

Abstract
Triple-negative breast cancer (TNBC) lacks significant expression of the estrogen receptor, the progesterone receptor, and of human epidermal growth factor receptor. It is the most aggressive and malignant of all breast cancers, and for which, there are currently no effective targeted therapies. We have shown previously that the RecQ helicase family member RECQL5 is essential for the proliferation and survival of TNBC cells; however, the mechanism of its involvement in cell viability has not been shown. Here, we report that the expression of RecQ family helicases, including RECQL5, is regulated by the deubiquitinase USP28. We found using genetic depletion or a small molecule inhibitor that like RECQL5, USP28 is also essential for TNBC cells to proliferate in vitro and in vivo. Compromising the function of USP28 by shRNA knockdown or the inhibitor caused TNBC cells to arrest in S/G2 phases, concurrent with DNA-damage checkpoint activation. We further showed that the small molecule inhibitor of USP28 displayed anti-tumor activity against xenografts derived from TNBC cells. Our results suggest that USP28 could be a potential therapeutic target for triple negative breast cancer.
Keywords: USP28, triple-negative breast cancer, deubiquitinase, RecQ family helicases, RECQL5
Abbreviations: BLM, bloom syndrome gene product; DUB, deubiquitin enzyme; TNBC, triple-negative breast cancer; USP, Ubiquitin-specific cysteine protease; WRN, werner syndrome gene product
Triple-negative breast cancer (TNBC) lacks significant expression of the estrogen receptor (ER), the progesterone receptor (PR), and of human epidermal growth factor receptor 2 (1). Although it is only 10 to 20% of all breast cancers, TNBC is more aggressive and poorer in prognosis than other breast malignancies. Gene expression profiling of TNBC showed enrichment of genes involved in DNA damage checkpoint response (2, 3), which is consistent with the finding of higher levels of endogenous DNA damage in TNBC cells in vitro and in vivo (4, 5). Moreover, about 20% of the TNBC patients carry mutation in BRCA1/2 (6, 7). Hormonal and targeted therapies are available for ER positive and human epidermal growth factor receptor 2 positive breast cancers, whereas triple TNBC currently lacks targeted therapies (except BRCA1/2 mutated ones) and relies on chemotherapy (8).
USP28 is a ubiquitin-specific cysteine protease (USP) and has been shown to play an important role in maintaining the stability of a large number of proteins including c-MYC (9), LSD1 (10), HIF1alpha (11), Notch1 (12), 53BP1 (13), and CLASPIN (14). Almost all these substrates, especially c-MYC, have been demonstrated to contribute significantly to the initiation and progression of various types of cancers. Consistent with that, Usp28 was shown to help the tumorigenesis of colorectal cancer in mice (12). More recently, USP28 was shown to be required by squamous cell carcinoma due to its function in maintaining the stability of Np63 (15).
Here, we report that USP28 is required for the viability of TNBC cells, and this requirement is not only associated with its function in maintaining the stability of c-MYC protein but more importantly with its role in protecting RECQL5 and other members of the RecQ family helicases from proteasome-mediated degradation. The RecQ family helicases are required for TNBC cells to deal with the elevated levels of endogenous DNA damage resulted from replication stress (5, 16). As a result, compromising the function of USP28 causes TNBC cells to arrest in S/G2 phases with DNA damage checkpoint activation. We further showed that a small molecule inhibitor of USP28 displayed antitumor activity against xenografts derived from TNBC cells. Our results point USP28 as a therapeutic target for TNBC.
Results
USP28 regulates the stability of RECQL5
We showed previously that the function of RECQL5 was critical in TNBC cells (5). To search for its regulators, we found that USP28 played a role in regulating its stability. As shown in Figure 1A, depletion of USP28 led to a significant decrease in the levels of RECQL5 in TNBC (MDA-MB 231), non-TNBC (T47D), and 293T cells. Furthermore, the other members of the RecQ helicase family, namely the Bloom syndrome gene product (BLM), the Werner syndrome gene product (WRN), and RECQL4, all depended on the function of USP28 to maintain their levels (Fig. 1B). The only exception is RECQL1, which seemed unaffected by USP28 status. To determine if USP28 regulates RECQL5 stability, we first measured the half-life of the helicase in control and USP28-depleted cells. As shown in Figure 1, C and D, depletion of USP28 greatly reduced the half-life of RECQL5. On the other hand, the overexpression of USP28 made the helicase much more stable (Fig. 1, E and F). As expected, overexpression of a protease-dead version of USP28 (C171A) could not stabilize RECQL5 (Fig. 1, E and F).
Figure 1.

USP28 regulates RecQ helicases.A, immunoblot analysis of RECQL5 in MDA-MB-231, T47D, and 293T cells depleted of USP28. B, immunoblot analysis of RecQ family helicases in MDA-MB-231 cells depleted of USP28. C, analysis of RECQL5 stability in a cycloheximide chase experiment. 293T cells were transfected with Flag-RECQL5 first and then depleted of USP28 expression, treated with 100 μg/ml cycloheximide (CHX), and harvested at different time points for analysis. D, quantification of Flag-RECQL5 levels normalized to ACTIN in (C). E, overexpression of USP28 stabilizes RECQL5. 293T cells with USP28 or USP28C171A overexpression were subjected to the same CHX chase experiment as in (C) except the endogenous RECQL5 was examined. F, quantification of RECQL5 levels normalized to Tubulin in (E). BLM, bloom syndrome gene product; USP, Ubiquitin-specific cysteine protease; WRN, werner syndrome gene product.
USP28 interacts with and deubiquitinates RECQL5
The results above suggest that USP28 is a deubiquitin enzyme (DUB) for RECQL5. To demonstrate that, we first set out to determine if the two proteins interact with each other. As shown in Figure 2A, immunoprecipitating RECQL5 could bring down USP28 and vice versa. Using deletion constructs (Fig. S1, A–D), we found that RECQL5 interacts via the domain between Zn2+ binding site and KIX with USP28, whereas the C-terminal domain of USP28 is required for its interaction with RECQL5.
Figure 2.

USP28 is a DUB for RECQL5.A, USP28 interacts with RECQL5. 293T cells were transfected with Flag-RECQL5 and the cell lysates were prepared. Flag-RECQL5 was immunoprecipitated with FLAG-M2 beads and USP28 with anti-USP28 antibodies from the cell lysates. The precipitates were then subjected to immunoblotting analysis. B, depleting USP28 expression enhances RECQL5 ubiquitination. 293T cells with ectopic expression of Flag-RECQL5 were transduced with shUSP28 or control shRNA, treated with 10 μM MG132 for 8 h, and immunoprecipitated with FLAG-M2 beads. The precipitates were then subjected to immunoblotting analysis with antibodies against ubiquitin, Flag, and USP28. C, overexpression of USP28 reduces RECQL5 ubiquitination. 293T cells were transfected with Flag-RECQL5, HA-ubiquitin (HA-Ub), and USP28 WT or USP28 C171A, treated with 10 μM MG132 for 8 h, and immunoprecipitated with FLAG-M2 beads. The precipitates were then subjected to immunoblotting analysis with antibodies against HA, Flag, and USP28. USP, Ubiquitin-specific cysteine protease.
Next, we looked at the levels of ubiquitin attached to RECQL5 to see if they were affected by USP28 status. Flag-tagged RECQL5 and shRNA against USP28 were expressed in 293T cells, and the cells were then treated with and without MG132 to block proteasome activities. The immunoprecipitates obtained with anti-Flag antibodies from these cells under denaturing conditions were separated in a SDS-gel and blotted for detection of ubiquitin. It is clear that the depletion of USP28 resulted in much higher levels of ubiquitin associated with RECQL5, when the cells were treated with MG132 (Fig. 2B). Otherwise, the ubiquitinated RECQL5 was degraded.
Opposite to depleting USP28, the overexpression resulted in a decrease of ubiquitination of RECQL5 (Fig. 2C), and as expected, overexpression of USP28 C171A did not cause such a decrease (Fig. 2C). Taken together, these results demonstrate that USP28 is the DUB that prevents excess ubiquitination and degradation of RECQL5 and likely other RecQ helicases as well.
USP28 is required for the proliferation of triple-negative breast cancer cells
To determine whether USP28 is like RECQL5 also critical for the proliferation of TNBC cells, we depleted USP28 expression in a number of TNBC cell lines. Indeed, knocking down the expression of USP28 could block the proliferation of MDA-MD-231, HCC1937, and HCC1806 (Fig. 3A) as well as MDA-MB-468, SUM159, and HCC38 (Fig. S2, A–C), but could block neither the proliferation of MCF7 (Fig. S2D) nor that of MCF10A cells (Fig. S2E). To rule out possible off-target effects of the shRNAs used, we overexpressed USP28 in SUM159 cells (Fig. 3B) after shRNA expression, and after that, we performed colony formation assays. As shown in Figure 3C, the reexpression of USP28 could rescue the growth inhibition effects caused by the shRNAs. Further, we synthesized the reported USP28 inhibitor AZ1 (17) and used it to treat TNBC cells. In HCC1806 cells, a 48-h AZ1 treatment resulted in a dose-dependent suppression of proliferation (Fig. 3, D–F), and similar results were obtained with MDA-MB-231 and HCC1937 (Fig. 3F).
Figure 3.

USP28 sustains the proliferation of TNBC cells.A, growth Analysis of MDA-MB-231, HCC1806, and HCC1937 cells depleted of USP28 expression via two independent shRNAs targeting the 3′-UTR region. B, immunoblot analysis of USP28 levels in SUM159 cells with USP28 overexpression first and then depleted of endogenous USP28 expression via the same shRNAs in (A). C, colony formation assay of the SUM159 cells from (B). D, representative images and (E) quantification of HCC1806 cells treated with 0, 10, 15, or 20 μM AZ1 for 48 h. The scale bar represents 400 μm. F, cell viability assay of MDA-MB-231, HCC1937, and HCC1806 upon treatment with 0, 10, 15, 20, 25, or 30 μM AZ1 for 48 h. TNBC, triple-negative breast cancer; USP, Ubiquitin-specific cysteine protease.
To demonstrate that USP28 is required for the proliferation of TNBC cells in vivo, we depleted its expression in HCC1806 cells via the two shRNAs above and inoculated the cells in female nude mice. As expected, these cells could not grow as xenografts (Fig. 4A). Further, we used AZ1 to treat mice carrying HCC1806 xenografts. As an USP28 inhibitor, AZ1 showed anti-TNBC tumor activities (Fig. 4B). These results demonstrate that UPS28 is required by the TNBC cells for proliferation in vitro and in vivo.
Figure 4.

Targeting USP28 impedes the growth of HCC1806 xenografts.A, HCC1806 cells infected with shNC or shUSP28 lentiviruses were innoculated subcutaneously into female Balb/c nude mice. The tumors were collected 3 weeks after the inoculation. The tumor weight and size were measured. The data are mean ± SD (n = 5 per group). Student’s t test: ∗∗, p < 0.01; ∗∗∗, p < 0.001. B, HCC1806 cells were innoculated subcutaneously into female Balb/c nude mice. When the average tumor size reached 100 mm3, the mice were divided randomly into two groups (n = 7 per group). One group were treated with vehicle, another with AZ1 (200 mg/kg, oral gavage) every day. The tumor sizes were measured every 3 days, and the tumors were harvested 12 days after the treatment. The data are mean ± SD. Student’s t test: ∗, p < 0.05. USP, Ubiquitin-specific cysteine protease.
The requirement of USP28 in TNBC cells could not be bypassed with MYC overexpression
USP28 was shown previously to regulate the stability of c-MYC protein (9). We therefore set out to determine if it does so in TNBC cells as well. As shown in Figure 5A, depleting USP28 did cause decreases in c-MYC levels in TNBC cells. However, c-MYC levels did not decline in MCF7 cells (Fig. 5A), which is consistent with the finding that USP28 loss of function through shRNA-mediated depletion had no effect on their proliferation (Fig. S2D). Thus, the decreases in c-MYC levels resulted from the depletion of USP28 might underlie the requirement of the DUB in these cells, and the regulation on RecQ helicases might not be as important in terms of TNBC cell proliferation. To determine if that was the case, we first analyzed the cell cycle distribution in the USP28 depleted cells. Given the function of c-MYC in promoting G1/S transition, we expected that the USP28-depleted TNBC cells would show a G1 arrest. However, we instead saw an increase in G2/M fraction, whereas MCF7 cells did not show any cell cycle disturbance (Fig. 5B). In both MDA-MB-231 and HCC1937 cells, S phase fraction also increased upon USP28 depletion (Fig. 5B). To show the cell cycle disturbance further, we synchronized USP28-depleted MDA-MB-231 cells with single thymidine treatment and then released them into fresh media. As shown in Figure S3, the control cells had returned to G1 after 18 h of release, whereas the USP28-depleted cells were still stuck at G2/M.
Figure 5.

USP28 depletion destabilizes c-MYC and induces cell cycle arrest in TNBC cells.A, immunoblot analysis of c-MYC in MDA-MB-231, HCC1806, HCC1937, and MCF7 cells infected with lentiviruses expressing control or USP28 shRNAs. B, cell cycle distribution in MDA-MB-231, HCC1937, and MCF7 cells with or without USP28 depletion. The percentage of the cells in each cell cycle phase were calculated with ModFit LT software. C, overexpression of c-MYC-T58A fails to rescue the growth inhibition induced by USP28 depletion in MDA-MB-231 cells. The expression of c-MYC (endogenous and exogenous T58A mutant) as well as USP28 was analyzed with immunoblotting, and the cells were subjected to growth curve analysis. TNBC, triple-negative breast cancer; USP, Ubiquitin-specific cysteine protease.
The results above suggest that USP28 regulate more than c-MYC in TNBC cells to help their proliferation. To demonstrate that, we next sought to determine if reexpressing c-MYC could rescue USP28 depletion. However, we were unable to express WT c-MYC in USP28-depleted cells, as the WT c-MYC protein relies on USP28 for stability. To circumvent that, we expressed c-MYCT58A, a mutant form resistant to ubiquitination catalyzed by FBXW7 (18, 19). c-MYCT58A was overexpressed in MDA-MB-231 cells first, and USP28 expression was subsequently depleted via shRNA (Fig. 5C). Cell proliferation assay on these cells demonstrated that restoring c-MYC expression could not prevent these cells from losing their ability to proliferate because of USP28 depletion (Fig. 5C). However, the exogenously expressed c-MYCT58A did not cause an increase in the rate of proliferation neither (Fig. 5C), which made us wonder whether the c-MYCT58A expressed was functional. To rule that out, we expressed the same c-MYCT58A in human normal diploid fibroblasts IMR90. Both WT c-MYC and c-MYCT58A could enhance the proliferation of IMR90 cells, and c-MYCT58A showed a stronger effect than the WT (Fig. S4, A and B). Thus, the c-MYCT58A expression construct was functional. It is likely that MDA-MB-231 cells express already high enough levels of c-MYC, and thus further increasing its expression levels would not be able to further stimulate proliferation. Taken together, these results indicate that the reduction in c-MYC stability resulted from USP28 depletion is not the whole reason for TNBC cells to cease proliferation, and USP28-mediated stabilization of RecQ helicases may just be as important as c-MYC.
Increased levels of endogenous DNA damage in USP28-compromised TNBC cells
Compromising RECQL5 in TNBC cells causes increases in the levels of DNA damage (5). We therefore decided to examine DNA damage levels in USP28-depleted TNBC cells. Staining of γH2AX showed that, indeed, the endogenous DNA damage levels increased significantly from 30% in the control to more than 60% of γH2AX positive cells when USP28 was depleted (Fig. 6A). Consistent with that, the level of phosphorylated CHK1 also increased significantly (Fig. 6B), indicating the activation of DNA damage checkpoint. USP28 was reported to regulate the stability of CLASPIN (14), which is involved in DNA damage response in S phase. However, the levels of CLASPIN did not change upon USP28 depletion (Fig. 6C), indicating that USP28 does not regulate CLASPIN in TNBC cells. In addition, AZ1 treatment recapitulated the effects of USP28 depletion on RecQ family helicases and the activation of CHK1 (Fig. 6D).
Figure 6.
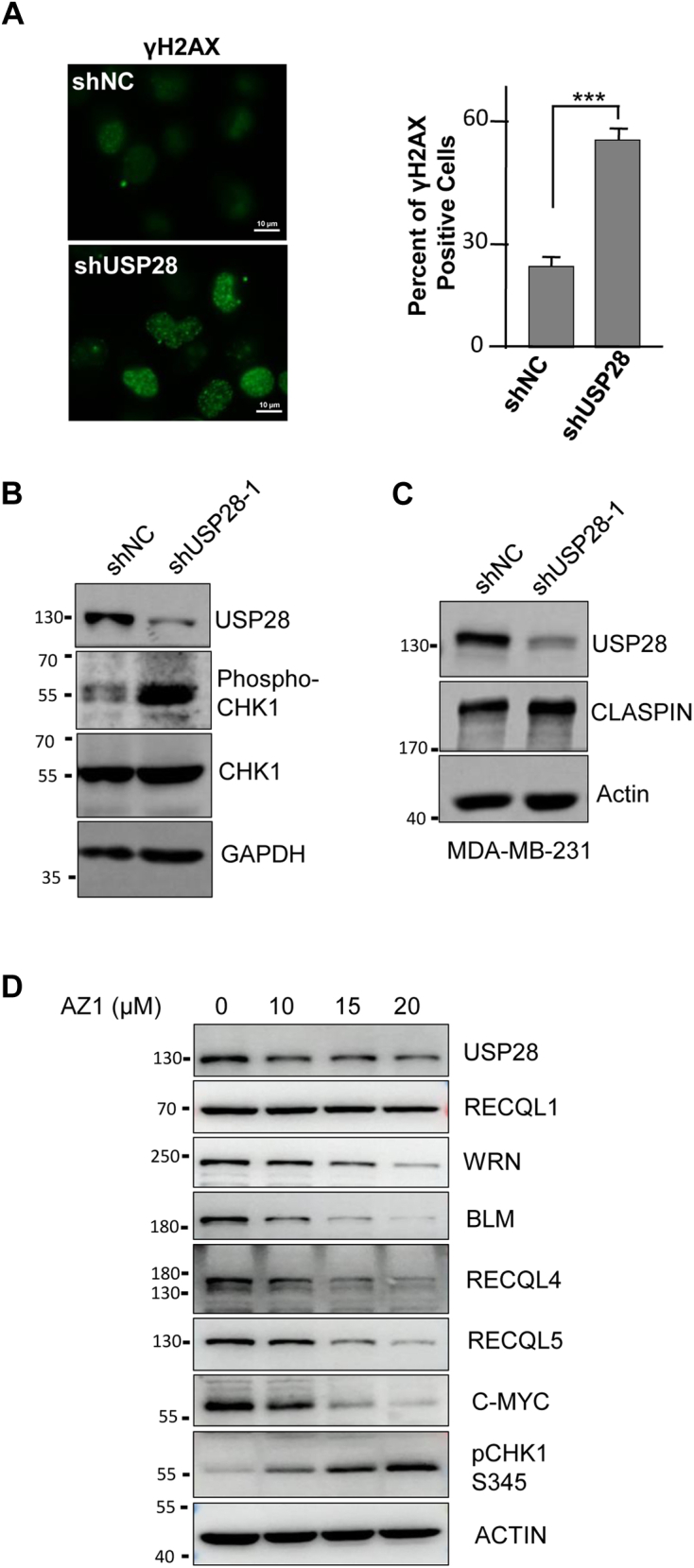
USP28 inhibition induces accumulation of DNA damage and activation of DNA damage checkpoint in TNBC cells.A, immunofluorescence staining and quantification of DNA damage marker γH2AX foci in USP28-depleted MDA-MB-231 cells. Student’s t test: ∗∗∗, p < 0.001. The scale bar represents 10 μm. B, immunoblot analysis of CHK1 in MDA-MB-231 cells depleted of USP28. C, immunoblot analysis of CLASPIN in MDA-MB-231 cells depleted of USP28. D, immunoblot analysis of relative proteins in HCC1806 cells treated with USP28 inhibitor AZ1 for 48 h. BLM, bloom syndrome gene product; TNBC, triple-negative breast cancer; USP, Ubiquitin-specific cysteine protease; WRN, werner syndrome gene product
Thus, much like compromising RECQL5 (5), compromising USP28 also led to increased DNA damage, checkpoint activation, and G2/M arrest.
Other members of the RecQ family helicases are also required for the proliferation of TNBC cells
Given the replication stress that TNBC cells face and the function of RecQ helicase in DNA metabolism, it is highly likely that like RECQL5, the rest four helicases are also essential in TNBC cells. To determine if that is the case, we depleted BLM, WRN, RECQL1, and RECQL4 expression (Fig. S5A) with two different shRNAs in TNBC cells MDA-MB-231 (Fig. 7A), HCC1806 (Fig. S5B), and HCC1937 (Fig. S5C). All three TNBC cell lines ceased proliferating upon knocking down each one of the RecQ family helicases. However, depleting these RecQ helicases in U2OS cells had little impact on their proliferation, except RECQL4, where its depletion caused some slowdown but certainly not cessation of proliferation (Fig. S5, D and E). Further, the overexpression of RecQL5 could not rescue USP28-depletion in TNBC (HCC1806) cells (Fig. S6, A and B). This result indicates that the four RecQ helicases regulated by USP28 are all important for the proliferation of TNBC cells, not just RECQL5.
Figure 7.

RECQ family helicases are required for the proliferation of TNBC cells.A, the growth curve analysis of MDA-MB-231 cells depleted of RECQ helicases. Each helicase was knocked down via two independent shRNAs. B, a schematic model illustrating the function of USP28 in helping replication stress relief in TNBC cells through RecQ family helicases, thereby maintaining the viability of TNBC cells. BLM, bloom syndrome gene product; TNBC, triple-negative breast cancer; USP, Ubiquitin-specific cysteine protease; WRN, werner syndrome gene product.
Discussion
TNBC is the more aggressive form amid all breast cancers and currently lacks effective therapies. We have shown previously that TNBC cells suffer unusually high levels of replication stress that require the function of RECQL5 to relieve (5). To look for the regulators of RECQL5, we identified USP28. We show here that USP28 is a DUB for RECQL5, and it regulates not only RECQL5, but also BLM, WRN, and RECQL4 (Fig. 1B). Like RECQL5, compromising the function of USP28 through shRNA-mediated depletion of expression or through small molecule inhibition also blocked the proliferation of TNBC cells in vitro and in vivo, which might reflect its regulation on RecQ helicases. However, the best-known substrate of USP28 is c-MYC (9), and c-MYC is well-known to be essential in promoting proliferation. Therefore, we reexpressed c-MYC in USP28-depleted TNBC cells to see if that could rescue the growth of these cells. Not surprisingly, it could not (Fig. 5C).
The depletion of USP28 actually led to more DNA damage in TNBC cells (Fig. 6A), despite the fact that DUB has been shown not to play any significant roles in DNA damage responses (13). In addition, we did not see destabilization of CLASPIN upon USP28 depletion in TNBC cells but did observe enhanced activation of CHK1, indicating DNA damage checkpoint activation. Consistent with that, depleting USP28 led to G2/M as well as intra-S phase arrests in TNBC cells (Fig. 5B). These results are very similar to those caused by depleting RECQL5 (5). Thus, stabilizing RECQL5 and the other RecQ helicases by USP28 is critical for TNBC cells to deal with their elevated levels of replication stress and thus their survival (Fig. 7B). However, one must bear in mind that USP28 regulates a large number of proteins (including c-MYC) implicated in the proliferation and survival of tumor cells, and the USP28-RecQ axis is only part of the action. Nonetheless, our results suggest that USP28 is a therapeutic target for triple negative cancer.
Experimental procedures
Cell culture
MDA-MB-231, MDA-MB-468, HCC1806, HCC38, SUM159, T47D, MCF7, MCF10A, and IMR90 cell lines were purchased from American Type Culture Collection. HCC1937 and 293T cells were obtained from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences. The above cells were cultured in DMEM, RPMI-1640, or F12 Medium with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37 °C. Dulbecco’s modified Eagle’s medium (DMEM), RPMI-1640 Medium, F12 Medium, and fetal bovine serum were purchased from Gibco, and Thymidine was purchased from Selleck.
Plasmids and lentiviruses
Plasmids used in the work were generated through standard cloning methods. shRNAs were constructed in pLKO.1 with following sequences:
| Gene | Sequence |
|---|---|
| Negative control | 5′-TTCTCCGAACGTGTCACGT-3′ |
| shUSP28-1 | 5′-GCACAGAAGTTCGTTGTCATA-3′ |
| shUSP28-2 | 5′-GACTGAAGATCATCCATTAAT-3′ |
| shRECQL1-1 | 5′-GCACATGCTATTACTATGCAA-3′ |
| shRECQL1-2 | 5′-GCCCTCAAACACTGAAGATTT-3′ |
| shWRN-1 | 5′-GCTGGCAATTACCAGAACAAT-3′ |
| shWRN-2 | 5′-GAGGGTTTCTATCTTACTAAA-3′ |
| shBLM-1 | 5′-GACGCTAGACAGATAAGTTTA-3′ |
| ShBLM-2 | 5′-TCACAAGGAATGAGAAATA-3′ |
| shRECQL4-1 | 5′-CTAGGAAGAGCCTCATCTAAG-3′ |
| shRECQL4-2 | 5′-CGGCTCAACATGAAGCAGAAA-3′ |
The USP28 cDNA (WT or C171A mutant) and RECQL5 cDNA (full length or deletion mutants) were cloned into lentiviral vector pHAGE. The USP28 full length or deletion mutants were cloned into pIRES-S-tag-Flag-tag. The c-MYC cDNA (WT or T58A mutant) were cloned into pCMV.
Lentiviruses-carrying overexpression, knockdown, or knockout elements were produced in the lab and used to infect the above cell lines with multiplicity of infection >1. The infected cells were selected with puromycin treatment (4 μg/ml for 2 days).
Assays for cell proliferation
For MTS assay, after lentiviral infection and selection, the cells were trypsinized and reseeded in 96-well plates at a density of 3000 cells/well and cultured for the indicated time. At the end of incubation, the number of viable cells was analyzed using a colorimetric assay (MTS). Briefly, 20 μl of MTS was added to 100 μl fresh complete culture medium in each well, and the cells were incubated for 2 h before the absorbance of the formazan product at 490 nm was measured.
Fluorescence activated cell sorting
The cells were trypsinized and washed once with cold PBS. For cell cycle analysis, the cells were fixed in 70% ice-cold EtOH, spun down, washed with cold PBS, and incubated in PBS containing propidium iodide (50 μg/ml) and RNase A (50 μg/ml) for 30 min at room temperature. The propidium iodide-stained single cell suspension was analyzed on a BD LSRFortessa SORP Flow Cytometer (BD Biosciences). ModFit LT software (Verity Software House) was used to analyze the DNA pattern and cell cycle stages.
Antibodies
The antibodies used in this study were as follows: USP28 (17707-1-AP, 1:1000 WB, Proteintech); RECQL1 (A300-450A, 1:1000 WB, Bethyl Lab, Montgomery); WRN (4666S,1:1000 WB,Cell Signaling); BLM (A300-110A,1:1000 WB, Bethyl Lab); RECQL4 (17008-1-AP,1:1000 WB, Proteintech); RECQL5 (A302-520A, 1:2000 WB, Bethyl Lab); Flag (F3165, 1:2000, Sigma); Ub (SC-8017, 1:200 WB, Santa Cruz Biotechnology); γH2AX (05-636, 1:500 IF, Millipore); Claspin (2800S, 1:1000 WB, Cell Signaling); c-MYC (SC-40, 1:200 WB, Santa Cruz); Phospho-Chk1-Ser345 (2348, 1:1000 WB, Cell Signaling); CHK1 (ab32531, 1:1000 WB, Abcam); GAPDH (60004-1-1g, 1:5000 WB, Proteintech); Tubulin (66240-1-1g, 1:5000 WB, Proteintech); ACTIN (66009-1-1g, 1:3000 WB, Proteintech). The secondary antibodies conjugated to horseradish peroxidase for Western blot and the secondary antibodies anti-mouse, -goat or -rabbit Alexa fluor 488 or 594 for immunofluorescence staining were purchased from Jackson ImmunoResearch Laboratories. siRNAs were synthesized by GenePharma.
Western blotting analysis
The cells were lysed with RIPA buffer (Applygen Technologies Inc) supplemented with a protease inhibitor cocktail (Roche Diagnostics). The Western blots were processed according to standard procedures and analyzed with Chemiluminescent imaging system LAS 500 (GE Healthcare).
Immunoprecipitation
HEK293T cells cultured in 10 cm dish were transfected with 5 μg Flag-RECQL5 using PEI (Polysciences Inc). After 48 h, harvest the cells and lysate them in NETN lysis buffer. The Flag M2 beads (Sigma) were washed with NETN lysis buffer. The protein A/G beads were washed and then mixed with Rabbit IgG as negative control or USP28 antibodies. About 2 mg of total protein from the cell lysates were incubated with the beads prepared above for 3 h at 4 °C. The bound proteins were denatured by adding 50 μl SDS loading buffer and boiling for 5 min and then analyzed by immunoblotting.
In vivo ubiquitination assay
HEK293T cells were transfected with Flag-RECQL5 and other indicated plasmids for 48 h after the treatment with 10 μM MG132 for 8 h. Lyse the cells in denaturing buffer containing 1%SDS and 1% sodium deoxycholate, vortex vigorously for 15–30 min, boil the cell lysates at 100 °C for 10 min, and then add 5× – 9× volumes of NETN buffer. The cell lysates were incubated with Flag M2 beads overnight at 4 °C after washing and Western blotting analysis.
Immunostaining
The cells after the indicated treatment were plated on coverslips, fixed with 4% paraformaldehyde for 15 min, permeabilized in PBS containing 0.5% Triton X-100 for 5 min, and blocked with 5% bovine serum albumin in PBS for 1 h at room temperature, after incubation with primary antibodies at 4 °C overnight. After three washes in PBS, the coverslips were incubated with secondary antibodies for 20 min at 37 °C. All images were taken on a Nikon Ni-E microscope (Nikon Corporation), with identical exposure times for each sample.
Tumor xenograft
2 × 106 HCC1806 cells were infected with lentiviruses expressing USP28 or control shRNAs and used to inoculate BALB/c nude mice the left inguen. The mice are purchased at 6–8 weeks of age from Beijing Vital River Laboratory Animal Technology Co, Ltd. All animals were kept in an environmentally controlled facility and given free access to water and a standard diet. Three weeks after the inoculation, the tumors were removed, and the tumor weight were measured. To evaluate the antitumor effect of AZ1, HCC1806 cells similarly inoculated and allowed to grow for 10 days when the tumor size had reached 100 mm3. The tumor-bearing mice were then randomized into two groups and dosed daily by oral gavage at 200 mg/kg of AZ1 or the vehicle. The tumor growth was monitored every 3 days using vernier caliper. Two weeks after the treatment, the mice were sacrificed. The tumors were excised out and weighed. All animal experiments were performed according to the guidelines approved by the Animal Care and Use Committee of the First Affiliated Hospital of Zhejiang University.
Statistics
Statistics analyses were performed with GraphPad Prism9.0 and ImageJ. Unpaired two-tailed Student’s t test was used to analyze the significance between two groups. p < 0.05 was considered as statistical significance (∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001).
Data availability
All data are contained within this article and available from the corresponding author on reasonable request.
Supporting information
This article contains supporting information.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank the core facilities in National Center for Protein Sciences (Beijing) and Zhejiang Provincial Key Laboratory of Pancreatic Disease for experimental support. The authors thank people in Zhang lab for helpful discussions and suggestions throughout the work. This work was supported by grants from the National Natural Science Foundation of China (81773032), the National Key R&D Program of China (2018YFA0507500).
Author contributions
J. W., J. J., S. H., P. Z., and J. P. conceptualization; J. W., H. M., L. W., X. Z., L. T., and J. P. methodology; J. W. and J. P. investigation; Y. D. software; Y. D. validation; H. M., L. W., X. Z., and L. T. resources; J. J. and S. H. visualization; P. Z. and J. P. supervision; P. Z. and J. P. writing–original draft.
Funding and additional information
J. W. was supported by a grant from the National Natural Science Foundation of China (81600458).
Edited by Patrick Sung
Supporting information
References
- 1.de Ruijter T.C., Veeck J., de Hoon J.P., van Engeland M., Tjan-Heijnen V.C. Characteristics of triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2011;137:183–192. doi: 10.1007/s00432-010-0957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reis-Filho J.S., Tutt A.N. Triple negative tumours: A critical review. Histopathology. 2008;52:108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- 3.Lehmann B.D., Bauer J.A., Chen X., Sanders M.E., Chakravarthy A.B., Shyr Y., Pietenpol J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartkova J., Tommiska J., Oplustilova L., Aaltonen K., Tamminen A., Heikkinen T., Mistrik M., Aittomaki K., Blomqvist C., Heikkila P., Lukas J., Nevanlinna H., Bartek J. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng J., Tang L., Cai M., Chen H., Wong J., Zhang P. RECQL5 plays an essential role in maintaining genome stability and viability of triple-negative breast cancer cells. Cancer Med. 2019;8:4743–4752. doi: 10.1002/cam4.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lakhani S.R., Van De Vijver M.J., Jacquemier J., Anderson T.J., Osin P.P., McGuffog L., Easton D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002;20:2310–2318. doi: 10.1200/JCO.2002.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Turner N., Tutt A., Ashworth A. Hallmarks of 'BRCAness' in sporadic cancers. Nat. Rev. Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 8.Shiu K.K., Tan D.S., Reis-Filho J.S. Development of therapeutic approaches to 'triple negative' phenotype breast cancer. Expert Opin. Ther. Targets. 2008;12:1123–1137. doi: 10.1517/14728222.12.9.1123. [DOI] [PubMed] [Google Scholar]
- 9.Popov N., Wanzel M., Madiredjo M., Zhang D., Beijersbergen R., Bernards R., Moll R., Elledge S.J., Eilers M. The ubiquitin-specific protease USP28 is required for MYC stability. Nat. Cell Biol. 2007;9:765–774. doi: 10.1038/ncb1601. [DOI] [PubMed] [Google Scholar]
- 10.Wu Y., Wang Y., Yang X.H., Kang T., Zhao Y., Wang C., Evers B.M., Zhou B.P. The deubiquitinase USP28 stabilizes LSD1 and confers stem-cell-like traits to breast cancer cells. Cell Rep. 2013;5:224–236. doi: 10.1016/j.celrep.2013.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flugel D., Gorlach A., Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119:1292–1301. doi: 10.1182/blood-2011-08-375014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diefenbacher M.E., Popov N., Blake S.M., Schulein-Volk C., Nye E., Spencer-Dene B., Jaenicke L.A., Eilers M., Behrens A. The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J. Clin. Invest. 2014;124:3407–3418. doi: 10.1172/JCI73733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knobel P.A., Belotserkovskaya R., Galanty Y., Schmidt C.K., Jackson S.P., Stracker T.H. USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double-strand break metabolism. Mol. Cell. Biol. 2014;34:2062–2074. doi: 10.1128/MCB.00197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang D., Zaugg K., Mak T.W., Elledge S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 15.Prieto-Garcia C., Hartmann O., Reissland M., Braun F., Fischer T., Walz S., Schulein-Volk C., Eilers U., Ade C.P., Calzado M.A., Orian A., Maric H.M., Munch C., Rosenfeldt M., Eilers M., et al. Maintaining protein stability of Np63 via USP28 is required by squamous cancer cells. EMBO Mol. Med. 2020;12 doi: 10.15252/emmm.201911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kok Y.P., Guerrero Llobet S., Schoonen P.M., Everts M., Bhattacharya A., Fehrmann R.S.N., van den Tempel N., van Vugt M. Overexpression of Cyclin E1 or Cdc25A leads to replication stress, mitotic aberrancies, and increased sensitivity to replication checkpoint inhibitors. Oncogenesis. 2020;9:88. doi: 10.1038/s41389-020-00270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrigley J.D., Gavory G., Simpson I., Preston M., Plant H., Bradley J., Goeppert A.U., Rozycka E., Davies G., Walsh J., Valentine A., McClelland K., Odrzywol K.E., Renshaw J., Boros J., et al. Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem. Biol. 2017;12:3113–3125. doi: 10.1021/acschembio.7b00334. [DOI] [PubMed] [Google Scholar]
- 18.Yada M., Hatakeyama S., Kamura T., Nishiyama M., Tsunematsu R., Imaki H., Ishida N., Okumura F., Nakayama K., Nakayama K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welcker M., Orian A., Jin J., Grim J.E., Harper J.W., Eisenman R.N., Clurman B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. U. S. A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within this article and available from the corresponding author on reasonable request.