

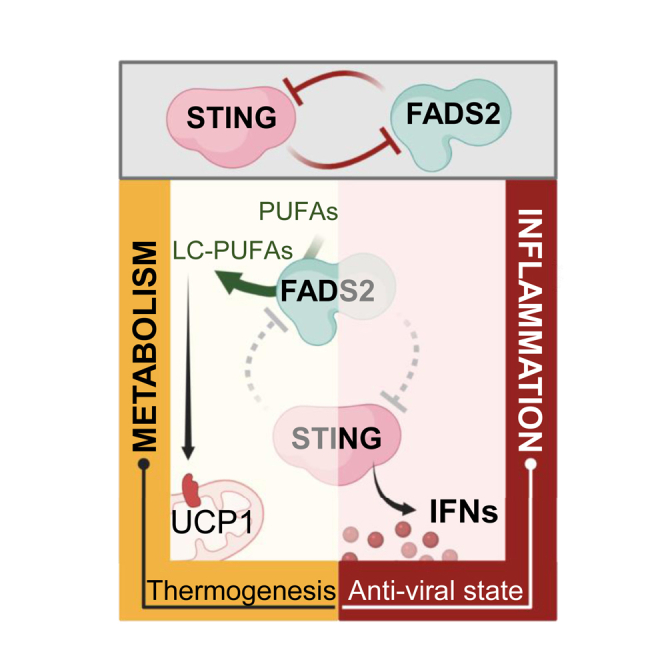
Summary
Concerted alteration of immune and metabolic homeostasis underlies several inflammation-related pathologies, ranging from metabolic syndrome to infectious diseases. Here, we explored the coordination of nucleic acid-dependent inflammatory responses and metabolic homeostasis. We reveal that the STING (stimulator of interferon genes) protein regulates metabolic homeostasis through inhibition of the fatty acid desaturase 2 (FADS2) rate-limiting enzyme in polyunsaturated fatty acid (PUFA) desaturation. STING ablation and agonist-mediated degradation increased FADS2-associated desaturase activity and led to accumulation of PUFA derivatives that drive thermogenesis. STING agonists directly activated FADS2-dependent desaturation, promoting metabolic alterations. PUFAs in turn inhibited STING, thereby regulating antiviral responses and contributing to resolving STING-associated inflammation. Thus, we have unveiled a negative regulatory feedback loop between STING and FADS2 that fine-tunes inflammatory responses. Our results highlight the role of metabolic alterations in human pathologies associated with aberrant STING activation and STING-targeting therapies.
Keywords: STING, cGAS, FADS2, delta-6 Desaturase, polyunsaturated fatty acids, interferon responses, nucleic acid immunity, inflammation, metabolism, cytosolic DNA
Graphical abstract
Highlights
-
•
STING inhibits FADS2-dependent desaturation of PUFAs and LC-PUFAs
-
•
STING activation leads to upregulation of FADS2-associated desaturase activity
-
•
STING agonists activate FADS2-dependent PUFA and LC-PUFA desaturation
-
•
PUFAs inhibit STING-dependent inflammatory responses
The stimulator of interferon genes (STING) is a central regulator of nucleic acid-associated inflammatory responses. Here, Vila et al. discover that STING regulates polyunsaturated fatty acid (PUFA) metabolism, and in turn, PUFAs inhibit STING-dependent inflammation. This cross-regulation is central to the maintenance of metabolic homeostasis.
Introduction
The endoplasmic reticulum (ER) resident stimulator of interferon genes (STING) adaptor protein is central to the mounting of inflammatory responses in the presence of pathological nucleic acids, including double-stranded DNA (dsDNA) (Ishikawa et al., 2009). Indeed, aberrant dsDNA accumulation under stress conditions (Bai et al., 2017; King et al., 2017) or following pathogen infections (Gao et al., 2013a) can be detected by the cyclic GMP-AMP (cGAMP) synthase (cGAS) (Sun et al., 2013), which catalyzes the production of cGAMP (Ablasser et al., 2013; Gao et al., 2013b). This second messenger interacts with STING (Zhang et al., 2013), promoting conformational changes (Shang et al., 2019), translocation toward the Golgi apparatus (Ishikawa et al., 2009), and recruitment of the tank-binding kinase 1 (TBK1) and transcription factors such as the interferon regulatory factor 3 (IRF3) (Liu et al., 2015). TBK1-dependent phosphorylation of STING and IRF3 ensues, orchestrating the production of inflammatory cytokines and type I interferons (IFNs) (Ishikawa et al., 2009). Following activation, STING is degraded to ensure resolution of inflammatory signaling (Konno et al., 2013).
The central role of STING in the initiation of cytosolic nucleic acid-associated inflammatory responses has attracted tremendous biomedical interest (Le Naour et al., 2020). Indeed, dysregulations of STING-associated signaling have been reported in a vast array of human pathologies, including type I interferonopathies (Crow and Casanova, 2014) or cancer (Zhu et al., 2019), prompting the development of STING-targeting therapeutics. However, several recent reports highlight that, while immune cells express high STING levels, some other cell types express low STING (Sun et al., 2009; Thomsen et al., 2016). Furthermore, STING levels are also heterogeneous among human populations; for example, the R71H-G230A-R293Q-STING (HAQ-STING) haplotype (Jin et al., 2011) is expressed at lower levels than wild-type (WT)-STING (Walker et al., 2020). This raises questions concerning the impact of systemic administration of STING agonists in these contexts of low STING levels, in particular in the light of non-immune cells playing a central role in the regulation of organ-specific inflammation (Krausgruber et al., 2020). Additionally, the consequences on homeostasis of post-activation STING degradation (Konno et al., 2013) that may result from persistent agonist treatment are unknown.
A link between STING activation and metabolic dysfunction has recently emerged. Indeed, STING-associated cytokine production can be induced by high-fat diets (HFDs) and alter metabolic homeostasis (Bai et al., 2017). In addition, TBK1 and IRF3 activation, both downstream effectors of STING, also regulate central metabolic pathways (Kumari et al., 2016; Zhao et al., 2018), including fatty acid metabolism (Tang et al., 2021). Conversely, fatty acid biosynthesis pathways have been proposed to participate in controlling antiviral responses through modulation of the expression of interferon-stimulated genes (Isg) (Kanno et al., 2021). While these studies highlight a role of STING activation and ensuing activation of downstream effectors in the maintenance of metabolic homeostasis, there is as of today no indication of a direct role of STING in metabolism. Notwithstanding, evolutionary studies indicate the conservation of Sting in prokaryotes (Kranzusch et al., 2015), suggesting an ancient role in host defense against bacteriophages, prior to the evolution of the mammalian innate immune system (Morehouse et al., 2020). Furthermore, STING activation in T cells has been shown to hamper T cell proliferation, independently of its role in innate immune regulation (Cerboni et al., 2017). Thus, mounting evidence suggests that STING harbors immune-unrelated functions.
Here, we investigated the role of STING in the maintenance of metabolic homeostasis. We reveal that STING regulates PUFA metabolism through inhibition of the fatty acid desaturase 2 (FADS2)-associated delta-6 desaturase (Δ6D) activity. We further show that PUFAs inhibit STING, thus revealing a negative regulatory feedback loop between STING and FADS2. Finally, we show that STING agonists directly activate FADS2-dependent PUFA desaturation, highlighting that STING-targeting strategies may lead to metabolic alterations. Therefore, we reveal a central role of STING in the maintenance of metabolic homeostasis.
Results
STING regulates metabolic homeostasis in vivo
To access STING function in the absence of inflammation, given the tight interconnection between metabolic and immune pathways (Brestoff and Artis, 2015; Buck et al., 2017), we questioned the impact of Sting ablation on metabolic parameters. Under normal diet, WT and Sting−/− mice did not exhibit spontaneous inflammation (Figure 1A) or differences in body weight (Figure 1B) and composition (Figure 1C). Yet Sting−/− mice presented increased food intake (Figure 1D) and improved insulin-independent (Figures 1E and S1A) glucose management (Figure 1F). Furthermore, quantitative analysis of fluorodeoxyglucose ([18F]-FDG) showed faster extraction from the blood of Sting−/− mice compared to WT mice (p = 0.0003) (Figure 1G). This indicates that Sting-deficient mice uptake more glucose compared to WT mice. Interestingly, these changes were coupled to decreased hepatic gluconeogenesis (Figure S1B).
Figure 1.

STING deficiency leads to global metabolic improvement
(A) Cxcl10, Tnf-α, and Il6 mRNA levels were measured in liver, muscle, and visceral adipose tissue (VAT) from Sting+/+ (n = 5) and Sting−/− (n = 5) mice.
(B) Body weight of Sting+/+ (n = 32) and Sting−/− (n = 34) mice under normal chow diet.
(C) Body composition of Sting+/+ (n = 6) and Sting−/− (n = 6) mice was assessed by EchoMRI.
(D) Food intake of Sting+/+ (n = 8) and Sting−/− (n = 9) mice.
(E) Insulin tolerance test (ITT) in Sting+/+ (n = 14) and Sting−/− (n = 15) mice. Left: glycemia (mg/dL) over time, following a bolus of insulin. Right: area above the curve.
(F) Glucose tolerance test (GTT) was performed in Sting+/+ (n = 25) and Sting−/− (n = 24) mice. Left: glycemia (mg/dL) over time, following a bolus of glucose. Right: area under the curve (AUC).
(G) [18F]-FDG clearance rates were measure over time in Sting+/+ (n = 5) and Sting−/− (n = 5) mice and are expressed as percent (%) injected dose (ID) per mL. p = 0.0003.
(H) Oxygen consumption of Sting+/+ (n = 6) and Sting−/− (n = 6) mice as determined in metabolic chambers.
(I) Energy expenditure during day (white) and night (gray) was determined as in (H). p value was determined by one-way ANOVA.
(J) Daily profile of voluntary running-wheel activity of Sting+/+ (n = 11) and Sting−/− (n = 10) mice.
(K) [18F]-FDG concentration in the subcutaneous (SAT), visceral (VAT), and brown adipose tissue (BAT) from Sting+/+ (n = 7) and Sting−/− (n = 7) mice was measured at 45 min post-injection of [18F]-FDG. Results are expressed as %ID/g of tissue.
(L) Pgc1α, Cidea, Dio2, and Ucp1 mRNA levels in the VAT and BAT of Sting+/+ (n = 4-6) and Sting−/− (n = 5) in mice.
(M) Rectal temperature of Sting+/+ (n = 13) and Sting−/− (n = 14) mice under normal chow diet.
(N) GTT was performed in Sting+/+ (n = 7) and Sting−/− (n = 9) mice under high-fat diet (HFD 60%) for 6 (6w) and 20 weeks (20w). Left: glycemia (mg/dL) over time, following a bolus of glucose. Right: AUC.
(O) Rectal temperature of Sting+/+ (n = 7) and Sting−/− (n = 9) mice after 6 or 20 weeks of HFD.
(P) Survival curve of Sting+/+ (n = 6) and Sting−/− (n = 7) mice under HFD.
All graphs present means ± SEM. p values were determined by Student’s t test, unless otherwise stated. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Related to Figure S1.
Because Sting-deficient mice uptake more glucose without change in body weight, we questioned the utilization of excess substrate. Indirect calorimetry measurements showed that Sting−/− mice consume more oxygen (Figure 1H) and present higher energy expenditure during the light phase (Figure 1I), in the absence of change in locomotor circadian rhythm (Figure S1C) or spontaneous locomotor activity (Figure 1J). Analysis of [18F]-FDG biodistribution in major glucose-consuming organs showed no change in the brain, liver, and muscle (Figure S1D), while revealing increased glucose uptake in the visceral adipose tissue (VAT), the brown adipose tissue (BAT), and a tendency (p = 0.08) in the subcutaneous adipose tissue (SAT) (Figure 1K), reflecting increased metabolic activity in adipose tissues. Accordingly, analysis of thermogenic gene expression showed increased peroxisome proliferator-activated receptor-gamma coactivator-1alpha (Pgc1α), cell death activator (Cidea), and iodothyronine deiodinase 2 (Dio2) in the BAT and of Pgc-1α and uncoupling protein 1 (Ucp1) in the VAT (Figure 1L), coupled to increased Ucp1 protein levels in the VAT (Figure S1E). These changes were accompanied by increased rectal temperature in Sting-deficient mice (Figure 1M), supporting increased thermogenesis in absence of Sting, and further reflecting the activation of adipose tissues’ browning pathways (Seale et al., 2007). Thus, altogether, Sting-deficient mice presented spontaneous activation of thermogenesis, implying that STING inhibits thermogenic programs in adipose tissues.
We next questioned whether the metabolic changes observed under normal chow diet are conserved under HFD. Metabolic phenotyping of Sting-deficient mice under HFD showed that absence of Sting leads to increased glucose clearance (Figure 1N) and increased food intake (Figure S1F), despite similar body weight (Figures S1G and S1H). Similar to what was observed in mice under normal chow diet, Sting-deficient mice presented increased rectal temperature, indicative of increased thermogenesis (Figure 1O). Finally, consistent with improved metabolic parameters in the absence of Sting, the survival of Sting−/− mice under HFD was increased compared to WT mice (Figure 1P). Altogether, these data show that STING deficiency confers a metabolic advantage that is conserved under metabolic stress.
To verify that the metabolic modifications observed in Sting−/− mice are independent of the canonical function of Sting in activating pro-inflammatory pathways, we next performed metabolic phenotyping of cGas−/− mice (Figures S1I–S1K) and conditional myeloid cell-specific Sting−/− mice (Figures S1L–S1N). Monitoring of the body weight, glucose management, and rectal temperature of these mice under normal chow diet showed no significant modification. This confirms that metabolic alterations witnessed in Sting-deficient mice are independent of its canonical innate immune function. Therefore, absence of STING is sufficient to cause global metabolic changes in vivo, suggesting that STING plays a central role in the maintenance of metabolic homeostasis.
STING interacts with the fatty acid desaturase 2
To identify the molecular mechanism through which Sting regulates metabolic homeostasis, we performed tandem-affinity purification of Flag- and HA-tagged Sting (F/HA-Sting) stably expressed in mouse embryonic fibroblast (MEF) knockout for Sting (MEFSting−/−). Immunopurified material was either silver-stained (Figure 2A) or analyzed by mass spectrometry (MS) to identify Sting protein partners. Besides known Sting interactors, this approach revealed a large number of proteins involved in metabolic pathways (Table 1), notably including the Δ6D enzyme, also known as Fads2, encoded by the Fads2 gene (Park et al., 2015). FADS2/Δ6D is the first rate-limiting enzyme involved in the desaturation of the linoleic acid (LA [18:2n-6, or omega-6]) and α-linolenic acid (ALA [18:3n-3, or omega-3]) PUFAs (Nakamura and Nara, 2004) to generate long-chain PUFAs (LC-PUFAs) (Figure 2B). A simplified view is that the Δ6D activity encoded by Fads2 and the delta-5 desaturase (Δ5D) activity encoded by Fads1 (Leonard et al., 2000) are required to generate the eicosapentaenoic acid (EPA) LC-PUFA. Fads2 is also required to generate docosahexaenoic acid (DHA) LC-PUFAs from EPA, but also to generate dihomo-γ-linolenic acid (DGLA) from LA (Nakamura and Nara, 2004). Desaturation of DGLA into arachidonic acid (AA) requires the Δ5D activity encoded by Fads1 (Leonard et al., 2000). Dedicated enzymes promote both elongation in between desaturation steps and further processing of LC-PUFAs into oxylipins that influence numerous physiological processes and that are known to bear both pro- and anti-inflammatory properties (Gabbs et al., 2015). This raised the hypothesis that FADS2 interaction with STING may be responsible for metabolic improvements witnessed in the absence of STING (Figures 1 and S1).
Figure 2.

STING interacts with FADS2 in the absence of pro-inflammatory stimulation
(A) Left: experimental scheme. Right: silver-staining of immunopurified Flag- and HA-tagged Sting (F/HA-Sting) separated on SDS-PAGE. Numbers on the left: molecular weight in kDa.
(B) Simplified schematic representation of the LA (omega-6 [Ω6], yellow) and ALA (omega-3 [Ω3], blue) fatty acid (FA) desaturation pathway, leading to the generation of LC-PUFAs. Where Fads2 is indicated is where the Fads2-dependent delta-6 desaturase (Δ6D) activity is required. Where Fads1 is indicated is where the Fads1-dependent delta-5 desaturase (Δ5D) activity is required. ALA, α-linolenic acid; LA, linoleic acid; DGLA, dihomo-γ-linolenic acid; AA, arachidonic acid; EPA, eicosapentaenoic acid; DHA, docosahexaenoic acid.
(C) Flag immunoprecipitation was performed on whole-cell extracts (WCEs) from MEFSting−/− expressing F/HA-Sting or not. Inputs and eluates from Flag-immunoprecipitated F/HA-Sting were analyzed by western blot (WB) using indicated antibodies.
(D) Immunoprecipitation was performed using a Sting-specific antibody on WCEs from MEF and MEFSting−/−. Inputs and immunoprecipitated material were analyzed by WB using indicated antibodies.
(E) As in (D), except that WCEs from MEFSting−/− expressing F/HA-Sting or not were used.
(F) Flag immunoprecipitation was performed on WCEs from MEFs expressing F-Fads2 or not. Inputs and eluates from Flag-immunoprecipitated Flag-Fads2 (F-Fads2) were analyzed by WB using indicated antibodies.
(G) Immunofluorescence analysis of MEFSting−/− stably expressing F/HA-Sting using anti-Fads2 and anti-HA antibodies and DAPI nuclear staining. BF, bright field. Pearson’s correlation coefficient values for co-localization of Fads2 and Sting in MEF cells. Pearson’s correlation coefficients were calculated from four randomly selected areas.
(H) As in (C), except that WCEs from MEFSting−/−, MEFs expressing F/HA-Sting, or MEFs expressing Flag-tagged STING (STING-F) or HAQ-STING (HAQ-STING-F) were used.
(I) As in (C), except that WCEs from MEFs expressing F/HA-WT-Sting or F/HA-StingΔTM were used.
(J) As in (G), except that cells were stimulated (DMXAA) or not (DMSO) for 30 min prior to immunofluorescence analysis. Pearson’s correlation coefficient values for co-localization of Fads2 and Sting in MEF cells treated with DMXAA or not. Pearson’s correlation coefficients were calculated from four randomly selected areas in each group.
All graphs present means ± SEM. p values were determined by Student’s t test. ∗∗p < 0.01.
Table 1.
Major protein partners of STING identified by mass spectrometry
| Uniprot reference | Gene symbol | MW | Mocka | IPb |
|---|---|---|---|---|
| sp|Q9WUN2|TBK1_MOUSE | Tbk1 | 83.37 | 0 | 8 |
| sp|Q9Z0R9|FADS2_MOUSE | Fads2 | 52.35 | 0 | 10 |
| sp|Q920L1|FADS1_MOUSE | Fads1 | 52.29 | 0 | 6 |
| sp|Q9D6K9|CERS5_MOUSE | Cers5 | 48.13 | 0 | 5 |
| sp|Q9CY27|TECR_MOUSE | Tecr | 36.07 | 0 | 5 |
| sp|Q8VCH6|DHC24_MOUSE | Dhcr24 | 60.07 | 0 | 4 |
| sp|Q6NVG1|LPCT4_MOUSE | Lpcat4 | 57.11 | 0 | 4 |
| sp|Q8K2C9|HACD3_MOUSE | Hacd3 | 43.10 | 0 | 2 |
| sp|P47740|AL3A2_MOUSE | Aldh3a2 | 53.94 | 0 | 2 |
To verify the interaction between FASD2 and STING, we first performed Flag or HA immunoprecipitation of F/HA-Sting from MEF, coupled to elution using an excess of Flag or HA peptides, respectively. Western blot (WB) analysis of input and eluates showed an enrichment of Fads2 following F/HA-Sting immunoprecipitation (Figures 2C and S2A). Second, we performed immunoprecipitation from WT-MEFs and MEFSting−/−, or from MEFSting−/− and MEFSting−/− stably expressing a WT-Sting allele, using a Sting-specific antibody. WB analysis of input and immunoprecipitates showed co-immunoprecipitation of Fads2 with Sting (Figures 2D and 2E). Conversely, Flag immunoprecipitation of Flag-tagged Fads2 (F-Fads2) from MEFs allowed co-immunoprecipitation of Sting (Figure 2F). Next, we performed immunofluorescence-based analysis of Sting and Fads2 localization in MEFs. This showed that Sting and Fads2 colocalize in perinuclear regions corresponding to the ER (Figures 2G and S2B), which is in agreement with their reported subcellular distribution (Ishikawa and Barber, 2008; Park et al., 2015). These data strongly suggest that Sting and Fads2 interact in the ER in the absence of pro-inflammatory stimulation. To assess whether this interaction is reproducible with human STING, we next performed Flag-immunoprecipitation experiments using Flag-tagged human WT- (WT-STING-F) or HAQ-STING (HAQ-STING-F) expressed in MEFs. We thereby observed that WT-STING-F and HAQ-STING-F interact with Fads2 (Figure 2H). Similarly, endogenous STING immunoprecipitation performed in the THP-1 human myeloid cell line that expresses the HAQ-STING allele (Diner et al., 2013) showed interaction with FADS2 (Figure S2C). Finally, to identify the domain of Sting involved in mediating interaction with Fads2, we performed immunoprecipitation experiments in MEFs expressing a Flag- and HA-tagged Sting truncation mutant that does not possess the transmembrane domains required for Sting anchoring to the ER (Sun et al., 2009) (F/HA-StingΔTM). WB analysis revealed that F/HA-StingΔTM does not interact with Fads2 (Figure 2I). Altogether, these data show that the interaction of STING with FADS2 takes place in the absence of pro-inflammatory stimulation and is conserved in murine and human cell lines.
Finally, we questioned whether activation of STING, which is reported to promote its re-localization to the Golgi apparatus, alters interaction with FADS2. Immunofluorescence analysis of Sting and Fads2 localization in MEFs after stimulation with the 5,6-dimethylxanthenone-4-acetic acid (DMXAA) Sting agonist showed the expected formation of Sting clusters (Corrales et al., 2015) from which Fads2 is excluded (Figure 2J). Thus, altogether our data show that the interaction of STING with FADS2 is decreased upon STING activation.
STING inhibits fatty acid desaturase 2-associated desaturation activity
PUFAs and derivatives are strong activators of transcription factors playing a major role in glucose and lipid metabolism (Clarke and Jump, 1994) and upregulate the expression of thermogenic genes such as Pgc1α and PR domain-containing 16 (Prdm16) (Fan et al., 2019; Seale et al., 2007) (Figure S3A). Since STING interacts with FADS2, this raised the possibility that STING modulates FASD2-associated Δ6D activity, thereby controlling PUFA, LC-PUFA, and derivative levels, which would in turn promote the metabolic alterations observed in Sting−/− mice (Figures 1 and S1). We therefore explored the impact of STING on FADS2 activity. We used liquid chromatography coupled to MS (LC-MS) to quantify PUFAs, LC-PUFAs, and their derivatives in liver and VAT samples from WT and Sting−/− mice. Partial least-squares discriminant analysis (PLS-DA) of LC-MS data showed that WT and Sting−/− liver and VAT are significantly different (Figures S3B and S3C). Correlation analysis showed a shift in PUFA content, leading to accumulation of omega-3 derivatives (Figures S3D and S3E). Although no significant shift in the total omega-6/omega-3 ratio was measured in mice liver samples (Figure 3A), quantification of the total amount of derivatives from the main PUFA families showed that absence of Sting leads to significantly increased levels of DGLA and DHA derivatives (Figure 3B), coupled to increased estimated Fads2-associated desaturase activity in Sting−/− mice (Figure 3C). Similar increase in LC-PUFAs that may derive from Fads2-associated desaturation, and their derivatives, was measured in MEFSting−/−, but not in MEFcGas−/−, compared to WT-MEFs (Figures 3D and 3E). Importantly, Pgc1α and Prdm16 mRNA levels were upregulated in Sting−/−, but not in cGas−/− MEFs (Figure 3F), suggesting that changes in Fads2-dependent desaturase activity and PUFA pools and derivatives drive activation of thermogenesis programs in the absence of Sting.
Figure 3.

STING modulates PUFA pools
(A) Ratio between total omega-6 (Ω6) and omega-3 (Ω3) PUFAs, LC-PUFAs, and derivatives measured in Sting+/+ (n = 5) and Sting−/− (n = 5) mice liver using LC-MS.
(B) Sum of indicated PUFAs and LC-PUFAs and respective derivatives in samples analyzed as in (A).
(C) Fads1- and Fads2-associated enzymatic activities, respectively Δ5D and Δ6D, were estimated by calculating the substrate/product ratio of PUFAs and LC-PUFAs measured in (A).
(D) Ratio between total omega-6 and omega-3 PUFAs and derivatives measured in WT-MEF, MEFSting−/−, and MEFcGas−/− (n = 4–5) using LC-MS.
(E) Sum of indicated PUFAs, LC-PUFAs, and respective derivatives in samples analyzed as in (D).
(F) qRT-PCR analysis of Prdm16 and Pgc1a mRNA levels in WT-MEF, MEFSting−/−, and MEFcGas−/− (n = 4).
(G) WCEs from WT-MEF, MEFSting−/−, and MEFSting−/− stably expressing F/HA-WT-Sting or F/HA-StingΔTM were analyzed by WB using indicated antibodies.
(H) Prdm16, Pgc1α, and Ucp1 mRNA levels in WT-MEF, MEFSting−/−, and MEFSting−/− stably expressing F/HA-WT-Sting or F/HA-StingΔTM (n = 3).
(I) Prdm16, Pgc1α, and Ucp1 mRNA levels in WT-MEF and MEFSting−/− treated or not with the sc26196 Fads2 inhibitor for 48 h (n = 4).
(J) GTT was performed in Sting+/+ (n = 4) and Sting−/− mice under standard (STD) (n = 8) or low omega-3 (Ω3L) diet (n = 7) for 4 weeks.
(K) AUC before and after 4 weeks of STD or Ω3L diet in Sting−/− mice was calculated from data in (J).
(L) Rectal temperature of Sting+/+ and Sting−/− mice from (J).
(M) Sting inhibits Fads2. Consequently, absence of Sting leads to increased Fads2-dependent desaturation of PUFAs and LC-PUFAs, driving the activation of thermogenic program genes and thermogenesis in vivo.
All graphs present means ± SEM. p values were determined by Student’s t test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Related to Figure S3.
To confirm that Sting-dependent inhibition of Fads2 is responsible for repression of thermogenic programs, we performed complementation experiments in which we re-expressed WT-Sting or StingΔTM in MEFSting−/− (Figure 3G). Analysis of thermogenesis gene expression showed that re-expression of WT-Sting, but not of StingΔTM, led to decreased Prdm16, Pgc1α, and Ucp1 levels (Figure 3H). In addition, treatment of WT-MEF and MEFSting−/− with a Fads2 inhibitor (sc26196) led to a significant decrease of the expression of thermogenic genes in MEFSting−/− (Figure 3I), supporting that increased thermogenic gene expression witnessed in the absence of Sting is dependent on Fads2. Accordingly, feeding Sting−/− mice with low omega-3 diet led to deteriorated glucose management and decreased rectal temperature compared to standard diet (Figures 3J–3L), accompanied by decreased Ucp1 levels (Figure S3F). Thus, withdrawal of omega-3 substrates in vivo decreases the metabolic advantage conferred by Sting ablation. Therefore, our data establish that STING inhibits FADS2 and that the absence of STING is sufficient to lead to upregulated FADS2-dependent desaturase activity, leading to increased thermogenic program activation (Figure 3M).
STING activation promotes FADS2-dependent desaturation
We have shown that STING activation disrupts its interaction with FADS2 (Figure 2J). In addition, following acute activation Sting is degraded (Konno et al., 2013). Thus, STING activation can be expected to alleviate the STING-dependent block on FADS2. To test this hypothesis, we measured PUFA, LC-PUFA, and derivative levels upon acute Sting activation. We used dsDNA transfection in 293T cells to activate Sting-dependent signaling (Figure 4A) and Sting degradation (Figure 4B), prior to analysis of PUFAs, LC-PUFAs, and derivatives. Similar to what was observed upon Sting ablation, we observed an increase in PUFAs that may derive from FADS2-dependent desaturation, and in particular omega-3 derivatives (Figure 4C), without significant shift in the omega-6/omega-3 balance (Figure 4D), accompanied by increased expression of Pgc1α and Prdm16 (Figure S4A). Intriguingly, we also observed decreased FADS2 levels following Sting activation (Figures 4B and S4B), in the absence of modification of FADS1 protein levels (Figure S4B), independently of type I IFN production (Figures S4C and S4D) and response to IFN (Figures S4E and S4F). This corroborates previous reports that FADS2 levels are decreased following its over-activation (Ralston et al., 2015) and further supports that FADS2 is responsible for the monitored changes in PUFA pools.
Figure 4.

STING activation promotes FADS2-dependent desaturation
(A) Ifnβ and Cxcl10 mRNA levels in 293T cells transfected or not with dsDNA for 24 h (n = 3).
(B) WCEs of cells treated as in (A) were analyzed by WB using indicated antibodies.
(C) Sum of indicated PUFAs, LC-PUFAs, and derivatives in samples prepared as in (A).
(D) Ratio between total omega-6 and omega-3 PUFAs and derivatives in samples from (C).
(E) Ifnβ and Cxcl10 mRNA levels in WT and Trex1-deficient MEF cells (n = 3).
(F) WCEs of WT and Trex1-deficient MEF cells were analyzed by WB using indicated antibodies.
(G) Sum of indicated PUFAs, LC-PUFAs, and derivatives in WT and Trex1-deficient MEF cells.
(H) Ratio between total omega-6 and omega-3 PUFAs and derivatives in samples from (G).
All graphs present means ± SEM. p values were determined by Student’s t test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, we monitored PUFA, LC-PUFA, and derivative levels in a model of chronic Sting activation. To this aim, we used MEF knockout of the three prime exonuclease 1 (MEFTrex1−/−) (Ablasser et al., 2014) (Figure 4E), in which upregulation of Ucp1 has been reported (Hasan et al., 2017). We observed decreased Fads2 levels in the absence of a change in Fads1 levels (Figure 4F), together with accumulation of products of Fads2-associated desaturase activity (Figure 4G). In contrast to what we observed upon acute Sting activation, we also measured an increase of omega-6 derivatives accompanied by an increase in the omega-6/omega-3 ratio (Figure 4I). Such activation of omega-6 derivatives likely feeds chronic inflammation (Innes and Calder, 2018). This supports that Sting activation promotes increased Fads2-dependent desaturase activity. Yet in the context of Trex1 deficiency, increased PUFA and LC-PUFA desaturation is also witnessed in the absence of decreased Sting levels. This implies that decreased STING levels are not the sole parameter regulating PUFA and LC-PUFA desaturation upon STING activation.
STING agonists activate FADS2-dependent desaturation
To investigate the molecular mechanism through which STING activation may promote FADS2-dependent PUFA and LC-PUFA desaturation, independently of STING degradation, we treated WT-MEF, MEFcGas−/−, and MEFSting−/− with the DMXAA Sting agonist prior to analysis of Fads2, PUFA, LC-PUFA, and derivative levels. Treatment with DMXAA led to decreased Fads2 levels (Figure 5A), accompanied by increased PUFA, LC-PUFA, and derivative levels (Figure 5B), regardless of the expression of cGas or Sting. In contrast, dsDNA transfection-induced Fads2 degradation required the presence cGas (Figure 5C). These data support that sole activation of Sting is sufficient to promote Fads2 activation. However, the requirement for cGAS in dsDNA-dependent FADS2 degradation raises the tempting hypothesis that dinucleotides regulating STING may directly regulate FADS2.
Figure 5.

STING agonists promote FADS2-dependent desaturation
(A) WCEs from WT, Sting−/−, or cGas−/− MEFs stimulated or not with DMXAA for 2 h were analyzed by WB using indicated antibodies.
(B) Sum of omega-6 and omega-3 PUFAs, LC-PUFAs, and derivatives measured in MEFs stimulated or not with DMXAA for 6 h.
(C) WCEs from WT, Sting−/−, or cGas−/− MEFs stimulated or not with dsDNA for 6 and 24 h were analyzed by WB using indicated antibodies.
(D) Molecular docking of cGAMP, DMXAA, and 6 PUFAs to STING (top) or FADS2 (bottom). Color coding for ligand: LA in blue, ALA in green, AA in orange, DHA in turquoise, DGLA in brown, and EPA in magenta.
(E) Binding of Flag-purified F-Fads2 or F/HA-Sting to cGAMP was analyzed by WB using anti-Sting or anti-Fads2 antibodies.
(F) Prdm16 and Ucp1 mRNA levels in MEFs stimulated or not with DMXAA for 3 h in presence or not of the sc26196 Fads2 inhibitor (n = 3).
(G) Ucp1 mRNA levels in the VAT of Sting−/− mice treated or not with DMXAA for 4 weeks (n = 5).
(H) Ucp1 protein levels in the VAT of Sting−/− mice treated or not with DMXAA for 4 weeks. Representative WB, n = 3.
(I) Sting agonist promotes Fads2-dependent desaturation of PUFAs, leading to increased LC-PUFAs and derivatives, thereby driving the activation of thermogenic program genes and thermogenesis in vitro and in vivo.
All graphs present means ± SEM. p values were determined by Student’s t test. ∗∗p < 0.01, ∗∗∗p < 0.001.
Related to Figure S5.
To test this hypothesis, we performed in silico docking experiments of cGAMP and DMXAA into Fads2. To this aim, we used the resolved crystal of Sting in complex with cGAMP (PDB: 6WD4) and the molecular model of Fads2 as starting biological systems. This predicted that cGAMP and DMXAA can dock into Fads2, adopting similar conformation (Figure 5D, left and central panels) and achieving analogous interactions and docking energies (Figure S5A; Table S1). This supports that Sting agonists are likely ligands of Fads2. In vitro binding assay, using streptavidin bead-immobilized biotinylated cGAMP and immunopurified Fads2 or Sting, showed that cGAMP recruits Fads2 (Figure 5E).
Next, we tested whether DMXAA can induce Fads2-specific metabolic alterations. To this aim, we treated WT-MEFs with DMXAA in the presence or absence of a Fads2 inhibitor. DMXAA treatment induced an upregulation of Prdm16 and Ucp1 that is decreased upon inhibition of Fads2 (Figure 5F). Furthermore, treatment with DMXAA also led to increased Ucp1 mRNA and protein levels in the VAT of Sting−/− mice (Figures 5G and 5H), demonstrating that DMXAA induces Fads2-dependent metabolic alterations in vivo. Altogether, these data establish that STING agonists are activators of FADS2 (Figure 5I).
cGAMP and PUFAs orchestrate the crosstalk between FADS2 and STING
Intriguingly, molecular docking analysis between PUFAs, LC-PUFAs, and Sting predicts that PUFAs and LC-PUFAs can dock to the cGAMP-binding domain of Sting (Figure 5D, right panels). Indeed, cGAMP, DMXAA, PUFAs, and LC-PUFAs all adopt similar conformations in in silico docking experiments performed with Sting or Fads2 (Figures S5A and S5B), achieving analogous binding energies (Table S1). Furthermore, Sting and Fads2 showed strong structural similarity, both with regard to the 3D arrangement of the docking site anatomy and in terms of hydrophobicity, electrostatics, and solvent accessibility (Figures S5A and S5B). Altogether, this suggests that PUFAs and LC-PUFAs are potential ligands of Sting, further predicting that they may modulate Sting activation.
We thus tested the impact of ALA and LA PUFAs on DMXAA-induced Sting activation in MEFs. Both treatments decreased DMXAA-dependent phosphorylation of Sting, Irf3, and Tbk1 (Figure 6A) and decreased expression of Ifnβ and tested Isgs (Figure 6B). Importantly, treatment with the oleic acid (OA) omega-9 fatty acid did not alter DMXAA-induced Tbk1 phosphorylation (Figure 6C). Similarly, treatment with ALA inhibited dsDNA-induced activation of Sting signaling (Figure 6D) but did not alter poly(I:C)-dependent, Sting-independent Irf3 phosphorylation (Figure S6A), attesting to the specificity of PUFA-mediated Sting inhibition. Additionally, treating Trex1-deficient MEFs with ALA led to decreased Ifnβ, Cxcl10, and Isg15 mRNA levels (Figure S6B). Finally, we preformed Flag immunoprecipitation of F/HA-Sting stably expressed in MEFs, following treatment with DMXAA, in the presence or absence of ALA. This showed that treatment with ALA decreases DMXAA-induced association of Sting with Tbk1 and pTbk1, reflecting decreased assembly of the Sting signalosome (Figure 6E). Thus, PUFAs promote dissociation of the Sting signalosome, supporting that ALA directly inhibits Sting.
Figure 6.

Fads2 and PUFAs regulate Sting activation
(A) WCEs from WT-MEFs treated or not with DMXAA, in the presence or absence of 50 μM ALA or LA for 2 or 6 h, were analyzed by WB using indicated antibodies.
(B) Ifnβ, Cxcl10, and Isg15 mRNA levels in cells treated as in (A) (n = 3).
(C) WT-MEFs were treated or not with DMXAA in the presence or not of 50 μM ALA or OA for 3 h prior to analysis by WB using indicated antibodies.
(D) WT-MEFs were transfected or not with dsDNA in the presence or absence of 50 μM ALA for 6 h prior to analysis by WB using indicated antibodies.
(E) Flag-immunoprecipitation was performed from MEFSting−/− stably expressing F/HA-Sting or not, following treatment with DMXAA or not in combination with ALA for 3 h. Input and eluates were analyzed by WB using indicated antibodies.
(F) MEFs expressing stably expressing shScr or shFads2 were treated or not with 50 μM ALA for 6 h prior to analysis by WB using indicated antibodies.
(G) WCEs from MEFs expressing shScr or shFads2 transfected or not with dsDNA for 6 h were analyzed by WB using indicated antibodies.
(H) Ifnβ and Cxcl10 mRNA levels in cells expressing shScr or shFads2 after stimulation or not with dsDNA for 6 h (n = 4).
(I) WT-MEFs were infected with HSV-KOS64 for 16 h prior to analysis of Ifnβ, Cxcl10, and Isg15 mRNA levels.
(J) As in (I), except that Prdm16, Pgc1α, and Ucp1 mRNA levels were analyzed.
(K) WT-MEFs were infected with HSV-KOS64 in presence or not of a Fads2 inhibitor (sc26196). Infection is presented as mean plaque number per cm2.
(L) WT-MEFs were infected with HSV-KOS64 in presence or not of 10 μM ALA. Infection is presented as mean plaque number per cm2.
(M) PUFAs and FADS2 inhibit STING.
All graphs are means ± SEM from at least 3 independent experiments. p values were determined by Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Related to Figure S6.
We next assessed the impact of antagonizing Fads2 on Sting activation. First, we observed that using a Fads2-targeting short hairpin RNA (shRNA) in MEFs, which does not affect Fads1 protein levels (Figure S6C), led to the expected decrease of Fads2 byproducts (Figure S6D). This was accompanied by increased basal pTbk1 levels (Figure 6F, compare lane 1 to 2), which are decreased upon complementation with ALA (compare lane 2 to 4). Similarly, complementing Fads2 knockdown MEFs with an shRNA-resistant Fads2 allele led to decreased hallmarks of Sting activation (Figure S6E). In addition, knockdown of Fads2 led to increased dsDNA-induced Sting activation, as attested by increased pIrf3 levels (Figure 6G) and Ifnβ and Cxcl10 expression (Figure 6H). Similar increase of dsDNA- and DMXAA-induced Sting activation was measured upon pharmacological inhibition of Fads2 (Figures S6F–S6H). Thus, PUFA derivatives generated by FASD2-desaturase activity inhibit STING.
Finally, to assess if the crosstalk between Fads2 and Sting impacts viral infections, we used the herpes simplex virus type I (HSV-1) DNA virus, known to promote Sting activation. Challenge with HSV-1 led to decreased Fads2 protein levels in vitro (Figure S6I) and in vivo (Figures S6J and S6K), likely reflecting Fads2 activation. Accordingly, HSV-1 infection of MEFs led to the expected increased expression of Ifnβ, Cxcl10, and Isg15 (Figure 6I), accompanied by increased expression of Prdm16, Pgc1α, and Ucp1 (Figure 6J). Antagonizing Fads2 in MEFs, using shRNAs or the sc26196 Fads2 inhibitor, led to decreased infection by HSV-1 (Figures 6K and S6L). Reciprocally, treatment with ALA led to increased infection by HSV-1 (Figures 6L and S6M). Interestingly, similar experiments performed with the B.1.1.7 (Alpha) and B.1.351 (Beta) variants of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) RNA virus showed no effect of Fads2 inhibition (Figure S6N). This supports that FADS2 and PUFAs inhibit STING (Figure 6M) and is crucial for the establishment of an antiviral state controlling DNA virus infection. Thus, inhibition of STING by FADS2 and PUFAs can modulate the establishment of an antiviral state that hinders HSV-1 infection.
Discussion
Altogether, we uncover a central role of STING in the regulation of metabolic homeostasis, independently of its reported innate immune function. Indeed, we show that STING inhibits FADS2-dependent PUFA and LC-PUFA desaturation. Absence of STING thus increases the levels of PUFAs, LC-PUFAs, and derivatives from the omega-3 branch that are known to improve glucose handling (Sirtori and Galli, 2002), prevent obesity-associated glucose intolerance (Belchior et al., 2015; Derosa et al., 2016), and increase cardiovascular protection (González-Périz et al., 2009). In agreement, we found that absence of Sting leads to better survival of mice under HFD. Under normal diet, Sting-deficient mice showed improved global metabolic profiles while lowering omega-3 from mice diet decreased such induction. Furthermore, omega-3 fatty acids can promote WAT browning (Fernández-Galilea et al., 2020), a process in which increased Ucp1, Pgc-1α, and Prdm16 expression is crucial (Ghandour et al., 2018). We show that absence of STING promotes FADS2-dependent induction of these thermogenic factors, demonstrating that metabolic remodeling in the absence of STING requires FADS2. In addition, we show that STING agonists can directly interact with FADS2 and promote FADS2-associated Δ6D activity, establishing FADS2 as a direct target of cGAMP and DMXAA. This implies that in pathologies presenting with chronic STING activation, STING-dependent modulation of PUFA and LC-PUFA desaturation may feed metabolic comorbidities such as dyslipidemia (Lira et al., 2014). Conversely, small molecules targeting STING (Haag et al., 2018; Wu et al., 2020) can be expected to alter FADS2-dependent desaturation of PUFAs and LC-PUFAs and downstream production of inflammatory lipid mediators. Such side effects of STING-targeting molecules are critical to consider in therapeutic strategies aiming to boost STING activity (Galon and Bruni, 2019).
That PUFAs can inhibit STING reveals a previously unappreciated link between fatty acid metabolism and innate immune responses. Indeed, there are indications that diet intervention and modulation of omega-3 or omega-6 intake can impact immune and antiviral responses (DiNicolantonio and O’Keefe, 2018), although the involved molecular mechanism remains poorly understood. While STING inhibition by PUFAs may serve in the resolution of STING-dependent inflammation, dietary habits that impact the substrates provided to FADS2 for desaturation (Galland, 2010; Tosi et al., 2014) may also directly influence STING activation and alter immune function (Sen et al., 2019). In addition, we show that modulating FADS2 impacts the establishment of an antiviral state. This is in agreement with previous reports that PUFA levels can both influence (Berra et al., 2017) and be influenced by HSV-1 infection (Zhang et al., 2020).
The increased production of lipid mediators, byproducts of FADS2 activation, appears as part of the physiological response after nucleic acid sensing. This suggests a non-transcriptional role of STING in innate immunity that remains to be explored. In addition, recent reports point out a role of FADS2 (Yamane et al., 2021) and STING in promoting ferroptosis (Jia et al., 2020; Li et al., 2021). Further study of the crosstalk between STING, FADS2, and PUFA production may unveil a coordinated role in ferroptosis during infections or in homeostasis. Additionally, it would be important to assess the coordination of PUFA metabolism and inflammatory responses in cases where STING levels are physiologically low, such as in humans bearing the HAQ-STING haplotype (Walker et al., 2020). In particular, our data suggest that HAQ-STING would recruit more FADS2 than WT-STING. This could allow efficient regulation of FADS2 in the presence of lower levels of STING.
Altogether, our findings offer unprecedented insight into the crosstalk between innate immune processes and metabolic regulation. Targeting this crosstalk in pathologies presenting with chronic inflammation bears the promise to alleviate associated comorbidities.
Limitations of study
The crystal structure of FADS2 is only partially resolved. Therefore, our predictions of binding cGAMP, DMXAA, and PUFAs to FADS2 may be limited.
Identification of the Sting interactome was performed in murine cells. Proteomics analysis of human STING interactome could have strengthened the conservation of the FADS2-STING interaction in human cells.
The estimation of the Δ6D activity was performed using DGLA over LA ratios. However, this ratio also comprises an elongation step in addition to the desaturation step. To precisely calculate the activity, GLA over LA ratios should have been calculated. However, LC-MS analysis as performed in the present study did not allow detection of GLA. Adaptation of the LC-MS analysis could have allowed a more precise estimation.
The effect of ALA and FADS2 inhibition was monitored only in the acute phase of infection. The effects of such treatment, beyond the primary impact on STING-dependent interferon levels, may yield different results. Additionally, long-term treatment with Fads2 inhibitors may yield toxicity in vitro and in vivo.
Finally, the study could have benefited from analysis of a potential additive effect of FADS2 activation by STING degradation and direct FADS2 activation by STING agonists. A FADS2 mutant unresponsive to STING agonists would have answered this question. However, the interaction points between PUFAs and STING agonists and FADS2 overlap, precluding such analyses.
STAR★Methods
Key resources table
| REAGENT/ RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA | BioLegend | Cat# 901501; RRID:AB_2565006 |
| Flag M2 | Sigma Aldrich | #F1804-200UG; RRID:AB_262044 |
| HSP90 | Cell Signaling | #C45G5; RRID:AB_2233307 |
| GAPDH | Proteintech | 60004-1-Ig; RRID:AB_2107436 |
| FADS2 | Thermofischer | #PA587765; RRID:AB_2804393 |
| FADS1 | Proteintech | 10627-1-AP; RRID:AB_2231403 |
| Calnexin | Millipore | #AB2301; RRID:AB_10948000 |
| pIRF3 | Cell Signaling | #4947 (4D4G); RRID:AB_823547 |
| IRF3 | Cell Signaling | #11904 (D6I4C); RRID:AB_2722521 |
| IRF3 | Cell Signaling | #4302 (D83B9); RRID:AB_1904036 |
| pTBK1 | Cell Signaling | #5483 (D52C2); RRID:AB_10693472 |
| TBK1 | Cell Signaling | #3504 (D1B4); RRID:AB_2255663 |
| pSTING | Cell Signaling | #72971 (D8F4W); RRID:AB_2799831 |
| STING | Cell Signaling | #13647 (D2P2F); RRID:AB_2732796 |
| TREX-1 | Santa Cruz Biotechnology | sc133112; RRID:AB_2208802 |
| cGAS | Cell Signaling | #31659 (D3080); RRID:AB_2799008 |
| UCP1 | Sigma | U6382; RRID:AB_261838 |
| GFP | Abcam | ab290; RRID: AB_303395 |
| IFNAR | Abcam | ab124764; RRID:AB_10972855 |
| Horse anti-mouse IgG | Cell Signaling | #7076; RRID:AB_330924 |
| Mouse Anti-rabbit IgG | Cell Signaling | #7074; RRID:AB_2099233 |
| Mouse Anti-rabbit IgG (Conformation Specific) | Cell Signaling | #3678; RRID:AB_1549606 |
| Goat anti-Rabbit IgG (H+L), Alexa Fluor 488 | Thermoficher | #R37116; RRID:AB_2556544 |
| Goat anti-Mouse IgG (H+L) Alexa Fluor 594 | Thermoficher | #R37121; RRID:AB_2556549 |
| Donkey anti-human IgG coupled to AlexaFluor 647 | Invitrogen | #A-21447; RRID:AB_141844 |
| Bacterial and virus strains | ||
| HSV KOS-64-GFP strain | Gift from Soren.R. Paludan | N/A |
| Alpha (B.1.1.7) variant of SARS-CoV-2 | Olivier Schwartz | N/A |
| Beta (B.1.351) variant of SARS-CoV-2 | Olivier Schwartz | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | Lonza | BE12-614F |
| RPMI 1640 | GIBCO | 21875034 |
| OPTIMEM | GIBCO | 51985 |
| FBS | Eurobio | CVFSVF00-01 |
| Human serum | Sigma | H4522 |
| L-Glutamine | GIBCO | A2916801 |
| Penicillin-streptomycin | GIBCO | 10378016 |
| 0.25% trypsin EDTA | GIBCO | 25200-056 |
| Sc26196 | Santa Cruz Biotechnology | sc-361350 |
| DMXAA | Invivogen | Tlrl-dmx |
| polyIC | Invivogen | tlrl-pic |
| Linoleic acid | Sigma | L1376 |
| Linolenic acid | Sigma | L2376 |
| Oleic acid | Sigma | O7501 |
| DMSO | Sigma | D2650 |
| FFA free BSA | Sigma | A7030 |
| Red ANTI-FLAG M2 Affinity Gel | Sigma | F2426 |
| 3X Flag peptide | Sigma | F4799 |
| HA probe | Santa Cruz Biotechnology | Sc-7392 |
| HA peptide | Thermo Scientific | 26184 |
| Trizol | Thermofisher | 15596018 |
| super script IV | Thermofisher | 18090050 |
| TB Green Premix Ex Taq | TAKARA | RR420W |
| Rnase out | Thermofisher | 10777019 |
| dNTP mix 10mM | Thermofisher | 18427013 |
| Kit TURBO DNA-free | Thermofisher | AM1907 |
| protein assay dye reageant | Biorad | 5000006 |
| TWEEN 80 | Sigma | P4780-100ML |
| PEG300 | Sigma | 8074841000 |
| Jet Prime tranfection kit | Ozyme | POL114-75 |
| Accu-Chek PERFORMA | Roche | N/A |
| Rectal probe for mice | Kent Scientific | RET-3 |
| Insulin | Sigma | I9278 |
| Glucose | Sigma | G7021 |
| Pyruvate | Sigma | P2256 |
| Critical commercial assays | ||
| Mouse Insulin ELISA | ALPCO Diagnostics | 80-INSMSU-E01 |
| Silver Quest | Invitrogen | LC6070 |
| Trans-Blot Turbo Transfer Pack 0.2 μm Nitrocellulose Midi | Biorad | 1704159 |
| Novex WedgeWell 10%, Tris-Glycine | Thermofisher | XP00105BOX |
| Novex WedgeWell 12%, Tris-Glycine | Thermofisher | XP00125BOX |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermofisher | 34577 |
| GenElute Total RNA Purification Kit | Sigma | RNB200 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermofisher | 34094 |
| Deposited data | ||
| MS/MS Data | This paper | https://doi.org/10.17632/23drspzh3s.1 |
| Experimental models: Cell lines | ||
| MEF WT, Sting−/−& cGas−/− | Soren R. Paludan Lab at Aarhus University, Aarhus | N/A |
| MEF WT & Trex−/− | Jan Rehwinkel Lab at Oxford University, Oxford | N/A |
| Caco2 | Clone TC7; provided by Nolwenn Jouvenet’s lab at Institut Pasteur, Paris | RRID:CVCL_0025 |
| T98G | Caroline Goujon Lab at IRIM, Montpellier | RRID:CVCL_0556 |
| THP1 | ATCC | RRID:CVCL_0006 |
| Vero | Raphael Gaudin, IRIM,Montpellier | RRID:CVCL_0059 |
| 293T | ATCC | RRID:CVCL_0063 |
| Experimental models: Organisms/strains | ||
| Sting deficient mice | Lei Jin, Albany Mecical College, USA | Tmem173 < tm1Camb > |
| Transgenic FLP recombinase | PCEA, Montpellier, France | Tg(CAG-Flpo)1Afst |
| Transgenic LysM-Cre mice | Michael Hahne, IGMM,France | Lyz2tm1(cre)Ifo/J |
| Cgas deficient mice | EMMA consortium | Mb21d1tm1a(EUCOMM)Hmgu |
| Oligonucleotides | ||
| Primers for gene expression analysis, see Table S3 | N/A | N/A |
| Guide RNAs for the CRISPR-Cas9 system, see Table S3 | Gift from C. Goujon and B. Bonaventure, Montpellier, IRIM | N/A |
| Software and algorithms | ||
| Prism Software | GraphPad | Version 9.1 |
| FlowJo software | N/A | 10.7.1 version |
| ImageJ | N/A | N/A |
| CLAMS software | Oxymax | Columbus Instruments |
| Other | ||
| CHOW diet | Safe diet | A04 |
| HFD 60% diet | Safe diet | 292HF |
| OMEGA STD diet; See Table S2 | Safe diet | Fat controlled custom diet for Mice U8978 v209 |
| OMEGA3L diet; See Table S2 | Safe diet | Fat controlled custom diet for Mice U8978 v211 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dr. Nadine Laguette (nadine.laguette@igh.cnrs.fr).
Materials availability
Information and requests for resources and reagents should be directed to Dr. Nadine Laguette (nadine.laguette@igh.cnrs.fr).
Experimental model and subject details
Animals
Animal protocols were performed in accordance with French and European Animal Care Facility guidelines. All experiments were approved by the Animal Welfare and Ethical Review Body of Languedoc-Roussillon. Housing and experimental procedures were approved by the French Agriculture and Forestry Ministry (A34-172-13 & 15040-2018050214043878). Animal facility have a 12 h light-dark cycle and a controlled housing temperature at 22°C. Males from 8 to 14 weeks of age were used for this study. Mice were fed on maintenance chow diet (Safe diet, SAFE A04), except stated otherwise. Mice were bred and housed under a Specific Pathogen Free (SPF) status, with health status checks every three months. The majority of subjects from the study are the result of in-house mating and littermate controls were used. Five age-matched C57BL/6J WT male were imported into the facility to complete experimental groups for 18FGD ex vivo distribution analysis. Mice Sting deficient (Tmem173 < tm1Camb > ; under C57BL/6J background) were provided by Pr. Lei Jin (Jin et al., 2013). cGAS deficient mice line (Mb21d1tm1a(EUCOMM)Hmgu; under C57BL/6N background) was purchased from the EMMA consortium (Strasbourg, France). Tmem173 < tm1Camb > mice (conditional ready) were crossed with mice expressing FLP recombinase (Tg(CAG-Flpo)1Afst; under C57BL/6 background) to obtain Sting floxed mice. Specific Tmem173 knock out was obtained by crossing homozygous Tmem173 flox mice (Stingfl/fl) with transgenic LysM-Cre mice (Lyz2tm1(cre)Ifo/J; under C57BL/6 background; gift from Michael Hahne, IGMM, Montpellier) expressing the Cre recombinase under the control of a myeloid gene promoter.
Cells and cell cultures
THP-1 (sex: male), 293T (sex: unknown), T98G (sex: male), Caco2 (sex: male), Vero (sex: unknown), WT-MEF (sex: unknown), MEFSting−/− (sex: unknown), MEFcGas−/− (sex: unknown) were maintained in DMEM or RPMI supplemented with 10% Fetal Bovine Serum (FBS), 1% Penicillin/Streptomycin and 1% Glutamine. Cells were maintained at a temperature of 37°C under 5% of CO2. The HAQ-STING was a gift from J. Lei. StingΔTM was constructed by deleting the amino acids 1-149 of Sting. MEFSting−/− overexpressing FLAG- and HA-tagged Sting (F/HA-WT-Sting) or (F/HA-StingΔTM) were generated by transducing MEFSting−/− with retroviral particles packaging the pOZ-F/HA-Sting or pOZ-F/HA-StingΔTM construct and selection with puromycin. WT-MEF overexpressing FLAG-tagged Fads2 (Flag-Fads2) were generated by transducing WT-MEF with retroviral particles packaging the pOZ-Flag-Fads2 construct and selection with puromycin. The T98GIRF3−/−, T98GIFNAR1−/− and control knockout cell lines were generated using LenticrisprV2-GFP system (Plasmid 589 #82416) and cell sorting using a BD FACS Melody. Pooled cells were subsequently amplified and IRF3 and IFNAR1 invalidation verified by Western Blot.
Method details
Mouse studies
Body weight was measured at 8 weeks of age. Body composition (Fat and lean mass) was evaluated by quantitative nuclear magnetic resonance imaging (EchoMRI 3-in-1 system; Echo Medical Systems, Houston, Texas) before metabolic chambers experiments. Metabolic rates in mice were measured by indirect calorimetry using a Comprehensive Lab Animal Monitoring System (CLAMS, Columbus Instruments). Briefly, mice were housed individually in metabolic chambers with free access to water and food for 2 days for acclimatization before animals were returned to metabolic chambers and monitored for the next day for oxygen consumption (VO2), carbon dioxide production (VCO2), and food intake. Energy expenditure was calculated using the formula energy expenditure (EE) = (3.815 + 1.232 VO2/VCO2) × VO2, and normalized to lean body mass. For food intake study, mice were housed individually and after 24 h of acclimatization, food consumption was measured twice over 48 h. Body temperature was assessed in mice using a RET-3 rectal probe (Kent Scientific). ITTs and GTTs were performed as previously described (Vila et al., 2014). Briefly, mice were fasted for 6 h with free access to drinking water. For ITTs, insulin was administered intraperitoneally (0.4 mU/g of mice fed normal chow diet), and blood glucose was measured at various times after injection from the tip of the tail with a Glucometer (Accu-Chek, Roche). For GTTs, d-glucose was administered intraperitoneally (2 g/kg of mice for normal CHOW diet experiment or 1 g/kg for HFD experiment), and blood glucose levels were monitored. At 15 min after glucose injection, blood was collected for insulin quantitation. Serum insulin concentrations were determined by ELISA (Mouse Ultrasensitive Insulin ELISA, ALPCO Diagnostics). For PTT, mice were fasted for 18 h, followed by intraperitoneal injection of pyruvate (2 g/kg of mice), and blood glucose levels were monitored. To measure spontaneous locomotion and circadian behavior, mice were housed individually in cages equipped with a running wheel. Voluntary activity was measured as running wheel revolutions recorded in one-minute bins and analyzed with the ClockLab software (Actimetrics). Circadian behavior was accessed during a period of light/dark cycle and under constant darkness as previously described (Abitbol et al., 2017). HSV-1 brain infections in mice were conducted as previously described (Reinert et al., 2021). For DMXAA (Invivogen) treatment in vivo, mice were injected intraperitoneally at a dose of 10 mg/kg twice a week in 5%DMSO/30%PEG-300/1%Tween80 for 4 weeks. For Low Omega-3 diet (Safe diet) experiment, GTTs (2 g/kg of mice) were performed before and after 4 weeks of diet. Rectal temperature and mRNA levels analyzed was performed after 6 weeks of diet.
Measure of blood glucose decrease by [18F]-FDG quantification
Experiment was approved by the local Experimental Animal Ethics Committee of the BUC-CMMI (ref. CMMI-2018-01) and was conducted in compliance with the Belgian Royal Decree of 29 May 2013 on the protection of laboratory animals. 5 Sting−/− mice and 5 littermates wild-type (WT) mice were fasted for 15 h – 20 h. 0.0057-0.0281 μL of blood aliquots (based on the weight of the blood samples that were measured and converted in μL by a μg-μL calibration line) were then collected on the same mice before and after 10 min, 20 min, 30 min, 45 min and 1 h post-intravenous injection of 8.90-12.15 MBq of [18F]-FDG per mouse. Glycemia rates (mg/dL) were monitored on each blood sample. The radioactivity present in each blood sample was measured by gamma counting. Unweighted least-squares regression with automatic outlier removal was used to fit the FDG blood uptake data with a monoexponential curve. The y-intercept was constrained to a shared value for both datasets. An extra sum-of-squares F test was used to check whether the exponent and asymptotic value differ between WT and Sting−/− mice.
Ex vivo quantification of [18F]-FDG in adipose tissues
Ex vivo biodistribution studies were assessed 45 min after the injection of [18F]-FDG after the end of imaging. Subcutaneous fat, visceral fat and brown fat were recovered and weighed. The radioactivity present in each sample was measured by gamma counting. Ex vivo biodistribution values (in %ID/g) between the two groups were compared for each relevant organ or tissue by t tests; Grubbs’ test was performed to remove outlier’s values.
Gamma counting
Biological samples radioactivity counting was performed during 1 min per sample with a gamma counter calibrated for the [18F] measurement (Automatic Gamma Counter Wizard2 2480, Perkin Elmer, USA). Data were expressed in counts per minute (CPM) and corrected for the radioactive background and the [18F] decay. All values expressed in CPM were converted in Bq according to the CPM-Bq calibration of the gamma counter for [18F]. Results were expressed in %Injected Dose (ID)/organ and %Injected Dose (ID)/g organ.
Diets
At the age of 8 weeks mice were fed with HFD (60% energy as fat, Safe Diet). Low Omega-3 diet (Safe diet; Ω3/Ω6 = 1/43) or corresponding Standard diet (Safe diet; Ω3/Ω6 = 1/14) was given at libitum at the age of 8 weeks for 4 weeks.
RNA Extraction and Real-Time PCR
Total RNA from tissues was extracted with Trizol reagent (Invitrogen) and RNA extraction kit (Sigma). RNA was quantified with a Nanodrop spectrophotometer (ND-1000, Nanodrop Technologies). RNA (1-2 μg) was reverse transcribed using SuperScript IV reverse transcriptase (Invitrogen). Expression of specific mRNAs was determined with a LightCycler (Roche) using the SYBR green PCR master mix (Takara). Reactions were performed in duplicate, and relative amounts of cDNA were normalized to Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and/or heat shock protein 90 (Hsp90). RT-qPCR primer sequences are:
Human:
GAPDH: F-CTGGCGTCTTCACCACCATGG; R-CATCACGCCACAGTTTCCCGG;
IFNβ: F-GAATGGGAGGCTTGAATACTGCCT; R-TAGCAAAGATGTTCTGGAGCATCTC;
Mouse:
Gapdh: F-TTCACCACCATGGAGAAGGC; R-GGCATCGACTGTGGTCATGA;
Ifnβ: F-CTGCGTTCCTGCTGTGCTTCTCCA; R-TTCTCCGTCATCTCCATAGGGATC;
Cxcl10: F-ATGACGGGCCAGTGAGAATG; R-TCAACACGTGGGCAGGATAG;
Isg15: F-GTGCTCCAGGACGGTCTTAC; R-CTCGCTGCAGTTCTGTACCA;
Hsp90: F-GTCCGCCGTGTGTTCATCAT; R-GCACTTCTTGACGATGTTCTTGC;
Tnfα: F-CTGTAGCCCACGTCGTAGC; R-TTGAGATCCATGCCGTTG;
Il-6: F-GACTTCCATCCAGTTGCCTTCT; R-TCCTCTCCGGACTTGTGAAGTA
Ucp1: F-CCTGCCTCTCTCGGAAACAA;R-TGTAGGCTGCCCAATGAACA;
Pgc1α: F- AAAGGATGCGCTCTCGTTCA; R- GGAATATGGTGATCGGGAACA
Prdm16: F-CAGCACGGTGAAGCCATTC; R-GCGTCGATCCGCTTGTG
Cidea: F- ;R-TGCTCTTCTGTATCGCCCAGT; F-GCCGTGTTAAGGAATCTGCTG
Dio2: F-GCTTACGGGGTAGCCTTTGA; R-CCAGCCAACTTCGGACTTCT
Western blot analysis
Tissues were homogenized using a Fastprep apparatus (MP) in a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP40, 0.5% Sodium deoxycholate, 0.1% SDS, 10 μl/mL protease inhibitor, 10 μl/mL phosphatase I inhibitor, and 10 μl/mL phosphatase II inhibitor. Cells were lysed in 5 packed cell volume of TENTG-150 [20 mM tris-HCl (pH 7.4), 0.5 mM EDTA, 150 mM NaCl, 10 mM KCl, 0.5% Triton X-100, 1.5 mM MgCl2, and 10% glycerol, supplemented with 10 mM β-mercaptoethanol, 0.5 mM PMSF and 1x phosphatase inhibitor] for 30 min at 4°C. Tissue and cell lysates were centrifuged at 14,000 g for 30 min at 4°C, and supernatants were stored at −80°C. Solubilized proteins (20-30 μg) from tissues or cells were run on 10% or 12% SDS-PAGE gels (Invitrogen Novex Tris-glycine) transferred onto nitrocellulose membrane (Biorad Trans blot turbo) and incubated with primary antibodies. Primary antibodies used include: anti-phospho IRF3 (1:500; Cell Signaling 4D4G), anti-IRF3(1:1000; Cell Signaling D6I4C), anti-phospho TBK1 (1:1000; Cell Signaling D52C2), anti-TBK1 (1:1000; Cell Signaling D1B4), anti-STING (1:1000; Cell Signaling D2P2F), anti-phospho STING (1:1000; Cell Signaling D8F4W), anti-glyceraldehyde-phosphate dehydrogenase (GAPDH; 1:5000; Proteintech Europe 60004-1-Ig), anti-TREX-1 (1:250; Santa Cruz Biotechnology C-11 sc133112), mouse specific anti-cGAS (1:1000; Cell Signaling D3080), anti-Ucp1 (1:1000; Sigma U6382), anti-Flag (1:1000; Sigma F1804), anti-HA (1:1000; Biolegend 901501) anti-HSP90 (1:1000; Cell Signaling C45G5), Mouse Anti-rabbit IgG (1:1000; Cell Signaling 3678), anti-FADS1 (1:1000; Proteintech), and anti-FADS2 (1:10000; Invitrogen PA5-87765). All secondary antibodies (Cell Signaling) were used at 1:2000 dilution. Immunoreactive proteins were visualized by chemiluminescence (SuperSignal West Pico or femto Thermo Scientific).
Cell treatment and transfection
Cells were transfected with JetPrime transfection reagent (Polyplus) at 1:2 ratio with 2 μg/mL double strand DNA (dsDNA) or Poly(I:C). DMXAA (Invivogen) was used at 100 or 200 μM in Opti-MEM (GIBCO). Sc26196 (Santa Cruz) was used at 4 μM. Cells were treated with 50 or 100μM of ALA (Sigma, L2376) or LA (Sigma, L1376) in Opti-MEM containing 2% (w/v) fatty-acid-free bovine serum albumin (Sigma, A7030).
HSV-KOS64 amplification
The HSV KOS-64-GFP strain was a gift from S. Paludan. The virus was amplified in Vero cells. Briefly, Vero cells were plated in T175 and infected with HSV KOS-64-GFP during 30 min. Media was subsequently replaced and cells were collected 72 h post infection for viral extraction using 3 freeze-thaw cycles. Two centrifugations steps were performed and concentrated virus was resuspended before storage at −80°C.
HSV-KOS64 infection
104 cells were seeded per well in 96-well plates, for plaque number measurement, or 2x105 per well in 6-well plates, for gene expression or western blot. 24 h later cells were infected with HSV KOS-64-GFP for 90 min in presence or not of Fads2 inhibitor (4 μM), or in presence of not of ALA (50 μM). For plaque number assessment, medium was replaced with DMEM supplemented with 2% human serum. Sixteen h later, medium was replaced with DMEM supplemented with 10% FBS medium for additional 32 h. Cells were fixed with 4% paraformaldehyde (PFA) prior to staining with crystal violet. Plaque were counted and scored visually according to area size into 4 classes (from small: 1, to big: 4). For gene expression or western blot analysis, cells were infected with HSV KOS-64-GFP for 16 h prior cell harvest.
SARS-CoV-2 infection
Human colorectal adenocarcinoma cells Caco-2 (clone TC7; provided by Nolwenn Jouvenet’s lab at Institut Pasteur, Paris) were seeded and grown in Dulbecco’s Modified Eagle Medium (DMEM; GIBCO) supplemented with 10% FBS (GIBCO), 100 U/mL penicillin (GIBCO), 100 μg/mL streptomycin (GIBCO) and 10 mM HEPES buffer (Sigma). Cells were incubated in the presence of increasing doses of the sc26196 Fads2 inhibitor for 4 h at 37°C. Cells were then exposed to the Alpha (B.1.1.7) or Beta (B.1.351) variants of SARS-CoV-2 at MOI 0.01. Forty-eight h post-infection, cells were fixed in a 4% PFA solution and stained for the Spike protein (S) by using a human monoclonal anti-S (mAb102, cloned in the Hugo Mouquet’s lab from S-specific blood memory B cells of COVID-19 convalescent individuals) as a primary antibody and a donkey anti-human IgG coupled to AlexaFluor 647 (Invitrogen) as a secondary antibody. Data were acquired by flow cytometry using a Attune NxT (Thermo Fisher) flow cytometer, and analyzed using the FlowJo software (10.7.1 version).
Immunofluorescence
2x105 cells were seeded on glass coverslips in 6 well plates. Cells were treated prior to fixation with cold methanol. Cells were blocked with PBS supplemented with 5% BSA and 0.1% Tween for 30 min at room temperature. Coverslips were incubated with the primary antibodies in PBS containing 0.1% Tween (PBS-T) at 37°C for 45 min. Primary antibodies used are: anti-FADS2 (Thermofischer #PA587765) used at 1:20 dilution, anti-HA (BioLegend Cat# 901501) used at 1:50 dilution, and anti-Calnexin (Sigma Aldrich #AB2301) used at 1:250 dilution. Next, slides were incubated at 37°C with both Alexa Fluor 488 coupled goat anti-Rabbit IgG, (Thermofischer #R37116), and Alexa Fluor 594-coupled goat anti-Mouse IgG (Thermofischer #R37121). Cells were stained with DAPI and mounted in anti-fade Vectashield.
Immunoprecipitation and mass spectrometry analysis
MEFSting−/− overexpressing F/HA- Sting were lysed in 5 packed cell volume of TENTG-150. The first immunoprecipitation used an anti-FLAG antibody, followed by the elution using an excess of FLAG peptide (Sigma). Eluates were subsequently used as input material for immunoprecipitation using an anti-HA antibody. Sting protein partners were eluted using an excess of HA peptide (Thermo Scientific). Part of the FLAG- and HA-immunoprecipitated material was silver-stained and the remainder Coomassie-stained. Portions of the Coomassie-stained gel were excised and analyzed by Mass Spectrometry.
In vitro pull-down using biotinylated cGAMP
Pull-down was carried out using 30 μl (0.3mg) of MyOne Streptavidin C1 Dynabeads per condition. An excess of Biotin or cGAMP was coupled to beads according to the manufacturer’s instructions. 30 μl of Flag-immunoprecipitated Fads2 or Sting was incubated with beads, on ice for 30 min in low-binding tubes (Axygen). Three consecutive washes were performed in 20 mM tris-HCl (pH 7.4), 10 mM KCl, 0.5% Triton, 150 mM NaCl, 10% glycerol, 1.5 mM MgCl2 and 10 mM β-mercaptoethanol, and 0.5 mM PMSF. Tubes were changed at first and last washes. Bound material was eluted in 30 μl of Laemmli buffer.
RNA interference
shRNA targeting Fads2 (Clone ID: NM_019699.1-487s1c1) and scramble (SHC016) were purchased from Sigma-Aldrich. shRNA-expressing lentiviral particles were produced by co-transfection of 2 × 106 293T cells with 5 μg of shFads 2,5 μg of psPAX2 (Gag-Pol), and 1 μg of pMD2G (Env), using the standard calcium-phosphate transfection protocol. Viral particles were harvested 48 h after transfection, filtered with 0.45 μM filters, and used for transduction. For knockdown of Fads2, 106 MEF cells were seeded 24 h before transduction. Medium was replaced 10 h after transduction, and 1.5 μg/mL puromycin selection was performed 72 h later.
Measurement of PUFAs and oxilipins in biological samples
Snap-frozen tissues samples or 3 × 106 cells were crushed and solubilized (sonicated) in 1 mL of methanol (Wako, Tokyo, Japan). The samples were then spiked with following 16 internal standards (at 30 μM final concentration): tetranor-PGEM-d6, 6-keto-PGF1a-d4, TXB2-d4, PGF2a-d4, PGE2-d4, PGD2-d4, LTC4-d5, LTB4-d4, 15-HETE-d8, 12-HETE-d8, 5-HETE-d8, PAF-d4, OEA-d4, EPA-d5, DHA-d5, DHA-d5 and AA-d8 (all from Cayman Chemicals, Ann Arbor, MI, USA). The samples were then sonicated for 10 s, then vortexed for 2 min and further incubated for 2 h at 4°C. Next, the samples were centrifugated at 15.000 g for 10 min, the supernatant was removed and diluted with 4 mL of 0.1% formic acid. The mix was briefly vortexed and loaded on the solid phase extraction (SPE) column Strata-X (Phenomenex, Torrance, CA, USA) in 1 mL steps. Before loading the sample, the SPE column was washed with 1 mL methanol and equilibrated with 2 mL of 0.1% formic acid. Following sample loading, the SPE column was washed with 1 mL 0.1% formic acid, 1 mL 15% ethanol and the compounds were eluted in 250 μL methanol. The eluate was lyophilized and then solubilized in 20 μL methanol, of which 5 μL sample were injected on UPLC coupled to triple-quadrupole MS (LCMS-8050). The sample was separated using analytical column Kinetex C8 (Cat. # 00F-4497-AN; Phenomenex), mobile phase A 0.1% formic acid (Sigma Aldrich) in water and mobile phase B acetonitrile (Sigma Aldrich). The flow rate was set at 0.4 mL/min, column oven temperature at 40°C. The gradient was: 0 min 90% A, 5 min 75% A, 10 min 65% A, 20 min 25% A, 20.1 – 25 min 5% A, 25.1 min 90% A. MS setting, data acquisition and data analysis were performed according to manufacturer instructions for analyzing Lipid Mediators version 2.0 (Cat. # 225-24873A, Shimadzu).
LC-MS data analysis
Following visual inspection, integration and calculation of peak surface area for all identified compounds, the data were normalized using the internal standards. As outlined in the Lipid Mediators version 2.0 manual, individual compounds were repartitioned in 17 different groups, which were then normalized with 17 internal standards outlined above. Next, for each sample we calculated total metabolite load (TML), which was equal to the sum of individual metabolites measured in the respective sample. For TML calculation, missing values for individual metabolites were imputed by assigning the least possible peak area found in the dataset. The imputation was based on the hypothesis that as long as a given metabolite was present in some of the samples, its absence in other samples was due to the limit of detection of the analysis. All values for all metabolites within a sample were normalized with respect to TML. Once the normalized quantities were calculated, the data were imported in R (4.0.2) [R Core Team (2020)] and analyzed using the MetaboAnalyst package (4.0) (Chong et al., 2019).
Estimation of Fads1- and Fads2-associated enzymatic activities
Delta-5 desaturase (Δ5D) and D6D (Δ6D) activities, corresponding respectively to Fads1 and Fads2 activities, were estimated using product-to-precursor fatty acid ratios. Precisely, arachidonic acid (AA)/dihomogamma-linolenic acid (DGLA) was used to estimate Δ5D, and DGLA/linoleic acid (LA) for Δ6D.
Homology Modeling
The homology modeling of the FADS2 was performed using the Molecular Operating Environment (MOE) Suite (Warde-Farley et al., 2010). The 3OZZ RCSB entry was used as template, which is the crystal structure of the Bos taurus cytochrome b5 core-swap mutant. The 3D models were subsequently energetically optimized the AMBER10 forcefield as it is implemented in the MOE Suite. Finally, all 3D models were assessed for their folding via the protein and geometry check of MOE Suite.
Molecular docking
The docking module of MOE was used for the docking of the 6 PUFAs or cGAMP/DMXAA to STING and the FADS2 model. The 6 PUFAs that were used were namely: linoleic acid (LA), α-linolanic acid (ALA), dihomo-γ-linolenic acid (DGLA), arachidonic acid (AA), docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA). A fast Fourier transformation (FFT) pipeline is utilized by MOE for the docking experiment. The overall score is influenced by the model’s packing, electrostatic, solvation and hydrophobic energies. Transient complexes of proteins are kept in a local database and their contact propensities are statistically used for docking. The top hits of the docking experiment were energetically optimized using energy minimization pipelines to relieve the models from any residual geometrical strain. Finally, the Drugster suite was used to perform a final and rapid energy minimization step using AMBER99 forcefield (Vilar et al., 2008), while solvated using an implicit Generalized Born (GB) water model.
Molecular dynamics
The interaction pattern and overall fold of the final complexes of each one of the 6 PUFAs or cGAMP/DMXAA to either STING and FADS2 model, were subjected to exhaustive molecular dynamics simulations using the DrugOn suite. Molecular dynamics simulations were executed in an explicitly SPC water solvated periodic cube system. Counter-ions were used as required to neutralize the molecular system. Each biological system was subjected to a hundred nanoseconds (100 ns) of molecular dynamics at 300K and at 1 fs step size. The molecular trajectory of each simulation was then imported into a local database for further analysis (Vlachakis et al., 2013).
Quantification and statistical analysis
Image processing and quantification
Images for immunofluorescence experiment were acquired on the Zeiss apotome Microscope with a 63X oil objective. Co-localization analysis was performed using JACoP plugin in ImageJ.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism software. Prior to analyzing the statistical significance of differences between groups, we tested data distribution using a Shapiro-Wilk test. To compare data from two groups/conditions, a standard paired or unpaired two-tailed Student’s t test was performed. In vivo energy expenditure P value was determined by One-way Anova. For in vivo values, a Grubbs test was applied to determine outliers. Results were considered significant when p < 0.05. The number of replicates (including number of animals used in each experiment) are indicated in the figures and/or figure legends. All data are expressed as mean ± SEM. The statistical parameters can be found in the figures and the figure legends. p ≤ 0.05 was considered significant (∗), p ≤ 0.01 (∗∗), and p ≤ 0.001 (∗∗∗).
Acknowledgments
We thank M. Benkirane, G. Cavalli, J. Déjardin, and B. de Massy for discussions and comments. We thank C. Goujon and B. Bonaventure for CRISPR/Cas9 gRNA sequences. We acknowledge the SIRIC Montpellier Cancer grant (INCa_Inserm_DGOS_12553); Metamus-RAM and iExplore-RAM animal facilities; the Laboratoire de Mesures Physiques of the University of Montpellier for access to the MS instruments; the MRI imaging facility, member of the national infrastructure France-BioImaging infrastructure supported by the French National Research Agency (ANR-10-INBS-04, “Investments for the future”); and Ross Tomaino from the Taplin Mass Spectrometry Facility of Harvard Medical School for MS analysis. We thank T. Emilien, A. Sedda, C. de Maeseneire, N. Passon, and C. Van Heymbeek for their contribution. We thank the Cyclotron team from the Erasme Hospital, Brussels, Belgium, for the FDG provision. Work in N.L.’s laboratory is supported by the European Research Council (ERC-Stg CrIC: 637763, ERC-PoC DIM-CrIC: 893772), la Ligue pour la Recherche contre le Cancer, and the Agence Nationale de Recherche sur le Sida et les Hépatites Virales (ANRS: ECTZ117448). H.C. is supported by a PhD fellowship from la Ligue pour la Recherche contre le Cancer. C.T. is supported by the Merck Sharp and Dohme Avenir (MSD-Avenir – GnoSTic) program and an ANRS fellowship (ECTZ119088). J.M. is supported by a Conventions Industrielles de Formation par la Recherche (CIFRE) fellowship from the Agence Nationale de Recherche Technologie (ANRT). A.S. is supported by the ERC-PoC DIM-CrIC (893772). I.K.V. is supported by the ERC-Stg CrIC (637763) and the Fondation pour la Recherche Médicale (ARF20170938586). Work in S.R.P.’s laboratory is supported by the European Research Council (ERC-AdG ENVISION; 786602), the Novo Nordisk Foundation (NNF18OC0030274), and the Lundbeck Foundation (R198-2015-171 and R268-2016-3927). Work in A.T.’s laboratory is supported by a SIRIC Montpellier Cancer grant (INCa_Inserm_DGOS_12553), the Fondation de France (grant no. 00078461), and a LabEx MabImprove Starting Grant. X.B. is supported by ANR GH-gen (ANR-18-CE14-0017). The Center for Microscopy and Molecular Imaging (CMMI) is supported by the European Regional Development Fund (ERDF), the Walloon Region, the Fondation ULB, the Fonds Erasme, and Association Vinçotte Nuclear (AVN). G.D. is supported by the European Regional Development Fund (ERDF) and the Walloon Region. Work in O.S.’s lab is funded by Institut Pasteur, Urgence COVID-19 Fundraising Campaign of Institut Pasteur, ANRS, the Vaccine Research Institute (ANR-10-LABX-77), Labex IBEID (ANR-10-LABX-62-IBEID), ANR/FRM Flash Covid PROTEO-SARS-CoV-2, and IDISCOVR, Fondation pour la Recherche Médicale. Schematic representations were created with https://biorender.com/.
Author contributions
I.K.V. and N.L. conceived the study and designed experiments. I.K.V., H.C., A.S., M.S., C.T., L.S.R., E.T., S.H., J.M., X.B., D.V., A.T., M.H., G.D., and G.V.S. performed experiments. I.K.V., B.S., O.S., S.R.P., D.V., A.T., and N.L. supervised the study. L.J. provided mice and mice tissues samples. I.K.V., D.V., G.V.S., A.T., and N.L. analyzed data and prepared figures. I.K.V., D.V., A.T., and N.L. wrote the manuscript. All authors read and approved the final version of the manuscript.
Declaration of interests
J.M. is a joint PhD student in Azelead, a startup company, and the Laguette laboratory. All other authors declare no competing interests.
Published: January 4, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2021.12.007.
Contributor Information
Isabelle K. Vila, Email: isabelle.vila@igh.cnrs.fr.
Nadine Laguette, Email: nadine.laguette@igh.cnrs.fr.
Supplemental information
Data and code availability
-
•
Proteomics data are deposited in the Mendeley repository: https://doi.org/10.17632/23drspzh3s.1
-
•
No new code has been generated in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abitbol K., Debiesse S., Molino F., Mesirca P., Bidaud I., Minami Y., Mangoni M.E., Yagita K., Mollard P., Bonnefont X. Clock-dependent and system-driven oscillators interact in the suprachiasmatic nuclei to pace mammalian circadian rhythms. PLoS One. 2017;12:e0187001. doi: 10.1371/journal.pone.0187001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A., Goldeck M., Cavlar T., Deimling T., Witte G., Röhl I., Hopfner K.P., Ludwig J., Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A., Hemmerling I., Schmid-Burgk J.L., Behrendt R., Roers A., Hornung V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014;192:5993–5997. doi: 10.4049/jimmunol.1400737. [DOI] [PubMed] [Google Scholar]
- Bai J., Cervantes C., Liu J., He S., Zhou H., Zhang B., Cai H., Yin D., Hu D., Li Z., et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proc. Natl. Acad. Sci. USA. 2017;114:12196–12201. doi: 10.1073/pnas.1708744114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belchior T., Paschoal V.A., Magdalon J., Chimin P., Farias T.M., Chaves-Filho A.B., Gorjão R., St-Pierre P., Miyamoto S., Kang J.X., et al. Omega-3 fatty acids protect from diet-induced obesity, glucose intolerance, and adipose tissue inflammation through PPARγ-dependent and PPARγ-independent actions. Mol. Nutr. Food Res. 2015;59:957–967. doi: 10.1002/mnfr.201400914. [DOI] [PubMed] [Google Scholar]
- Berra A., Tau J., Zapata G., Chiaradia P. Effects of PUFAs in a mouse model of HSV-1 chorioretinitis. Ocul. Immunol. Inflamm. 2017;25:844–854. doi: 10.1080/09273948.2016.1184287. [DOI] [PubMed] [Google Scholar]
- Brestoff J.R., Artis D. Immune regulation of metabolic homeostasis in health and disease. Cell. 2015;161:146–160. doi: 10.1016/j.cell.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M.D., Sowell R.T., Kaech S.M., Pearce E.L. Metabolic instruction of immunity. Cell. 2017;169:570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerboni S., Jeremiah N., Gentili M., Gehrmann U., Conrad C., Stolzenberg M.C., Picard C., Neven B., Fischer A., Amigorena S., et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 2017;214:1769–1785. doi: 10.1084/jem.20161674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J., Wishart D.S., Xia J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinformatics. 2019;68:e86. doi: 10.1002/cpbi.86. [DOI] [PubMed] [Google Scholar]
- Clarke S.D., Jump D.B. Dietary polyunsaturated fatty acid regulation of gene transcription. Annu. Rev. Nutr. 1994;14:83–98. doi: 10.1146/annurev.nu.14.070194.000503. [DOI] [PubMed] [Google Scholar]
- Corrales L., Glickman L.H., McWhirter S.M., Kanne D.B., Sivick K.E., Katibah G.E., Woo S.R., Lemmens E., Banda T., Leong J.J., et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow Y.J., Casanova J.L. STING-associated vasculopathy with onset in infancy--a new interferonopathy. N. Engl. J. Med. 2014;371:568–571. doi: 10.1056/NEJMe1407246. [DOI] [PubMed] [Google Scholar]
- Derosa G., Cicero A.F., D’Angelo A., Borghi C., Maffioli P. Effects of n-3 PUFAs on fasting plasma glucose and insulin resistance in patients with impaired fasting glucose or impaired glucose tolerance. Biofactors. 2016;42:316–322. doi: 10.1002/biof.1277. [DOI] [PubMed] [Google Scholar]
- Diner E.J., Burdette D.L., Wilson S.C., Monroe K.M., Kellenberger C.A., Hyodo M., Hayakawa Y., Hammond M.C., Vance R.E. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNicolantonio J.J., O’Keefe J.H. Importance of maintaining a low omega-6/omega-3 ratio for reducing inflammation. Open Heart. 2018;5:e000946. doi: 10.1136/openhrt-2018-000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R., Koehler K., Chung S. Adaptive thermogenesis by dietary n-3 polyunsaturated fatty acids: emerging evidence and mechanisms. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids. 2019;1864:59–70. doi: 10.1016/j.bbalip.2018.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Galilea M., Félix-Soriano E., Colón-Mesa I., Escoté X., Moreno-Aliaga M.J. Omega-3 fatty acids as regulators of brown/beige adipose tissue: from mechanisms to therapeutic potential. J. Physiol. Biochem. 2020;76:251–267. doi: 10.1007/s13105-019-00720-5. [DOI] [PubMed] [Google Scholar]
- Gabbs M., Leng S., Devassy J.G., Monirujjaman M., Aukema H.M. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv. Nutr. 2015;6:513–540. doi: 10.3945/an.114.007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galland L. Diet and inflammation. Nutr. Clin. Pract. 2010;25:634–640. doi: 10.1177/0884533610385703. [DOI] [PubMed] [Google Scholar]
- Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- Gao D., Wu J., Wu Y.T., Du F., Aroh C., Yan N., Sun L., Chen Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P., Ascano M., Wu Y., Barchet W., Gaffney B.L., Zillinger T., Serganov A.A., Liu Y., Jones R.A., Hartmann G., et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandour R.A., Colson C., Giroud M., Maurer S., Rekima S., Ailhaud G., Klingenspor M., Amri E.Z., Pisani D.F. Impact of dietary ω3 polyunsaturated fatty acid supplementation on brown and brite adipocyte function. J. Lipid Res. 2018;59:452–461. doi: 10.1194/jlr.M081091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Périz A., Horrillo R., Ferré N., Gronert K., Dong B., Morán-Salvador E., Titos E., Martínez-Clemente M., López-Parra M., Arroyo V., Clària J. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 2009;23:1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag S.M., Gulen M.F., Reymond L., Gibelin A., Abrami L., Decout A., Heymann M., van der Goot F.G., Turcatti G., Behrendt R., Ablasser A. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559:269–273. doi: 10.1038/s41586-018-0287-8. [DOI] [PubMed] [Google Scholar]
- Hasan M., Gonugunta V.K., Dobbs N., Ali A., Palchik G., Calvaruso M.A., DeBerardinis R.J., Yan N. Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc. Natl. Acad. Sci. USA. 2017;114:746–751. doi: 10.1073/pnas.1611113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes J.K., Calder P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fatty Acids. 2018;132:41–48. doi: 10.1016/j.plefa.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M., Qin D., Zhao C., Chai L., Yu Z., Wang W., Tong L., Lv L., Wang Y., Rehwinkel J., et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020;21:727–735. doi: 10.1038/s41590-020-0699-0. [DOI] [PubMed] [Google Scholar]
- Jin L., Xu L.G., Yang I.V., Davidson E.J., Schwartz D.A., Wurfel M.M., Cambier J.C. Identification and characterization of a loss-of-function human MPYS variant. Genes Immun. 2011;12:263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L., Getahun A., Knowles H.M., Mogan J., Akerlund L.J., Packard T.A., Perraud A.L., Cambier J.C. STING/MPYS mediates host defense against Listeria monocytogenes infection by regulating Ly6C(hi) monocyte migration. J. Immunol. 2013;190:2835–2843. doi: 10.4049/jimmunol.1201788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T., Nakajima T., Yokoyama S., Asou H.K., Sasamoto S., Kamii Y., Hayashizaki K., Ouchi Y., Onodera T., Takahashi Y., et al. SCD2-mediated monounsaturated fatty acid metabolism regulates cGAS-STING-dependent type I IFN responses in CD4+ T cells. Commun. Biol. 2021;4:820. doi: 10.1038/s42003-021-02310-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King K.R., Aguirre A.D., Ye Y.X., Sun Y., Roh J.D., Ng R.P., Jr., Kohler R.H., Arlauckas S.P., Iwamoto Y., Savol A., et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017;23:1481–1487. doi: 10.1038/nm.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H., Konno K., Barber G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch P.J., Wilson S.C., Lee A.S., Berger J.M., Doudna J.A., Vance R.E. Ancient origin of cGAS-STING reveals mechanism of universal 2′,3′ cGAMP signaling. Mol. Cell. 2015;59:891–903. doi: 10.1016/j.molcel.2015.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausgruber T., Fortelny N., Fife-Gernedl V., Senekowitsch M., Schuster L.C., Lercher A., Nemc A., Schmidl C., Rendeiro A.F., Bergthaler A., Bock C. Structural cells are key regulators of organ-specific immune responses. Nature. 2020;583:296–302. doi: 10.1038/s41586-020-2424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari M., Wang X., Lantier L., Lyubetskaya A., Eguchi J., Kang S., Tenen D., Roh H.C., Kong X., Kazak L., et al. IRF3 promotes adipose inflammation and insulin resistance and represses browning. J. Clin. Invest. 2016;126:2839–2854. doi: 10.1172/JCI86080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Naour J., Zitvogel L., Galluzzi L., Vacchelli E., Kroemer G. Trial watch: STING agonists in cancer therapy. OncoImmunology. 2020;9:1777624. doi: 10.1080/2162402X.2020.1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard A.E., Kelder B., Bobik E.G., Chuang L.T., Parker-Barnes J.M., Thurmond J.M., Kroeger P.E., Kopchick J.J., Huang Y.S., Mukerji P. cDNA cloning and characterization of human Delta5-desaturase involved in the biosynthesis of arachidonic acid. Biochem. J. 2000;347:719–724. [PMC free article] [PubMed] [Google Scholar]
- Li C., Liu J., Hou W., Kang R., Tang D. STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusion. Front. Cell Dev. Biol. 2021;9:698679. doi: 10.3389/fcell.2021.698679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira F.S., Rosa Neto J.C., Antunes B.M., Fernandes R.A. The relationship between inflammation, dyslipidemia and physical exercise: from the epidemiological to molecular approach. Curr. Diabetes Rev. 2014;10:391–396. doi: 10.2174/1573399810666141122210135. [DOI] [PubMed] [Google Scholar]
- Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y.T., Grishin N.V., Chen Z.J. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- Morehouse B.R., Govande A.A., Millman A., Keszei A.F.A., Lowey B., Ofir G., Shao S., Sorek R., Kranzusch P.J. STING cyclic dinucleotide sensing originated in bacteria. Nature. 2020;586:429–433. doi: 10.1038/s41586-020-2719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M.T., Nara T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004;24:345–376. doi: 10.1146/annurev.nutr.24.121803.063211. [DOI] [PubMed] [Google Scholar]
- Park H.G., Park W.J., Kothapalli K.S., Brenna J.T. The fatty acid desaturase 2 (FADS2) gene product catalyzes Δ4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J. 2015;29:3911–3919. doi: 10.1096/fj.15-271783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston J.C., Matravadia S., Gaudio N., Holloway G.P., Mutch D.M. Polyunsaturated fatty acid regulation of adipocyte FADS1 and FADS2 expression and function. Obesity (Silver Spring) 2015;23:725–728. doi: 10.1002/oby.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinert L.S., Rashidi A.S., Tran D.N., Katzilieris-Petras G., Hvidt A.K., Gohr M., Fruhwürth S., Bodda C., Thomsen M.K., Vendelbo M.H., et al. Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production. J. Clin. Invest. 2021;131:e136824. doi: 10.1172/JCI136824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P., Kajimura S., Yang W., Chin S., Rohas L.M., Uldry M., Tavernier G., Langin D., Spiegelman B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T., Rodriguez B.L., Chen L., Corte C.M.D., Morikawa N., Fujimoto J., Cristea S., Nguyen T., Diao L., Li L., et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9:646–661. doi: 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang G., Zhang C., Chen Z.J., Bai X.C., Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. 2019;567:389–393. doi: 10.1038/s41586-019-0998-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirtori C.R., Galli C. N-3 fatty acids and diabetes. Biomed. Pharmacother. 2002;56:397–406. doi: 10.1016/s0753-3322(02)00255-x. [DOI] [PubMed] [Google Scholar]
- Sun W., Li Y., Chen L., Chen H., You F., Zhou X., Zhou Y., Zhai Z., Chen D., Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. USA. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang P., Virtue S., Goie J.Y.G., Png C.W., Guo J., Li Y., Jiao H., Chua Y.L., Campbell M., Moreno-Navarrete J.M., et al. Regulation of adipogenic differentiation and adipose tissue inflammation by interferon regulatory factor 3. Cell Death Differ. 2021;28:3022–3035. doi: 10.1038/s41418-021-00798-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M.K., Nandakumar R., Stadler D., Malo A., Valls R.M., Wang F., Reinert L.S., Dagnaes-Hansen F., Hollensen A.K., Mikkelsen J.G., et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology. 2016;64:746–759. doi: 10.1002/hep.28685. [DOI] [PubMed] [Google Scholar]
- Tosi F., Sartori F., Guarini P., Olivieri O., Martinelli N. Delta-5 and delta-6 desaturases: crucial enzymes in polyunsaturated fatty acid-related pathways with pleiotropic influences in health and disease. Adv. Exp. Med. Biol. 2014;824:61–81. doi: 10.1007/978-3-319-07320-0_7. [DOI] [PubMed] [Google Scholar]
- Vila I.K., Badin P.M., Marques M.A., Monbrun L., Lefort C., Mir L., Louche K., Bourlier V., Roussel B., Gui P., et al. Immune cell Toll-like receptor 4 mediates the development of obesity- and endotoxemia-associated adipose tissue fibrosis. Cell Rep. 2014;7:1116–1129. doi: 10.1016/j.celrep.2014.03.062. [DOI] [PubMed] [Google Scholar]
- Vilar S., Cozza G., Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008;8:1555–1572. doi: 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Vlachakis D., Tsagrasoulis D., Megalooikonomou V., Kossida S. Introducing Drugster: a comprehensive and fully integrated drug design, lead and structure optimization toolkit. Bioinformatics. 2013;29:126–128. doi: 10.1093/bioinformatics/bts637. [DOI] [PubMed] [Google Scholar]
- Walker M.M., Kim S., Crisler W.J., Nguyen K., Lenz L.L., Cambier J.C., Getahun A. Selective loss of responsiveness to exogenous but not endogenous cyclic-dinucleotides in mice expressing STING-R231H. Front. Immunol. 2020;11:238. doi: 10.3389/fimmu.2020.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warde-Farley D., Donaldson S.L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C.T., et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214-20. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.J., Zhao L., Hu H.G., Li W.H., Li Y.M. Agonists and inhibitors of the STING pathway: potential agents for immunotherapy. Med. Res. Rev. 2020;40:1117–1141. doi: 10.1002/med.21649. [DOI] [PubMed] [Google Scholar]
- Yamane D., Hayashi Y., Matsumoto M., Nakanishi H., Imagawa H., Kohara M., Lemon S.M., Ichi I. FADS2-dependent fatty acid desaturation dictates cellular sensitivity to ferroptosis and permissiveness for hepatitis C virus replication. Cell Chem. Biol. 2021 doi: 10.1016/j.chembiol.2021.07.022. Published online September 11, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Shi H., Wu J., Zhang X., Sun L., Chen C., Chen Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Hu Z., Wang K., Yang L., Li Y., Schlüter H., Yang P., Hong J., Yu H. Lipidomic profiling of virus infection identifies mediators that resolve herpes simplex virus-induced corneal inflammatory lesions. Analyst (Lond.) 2020;145:3967–3976. doi: 10.1039/d0an00263a. [DOI] [PubMed] [Google Scholar]
- Zhao P., Wong K.I., Sun X., Reilly S.M., Uhm M., Liao Z., Skorobogatko Y., Saltiel A.R. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018;172:731–743.e12. doi: 10.1016/j.cell.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., An X., Zhang X., Qiao Y., Zheng T., Li X. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer. 2019;18:152. doi: 10.1186/s12943-019-1087-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Proteomics data are deposited in the Mendeley repository: https://doi.org/10.17632/23drspzh3s.1
-
•
No new code has been generated in this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.