

This study identifies race-specific genetic associations with breast cancer risk of recurrence scores and suggests mediation of these associations by PAM50 subtype and expression, with implications for clinical interpretation of these scores.
Abstract
Continuous risk of recurrence scores (CRS) based on tumor gene expression are vital prognostic tools for breast cancer. Studies have shown that Black women (BW) have higher CRS than White women (WW). Although systemic injustices contribute substantially to breast cancer disparities, evidence of biological and germline contributions is emerging. In this study, we investigated germline genetic associations with CRS and CRS disparity using approaches modeled after transcriptome-wide association studies (TWAS). In the Carolina Breast Cancer Study, using race-specific predictive models of tumor expression from germline genetics, we performed race-stratified (N = 1,043 WW, 1,083 BW) linear regressions of three CRS (ROR-S: PAM50 subtype score; proliferation score; ROR-P: ROR-S plus proliferation score) on imputed tumor genetically regulated tumor expression (GReX). Bayesian multivariate regression and adaptive shrinkage tested GReX-prioritized genes for associations with tumor PAM50 expression and subtype to elucidate patterns of germline regulation underlying GReX-CRS associations. At FDR-adjusted P < 0.10, 7 and 1 GReX prioritized genes among WW and BW, respectively. Among WW, CRS were positively associated with MCM10, FAM64A, CCNB2, and MMP1 GReX and negatively associated with VAV3, PCSK6, and GNG11 GReX. Among BW, higher MMP1 GReX predicted lower proliferation score and ROR-P. GReX-prioritized gene and PAM50 tumor expression associations highlighted potential mechanisms for GReX-prioritized gene to CRS associations. Among patients with breast cancer, differential germline associations with CRS were found by race, underscoring the need for larger, diverse datasets in molecular studies of breast cancer. These findings also suggest possible germline trans-regulation of PAM50 tumor expression, with potential implications for CRS interpretation in clinical settings.
Significance:
This study identifies race-specific genetic associations with breast cancer risk of recurrence scores and suggests mediation of these associations by PAM50 subtype and expression, with implications for clinical interpretation of these scores.
Introduction
Tumor expression–based molecular profiling has improved clinical classification of breast cancer (1–3). One tool is the PAM50 assay, which integrates tumor expression of 50 genes (derived from a set of 1,900 subtype-specific genes identified in microarray studies) to determine PAM50 intrinsic molecular subtypes: luminal A (LumA), luminal B (LumB), HER2-enriched, basal-like, and normal-like (1, 4). Intrinsic molecular subtypes are strong prognostic factors for breast cancer outcomes, including recurrence and mortality. For instance, basal-like breast cancer has substantially higher recurrence and mortality risk compared with LumA breast cancer (5–8). In recent years, continuous risk of recurrence scores (CRS) have gained traction as a potential clinical tool that encapsulates prognostic differences of breast cancer intrinsic molecular subtypes into a singular measure that can be used to guide treatment decisions. CRS include ROR-S, proliferation score, ROR-P, and ROR-PT (1, 9). ROR-P, for instance, is determined by combining ROR-S (PAM50 tumor expression-based subtype score) and proliferation score (tumor expression of 11 PAM50 genes). ROR-PT further integrates ROR-P with information on tumor size. Studies show that CRS offer significant prognostic information beyond clinical variables (e.g., nodal status, tumor grade, age, hormonal therapy), improve adjuvant treatment decisions, and offer robust risk stratification for distant (5–10 years after diagnosis) recurrence (10–12).
In the Carolina Breast Cancer Study (CBCS), Black women (BW) with breast cancer have disproportionately higher CRS than White women (WW; ref. 9), and similar disparities have been noted in Oncotype Dx recurrence score (9, 13). Systemic injustices, like disparities in health care access, explain a substantial proportion of breast cancer outcome disparities (14–17). Recent studies additionally suggest that germline genetic variation is associated with breast cancer outcomes, and these associations vary across ancestry groups (18–21). In The Cancer Genome Atlas (TCGA), BW had substantially higher polygenic risk scores for the more aggressive estrogen receptor (ER)-negative subtype than WW, suggesting differential genetic contributions for susceptibility for breast cancer, especially ER-negative breast cancer (21). In a transcriptome-wide association study (TWAS) of breast cancer mortality, germline-regulated gene expression (GReX) of four genes was associated with mortality among BW and gene expression for no genes was associated among WW (18). However, the role of germline genetic variation in recurrence, CRS, and CRS disparity remains a critical knowledge gap. Studying genetic associations with breast cancer outcomes in BW is necessary to ensure advancements in breast cancer genetics are not limited to or generalizable in only White populations, thus aiding in decreasing health disparities.
As racially diverse genetic datasets typically have small samples of BW, gene-level association tests can increase study power. These approaches include TWAS, which integrates relationships between SNPs and gene expression with genome-wide association studies (GWAS) to prioritize gene-trait associations (22, 23). TWAS aids in interpreting genetic associations by mapping significant GWAS associations to tissue-specific expression of individual genes. In cancer applications, TWAS has identified susceptibility genes at loci previously undetected through GWAS, highlighting its improved power and interpretability (24–26). Previous studies show that stratification of the entire TWAS (model training, imputation, and association testing) is preferable in diverse populations, as models may perform poorly across ancestry groups and methods for TWAS in admixed populations are unavailable (18, 27).
Here, using data from the CBCS, which includes a large sample of Black patients with breast cancer with tumor gene expression data, we study race-specific germline genetic associations for CRS using a gene-based association testing approach that borrows from TWAS methodology. CRS included in this study are ROR-S, proliferation score, and ROR-P. Using race-specific predictive models for tumor expression from germline genetics, we identify sets of GReX-prioritized genes (i.e., genes whose GReX is associated with CRS) across BW and WW. We additionally investigate ROR-P specific GReX-prioritized genes for associations with PAM50 subtype and subtype-specific tumor gene expression to elucidate germline contributions to PAM50 subtype, and how these mediate GReX-prioritized gene and CRS associations. Unlike previous studies that correlated tumor gene expression (as opposed to germline-regulated tumor gene expression) with subtype or subtype-specific tumor gene expression, TWAS enables directional interpretation of observed associations (22, 23).
Materials and Methods
Data collection
Study population
The CBCS is a population-based study of North Carolina (NC) patients with breast cancer, enrolled in three phases; study details have been described previously (28, 29). Patients ages 20 to 74 were identified using rapid case ascertainment with the NC Central Cancer Registry with randomized recruitment to oversample self-identified Black and young women (ages 20–49; refs. 9, 29). Demographic and clinical data (age, menopausal status, body mass index, hormone receptor status, tumor stage, study phase, recurrence) were obtained through questionnaires and medical records. The study was approved by the Office of Human Research Ethics at the University of North Carolina at Chapel Hill (Chapel Hill, NC), and written informed consent was obtained from each participant.
CBCS genotype data
Genotypes were assayed on the OncoArray Consortium's custom SNP array (Illumina Infinium OncoArray; ref. 30) and imputed using the 1000 Genomes Project (Phase III) as a reference panel for two-step phasing and imputation using SHAPEIT2 and IMPUTEv2 (31–34). The DCEG Cancer Genomics Research Laboratory conducted genotype calling, quality control, and imputation (30). We excluded variants with less than 1% minor allele frequency and deviations from Hardy–Weinberg equilibrium at 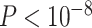 (35, 36). We intersected genotyping panels for BW and WW samples, resulting in 5,989,134 autosomal variants and 334,391 variants on the X chromosome (37). We only consider the autosomal variants in this study.
(35, 36). We intersected genotyping panels for BW and WW samples, resulting in 5,989,134 autosomal variants and 334,391 variants on the X chromosome (37). We only consider the autosomal variants in this study.
CBCS gene expression data
Paraffin-embedded tumor blocks were assayed for gene expression of 406 breast cancer–related and 11 housekeeping genes using NanoString nCounter at the Translational Genomics Laboratory at UNC-Chapel Hill (4, 9). These 406 breast cancer–related genes include genes part of the PAM50, P53, E2, IGF, and EGFR signatures, among others (Supplementary Table S1). As described previously, we eliminated samples with insufficient data quality using NanoStringQCPro (18, 38), scaled distributional difference between lanes with upper quartile normalization (39), and removed two dimensions of unwanted technical and biological variation, estimated from housekeeping genes using RUVSeq (39, 40). The current analysis included 1,199 samples with both genotype and gene expression data (628 BW, 571 WW).
Statistical analysis
Overview of GReX and TWAS
We adopted TWAS methodology to construct GReX (exposure of interest in this study). GReX for a given gene represents the portion of tumor expression explained by cis-genetic regulation; GReX was constructed for the aforementioned set of breast cancer–related genes (Supplementary Table S1). Briefly, TWAS integrates expression data with GWAS to prioritize gene-level germline-trait associations through a two-step analysis (Fig. 1A and B). First, using germline and transcriptomic data, we trained predictive models of tumor gene expression using all SNPs within 0.5 Megabase of the gene (18, 23). Second, we used these models to impute the GReX of a gene by multiplying the SNP-gene weights from the predictive model with the dosages of each SNP. Associations between GReX (for a given gene) and trait (CRS, for instance) in regression analyses identify gene-trait relationships that are a consequence of germline variation. If sufficiently heritable genes are assayed in the correct tissue, TWAS-based GReX analyses increase power to detect germline-trait associations and aids interpretability of results, as associations are mapped from germline genetics to individual genes (23, 41).
Figure 1.

Schematic of study analytic approach. A, In CBCS, constructed race-stratified predictive models of tumor gene expression from cis-SNPs. B, In CBCS, imputed GReX at individual level using genotypes and tested for associations between GReX and CRS in race-stratified linear models; only GReX of genes with significant cis-h2 and high cross-validation performance (R2 > 0.01 between observed and predicted expression) considered for race-stratified association analyses. C, Follow-up analyses on GReX-prioritized genes (i.e., genes whose GReX were significantly associated with CRS at FDR < 0.10). In race-stratified models, PAM50 SCCs and PAM50 tumor expressions were regressed against GReX-prioritized genes under a Bayesian multivariate regression and multivariate adaptive shrinkage approach.
GReX analysis of CRS in CBCS
We adopted techniques from FUSION to train predictive models of tumor expression from cis-germline genotypes (18, 23). Motivated by strong associations between germline genetics and tumor expression in CBCS (18), for genes with non-zero cis-heritability at nominal 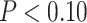 , we trained predictive models for covariate-residualized tumor expression with all cis-SNPs within 0.5 Megabase using linear mixed modeling or elastic net regression (Supplementary Materials and Methods; refs. 42, 43). Here, we used the 628 BW samples and 571 WW samples with both genotype and expression data to train these race-specific expression models. We selected models with 5-fold cross-validation adjusted
, we trained predictive models for covariate-residualized tumor expression with all cis-SNPs within 0.5 Megabase using linear mixed modeling or elastic net regression (Supplementary Materials and Methods; refs. 42, 43). Here, we used the 628 BW samples and 571 WW samples with both genotype and expression data to train these race-specific expression models. We selected models with 5-fold cross-validation adjusted 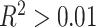 between predicted and observed expression values, resulting in 59 and 45 models for WW and BW, respectively. Further details on these models, including heritability and cross-validation performance are available at Supplementary Table S2. These models also showed sufficiently strong predictive performance in external validation using TCGA data (18).
between predicted and observed expression values, resulting in 59 and 45 models for WW and BW, respectively. Further details on these models, including heritability and cross-validation performance are available at Supplementary Table S2. These models also showed sufficiently strong predictive performance in external validation using TCGA data (18).
Using only germline genetics as input, we imputed GReX in 1,043 WW and 1,083 BW, respectively, in CBCS. For samples not present in the training dataset, we multiplied the SNP weights from the predictive models with the SNP dosages to construct GReX. For samples in both the training and imputation datasets, GReX was imputed via cross-validation to minimize data leakage. We tested GReX for associations with ROR-S, proliferation score, and ROR-P using multiple linear regression adjusted for age, ER status, tumor stage, and study phase (1). We corrected for test-statistic bias and inflation using a Bayesian bias and inflation adjustment method bacon, as TWAS are prone to bias and inflation of test statistics (44). We then adjusted for multiple testing using the Benjamini–Hochberg procedure (44, 45). As a comparison for the germline effect of GReX-prioritized genes, we additionally assessed the effect of total (germline-regulated and posttranscriptional) tumor expression of those GReX-prioritized genes on CRS using similar linear models. We were underpowered to study time to recurrence, as recurrence data were collected only in CBCS phase III (635 WW, 742 BW with GReX and recurrence data; 183 WW, 283 BW with tumor expression and recurrence data). For significant GReX-prioritized genes for CRS (FDR-adjusted P < 0.10), we conducted follow-up permutation tests: we shuffle the SNP-gene weights in the predictive model 5,000 times to generate a null distribution and compare the original GReX-CRS associations to this null distribution. This permutation test assessed whether the GReX association provides more tissue-specific expression context, beyond any strong SNP-CRS associations at the genetic locus (23).
PAM50 assay and ROR-S, proliferation score, and ROR-P calculation
As described previously (1), using partition-around-medoid clustering, we calculated the correlation with each subtype's centroid for study individuals based on PAM50 expressions (10 PAM50 genes per subtype). The largest subtype-centroid correlation defined the individual's molecular subtype. ROR-S was determined via a linear combination of the PAM50 subtype-centroid correlations (SCC); the coefficients to the PAM50 SCCs in the linear combination are positive for LumB, HER2-enriched, and basal-like and negative for LumA (1). Proliferation score was computed using log-scale expression of 11 PAM50 genes, while ROR-P was computed by combining ROR-S and proliferation score.
Assignment of PAM50 gene to subtype was based on PAM50 gene centroid values for each subtype; a PAM50 gene is assigned to the subtype with the largest positive centroid value. Subtype assignment through this “greedy algorithm” are specific to this study and represent a simplified reality (e.g., ESR1 classified as part of LumA subtype only even though ESR1 expression correlates with both LumA and to a slightly lesser degree LumB subtype). Moreover, subtype assignment for this portion of analyses was conducted only for visual comparison of patterns of associations between GReX-prioritized genes and PAM50 tumor gene expressions (i.e., subtype assignment in this portion of analyses had no bearing on continuous ROR score calculations or subtype-centroid correlations).
Bayesian multivariate regressions and multivariate adaptive shrinkage
As noted previously (1), CRS are functions of PAM50 SCCs and gene expression profiles. Thus, we followed up on CRS-associated GReX-prioritized genes by studying their associations with PAM50 SCCs and gene expression. We assessed GReX-prioritized genes (for ROR-P) in relation to SCCs and PAM50 tumor gene expression (Fig. 1C). Importantly, consistent with the original formulation of ROR-S, we did not consider normal-like subtype and normal-like subtype specific genes; subtype-specific genes were determined using a greedy assignment algorithm, described in the previous section. This classification scheme offers analytic simplicity but is an oversimplification for some PAM50 genes. We found that none of our GReX-prioritized genes were within 1 Megabase of PAM50 genes and that most GReX-prioritized genes were not on the same chromosome as PAM50 genes (Supplementary Table S3).
Existing gene-based mapping techniques for trans-expression quantitative trait loci (eQTL; SNP and gene are separated by more than 1 Megabase) mapping include trans-PrediXcan and GBAT (46, 47). We employed Bayesian multivariate linear regression (BtQTL) to account for correlation in multivariate outcomes (SCCs and PAM50 gene expression) in association testing. BtQTL improves power to detect significant trans-associations, especially when considering multiple genes with highly correlated (>0.5) expression (Supplementary Figs. S1 and S2). Finally, we conducted adaptive shrinkage on BtQTL estimates using mashr, an empirical Bayes method to estimate patterns of similarity and improve accuracy in association tests across multiple outcomes (48). mashr outputs revised posterior means, SDs, and corresponding measures of significance (local false sign rates, or LFSR).
Associations of genetic ancestry and race with tumor expression and GReX of GReX-prioritized genes
Prior studies using CBCS have reported concordance between self-reported race and genetic ancestry (first principal component of combined genotype matrix; ref. 18). In an effort to further contextualize CRS associations across race and to disentangle race from genetic ancestry in our study population (specifically, whether race, which captures both genetic ancestry and socioeconomic context, is a proxy for genetic ancestry in our study population), we investigated: (i) association between genetic ancestry and tumor expression of GReX-prioritized genes; (ii) association between genetic ancestry and GReX of GReX-prioritized genes; (iii) association between race and tumor expression of GReX-prioritized genes; (iv) association between race and GReX of GReX-prioritized genes. Genetic ancestry was computed by aggregating across local ancestry, as determined through the RFMix pipeline (49).
Availability of data and materials
Expression data from CBCS are available on NCBI Gene Expression Omnibus with accession number GSE148426. CBCS genotype datasets analyzed in this study are not publicly available as many CBCS patients are still being followed and accordingly CBCS data are considered sensitive; the data are available from M.A. Troester upon reasonable request. Supplementary Data include summary statistics for eQTL results, tumor expression models, and relevant R code for training expression models in CBCS and are freely available at https://github.com/bhattacharya-a-bt/CBCS_TWAS_Paper/. Scripts utilized in this analysis are provided at https://github.com/APUNC/CBCS-Risk-of-Recurrence-Paper.
Results
Race-specific associations between GReX and CRS
We performed race-specific GReX analysis for CRS to investigate the role of germline genetic variation in CRS and CRS racial disparity. We identified eight genes (MCM10, FAM64A, CCNB2, MMP1, VAV3, PCSK6, NDC80, MLPH), eight genes (MCM10, FAM64A, CCNB2, MMP1, VAV3, NDC80, MLPH, EXO1), and 10 genes (MCM10, FAM64A, CCNB2, MMP1, VAV3, PCSK6, GNG11, NDC80, MLPH, EXO1) whose GReX was associated with ROR-S, proliferation, and ROR-P, respectively, in WW, and 1 gene (MMP1) whose GReX was associated with proliferation and ROR-P in BW at FDR-adjusted P < 0.10 (Fig. 2A and B). No associations were detected between GReX and ROR-S among BW. We refer to genes with statistically significant GReX analysis associations (FDR-adjusted P < 0.10) as GReX-prioritized genes. Among these identified genes, only genes that are not part of the PAM50 panel (i.e., excluding NDC80, MLPH, EXO1) were considered in downstream permutation and GReX-prioritized gene follow-up analyses (Fig. 1C), as we wished to focus investigation on relationship between non-PAM50 GReX-prioritized genes and PAM50 (tumor) genes. Supplementary Figure S3 shows results from a sensitivity analysis comparing the effect sizes for the GReX-CRS associations within samples used in training, not used in training, and the overall associations using all training and non-training samples. In general, we see concordance in the direction of association across these three splits of data, though some of the associations detected within only training or non-training samples intersect the null.
Figure 2.

Permutation tests and associations between GReX-prioritized genes and CRS for WW and BW. A, Effect estimates correspond to change in ROR-S, proliferation score, and ROR-P per one SD increase in GReX-prioritized gene expression (i.e., one SD increase in GReX of gene). Triangle, WW; circle, BW. B, Boxplots correspond to null distributions (left, shuffled GReX-sample labels; right, random set of genes) of covariates-residualized R2 for regressions of CRS on GReX-prioritized genes. Null distributions are provided for 10,000 permutations of the GReX-sample labels and 10,000 random sets of genes. Dashed horizontal lines correspond to observed covariates-residualized R2.
Among WW, increased GReX of MCM10, FAM64A, CCNB2, and MMP1 were associated with higher CRS while increased GReX of VAV3, PCSK6, and GNG11 were associated with lower CRS (Fig. 2A).
Among BW, increased GReX of MMP1 was associated with lower CRS (proliferation, ROR-P, but not ROR-S; Fig. 2A). Supplementary Figure S4 shows the nominal differences in eQTL architecture across BW and WW for these genes. In particular, for MMP1, we found differences in the standardized effects across WW and BW: a sizable proportion of shared eQTLs had discordant effects across WW and BW (Supplementary Fig. S5). The linkage disequilibrium structure for eQTLs differed across WW and BW, with eQTL effect size peaks [−log10P: 4.73 (WW); 3.17 (BW)] at differing genomic locations (Supplementary Fig. S5).
Briefly, to contextualize the functions of these GReX-prioritized genes, MCM10 is involved in DNA replication, FAM64A and CCNB2 are implicated in progression and regulation of the cell cycle, and MMP1, like the broader MMP family, is involved in the breakdown of the extracellular matrix (50–54). GNG11 and VAV3 are involved in signal transduction: GNG11 as a component of a transmembrane G-protein and VAV3 as a guanine nucleotide exchange factor for GTPases (55, 56).
Associations between tumor expression of GReX-prioritized genes and CRS were concordant, in terms of direction of association to germline-only effects among WW; findings were discordant among BW where higher tumor expression of MMP1 was associated with higher CRS (Table 1; Supplementary Table S4). We found differences in the pattern of associations between genetic ancestry and race with tumor expression and GReX of GReX-prioritized genes (Supplementary Fig. S6). For instance, while higher African ancestry was associated with higher tumor expression of MCM10, higher African ancestry was instead associated with lower GReX of MCM10.
Table 1.
Race-specific associations between GReX of GReX-prioritized genes and CRS. Effect estimates correspond to change in CRS per one SD increase in GReX, adjusted for age, ER status, stage, and CBCS study phase. Ninety-five percent confidence intervals of effect sizes are provided. All GReX-prioritized gene and CRS pairs shown here showed overall association FDR-adjusted P < 0.10 and FDR-adjusted permutation P < 0.05 (across 5,000 permutations of the SNP-gene weights). We also provide signatures that include these genes as reference (Supplementary Table S1).
| WW (N = 1,043) | BW (N = 1,083) | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Signature | ROR-S | Proliferation | ROR-P | ROR-S | Proliferation | ROR-P |
| MCM10 | IGF | 3.03 (1.73, 4.33) | 0.06 (0.03, 0.08) | 3.11 (1.72, 4.50) | — | — | — |
| FAM64A | IGF | 2.57 (1.28, 3.86) | 0.05 (0.02, 0.07) | 2.64 (1.26, 4.02) | — | — | — |
| CCNB2 | Estradiol | 2.69 (1.40, 3.98) | 0.05 (0.02, 0.08) | 2.71 (1.33, 4.09) | — | — | — |
| MMP1 | Estradiol | 2.73 (1.45, 4.01) | 0.05 (0.02, 0.07) | 2.58 (1.21, 3.96) | −1.84 (−3.12, −0.56) | −0.04 (−0.07, −0.02) | −2.21 (−3.56, −0.87) |
| VAV3 | Other | −2.22 (−3.51, −0.93) | −0.04 (−0.07, −0.02) | −2.40 (−3.79, −1.03) | — | — | — |
| PCSK6 | IGF | −2.16 (−3.45, −0.88) | −0.03 (−0.06, 0.00) | −1.88 (−3.25, −0.50) | — | — | — |
| GNG11 | Claudin-low | −1.27 (−2.56, 0.02) | −0.02 (−0.05, 0.00) | −1.42 (−2.80, −0.05) | — | — | — |
Permutation testing provides context to GReX-prioritized gene and CRS associations
To assess the statistical significance for the observed variance in CRS explained by significant GReX-prioritized genes, we conducted two permutation analyses. First, we assessed the per-gene significance of the GReX-CRS associations, conditional on the SNP-trait effects at the locus, by generating a null distribution for the GReX-CRS association via shuffling the SNP-gene weights from the predictive models 5,000 times. We generated a permutation P value for the GReX-CRS association by comparing with this null distribution. Here, we found that all GReX-CRS associations showed significance in permutation testing at FDR-adjusted P < 0.05 (Table 1). These per-GReX-prioritized gene permutation tests show that GReX (of GReX-prioritized genes) adds more context beyond the genetic architecture at the locus and provide evidence that germline genetics to GReX-prioritized gene expression relationship mediates, to some level, the complex genetic effects on CRS.
Next, we quantified the percent variance explained of CRS by the GReX-prioritized genes, in aggregate, by calculating the model adjusted R2 for a regression of covariate-residualized CRS on GReX all GReX-prioritized genes. To context these model adjusted R2, we conducted two permutation tests. First, we permuted the sample labels for covariate-residualized CRS 10,000 times and computed the model adjusted R2 at each iteration to generate a null distribution for adjusted R2 between GReX-prioritized genes and CRS. Across WW and BW, the observed R2 of GReX-prioritized genes against CRS (7%–10% among WW and 1% among BW) were statistically significant against the respective null distributions (P values and distributions in Fig. 2B).To further contextualize the proportion of variance in CRS explained by GReX-prioritized genes, we computed race-specific heritability estimates using GCTA (57). Given the limited sample size for which CRS data were available, we computed the heritability based on typed SNPs. Moreover, heritability estimates for CRS were stratified by race. Among WW, heritability ranged from 0.13 (SE: 0.23) for ROR-S to 0.21 (SE: 0.23) for proliferation score. Among BW, heritability was much lower and ranged from 0.01 (SE: 0.12) for proliferation score to 0.02 (SE: 0.14) for ROR-P. However, we note that heritability estimates from GCTA were imprecise due to limited sample size. Permutation tests for analyses of tumor expression of GReX-prioritized genes and CRS are available in Supplementary Fig. S7.
Second, we wanted to assess whether the GReX of these sets of GReX-prioritized genes (7 in WW and 1 in BW) explained more of the variance in CRS than the GReX of a randomly selected set of genes of the same size. Previous studies have shown that the tumor expression of a set randomly selected genes is likely to be predictive of breast cancer outcomes; we wished to investigate this phenomenon on the GReX level (58, 59). Over 10,000 repetitions, we randomly selected 7 and 1 genes in WW and BW subjects, respectively, ran a multivariable regression, and calculated the model adjusted R2 to generate another null distribution. Here again, we found that the true model R2 outperformed the null distribution, all showing permutation P < 0.05 in these settings (Fig. 2B). These permutation tests show that our GReX-prioritized genes, taken together, appreciably explain differences in CRS.
Associations between GReX-prioritized genes and PAM50 subtype correlations and gene expression
As CRS are constructed from PAM50 subtype-specific correlations and gene expression profiles, we further studied associations between GReX of GReX-prioritized genes and PAM50 SCCs and gene expression to understand how PAM50 subtype and gene expression mediate GReX-prioritized gene and CRS associations. Among WW, a one SD increase in FAM64A and CCNB2 GReX resulted in significantly increased basal-like SCC while an identical increase in VAV3, PCSK6, and GNG11 GReX resulted in significantly increased LumA SCC. The magnitude of increase in correlation for respective subtypes per GReX-prioritized gene was approximately 0.05, and most estimates had credible intervals that did not intersect the null. Among WW, associations between HER2-like SCC and GReX-prioritized genes followed similar patterns to associations for the basal-like subtype, although associations for HER2 were more precise (Fig. 3A). We found predominantly null associations between GReX-prioritized genes and LumB SCC among WW (Fig. 3A). Unlike in WW, for BW, an increase in MMP1 GReX was not associated with LumA, HER2, or basal-like SCCs. Instead, among BW, MMP1 GReX was significantly negatively associated with LumB SCC. Estimates from univariate regressions are provided in Supplementary Tables S5–S8.
Figure 3.

Associations between GReX-prioritized genes and PAM50 SCCs and gene expression. A, Among BW (top) and WW (bottom), associations between GReX-prioritized genes and PAM50 SCCs using Bayesian multivariate regression and multivariate adaptive shrinkage. Effect estimates show change in SCCs (range, −1 to 1) for one SD increase in GReX-prioritized gene GReX. Circle, triangle, and square denote corresponding LFSR intervals for effect sizes. B, Heatmap of change in log2-normalized PAM50 tumor expression for one SD increase in GReX-prioritized gene GReX. *, **, *** denote FDR intervals for effect sizes.
For both WW and BW, the pattern of associations between GReX-prioritized genes and PAM50 tumor expression were predominantly congruent with observed associations between GReX-prioritized genes and PAM50 SCCs as well as GReX-prioritized genes and CRS (Fig. 3; Supplementary Tables S9–S12). In WW, a one SD increase in CCNB2 GReX was associated with significantly increased ORC6L, PTTG1, and KIF2C (basal-like genes) expression and UBE2T and MYBL2 (LumB genes) expression. In contrast, a one SD increase in PCSK6 GReX significantly increased BAG1, FOXA1, MAPT, and NAT1 (LumA genes) expression (Fig. 3B). While increased MMP1 GReX was associated with significantly increased expression of ORC6 L (basal-like gene), MYBL2, and BIRC5 (LumB genes) among WW, this was not the case among BW. Instead, increased MMP1 GReX among BW was significantly associated with increased expression of SLC39A6 (LumA gene) and decreased expression of ACTR3B, PTTG1, and EXO1 (basal-like genes; Fig. 3B). Associations between GReX-prioritized genes and PAM50 genes provide a granular, gene interaction level view into the mediation of the GReX-prioritized gene and CRS association, suggesting that trans-regulation of subtype-specific PAM50 genes by GReX-prioritized genes in breast tumors could be a possible contributor to subtypes and, subsequently, CRS and recurrence.
Discussion
Through a GReX analysis, we identified 7 and 1 genes among WW and BW, respectively, for which genetically regulated breast tumor expression was associated with CRS and underlying PAM50 gene expression and subtype. Among WW, these 7 GReX-prioritized genes explained between 7% and 10% of the variation in CRS, a large and statistically significant proportion of variance. Among BW, the singular GReX-prioritized gene explained a statistically significant approximately 1% of the variation in proliferation score and ROR-P. The magnitudes of these estimates were concordant with race-specific heritability estimates for CRS (13%–21% for WW; 1%–2% or BW) in this study population and suggest higher germline genetic contribution to CRS among WW compared with BW and as substantial contribution of GReX-prioritized genes to race-specific CRS heritability. There are two key novel aspects to this study. First, existing literature on associations between tumor gene expression and recurrence (for which CRS are a proxy) cannot distinguish between genetic and non-genetic components of effect (60), whereas, here, we estimate the contribution of the genetic component. Second, GReX analysis allows directional interpretation of observed associations that are not possible when correlating tumor gene expression and recurrence. For instance, prior studies report CCNB2 is upregulated in triple-negative breast cancers (TNBC) but were unable to determine whether increased CCNB2 expression contributes to development or maintenance of TNBC or is part of the molecular response to cancer progression (61, 62). In contrast, GReX is a function of only genetic variation. As such, we can confidently rule out that differences in CCNB2 GReX are not direct consequences of subtype (and by extension recurrence); however, our observed associations of CCNB2 GReX and subtype suggest a potential directional relationship for further study. Thus, GReX analysis allows a directional, potentially causal interpretation, subject to effective control for population stratification, minimal horizontal pleiotropy, and assumptions of independent assortment of alleles (22, 23).
Our GReX-prioritized gene and subtype associations among WW are consistent with literature on the association between tumor (i.e., genetic and non-genetic) expression of our GReX-prioritized genes and subtype. Prior investigations in cohorts of primarily European ancestry have reported that MCM10, FAM64A, and CCNB2 expression is higher in ER-negative compared with ER-positive tumors (61–63). In studies that compared triple-negative and non–triple-negative subtypes, higher MCM10, FAM64A, and CCNB2 expression was detected in triple-negative breast cancer (61, 62). Histologically, HER2-enriched and basal-like subtypes are typically ER negative, and triple negatives are similar to basal-like subtypes (9, 64). Moreover, our findings among WW that GReX of PCSK6 and VAV3 associated with LumA subtype and LumA-specific gene expression are also consistent with previous results of PCSK6 and VAV3 upregulation in ER-positive subtypes (65, 66). Importantly, our associations suggest directional mechanisms: from germline variation, to GReX of GReX-prioritized gene, and ultimately, to subtype.
Presently, little is known about germline genetic regulation of PAM50 tumor expression. In CBCS, we found that tumor expression of most PAM50 genes is not cis-heritable (18). Instead, observed GReX-prioritized gene and PAM50 gene expression associations may implicate trans-gene regulation of the PAM50 signature. For instance, we found that VAV3 GReX is significantly positively associated with tumor expression of BAG1, FOXA1, MAPT, and NAT1 and nominally with increased tumor ESR1 expression, all of which correspond well with LumA signature. Such trans-genic regulation signals, especially in the case of ESR1, pose significant clinical and therapeutic implication if confirmed under experimental conditions. For example, VAV3 has been shown to activate RAC1, which upregulates ESR1 (67, 68), but such mechanistic evidence is sparse for other putative GReX-prioritized gene to PAM50 associations. More generally, two of the GReX-prioritized genes among WW have been found to activate transcription factors; FAM64A enhances oncogenic NFκB signaling while both FAM64A and PCSK6 activate oncogenic STAT3 signaling (69–71).
Interestingly, we found MMP1 GReX has divergent associations with CRS across race. There are a few potential explanations. While heritability and proportion of variance in MMP1 expression were similar across WW and BW predictive models, we found that the range of MMP1 GReX was manifold among WW than BW. Potential differences in influence of germline genetics on tumor expression and CRS by race could be an artifact of divergent somatic or epigenetic factors that CBCS has not assayed (72–75). Second, while studies generally report that MMP1 tumor expression is higher in triple-negative and basal-like breast cancer, one study reported that MMP1 expression in tumor cells does not significantly differ by subtype (76–78). Instead, Boström and colleagues reported that MMP1 expression differs in stromal cells of patients with different subtypes (78). There is evidence to suggest that tumor composition, including stromal and immune components, may influence breast cancer progression in a subtype-specific manner. Future studies should consider expression predictive models that integrate greater detail on tumor cell–type composition to disentangle potential race-specific tumor composition effects on race-specific GReX associations (79, 80).
In this study, race (derived from self-report) captures genetic ancestry and additionally, socioeconomic context. Prior investigations using CBCS data have reported concordance between self-reported race and the first principal component of the combined (i.e., WW and BW) genotype matrix. In our analysis of local ancestry–derived global ancestry estimates and self-reported race, we found a similar, high level of concordance. In the absence of available methods that allow stratification or adjustments based on genetic ancestry across the GReX analytic framework, the use of race as a stratifying variable is intended to serve as a proxy for stratification by genetic ancestry. We acknowledge the limitation that race may not be a viable proxy across other populations outside CBCS, and that it is challenging to parse effects seen across race into effects of genetic ancestry and effects of socioeconomic context.
We found marked differences in the pattern of associations between genetic ancestry and race with tumor expression and GReX of GReX-prioritized genes, highlighting potential differences in contributions of germline and non-germline components to tumor expression across European and African ancestry groups. One particular example is MCM10. In the literature, higher MCM10 tumor expression is correlated with basal-like subtype, which is more prevalent among BW. The spectrum of our observations suggests that higher MCM10 tumor expression is associated with basal-like subtype across both BW and WW, but that the germline-regulated component of this expression may be stronger among WW. Similar patterns were seen for FAM64A and CCNB2. Analyses by race instead of genetic ancestry yielded associations similar in magnitude and direction. Racial differences in non-germline components of tumor expression, including tumor methylation and somatic alternations, may partly explain race-specific differences in GReX-prioritized genes (18, 72–75, 81, 82). Other factors that warrant further investigation include potential greater contribution of trans-regulation in tumor gene expression in BW (methods for capturing trans-regulation in gene expression predictive models are not as well developed as those for cis-regulation; refs. 18, 83). These factors should be investigated further as transcriptomic and epigenomic datasets for racially diverse cohorts of patients with breast cancer become available.
There are a few limitations to this study. First, as CBCS used a Nanostring nCounter probeset for mRNA expression quantification of genes relevant for breast cancer, we could not analyze the whole human transcriptome. While this probeset may exclude several cis-heritable genes, CBCS contains one of the largest breast tumor transcriptomic datasets for Black women, allowing us to build well-powered race-specific predictive models, a pivotal step in ancestry-specific GReX analysis. Second, CBCS lacked data on somatic amplifications and deletions, inclusion of which could enhance the performance of predictive models of tumor expression (84). Third, as recurrence data were collected in a small subset with few recurrence events, we were unable to make a direct comparison between CRS and recurrence results, which may affect clinical generalizability. However, to our knowledge, CBCS is the largest resource of PAM50-based CRS data.
Our analysis provides evidence of race-specific putative germline associations to CRS, mediated through associations between genetically regulated tumor expression of GReX-prioritized genes and PAM50 expressions and subtype. This work underscores the need for larger and more diverse cohorts for genetic epidemiology studies of breast cancer. Future studies should consider subtype-specific genetics (i.e., stratification by subtype in predictive model training and association analyses) to elucidate heritable gene expression effects on breast cancer outcomes both across and within subtype, which may yield further hypotheses for more fine-tuned clinical intervention.
Authors' Disclosures
C.M. Perou reports personal fees from Bioclassifier LLC outside the submitted work; in addition, C.M. Perou has a patent for U.S. Patent No. 12,995,459 issued, licensed, and with royalties paid from Bioclassifier. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Methods, Figures S1-S7, and References
Supplementary Tables S1-S12
Acknowledgments
This work was supported by Susan G. Komen for the Cure for CBCS study infrastructure. Funding was provided by the NIH, NCI P01-CA151135, P50-CA05822, and U01-CA179715 to A.F. Olshan, C.M. Perou, and M.A. Troester. A. Patel is supported by T32ES007018. M.I. Love is supported by R01-HG009937, R01-MH118349, P01-CA142538, and P30-ES010126. The Translational Genomics Laboratory is supported in part by grants from the NCI (3P30CA016086) and the University of North Carolina at Chapel Hill University Cancer Research Fund. Genotyping was done at the DCEG Cancer Genomics Research Laboratory using funds from the NCI Intramural Research Program. This content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funder had no role in study design, data collection, analysis or interpretation, or writing of the article.
Funding for BCAC came from: Cancer Research UK (grant numbers C1287/A16563, C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the European Union's Horizon 2020 Research and Innovation.
Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST, respectively), the European Community's Seventh Framework Programme under grant agreement no. 223175 (HEALTHF2-2009-223175; COGS), the NIH (CA128978) and Post-Cancer GWAS initiative [1U19CA148537, 1U19 CA148065-01 (DRIVE), and 1U19 CA148112—the GAME-ON initiative], the Department of Defence (W81XWH-10-1-0341), and the Canadian Institutes of Health Research CIHR) for the CIHR Team in Familial Risks of Breast Cancer (grant PSR-SIIRI-701). All studies and funders as listed in Michailidou and colleagues (2013 and 2015) and in Guo and colleagues (2015) are acknowledged for their contributions.
The authors thank the Carolina Breast Cancer Study participants and volunteers. They also thank Colin Begg, Jianwen Cai, Katherine Hoadley, Yun Li, and Bogdan Pasaniuc for valuable discussion during the research process. The authors thank Erin Kirk and Jessica Tse for their invaluable support during the research process. They thank the DCEG Cancer Genomics Research Laboratory and acknowledge the support from Stephen Chanock, Rose Yang, Meredith Yeager, Belynda Hicks, and Bin Zhu.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
A. Patel: Formal analysis, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. M. García-Closas: Resources, data curation, funding acquisition, project administration, writing–review and editing. A.F. Olshan: Resources, data curation, supervision, funding acquisition, investigation, writing–review and editing. C.M. Perou: Resources, data curation, supervision, funding acquisition, project administration, writing–review and editing. M.A. Troester: Conceptualization, resources, data curation, supervision, funding acquisition, investigation, methodology, writing–review and editing. M.I. Love: Conceptualization, resources, software, supervision, funding acquisition, investigation, methodology, writing–review and editing. A. Bhattacharya: Conceptualization, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallden B, Storhoff J, Nielsen T, Dowidar N, Schaper C, Ferree S, et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med Genomics 2015;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med 2004;351:2817–26. [DOI] [PubMed] [Google Scholar]
- 4. Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 2008;26:317–25. [DOI] [PubMed] [Google Scholar]
- 5. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the carolina breast cancer study. JAMA 2006;295:2492–502. [DOI] [PubMed] [Google Scholar]
- 6. O'Brien KM, Cole SR, Tse CK, Perou CM, Carey LA, Foulkes WD, et al. Intrinsic breast tumor subtypes, race, and long-term survival in the carolina breast cancer study. Clin Cancer Res 2010;16:6100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shim HJ, Kim SH, Kang BJ, Choi BG, Kim HS, Cha ES, et al. Breast cancer recurrence according to molecular subtype. Asian Pac J Cancer Prev 2014;15:5539–44. [DOI] [PubMed] [Google Scholar]
- 8. van Maaren MC, de Munck L, Strobbe LJA, Sonke GS, Westenend PJ, Smidt ML, et al. Ten-year recurrence rates for breast cancer subtypes in the Netherlands: a large population-based study. Int J Cancer 2019;144:263–72. [DOI] [PubMed] [Google Scholar]
- 9. Troester MA, Sun X, Allott EH, Geradts J, Cohen SM, Tse CK, et al. Racial differences in PAM50 subtypes in the carolina breast cancer study. J Natl Cancer Inst 2018;110:176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dowsett M, Sestak I, Lopez-Knowles E, Sidhu K, Dunbier AK, Cowens JW, et al. Comparison of PAM50 risk of recurrence score with oncotype DX and IHC4 for predicting risk of distant recurrence after endocrine therapy. J Clin Oncol 2013;31:2783–90. [DOI] [PubMed] [Google Scholar]
- 11. Sestak I, Buus R, Cuzick J, Dubsky P, Kronenwett R, Denkert C, et al. Comparison of the performance of 6 prognostic signatures for estrogen receptor-positive breast cancer: a secondary analysis of a randomized clinical trial. JAMA Oncol 2018;4:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ohnstad HO, Borgen E, Falk RS, Lien TG, Aaserud M, Sveli MAT, et al. Prognostic value of PAM50 and risk of recurrence score in patients with early-stage breast cancer with long-term follow-up. Breast Cancer Res 2017;19:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albain KS, Gray RJ, Makower DF, Faghih A, Hayes DF, Geyer CE, et al. Race, ethnicity and clinical outcomes in hormone receptor-positive, HER2-negative, node-negative breast cancer in the randomized TAILORx trial. J Natl Cancer Inst 2020;113:390–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reeder-Hayes KE, Anderson BO. Breast cancer disparities at home and abroad: a review of the challenges and opportunities for system-level change. Clin Cancer Res 2017;23:2655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Durham DD, Robinson WR, Lee SS, Wheeler SB, Reeder-Hayes KE, Bowling JM, et al. Insurance-based differences in time to diagnostic follow-up after positive screening mammography. Cancer Epidemiol Biomarkers Prev 2016;25:1474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wheeler SB, Reeder-Hayes KE, Carey LA. Disparities in breast cancer treatment and outcomes: biological, social, and health system determinants and opportunities for research. Oncologist 2013;18:986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ko NY, Hong S, Winn RA, Calip GS. Association of insurance status and racial disparities with the detection of early-stage breast cancer. JAMA Oncol 2020;6:385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhattacharya A, García-Closas M, Olshan AF, Perou CM, Troester MA, Love MI. A framework for transcriptome-wide association studies in breast cancer in diverse study populations. Genome Biol 2020;21:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Escala-Garcia M, Guo Q, Dörk T, Canisius S, Keeman R, Dennis J, et al. Genome-wide association study of germline variants and breast cancer-specific mortality. Br J Cancer 2019;120:647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muranen TA, Khan S, Fagerholm R, Aittomäki K, Cunningham JM, Dennis J, et al. Association of germline variation with the survival of women with BRCA1/2 pathogenic variants and breast cancer. NPJ Breast Cancer 2020;6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huo D, Hu H, Rhie SK, Gamazon ER, Cherniack AD, Liu J, et al. Comparison of breast cancer molecular features and survival by african and european ancestry in the cancer genome atlas. JAMA Oncol 2017;3:1654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 2015;47:1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 2016;48:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhong J, Jermusyk A, Wu L, Hoskins JW, Collins I, Mocci E, et al. A transcriptome-wide association study identifies novel candidate susceptibility genes for pancreatic cancer. J Natl Cancer Inst 2020;112:1003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu L, Shi W, Long J, Guo X, Michailidou K, Beesley J, et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat Genet 2018;50:968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mancuso N, Gayther S, Gusev A, Zheng W, Penney KL, Kote-Jarai Z, et al. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat Commun 2018;9:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keys KL, Mak ACY, White MJ, Eckalbar WL, Dahl AW, Mefford J, et al. On the cross-population generalizability of gene expression prediction models. PLoS Genet 2020;16:e1008927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hair BY, Hayes S, Tse CK, Bell MB, Olshan AF. Racial differences in physical activity among breast cancer survivors: implications for breast cancer care. Cancer 2014;120:2174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Newman B, Moorman PG, Millikan R, Qaqish BF, Geradts J, Aldrich TE, et al. The Carolina Breast Cancer Study: integrating population-based epidemiology and molecular biology. Breast Cancer Res Treat 1995;35:51–60. [DOI] [PubMed] [Google Scholar]
- 30. Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The OncoArray consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol Biomarkers Prev 2017;26:126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet 2014;10:e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods 2011;9:179–81. [DOI] [PubMed] [Google Scholar]
- 34. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wigginton JE, Cutler DJ, Abecasis GR. A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 2005;76:887–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bhattacharya A, Hamilton AM, Furberg H, Pietzak E, Purdue MP, Troester MA, et al. An approach for normalization and quality control for NanoString RNA expression data. Brief Bioinform 2020;22:bbaa163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ding B, Cao C, Li Q, Wu J, Long Q. Power analysis of transcriptome-wide association study. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Endelman JB. Ridge regression and other kernels for genomic selection with R package rrBLUP. The Plant Genome 2011;4. [Google Scholar]
- 43. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 44. van Iterson M, van Zwet EW, Heijmans BT. Controlling bias and inflation in epigenome- and transcriptome-wide association studies using the empirical null distribution. Genome Biol 2017;18:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Methodol 1995;57:289–300. [Google Scholar]
- 46. Wheeler HE, Ploch S, Barbeira AN, Bonazzola R, Andaleon A, Siahpirani FA, et al. Imputed gene associations identify replicable trans-acting genes enriched in transcription pathways and complex traits. Genet Epidemiol 2019;43:596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu X, Mefford JA, Dahl A, He Y, Subramaniam M, Battle A, et al. GBAT: a gene-based association test for robust detection of trans-gene regulation. Genome Biol 2020;21:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Urbut SM, Wang G, Carbonetto P, Stephens M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat Genet 2019;51:187–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maples BK, Gravel S, Kenny EE, Bustamante CD. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet 2013;93:278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Watase G, Takisawa H, Kanemaki MT. Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Curr Biol 2012;22:343–9. [DOI] [PubMed] [Google Scholar]
- 51. Zhao W-M, JA C, Seki A, X-l C, Yates JR, Fang G. RCS1, a substrate of APC/C, controls the metaphase to anaphase transition. Proc Natl Acad Sci U S A 2008;105:13415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Daldello EM, Luong XG, Yang C-R, Kuhn J, Conti M. Cyclin B2 is required for progression through meiosis in mouse oocytes. Development 2019;146:dev172734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Draetta G, Luca F, Westendorf J, Brizuela L, Ruderman J, Beach D. Cdc2 protein kinase is complexed with both cyclin A and B: evidence for proteolytic inactivation of MPF. Cell 1989;56:829–38. [DOI] [PubMed] [Google Scholar]
- 54. Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol 2007;8:221–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rao S, Lyons LS, Fahrenholtz CD, Wu F, Farooq A, Balkan W, et al. A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene 2012;31:716–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hossain MN, Sakemura R, Fujii M, Ayusawa D. G-protein gamma subunit GNG11 strongly regulates cellular senescence. Biochem Biophys Res Commun 2006;351:645–50. [DOI] [PubMed] [Google Scholar]
- 57. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol 2011;7:e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shimoni Y. Association between expression of random gene sets and survival is evident in multiple cancer types and may be explained by sub-classification. PLoS Comput Biol 2018;14:e1006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parada H Jr, Sun X, Fleming JM, Williams-DeVane CR, Kirk EL, Olsson LT, et al. Race-associated biological differences among luminal A and basal-like breast cancers in the Carolina Breast Cancer Study. Breast Cancer Res 2017;19:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 2013;18:123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang C, Han Y, Huang H, Min L, Qu L, Shou C. Integrated analysis of expression profiling data identifies three genes in correlation with poor prognosis of triple-negative breast cancer. Int J Oncol 2014;44:2025–33. [DOI] [PubMed] [Google Scholar]
- 63. Mahadevappa R, Neves H, Yuen SM, Jameel M, Bai Y, Yuen HF, et al. DNA Replication Licensing Protein MCM10 Promotes Tumor Progression and Is a Novel Prognostic Biomarker and Potential Therapeutic Target in Breast Cancer. Cancers 2018;10:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hagemann IS. Molecular Testing in Breast Cancer: A Guide to Current Practices. Arch Pathol Lab Med 2016;140:815–24. [DOI] [PubMed] [Google Scholar]
- 65. Thakkar AD, Raj H, Chakrabarti D, Ravishankar , Saravanan N, Muthuvelan B, et al. Identification of gene expression signature in estrogen receptor positive breast carcinoma. Biomark Cancer 2010;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Aguilar H, Urruticoechea A, Halonen P, Kiyotani K, Mushiroda T, Barril X, et al. VAV3 mediates resistance to breast cancer endocrine therapy. Breast Cancer Res 2014;16:R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zeng L, Sachdev P, Yan L, Chan JL, Trenkle T, McClelland M, et al. Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol Cell Biol 2000;20:9212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rosenblatt AE, Garcia MI, Lyons L, Xie Y, Maiorino C, Désiré L, et al. Inhibition of the Rho GTPase, Rac1, decreases estrogen receptor levels and is a novel therapeutic strategy in breast cancer. Endocr Relat Cancer 2011;18:207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu Z-S, Zhang H-X, Li W-W, Ran Y, Liu T-T, Xiong M-G, et al. FAM64A positively regulates STAT3 activity to promote Th17 differentiation and colitis-associated carcinogenesis. Proc Natl Acad Sci U S A 2019;116:10447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jiang H, Wang L, Wang F, Pan J. Proprotein convertase subtilisin/kexin type 6 promotes in vitro proliferation, migration and inflammatory cytokine secretion of synovial fibroblast-like cells from rheumatoid arthritis via nuclear-κB, signal transducer and activator of transcription 3 and extracellular signal regulated 1/2 pathways. Mol Med Rep 2017;16:8477–84. [DOI] [PubMed] [Google Scholar]
- 71. Jiang L, Ren L, Zhang X, Chen H, Chen X, Lin C, et al. Overexpression of PIMREG promotes breast cancer aggressiveness via constitutive activation of NF-κB signaling. EBioMedicine 2019;43:188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shang L, Smith JA, Zhao W, Kho M, Turner ST, Mosley TH, et al. Genetic architecture of gene expression in european and african americans: an eQTL mapping study in GENOA. Am J Hum Genet 2020;106:496–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang S, Dorsey TH, Terunuma A, Kittles RA, Ambs S, Kwabi-Addo B. Relationship between tumor DNA methylation status and patient characteristics in African-American and European-American women with breast cancer. PLoS One 2012;7:e37928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Conway K, Edmiston SN, Tse CK, Bryant C, Kuan PF, Hair BY, et al. Racial variation in breast tumor promoter methylation in the carolina breast cancer study. Cancer Epidemiol Biomarkers Prev 2015;24:921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen Y, Sadasivan SM, She R, Datta I, Taneja K, Chitale D, et al. Breast and prostate cancers harbor common somatic copy number alterations that consistently differ by race and are associated with survival. BMC Med Genomics 2020;13:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang QM, Lv L, Tang Y, Zhang L, Wang LF. MMP-1 is overexpressed in triple-negative breast cancer tissues and the knockdown of MMP-1 expression inhibits tumor cell malignant behaviors in vitro. Oncol Lett 2019;17:1732–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McGowan PM, Duffy MJ. Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Ann Oncol 2008;19:1566–72. [DOI] [PubMed] [Google Scholar]
- 78. Boström P, Söderström M, Vahlberg T, Söderström KO, Roberts PJ, Carpén O, et al. MMP-1 expression has an independent prognostic value in breast cancer. BMC Cancer 2011;11:348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol 2015;7:1120–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. González LO, Corte MD, Junquera S, González-Fernández R, del Casar JM, García C, et al. Expression and prognostic significance of metalloproteases and their inhibitors in luminal A and basal-like phenotypes of breast carcinoma. Hum Pathol 2009;40:1224–33. [DOI] [PubMed] [Google Scholar]
- 81. Gravel S. Population genetics models of local ancestry. Genetics 2012;191:607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nelson D, Kelleher J, Ragsdale AP, Moreau C, McVean G, Gravel S. Accounting for long-range correlations in genome-wide simulations of large cohorts. PLoS Genet 2020;16:e1008619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bhattacharya A, Li Y, Love MI. MOSTWAS: Multi-Omic strategies for transcriptome-wide association studies. PLoS Genet 2021;17:e1009398. doi: 10.1371/journal.pgen.1009398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xia Y, Fan C, Hoadley KA, Parker JS, Perou CM. Genetic determinants of the molecular portraits of epithelial cancers. Nat Commun 2019;10:5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods, Figures S1-S7, and References
Supplementary Tables S1-S12
Data Availability Statement
Expression data from CBCS are available on NCBI Gene Expression Omnibus with accession number GSE148426. CBCS genotype datasets analyzed in this study are not publicly available as many CBCS patients are still being followed and accordingly CBCS data are considered sensitive; the data are available from M.A. Troester upon reasonable request. Supplementary Data include summary statistics for eQTL results, tumor expression models, and relevant R code for training expression models in CBCS and are freely available at https://github.com/bhattacharya-a-bt/CBCS_TWAS_Paper/. Scripts utilized in this analysis are provided at https://github.com/APUNC/CBCS-Risk-of-Recurrence-Paper.