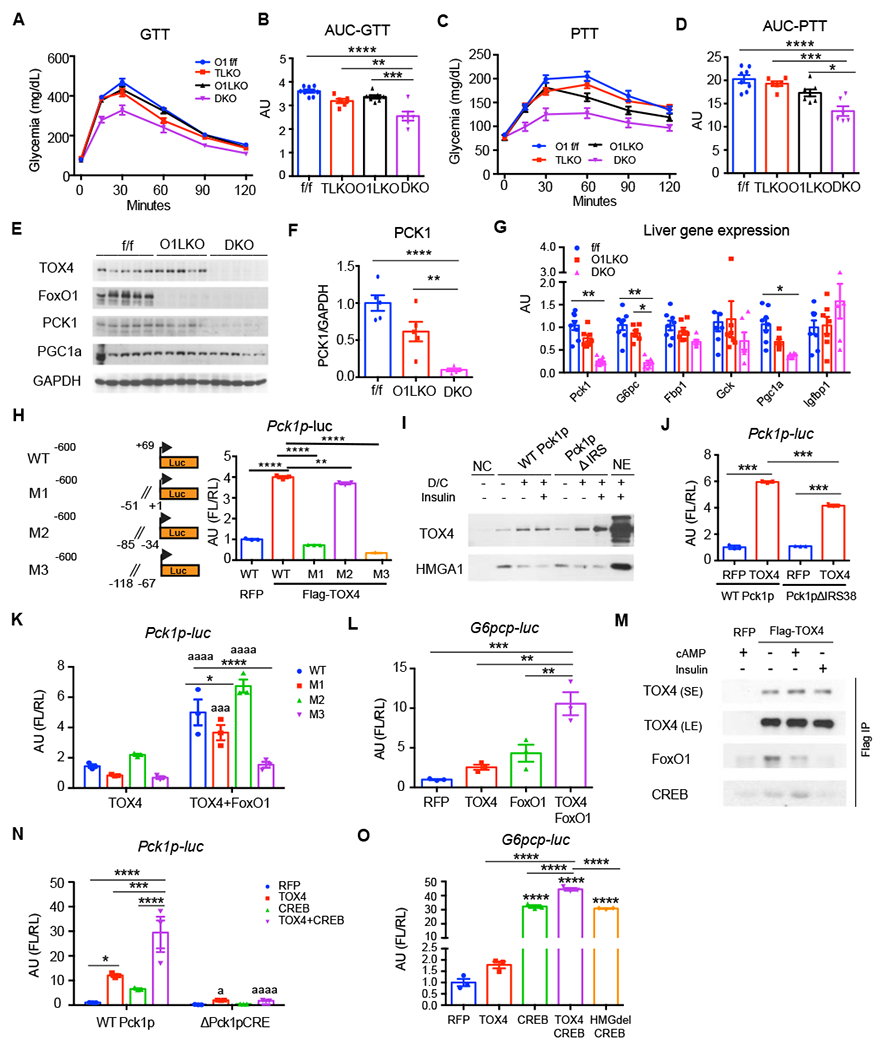
(A-B) GTT (A) and their Area Under the Curve (AUC) (B).
(C-D) PTT (C) and their AUC (D) in 17-week-old male FoxO1 f/f (n=8), TOX4 LKO (n=6), FoxO1 LKO (O1LKO, n=7) and TOX4/FoxO1 double LKO (DKO, n=6) mice.
(E-G) WB (E) and quantification (F), and liver mRNA expression (G) in liver from 4-hr-fasted mice.
(H) Identification of TOX4 binding sites on Pck1p. Dual luciferase assay in 293 cells transfected with WT or mutant Pck1p-FL (M1,M2,M3) constructs in the presence of pCMV-RL, RFP or TOX4. Left panel shows a diagram of the four constructs (n=3).
(I) Western blot of WT Pck1p or Pck1pΔIRS pulldown NE from WT primary hepatocytes.
(J) Luciferase assays of WT Pck1p or Pck1p ΔIRS38 in 293 cells transfected with RFP and TOX4 (n=3).
(K-L) Luciferase assays of WT and mutant Pck1p (K) and G6pcp (L) in the presence of TOX4 or TOX4 plus FoxO1 in H4IIE cells. All panels were normalized to RFP/WT Pck1p.
(M) Western blot of eluates from Flag-immunoprecipitation (Scott et al.) and input from RFP or Flag-TOX4-transfected AML12 cells after 30min treatment with cAMP or insulin.
(N-O) Luciferase assays of TOX4, TOX4HMGdel, CREB and their combination in co-transfections with Pck1p and ΔPck1pCRE (N) and G6pcp (O)(n=3) in 293 cells.
*/a p<0.05, **p<0.01, ***/aaa p<0.001, ****/aaaa p<0.0001 by one-way ANOVA in B-H, L and O, 2-way ANOVA in K and N, or 2-tailed student’s t-test in J as compared with f/f mice, WT or RFP or TOX4 group. Data are presented as means ± SEM.
To investigate mechanisms underlying the regulation of HGP genes, we performed gene expression and promoter assays. First, we mapped TOX4 binding sites on Pck1p using site-directed mutagenesis. We generated three mutants carrying 50bp deletions proximal to the transcription start site: M1 (−51 to −1bp deletion), M2 (−84 to −35bp deletion), and M3 (−117 to −68bp deletion) (Figure 4H). Compared with WT Pck1p, M1 and M3 failed to be induced by TOX4, while M2 showed an almost intact response (Figure 4H), suggesting that TOX4 binds to cis-acting elements between −117 and −68bp, and −51 to −1bp to regulate Pck1 transcription.
Next, we used pulldown assays of a biotin-labeled Pck1p in primary hepatocytes to determine binding of TOX4 or FoxO1 to this cis-acting element. Binding was induced by D/C and modestly suppressed by insulin (Figure 4I). Deletion of the insulin response element (IRE) had no effect on TOX4 binding, but completely abolished D/C-induced FoxO1 binding (Wang et al., 2019). However, reporter assays showed that TOX4-mediated activation of Pck1 decreased by ~30% after deleting the IRE (−433 to −396 bp) (Figure 4J). These data indicate that FoxO1 and TOX4 bind to different cis-acting elements, but FoxO1 appears to be required for full induction of TOX4-dependent activity. We next performed Pck1p- and G6pcp-luciferase assays following co-transfection of TOX4 and FoxO1. Compared with single transfection, TOX4 and FoxO1 co-transfection showed additive effects on Pck1 (Figure 4K) and G6pc reporter gene transcription (Figure 4L). This may explain the additive effects observed in DKO mice.
Given the role of TOX4 in regulating Gcgr mRNA levels, we asked whether it is involved in the cAMP response. To this end, we performed co-immunoprecipitation assays using transfection of Flag-TOX4 in 293 cells, and found endogenous CREB and FoxO1 in TOX4 immunoprecipitates (Figure 4M, S6G). The interaction between FoxO1 and TOX4 was independent of cAMP treatment but was inhibited by insulin (Figure 4M). In contrast, the interaction between CREB and TOX4 was increased upon cAMP exposure and suppressed by insulin. A TOX4 binding site (−117bp to −68bp) on Pck1p overlaps with the CREB response element (CRE, −101bp to −80bp). Co-expression of TOX4 and CREB showed additive effects on the transcriptional regulation of both Pck1p and G6pcp (Figure 4N–O). Deletion of the CRE consensus sequence (−83bp to −94bp) abolished the effect of either TOX4 and/or CREB (Figure 4N). Thus TOX4 regulate HGP at least in part through interactions with CREB.
TOX4 regulates hepatic glucose metabolism independent of insulin receptor
FoxO1 is regulated by insulin receptor signaling (Matsumoto et al., 2007; O. Sullivan et al., 2015; Titchenell et al., 2015). If TOX4 acted independently of FoxO1, we would also expect it to be non-epistatic with insulin receptor. To answer this question, we ablated both genes in the liver of mice (LIRTDKO) (Figure S7A–B). Fasting glucose decreased ~10% in LIRKO , and ~30% in LIRTDKO mice compared to controls, despite similar body weights (Figure 5A–B). Consistent with these data, glucose levels were lower at early time points during pyruvate challenge in LIRTDKO mice, but not in LIRKO mice (Figure 5C and Figure S7C–D). In contrast, the impaired glucose tolerance of LIRKO mice was not improved by ablating TOX4 (Figure 5D–E). Moreover, TOX4 deletion further increased the elevated insulin levels of LIRKO mice (Figure 5F). The decreased PAS staining in 4hr-fasted livers indicated reduced glycogen content in LIRTDKO compared with LIRKO mice (Figure S7E), which may also contribute to the decrease of fasting glucose levels.
Figure 5. TOX4 regulates hepatic glucose metabolism independent of insulin receptor.
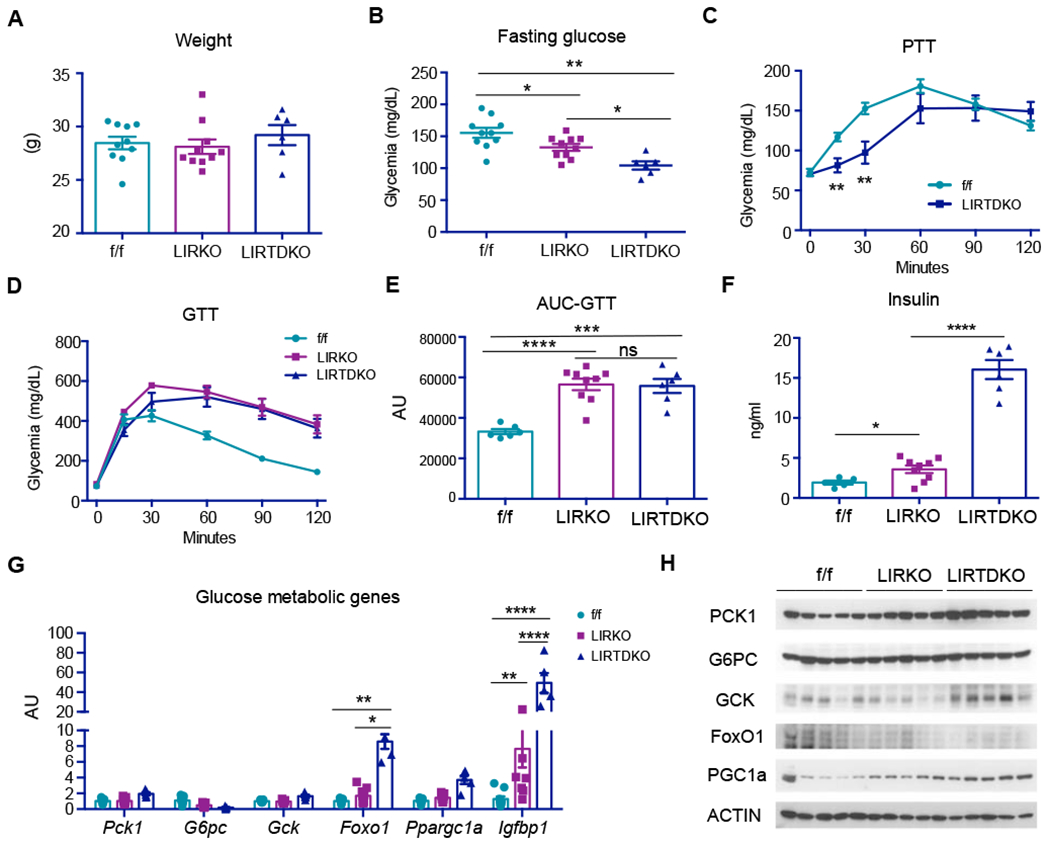
(A-B) Body weight (A) and 4-hr-fasting glucose (B) in 14- to 15-week-old male IR or TOX4 f/f (n=10), LIRKO (n=10) and liver-specific IR/TOX4 DKO (LIRTDKO, n=6) mice.
(C) PTT in 12- to 13-week-old TOX4/IR f/f (n=6) and LTIRDKO (n=9) mice.
(D) GTT, and (E) AUC in 14-week-old male IR f/f (n=6), LIRKO (n=9), LIRTDKO (n=6).
(F) 4-hr-fasting insulin of mice in (D).
(G) Liver gene expression as determined by QPCR.
(H) WB of selected liver proteins involved in glucose metabolism.
* p<0.05, **p<0.01, *** p<0.001, **** p<0.0001 by one-way ANOVA analysis in A-B, E-F; 2-tailed Student’s t-test in C and 2-way ANOVA in G. ns: no significant difference. Data are presented as means ± SEM.
Liver gene expression revealed that both Foxo1 and Igfbp1 were greatly increased in LIRTDKO mice (Figure 5G). PCK1, GCK and PGC1a levels also increased in LIRTDKO liver (Figure 5H). These data are also consistent with an impaired counterregulatory response. Thus, unlike FoxO1 deletion, TOX4 deletion has either no effect or worsens the LIRKO phenotype, consistent with a parallel pathway of metabolic regulation.
TOX4 knockdown improves glucose metabolism in diet-induced obese mice
Increased HGP contributes to fasting hyperglycemia in T2D (Han et al., 2016). Thus, we asked whether TOX4 contributes to this process. Indeed, genetic evidence links TOX4 with the glycemic trait in T2D (Figure S8). We measured TOX4 levels in liver biopsies from diabetic NAFLD patients and found a ~10-fold increase compared with healthy controls (Figure 6A–B). G6PC levels were elevated in these patients, implicating a potential increase of gluconeogenesis. Interestingly, p-AKT and total FoxO1 levels were also increased. The former is presumably a consequence of hyperinsulinemia, and is a likely cause of the increased FoxO1 levels in these patients, similar to what has been found in non-diabetic NAFLD/Non-alcoholic steatohepatitis (NASH) (Valenti et al., 2008). This induction of TOX4 was recapitulated in HFD-induced obese (DIO) mice (Figure 6C–E).
Figure 6. TOX4 knockdown improves glucose metabolism in diet-induced obese mice.
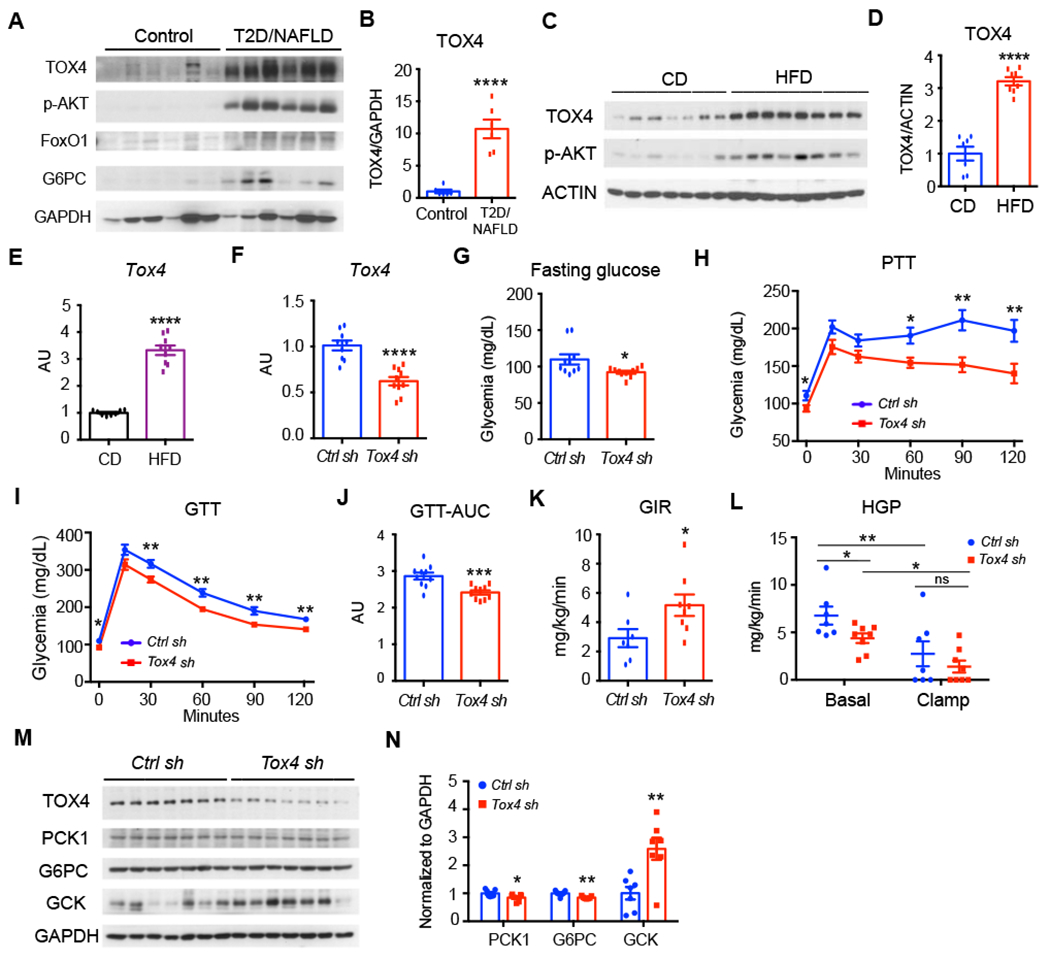
(A-B) TOX4 WB (A) and quantification (B) in human liver biopsies.
(C-E) Liver TOX4 WB (C) and quantification (D) and Tox4 mRNA (E) in 13-week-old WT male mice on chow (n=7) or HFD for 5 weeks after a 4-hr fast (n=8).
Ad-shRNA mediated TOX4 silencing in 13-week-old WT male mice fed with HFD for 5 weeks.
(F) Liver Tox4 mRNA.
(G) 16-hr-fasting glucose.
(H) PTT at day 10 post adenovirus injection. n=10 for each group.
(I-J) GTT at day 6 post adenovirus injection and their AUC (J). n=10 for each group.
(K-L) Euglycemic-hyperinsulinemic clamps at day 7 post-transduction. Glucose infusion rate (GIR) (K) , and hepatic glucose production (HGP) (L) in mice administered Ctrl sh- or Tox4 sh-adenovirus (n=7, 8 respectively).
(M-N) WB (M) and quantification (N) of liver extracts in HFD mice following administration of Ctrl sh- or Tox4 sh.
* p<0.05, ** p<0.01, *** p<0.001, p<0.0001 by 2-tailed student’s test in (B-K, O) or 2-way ANOVA (L). ns: no significant difference. Data are presented as means ± SEM.
Thus, we asked whether inhibiting TOX4 expression improves glucose metabolism in insulin-resistant, obese mice. To this end, we fed C57BL/6J male mice HFD for four weeks to induce insulin resistance and hyperglycemia, then administered Ctrl-sh or Tox4-sh adenovirus. After 2 weeks, liver Tox4 mRNA decreased by ~40% (Figure 6F). This decrease was associated with a ~20% decrease of fasting glucose (Figure 6G), lower glucose levels following pyruvate administration (Figure 6H) and improved glucose tolerance (Figure 6I–J). In contrast, body or liver weight (Figure S9A–B), as well as plasma insulin, triglyceride, total cholesterol, NEFA and ALT levels were unchanged (Figure S9C–G). Accordingly, liver glycogen content also showed no difference (Figure S9H).To further explore the effects of TOX4 inhibition on insulin sensitivity and HGP, we performed euglycemic-hyperinsulinemic clamps in DIO mice one week after administration of Ctrl-sh or Tox4-sh. TOX4 inhibition was associated with a ~60% increase of the glucose infusion rate (Figure 6K). Basal HGP was decreased by ~40% following TOX4 inhibition, and was suppressed to the same extent in both groups of animals by infusing insulin at 2.5mU/kg/min (Figure 6L). The mRNA levels of gluconeogenic genes—Pck1, G6pc and Fbp1 did not show any difference (Figure S9I). Western blots showed that TOX4 inhibition decreased PCK1 and G6PC by ~20%, while increasing GCK 2- to 3-fold (Figure 6M–N). These data provide evidence for a role of TOX4 in hepatic glucose metabolism in mice and a potential link to human NAFLD and T2D.
Silencing Tox4 ameliorates hyperglycemia and glucose intolerance in db/db mice
Next, we studied a genetic model of obese T2D, db/db mice. Tox4 mRNA and protein levels increased ~50% in these mice compared with age-matched C57BL/6J mice (Figure 7A–C), consistent with the data in HFD mice. We injected Tox4-sh adenovirus in 12-week-old db/db mice, and 12 days after transduction we saw a 60% reduction of TOX4 levels in liver (Figure 7D), but not in muscle (Figure S10A). This procedure did not affect body weight (Figure S10B), but increased liver weight (Figure S9C) compared with animals administered Ctrl-sh. We saw a ~50% decrease of ad libitum-fed and 4hr-fasted glycemia following TOX4 inhibition (Figure 7E) accompanied by improved glucose tolerance (Figure 7F, G). Plasma insulin and lipid (Figure S10D–F) were unaltered while ALT level was slightly increased (Figure S10G). Though insulin sensitivity in these mice was unchanged, glucoses levels decreased substantially in Tox4-sh treated db/db mice (Figure 7H). PCK1 and G6PC levels decreased (Figure 7I, J), and GCK1 levels increased in livers of db/db mice treated with Tox4-sh. Analyses of mRNA expression showed significant decreases of Fbp1 and Ppargc1a (Figure 7K). Histochemistry and glycogen measurement did not reveal changes to glycogen or lipid content (Figure 7L, Figure S10H). Collectively, these data show that inhibiting hepatic TOX4 reduced hyperglycemia and improved glucose tolerance in db/db mice.
Figure 7. Silencing Tox4 ameliorates hyperglycemia and glucose intolerance in db/db mice.
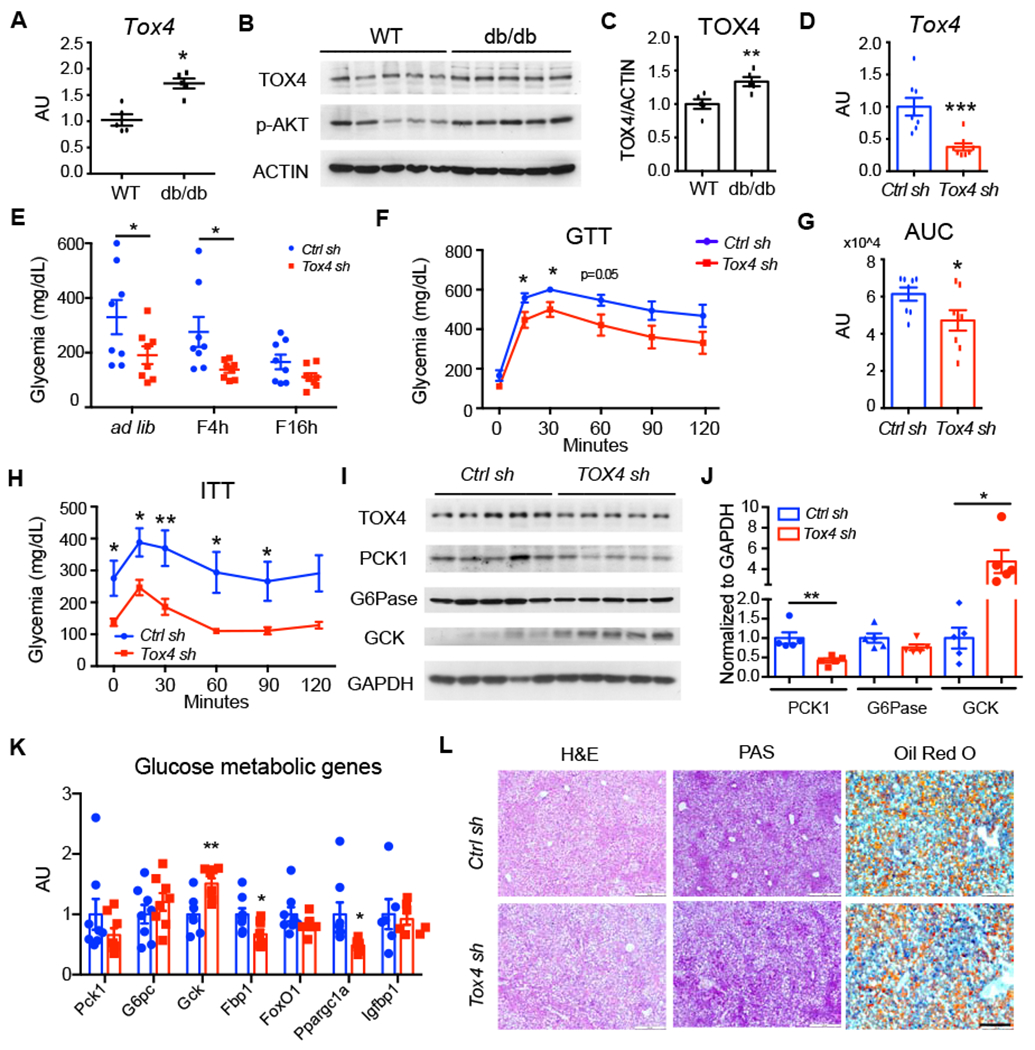
(A-C) Liver Tox4 mRNA (A) and WB (B) with quantification (C) in 4-hr-fasted, 12-wk-old male WT and db/db mice.
(D-G) TOX4 inhibition in 12-week-old db/db male mice. Liver Tox4 mRNA (D); Blood glucose (E) (* p<0.05 by 2-way ANOVA); GTT (F) and AUC (G) at day 4 post viral injection; n=8 for each group.
(H) ITT at day 7 post viral injection. n=8 for each group.
(I-J) Hepatic protein levels were determined by WB (I) and quantification (J).
(K) Liver mRNA expression in mice killed at day 10 post virus transduction.
(L) Hematoxylin and eosin (H&E), PAS, and Oil Red O staining of liver sections from Ctrl sh and Tox4 sh treated db/db mice. Scale bar, 200 μm.
* p<0.05, ** p<0.01, *** p<0.001 compared with WT or Ctrl sh-treated mice, 2-tailed Student’s t-test. Data are presented as means ± SEM.
Discussion
Key conclusions of our work are: (i) TOX4 was hitherto unknown to be activated by cAMP and dexamethasone, and to participate in the transcriptional regulation of gluconeogenic genes. (ii) Partial or complete loss-of-function of Tox4 reduced HGP in vitro and in vivo. (iii) TOX4 and FoxO1 regulate PCK1 expression through different cis-acting DNA elements. (iv) TOX4 and CREB interact with each other and cooperatively regulate PCK1 expression through the CRE. (v) Combined ablation of hepatocyte TOX4 and FoxO1 yields additive effects on HGP and glucose tolerance. (vi) Consistent with this observation TOX4 ablation failed to reverse, and partly worsened the phenotype due to insulin receptor ablation. Thus, the TOX4 pathway should be viewed as largely distinct from the insulin receptor pathway. (vii) TOX4 levels are elevated in human T2D/NAFLD and insulin-resistant mouse models, suggesting a pathological role in T2D. (viii) TOX4 inhibition in DIO and db/db mice ameliorates hyperglycemia and improves glucose metabolism, consistent with the possibility that TOX4 increases HGP during the progression of T2D.
In addition to gluconeogenesis, ablation of TOX4 in hepatocytes increased Gck, Hk1 and Hk3, indicating a potential role of TOX4 in glycolysis. Increased glycolysis may also contribute to increasing glucose tolerance and lowering glycemia following TOX4 loss-of-function. Moreover, RNAseq data highlight the potential effects of TOX4 on lipid metabolism. Genome-wide ChIP-seq will be required to build a TOX4 regulome and fully understand TOX4 function. Furthermore, it remains unclear how TOX4 is activated. We have not seen nuclear exclusion as a mechanism of TOX4 regulation; rather, it appears to be modulated via fasting/refeeding-dependent changes in protein levels. Among the various possibilities, co-factor exchange may be required to achieve differential transcriptional regulation by TOX4 upon hormone exposure; or there may be direct post-translational modifications of TOX4.
Pck1 appears to be a striking example of additivity of the FoxO1 and TOX4 pathways. Consistent with data by Burgess and associates, the effects of TOX4 on HGP in primary hepatocytes and liver paralleled the levels of PCK1 inhibition (Burgess et al., 2007). However, it’s possible that additional TOX4 targets participate in regulating HGP, primarily Gcgr. Liver-specific Gcgr KO mice exhibit fasting hypoglycemia, increased glucose tolerance and insulin sensitivity, similar to TOX4 LKO mice (Longuet et al., 2013). Furthermore, TOX4 and CREB interact through the CRE to regulate Pck1 and G6pc transcription. Therefore, TOX4 may regulate HGP through the GCGR-CREB axis., which will require further investigation. Acute inhibition of TOX4 has a stronger effect on HGP than complete somatic ablation. This observation is reminiscent of our studies of FoxO1 in liver. In that instance too, the induced knockout in adult mice has more marked effects on glucose metabolism than the constitutive somatic ablation (Kitamoto et al., 2021). We have defined a mechanism of transcriptional “resiliency” of gluconeogenic genes mediated, in the case of FoxO1, by PPARα.
The regulation of HGP, and the mechanism(s) driving its increase in T2D are complex. No animal model can be said to faithfully recapitulate the human disease process, partly because the latter is likely heterogeneous and dynamic, i.e. it changes over the course of the disease depending on patient features, degree of glycemic control, and therapies employed (Monnier et al., 2007). Rodent models are a compromise choice: the benefits of being able to discover genes and manipulate their functions being offset by the more “hepatocentric” nature of murine glucose metabolism (Lauro et al., 1998), and by the glaring differences in lipid profiles (Lin and Accili, 2011). We were therefore gratified to see that human liver biopsies in T2D patients confirmed not only the increase of TOX4 predicted based on the db/db and diet-induced diabetic rodent models, but also the increase of FoxO1 levels that would be predicted based on the underlying insulin resistance (Valenti et al., 2008), as well as increased G6PC levels. The heterogeneity of human diabetes, the fact that we measured protein and not RNA levels, as well as the fact that our patients did not undergo preparation for bariatric procedures may explain why our data differ from Samuel and colleagues (Samuel et al., 2009).
Bearing in mind the obvious species-specific differences, it should be recalled that in 2005 we made the surprising observation that mice with functional insulin signaling in liver, but not in other tissues, were resistant to insulin suppression of HGP (Okamoto et al., 2005). We repeated this experiment using a different approach in 2011 and still obtained the same result, which we linked to the failure of insulin to suppress FFA (Lin et al., 2011). Similar data emphasizing the role of indirect mechanisms on insulin control of HGP have been obtained more recently by Birnbaum and coworkers (Titchenell et al., 2015). However, one should be careful not to misconstrue these data as indicating that hepatic insulin signaling “doesn’t matter”. For example, Unterman and colleagues found that combined insulin receptor/FoxO1 ablation affects both direct and indirect actions of insulin on HGP (O. Sullivan et al., 2015). Moreover, regardless of the direct effects of insulin on gluconeogenic genes, the hormone affects lipid, lipoprotein, and cholesterol metabolism in hepatocytes in ways that prime the liver to produce more glucose (Haeusler et al., 2014). Finally, insulin resistance begets an increased glucagon “tone” that can promote HGP (Samuel and Shulman, 2016). Given the dominant effect of insulin vs. glucagon in regulating HGP (Exton and Park, 1968), it’s difficult to envision that the latter can increase without a permissive effect of impaired hepatocyte insulin signaling. More likely, the pathogenic process of increased HGP in T2D should be viewed as the combination of these processes. Ultimately, it should be remembered that none of the new T2D therapeutics affects HGP directly, and metformin remains the only agent to reduce HGP. Thus, this is an unmet treatment need, and as such should be viewed as a research priority.
Limitations of Study
Our study demonstrates an important role of TOX4 in regulating hepatocyte glucose production in parallel to the canonical FoxO1-IR pathway. It remains to be determined how TOX4 integrates other metabolic regulators in the liver transcriptional network. With a focus on gluconeogenesis here, RNA-seq analysis of TOX4 KD liver highlighted its potential effects on the GCGR pathway, inflammatory response and lipid metabolism, all of which will require further investigation. The paradoxical rise of insulin levels in LIRTDKO mice may be related to altered glucagon signaling or to the inflammatory response. We don’t know why the effects of TOX4 ablation on HGP were blunted in vivo compared with primary hepatocytes or following acute knockdown vs. constitutive somatic ablation of the gene. We suggest two potential explanations: compensation by indirect mechanisms of HGP control, as well as transcriptional resiliency of HGP genes (Kitamoto et al., 2021; Wang et al., 2019). Whether TOX4 actions on other hepatic metabolic processes, such as lipid turnover, contribute to these findings remains to be determined. Hyperinsulinemic-euglycemic clamps will further elucidate the metabolic role of TOX4.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagent and resource sharing should be directed to and will be fulfilled by the Lead Contact, Dr. Domenico Accili (da230@cumc.columbia.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data availability
The RNA-seq data have been deposited in the NCBI’s Gene Expression Omnibus (GEO GSE184239) and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Studies.
C57BL/6J (#000664), db/db (B6.129P2(Cg)-Lepr tm1.1Rck/J, #019377), FLP0-10 (# 011065), Albumin-Cre (Alb-Cre, #003574), InsR loxP (#00695) were from the Jackson Laboratories, Tox4Tm1a(KOMP)Mbp from KOMP (UC DAVIS), and FoxO1 fl/fl mice have been described (Matsumoto et al., 2007). Animals were housed in a 12-hr light/dark cycle (7AM/7PM) barrier facility with free access to water and food. High-fat diet (HFD, 60 kcal% Fat) was from Research Diets, Inc. (New Brunswick, NJ). Adenovirus Ctrl- or Tox4-shRNA, Ad-GFP or Ad-TOX4 (2.5x108 particles/gram) were administered by tail vein injection to 8-10-wk-old C57BL/6J, 12-week-old db/db, or 13-week-old C57BL/6J male mice after 5 weeks of HFD. Metabolic tests and hyperinsulinemic-euglycemic clamps were performed within 12 days of injection (Langlet et al., 2017). All animal studies were approved by and overseen by Columbia University Institutional Animal Care and Use Committee (IACUC).
Human liver samples.
The first set of liver biopsies used in this study has been described (Wang et al., 2020; Wang et al., 2016). Normal and diseased human livers were obtained through the Liver Tissue Cell Distribution System in Minnesota (Table S1). All six NAFLD subjects were diagnosed with fatty liver without steatohepatitis, four were also diagnosed with T2D, while the other two underwent gastric bypass surgery for morbid obesity (BMI = 32.3 and 44). Six age-matched subjects with normal liver function were selected as controls. Human studies were approved by the Columbia University Institutional Review Board and were conducted in accordance with National Institutes of Health and institutional guidelines for human subject research.
METHODS DETAILS
Primary hepatocytes studies.
Primary hepatocytes were isolated from 8- to 12-week-old male C57BL/6J (WT), TOX4 f/f, TOX4 LKO mice as described (Langlet et al., 2017). We anesthetized mice with ketamine and xylazine (ketamine 100 mg/kg IP, xylazine 10 mg/kg, i.p. injection). We clamped the supradiaphragmatic inferior vena cava (IVC), catheterized the inferior vena cava with a 24-gauge catheter (Exel international) and infused 50 ml perfusion solution (1x HBSS+0.5mM EGTA, Life Technologies) followed by 100 ml type IV collagenase solution (Worthington Biochemicals, 75mg/100ml perfusion solution: 500ml Medium 199 with 20mM HEPES+1% BSA+100 units/ml Penicillin-streptomycin+10 μg/ml Gentamycin). Following cell dissociation, we filtered cells with 100-μm mesh cell strainers, and wash 2x with hepatocyte plating medium(HPM): 500 ml M199 medium+10% FBS+ 100 units/ml Penicillin-streptomycin+10 μg/ml Gentamycin (Thermo Fisher Scientific). We further purified hepatocytes using Percoll (Sigma) gradient centrifugation: Mix 12ml cell suspension with 8ml 1xPBS Percoll solution and centrifuge at 700 rpm for 10min to collect cell pellets. Then, we suspended hepatocytes at 5x105 cells/ml in HPM. The Tox4 shRNA sequence is: AGCCAGTTGACCACTATTGATCTCGAGATCAATAGTGGTCAACTGGCT. Mouse Tox4 shRNA sequence was amplified and sub-cloned into shuttle vector pEQU6, then recombined with U6 promoter in a adenovirus vector for generating the adenovirus. Mouse Flag-TOX4 sequence was cloned into adenovirus vector with CMV promoter for generating adenovirus (Welgen). Ad-GFP was from Welgen (#V1020). 2-hr after plating, cells were transduced with Ad -Control shRNA (#V1050), Ad-Tox4 shRNA, Ad-GFP or Ad-Flag-TOX4 at MOI=10 or transfected with 25nM control (AM4611) or Tox4 siRNA mix (#4390771, s114086 and s114087, Thermo Fisher Scientific) using the Viromer BLUE reagent (Lipocalyx, Germany) following its instruction. After 48-hr, cells were incubated in M199 medium supplemented with Penicillin-streptomycin (100 U/ml) and Gentamycin (10 μg/ml) overnight, then switched to medium supplemented with either vehicle (0.002% methanol), or 1 μM dexamethasone and 0.1 mM 8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt (cAMP, Sigma #C3912) (D/C) for 6 hr, followed by the addition of 100 nM insulin for 2 or 6 hr. For Glucose production assay, serum-free medium was replaced with glucose production medium (glucose-free DMEM supplemented with 1% BSA, 3.3 g/L NaHCO3, 20 mmol/L calcium lactate, and 2 mmol/L sodium pyruvate, Penstrep). Cells were incubated with 0.5ml glucose production medium supplemented with vehicle, D/C or D/C plus insulin for 6 hr. Culture medium was collected and its glucose concentration was measured via peroxidase-glucose oxidase assay (Sigma) and normalized to protein content (Langlet et al., 2017).
DNA pulldown and Nano-LC/MS.
Primary hepatocytes were fractionated using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (#78835, Thermo Fisher Scientific). Biotin-labeled Pck1p and ΔIRS Pck1p DNA pulldown followed by mass spectrometry has been described (Wang et al., 2019). DNA pulldown samples were also analyzed by western blot.
Luciferase reporter assays.
RFP, pCMV-FoxO1, pGL3-Pck1p and pGL3-G6pcp (Wang et al., 2019) and pGL3-Gckp have been described (Langlet et al., 2017). We purchased pcDNA3.1 Flag-TOX4 (General Biosystems, Morrisville, NC), pCF CREB (Addgene #22968), pCMV-HNF4a (Origene, #MR227662). pcDNA3.1 Flag-TOX4 NLSdel mutant, Flag-TOX4HMGdel mutant, pGL3-Pck1p M1-M3, pGL3-ΔPck1pIRS38, pGL3-ΔPck1pCRE (−83bp to −94bp deletion) and TOX4HMGdel were generated using Q5 site-directed mutagenesis (New England Biolabs). The following primers were used: Tox4NLSdel for: AAAGACCCAAATGAACCTC; Tox4NLSdel rev: GGGAGCCTTTTGCTTTTTC; HMGdel-for: GACAACCAGGAATGCCAG; HMGdel-rev: TTCATTTGGGTCTTTCTTTTTTC; M1 (Pck1p −50bp to −1bp deletion) Pck1p-50del for: ACAGTTGGCCTTCCCTCTGGG; Pck1p-50del rev: GCCCTGCCCCTCAGCTGG; pGL3-Pck1p M2(Pck1p −84bp to −34bp deletion) Pck1p-84del for: TAGTATTTAAAGCAAGGAGGGCG; Pck1p-84del rev: CTGACGTAAGGGGCAGGC; Pck1p-117del for: CAGCTGAGGGGCAGGGCT; M3(−117bp to −68bp deletion) Pck1p-117del rev: ATGGTCAGCACGGTTTGGAACTG; 38IRSdel-for-CCACCGGCACACAAAATGTG and 38IRSdel-rev-TCCCACGGCCAAAGGTCA; CREdel-for: GCGAGCCTCCGGGTCCAG; CREdel-rev: GGGCAGGCCTTTGGATCATAGC. 1 x 106 HEK293/H4IIE cells (ATCC #CRL-1537 and #CRL1548) were seeded on each well of a 12-well plate. 200 ng of each DNA (pGL3 luciferase constructs or Flag-TOX4 or its mutants, CREB, HNF4a or RFP), and 20 ng of pRL-CMV plasmids were mixed with lipofectamine 3000 reagent in 0.1 ml OptiMEM (Thermofisher) and added to each well after incubation at RT for 15 min (Wang et al., 2019). After 36 hr we aspirated the culture medium, washed with PBS, and lysed cells in 0.35 ml 1x passive lysis buffer (Promega, #E1910). We performed dual luciferase assays (Promega, #E1910), and acquired signals with a microplate luminometer (Orion L). Plasmids are available upon request.
Immunoprecipitation and Western Blot.
5-6x106 AML12 cells were transfected with 20 μg Flag-TOX4 or RFP plasmids with lipofectamine 3000 (Thermo Fisher Scientific) for 36 hrs. Cells were deprived of serum for 4 hrs and treated with 0.1 mM cAMP or 100 nM insulin for 30 min. Cell lysates were harvested in 1ml Pierce™ IP lysis buffer (#87787, Thermo Fisher Scientific) plus Halt protease and phosphatase inhibitor cocktail (#78441, Thermo Fisher Scientific) from each well and sonicated in ice-water for 5 min. Supernatant was collected after centrifugation at 14,000 rpm for 10min at 4°C and protein concentration was determined by Pierce™ BCA assay (#23225, Thermo Fisher Scientific). 1 mg protein was incubated with 50 μl EZview Red Anti-Fl_AG M2 affinity gel (#F2426, Sigma) with overnight shaking at 4°C. Beads were washed 3x with 500 μl cold TBS (50 mM Tris HCl, 150 mM NaCl, pH7.4) and processed for SDS-PAGE.
Primary hepatocytes and liver were lysed in buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 2% Nonidet P-40, 1 mM EDTA, pH 8.0, 10% glycerol, 0.5% sodium deoxycholate, 0.2% semi-dehydroascorbate) supplemented with halt protease and phosphatase inhibitor. Livers were homogenized using Ika Ultra-Turrax dispersers T-10 basic (Sigma) and lysates sonicated in ice-cold water for 5 min. Liver and cell lysates were centrifuged at 14,000 rpm at 4°C for 10min. 30 μg protein was mixed with 6x SDS sample buffer (#BP-11R, Boston BioProducts) and boiled for 5min before loading on SDS-PAGE. Biorad wet transfer system was used. PVDF membranes were blocked with 5% dry milk in TBST (TBS+0.05% Tween 20) further incubated with primary antibodies (dilute in TBST with 3% BSA+0.05% NaN3) at 4°C overnight. Membrane was washed 4x in TBST with shaking for 10 min prior to incubation with secondary antibodies (dilution 1:2,000 in TBST) for additional 2 hr at room temperature (RT). Immune complexes were washed 4x with TBST with shaking for 10min at RT. Membranes were further reacted with Pierce™ ECL western blotting substrate and exposed. Films were scanned and saved as JPEGs. Western blot was quantified by Image J.
RNA isolation and QPCR analysis.
Primary hepatocytes or frozen liver were lysed in 1ml TRIzol. RNA were further purified using RNeasy mini kit (Qiagen). For reverse transcription, we used qScript cDNA synthesis kit (QuantaBio). 1 μg RNA was used for each reaction. The 20 μl cDNA solution was diluted with RNase-free water to 200 μl final volume. Go-tag qPCR master mix (Promega, Madison, WI) was used for following QPCR analysis. Cyclophilin A was used as the reference gene. Gene expression levels were calculated using the 2−ΔΔCt method and presented as relative expression levels as arbitrary units (AU). Primer information is available upon request.
RNA-sequencing and analysis
The RNA-sequencing was performed by Columbia Genome Center. Poly-A pull-down was used to enrich mRNA from total liver RNA and library construction with Illumina TruSeq. Libraries were sequenced using Illumina NovaSeq 6000. Kallisto pipeline was used to quantify transcript abundance (Bray et al., 2016). To find Tox4 targets, we employed the R package limma (v3.44.3) to identify differentially expressed genes (DEG) between Ctrl sh and Tox4 sh with a |log2(Fold Change)| > 0.5 and Adjusted P-value <0.1 (Ritchie et al., 2015). We conducted the functional enrichment with a cutoff of FDR < 0.1 for upregulated and downregulated genes by gprofiler2 (v0.2.0), separately (Raudvere et al., 2019).
Metabolic tests.
For GTT and PTT, mice were fasted (food deprivation) for 16 hr. Glucose and body weight were recorded. Mice were injected intraperitoneally with 2 g/kg body weight of glucose or pyruvate in PBS for GTT and PTT, respectively. Glucose was measured by Contour Next ONE glucometer (Amazon) at 15, 30, 60, 90 and 120 min. For ITT, mice were fasted for 4 hr starting at 10 am. Fasting glucose and body weight were recorded, mice were injected with 0.75 IU/kg body weight of insulin and glucose was measured at 15, 30, 60, 90 and 120 min. Euglycemic-hyperinsulinemic clamps were performed in mice (on HFD for 5 weeks) one week after adenovirus administration. Studies were performed in conscious, unrestrained, catheterized mice for 120 min as previously described (Ayala et al., 2011) and conducted by the Mouse Metabolic Function and Phenotyping Core at Columbia University. Insulin was determined by Mercodia ELISA kit. Plasma TG, total cholesterol and NEFA were measured by Infinity Triglycerides Liquid Stable Reagent (#TR22421), Wako Diagnostic Total Cholesterol E kit (#NC9138103) and NEFA linearity material (#NC9349895). Plasma ALT levels were measured by ALT/SGPT color endpoint kit (Teco Diagnostics, #A526120). Liver glycogen content were measured using the Glycogen Assay kit (Sigma, #MAK016).
Histology.
Livers were fixed with 10% formalin (Fisher Scientific) overnight, further dehydrated in 70% EtOH for 24 hr, embedded in Paraffin and cut into 5 μm sections. Sections were further processed for Hematoxylin and eosin (H&E) and Periodic Acid-Schiff (PAS) staining. For Oil Red O staining, livers were fixed with 4% PFA (Fisher Scientific) for 2hr at 4°C dehydrated in 30% sucrose PBS solution for 16 hr, embedded in Tissue-Tek O.C.T (Sakura) and frozen at −80°C. 5μm frozen sections were cut and stained with Oil Red O solution (Sigma). Stained sections were imaged using Olympus IX70 microscope and captured by DP74 camera using 10x objective lens.
Statistical analysis.
Each experiment was replicated at least three times for each condition. Liver MS was carried out once. Statistical analyses were performed using Prism 6.0 software (Graph Pad). We used two-tailed Student’s t-test for comparison between groups, one-way ANOVA for comparisons among three or more groups and two-way ANOVA to examine effects of two variables. P<0.05 is used to declare statistical significance. All data were presented as means ± SEM (standard error).
Supplementary Material
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–7 and S1–S10.
KEY RESOURCES TABLE
| REAGENT or RESCOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TOX4 | Bethyl Laboratories | Cat#A304-873A |
| Rabbit monoclonal anti-p RNA Pol II CTD (EPR19015) | Abcam | Cat#193467 |
| Rabbit monoclonal anti-FoxO1 (C29H4) | Cell signaling technology | Cat#2880S |
| Rabbit polyclonal anti-p-AKT (Ser473) | Cell signaling technology | Cat#9271S |
| Rabbit polyclonal anti-G6Pase | Abcam | Cat#ab93857 |
| Rabbit monoclonal anti-PCK1(D12F5) | Cell signaling technology | Cat#12940S |
| Rabbit polyclonal anti-PCK1 | Abcam | Cat#ab70358 |
| Rabbit monoclonal anti-FBP1 (D2T7F) | Cell signaling technology | Cat#59172 |
| Rabbit monoclonal anti-ACTIN (8H10D10) | Cell signaling technology | Cat#3700 |
| Rabbit monoclonal anti-Lamin A/C (EPR4100) | Abcam | Cat#ab108595 |
| Rabbit monoclonal anti-HMGA1 (EPR7839) | Abcam | Cat#ab129153 |
| Rabbit polyclonal anti-GAPDH | Abcam | Cat#ab9485 |
| Rabbit polyclonal anti-Phospho-FoxO1 (Ser256) | Cell signaling technology | Cat#9461 |
| Rabbit monoclonal anti-Phospho-CREB (Ser133)(87G3) | Cell signaling technology | Cat#9198 |
| Rabbit monoclonal anti-CREB (48H2) | Cell signaling technology | Cat#9197 |
| Rabbit polyclonal anti-PGC1a | Abcam | Cat#ab54481 |
| Rabbit monoclonal anti-IR β (4B8) | Cell signaling technology | Cat#3025 |
| Mouse monoclonal anti-GCK(G6) | Santa Cruz Biotechnology | Cat#sc-17819 |
| Adenovirus | ||
| Ad-TOX4 shRNA | This paper | N/A |
| Ad-Control shRNA | Welgen | Cat#V1050 |
| Ad-TOX4 | This paper | N/A |
| Ad-GFP | Welgen | Cat#V1020 |
| Biological samples | ||
| Human liver samples | (Wang et al., 2016) | |
| Chemicals, peptides, and recombinant proteins | ||
| 8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic monophosphate sodium salt | Sigma | Cat#C3912 |
| Dexamethasone | Sigma | Cat#4902 |
| Glucose oxidase/peroxidase reagent | Sigma | Cat#G3660 |
| o-Dianisidine dihydrochloride | Sigma | Cat#D3252 |
| Insulin aspart injection 100 Units/ml | Novalog | N/A |
| Opti-MEM | ThermoFisher Scientific | Cat#31985070 |
| Lipofectamine 3000 | Thermofisher Scientific | Cat#L3000015 |
| Go-Tag qPCR master mix | Promega | Cat#A6002 |
| Viromer BLUE reagent | Lipocalyx (Germany) | Cat#VB-01LB-00 |
| TRIzol™ Reagent | Fisher Scientific | Cat#15596018 |
| Critical Commercial Assays | ||
| Q5®Site-directed mutagenesis kit | NEB | Cat#E0554S |
| Rneasy mini kit | QIAGEN | Cat#74106 |
| qScript cDNA synthesis Kit (QuantaBio) | VWR | Cat#101414-100 |
| Insulin ELISA kit | Mercodia | Cat#10-1113-01 |
| Teco Diagnostics ALT/SGPT Color endpoint kit | Fisher Scientific | Cat#NC9511694 |
| Glycogen Assay Kit | Sigma | Cat#MAK016 |
| Wako Diagnostics Total Cholesterol E 50/kit | Fisher Scientific | Cat#NC9138103 |
| HR Series NEFA-HR(2) Color Reagent B | Fisher Scientific | Cat#991-34891 |
| HR Series NEFA-HR(2) Solvent A | Fisher Scientific | Cat#995-34791 |
| HR Series NEFA-HR(2) Color Reagent A | Fisher Scientific | Cat#999-34691 |
| HR Series NEFA-HR(2) Solvent B | Fisher Scientific | Cat#993-35191 |
| Infinity Triglyceride kit | Fisher Scientific | Cat#TR22421 |
| Dual-Luciferase® Reporter Assay System | Promega | Cat#E1960 |
| BCA assay | Thermofisher Scientific | Cat#23227 |
| Pierce™ ECL Western Blotting Substrate | Thermofisher Scientific | Cat#PI32209 |
| Deposited data | ||
| RNA-seq data of TOX4 knockdown liver samples | GEO | GSE184239 |
| Experimental Models: Cell lines | ||
| 293 cells | ATCC | Cat#CRL-1573 |
| AML12 cells | ATCC | Cat#CRL-2254 |
| H4IIE cells | ATCC | Cat#CRL-1548 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | JAX | Stock#000664 |
| db/db (B6.129P2(Cg)-Lepr Tm1.1Rck/J | JAX | Stock#019377 |
| FLP0-10 | JAX | Stock#011065 |
| Albumin-Cre | JAX | Stock#003574 |
| InsR loxP | JAX | Stock#00695 |
| Tox4Tm1a(KOMP)Mbp | MMRRC | Stock#050190-UCD |
| FoxO1 LoxP | (Matsumoto et al., 2007) | |
| Oligonucleotides | ||
| Ctrl siRNA | Thermo Fisher Scientific | Cat# AM4611 |
| Tox4 siRNA 1 | Thermo Fisher Scientific | Cat#4390771-s114086 |
| Tox4 siRNA 2 | Thermo Fisher Scientific | Cat#4390771-s114086 |
| Recombinant DNA | ||
| pcDNA3.1 Myc-His(-)A-Flag-TOX4 | This paper | N/A |
| pcDNA3.1TOX4NLSdel | This paper | N/A |
| pcDNA3.1TOX4HMGdel | This paper | N/A |
| pGL3-G6pcp | (Wang et al., 2019) | |
| pGL3-Pck1p | (Wang et al., 2019) | |
| pGL3-Pck1pΔIRS | (Wang et al., 2019) | |
| pGL3-Pck1pΔIRS38 | This paper | N/A |
| pGL3-ΔPck1pCRE | This paper | N/A |
| pGL3-Gckp | (Langlet et al., 2017) | |
| pGL3-Pck1p-M1 | This paper | N/A |
| pGL3-Pck1p-M2 | This paper | N/A |
| pGL3-Pck1p-M3 | This paper | N/A |
| WT FoxO1 | (Langlet et al., 2017) | |
| pCMV-HNF4a | Origene | Cat#MR227662 |
| pCF-CREB | Addgene | Cat#22968 |
| Software and algorithms | ||
| PRISM | GraphPad Software | Version 6 |
| Image J | NIH | https://imagej.nih.gov/ij |
| Other | ||
| High fat diet (60 kcal% fat) | Research Diets | Cat#D12492 |
1. Highlights:
Discovery of TOX4 as a hormone-responsive transcription factor acting on hepatic Pck1
Inhibition of hepatic TOX4 reduces gluconeogenesis and improves glucose tolerance
Silencing hepatic TOX4 ameliorates hyperglycemia in DIO and db/db mice
Effects of TOX4 are additive to FoxO1 and independent of IR
Acknowledgement
We thank Ira Tabas for sharing human liver samples, Takumi Kitamoto for helpful discussions; Ana Flete and Thomas Kolar for help with mouse studies; Lina Xu and Xi Sun of the Diabetes and Research Center Core for histology analysis, the Liver Tissue Cell Distribution System funded by NIH Contract #HHSN276201200017C. This research was supported by DK58282, DK63618, 7T32DK00755925, K01 DK123199, and P30CA013696.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests. D.A. is a founder, director, and chair of the SAB of Forkhead Biotherapeutics, Corp.
References
- Ayala JE, Bracy DP, Malabanan C, James FD, Ansari T, Fueger PT, McGuinness OP, and Wasserman DH (2011). Hyperinsulinemic-euglycemic clamps in conscious, unrestrained mice. J Vis Exp 57, 3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Burgess SC, He T, Yan Z, Lindner J, Sherry AD, Malloy CR, Browning JD, and Magnuson MA (2007). Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab 5, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA, and White MF (2008). Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab 8, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton DS, Ramnanan CJ, Grueter CA, Johnson KM, Lautz M, Neal DW, Williams PE, and Cherrington AD (2009). Effects of insulin on the metabolic control of hepatic gluconeogenesis in vivo. Diabetes 58, 2766–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erion DM, Ignatova ID, Yonemitsu S, Nagai Y, Chatterjee P, Weismann D, Hsiao JJ, Zhang D, Iwasaki T, Stark R, et al. (2009). Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metab 10, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exton JH, and Park CR (1968). Control of gluconeogenesis in liver. II. Effects of glucagon, catecholamines, and adenosine 3′,5′-monophosphate on gluconeogenesis in the perfused rat liver. J Biol Chem 243, 4189–4196. [PubMed] [Google Scholar]
- Gottmann P, Ouni M, Zellner L, Jahnert M, Rittig K, Walther D, and Schurmann A (2020). Polymorphisms in miRNA binding sites involved in metabolic diseases in mice and humans. Sci Rep 10, 7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granner DK (2015). In pursuit of genes of glucose metabolism. J Biol Chem 290, 22312–22324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR, Kammoun HL, Febbraio MA, Gutierrez-Juarez R, Kurland IJ, et al. (2014). Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nature commun 5, 5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Kaestner KH, and Accili D (2010). FoxOs function synergistically to promote glucose production. J Biol Chem 285, 35245–35248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han HS, Kang G, Kim JS, Choi BH, and Koo SH (2016). Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med 48, e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Sabet A, Djedjos S, Miller R, Sun X, Hussain MA, Radovick S, and Wondisford FE (2009). Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 137, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. (2001). CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183. [DOI] [PubMed] [Google Scholar]
- Jackness C, Karmally W, Febres G, Conwell IM, Ahmed L, Bessler M, McMahon DJ, and Korner J (2013). Very low-calorie diet mimics the early beneficial effect of Roux-en-Y gastric bypass on insulin sensitivity and beta-cell Function in type 2 diabetic patients. Diabetes 62, 3027–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto T, Kuo T, Okabe A, Kaneda A, and Accili D (2021). An integrative transcriptional logic model of hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, et al. (2005). The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111. [DOI] [PubMed] [Google Scholar]
- Kubota N, Kubota T, Itoh S, Kumagai H, Kozono H, Takamoto I, Mineyama T, Ogata H, Tokuyama K, Ohsugi M, et al. (2008). Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell Metab 8, 49–64. [DOI] [PubMed] [Google Scholar]
- Langlet F, Haeusler RA, Linden D, Ericson E, Norris T, Johansson A, Cook JR, Aizawa K, Wang L, Buettner C, et al. (2017). Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 171, 824–835 e818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro D, Kido Y, Castle AL, Zarnowski MJ, Hayashi H, Ebina Y, and Accili D (1998). Impaired glucose tolerance in mice with a targeted impairment of insulin action in muscle and adipose tissue. Nat Genet 20, 294–298. [DOI] [PubMed] [Google Scholar]
- Lee JH, You J, Dobrota E, and Skalnik DG (2010). Identification and characterization of a novel human PP1 phosphatase complex. The Journal of biological chemistry 285, 24466–24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GF, Vranic M, Harley P, and Giacca A (1997). Fatty acids mediate the acute extrahepatic effects of insulin on hepatic glucose production in humans. Diabetes 46, 1111–1119. [DOI] [PubMed] [Google Scholar]
- Lin HV, and Accili D (2011). Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HV, Ren H, Samuel VT, Lee HY, Lu TY, Shulman GI, and Accili D (2011). Diabetes in mice with selective impairment of insulin action in Glut4-expressing tissues. Diabetes 60, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I, de Chavez V, Vuguin PM, Charron MJ, Powers AC, et al. (2013). Liver-specific disruption of the murine glucagon receptor produces alpha-cell hyperplasia: evidence for a circulating alpha-cell growth factor. Diabetes 62, 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, Ahima RS, Ueki K, Kahn CR, and Birnbaum MJ (2012). Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med 18, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, and Accili D (2007). Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6, 208–216. [DOI] [PubMed] [Google Scholar]
- Monnier L, Colette C, Dunseath GJ, and Owens DR (2007). The loss of postprandial glycemic control precedes stepwise deterioration of fasting with worsening diabetes. Diabetes Care 30, 263–269. [DOI] [PubMed] [Google Scholar]
- O. Sullivan I Zhang W, Wasserman DH, Liew CW, Liu J, Paik J, DePinho RA, Stolz DB, Kahn CR, Schwartz MW, et al. (2015). FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun 6, 7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Obici S, Accili D, and Rossetti L (2005). Restoration of liver insulin signaling in Insr knockout mice fails to normalize hepatic insulin action. J. Clin. Invest 115, 1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47, W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L (2014). Energy metabolism in the liver. Compr Physiol 4, 177–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Beddow SA, Iwasaki T, Zhang XM, Chu X, Still CD, Gerhard GS, and Shulman GI (2009). Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with Type 2 Diabetes. Proc. Natl. Acad. Sci. USA 106, 12121–12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J. Clin. Invest 126, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Chu Q, Monks BR, and Birnbaum MJ (2015). Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun 6, 7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, Chen H, Monks BR, Chen J, Rabinowitz JD, et al. (2016). Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab 23, 1154–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti L, Rametta R, Dongiovanni P, Maggioni M, Fracanzani AL, Zappa M, Lattuada E, Roviaro G, and Fargion S (2008). Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes 57, 1355–1362. [DOI] [PubMed] [Google Scholar]
- Vanheer L, Song J, De Geest N, Janiszewski A, Talon I, Provenzano C, Oh T, Chappell J, and Pasque V (2019). Tox4 modulates cell fate reprogramming. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, and Luscombe NM (2009). A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10, 252–263. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu Q, Kitamoto T, Hou J, Qin J, and Accili D (2019). Identification of Insulin-Responsive Transcription Factors That Regulate Glucose Production by Hepatocytes. Diabetes 68, 1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, et al. (2020). Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab 31, 969–986 e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, et al. (2016). Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab 24, 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. (2001). Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–7 and S1–S10.
Data Availability Statement
The RNA-seq data have been deposited in the NCBI’s Gene Expression Omnibus (GEO GSE184239) and are publicly available as of the date of publication.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.