

Abstract
Glioma‐initiating cells (GICs), a major source of glioblastoma recurrence, are characterized by the expression of neural stem cell markers and the ability to grow by forming nonadherent spheres under serum‐free conditions. Bone morphogenetic proteins (BMPs), members of the transforming growth factor‐β family, induce differentiation of GICs and suppress their tumorigenicity. However, the mechanisms underlying the BMP‐induced loss of GIC stemness have not been fully elucidated. Here, we show that paired related homeobox 1 (PRRX1) induced by BMPs decreases the CD133‐positive GIC population and inhibits tumorigenic activity of GICs in vivo. Of the two splice isoforms of PRRX1, the longer isoform, pmx‐1b, but not the shorter isoform, pmx‐1a, induces GIC differentiation. Upon BMP stimulation, pmx‐1b interacts with the DNA methyltransferase DNMT3A and induces promoter methylation of the PROM1 gene encoding CD133. Silencing DNMT3A maintains PROM1 expression and increases the CD133‐positive GIC population. Thus, pmx‐1b promotes loss of stem cell‐like properties of GICs through region‐specific epigenetic regulation of CD133 expression by recruiting DNMT3A, which is associated with decreased tumorigenicity of GICs.
Keywords: BMP, cancer‐initiating cell, CD133, DNA methyltransferase, glioblastoma, PRRX1
BMPs induce differentiation and apoptosis of glioma‐initiating cells (GICs). Here, we identified PRRX1 as a target of BMPs in GICs. Of the two splice isoforms, only pmx‐1b induced GIC differentiation. Through interaction with DNMT3A, pmx‐1b induced the methylation of the PROM1 gene promoter and suppressed the CD133+ GIC population. Thus, site‐specific epigenetic regulation plays a critical role in the differentiation of GICs.
Abbreviations
- ALK
activin receptor‐like kinase
- bFGF
basic fibroblast growth factor
- BMP
bone morphogenetic protein
- BMPR
BMP receptor
- ChIP
chromatin immunoprecipitation
- DNMT
DNA methyltransferase
- EGF
epidermal growth factor
- EGFP
enhanced green fluorescent protein
- GIC
glioma‐initiating cell
- HGCC
Human Glioblastoma Cell Culture
- hOPC
human oligodendrocyte progenitor cell
- NSC
neural stem cell
- PCR
polymerase chain reaction
- PI3K
phosphoinositide 3’-kinase
- pmx1
paired mesoderm homeobox 1
- PRRX1
paired related homeobox 1
- qRT-PCR
quantitative real-time PCR
- shRNA
short hairpin RNA
- TGF-β
transforming growth factor‐β
1. Introduction
Glioblastoma is the most malignant form of adult brain tumors. Despite recent advances and new treatment strategies, the median survival of patients with glioblastoma remains <20 months [1, 2, 3]. Recent efforts to target abnormalities in the signaling pathways in glioblastoma cells have not been successful in improving treatment efficacy [4, 5].
Glioma‐initiating cells (GICs) play central roles in the initiation, progression, recurrence, tumor heterogeneity, and drug and radiation resistance of glioblastoma [6, 7, 8]. GICs are cellular subpopulations of glioblastoma that express neural stem and progenitor cell markers, such as PROM1 (encoding CD133), OLIG2, and SOX2 [9, 10]. CD133 is a cell surface glycoprotein expressed on neural stem cells (NSCs), and its expression decreases as cells differentiate. CD133 is thus a stem cell marker that is frequently used to isolate GICs [6, 7]. Moreover, loss of function of CD133 has been shown to impair self‐renewal and tumorigenic capacity of GICs [11], suggesting critical roles of CD133 in the pathogenesis of glioblastoma.
Members of the transforming growth factor‐β (TGF‐β) family are multifunctional proteins, involved in pathogenesis of various diseases, including cancer [12, 13]. Bone morphogenetic proteins (BMPs), members of the TGF‐β family, regulate the growth and differentiation of a wide variety of cells, including different types of cancer cells [14]. BMPs activate BMP receptor–SMAD signaling pathways and non‐SMAD pathways [15]. BMP signaling regulates normal embryonic neural development through the activation of BMP type I receptors, including activin receptor‐like kinase (ALK)‐2, ALK‐3/BMP receptor type IA (BMPRIA), and ALK‐6/BMPRIB. BMPs inhibit the tumorigenic potential of human GICs by inducing cell differentiation, cell cycle arrest, and apoptosis [16, 17, 18, 19, 20] and render GICs more susceptible to conventional chemotherapies [21]. Expression of BMP4 is decreased in high‐grade glioma compared to low‐grade glioma, and patients with high BMP4 expression show significantly better prognosis than those with low BMP4 expression [22]. Thus, BMP signaling components are considered important biomarkers and potential therapeutic targets for glioblastoma.
Antitumorigenic activities of BMPs on GICs have been reported by many investigators [21, 23]; however, some GICs respond to BMP treatment, whereas others escape the differentiation‐inducing effect of BMP and fail to undergo terminal cell cycle arrest despite active BMP signaling [24, 25]. A fraction of genes that are silenced during the course of BMP‐induced astrocyte differentiation of NSCs fail to be downregulated in the latter population of GICs [24]. Thus, considering the heterogeneity of glioblastoma cells, the mechanisms by which BMPs affect GICs should be further investigated.
Various molecules have been reported to function as downstream components in the BMP signaling pathways in glioblastoma. Transcription factors such as Snail, p21WAF1/CIP1, and Distal‐less homeobox 2 have been shown to be induced by BMP signaling and play tumor suppressive roles in glioblastoma [19, 20, 26, 27]. In addition, a decrease in cyclin D1 is involved in growth inhibition and the induction of Bax and inhibition of Bcl‐2 and Bcl‐xL in apoptosis [18]. Moreover, certain extracellular or cell surface molecules regulate the action of BMPs in glioblastoma. Gremlin1, an extracellular antagonist of BMPs, is secreted by GICs and maintains their stem cell‐like properties by inhibiting endogenous BMP signaling [19]. We showed that, among the three BMP type I receptors expressed in GICs, ALK‐2 plays a major role in apoptosis regulation [20] and that cooperation with EphA6 tyrosine kinase receptor sensitizes some resistant GICs for BMP‐induced apoptosis [28]. However, because of the heterogeneity of glioblastoma cells, certain other BMP target molecules may also be critically involved in differentiation and apoptosis of GICs, and it remains to be elucidated how the stem cell‐like properties of GICs are regulated by BMPs.
By analyzing RNA‐sequencing (RNA‐seq) data [20], we have identified certain genes specifically induced by BMPs in GICs. We show that paired related homeobox 1 (PRRX1; also known as pmx1 (paired mesoderm homeobox 1)) is induced by BMP‐4 stimulation and promotes differentiation of GICs by decreasing the CD133‐positive cell population. Intriguingly, the DNA methyltransferase 3A (DNMT3A) interacts with one of the splice isoforms of PRRX1 and induces methylation of the PROM1 gene promoter, leading to a decrease in the CD133‐positive GIC population. Thus, our findings suggest a critical role of PRRX1 induction by BMP‐4 in regulation of the stem cell‐like properties of GICs through epigenetic regulation of the PROM1 gene by DNA methyltransferase.
2. Materials and methods
2.1. Cell culture and reagents
TGS‐01 and TGS‐04 cells were obtained during surgery from consenting patients with glioblastoma multiforme (grade IV), as approved by the Institutional Review Board at the University of Tokyo Hospital (characteristics of the cells are listed in Table S1), as described previously [20, 29], and were cultured in Dulbecco’s Eagle’s medium/F12 containing B27 supplement (Thermo Fisher Scientific, Waltham, MA, USA) and 20 ng·mL−1 of epidermal growth factor (EGF, PeproTech, Cranbury, NJ, USA) and basic fibroblast growth factor (bFGF, PeproTech). The use of patient‐derived glioma cells (TGS‐01 and TGS‐04) was approved by the Research Ethics Committee of the Graduate School of Medicine, The University of Tokyo. Patient‐derived glioblastoma cells (U3005MG and U3024MG), obtained in accordance with protocols approved by the regional ethical review board, were acquired from the Human Glioblastoma Cell Culture (HGCC) resource (https://www.hgcc.se) at the Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala, Sweden [30]. U3005MG and U3024MG were cultured in Dulbecco’s Eagle’s medium/F12 and Neurobasal medium (Thermo Fisher Scientific), enriched with B‐27 supplement, N‐2 supplement (Thermo Fisher Scientific), and 20 ng·mL−1 of EGF and bFGF. Recombinant human BMP‐4 was purchased from R&D Systems (Minneapolis, MN, USA) and used at 30 ng·mL−1. The antibodies used were as follows: anti‐PRRX1 (ab67631, Abcam, Cambridge, UK), anti‐PRRX1 (NBP1‐06067, Novus Biological, Centennial, CO, USA), anti‐PRRX1 C‐terminal (ab174201, Abcam), anti‐α‐tubulin (T6199, Sigma‐Aldrich, Merck, Burlington, MA, USA), anti‐FLAG M2 (F3165, Sigma‐Aldrich, Merck), anti‐CD133/2 (293C2, Miltenyi Biotec, Auburn, CA, USA), IgG2b (IS6‐11E5.11, Miltenyi Biotec), anti‐DNMT3A (ab2850 for immunoblotting and ab13888 for immunoprecipitation, Abcam), anti‐SMAD1 (9743, Cell Signaling Technology, Danvers, MA, USA), anti‐phospho‐SMAD1/5 (9516, Cell Signaling Technology), mouse IgG1 isotype control (MAB002, R&D Systems), and normal rabbit IgG (sc‐2027, Santa Cruz, Dallas, TX, USA).
2.2. Quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis
Total RNA was extracted from GICs using RNeasy Mini Kit (Qiagen, Hilden, Germany). Complementary DNA was synthesized using a PrimeScript II First Strand cDNA Synthesis Kit (Takara Bio, Shiga, Japan) or an iScript cDNA Synthesis Kit (Bio‐Rad, Hercules, CA, USA). Gene expression levels were quantified on Step One Plus Real‐Time PCR Systems (Thermo Fisher Scientific) or CFX Connect Real‐Time PCR Detection System (Bio‐Rad) with FastStart Universal SYBR Green Master Mix (Roche Applied Science, Basel, Switzerland) or KAPA SYBR FAST qPCR Kit Master Mix (Kapa Biosystems, London, UK). Primer sets are listed in Table S2.
2.3. Plasmid construction
Enhanced green fluorescent protein (EGFP), FLAG‐tagged PRRX1 pmx‐1a and pmx‐1b, and firefly luciferase (Promega, Madison, WI, USA) cDNAs were cloned into pENTR201 vector (Thermo Fisher Scientific), and recombination between pENTR201 and CSII‐EF‐RfA vectors was catalyzed by LR clonase (Thermo Fisher Scientific). DNA sequences encoding short hairpin RNAs (shRNAs) were inserted into pENTR4‐H1 vector, and LR reaction between pENTR4‐H1 and CS‐RfA‐CMV‐PuroR vectors was performed. The sequences targeted by shRNAs are listed in Table S3. PROM1 promoter P1 (−1900 to −1) was cloned as previously described [31]. The PROM1 promoter P1 and firefly luciferase (Fluc) genes were subcloned between SalII and XbaI sites of the pENTR4‐H1 vector to generate pENTR4‐PROM1‐Fluc. The EGFP gene on CS‐RfA‐CG was replaced by the renilla luciferase gene to generate CS‐RfA‐CMV‐Rluc, and LR reaction between the pENTR4‐PROM1‐Fluc and CS‐RfA‐CMV‐Rluc vectors was performed. PROM1 promoter P1 (−1010 to −1) was subcloned as a deletion mutant. Mutagenesis of the putative PRRX1‐binding sites on PROM1 promoter P1 was introduced by site‐specific PCR with the oligonucleotides shown in Table S4.
2.4. Lentiviral transduction
The indicated vector plasmids, pCAG‐HIVgp and pCMV‐VSV‐G‐Rev, were transduced into HEK293FT cells using Lipofectamine 2000 (Thermo Fisher Scientific). Lentiviral particles were concentrated with Lenti‐X Concentrator (Takara Bio). Lentiviral vectors, pCSII‐EF‐RfA, pENTR4‐H1, pCAG‐HIVgp, and pCMV‐VSV‐G‐Rev, were gifts from late H. Miyoshi (formerly RIKEN, Saitama, Japan).
2.5. Luciferase reporter assay
Cells stably expressing firefly and renilla luciferase were lysed, and luciferase activities were determined using a Dual‐luciferase Reporter Assay System (Promega) on Mithras LB940 (Berthold Technologies, Bad Wildbad, Germany). The values of firefly luciferase activity were normalized to those of renilla luciferase activity.
2.6. Sphere formation assay
TGS‐01 or TGS‐04 cells were seeded on ultra‐low attachment microplates (Corning, Kennebunk, ME, USA) at a density of 1–50 cells per well by using Cell Sorter SH800 Series (Sony Corporation, Tokyo, Japan) and then cultured for 7 days. Cell masses with more than 50 µm in diameter were considered as spheres.
2.7. Immunoblot analysis
Cells were lysed with a buffer containing 50 mm Tris/HCl (pH 8.0), 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Triton X‐100. Cleared cell lysates were subjected to SDS/PAGE and transferred onto a Fluoro Trans W Membrane (Pall, East Hills, NY, USA). Immunoblotting was performed using the indicated antibodies, and chemiluminescence was detected using ImageQuant LAS4000 (Fujifilm, Tokyo, Japan).
2.8. Immunoprecipitation followed by immunoblotting
For immunoprecipitation, cells were lysed with a lysis buffer (20 mm Tris/HCl (pH 7.4), 10 mm KCl, 10 mm MgCl2, 2 mm EDTA, 10% glycerol, 1% Triton X‐100, 2.5 mm β‐glycerophosphate, 1 mm NaF, 1 mm dithiothreitol, and cOmplete Protease Inhibitor Cocktail (Sigma‐Aldrich, Merck)), followed by resolubilization by the addition of NaCl at a final concentration of 420 mm [32]. Antibodies bound to Dynabeads M‐280 sheep anti‐mouse IgG or Dynabeads Protein A (Thermo Fisher Scientific) were mixed with cell lysates at 4 °C. Dynabeads were washed with RIPA buffer containing 50 mm Tris/HCl (pH 8.0), 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, and 1% Nonidet P‐40 and boiled at 95 °C for 5 min. Samples were then subjected to SDS/PAGE, followed by immunoblot analysis.
2.9. Chromatin immunoprecipitation (ChIP)
Cells were fixed with 1% formaldehyde for 10 min, whereafter glycine was added to a final concentration of 0.125 m to stop the reaction. After washing with ice‐cold phosphate‐buffered saline, the cells were scraped, pelleted, and resuspended in sonication/elution buffer (50 mm Tris/HCl (pH 8.1), 1% SDS, and 10 mm EDTA, containing cOmplete Protease Inhibitor Cocktail). After sonication, a solution containing the complex of anti‐FLAG antibody and Dynabeads M‐280 sheep anti‐mouse IgG, mixed with immunoprecipitation buffer (20 mm Tris/HCl (pH 8.0), 150 mm NaCl, 2 mm EDTA, 1% Triton X‐100, and cOmplete Protease Inhibitor Cocktail), was added, followed by centrifugation at 4 °C. The beads were then washed with washing buffer (50 mm HEPES‐KOH (pH 7.0), 500 mm LiCl, 1 mm EDTA, 0.7% sodium deoxycholate, and 1% Nonidet P‐40) and TE buffer (10 mm Tris/HCl (pH 8.0), 1 mm EDTA (pH 8.0)), and DNA was eluted in the sonication/elution buffer at 65 °C for 16 h. Genomic DNA was then extracted with a PCR purification kit (Qiagen), and DNA fragments were analyzed using the primers shown in Table S5, FastStart Universal SYBR Green Master Mix (Roche Applied Science, Penzberg, Germany), and Step One Plus Real‐Time PCR Systems.
2.10. Flow cytometric analysis
Cells were dissociated and labeled with a phycoerythrin‐conjugated anti‐CD133/2 (293C2) antibody (Miltenyi Biotec). The expression of CD133 on cell surfaces was analyzed on BD LSR II Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA) or Cell Sorter SH800 Series (Sony Corporation) with FlowJo (BD Biosciences).
2.11. Mouse tumor model
All animal experiments were approved by and carried out according to the guidelines of the Animal Ethics Committee of the Graduate School of Medicine, The University of Tokyo. A total of 5 × 104 viable cells were injected over bregma 2 mm to the right of the sagittal suture and 3 mm below the surface of the skulls of 5‐week‐old female BALB/c nu/nu mice (Sankyo Lab Service, Tokyo, Japan). For in vivo bioluminescence imaging, D‐luciferin (Promega) was injected intraperitoneally and photons were detected using NightOwl LB983 (Berthold Technologies). The mice were euthanized when a weight loss > 20% or neurological signs such as hemiparesis were observed.
2.12. DNA methylation analysis
Genomic DNA was isolated from TGS‐01 and TGS‐04 cells, and bisulfite treatment was performed using an EpiTect Bisulfite Conversion Kit (Qiagen). Using the bisulfite‐treated genomic DNA as a template, PCR was carried out using EpiTaq HS (Takara Bio). The PCR products were analyzed by sequencing or agarose gel electrophoresis. The primers used for the PCR are listed in Tables S6 and S7.
2.13. Statistical analysis
Student’s t‐test, Tukey’s test, and the log‐rank test were performed using statistical software r (http://www.R‐project.org).
3. Results
3.1. PRRX1 is induced by BMP‐4 and directly regulates the PROM1 promoter in GICs
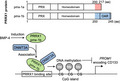
As we previously reported, BMP‐4 induces differentiation and apoptosis of GICs [20]. Consistently, neurosphere formation by patient‐derived GICs, TGS‐01, in a serum‐free condition, was suppressed by BMP‐4 stimulation (Fig. 1A). BMP‐4 reduced the expression of glioma stem cell marker mRNAs, including PROM1, OLIG2, and SOX2, in TGS‐01 as well as in TGS‐04 cells (Fig. 1B). To address the mechanisms underlying BMP‐induced differentiation of GICs, we screened for genes regulated by BMP‐4 in GICs using an RNA‐seq analysis of TGS‐01 cells treated with or without BMP‐4 [20]. Among the genes induced by BMP‐4 in TGS‐01 cells, we focused our study on the function of the gene encoding PRRX1, because PRRX1 has been reported to play critical roles in normal mouse NSCs [33].
Fig. 1.
BMP‐4 induces differentiation of GIC and upregulates the expression of PRRX1. TGS‐01 and TGS‐04 cells were treated with or without BMP‐4 (30 ng·mL−1) for 72 h, except for panel (A). (A) Failure of sphere formation of TGS‐01 cells in the presence of BMP‐4 for 5 days. Scale bars: 200 µm. (B) Downregulation of glioma stem cell markers (n = 3 biological replicates). (C) A scheme of the splice isoforms of PRRX1. (D) Induction of the expression of two splice isoforms of PRRX1 in TGS‐01 and TGS‐04 cells by BMP‐4 stimulation (n = 3 biological replicates). (E) ChIP‐qPCR analysis of the binding of pmx‐1a and pmx‐1b on the PROM1 promoter in TGS‐01 cells stably expressing FLAG‐tagged PRRX1 proteins (n = 4 biological replicates). (F) Relative promoter activity measured in TGS‐01 cells treated with or without BMP‐4 by dual‐luciferase assay (n = 3 biological replicates). The PROM1 promoter was subcloned, and the AT‐rich sequence of PRRX1‐binding sites was deleted or converted into a GC‐rich sequence. The graphs in panels (B) and (D–F) represent mean ± SD of biological replicates. The P‐values were determined by Student’s t‐test (B) or Tukey’s test (D–F).
PRRX1 occurs as two isoforms, that is, pmx‐1a and pmx‐1b, that are produced by alternative splicing. The first 199 amino acid residues have identical sequences, but the C‐terminal regions differ between the two isoforms (Fig. 1C). The longer isoform of 245 amino acid residues, pmx‐1b (also known as PRRX1A, encoded by pmx‐1b, NM_022716), has an OAR (otp, aristaless, and rax) domain in its C‐terminal region (amino acid residues 200‐245), whereas the shorter isoform of 217 amino acid residues, pmx‐1a (also known as PRRX1B, encoded by pmx‐1a, NM_006902), lacks this domain but contains a repressor domain (amino acid residues 200‐217) [34]. qRT‐PCR analysis revealed that both isoforms were upregulated in TGS‐01 and TGS‐04 by BMP‐4 stimulation (Fig. 1D).
The PROM1 promoter has two highly conserved putative PRRX1‐binding sites (Fig. S1A). ChIP‐qPCR analysis using TGS‐01 cells stably expressing pmx‐1a or pmx‐1b isoforms revealed that both isoforms of PRRX1 bound to the PROM1 promoter regions containing the two predicted PRRX1‐binding sites (Fig. 1E). Next, luciferase reporter assays were performed to investigate whether PRRX1 regulates PROM1 promoter activity. Deletion of or mutations in the PRRX1‐binding sites in the PROM1 promoter showed enhanced promoter activity in TGS‐01 and TGS‐04 cells, which was not significantly affected by BMP‐4 treatment (Fig. 1F, Fig. S1B). Of the two PRRX1‐binding sites, the distal binding site (region −1593 to −1560) was more important than the proximal binding site (region −1033 to −1003). These observations suggest that PRRX1 suppresses the PROM1 promoter activity by binding to this region.
3.2. Silencing PRRX1 maintains the neurosphere‐forming ability of GICs and induces the acquisition of GIC properties
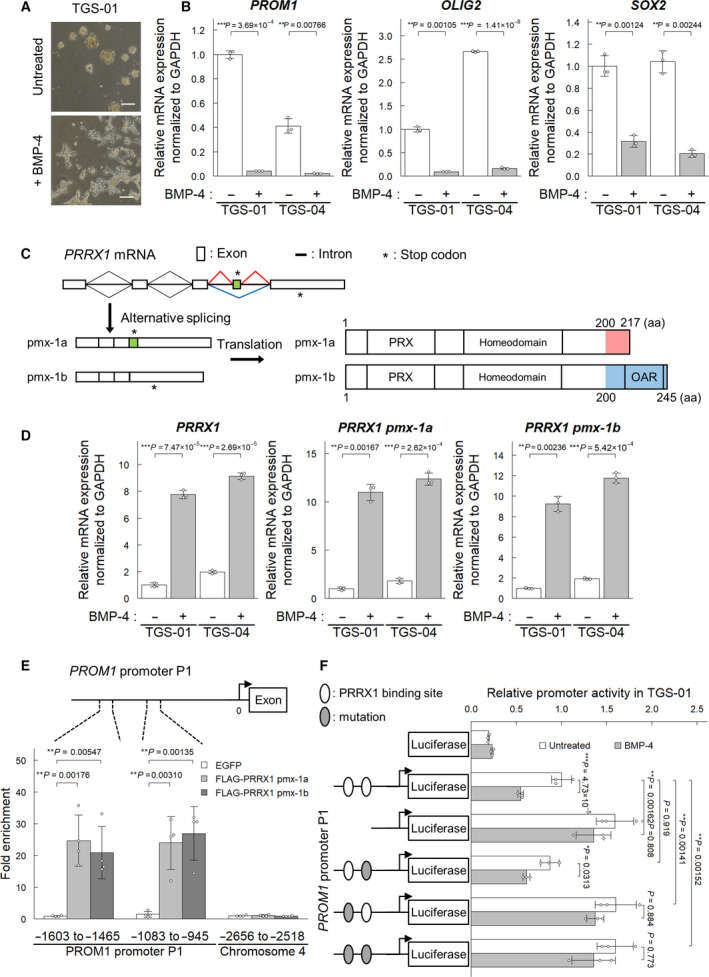
To address whether PRRX1 plays a role in maintaining stem cell‐like properties of GICs, we silenced PRRX1 expression in GICs and examined their neurosphere formation. Lentiviral‐mediated shRNA reduced the expressions of PRRX1 mRNA and protein in TGS‐01 and TGS‐04 cells (Fig. 2A,B and Fig. S2A,B). While the neurosphere formation of TGS‐01 cells expressing nontargeting control shRNA was suppressed by BMP‐4 stimulation, TGS‐01 cells expressing PRRX1 shRNA maintained the ability to form neurospheres even in the presence of BMP‐4, although the size of spheres decreased by knockdown of PRRX1 in the absence of BMP‐4 stimulation (Fig. 2C). In TGS‐04 cells, knockdown of PRRX1 resulted in the increased ability of GICs to form neurospheres in the presence and absence of BMP‐4; similar to TGS‐01 cells, the size of spheres decreased by knockdown of PRRX1 in the absence of BMP‐4 stimulation in TGS‐04 cells (Fig. S2C).
Fig. 2.
PRRX1 is required for BMP‐induced loss of the CD133‐positive GIC population. TGS‐01 cells expressing shRNA were treated for 72 h with or without BMP‐4 (30 ng·mL−1), except for panel (C). (A, B) Knockdown of PRRX1 mRNA (A) and protein (B) by PRRX1 shRNA in TGS‐01 cells (n = 4 biological replicates in (A)). Representative data of the two independent experiments are shown (B). (C) Regulation of sphere‐forming ability by knockdown of PRRX1. Phase‐contrast images showing the morphology of shRNA‐expressing TGS‐01 cells treated with or without BMP‐4 for 5 days under serum‐free condition (left). Scale bars: 200 µm. Sphere‐forming ability was determined by limiting dilution assay (middle, n = 3 independent experiments), and the sizes of spheres (cultured at the initial density of 50 cells per well) were determined (right, n = 21–48 from 3 independent experiments). Cells were treated with or without BMP‐4 (30 ng·mL−1) for 7 days. (D) Regulation of PROM1 mRNA by silencing of PRRX1 in TGS‐01 (n = 4 biological replicates). (E) CD133 expression on the surface of TGS‐01 cells was evaluated by flow cytometric analysis (n = 3 biological replicates). The graphs in panels (A) and (C–E) represent mean ± SD of biological replicates. The P‐values were determined by Tukey’s test (A, C–E).
We next examined the effect of PRRX1 knockdown on the expression levels of glioma stem cell markers. The expression of PROM1 mRNA encoding CD133 was upregulated in GICs expressing PRRX1 shRNA compared to those expressing control shRNA (Fig. 2D). In contrast, silencing of PRRX1 in GICs only showed weak or nonsignificant effects on OLIG2 or SOX2 expression, suggesting that the effects of BMP‐4‐induced PRRX1 on the expression of stem cell markers are specific to some markers, such as PROM1 (Fig. S2D,E). Consistent with the increase in PROM1 mRNA expression, flow cytometric analysis revealed that the CD133‐positive population was increased by PRRX1 silencing in TGS‐01 and TGS‐04 cells even in the presence of BMP‐4 (Fig. 2E and Fig. S2F).
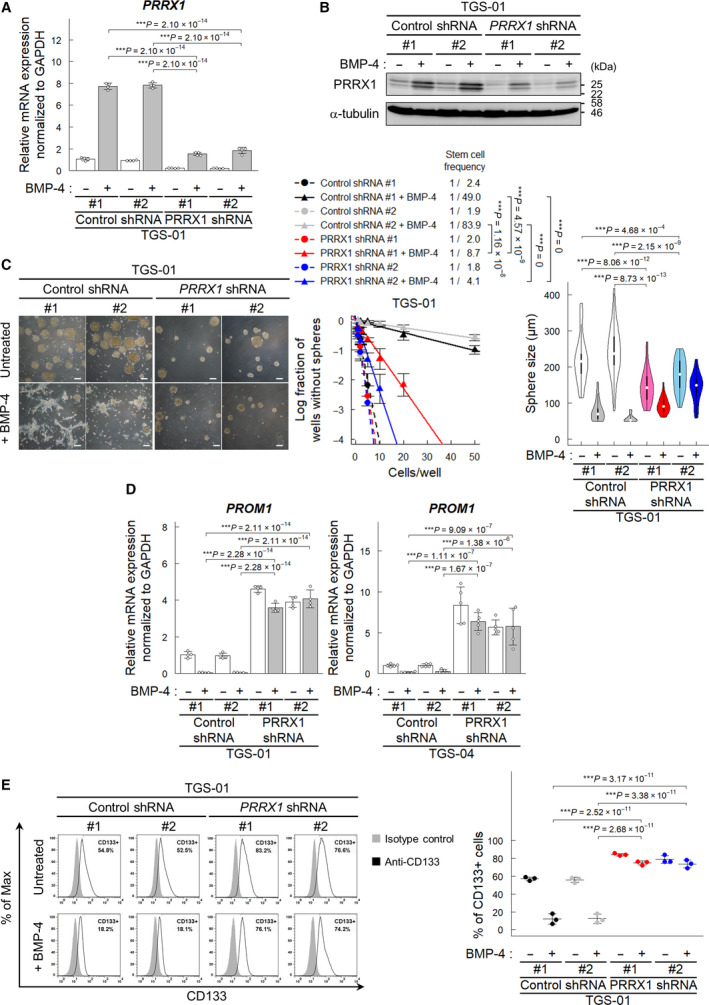
To address whether PRRX1 expression in GICs affects their tumorigenic activity, GICs expressing PRRX1 shRNA, or nontargeting control shRNA, together with firefly luciferase were injected intracranially into nude mice. In vivo bioluminescent imaging of orthotopic tumors revealed that silencing PRRX1 in TGS‐01 cells accelerated tumor growth (Fig. 3A) and consistently shortened the survival of the mice (Fig. 3B).
Fig. 3.
Effect of PRRX1 knockdown in TGS‐01 cells in vivo. Intracranial proliferation of GICs expressing firefly luciferase and control or PRRX1 shRNA (n = 7 mice for each group). (A) In vivo bioluminescent imaging analysis was performed 2 weeks after intracranial injection. (B) Kaplan–Meier survival curves of mice injected with TGS‐01 cells expressing control or PRRX1 shRNA. The graph represents mean ± SD of biological replicates (A). The P‐values were determined by Tukey’s test (A) or by two‐tailed log‐rank test and then adjusted by Bonferroni correction (B).
3.3. The PRRX1 pmx‐1b isoform plays an essential role in impairment of GIC properties
Distinct functions have been reported for the two isoforms of PRRX1 in certain cancers, as well as in human oligodendrocyte progenitor cells (hOPCs) [34, 35, 36, 37]. To investigate whether the isoforms have functional differences in GICs, the pmx‐1b isoform alone was knocked down in TGS‐01 and TGS‐04 cells by shRNA (Fig. 4A and Fig. S3A). As observed in GICs expressing shRNA against both isoforms of PRRX1, GICs expressing the pmx‐1b shRNA formed neurospheres even in the presence of BMP‐4 with significantly increased stem cell frequency (Fig. 4B and Fig. S3B). Moreover, silencing the pmx‐1b isoform induced PROM1 expression and increased the CD133‐positive population even in the presence of BMP‐4 (Fig. 4C,D and Fig. S3C,D). To clarify the effect of the pmx‐1b isoform on in vivo tumorigenicity, TGS‐01 cells expressing shRNA against the pmx‐1b isoform were injected intracranially into nude mice. In a similar way to the silencing of PRRX1, knockdown of the pmx‐1b isoform alone in TGS‐01 cells enhanced their tumorigenic activity and shortened the survival of the mice (Fig. 4E,F). These results suggest that the pmx‐1b isoform of PRRX1 is essential for BMP‐induced differentiation of GICs.
Fig. 4.
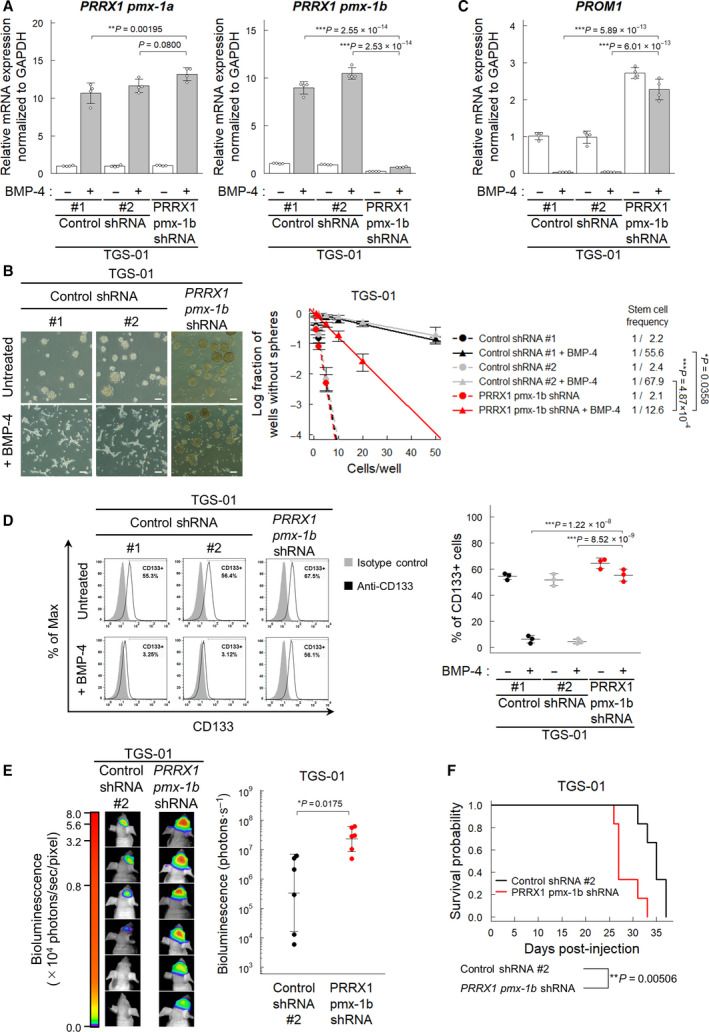
The PRRX1 pmx‐1b isoform is important for the decrease in the CD133‐positive population of GICs. TGS‐01 cells were treated with or without BMP‐4 (30 ng·mL−1) for 72 h, except for panel (B). (A) Knockdown of PRRX1 pmx‐1b by a shRNA specific against the pmx‐1b isoform in TGS‐01 cells (n = 4 biological replicates). (B) Phase‐contrast images showing the morphology of shRNA‐expressing TGS‐01 cells treated with or without BMP‐4 (30 ng·mL−1) for 5 days under the serum‐free condition (left). Scale bars: 200 µm. Sphere‐forming ability was determined by limiting dilution assay (right, n = 3 independent experiments). Cells were treated with or without BMP‐4 (30 ng·mL−1) for 7 days. (C) Upregulation of PROM1 mRNA by treatment with shRNA against the PRRX1 pmx‐1b isoform in TGS‐01 cells (n = 4 biological replicates). (D) Surface expression of CD133 in TGS‐01 cells evaluated by flow cytometric analysis (n = 3 biological replicates). (E, F) Intracranial proliferation of GICs expressing firefly luciferase and control or pmx‐1b shRNA (n = 6 mice for each group). The in vivo bioluminescent imaging analysis was performed 2 weeks after the intracranial injection (E). Kaplan–Meier survival curves of mice injected with TGS‐01 cells expressing control or pmx‐1b shRNA (F). The graphs in panels (A–E) represent mean ± SD of biological replicates. The P‐values were determined by Tukey’s test (A–D), Student’s t‐test (E), or two‐tailed log‐rank test (F).
3.4. The PRRX1 pmx‐1b isoform depletes neurosphere‐forming ability of GICs and induces loss of GIC properties
To test whether PRRX1 induction is sufficient for loss of stem cell‐like properties in GICs, we examined the effect of ectopic expression of PRRX1 on neurosphere formation and glioma stem cell marker expression. Enforced expression of either pmx‐1a or pmx‐1b suppressed the neurosphere‐forming ability of TGS‐01 cells, whereas only pmx‐1b, but not pmx‐1a, suppressed the neurosphere‐forming ability of TGS‐04 cells (Fig. 5A,B). Inhibition of sphere formation of TGS‐01 cells by the ectopic expression of pmx‐1a may not be due to loss of the stem cell‐like phenotype, because only the pmx‐1b isoform, but not the pmx‐1a isoform, drastically reduced the expression levels of PROM1 in both TGS‐01 cells and TGS‐04 cells (Fig. 5C). We also confirmed that the ectopic expression of only pmx‐1b, but not pmx‐1a, depleted the CD133‐positive populations of TGS‐01 and TGS‐04 cells (Fig. 5D).
Fig. 5.
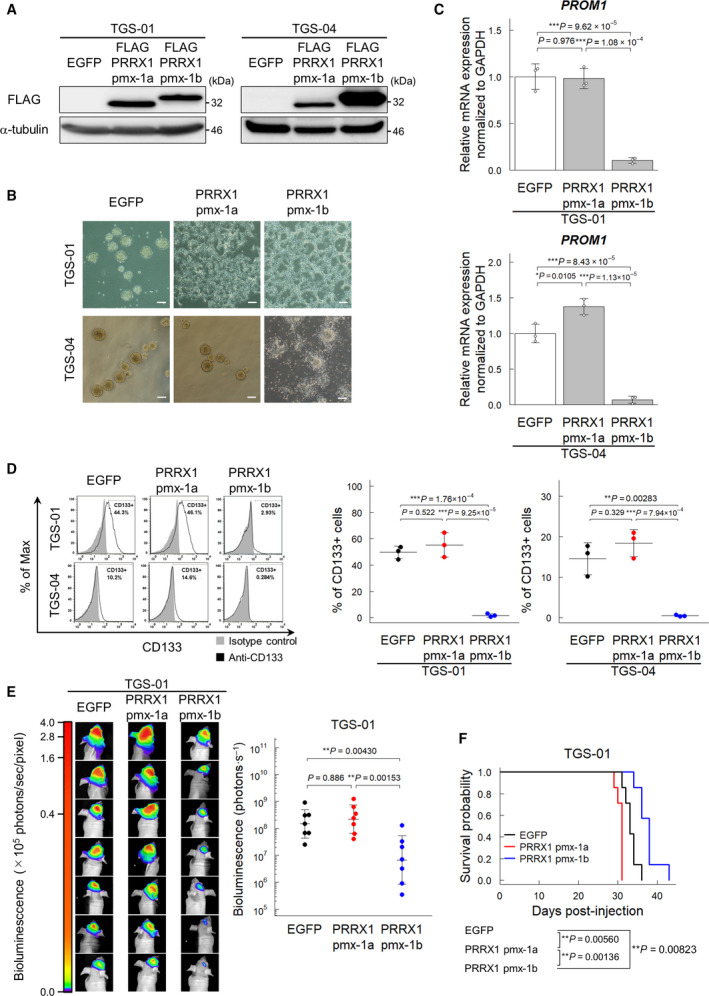
The PRRX1 pmx‐1b isoform reduces the CD133‐positive population of GICs. Cells were treated with or without BMP‐4 (30 ng·mL−1) for 72 h, except for panel (B). (A) Immunoblot analysis of enforced expression of FLAG‐tagged pmx‐1a or pmx‐1b in TGS‐01 and TGS‐04 cells. (B) Phase‐contrast images showing the morphology of EGFP‐ and FLAG‐pmx‐1a‐ or FLAG‐pmx‐1b‐expressing TGS‐01 and TGS‐04 cells treated with or without BMP‐4 (30 ng·mL−1) for 5 days under serum‐free condition. Scale bars: 200 µm. (C) Downregulation of PROM1 mRNA by ectopic expression of the PRRX1 pmx‐1b isoform in TGS‐01 and TGS‐04 cells (n = 3 biological replicates). (D) Surface expression of CD133 in TGS‐01 and TGS‐04 cells evaluated by flow cytometric analysis. Ectopic expression of the PRRX1 pmx‐1b decreased the CD133‐positive population (n = 3 biological replicates). (E, F) In vivo tumorigenic activity of GICs expressing firefly luciferase and EGFP, pmx‐1a, or pmx‐1b (n = 7 mice for each group). The in vivo bioluminescent imaging analysis was performed 2 weeks after intracranial injection (E). Kaplan–Meier survival curves of mice injected with TGS‐01 cells expressing EGFP, pmx‐1a, or pmx‐1b (F). The graphs in panels (C–E) represent mean ± SD of biological replicates. The P‐values were determined by Tukey’s test (C–E). In panel (F), the P‐value was determined by two‐tailed log‐rank test and then adjusted by Bonferroni correction.
To examine the effect of expression of PRRX1 isoforms on tumorigenicity, TGS‐01 cells ectopically expressing pmx‐1a, pmx‐1b, or EGFP, together with firefly luciferase, were injected intracranially into nude mice. Ectopic expression of the pmx‐1b isoform suppressed tumor growth and significantly prolonged the survival of the mice, while the expression of the pmx‐1a isoform shortened the life span of the mice (Fig. 5E,F). These findings indicate that the PRRX1 pmx‐1b isoform plays a crucial role in BMP‐induced loss of stem cell‐like properties in GICs.
To investigate the generality of the effects of PRRX1 on PROM1 expression, we used two other GICs, U3005MG and U3024MG cells, that were obtained from the HGCC resource [30]. BMP‐4 induced the pmx‐1a and pmx‐1b isoforms of PRRX1, and silencing of PRRX1 or pmx‐1b strongly induced PROM1 expression in U3005MG and U3024MG cells as well as in TGS‐01 and TGS‐04 cells (Fig. S4A,B). Moreover, ectopic expression of only pmx‐1b, but not pmx‐1a, reduced the expression levels of PROM1 in U3005MG and U3024MG cells (Fig. S4C).
3.5. DNMT3A is involved in the reduction of CD133 expression after BMP‐4 stimulation
A CpG island is present in the vicinity of PRRX1‐binding sites in the PROM1 promoter P1 region. Although the CpG sites in the PROM1 promoter P1 region are hypomethylated in CD133‐positive glioblastoma cells, they are highly methylated in CD133‐negative glioblastoma and in normal neural tissues [38]. Since PRRX1 directly regulates CD133 expression in GICs, we examined whether BMP‐4 stimulation affects DNA methylation in the PROM1 promoter. BMP‐4 increased the levels of DNA methylation in the PROM1 promoter P1 region in TGS‐01 and TGS‐04 cells (Fig. 6A,B). Moreover, the ectopic expression of pmx‐1b, but not pmx‐1a, increased DNA methylation levels in TGS‐01 and TGS‐04 cells (Fig. 6C,D).
Fig. 6.
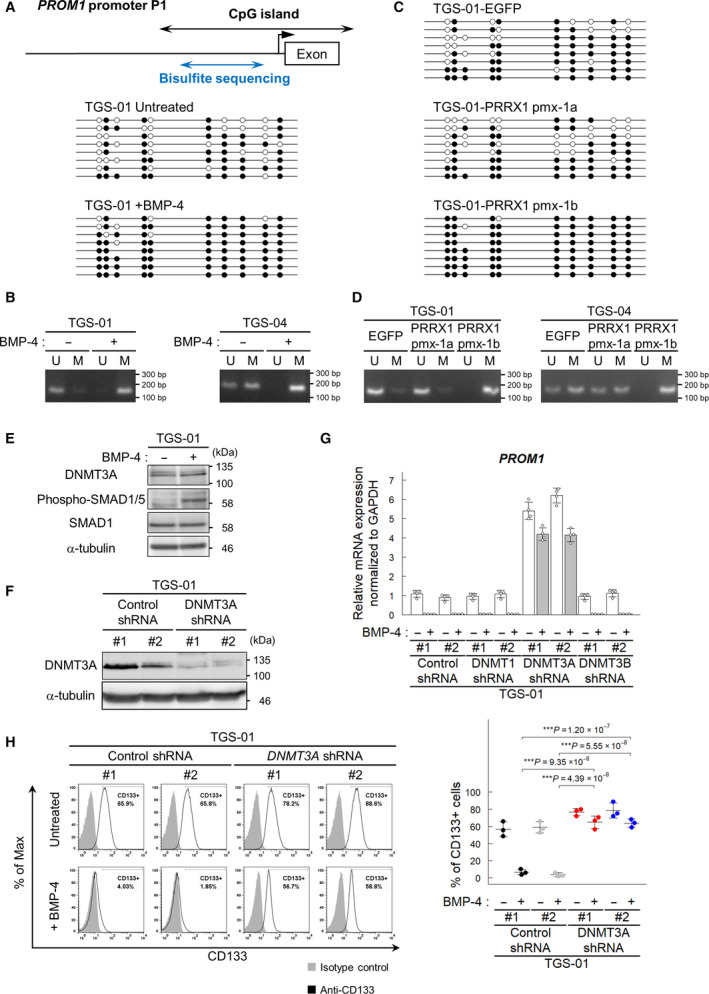
DNMT3A is involved in the reduction of CD133 expression by BMP signaling. (A, B) DNA methylation of the PROM1 P1 promoter region by BMP‐4. Cells were treated with BMP‐4 (30 ng·mL−1) for 72 h. U, unmethylated and M, methylated. (C, D) DNA methylation of the PROM1 P1 promoter region by ectopic expression of pmx‐1a or pmx‐1b. (E, F) Immunoblot analysis of the expression of DNMT3A protein in TGS‐01 cells treated with or without BMP‐4 (30 ng·mL−1) for 72 h (E) and with or without downregulation of DNMT3A by shRNA in TGS‐01 cells (F). (G) Screening for DNA methyltransferases involved in the expression of PROM1. DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) were knocked down by shRNAs in TGS‐01 cells, and expression levels of PROM1 (encoding CD133) were quantified by real‐time qRT‐PCR. The cells were treated with or without BMP‐4 (30 ng·mL−1) for 72 h (n = 4 biological replicates). (H) Surface expression of CD133 evaluated by flow cytometric analysis. TGS‐01 cells expressing control or DNMT3A shRNA were treated for 72 h with or without BMP‐4 (30 ng·mL−1). The graphs in panels (G, H) represent mean ± SD of biological replicates. The P‐values were determined by Tukey’s test.
To examine which DNMTs are involved in BMP‐induced DNA methylation in the PROM1 promoter P1 region in GICs, each of the three DNMTs was knocked down by shRNA in TGS‐01 and TGS‐04 cells (Fig. 6E,F, Fig. S5A–D). BMP‐4 stimulation did not strongly affect DNMT3A expression in TGS‐01 and TGS‐04 cells (Fig. 6E and Fig. S5A–D). Of the three DNMTs in mammals, silencing the expression of DNMT3A, but not that of DNMT1 or DNMT3B, induced the expression of PROM1 (Fig. 6G and Fig. S5E). The downregulation of DNMT3A expression by shRNA resulted in an increase in the CD133‐positive population even in the presence of BMP‐4 stimulation (Fig. 6H and Fig. S5F). These findings suggest that DNMT3A is critically involved in the decrease in the CD133‐positive population.
3.6. The PRRX1 pmx‐1b isoform cooperates with DNMT3A to downregulate CD133 expression
The pmx‐1a and pmx‐1b isoforms of PRRX1 can bind to the PROM1 promoter P1 region (Fig. 1E). However, these two isoforms, which have different C‐terminal regions, showed differential effects on the transcription of PROM1 (Fig. 4C, Figs S3C and S4C). We thus examined whether the two isoforms of PRRX1 physically interact with DNMT3A. Immunoprecipitation of FLAG‐tagged PRRX1 followed by immunoblotting with a DNMT3A antibody revealed that pmx‐1b, but not pmx‐1a, interacted with DNMT3A (Fig. 7A). Moreover, endogenous interaction between the DNMT3A and pmx‐1b proteins was observed in the presence and absence of BMP‐4 stimulation in TGS‐01 cells (Fig. 7B).
Fig. 7.
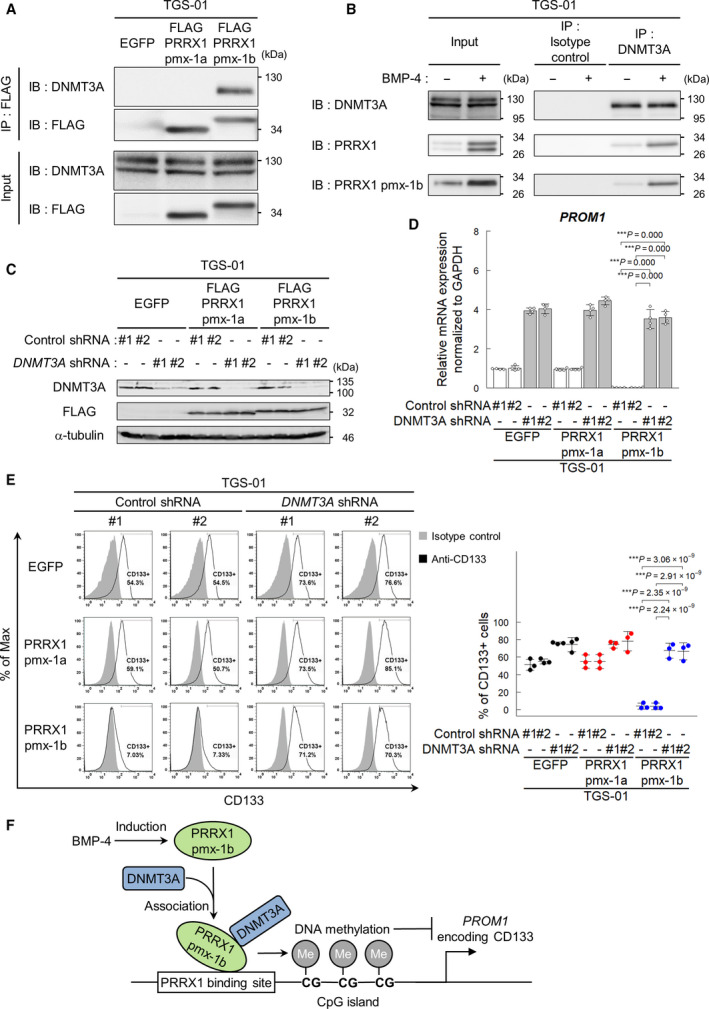
DNMT3A is required for downregulation of CD133 expression by PRRX1 pmx‐1b isoform. (A, B) Immunoblot analysis to examine the physical interaction between DNMT3A and PRRX1. In TGS‐01 cells expressing FLAG‐tagged PRRX1, DNMT3A was co‐immunoprecipitated with FLAG‐tagged PRRX1 pmx‐1b, but not with FLAG‐tagged pmx‐1a (A). In TGS‐01 cells treated for 72 h with or without BMP‐4 (30 ng·mL−1), endogenous PRRX1 pmx‐1b was co‐immunoprecipitated with DNMT3A (B). Representative data of the three independent experiments are shown (A, B). (C–E) DNMT3A shRNA and FLAG‐tagged PRRX1 were transduced into TGS‐01. Immunoblot analysis showing the expression of DNMT3A shRNA and FLAG‐tagged PRRX1 in TGS‐01 cells (C). Expression of PROM1 in TGS‐01 cells expressing DNMT3A shRNA and PRRX1 (n = 4 biological replicates) (D). CD133 expression on cell surface evaluated by flow cytometric analysis in TGS‐01 cells (n = 3 biological replicates) (E). The graphs in panels (D, E) represent mean ± SD of biological replicates. The P‐values were determined by Tukey’s test (D, E). (F) A scheme showing the regulation of CD133 expression by PRRX1 pmx‐1b and DNMT3A regulated by BMP stimulation. Of the two splice isoforms, only the longer isoform of PRRX1, pmx‐1b, induces the differentiation of GICs. Through functional cooperation with DNMT3A, pmx‐1b induces the promoter methylation of the key stem cell marker gene PROM1 by recruitment of DNMT3A and suppresses the CD133‐positive GIC population.
To study how pmx‐1b and DNMT3A cooperatively regulate the transcription of PROM1, DNMT3A shRNA and PRRX1 were transduced in GICs (Fig. 7C and Fig. S6A). Silencing of DNMT3A increased the expression of PROM1 mRNA and CD133‐positive populations of TGS‐01 and TGS‐04 cells (Fig. 6G,H and Fig. S5E,F). Although the ectopic expression of pmx‐1b, but not of pmx‐1a, decreased PROM1 mRNA expression and the CD133‐positive population, the effects of pmx‐1b were drastically attenuated by silencing of DNMT3A (Fig. 7D,E and Fig. S6B,C). These findings reveal that, upon interaction with the pmx‐1b isoform, DNMT3A decreases the CD133‐positive population of GICs through epigenetic regulation of the PROM1 promoter by DNMT3A (Fig. 7F).
4. Discussion
BMPs play pivotal roles in the growth, differentiation, and function of normal neural cells, and BMP signaling regulates the differentiation of normal NSCs [39, 40]. Although many studies have shown that BMP signaling induces growth arrest, apoptosis, and differentiation of GICs [23], GICs show heterogeneous responses to BMPs, and certain populations of GICs escape the differentiation‐inducing effects of BMPs [25]. Several BMP‐induced molecules have been shown to be involved in the induction of cell cycle arrest, apoptosis, and cell differentiation of GICs [18, 19, 20, 26, 27], but less is known about the BMP target molecules that are involved in the regulation of the stem cell‐like properties of GICs.
Through RNA‐seq analysis using GICs treated with or without BMP‐4, we identified PRRX1 as a BMP target gene in human GICs. PRRX1 is a homeobox transcription factor containing the paired‐type DNA‐binding homeodomain, which is essential for normal development. PRRX1 is widely expressed in various cells, including undifferentiated mesenchymal cells, and Prrx1‐deficient mice show perinatal death with skeletal malformations [41]. In the development of the nervous system, PRRX1 is involved in the maintenance of self‐renewal of NSCs in cooperation with SOX2, and depletion of Prrx1 expression in cultured adult mouse NSCs results in attenuation of their self‐renewal ability [33]. Moreover, the knockdown of Prrx1 expression in NSCs induces the expression of genes involved in neuronal and glial differentiation.
Although PRRX1 plays a critical role in the maintenance of the stem cell‐like properties of normal NSCs [33], it also suppresses proliferation and migration of some neuronal progenitor cells. PRRX1 has been reported to upregulate the expression of cyclin dependent kinase inhibitors, including p21WAF1/CIP1 and p15INK4B, and induce the cell cycle arrest of hOPCs [37]. Interestingly, both isoforms of PRRX1 suppressed hOPC proliferation, whereas only pmx‐1b, but not pmx‐1a, reduced cell migration of hOPCs. In addition to human GICs, BMP treatment increased both pmx‐1a and pmx‐1b in hOPCs.
In the present study, we showed that silencing of the PRRX1 expression maintained the stem cell‐like phenotype of GICs. PRRX1 bound to the PROM1 promoter and suppressed the expression of CD133. In contrast, predicted PRRX1‐binding sites were not detected in the promoter regions of OLIG2 and SOX2 (data not shown). Thus, the effects of PRRX1 on normal and malignant neural stem or progenitor cells may thus be cell‐ and context‐dependent, possibly through the interaction with other transcription factors. To confirm that the effects of PRRX1 on PROM1 expression are not limited to TGS‐01 and TGS‐04 cells, we also used U3005MG and U3024MG cells obtained from the HGCC resource, which are maintained under serum‐free conditions to preserve the GIC characteristics [30] and respond to BMP‐4 stimulation (data not shown). Consistent with TGS‐01 and TGS‐04 cells, the PRRX1 pmx‐1b isoform suppressed PROM1 expression in U3005MG and U3024MG cells (Fig. S4).
CD133‐positive cells in human glioblastoma have two key properties of cancer‐initiating cells, that is, the ability to self‐renew and to recapitulate tumor heterogeneity through cell differentiation [42]. Some reports suggested that CD133 is not a universal marker of GICs, and instances of CD133‐negative cells exhibiting similar self‐renewal properties as CD133‐positive cells have also been reported [6, 43, 44, 45, 46]. However, knockdown of PROM1/CD133 leads to the disruption of self‐renewal and tumorigenic abilities of neurosphere‐derived human glioblastoma cells, and the re‐expression of CD133 restores the stem cell‐like phenotype in PROM1/CD133‐silenced glioblastoma cells, suggesting that CD133 plays an important role in the maintenance of stem cell‐like properties of GICs [11].
Yan et al. reported that a CD133‐related gene expression signature obtained by gene expression profiling experiments using DNA microarrays resembles human embryonic stem cells and glioblastoma stem cells. Moreover, the CD133 gene expression signature identifies aggressive subpopulations of glioblastoma [47]. Phosphorylation of Tyr828 in the cytoplasmic domain of CD133 mediates interaction with the p85 subunit of phosphoinositide 3’‐kinase (PI3K), leading to activation of the PI3K‐Akt pathway in GICs [48]. Thus, silencing PROM1/CD133 expression suppresses the activity of the PI3K‐Akt pathway, resulting in attenuation of the self‐renewal and tumorigenicity of GICs. In agreement with these findings, our data demonstrated that decreased CD133 expression by the knockdown of PRRX1 expression resulted in loss of neurosphere‐forming ability and tumorigenic activity of GICs.
The two major isoforms of PRRX1, pmx‐1a (PRRX1B) and pmx‐1b (PRRX1A), are generated by alternative splicing and have distinct functions [35, 36, 37]. The C‐terminal region of pmx‐1b containing the OAR domain can mask transcription activation, whereas the C‐terminal of pmx‐1a contains a repression domain [34]. During the development and dissemination of pancreatic ductal adenocarcinoma, pmx‐1b stimulates mesenchymal–epithelial transition, tumor differentiation, and metastatic colonization of the tumor, whereas pmx‐1a facilitates epithelial–mesenchymal transition, tumor de‐differentiation, and invasion of tumor cells [36].
In the present study, we showed that of the two major isoforms of PRRX1, pmx‐1b plays a major role in the induction of GIC differentiation. Both pmx‐1a and pmx‐1b are expressed in the GICs used in the present study, and BMP‐4 stimulation induced both isoforms (Fig. 1D). Silencing of pmx‐1b alone mimicked the effect of knockdown of both isoforms (Fig. 4, Figs S3 and S4A,B). On the other hand, the enforced expression of pmx‐1a failed to decrease the population of CD133‐positive GICs (Fig. 5D), and inoculation of TGS‐01 cells expressing pmx‐1a resulted in shorter survival of mice than inoculation with control GICs expressing EGFP (Fig. 5F).
Knockdown of PRRX1 was reported to attenuate the invasiveness and neurosphere‐forming ability of glioblastoma cell lines, including T98 and U251MG, in a serum‐containing condition [49]. PRRX1 was also reported to transactivate dopamine D2 receptor in GICs cultured in serum‐free conditions and maintain the tumorigenic activity of GICs in vivo [50]. Functions of the two PRRX1 isoforms were not investigated in these studies [49, 50]. In the GICs used in the present study, expression of dopamine D2 receptor was not induced by BMP signaling nor suppressed by PRRX1 knockdown (data not shown). PRRX1 may exhibit different functions in GICs, possibly depending on cell culture conditions, cell differentiation status, and balance of the expression of the two PRRX1 isoforms.
DNA methylation is a highly regulated process in normal cells, whereas methylation profiles are drastically altered in various cancer cells [51]. Compared to normal cells, cancer cells show global hypomethylation of genomic DNA and specific hypermethylation of the promoters of various tumor suppressor genes. DNA methylation is induced by three different DNA methyltransferases in mammals; DNMT3A and DNMT3B are involved in the de novo methylation of CpG islands, whereas DNMT1 is mainly responsible for the maintenance of established DNA methylation patterns. Although DNA methyltransferases play critical roles in differentiation of various stem cells, including embryonic stem cells, neural stem cells, and hematopoietic stem cells [52, 53], it is not fully understood how they are recruited to chromatin and induce methylation of particular sites. Mutations in the DNMT3A gene are often observed in hematological malignancies, but less frequently in other cancers. In contrast, aberrant expression of DNMTs, for example, high expression of DNMT1 and DNMT3B [54] and low expression of DNMT3A [55], has been reported in glioblastoma. DNMT3A was shown to play a crucial role in the proliferation and differentiation of mouse NSCs [53, 56]; however, little is known about how DNMTs regulate the differentiation of glioblastoma cells.
In the present study, we showed that, among the three DNA methyltransferases, the silencing of DNMT3A maintained the expression of PROM1 and the number of CD133‐positive glioblastoma cells, even in the presence of BMP‐4 (Fig. 6 and Fig. S5). Co‐immunoprecipitation analysis demonstrated that the pmx‐1b, but not the pmx‐1a isoform, interacted with DNMT3A (Fig. 7A,B). The functional interaction between pmx‐1b and DNMT3A may thus facilitate DNA methylation of the PROM1 gene promoter. DNMT1 and DNMT3A have been shown to interact with the transcription factor ZEB1 and HMGA2, respectively, and maintain the DNA methylation of the CDH1 promoter in normal and malignant breast epithelial cells during epithelial–mesenchymal transition [57, 58]. Together, these findings suggest that DNA methylation may be induced only in certain genomic regions through recruitment of DNA methyltransferases by DNA‐binding proteins.
Using the public database TCGA, we analyzed the expression of genes studied in the present study, including PRRX1, PROM1, and DNMT3A, to investigate possible correlations between the expression of these genes, the brain tumor grade, and patient prognosis. However, we failed to find any clear tendency of correlations by these analyses. This may be because the populations of GICs are small, and their characteristics may thus not be reflected in the gene expression of the brain tumors as a whole. In addition, the mechanism proposed in the present study may be primarily related to CD133‐positive stem‐like populations.
Epigenetic gene silencing is induced by modifications of histones, DNA methylation, and other components of chromatin. Histone modification by polycomb repressive complex 2, through its catalytic subunit EZH2, induces the epigenetic silencing of the BMPR1B/ALK6 gene, leading to escape from the BMP‐induced differentiation of glioblastoma cells [17]. Our findings reveal another important mechanism of the epigenetic regulation of GIC differentiation by BMPs. Since recruitment of DNMT3A by the pmx‐1b isoform of PRRX1 leads to GIC differentiation through the epigenetic regulation of PROM1 gene expression, pmx‐1b and DNMT3A are potentially interesting targets for the treatment of glioblastoma.
5. Conclusion
The present study reveals a mechanism of differentiation induction of GICs by BMPs. Through recruitment of DNMT3A to the PROM1 promoter, the longer splice isoform of PRRX1, induced by BMP signaling, enhances the promoter methylation of the key stem cell marker gene PROM1 and drives differentiation of GICs.
Conflict of interest
Satoshi Sakai is currently an employee of Asahi Kasei Pharma Corporation. The other authors declare no competing or financial interests.
Author contributions
RT, KM, and C‐HH designed the study and wrote the manuscript. RT performed most of the experiments. TT, NT, and BW provided essential experimental materials. TT, CI, AK, SS, ER, DK, MM, and BW participated in experimental design and data analyses. All authors participated in writing, review, and revision of the manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.13051.
Supporting information
Fig. S1. PRRX1 binds to the PROM1 promoter and negatively modulates its promoter activity.
Fig. S2. PRRX1 is required for BMP‐induced loss of the CD133‐positive GIC population.
Fig. S3. The PRRX1 pmx‐1b isoform is important for the decrease in the CD133‐positive population of GICs.
Fig. S4. Upregulation of PROM1 mRNA by treatment with shRNA for PRRX1 or the pmx‐1b isoform in U3005MG and U3024MG cells.
Fig. S5. DNMT3A is involved in the reduction of CD133 expression by BMP signaling.
Fig. S6. DNMT3A is required for the PRRX1 pmx‐1b isoform to downregulate CD133 expression.
Table S1. Characteristics of glioblastoma cells used in the present study.
Table S2. Primer sets for real‐time qPCR.
Table S3. Target sequences of shRNA.
Table S4. Oligonucleotides used for mutagenesis of the PRRX1‐binding sites.
Table S5. Primers used for ChIP‐qPCR.
Table S6. Primer sets used for bisulfite sequencing.
Table S7. Primer sets used for methylation‐specific PCR.
Acknowledgements
We thank late H. Miyoshi for the lentivirus vector system. We are grateful to members of the Heldin and Miyazono laboratories for helpful discussion. Funding was provided by grants from KAKENHI (grants‐in‐aid for scientific research) on Innovative Area on Integrated Analysis and Regulation of Cellular Diversity (grant number 17H06326) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT) and Scientific Research (S) (15H05774) from the Japan Society for the Promotion of Science (JSPS). KM was also supported by the Swedish Cancer Society (100452). C‐HH was supported by the Swedish Research Council (2015‐02757 and 2020‐01291) and the European Research Council (787472). RT was supported by JSPS Research Fellowship for Young Scientists (DC; 16J05225).
Contributor Information
Kohei Miyazono, Email: miyazono@m.u-tokyo.ac.jp.
Carl‐Henrik Heldin, Email: c-h.heldin@imbim.uu.se.
Data accessibility
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J & NABTT CNS Consortium (2010) Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res 16, 2443–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K et al. (2017) Effect of tumor‐treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 18, 2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, Schulz D, Simon M, Schlegel U, Senft C et al. (2015) A randomised, open label phase III trial with nimotuzumab, an anti‐epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer 51, 522–532. [DOI] [PubMed] [Google Scholar]
- 5. Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, Brandes AA, Taal W, Domont J, Idbaih A et al. (2017) Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med 377, 1954–1963. [DOI] [PubMed] [Google Scholar]
- 6. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD & Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432, 396–401. [DOI] [PubMed] [Google Scholar]
- 7. Singh SK, Clarke ID, Hide T & Dirks PB (2004) Cancer stem cells in nervous system tumors. Oncogene 23, 7267–7273. [DOI] [PubMed] [Google Scholar]
- 8. Vescovi AL, Galli R & Reynolds BA (2006) Brain tumour stem cells. Nat Rev Cancer 6, 425–436. [DOI] [PubMed] [Google Scholar]
- 9. Lathia JD, Mack SC, Mulkearns‐Hubert EE, Valentim CL & Rich JN (2015) Cancer stem cells in glioblastoma. Genes Dev 29, 1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu T, Xu H, Huang M, Ma W, Saxena D, Lustig RA, Alonso‐Basanta M, Zhang Z, O'Rourke DM, Zhang L et al. (2018) Circulating glioma cells exhibit stem cell‐like properties. Cancer Res 78, 6632–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brescia P, Ortensi B, Fornasari L, Levi D, Broggi G & Pelicci G (2013) CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 31, 857–869. [DOI] [PubMed] [Google Scholar]
- 12. Derynck R & Budi EH (2019) Specificity, versatility, and control of TGF‐β family signaling. Sci Signal 12, eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seoane J & Gomis RR (2017) TGF‐β family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol 9, a022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davis H, Raja E, Miyazono K, Tsubakihara Y & Moustakas A (2016) Mechanisms of action of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev 27, 81–92. [DOI] [PubMed] [Google Scholar]
- 15. Miyazono K, Kamiya Y & Morikawa M (2010) Bone morphogenetic protein receptors and signal transduction. J Biochem 147, 35–51. [DOI] [PubMed] [Google Scholar]
- 16. Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F & Vescovi AL (2006) Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour‐initiating cells. Nature 444, 761–765. [DOI] [PubMed] [Google Scholar]
- 17. Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S et al. (2008) Epigenetic‐mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma‐initiating cells. Cancer Cell 13, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Z, Sun L, Wang Y, Wu Z, Geng J, Miu W, Pu Y, You Y, Yang Z & Liu N (2011) Bone morphogenetic protein 4 inhibits cell proliferation and induces apoptosis in glioma stem cells. Cancer Biother Radiopharm 26, 77–83. [DOI] [PubMed] [Google Scholar]
- 19. Yan K, Wu Q, Yan DH, Lee CH, Rahim N, Tritschler I, DeVecchio J, Kalady MF, Hjelmeland AB & Rich JN (2014) Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev 28, 1085–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raja E, Komuro A, Tanabe R, Sakai S, Ino Y, Saito N, Todo T, Morikawa M, Aburatani H, Koinuma D et al. (2017) Bone morphogenetic protein signaling mediated by ALK‐2 and DLX2 regulates apoptosis in glioma‐initiating cells. Oncogene 36, 4963–4974. [DOI] [PubMed] [Google Scholar]
- 21. González‐Gómez P, Anselmo NP & Mira H (2014) BMPs as therapeutic targets and biomarkers in astrocytic glioma. Biomed Res Int 2014, 549742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bao Z, Zhang C, Yan W, Liu Y, Li M, Zhang W & Jiang T (2013) BMP4, a strong better prognosis predictor, has a subtype preference and cell development association in gliomas. J Transl Med 11, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xi G, Best B, Mania‐Farnell B, James CD & Tomita T (2017) Therapeutic potential for bone morphogenetic protein 4 in human malignant glioma. Neoplasia 19, 261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carén H, Stricker SH, Bulstrode H, Gagrica S, Johnstone E, Bartlett TE, Feber A, Wilson G, Teschendorff AE, Bertone P et al. (2015) Glioblastoma stem cells respond to differentiation cues but fail to undergo commitment and terminal cell‐cycle arrest. Stem Cell Rep 5, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Darmanis S, Gallant CJ, Marinescu VD, Niklasson M, Segerman A, Flamourakis G, Fredriksson S, Assarsson E, Lundberg M, Nelander S et al. (2016) Simultaneous multiplexed measurement of RNA and proteins in single cells. Cell Rep 14, 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Savary K, Caglayan D, Caja L, Tzavlaki K, Bin Nayeem S, Bergström T, Jiang Y, Uhrbom L, Forsberg‐Nilsson K, Westermark B et al. (2013) Snail depletes the tumorigenic potential of glioblastoma. Oncogene 32, 5409–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caja L, Tzavlaki K, Dadras MS, Tan E‐J, Hatem G, Maturi NP, Morén A, Wik L, Watanabe Y, Savary K et al. (2018) Snail regulates BMP and TGFβ pathways to control the differentiation status of glioma‐initiating cells. Oncogene 37, 2515–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raja E, Morikawa M, Nishida J, Tanabe R, Takahashi K, Seeherman HJ, Saito N, Todo T & Miyazono K (2019) Tyrosine kinase Eph receptor A6 sensitizes glioma‐initiating cells towards bone morphogenetic protein‐induced apoptosis. Cancer Sci 110, 3486–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K & Miyazono K (2009) Autocrine TGF‐β signaling maintains tumorigenicity of glioma‐initiating cells through Sry‐related HMG‐box factors. Cell Stem Cell 5, 504–514. [DOI] [PubMed] [Google Scholar]
- 30. Xie Y, Bergström T, Jiang Y, Johansson P, Marinescu VD, Lindberg N, Segerman A, Wicher G, Niklasson M, Baskaran S et al. (2015) The human glioblastoma cell culture resource: validated cell models representing all molecular subtypes. EBioMedicine 2, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sompallae R, Hofmann O, Maher CA, Gedye C, Behren A, Vitezic M, Daub CO, Devalle S, Caballero OL, Carninci P et al. (2013) A comprehensive promoter landscape identifies a novel promoter for CD133 in restricted tissues, cancers, and stem cells. Front Genet 4, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klenova E, Chernukhin I, Inoue T, Shamsuddin S & Norton J (2002) Immunoprecipitation techniques for the analysis of transcription factor complexes. Methods 26, 254–259. [DOI] [PubMed] [Google Scholar]
- 33. Shimazaki K, Clemenson GD Jr & Gage FH (2013) Paired related homeobox protein 1 is a regulator of stemness in adult neural stem/progenitor cells. J Neurosci 33, 4066–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Norris RA & Kern MJ (2001) The identification of Prx1 transcription regulatory domains provides a mechanism for unequal compensation by the Prx1 and Prx2 loci. J Biol Chem 276, 26829–26837. [DOI] [PubMed] [Google Scholar]
- 35. Reichert M, Takano S, von Burstin J, Kim SB, Lee JS, Ihida‐Stansbury K, Hahn C, Heeg S, Schneider G, Rhim AD et al. (2013) The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes Dev 27, 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takano S, Reichert M, Bakir B, Das KK, Nishida T, Miyazaki M, Heeg S, Collins MA, Marchand B, Hicks PD et al. (2016) Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev 30, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang J, Saraswat D, Sinha AK, Polanco J, Dietz K, O'Bara MA, Pol SU, Shayya HJ & Sim FJ (2018) Paired related homeobox protein 1 regulates quiescence in human oligodendrocyte progenitors. Cell Rep 25, 3435–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tabu K, Sasai K, Kimura T, Wang L, Aoyanagi E, Kohsaka S, Tanino M, Nishihara H & Tanaka S (2008) Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res 18, 1037–1046. [DOI] [PubMed] [Google Scholar]
- 39. Nakashima K, Takizawa T, Ochiai W, Yanagisawa M, Hisatsune T, Nakafuku M, Miyazono K, Kishimoto T, Kageyama R & Taga T (2001) BMP2‐mediated alteration in the developmental pathway of fetal mouse brain cells from neurogenesis to astrocytogenesis. Proc Natl Acad Sci USA 98, 5868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bonaguidi MA, Peng CY, McGuire T, Falciglia G, Gobeske KT, Czeisler C & Kessler JA (2008) Noggin expands neural stem cells in the adult hippocampus. J Neurosci 28, 9194–9204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin JF, Bradley A & Olson EN (1995) The paired‐like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev 9, 1237–1249. [DOI] [PubMed] [Google Scholar]
- 42. Wang JCY & Dick JE (2011) Cancer stem cells. In Cancer: Principles & Practice of Oncology ‐ Primer of the Molecular Biology of Cancer (DeVita VT, Lawrence TS & Rosenberg SA, eds), pp. 163–179. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 43. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U & Beier CP (2007) CD133+ and CD133‐ glioblastoma‐derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 67, 4010–4015. [DOI] [PubMed] [Google Scholar]
- 44. Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI, Jeon JW, Kim MH, Kang BG, Jung Y et al. (2008) Clinical and biological implications of CD133‐positive and CD133‐negative cells in glioblastomas. Lab Invest 88, 808–815. [DOI] [PubMed] [Google Scholar]
- 45. Nishide K, Nakatani Y, Kiyonari H & Kondo T (2009) Glioblastoma formation from cell population depleted of Prominin1‐expressing cells. PLoS One 4, e6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, Greve JM, Soriano RH, Gilmour LL, Rivers CS et al. (2010) A hierarchy of self‐renewing tumor‐initiating cell types in glioblastoma. Cancer Cell 17, 362–375. [DOI] [PubMed] [Google Scholar]
- 47. Yan X, Ma L, Yi D, Yoon JG, Diercks A, Foltz G, Price ND, Hood LE & Tian Q (2011) A CD133‐related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc Natl Acad Sci USA 108, 1591–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, Xu W, Cui C, Xing Y, Liu Y et al. (2013) Activation of PI3K/Akt pathway by CD133‐p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci USA 110, 6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sugiyama M, Hasegawa H, Ito S, Sugiyama K, Maeda M, Aoki K, Wakabayashi T, Hamaguchi M, Natsume A & Senga T (2015) Paired related homeobox 1 is associated with the invasive properties of glioblastoma cells. Oncol Rep 33, 1123–1130. [DOI] [PubMed] [Google Scholar]
- 50. Li Y, Wang W, Wang F, Wu Q, Li W, Zhong X, Tian K, Zeng T, Gao L, Liu Y et al. (2017) Paired related homeobox 1 transactivates dopamine D2 receptor to maintain propagation and tumorigenicity of glioma‐initiating cells. J Mol Cell Biol 9, 302–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klutstein M, Nejman D, Greenfield R & Cedar H (2016) DNA methylation in cancer and aging. Cancer Res 76, 3446–3450. [DOI] [PubMed] [Google Scholar]
- 52. Chen T, Ueda Y, Dodge JE, Wang Z & Li E (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol 23, 5594–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y & Sun YE (2010) Dnmt3a‐dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rajendran G, Shanmuganandam K, Bendre A, Muzumdar D, Goel A & Shiras A (2011) Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J Neurooncol 104, 483–494. [DOI] [PubMed] [Google Scholar]
- 55. Fanelli M, Caprodossi S, Ricci‐Vitiani L, Porcellini A, Tomassoni‐Ardori F, Amatori S, Andreoni F, Magnani M, De Maria R, Santoni A et al. (2008) Loss of pericentromeric DNA methylation pattern in human glioblastoma is associated with altered DNA methyltransferases expression and involves the stem cell compartment. Oncogene 27, 358–365. [DOI] [PubMed] [Google Scholar]
- 56. Wu Z, Huang K, Yu J, Le T, Namihira M, Liu Y, Zhang J, Xue Z, Cheng L & Fan G (2012) Dnmt3a regulates both proliferation and differentiation of mouse neural stem cells. J Neurosci Res 90, 1883–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fukagawa A, Ishii H, Miyazawa K & Saitoh M (2015) δEF1 associates with DNMT1 and maintains DNA methylation of the E‐cadherin promoter in breast cancer cells. Cancer Med 4, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tan E‐J, Kahata K, Idås O, Thuault S, Heldin C‐H & Moustakas A (2015) The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial‐to‐mesenchymal transition. Nucleic Acids Res 43, 162–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. PRRX1 binds to the PROM1 promoter and negatively modulates its promoter activity.
Fig. S2. PRRX1 is required for BMP‐induced loss of the CD133‐positive GIC population.
Fig. S3. The PRRX1 pmx‐1b isoform is important for the decrease in the CD133‐positive population of GICs.
Fig. S4. Upregulation of PROM1 mRNA by treatment with shRNA for PRRX1 or the pmx‐1b isoform in U3005MG and U3024MG cells.
Fig. S5. DNMT3A is involved in the reduction of CD133 expression by BMP signaling.
Fig. S6. DNMT3A is required for the PRRX1 pmx‐1b isoform to downregulate CD133 expression.
Table S1. Characteristics of glioblastoma cells used in the present study.
Table S2. Primer sets for real‐time qPCR.
Table S3. Target sequences of shRNA.
Table S4. Oligonucleotides used for mutagenesis of the PRRX1‐binding sites.
Table S5. Primers used for ChIP‐qPCR.
Table S6. Primer sets used for bisulfite sequencing.
Table S7. Primer sets used for methylation‐specific PCR.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.