

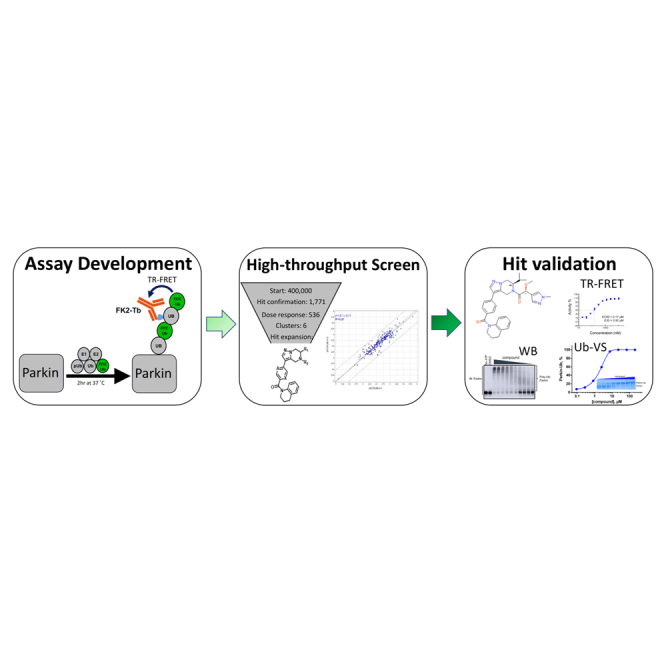
Summary
Pharmacological activation of the E3 ligase Parkin represents a rational therapeutic intervention for the treatment of Parkinson’s disease. Here we identify several compounds that enhance the activity of wildtype Parkin in the presence of phospho-ubiquitin and act as positive allosteric modulators (PAMs). While these compounds activate Parkin in a series of biochemical assays, they do not act by thermally destabilizing Parkin and fail to enhance the Parkin translocation rate to mitochondria or to enact mitophagy in cell-based assays. We conclude that in the context of the cellular milieu the therapeutic window to pharmacologically activate Parkin is very narrow.
Subject areas: Chemistry, Biochemistry, Biochemistry Applications
Graphical abstract
Highlights
-
•
A biochemical FRET-based HTS identified compounds that activate wildtype Parkin
-
•
A Ub-charging assay confirms results and suggests they act as PAMs to activate Parkin
-
•
Compounds do not enhance Parkin’s mito translocation or enhance mitophagy in cells
-
•
Data suggests a model of activation after the REP domain release, limiting pharmacology
Chemistry; Biochemistry; Biochemistry Applications
Introduction
Parkin (PARK2) is a cytosolic E3 ligase and PINK1 is a mitochondrial kinase, key players in the mitophagy pathway, whose loss may result in Parkinson’s disease. In healthy mitochondria, PINK1 kinase is imported into the mitochondrial matrix in a membrane potential-dependent manner. PINK1 is then cleaved by various matrix-resident proteases; the cleaved PINK1 is retro-translocated to the cytosol for N-end rule mediated degradation by the proteasome (Jin et al., 2010; Yamano and Youle, 2013). Upon mitochondrial stress and depolarization, PINK1 is stabilized in the outer mitochondrial membrane, where it first auto-activates by phosphorylation (Okatsu et al., 2012; Rasool et al., 2018) and then activates the mitophagy pathway by phosphorylating ubiquitin (Kane et al., 2014; Klosowiak et al., 2016; Koyano et al., 2014) to form phospho-ubiquitin (pUb) as well as Parkin to form phospho-Parkin (pParkin) (Kondapalli et al., 2012; Shiba-Fukishima et al., 2012). pUb acts as an activator of Parkin; its gradient, emanating from the site of PINK1 stabilization, promotes Parkin recruitment to damaged mitochondria (Shiba-Fukushima et al., 2014). Binding to pUb and phosphorylation of Parkin by PINK1 causes a conformational change in the enzyme that releases the autoinhibited state, so it can ubiquitinate nearby (Gladkova et al., 2018; Sauve et al., 2018). Thus, in healthy individuals, stabilized PINK1 acts as the sensor of damage, pUb acts as a signaling beacon that recruits Parkin to the mitochondrial surface, and phospho-Parkin ubiquitinates outer mitochondrial membrane proteins, ultimately leading to the removal of damaged mitochondria by mitophagy, a form of mitochondrial quality control.
Loss of function mutations in Parkin (Farrer et al., 2001; Kitada et al., 1998) and PINK1 (Valente et al., 2004), are a common cause of early onset autosomal Parkinson’s Disease (Bayne and Trempe, 2019). Postmortem analysis of PD and Lewy body disease brains has shown an accumulation of pUb in disease-relevant regions (Hou et al., 2018; Shiba-Fukushima et al., 2017). One prevailing hypothesis is that PD patients are unable to resolve the pUb beacon due to inadequate Parkin activation—they have excessive mitochondrial damage with limited turnover of this organelle. This mitochondrial damage results in the degeneration of substantia nigra neurons which are known to be particularly dependent on functional mitochondria (Surmeier et al., 2010; Malpartida et al., 2021). Enhancing Parkin activity directly with a small molecule activator in the presence of pUb is predicted to stimulate turnover of damaged mitochondria and potentially slow the progression of PD (Miller and Muqit, 2019).
A RING-in-between-RING (RBR) E3 ligase (Chaugule et al., 2011), Parkin catalyzes ubiquitin transfer through a 2-step reaction in which ubiquitin is first transferred to Parkin’s catalytic cysteine in RING2 and then transferred to the substrate (Seirafi et al., 2015; Trempe et al., 2013). Parkin is autoinhibited in the absence of PINK1 activation (Kane et al., 2014; Kazlauskaite et al., 2014). Two regulatory elements keep Parkin in an autoinhibited state: a ubiquitin-like (Ubl) domain which is phosphorylated by PINK1 and a unique RING0 domain and a repressor (REP) (Pao et al., 2016) region that suppresses ligase activity (Chaugule et al., 2011; Gladkova et al., 2018; Trempe et al., 2013)
As a first regulatory step, pUb binding triggers a conformational change that primes Parkin for activation by releasing the Ubl domain for phosphorylation by PINK1 (Sauve et al., 2018; Gladkova et al., 2018). The synergy of binding the activator pUb to form a more active conformation can be seen from the altered affinities that different Parkin forms have for pUb: pParkin binds pUb with 25-fold better affinity than unphosphorylated Parkin does in vitro (Kumar et al., 2015; Sauve et al., 2015).
As a second regulatory step, the REP binding region keeps Parkin from activating unnecessarily. A single site mutation in the REP binding region (W403A-Parkin) artificially causes Parkin to translocate faster to the mitochondrial membrane to stimulate the flux to mitophagy (Trempe et al., 2013; Tang et al., 2017). This mutation also activates Parkin in conjunction with pUb binding, and if pUb binding is impaired, for example by the H302A mutation, the W403A mutation can only partly rescue Parkin’s mitochondrial recruitment activity (Tang et al., 2017).
After a certain threshold level of Parkin activation, a positive feedforward loop is triggered that results in mitophagy (Ordureau et al., 2014; Shiba-Fukushima et al., 2014). Active Parkin links ubiquitin chains onto many mitochondrial membrane proteins (Sarraf et al., 2013); the ubiquitin is then phosphorylated by PINK1, recruiting and activating additional Parkin molecules. Eventually Parkin coats the mitochondria with ubiquitinated proteins, a signal which triggers their recognition by autophagy adaptors Optineurin and TBK1, among others (Harper et al., 2018), sending the damaged mitochondria for degradation in the lysosome. Over time, pParkin levels are reduced suggestive of PINK1/Parkin pathway attenuation via pParkin degradation (Pao et al., 2016)
We wished to identify small molecule compounds that activate WT-Parkin in a simulated in vitro reconstituted environment that mimics the cellular milieu when mitochondria are damaged. This approach represents a somewhat unexplored goal in drug discovery. While inhibitors of E3 ligases have been identified, reports of activators of E3 ligases have been less prevalent, although recently activators have been identified as leads that stabilize the active form of the HECT E6AP E3 ligase (Offensperger et al., 2020). We report here that we successfully screened a proprietary small molecule library in a miniaturized biochemical assay that measures autoubiquitination of Parkin in the presence of pUb. We identified and validated 536 Parkin-activating compounds at a hit rate of 0.13% (here and elsewhere termed ‘Parkin activators’). One series of activators was selected for medicinal chemistry optimization of compound properties and potency, resulting in compounds with nM Ec50 and 100% Emax (achieving the maximum effect on activity, as defined by W403A-Parkin). We demonstrate that these optimized compounds can also activate W403A-Parkin in the presence of pUb and can activate pParkin in the absence of pUb. However, in cells the compounds fail to enhance Parkin translocation to mitochondria or to enhance mitophagy upon mitochondrial depolarization. Our study suggests that the opportunity to pharmacologically activate Parkin in cells is very narrow; possibly compounds that speed up rate-limiting steps of Parkin activation are more likely to enhance the speed of Parkin translocation to mitochondria and thus enhance mitophagy.
Results
Establishing a biochemical assay to identify small molecule activators of Parkin
Mitochondria are polarized organelles and can easily concentrate lipophilic, positively charged compounds, which in turn can drive mitochondrial toxicity (Smith et al., 2012). Since the PINK1/Parkin pathway is a sensor of mitochondrial damage, we anticipated that a cell-based screen for Parkin activators would yield a large number of mitochondrial toxins and thereby a large number of false positives. Therefore, we leveraged the ability of Parkin to autoubiquitinate in vitro (Matsuda et al., 2006) and established a Time-resolved Förster resonance energy transfer (TR-FRET) based assay compatible with high-throughput screening.
We first set up a Parkin autoubiquitination reaction in the presence of UBE1 (as E1), UbcH7 (as E2) and unlabeled Ub spiked with FITC-labeled Ub to provide adequate substrate for the reaction while enabling later tracking in the FRET readout (Figure 1A). To quantify autoubiquitination, we added a Terbium (Tb) labeled anti-polyubiquitin antibody (FK2-Tb), which detects polyubiquitin chains generated by Parkin and readout the TR-FRET signal between FK2-Tb and FITC-labeled Ub. Since PINK1-phosphorylated ubiquitin (pUb) mediates the first step of Parkin activation in the cell (Harper et al., 2018), we included pUb in our reaction. Adding pUb to the reaction was sufficient to drive Parkin autoubiquitination (Figure S1A). In the presence of 1 μM pUb, we observed a significant FRET ratio signal driven by WT-Parkin (Figure 1B). The FRET ratio more than doubled when a molar equivalent of similar purity W403A-Parkin was employed, while the C431S-Parkin catalytically dead mutant showed lower FRET signal when compared to WT-Parkin. Consistently, W403A-Parkin accumulated in large molecular weight species in the reaction (Figure 1C).
Figure 1.

Primary assay development
(A) Schematic of the biochemical assay including detection by an FK2 antibody, which recognizes polyubiquitin and provides an FRET signal to the FITC-labeled ubiquitin. The components of the reaction are depicted with circles.
(B) Comparison of the FRET ratio of the WT-, W403A- and C431S-Parkin after 1 h incubation.
(C) Silver stain indicating that W403A-Parkin was more active than WT-Parkin in the presence of 1 μM pUb, as indicated by the disappearance of the Parkin band and the appearance of a high-molecular weight smear.
(D) Dependency of WT- (blue) and W403A-Parkin (red) activity on pUb concentration. The C431S-Parkin (yellow) is inactive at all pUb concentrations.
(E) Titration of pUb in TR-FRET assay with WT-Parkin (blue) at a 3 h reaction time and calculation of Ec50. Each data point represents 2 technical replicates.
(F) At a given time (i.e., 180 min), we can enable a large window between WT-Parkin (blue) and the W403A-Parkin positive control (red). Each data point represents 2 technical replicates. Statistical test performed: ANOVA with Dunnett’s MCT versus WT ∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. For panel B, error bars represent standard deviation with n = 3 technical replicates. See also Figure S1.
To further understand the assay, we quantitated the FRET signal response of the three forms of Parkin across a range of pUb concentrations (Figure 1D red W403A-Parkin, blue WT-Parkin, yellow C431S-Parkin). WT-Parkin showed a linear response to pUb, suggesting that pUb acts as an agonist in this activation reaction, and also suggesting that our assay would be biased toward identification of positive allosteric modulators. In comparison, W403A-Parkin starts at a higher basal level of activity in absence of pUb and responded to as little as 0.7 μM of pUb with a 3-fold increase in signal over the WT-Parkin and plateaued at that level due to exhaustion of the TR-FRET reaction. As expected, we observed no activity of C431S-Parkin at any concentration of pUb. Titrating the concentration of pUb required to activate WT-Parkin, we determined an EC50 of 6 μM at a time course of 2 h (Figure 1E). At a fixed concentration of pUb (5 μM), WT-Parkin generated FRET signal with linear kinetics, whereas W403A-Parkin, which starts at a higher basal level of activity in the absence of pUb, showed faster kinetics, reaching plateau at 100 min (Figure 1F). Therefore, W403A-Parkin accelerates the kinetics of Parkin autoubiquitination, and the reaction is enhanced with addition of pUb.
We also asked if the TR-FRET assay would be sensitive to the PINK1-phosphorylation status of Parkin. PINK1-phosphorylated Parkin represents the state of maximum activation of Parkin (Bayne and Trempe, 2019). We compared FRET signal of 50 nM pParkin to 50 nM W403A-Parkin in the presence of increasing concentrations of pUb, and observed that pParkin activity was independent of pUb, with maximal FRET signal even at 0 mM pUb (Figure S1B), confirming that PINK1-phosphorylation of Parkin alone can activate Parkin.
Taking all these observations together, we decided to establish a TR-FRET-based small molecule screen for compounds that activate unphosphorylated WT-Parkin in the presence of pUb. Since W403A-Parkin has been shown to enhance mitophagy upon mitochondrial stress, we hypothesized that a small molecule that binds in the W403 binding site, or other hotspot, might relieve Parkin autoinhibition, enhance activity in the biochemical assay, and translate to enhanced mitophagy in cells.
High-throughput screen and assay development to identify Parkin activators
We miniaturized the assay to 1,536 well plates and conducted a high-throughput screen using the primary assay with an incubation time of 2 h with pUb at the EC50 (5 μM) (Figure S2A;Table S1). A ∼400,000 proprietary compound library was screened against tag-less WT Parkin at a single dose (30 μM). W403A-Parkin was included as a positive control. The FRET signal obtained with W403A-Parkin was designated as 100% Emax for the WT-Parkin. The assay performed with an overall Z′ of 0.84 and assay window of 2.8 ± 0.6 with no obvious plate effects. We identified 1,771 compounds that increased % TR-FRET signal above 2 sigma from the mean signal in both replicates and performed a counter-screen assay in the primary assay format (WT-Parkin) as well as on the C431S-Parkin catalytically dead mutant. Counter-screen yielded 536 compounds (0.13% hit rate) that were selected for concentration response curves (CRC) with a maximum concentration of 200 μM, 1:2 dilutions steps in duplicate. We observed good correlation between duplicate EC50 values in the CRC (R2 = 0.97) (Figure S2B). This triage scheme yielded ∼200 compounds with EC50 ranging between 1 and 50 μM.
Next, we performed cluster analysis of the hits using a UNITY fingerprints algorithm to find groups or clusters (A–F) of compounds with similar structures and created a structural similarity map (Figure 2A). Molecules that lie on the boundary of the map represent singletons. The most promising clusters A and B contained hits with Emax> 75% and displayed some structure-activity relationship (SAR). Cluster D also contained hits with Emax> 75% but was not straightforward to advance in medicinal chemistry optimization. We also identified a singleton, BIO-1741376, a tetrahydropyrazolo pyrazine-containing scaffold (THPP) with EC50 = 14.5 μM and Emax = 76%. Representative concentration response curves of BIO-1741376 and representative hits from cluster A and B are shown in Figures 2D, 2B, and 2C, respectively and data in biological triplicate is depicted in Figure S2C.
Figure 2.

High-throughput screen for Parkin activators
(A) Structural similarity map based on Unity fingerprints demonstrating that there are more than 6 clusters of hits, with a singleton, BIO-1741376 (THPP series). Compounds with an Emax> 75% are designated as green circles and compounds with an Emax 45–75% are designated as orange circles. The lead THPP compound BIO-1741376 is identified as a singleton.
(B) Dose-response curve for cluster A representative compound, BIO-0030130. Each point on the curve represents n = 2 technical replicates. BIO-0030130 is a racemic mixture whose active enantiomer BIO-1953559 is detailed in Figure S2C.
(C) Dose-response curve for cluster B representative compound BIO-0550701; each point on the curve represents n = 2 technical replicates.
(D) Dose-response curve for THPP singleton, BIO-1741376; each point on the curve represents n = 2 technical replicates. Concentration response curves in n = 3 biological replicates for these compounds are shown in Figure S2C.
(E) Confirmation of FRET assay using Western blot with increasing concentrations of cluster A compound. Smears indicate polyubiquitination of Parkin, and were measured after 5 min reaction times, while TR-FRET reactions were carried out over a 2 h time course at 37°C.
(F) Same as E for cluster B representative compound BIO-0550701.
(G) Western blot demonstrating that cluster A and THPP compound BIO-1967174 not only autoubiquitinate Parkin, but can enhance the rate of monoubiquitination of Miro1 substrate to a level comparable to that of W403A-Parkin. Note that PRK8 antibody does not recognize W403A-Parkin as previously noted (Zhu et al., 2018). Western blots represent n = 2 biological replicates. See also Figure S2 and Table S1.
We then asked, using a representative set of compounds, if these HTS hits were able to stimulate Parkin autoubiquitination as visualized by Western blot analysis. We pre-incubated the compounds for 30 min at 37°C with E1, E2, and WT-Parkin, then added Ub, FITC-Ub, pUb and triggered the reaction with ATP. We let the reaction run for 5 min at 37°C and then loaded the products on an SDS-PAGE gel as a representative blot from 2 technical replicates (Figures 2E and 2F). The compounds promoted the appearance of large molecular weight species of Parkin in a concentration-dependent manner. We noted that there is a difference in each compound’s ability to reach the Emax after 5 min reaction time; cluster B compound BIO-0550701 appears to lag behind cluster A compound BIO-0030130, though both compounds reach Emax over the course of 3 h at 37°C in the TR-FRET (Figures 2B and 2C). Thus, we verified that the TR-FRET assay was detecting autoubiquitinated Parkin species and that these compounds were able to stimulate Parkin autoubiquitination.
Finally, we asked if this set of compounds could promote the Parkin-mediated ubiquitination of another substrate that was not Parkin itself. Miro1/RhoT1 is a component of the mitochondrial motor/adaptor complex and a substrate of Parkin (Wang et al., 2011; Shlevkov et al., 2016). We pre-incubated a representative compound from cluster A or cluster B with E1, E2, Parkin and His-Miro1 (186-591) for 30 min, then triggered the reaction with Ub, pUb, and ATP and stopped the reaction at 5, 10 or 30 min (Figure 2G) followed by Western blot analysis. We observed the appearance of mono-ubiquitinated forms of Miro1 as soon as 5 min, whereas DMSO only induced a slight increase in mono-ubiquitinated Miro1 forms, at 30 min. By 30 min, WT-Parkin activated by some of these compounds was as effective as W403A-Parkin in promoting Miro1 ubiquitination (Figure 2G).
We were surprised to see that while Parkin autoubiquitinates itself with a smear of ubiquitin-linked chains, the Miro1 soluble construct that was used in this assay was primarily mono-ubiquitinated, even when using highly active W403A-Parkin. This construct used in the assays includes two EF hands and cGTPase domains but lacks the C-terminal membrane domain. It has been previously shown that K572 within the C-terminal GTPase of hMiro1 is prioritized by Parkin and that a minimal monomeric substrate is only mono-ubiquitinated (Klosowiak et al., 2016). The authors suggest that the micro-environment of that lysine plays a key role in the recognition of this site by Parkin. The construct that we employed in this assay is the same as the hMiro1-BC form in the Klosowiak et al. study, and the results are consistent with a specific interaction with monomeric hMiro1 leading to preferential K572 targeting. Taken together, we concluded that the TR-FRET screen identified compounds that promote autoubiquitination of WT-Parkin and can stimulate its activity toward physiologically relevant targets.
Structure-activity relationship (SAR) study for the THPP compound series
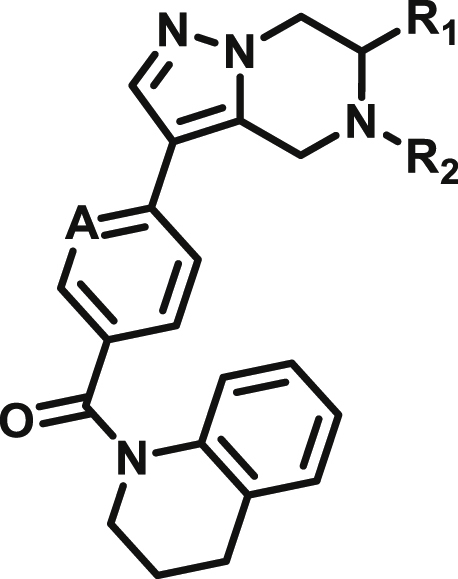
Based on this promising data and the discovery of several Parkin activator chemotypes, we turned our attention toward the chemical optimization of the HTS hits, with a focus on improving their physicochemical properties and biochemical potency using the Parkin autoubiquitination assay to drive structure-activity relationships (SAR). Despite promising SAR and the discovery of analogs with EC50< 1 μM, the progress for cluster A hits was halted due to the compounds’ likely poor CNS-penetrant properties, while the progress for cluster B hits was halted due to steep SAR or not being able to rationally enhance activity. On the other hand, the optimization of the singleton BIO-1741376 was successful, as we were able to improve its potency by >350-fold (Table 1).
Table 1.
THPP series potency and properties improvements through structural modification

aavg: average of Papp AB/BA. [r]: ratio.
Comparison of fully active THPP compound BIO-2007817, partially active enantiomer BIO-2007818, and fully inactive compound BIO-198542, all of which are highly soluble and low protein-binding compounds, with moderate permeability. EC50 and Emax were calculated after averaging technical replicates n = 2. See also Figure 3.
Starting from the original hit BIO-1741376, we were able to reach 100% Emax and improve potency 5-fold by replacing the acetamide with phenylacetamide (BIO-1953719) and changing the substitution at the core from (R)-Me to (R)-iPr (BIO-1966561) (Table 1). Notably, the enantiomer of BIO-1966561 (BIO-1966560) was found to be inactive. Additional methyl substitution at the phenylacetamide α-position not only enabled a slight potency improvement, while maintaining 100% Emax (BIO-1967173), but also demonstrated that the other 3 diastereomers were either weakly active (BIO-1967174) or inactive (BIO-1968010 and BIO-1968011). Through multiple rounds of library synthesis, we discovered that adding a methoxy group at the phenylacetamide α-position provided a 37x boost in potency, yielding BIO-2008550 with EC50 = 0.04 μM (Emax = 100%). However, despite being very potent in the biochemical assay, these analogs displayed poor solubility which might limit their effectiveness as tools for cell-based assays.
We then altered our strategy from a combinatorial and library-focused approach to a property-focused one to deliver analogs with decreased lipophilicity (ElogD7.4 in Table 1), which should translate to improved solubility. We determined that replacing the phenylacetamide from BIO-2008550 with a methyl-pyrazol acetamide moiety (BIO-2007817) resulted in a compound with lower ELogD7.4, which translated into excellent solubility with similar potency. Once again, the methoxy substitution at the α-position proved to be critical as the corresponding diastereomer BIO-2007818 was 300x less potent. Finally, introduction of a pyridine moiety near the tetrahydroquinoline amide enabled a further decrease in lipophilicity with an ELogD7.4 = 3.1 while maintaining EC50 below 0.2 μM (BIO-2007967). As measured by the average permeability in the MDR1-MDCK assay and based on their solubility and ElogD7.4, most of these compounds are expected to display sufficient cell permeability to test in cells.
To confirm that this enhanced activity was not solely an artifact of the TR-FRET readout, we performed a modified form of the activity assay with WT-Parkin with either Parkin itself or Miro1 soluble fragment as a substrate, and analyzed the ubiquitinated products by Western blots. We chose to compare BIO-2007817, BIO-2007818, and BIO-1984542 as representative examples of active, moderate, and inactive compounds, respectively. BIO-2007817 is the most potent compound of the series (Figure 3A). The compound stimulated Parkin autoubiquitination as measured by Western blots in a concentration-dependent manner (Figure 3B) and was also able to induce the appearance of monoubiquitinated forms of Miro1 (Figure 3C). Enantiomer BIO-2007818 showed 10x less potency in the TR-FRET assay (Figure 3D) and reduced ability to induce Parkin autoubiquitination as measured by Western blots (Figure 3E) or ubiquitination of Miro1 (Figure 3F). BIO-1984542 was inactive in all three assays (Figures 3G–3I).
Figure 3.

THPP series activity in biochemical assays
(A) BIO-2007817 structure and TR-FRET concentration response curve.
(B and C) (B) Parkin autoubiquitination and (C) Miro1 ubiquitination induced by increasing concentrations of BIO-2007817. W403A-Parkin is shown as a positive control.
(D) BIO-2007818 structure and concentration response curve.
(E and F)(E) Parkin autoubiquitination and (F) Miro1 ubiquitination induced by increasing concentrations of BIO-2007818. (G) BIO-1984542 structure and concentration response curve.
(H and I) (H) Parkin autoubiquitination and (I) Miro1 ubiquitination induced by increasing concentrations of BIO-1984542. Concentration response curves represent n = 2 technical replicates. Western blots represent n = 1 biological replicates. See also Table 1.
THPP compounds are positive allosteric modulators of Parkin
We next asked if the compounds act on Parkin or one of the other proteins in the biochemical reaction by setting up a minimal “charging” or “priming” assay to measure the first step of the Parkin reaction—conjugation of ubiquitin to C431. We hypothesized that an activator compound should be able to promote accessibility of the catalytic site of Parkin (C431). This active site is in the RING2 domain of Parkin and is buried in the autoinhibited conformation (Trempe et al., 2013). The site becomes exposed once pUb binds, triggering the release of the REP domain, enabling RING2 exposure (Gladkova et al., 2018). Ubiquitin-vinyl sulfone (Ub-VS) is a modified form of ubiquitin that reacts with the active-site cysteine residue in E3 ligases forming a covalent bond that increases the molecular weight of the enzyme by ∼8.5 kDa, which can be detected by SDS-PAGE (Borodovsky et al., 2001). We used this as a tool to measure the degree of accessibility of the catalytic site of Parkin, in the absence of E1 and E2 enzymes (Wauer and Komander, 2013; Park et al., 2017).
We incubated Ub-VS with N-terminally Gly-His6-Avi-tagged WT-Parkin (tagged Parkin), pUb and the selected THPP compounds for 2 h (note that no E1, E2, or ATP was present) before analyzing the reaction by SDS-PAGE. The highly active compound BIO-2007817, and to a lesser extent, BIO-2007818 induced the appearance of slower-migrating Parkin species in a concentration-dependent manner (Figure 4A). The structurally similar but inactive compound BIO-1984542 showed no significant activity in the assay. BIO-2007817 approached maximal efficacy at concentrations above 20 μM and the effect of the compound was consistently observed across several independent experiments (Figures 4B and S3). Thus, active compounds promote the accessibility of active-site C431, likely by directly acting on Parkin. Moreover, we observed excellent correlation in the rank-order potency of the compounds measured by the Ub-VS charging assay and the Parkin autoubiquitination assay.
Figure 4.

Activator compounds directly engage Parkin in the presence of pUb
(A) Gel shift upon formation of Ub-VS linked Parkin demonstrating that THPP compounds retain the same activity ranking in the Ub-VS charging assay as in the biochemical assay. Reactions were run for 2 h (WT-Parkin).
(B) Quantitation of the % charged with respect to compound concentration in (A). This panel represents n = 1 technical replicates, but n = 3 biological replicates is shown in Figure S3A and quantitated in Figure S3C. Activating mutant W403A-Parkin is also activated by compounds in the presence of pUb and with the same activity ranking as in the biochemical assay. Reactions were run for 1 h (W403A-Parkin).
(C and D) (C) Gel shift upon formation of Ub-VS linked W403A-Parkin after 1 hr incubation. (D) Quantitation of the % Parkin charged with respect to compound concentrations in (C). This panel represents n = 1 technical replicates, but n = 3 biological replicates is shown in Figure S3B and quantitated in Figure S3D. Compound activation hierarchy is preserved in both cases.
(E) C431S-Parkin is not responsive to activating compound BIO-2007817 over a range of pUb concentrations.
(F) At a fixed concentration of 200 μM BIO-2007817, WT-Parkin charging is steeply dependent on pUb concentrations with EC50 = 1.8 ± 0.4 μM.
(G) At a fixed concentration of 200 μM BIO-2007817, W403A-Parkin charging is dependent on pUb concentrations, and activation occurs at lower pUb concentrations in the presence of the compound, as evidenced by a reduction in EC50 for pUb (EC50-‘817 = 5.4 ± 0.3 μM vs EC50+‘817 = 1.6 ± 0.2 μM).
(H) Quantitation of % charged with respect to compound concentrations in (E).
(I) Quantitation of % charged with respect to pUb concentrations in (F).
(J) Quantitation of % charged with respect to pUb concentrations in (G). Each experiment in (E), (F) and (G) represents n = 1 technical and biological replicates. See also Figures S3, S4, and S6.
We next asked if we could further understand the mechanism by which these compounds act and whether they behave similar to the mechanism of W403A-Parkin which enables REP and RING2 release. We postulated that if the BIO-2007817 compound acted at the W403 binding site, W403A-Parkin mutant should be less susceptible to the compound and thus show no additive activation response by the compound. Conversely, if BIO-2007817 could further activate W403A-Parkin, its activity might represent a different step in the pathway and we might see an additive activation effect. N-terminally tagged W403A-Parkin was significantly more active than N-terminally tagged WT-Parkin in the Ub-VS assay in the absence of compounds, confirming that the W403A-Parkin mutant is in a partially active state (Figure 4C). Although the intrinsic activity of W403A-Parkin in the assay reduces the dynamic range of the assay, we observed that BIO-2007817, and to a lesser extent BIO-2007818, further stimulated exposure of the active-site C431 in the context of the W403A-Parkin mutant (Figure 4D). Therefore, we concluded that the THPP active compounds BIO-2007817 and BIO-2007818 likely activate Parkin within the activation cascade at a step that is distinct from the REP/RING2 release step.
What is the mechanism by which the compounds act? pUb is a required interactor with Parkin that fits the description of an agonist. Therefore, we considered whether the THPP actives may act as positive allosteric modulators (PAMs). A PAM is expected to shift the concentration response curves of the enzymatic reaction toward improved potency by making the enzyme less dependent on the concentration of the agonist for activity. To ask if our lead BIO-2007817 acted as a PAM, we incubated Ub-VS and BIO-2007817 with either N-terminally tagged WT-Parkin, N-terminally tagged W403A-Parkin, or catalytically dead N-terminally tagged C431S-Parkin in the presence of increasing concentrations of pUb (Figures 4E–4I). As expected of the catalytically inactive mutant, we observed no activity of C431S-Parkin in presence of BIO-2007817 at any concentration of pUb, confirming that the Ub-VS assay is a direct measurement of catalytic activity of Parkin (Figures 4E and 4H). When we incubated WT-Parkin with increasing concentrations of pUb, we observed a slight activation of Parkin at the highest concentrations of pUb (Figures 4F and 4I, open circles). However, BIO-2007817 shifted the response curve of WT-Parkin to lower agonist concentrations and increased the maximum response, which is consistent with a PAM mechanism (Figures 4F and 4I filled circles). Interestingly, when we tested the response of W403A-Parkin, BIO-2007817 acted more like a PAM-agonist, shifting the curve to the left, but is an agonist itself by inducing a small basal response, even at minimal concentrations of pUb (Figures 4G and 4J) (Kenakin, 2016). This is consistent with the observation that BIO-2007817 can act as an agonist for activated phospho-Parkin even in the absence of pUb (Figure S3F). Moreover, compared to W403A-Parkin, the steep dependence of WT-Parkin on pUb is suggestive of synergistic conformational changes at play; positive cooperativity is more pronounced when starting from an inactive state (WT-Parkin) versus a pre-sampled active conformational state (W403A-Parkin). Taken together, we conclude that BIO-2007817 acts independently of the REP domain release step and can act as a weak agonist even when the protein pre-samples the fully activated form.
Finally, we wanted to understand whether BIO-2007817 could mediate its activity by destabilizing the autoinhibited conformation using a thermal shift assay (Figure S4). Whereas the N-terminally tagged W403A-Parkin significantly alters the conformation of Parkin resulting in 4–5°C lower thermal melting temperature, the compounds did not induce similar thermal destabilization effects on N-terminally tagged WT Parkin. The ∼1.5°C shifts in temperature are close to the level of systematic error in the assay based on an n = 3 measurement. Furthermore, there is no correlation between the degree of destabilization and the level of activity in the biochemical assays. This provides additional support to the hypothesis that the compounds act in a mechanism that is distinct to the thermally destabilizing REP-release mechanism as represented by W403A-Parkin.
THPP compounds tested in cell-based assays of Parkin signaling
Given the improved potency and solubility of the advanced THPP compounds, we tested if the THPP compounds could activate Parkin in two cellular assay formats. The first assay relies on the observation that Parkin translocation to damaged mitochondria depends on Parkin’s catalytic activity (Trempe et al., 2013; Tang et al., 2017; Narendra et al., 2008). We created HeLa cell lines stably overexpressing eGFP-WT-Parkin, eGFP-W403A-Parkin or eGFP-C431A-Parkin. We then assessed the rate of Parkin translocation to mitochondria in the three lines (Figures 5A and S5A). When cells were stimulated with carbonyl cyanide 3-chlorophenylhydrazone (CCCP), WT-Parkin translocated with T50 (defined as time (in minutes) needed for 50% of the cells exhibiting Parkin translocated to mitochondria) of ∼90 min, consistent with previous literature (Tang et al., 2017). As expected, W403A-Parkin translocated significantly faster than WT-Parkin. We calculated an average T50 ∼40 min for W403A-Parkin, representing a 56% increase in translocation rate (Figure 5B). The catalytically dead C431A-Parkin mutant did not translocate, confirming previous results (Tang et al., 2017). Thus, this timecourse assay provides a good window to test the effect of the compounds on Parkin translocation.
Figure 5.

Compounds do not affect the rate of Parkin translocation to mitochondria nor the number of mitochondria within lysosomes
(A) The half-max time (T50) for W403A-Parkin translocation to mitochondria is within 40 min, the half-max time for WT- Parkin is 90 min, and the half-max time for C431A Parkin is never reached (inactive).
(B) The translocation time for W403A-Parkin is 56% faster than WT-Parkin.
(C) Calculated fold change in T50 relative to compound concentration for active (green), partially active (orange) and inactive (gray) THPP series compounds.
(D–F)(D) BIO-2007817, (E) BIO-2007818 and (F) BIO-1984542 (10 μM) translocation curves relative to control (DMSO with CCCP treatment).
(G) Baseline imaging for MitoQC in SH-SY5Y cells. MitoQC is a pH-sensitive mitochondrial fluorescent probe. 24 h of 5 μM Antimycin +10μM Oligomycin stimulates an increased level of mCherry puncta signal indicating mitolysosomes, whose pH is neutralized with BafA1.
(H) Quantitation of number of mitochondria in lysosomes under different conditions described in (G).
(I) At a fixed timepoint (8 h) there is no dose-dependent increase in mitolysosomes for the active compound (green) relative to the reduced activity compound (orange) or inactive compound (gray). In A, n = 2 technical replicates. In C, n = 3 biological replicates. In (D)–(F) n = 2 technical replicates and n = 3 biological replicates. See also Figure S5.
We hypothesized that if the active THPP compound BIO-2007817 was able to activate WT-Parkin in vitro to levels comparable to W403A-Parkin it should promote faster translocation of Parkin to mitochondria upon mitochondrial depolarization. We pretreated eGFP-WT-Parkin expressing cells for 1 h with BIO-2007817 and then triggered the translocation of Parkin to mitochondria by stimulation with CCCP. However, the addition of the active compound did not significantly change the rate of translocation of Parkin (Figures 5C and 5D). We also failed to observe any significant effects on Parkin translocation upon treatment with the least active THPP compounds (Figures 5C, 5E, and 5F).
Since the eGFP-Parkin translocation assay does not reflect the functional endpoint of Parkin pathway (mitophagy), and relies on overexpressed levels of the enzyme, we also tested the three compounds in an assay measuring the rate of mitophagy driven by endogenous Parkin. The MitoQC construct encodes a tandem mCherry-GFP tag fused to the mitochondrial targeting sequence of the outer mitochondrial membrane protein FIS1. The color of each mitochondria expressing MitoQC is influenced by the pH of its environment, as the GFP is quenched in the acidic environment of the lysosome. By evaluating the appearance of mCherry-only (red) puncta signal and the disappearance of yellow, one can track the movement of mitochondria to lysosomes. We created a stably expressing SH-SY5Y cell line expressing MitoQC and established an automated imaging and analysis pipeline to quantify the appearance of lysosomal mitochondria. Upon treatment with mitochondrial toxins antimycin and oligomycin (AO), we observed an increase in numbers of lysosomal mitochondria. This was completely abolished by treatment with Bafilomycin A1, which inhibits acidification of lysosomes (Figures 5G and 5H). Thus, we confirmed that our algorithms can quantify the flux of damaged mitochondria to the lysosome induced by mitochondrial damage.
To determine the assay window, we performed a timecourse experiment (Figure S5B). This experiment demonstrated that the number of lysosomal mitochondria induced by AO can be further increased when the cells are allowed to incubate for 12 h after AO treatment and prior to fixing/imaging. We hypothesized that a Parkin activator should enhance the flux through mitophagy and, therefore, significantly increase the number of mitochondria in the lysosome before reaching the plateau (or allow for the cells to reach the plateau sooner). Thus, we evaluated the effect of the THPP compounds after ∼12 h incubation in our cell line. THPP active compounds were pre-incubated with cells 1 h prior to AO treatment and kept in the well throughout the experiment. AO treatment increased the flux through mitophagy as expected. However, none of the biochemically active THPP compounds were found to be active above the AO stimulant levels in cell-based assays that measure mitophagy (Figure 5I).
Discussion
When we started this drug discovery campaign, we reasoned that activating wildtype Parkin could be therapeutic for idiopathic Parkinson’s disease patients based on the following observations: (1) Parkin is found in an insoluble form in the aged human brain and thus likely enzymatically dead (Tokarew et al., 2021). (2) Some studies suggest that insoluble Parkin is elevated in PD patients (Lavoie et al., 2005;Wang et al., 2005). (3) Furthermore, p-Ser65 Ub accumulation is found in the brains of sporadic PD patients (Hou et al., 2018), suggesting a dysfunction of the Pink1/Parkin pathway. We thus reasoned that in sporadic PD patients with wildtype Parkin, activation of the remaining soluble Parkin could enhance mitophagy and thus be beneficial. Whether single Parkin mutation (heterozygous) carriers could provide additional patient stratification and/or enrichment may be a more controversial point due to the ambiguity of robust genetic links to PD (Lubbe et al., 2021, Zhu et al., 2021). Improving genotyping to identify PD patients that carry more than one Parkin mutation may resolve this in the future. Complementing the genetics with more robust biochemical assays to measure Parkin activity in relevant tissues or fluids will be the best strategy to address this question of which patient will benefit the most from a Parkin targeted therapy.
In this study we show that (1) this TR-FRET-based Parkin autoubiquitination assay is scalable and amenable for high-throughput screening, (2) the assay identified chemical matter that activated Parkin in orthogonal biochemical assays measuring autoubiquitination and ubiquitination of Parkin substrate Miro1, (3) a structure-activity relationship (SAR) was observed upon modification of the THPP series of compounds with a clear enantiomeric preference, (4) THPP compound BIO-2007817 acts directly on WT-Parkin, is a PAM, and acts in a manner complementary to the mechanism of REP domain release, (5) the THPP active compound BIO-2007817 was not able to promote Parkin translocation to depolarized mitochondria in HeLa cells nor promote mitophagy in MitoQC SH-SY5Y cell lines.
In prior studies, several compounds have been described as modifying Parkin’s thermal stability and enhancing cellular mitophagy (Springer et al., 2018; Garofalo et al., 2017; Johnston and Garofalo, 2017; Shiba-Fukushima et al., 2020). These compounds were identified in phenotypic cell-based assays monitoring the end state of mitochondrial clearance. However, such assays are prone to identifying mitochondrial depolarizers, which can be mistaken for bona fide Parkin activators. We predict that many other published Parkin “activators” identified in cell-based screens are, in fact, mitochondrial depolarizers. Therefore, we decided to change strategies and identify Parkin activators using an in vitro biochemical assay as the primary assay followed by cell-based activity assays as secondary and tertiary assays.
We were successful in identifying chemical matter that activates WT Parkin biochemically using our hit-triage scheme. We report on the characterization of the THPP series of compounds, deemed most tractable and for which we observed a structure-activity relationship for potency and physicochemical properties improvement upon modification. Our mechanism of action studies using the Ub-VS assay suggest that the compounds activate WT-Parkin as PAMs. The evidence that the compounds appear to activate W403A-Parkin as a PAM-agonist or can activate pParkin in the absence of pUb suggests that they may act at a different stage of the activation process from REP-release. This conclusion is further supported by the observation that while the compounds act as PAMs they do not destabilize the autoinhibited form of Parkin in the same way that the W403A mutation does (Figure S4).
We note that there are several references in the Parkin literature that report that N-terminally tagging Parkin alters its activity (Matsuda et al., 2006; Burchell et al., 2012; Chaugule et al., 2011). In these studies the authors point out that addition of an N-terminal tag affixed to the Ubl domain appears to enhance pUb binding. In the experiments described here, we used tag-less WT-Parkin and W403A-Parkin for the HTS TR-FRET studies and used N-terminally tagged Parkin forms (Gly-His6-Avi tag) for the Ub-VS charging assays and nanoDSF studies; we did not observe that the tag measurably impacts these conclusions. First, N-terminally tagged WT-Parkin did not demonstrate any activity in the Ub-VS assay in the absence of pUb and its melting temperature was similar to that of tag-less WT-Parkin. Second, because the compounds appear to work on second step Ub-charging, not pUb binding, it seems less likely that the N-terminal tag will measurably impact the conclusion that the compounds are working at some step after REP domain release.
While we have tested the compounds’ ability to activate WT-Parkin in cells, we have not tested whether these small molecule activators might rescue some of the Parkin mutations carried by patients with autosomal recessive juvenile PD. Yi et al. have shown that introducing the W403A mutation in cis with PD pathological mutants rescued mitophagy after 4 h of CCCP treatment in Parkin-overexpressed U2OS cells (Yi et al., 2019). Most likely, these THPP PAMs will not rescue the same Parkin mutants because they do not behave similarly to W403A, but whether any of these pathological forms can be rescued remains to be tested and could shed further light on the mechanism of action.
However, the Parkin activator compounds that we identified do not increase Parkin translocation in cell-based assays. We have eliminated the trivial explanation that the compounds may not be cell-penetrant because they have an appropriate ElogD7.4 (<5) and overall average permeability (>100) to be passively permeable. The more active tool compound BIO-2007817 has an ElogD of 4.2 and reasonable average permeability (198 nm/s) which is the derived average of inward and outward permeability in the MDCK-MDR1 line. Most of the compounds have an average passive permeability >100, with the exception of BIO-1984542 with an average permeability of 44 (Table 1). None of the compounds demonstrated enhanced translocation of Parkin to mitochondria in the HeLa cell line where transport should not have had an adverse impact. To eliminate the possibility that the compounds might have been effluxed, BIO-2007817 was also tested in the HELA cells with and without valspodar (MDR1 inhibitor). No change in Parkin translocation rate was observed in the presence of valspodar, suggesting that the lack of compound effect on Parkin translocation is not likely due to MDR1 efflux.
For the MitoQC assay in SH-SY5Y cells, it is possible that efflux of the compound could have played a role in the results. For example, BIO-2007817 has an efflux ratio of 75 in the MDR1-MDCK cell line and an efflux ratio of 1.5 in the BCRP-MDCK line (Table 1). While MDR1 efflux may have played a role for this compound in the SH-SY5Y cells, we found that even compounds with moderate efflux (i.e., BIO-2008550 with efflux ratio 7.8 or BIO-1967173 with efflux ratio 4.1 in MDCK-MDR1 cells) showed no enhanced MitoQC activity. Also, historically high efflux ratios in the MDR1-MDCK cell line have not been found to correlate with poor cellular activity for any of our internal drug discovery programs. We conclude that the compounds should have been present at the site of action in cells.
Assuming that compounds are permeable, one possible explanation for the lack of cellular effect is that there are significant differences between a reconstituted biochemical assay and the complex cellular milieu with other binding partners that could explain the lack of cellular translatability. Another explanation is that the stimulus we are using to depolarize mitochondria is artificially more harsh and abrupt compared to the stress induced in vivo, and thus we have set up an unreasonably short window for compounds to act. But we favor a third hypothesis: that the molecular mechanism of activation we have identified for these small molecule tools does not thermally destabilize Parkin in the same manner as the W403A mutation, and therefore may not act in a manner conducive for enhancing mitophagy. Significant destabilization of autoinhibited Parkin may be required to trigger the threshold signal for mitophagy. Moreover, in the cell, pUb and PINK1 levels rise sharply with damage and once Parkin is fully activated by phosphorylation, the compounds may have little window of opportunity to activate further. Since the compounds do not enhance Parkin translocation to mitochondria, they also do not enhance the downstream Antimycin/Oligomycin-induced mitophagy (Figure 5). Based on these observations, we believe that there may be certain rate-determining steps in the Parkin activation pathway that would be critical for action by a small therapeutic molecule and the compounds identified are not acting at those rate-determining steps. More studies devoted to investigating how to target REP displacement specifically may present a more tractable therapeutic strategy.
Limitations of the study
The activating compounds’ binding sites on Parkin have not been specifically identified, despite efforts to co-crystallize with WT Parkin or perform SPR assays to measure affinity. Additional efforts to better understand how they alter the conformational mechanism of activation of this autoinhibited E3 ligase, include using HD exchange and NMR experiments. Also, we cannot completely exclude the possibility that poor cell accessibility is the culprit for lack of cell-based activity. Using an in-organello assay (Tang et al., 2017), one could lyse cells with depolarized mitochondria and isolate mitochondria to check for the effect of the activators on Parkin target-substrate ubiquitination, such as Mitofusin. Similar experiments would help understand the lack of cellular effect of the compounds.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Ser65 ubiquitin | Cell Signaling Technology | Cat#: 62802; RRID: AB_2799632 |
| Parkin (Prk8) | Cell Signaling Technology | Cat#: 4211; RRID: AB_2159920 |
| ubiquitinated proteins (FK2; unconjugated) | Sigma | Cat#: 04-263; RRID: AB_612093 |
| IRDye 800 CW goat-α-rabbit IgG secondary antibody | Li-Cor Biosciences | Cat#: 926-32211; RRID: AB_621843 |
| IRDye 680RD goat-α-mouse IgG secondary antibody | Li-Cor Biosciences | Cat#: 926-68070; RRID: AB_10956588 |
| Tb-α-ubiquitin-FK2 Antibody | Cisbio labelled Enzo antibody | Custom labelled Cat# BML-PW8810; RRID: AB_10541840 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco's phosphate-buffered saline (DPBS) | Thermo Fisher Scientific | Cat#: 14190144 |
| RIPA Buffer (10x) | Cell Signaling Technology | Cat#: 9806 |
| Dosium dodecyl sulfate solution (10%) | Millipore Sigma | Cat#: 71736 |
| TBS Tween-20 Buffer (TBS/T; 20x) | Thermo Fisher Scientific | Cat#: 28360 |
| Protease and phosphatase inhibitor cocktail | Thermo Fisher Scientific | Cat#: 78441 |
| MSD Blocker A | Meso Scale Discovery | Cat#: R93BA-4 |
| Read Buffer T with surficant | Meso Scale Discovery | Cat#: R92TC |
| 4x LDS Sample Buffer | Thermo Fisher Scientific | Cat#: B0007 |
| 10x Sample Reducing Reagent | Thermo Fisher Scientific | Cat#: B0009 |
| MOPS SDS Running Buffer | Thermo Fisher Scientific | Cat#: NP0001 |
| 4-12% bis-tris mini protein gel, 15-well | Thermo Fisher Scientific | Cat#: NW04125BOX |
| 4-12% bis-tris mdni protein gel, 26-well | Thermo Fisher Scientific | Cat#: NXP42026BOX |
| iBlot 2 transfer stacks, PVDF | Thermo Fisher Scientific | Cat#: IB24001 |
| SuperBlock (TBS) Blocking Buffer | Thermo Fisher Scientific | Cat#: 37535 |
| Attachment Factor Protein (1x) | Thermo Fisher Scientific | Cat#: S006100 |
| DMEM/F-12, no glutamine | Thermo Fisher Scientific | Cat#: 21331020 |
| DMEM, high glucose | Thermo Fisher Scientific | Cat#: 11965092 |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat#: 35050061 |
| Sodium Pyruvate (100 mM) | Thermo Fisher Scientific | Cat#: 11360070 |
| MEM Non-Essential Amino Acids Solution (100x) | Thermo Fisher Scientific | Cat#: 11140050 |
| Penicillin-Streptomycin (100x) | Thermo Fisher Scientific | Cat#: 15140122 |
| Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) | Millipore Sigma | Cat#: 215911-250MG |
| Antimycin A | Millipore Sigma | Cat#: A8674-25MG |
| Oligomycin A | Millipore Sigma | Cat#: 75351-5MG |
| NucBlue Live Cell Reagent (Hoechst 33342) | Thermo Fisher Scientific | Cat#: R37605 |
| NucBlue Fixed Cell Reagent (DAPI) | Thermo Fisher Scientific | Cat#: R37606 |
| Fetal Bovine Serum (FBS) | Sigma-Aldrich | Cat#: 12103C |
| Lipofectamine 3000 transfection reagent | Thermo Fisher Scientific | Cat#: L3000008 |
| pLenti6.3/V5-DEST Gateway cloning kit | Thermo Fisher Scientific | Cat#: V53306 |
| pLenti4/V5-DEST Gateway cloning kit | Thermo Fisher Scientific | Cat#: K498000 |
| QuikChange LightningTM mutagenesis kit | Agilent | Cat#: 210519 |
| PhosTagTM gels | Thermo Fisher Scientific | Cat#: NC0707348 |
| AnykDTM CritereonTM gels | BIO-RAD | Cat#: 5671125 |
| InstantBlue® Coomassie protein stain | Abcam | Cat#: ab119211 |
| BSA | Sigma Aldrich | Cat# A2153-100G |
| ATP Solution (100 mM) | Thermo Fisher | Cat# R0441 |
| UltraPure DNAse/RNase-Free Distilled Water | Thermo Fisher | Cat# 10977015 |
| UltraPure™ 0.5M EDTA, pH 8.0 | Thermo Fisher | Cat# 15575020 |
| LanthaScreen TR-FRET dilution buffer, 1X | Thermo Fisher | Cat# PV3574 |
| L-rhamnose | Millipore Sigma | Cat#: W373011 |
| E1: His6-ubiquitin E1 Enzyme (UBE1) | Boston Biochem | Cat# E-304 |
| E2: UbcH7/UBE2L3 | Boston Biochem | Cat# E2-640 |
| E3: Untagged Parkin E3 ubiquitin Ligase | Boston Biochem | Cat# E3-160 |
| Fluorescein-ubiquitin (F-Ub): ubiquitin N-Terminal Fluorescein | Boston Biochem | Cat# U-580 |
| ubiquitin: Human Recombinant ubiquitin | Boston Biochem | Cat# U-100H |
| Phospho-ubiquitin (pUb): Human Recombinant pSer65-Ubiquitin | Boston Biochem/in-house Biogen stocks | Cat# U-102 |
| Untagged W403A E3, hyperactive mutant Parkin | Boston Biochem | Cat# E3-162 |
| Untagged C431S E3, catalytically inactive mutant Parkin | Boston Biochem | Cat# E3-164 |
| Blasticidin | Thermo Fisher | A1113902 |
| Polybrene | Santa Cruz Biotechnology | sc-143220 |
| cOmpleteTM EDTA-free protease inhibitor cocktail | Millipore Sigma | Cat#: 5056489001 |
| AcTEV | Thermo Fisher Scientific | Cat#: 12575 |
| ubiquitin | R & D Systems | Cat#: U-100H |
| ubiquitin Vinyl sulfone (Ub-Vs) | R & D Systems | Cat#: U202050 |
| Experimental models: organisms/strains | ||
| HeLa | ATCC | CCL-2 |
| SH-SY5Y | ATCC | CRL-2266 |
| Recombinant DNA | ||
| pCMV-VSVG | RRID: Addgene_8454 | |
| pMDLg/pRRE | RRID: Addgene_122251 | |
| pRSSV-Rev | RRID: Addgene_12253 | |
| Other | ||
| 96-well High Bind SECTOR plates | Meso Scale Discovery | Cat# L15Xa |
| CellCarrier-96 Ultra Microplates | Perkin Elmer | Cat# 6055300 |
| HisTrap Crude FF column | Cytiva Lifesciences | Cat#: 17528601 |
| HiLoad 26/60 Superdex 200 pg | Cytiva Lifesciences | Cat#: 28989336 |
| HiPrep 26/10 desalting column | Cytiva Lifesciences | Cat#: GE17-5087-01 |
| MBPTrap column | Cytiva Lifesciences | Cat#: 28-9187-8 |
| HiTrap Q HP column | Cytiva Lifesciences | Cat#: 17115401 |
| Prometheus nanoDSF (Differential Scanning Fluorimetry) | Nanotemper | |
| 384-well Low Flange Black Flat Bottom Polystyrene NBS Microplate, without Lid, Nonsterile | Corning | Cat# 3575BC |
| 384W plate | Greiner | Cat# 784201 |
| ECHO Plate | Labcyte | Cat# LP-0200 |
| Adhesive Plate Foils | Thermo Fisher | Cat# AB0626 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be fulfilled by lead contact Laura Silvian (laura.silvian@biogen.com).
Materials availability
Some compounds, plasmids and cell lines are available for distribution.
Experimental model and subject details
Cell culture and cell line development
All tissue culture was carried out in a humidified incubator at 37°C and 5% CO2. HeLa cells were cultured in DMEM supplemented with 10% FBS, GlutaMAX, 1 mM sodium pyruvate, and 100 U/mL each penicillin and streptomycin. 293LV cells were cultured in the same media with additional supplemental NEAA. SH-SY5Y were cultured in DMEM/F12 supplemented with 10% FBS, GlutaMAX, 1 mM sodium pyruvate, and 100 U/mL each penicillin and streptomycin.
GFP-Parkin HeLa and MitoQC SH-SY5Y stable cell lines were generated by infection with lentivirus carrying the respective gene of interest in 8 μg/mL polybrene overnight. Following 48 h recovery in complete media, cells were selected with the appropriate antibiotic. Monoclonal lines were further isolated by limiting dilution. Fluorescent reporter expression and homogeneity were characterized by flow cytometry on an LSR II flow cytometer (BD Biosciences).
Method details
Parkin catalytic poly-auto-Ub TR-FRET assay
The first step in establishing an autoubiquitination assay for Parkin was to understand its dependence on pUb and time course, as our goal was to identify a window for further activation by a small molecule. All Parkin WT and mutant enzymes used in the TR-FRET and western blot assays were purchased from Boston Biochem and contain no N-terminal tag (see key resources table). The reaction was performed with various pUb concentrations at 2 h incubation and the EC50 was calculated. We chose experimental parameters to generate a window to identify hits while enabling both activators and PAMs to be identified.
The Parkin TR-FRET reaction was performed in 50 mM HEPES pH 7.4, 100 mM NaCl, 5 mM MgCl2, 0.01% Tween 20, 1% DMSO, 0.02% BSA. A 2X solution of E1, E2 and Parkin (40 nM E1, 800 nM E2 and 100 nM Parkin E3 in assay buffer) was added and incubated at 37°C for 30 min. Then, a 2X substrate mixture was added (760 nM Fluorescein-ubiquitin (F-Ub), 50 μM ubiquitin (Hou et al.), various concentrations of phospho-ubiquitin (pUb) from 0 to 37.5 μM and 2 mM ATP in assay buffer and incubated for 2 h at 37°C, at which point a 2X detection reagent mixture (30 nM Terbium-FK2 Antibody, 40 mM EDTA in TR-FRET dilution buffer) was added. 1 h after addition of the detection reagent, the plate was read in an EnVision reader with excitation at 340 nm and emission at 520 nm/495 nm. TR-FRET ratio was calculated as 520 nm/495 nm. The readout values (104× 520 nm/495 nm) for each pUb point were then normalized with the value of the 0% activity control and 100% activity control (measured using W403A-Parkin) on the same plate set to 0% activity and 100% activity, respectively. The EC50 value for pUb was calculated at 6 μM with Hill slope of 2.7.
The Parkin TR-FRET reaction at a fixed compound concentration was performed in 50 mM HEPES pH 7.4, 100 mM NaCl, 5 mM MgCl2, 0.01% Tween 20, 1% DMSO, 0.02% BSA. Compounds were diluted and prepared on an ECHO liquid handler and the plate was spun at 1000 rpm for 30 sec. A top compound concentration was set at 200 μM to identify weak activators. 0% activity control was established as WT-Parkin + DMSO + EC50 concentration of pUb (5 μM). 100% activity control was 50 nM W403A-Parkin + DMSO + EC50 concentration of pUb. A 2X concentration of the compounds were dispensed to the assay plates by ECHO. On top of the compounds, a 2X solution of E1, E2 and either WT-Parkin mixture (40 nM E1, 800 nM E2 and 100 nM WT-Parkin E3 in assay buffer) or W403A-Parkin mixture (40 nM E1, 800 nM E2 and 100 nM W403A-Parkin E3 in assay buffer) was added and incubated for 30 min at 37°C. Then, a 2X substrate mixture was added (760 nM Fluorescein-ubiquitin (F-Ub), 50 μM ubiquitin (Hou et al.), 10 μM phospho-ubiquitin (pUb) and 2 mM ATP in assay buffer and incubated for 3 h at 37°C, at which point a 2X detection reagent mixture (15 nM Tb-FK2 Antibody, 40 mM EDTA in TR-FRET dilution buffer) was added. 1 h after addition of the detection reagent, the plate was read in an EnVision reader with excitation at 340 nm and emission 520 nm/495nm. TR-FRET ratio was calculated as 520 nm/495 nm. The readout values (104× 520 nm/495 nm) for each compound point were then normalized with the value of the 0% activity control and 100% activity control on the same plate set to 0% activity and 100% activity, respectively.
| Activity% = (Well data −0% Activity) / (100% Activity - 0% Activity) × 100% |
To establish concentration curves, percent activity versus the compound concentration data were fit to the following 4 parameter logistic model (XLFIT5 205):
where UA and LA are upper and lower asymptotes. EC50 values (the concentration that leads to 50% maximal response) were generated from full concentration response curves fitted without fixing any of the 4 parameters and were typically failed if curve linear regression (CLR) ≥ 5 and compounds scheduled for re-test in the next cycle. In addition to EC50 values for full curves, EC35 values (the concentration that leads to 35% maximal response) were calculated from the fit parameters using the following equation
Compound clustering and analysis
A structural similarity map for all validated hits was generated to assemble the hits into groups with similar chemotypes. The default definition of Unity fingerprints is a 998 bit keyset and takes into account the presence or absence of structural features as well as the connectivity path between atoms to access similarity between molecular structures. These fingerprints can be used to characterize molecules and are used for substructure searching, clustering and diversity analysis. The distance between points in the map is a reflection of similarity between chemical matter and can be used for clustering, while the axis of the plot has no meaning. UNITY is distributed by Certara, Inc.
Protein expression and purification
Bacterial cloning and expression of Parkin was first described by Chaugule et al. (2011). For use in Ub-VS and nano DSF thermal shift assays, Parkin full length (1-465) coding sequence was optimized for bacterial expression with a His10-SUMO-TEV-His6-Avi tag at the N-terminus and a double stop codon at the C-terminus was cloned in-frame at the NcoI and XhoI sites of a pET28a vector (Novagen) for expression in Escherichia coli. W403A and C431S mutants were generated by polymerase chain reaction using the QuikChange LightningTM mutagenesis kit (Agilent, Santa Clara, CA) with the pet28[Parkin-pET28a] wild-type construct as the template. Pediculus humanus PINK1, PhPINK1 (codon optimized for bacterial expression), kinase domain (amino acids 128-575) N-terminally tagged with a His10-MBP-TEV sequence was cloned into a rhaB promoter-driven vector. Various reports have shown that N-terminally tagged Parkin is more active than untagged Parkin (Burchell et al., 2012; Chaugule et al., 2011; Matsuda et al., 2006). This could be due to pulling on, and releasing the autoinhibitory Ubl domain. We tested untagged (Boston Biochem – purchased) and tagged (in-house produced) Parkin in the TR-FRET assay and did not see a difference in activity.
WT-Parkin plasmid was transformed into TAP435 – an in-house modified protease-deficient BL21 (DE3) E. coli strain – and a single colony was then grown overnight at 37°C shaking at 225 rpm in LB supplemented with 50 μg/mL Kanamycin. Large scale cultures were inoculated with overnight cultures and incubated at 30°C shaking at 180 rpm until OD600 = 0.4 at which point the temperature was lowered to 16°C and shaking speed reduced to 160 rpm. Protein expression was induced at OD600 = 0.6–0.8 with 50 μM IPTG supplemented with 500 μM ZnCl2 and followed overnight at 16°C shaking with at 160 rpm. Cells were then harvested by centrifugation (5,653 × g for 20 min at 4°C) and resuspended in buffer A (50 mM Tris, 500 mM NaCl, 1 mM TCEP, pH 8.0) supplemented with 30 mM imidazole, Benzonase (Millipore) and cOmpleteTM EDTA-free protease inhibitor cocktail tablets (Roche, 6 mL of lysis buffer was used per g of cells) and lysed by Microfluidizer (15,000 psi for 3 passes). The soluble fraction, collected by centrifugation at 44,000 × g for 30 min at 4°C, was applied to a 5 mL pre-packed HisTrap Crude FF column (Cytiva Lifesciences) pre-equilibrated with buffer A supplemented with 30 mM imidazole. Following sample loading, the column was washed with 10 column volumes of buffer A supplemented with 30 mM imidazole and 1 mM ATP – to release nonspecifically bound chaperones of similar molecular weight to Parkin – and Parkin was eluted in the presence of buffer B (buffer A with 250 mM imidazole). The eluted protein was dialyzed overnight in a 30 kDa MWCO dialysis cassette (Amicon) against buffer C (25 mM Tris-HCl, 75 mM NaCl, 1 mM TCEP, pH 8.0) in the presence of 1:50 molar ratio AcTEV (ThermoFisher Scientific # 12575). Dialyzed protein was re-loaded onto to a 5 mL pre-packed HisTrap Crude FF column (Cytiva Lifesciences) equilibrated with buffer D (50 mM Tris-HCl, 500 mM NaCl, 1 mM TCEP, pH 8.0) and eluted with buffer E (buffer D with 50 mM imidazole). Pooled protein was concentrated, applied to a 26/600 Superdex 200 pg gel filtration column (Cytiva Lifesciences) equilibrated in 25 mM Tris-HCl, 150 mM NaCl, 1 mM TCEP, 10% glycerol, pH 7.5 and stored at −80°C. W403A and C431S mutant proteins were expressed and purified using the same protocol as WT-Parkin. Sample molecular weight and purity were confirmed by mass spectrometry.
PhPINK1 transformation, pre-cultures and large-scale culture setup was similar to that of Parkin. Protein expression was induced at OD600 = 0.6–0.8 with 0.2% (w/v) L-rhamnose (Sigma, #W373011) for 20 h at 18°C with shaking at 160 rpm. Cells were harvested, lysed (lysis buffer: buffer A with 30 mM imidazole, 10% (v/v) glycerol; supplemented as above with Benzonase and protease inhibitors), centrifuged and loaded onto HisTrap Crude FF column as detailed above. PhPINK1 eluted with buffer B was loaded onto a 5 mL pre-packed MBP Trap (Cytiva Lifesciences) column pre-equilibrated with 20 mM Tris,150 mM NaCl, 1 mM TCEP, 10%(v/v) glycerol, pH 7.8. PhPINK1, eluted in the presence of 30 mM maltose, was pooled, concentrated and loaded onto a 26/600 Superdex 200 pg gel filtration column (Cytiva Lifesciences) equilibrated in 20 mM Tris, 150 mM NaCl, 1 mM TCEP, 10% (v/v) glycerol, pH 7.4, and finally stored at −80°C. Sample molecular weight and purity were confirmed by mass spectrometry.
pUb was generated from purified Ub (Boston Biochem #U-100H) (Pao et al., 2016). After phosphorylation with PhPINK1 (0.7 mM Ub: 0.7 μM PhPINK1 were incubated for 18 h at 37°C in 20 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 5 mM ATP, 1 mM TCEP, pH 7.4), samples were buffer exchanged into 20 mM Tris pH 8.8 using a HiPrep 26/10 desalting column (Cytiva Lifesciences) and loaded onto pre-packed, tandem 1 mL MBPTrap (for PhPINK1 binding) and 5 mL HiTrapQ (for Ub vs pUb separation) columns. pUb was eluted with 50 mM Tris pH 7.4. pUb (90% phosphorylation efficiency) was confirmed for mono-phosphorylation by Phos-tagTM gels (Fujifilm Wako Chemicals, 12.5% acrylamide run for 1 h at 200V) and mass spectrometry.
In vitro ubiquitin vinyl sulfone (Ub-VS) assay
To assess the effect of compounds on Ub-VS-charging of Parkin at a fixed pUb concentration, 2 μM Parkin (either N-terminally tagged WT or W403A mutant) were incubated in the presence of 10 μM Ub-VS (R&D Systems #U202050), 3 μM pUb, at the following compound concentrations (0 μM, 0.09 μM, 0.27 μM, 0.8 μM, 2.5 μM, 7.4 μM, 22.2 μM, 66.7 μM, and 200 μM) in 50 mM HEPES, 150 mM NaCl, 1 mM TCEP, pH 8.0 (2% (v/v) DMSO final). Reactions were quenched by addition of reducing Laemmeli sample buffer following incubation at 25°C for 2 h (for WT-Parkin) or 1 h (for W403A-Parkin). Samples were loaded onto AnykDTM CriterionTM gels (Bio-rad), ran for 1 h at 120V, visualized with InstantBlue® Coomassie protein stain (Abcam # ab119211) and analyzed using ImageJ software (NIH, USA).
To investigate the role of pUb in Ub-VS-charging of Parkin in the presence or absence of compounds (Figure S4), assays were set up as follows: 2 μM Parkin (either unphosphorylated WT or W403A or C431A mutants), 10 μM Ub-VS (R&D Systems #U202050), pUb at the following concentrations (0 μM, 0.025 μM, 0.05 μM, 0.1 μM, 0.2 μM, 0.4 μM, 0.8 μM, 1.6 μM, 3.1 μM, 6.25 μM, 12.5 μM, 25 μM), compound (200 μM or DMSO) in 50 mM HEPES, 150 mM NaCl, 1 mM TCEP, pH 8.0 (2% (v/v) DMSO final) were incubated for 1 h at 25°C. Sample quenching, loading and analysis were done as detailed above.
Molecular cloning for mammalian expression
The codon-optimized sequence for GFP-Parkin was synthesized at Thermo Fisher Scientific based on the construct described by Trempe et al. (2013) (Addgene #45875), provided in pDONR221 and subsequently cloned into pLenti6.3 using Gateway cloning. GFP-Parkin-(W403A) and GFP-Parkin-(C431A) variants were also prepared at Thermo Fisher Scientific, and subsequently subcloned into pLenti6.3 using Gateway cloning. The codon-optimized sequence for MitoQC was similarly synthesized at Thermo Fisher Scientific based on the construct described by Allen et al.(2013), provided in pDONR221 and subsequently cloned into pLenti4 using Gateway cloning.
Lentivirus preparation
HEK 293FT were seeded into 150 mm dishes and recovered for 48 h, or until about 70% confluent. Cells were transfected with Lipofectamine 3000 and 15 μg each of lentivector containing the gene of interest and packaging plasmids pCMV-VSVG, pMDLg/pRRE, and pRSV-Rev under serum-free conditions for 6h before changing to antibiotic-free culture media. Conditioned media was harvested at 24 and 48 h post-transfection, and clarified by centrifugation at 3,000 × g for 5 min. Lentivirus was concentrated from clarified conditioned media by ultracentrifugation at 19,000 × g for 90 min, resuspended in 200 μL antibiotic-free culture media, and stored at −80°C.
Generation of cell lines
HeLa cells purchased from ATCC (Cat# CCL-2™) expressing GFP-Parkin wildtype and the variants W403A and C431A were generated by spin infection for 60 min at 800 ×g in media containing lentivirus plus 8 μg/mL polybrene. Following 48 h recovery in complete media, infected cells were selected with 10 μg/mL Blasticidin for 3 weeks. Expanded cultures underwent 3 rounds of FACS sorting (gating for top 10–20% of the GFP MFI) to select for a population of cells expressing Parkin wildtype or variants with similar GFP intensity.
Parkin autoubiquitination and Miro1 ubiquitination assays
Untagged Parkin used in FRET and Western blot assays was purchased from Boston Biotech/R&D systems. To determine concentration response curves, the Parkin autoubiquitination reaction was performed exactly as for the TR-FRET single point assay except that the volume of the reaction was scaled up to 40 μL, and the reaction was stopped after 5 min before addition of 1x SDS sample buffer. Miro1 ubiquitination assay was performed following the same protocol as the TR-FRET assay where Miro1 (250 nM final concentration) was included in step 1 (E1, E2, E3, and His-Miro1186-591– exposed to compounds for 30 min) followed by the step 2 addition for pUb, Ub and ATP to initiate the reaction. Then the mix was incubated for 20 min before addition of 1x SDS sample buffer. W403A Parkin was used as a positive control for Miro1 ubiquitination and followed the W403A TR-FRET protocol and was incubated for 30 min.
Parkin translocation assay
GFP-Parkin expressing HeLa cells were seeded at 1.6 × 104 cells per well in CellCarrier-96 Ultra plates pre-coated with Attachment Factor and allowed to recover for 24 h. Nuclei were stained with Hoechst 33342 for 30 min and media replaced. Cells were pre-treated for 1 h with compound before adding 20 μM CCCP and immediately imaged every 10 min for 3 h on an Opera Phenix high content screening system at 20x, with environmental controls set to 37°C and 5% CO2. For each condition, 9 fields were imaged in each of 4 replicate wells.
MitoQC
SH-SY5Y cells expressing the MitoQC construct were seeded at 1.6×104 cells per well in CellCarrier-96 Ultra plates pre-coated with Attachment Factor and allowed to recover for 72 h. Cells were pre-treated for 1 h with compound before adding 5 μM Antimycin and 10 μM Oligomycin or DMSO for an additional 8 h except where indicated otherwise. Cells were fixed by addition of 4% paraformaldehyde (PFA) solution in DPBS for a final concentration of 2% PFA, and incubation on ice for 30 min. Nuclei were stained with DAPI for 15 min. Fixed cells were imaged in DPBS on an Opera Phenix high content screening system at 64x. For each condition, 25 fields were imaged in each of 4 replicate wells.
Cell lysate preparation for immunoassay
Following treatment, cells were washed in DPBS and lysed in RIPA lysis buffer containing 0.1% SDS and 1x protease-phosphatase inhibitors. Lysates were normalized according to total protein determined by BCA assay (Bicinchoninic Acid Kit; sigma).
Western blotting
For Western blotting, lysates were prepared for SDS-PAGE with sample buffer and reducing reagent and heated at 70°C for 15 min. SDS-PAGE was carried out at 180V on 4–12% bis-tris gels with MOPS. Gels were transferred to PVDF membranes on iBlot2 gel transfer device using program 3. Blots were blocked with SuperBlock (Thermo Fisher Scientific) blocking buffer for 1 h before overnight incubation with primary antibodies at the following dilutions: pSer65-Ub 1:1,000; Parkin 1:1,000–1:5,000. IR Dye-conjugated secondary antibodies (1:10,000) were incubated for 1 h. Blots were imaged on an Odyssey CLx imager.
Lipophilicity determination of compounds at pH 7.4
Samples were thawed, centrifuged, and sonicated in a 40°C water bath to facilitate dissolution. Following sonication, a 5 μL aliquot of each of the 10 mM solutions was volumetrically diluted ten-fold to 1 mM in a 1:1 v/v methanol:water solution. The HPLC-logD assay was performed in singleton. An Agilent 1100 HPLC equipped with a micro-well plate autosampler, quaternary HPLC pump, and diode array detector was used for analysis. Each sample (5 μL) was injected onto the column (Supelcosil LC-ABZ, 5 μM 50 × 4.6mm) and eluted using a series of three isocratic methods, each with decreasing ratios of organic phase to aqueous phase; the ratios ranged from 70:30 organic:aqueous to 15:85 organic:aqueous. The isocratic methods used are dependent on the expected logD of the sample compound, with classifications of high (>3), mid-range (1–3), and low (<1) logD. The aqueous mobile phase was comprised of nitrogen-purged 20 mM MOPS pH 7.4 buffer in octanol-saturated water with 0.15% n-decylamine, while the organic mobile phase consisted of methanol with 0.25% octanol. Data were collected at 235, 255, 265, 275, and 310 nm. Results are reported using data obtained at 265 nm. Atenolol was included as a t0 internal standard and three assay standards were used for each of the three logD ranges as described above.
Using the retention times of each sample relative to the atenolol t0, a capacity factor k’ is obtained for each isocratic method’s organic phase concentration where k’ = (tr− t0)/t0. Taking the logarithm of each sample’s k’ values and extrapolating to 0% organic phase yields a log(k'w) value. This log(k'w) is then converted to a logD value using a generic calibration curve developed using compounds spanning from values ranging −1 < logD <6.1. The equation used for the conversion is logDoct = 1.1267 ∗ log k’ + 0.2075.
Solubility of compounds at pH 6.8
Samples were thawed, centrifuged, and sonicated in a 40°C water bath to facilitate dissolution. Each supplied 10 mM sample in DMSO was diluted 50-fold in each specified assay media in singleton using a Hamilton Starlet robotic liquid handler and placed directly in a Millipore solubility filter plate with 0.45 μm polycarbonate filter membrane for a final DMSO concentration of 2%. Each well was mixed by repeated pipetting and the filter plate was sealed. The dilutions were incubated at ambient temperature for 24 h on a rotary shaker at 200 rpm. Following incubation, the dilutions were vacuum-filtered and the filtrates were collected for analysis by Direct-Infusion CLND V. Filtrates were injected into the nitrogen detector for quantitation. The equimolar nitrogen response of the detector was calibrated using standards which span the dynamic range of the instrument from 0.08 to 4500 μg/mL nitrogen. The filtrates were quantified with respect to this calibration curve. The results are reported electronically in both μM and μg/mL.
Plasma protein binding of compounds
Samples were diluted to 125 μM (5 μL 10mM stock to 395 μL acetonitrile) in a 1 mL 96-well plate (Waters 186002481 Milford, MA). Frozen (rat, human) plasma (BIOIVT, Westbury, NY) was thawed and PBS buffer was warmed in a 37°C water bath. Samples were further diluted to 1 μM (8 μL 125 μM stock to 992 μL of plasma) in a 2 mL 96-well plate (Costar 3961) and mixed thoroughly. Internal standard (200 μL of 25 ng/mL solution of CPDPX (8-Cyclopentyl-1,3-dipropylxanthine, Sigma-Aldrich, C101) in 1:1 acetonitrile/methanol solvent solution) was transferred to a 1 mL 96-well plate and chilled on ice. Warmed PBS buffer (500 μL) was transferred to the white (open) side of the rapid equilibrium dialysis (RED) device (Thermo Scientific, Rockford IL, baseplate cat# 89811, insert cat# 89810) and 300 μL of the spiked plasma was added to the corresponding red ring (dialysis chamber) side of the RED device. From the remaining 1 μM stock, 50 μL (T = 0 h) was removed and placed into a crash plate containing 200 μL internal standard. Samples were matrix matched with 50 μL of blank buffer added to the crashed sample. The remaining 1 μM test article in plasma plate was covered with a lid and maintained at 37°C for 4 h. All the RED device plates were covered with a lid and transferred to a 37°C incubator with 5% CO2 environment and shaken at 200 rpm for 4 h.
After 4 h, 50 μL of sample (plasma or buffer sample) and 50 μL of the opposite blank matrix (add blank buffer to the plasma samples and blank plasma to the buffer samples) were transferred to crash plates containing 200 μL internal standard and mixed thoroughly. From 1 μM test article in plasma, 50 μL (T = 4 h) of each plasma sample was removed and placed into a crash plate. All samples were matrix matched, adding 50 μL of blank buffer to the crashed sample. Crashed plates were centrifuged at 3,900 rpm for 10 min at 4°C (Eppendorf Centrifuge 5810R, Hamburg, Germany).
Supernatant from the crash plates (30 μL) was transferred to 384-well plates containing 120 μL of 0.1 formic acid in 90:10 water:acetonitrile and injected into the LC/MS.
Calculation of %Free (Plasma Protein Bindingunbound) (% PPBunb)
PAR – Peak area ratio (PAR)
Fu = fraction unbound
Stability in plasma
When % stability outside the RED device is <50% then the %binding results are not reported (due to inaccuracies).
MDCK-MDR1 assessment of compound permeability in cells
MDCK-MDR1 (transfected Madin-Darby canine kidney cells, over-expressing P-gp/MDR1, licensed from Absorption Systems/NIH) cells were cultured and plated for a minimum of seven days according to the manufacturer’s recommendations. Once a cell flask became ∼95% confluent, all media was aspirated and washed once with 5 mL trypsin, then aspirated. ∼2 mL Trypsin (depending on size of flask) was added and incubated for 15–20 min until cells fully lifted and separated. Cells were removed from the flask using 10 mL of warmed culture media and dispensed into a sterile conical tube. Cells were centrifuged for 5 min at 1,500 rpm 4°C. Trypsin was aspirated off and pellet resuspended in fresh warm media. Using cell dye, live cells were counted and diluted to 0.28× 106 cells/mL. 75 μL of diluted cells were dispensed to the permeable insert of a Corning 96 transwell plate in the apical chamber and 250 μL of warmed culture media was added into the receiver well of the plate. Cell plates were placed in an incubator at 37°C and maintained for 7 days before use in assays. Media was changed twice before cells were used for assays. While changing media, the membrane should not be poked nor the cellular monolayer disturbed.
On day seven of cell culture in the transwell plate, the plates were washed (support and receiver wells) with a warm 1X dilution of Hanks’ Balanced Salt Solution (HBSS) with 1% HEPES (pH 7.4) for a total of three rinses. To confirm integrity of the monolayer after washing, the transepithelial electrical resistance (TEER) was measured with 300 μL of buffer in the receiver plate and 85 μL in the permeable support insert. TEER values prior to running assay had to be over 430 ohm∗cm2. TEER values must remain consistent for a post-assay evaluation.
10 mM compound stocks were diluted to 200 μM in dimethyl sulfoxide (DMSO). Compounds were placed on a Tecan liquid handler, where an additional dilution in warm 1X dilution of Hanks’ Balanced Salt Solution (HBSS) with 1% HEPES (p.H 7.4) brought the stock concentration to 1 μM. Transwell plates were then emptied of buffer prior to being placed on the Tecan. The liquid handler then added 85 μL of 1 μM compound to the permeable support of the A>B wells and 250 μL of buffer with 1%BSA to the receiver wells. Next, 260 μL of 1 μM solution was added to the receiver wells of the B>A plate and 75 μL of buffer with 1%BSA to the permeable support wells. For the T = 0 timepoint, 10 μL samples from both sets of donor wells were immediately removed and added to a 384-well plate with 40 μL of buffer with 1% BSA. The T = 0 timepoint was halted with the addition of 100 μL ice-cold acetonitrile with 25 ng/mL internal standard (CPDPEX 8 cyclopentyl-1,3- dipropylxanthine- Sigma-RBI) to those wells.
MDR1 plates were incubated 37°C on a shaker for 2 h. At 2 h, 10 μL samples from the donor wells were transferred into a 384-well plate with 40 μL buffer with 1.0% BSA to sample wells (the T = 120 timepoint). 50 μL samples from the receiver wells were put into a 384-well plate. Samples were quenched by addition of 100 μL of ice-cold acetonitrile with 25 ng/mL internal standard. Cell plates were washed a final time to remove compound. TEER values measured post-assay must be again over 430 ohm∗cm2. 384-well sample plates were spun; 50 μL of corresponding supernatant was transferred to a 384-well plate and mixed with 50 μL Millipore water. The samples were analyzed by multiple reaction monitoring on LC/MS/MS with or without RapidFire. The mobile phases were the following: A) 0.1% formic acid in distilled, ionized water, B) 0.1% formic acid in acetonitrile/methanol. Column or cartridge types were C8, C18, or Phenyl.
The substrate identification for Human MDR1 efflux transporter assay measures the bidirectional transport of test article in polarized cell monolayers. Apparent permeability is calculated bi-directionally using 96-well permeable support plates. LC-MS/MS raw data for a 384-well plate is returned as integrated analyte peak areas and IS areas used in the quench solution for the reaction. To correct for possible differences in analyte extraction from the reaction mixture, ionization and detection variability, the Analyte:Internal standard peak area ratio (PAR) is used for all data comparisons. All IS peak areas should ideally be within ±30% CV of the average of the IS peak areas obtained throughout a test plate. %CV>30%, especially at higher concentrations of NCE, may be indicative of ionization suppression or mass interference.
DQ/dt = permeability rate
C0 = initial concentration on the donor side
A = surface area of the filter
B>A = basolateral to apical
A>B = apical to basolateral
Vr = volume of the receiver compartment in cm3
Vd = volume of the donor compartment in cm3
C0 = calculated experimental concentration (Time = 0) of the dosing solution in μM
Crfinal = concentration of the receiver in μM at the end of the incubation period
Cdfinal = concentration of the donor in μM at the end of the incubation period
Thermal shift assay
To measure the thermal stability of Parkin in the presence or absence of compound, we determined the thermal unfolding melting point (Tm) of 2 μM N-terminally tagged WT-Parkin either in the presence of DMSO or 200 μM compound in reaction buffer (20 mM HEPES, 150 mM NaCl, 1 mM TCEP, 0.05% Tween-20, pH 7.4). Care was taken to match DMSO concentration to 2%(v/v) in all conditions. 10 μL reaction volume was used for each High-Sensitivity capillary. Change in fluorescence intensity was monitored at 350 nm and 330 nm with Nanotemper’s Prometheus nanoDSF (Differential Scanning Fluorimetry) in a temperature gradient from 20 to 95°C using a ramp of 1°C/min. Similarly, thermal unfolding of 2 μM N-terinally tagged W403A-Parkin in reaction buffer including 2% (v/v) DMSO was monitored. Experiments were repeated with n = 3 for all compounds.
Synthesis and compound QC
All solvents and chemicals used were reagent grade. Anhydrous solvents were purchased from Sigma-Aldrich and used as received. Analytical thin layer chromatography (TLC) and silica gel column chromatography were performed on Merck silica gel 60 (230–400 mesh). Removal of solvents was conducted by using a rotary evaporator and residual solvents were removed from non-volatile compounds using a vacuum manifold maintained at approximately 1 Torr. NMR spectra were recorded on a Bruker Advance 400 MHz, 500 MHz and 600 MHz NMR spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to residual undeuterated solvent as internal reference and coupling constants (J) are reported in hertz (Hz). Splitting patterns are indicated as follows: s = singlet; d = doublet; t = triplet; q = quartet; qn = quintet; dd = doublet of doublets; dt = doublet of triplets; tt = triplet of triplets; m = multiplet; br = broad peak. All yields reported are isolated yields. All final compounds were purified to ≥95% purity as determined by LC/MS analysis (using a lineal gradient of elution: 90% water in trifluoroacetic acid (containing 0.1% v/v)/10% CH3CN in trifluoroacetic acid (containing 0.1% v/v) to 10% water in trifluoroacetic acid (containing 0.1% v/v) and 90% CH3CN in trifluoroacetic acid (0.1% v/v) for 2 min and S2 then holding at 10% water in trifluoroacetic acid (0.1% v/v) and 90% CH3CN in trifluoroacetic acid (0.1% v/v) up to 3 min at a flowrate of 3 mL/min (injection volume 5μL and using a Waters Sunfire C18 3.5 mM 4.6 × 20mm IS column)). MS mode: MS:ESI+ scan range 100–1000 daltons. PDA detection 210–400 nm. Final compounds were analyzed using UPLC (Water’s Acquity (Waters Milford, MA)) coupled with an AB Sciex 6600 Triple-TOF mass spectrometer (AB Sciex Framingham, MA). A Water’s Acquity HSS T3 (1.7mm beads, 2.1 × 50mm) column was used for separation. Mobile phase A was water with 0.1% formic acid and mobile phase B was acetonitrile with 0.1% formic acid. The flow rate was 0.45 mL/min and the following gradient was used from 0–0.2 min 5% B and increased linearly to 65% B at 5 min and 90% at 6.1 min and remained there for 0.4 min, dropped back to 5% B over 0.1 min and remained there for 0.4 min. The mass spectrometer was operated in positive ion mode. An electrospray ionization source was used with the following parameters: Ionspray voltage floating 4500 V, ion source gas 1 50 (arbitrary units), ion source gas 2 50 (arbitrary units), curtain gas 30 (arbitrary units), and temperature 500°C.
General procedure A

ethyl (R)-1-(2-((tert-butoxycarbonyl)amino)-3-methylbutyl)-1H-pyrazole-5-carboxylate:
To a solution of triphenylphosphine (6.08 g, 23.19 mmol) in dry THF (75 mL) was added DIAD (4.33 g, 21.41 mmol, 4.20 mL) dropwise at 0°C. The solution was stirred at 0°C for 15 min resulting in the formation of a white precipitate. A solution of ethyl 1H-pyrazole-5-carboxylate (2.50 g, 17.84 mmol) and tert-butyl (R)-(1-hydroxy-3-methylbutan-2-yl) carbamate (3.99 g, 19.62 mmol) in dry THF (25 mL) was then added and the reacting mixture was stirred overnight at room temperature. Silica gel (40 g) was added and the solvent was removed under vacuum. The material was purified by column chromatography on silica gel (10–55% Acetone in heptane) to afford ethyl (R)-1-(2-amino-3-methylbutyl)-1H-pyrazole-5-carboxylate as a white solid. Yield: 5.15 g (89%); UPLC-MS (ESI)+m/z: 327 [M + H]+. 1H NMR (500MHz, CHLOROFORM-d) Shift = 7.48 (d, J = 1.8 Hz, 1H), 6.83 (d, J = 1.8 Hz, 1H), 4.80 (br d, J = 10.4 Hz, 1H), 4.69–4.53 (m, 2H), 4.43–4.22 (m, 2H), 3.94 (m, 1H), 1.90–1.71 (m, 1H), 1.39 (t, J = 7.0 Hz, 3H), 1.31 (s, 9H), 1.01 (d, J = 7.3 Hz, 3H), 0.99 (d, J = 7.3 Hz, 3H).
ethyl (R)-1-(2-amino-3-methylbutyl)-1H-pyrazole-5-carboxylate:
To a solution of ethyl (R)-1-(2-((tert-butoxycarbonyl)amino)-3-methylbutyl)-1H-pyrazole-5-carboxylate in 1,4-dioxane (30 mL) was added a solution of 4N HCl in dioxane (30 mL) at 0°C. The solution was stirred overnight at room temperature then concentrated under vacuum. The precipitate was triturated with Et2O (50 mL), filtered and washed with Et2O. Ethyl (R)-1-(2-amino-3-methylbutyl)-1H-pyrazole-5-carboxylate was obtained as a white solid. Yield: 3.57 g (88%); UPLC-MS (ESI)+m/z: 227 [M + H]+. 1H NMR (500MHz, DMSO-d6) Shift = 8.07 (br s, 3H), 7.70 (d, J = 1.8 Hz, 1H), 6.96 (d, J = 1.8 Hz, 1H), 4.83 (dd, J = 4.9, 14.7 Hz, 1H), 4.60 (dd, J = 8.2, 14.3 Hz, 1H), 4.38–4.24 (q, J = 7.0 Hz, 2H), 3.51 (m, 1H), 1.96–1.77 (m, 1H), 1.31 (t, J = 7.0 Hz, 3H), 1.04–0.92 (m, 6H).
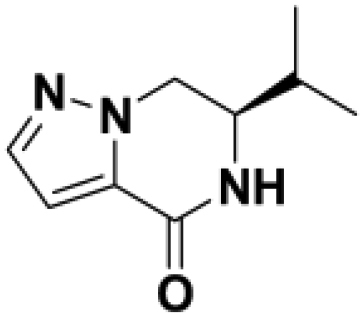
(R)-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one:
A solution of Ethyl (R)-1-(2-amino-3-methylbutyl)-1H-pyrazole-5-carboxylate (3.57 g, 13.64 mmol, Hydrochloride) in sat. NaHCO3 (100 mL) was stirred at room temperature for 48h. The solution was extracted with DCM (4×15 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated to afford (R)-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one as a white solid. Yield: 2.67 g (100%); UPLC-MS (ESI)+m/z: 180 [M + H]+. 1H NMR (500MHz, CHLOROFORM-d) Shift = 7.56 (d, J = 1.8 Hz, 1H), 6.88 (d, J = 1.8 Hz, 1H), 5.93 (br s, 1H), 4.45 (dd, J = 4.6, 12.5 Hz, 1H), 4.19 (dd, J = 9.8, 12.8 Hz, 1H), 3.73 (m, 1H), 1.94 (m, 1H), 1.07 (d, J= 6.7 Hz, 3H), 1.06 (d, J= 6.7 Hz, 3H).
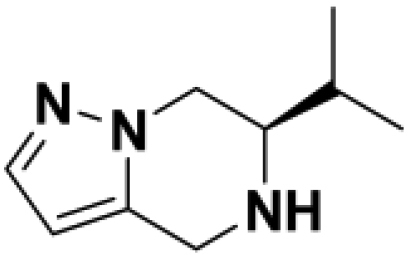
(R)-6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine:
In an oven dried flask, a solution of LiAlH4 (2.0 M in THF, 9.74 mL) was added dropwise to a solution of (R)-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazin-4(5H)-one (2.68 g, 14.98 mmol) in dry THF (40 mL) at 0°C under inert atmosphere. The solution was kept at 0°C for 15 min, then stirred at 65°C overnight. The reacting mixture was cooled to 0°C then carefully quenched with water (0.74 mL), 15% NaOH in water (0.74 mL) and water (3× 0.74 mL). The suspension was stirred at room temperature for 2h, then diluted in Et2O (100 mL) followed by addition of MgSO4 (5 g). After 20 min, the suspension was filtered and washed with Et2O. The organic layer was concentrated affording (R)-6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine as a white solid. Yield: 2.45 g (99%); UPLC-MS (ESI)+m/z: 166 [M + H]+. 1H NMR (400MHz, METHANOL-d4) Shift = 7.44 (d, J = 1.8 Hz, 1H), 6.10–6.00 (m, 1H), 4.25–4.13 (m, 2H), 3.98–3.91 (m, 1H), 3.80 (dd, J= 11.5, 11.5 Hz, 1H), 2.89 (ddd, J = 4.1, 6.6, 10.9 Hz, 1H), 1.93–1.79 (m, 1H), 1.10 (d, J = 7.0 Hz, 3H), 1.06 (d, J = 7.0 Hz, 3H).
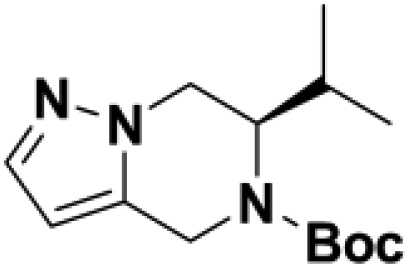
tert-butyl (R)-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate:
To a solution of (R)-6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazine (2.45 g, 14.83 mmol) and DIPEA (5.75 g, 44.48 mmol, 7.77 mL) in DCM (25 mL) was added tert-butoxycarbonyl tert-butyl carbonate (3.88 g, 17.79 mmol, 4.09 mL) at room temperature. The reacting mixture was stirred overnight at room temperature, then diluted in water followed by extraction with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (10–75% AcOEt in heptane) affording tert-butyl (R)-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate as a white solid. Yield: 3.75 g (95%); UPLC-MS (ESI)+m/z: 266 [M + H]+. 1H NMR (500MHz, METHANOL-d4) Shift = 7.49 (d, J = 1.8 Hz, 1H), 6.18 (d, J = 1.8 Hz, 1H), 5.03 (d, J = 17.1 Hz, 1H), 4.43 (d, J = 12.8 Hz, 1H), 4.27 (br, 2H), 4.11 (dd, J = 4.6, 13.1 Hz, 1H), 1.73–1.60 (m, 1H), 1.53 (s, 9H), 1.00 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
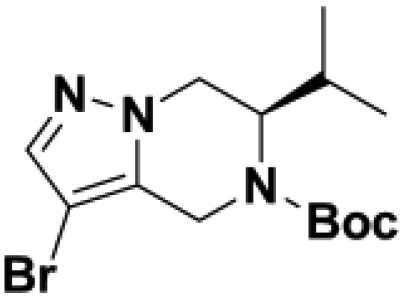
tert-butyl (R)-3-bromo-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate:
To a solution of tert-butyl (R)-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (1.05 g, 3.96 mmol) in DCM (25 mL) was added N-Bromosuccinimide (774.70 mg, 4.35 mmol) at room temperature. The reacting mixture was stirred overnight at room temperature then was concentrated. The residue was purified by column chromatography on silica gel (10–60% AcOEt in heptane) to afford tert-butyl (R)-3-bromo-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate as a white solid. Yield: 1.28 g (94%); UPLC-MS (ESI)+m/z: 344 [M + H]+. 1H NMR (500MHz, METHANOL-d4) Shift = 7.52 (s, 1H), 4.91 (m, 1H), 4.41 (d, J = 13.4 Hz, 1H), 4.37–4.01 (m, 3H), 1.74–1.58 (m, 1H), 1.54 (s, 9H), 0.99 (d, J = 6.1 Hz, 3H), 0.95 (d, J = 6.1 Hz, 3H).
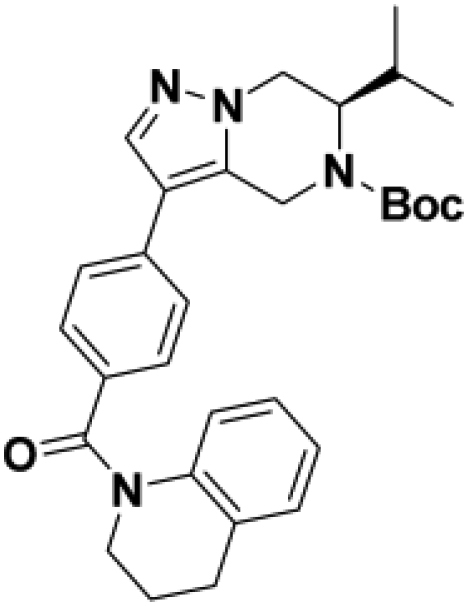
tert-butyl (R)-6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate:
A solution of tert-butyl (R)-3-bromo-6-isopropyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (200.00 mg, 580.97 mmol),[4-(3,4-dihydro-2H-quinoline-1-carbonyl)phenyl]boronic acid (179.65 mg, 639.07 mmol), potassium carbonate (240.89 mg, 1.74 mmol) and Pd(dppf)Cl2.CH2Cl2 (23.72 mg, 29.05 umol) in 1,4-dioxane:water (2:1, 5 mL) was stirred overnight at 80°C. The solution was cooled down, diluted in water followed by extraction with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (0–50% AcOEt in DCM) to afford tert-butyl (R)-6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate as a yellow solid containing some impurities. Yield: 201 mg; UPLC-MS (ESI)+m/z: 501 [M + H]+.
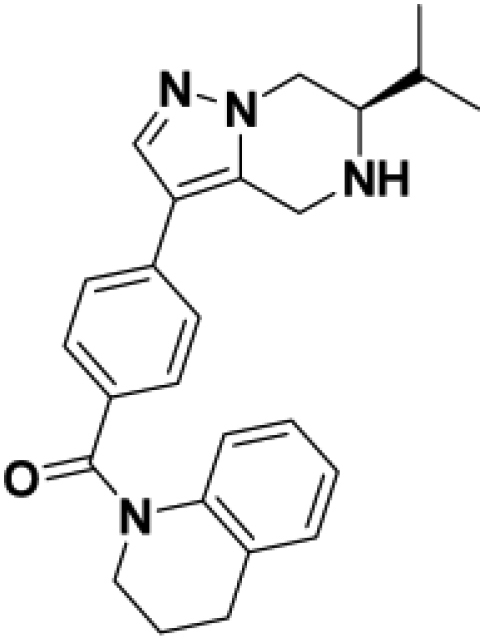
(R)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone:
A solution of tert-butyl (R)-6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (201 mg) was stirred overnight in a solution of 4N HCl in dioxane (15 mL). The volatiles were removed under vacuum. The residue was triturated with Et2O and filtered to afford (R)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone as an off-white solid. Yield: 101 mg (43%); UPLC-MS (ESI)+m/z: 401 [M + H]+. 1H NMR (500MHz, METHANOL-d4) Shift = 7.93 (s, 1H), 7.43 (d, J = 8.5 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H), 7.26–7.22 (m, 1H), 7.08–7.02 (m, 1H), 6.90 (t, J = 7.9 Hz, 1H), 6.76 (br s, 1H), 4.79 (d, J = 15.9 Hz, 1H), 4.70 (d, J = 15.9 Hz, 1H), 4.64 (dd, J = 4.6, 13.7 Hz, 1H), 4.30 (dd, J = 11.3, 13.7 Hz, 1H), 3.91 (t, J = 6.4 Hz, 3H), 2.90 (t, J = 6.7 Hz, 2H), 2.29 (m, 1H), 2.15–2.04 (m, 2H), 1.23 (d, J = 6.7 Hz, 3H), 1.19 (d, J = 6.7 Hz, 3H).

(S)-1-((R)-6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-methoxy-2-(1-methyl-1H-pyrazol-4-yl)ethan-1-one (BIO-2007817)
(R)-1-((R)-6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-methoxy-2-(1-methyl-1H-pyrazol-4-yl)ethan-1-one (BIO-2007818)
To a solution of (R)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone (100 mg, 249.68 mmol), 2-methoxy-2-(1-methylpyrazol-4-yl)acetic acid (46.74 mg, 274.65 mmol) in EtOAc (3 mL) was added triethylamine (363.00 mg, 3.59 mmol, 0.5 mL) and T3P (158.89 mg, 249.68 mmol, 1mL, 50% purity in DMF) and the mixture was stirred at 20°C for 2 h. The residue was poured in water (10 mL) and the aqueous phase was extracted with ethyl acetate (10 mL × 3). The combined organic layers were washed with brine (20 mL × 3), dried over Na2SO4, filtered and concentrated. The residue was purified by Prep-HPLC (Column: Welch Xtimate C18 150∗25mm∗5um; Condition: water(10mM NH4HCO3)-ACN; Begin B: 33; End B: 63; Gradient Time 10; 100%B Hold Time 2; FlowRate (mL/min): 25) to provide a mixture of both desired products (60 mg, 108.56 umol, 43.48% yield) as a white solid. The product was purified by chiral SFC (Condition: 0.1%NH3H2O/EtOH; Column: DAICEL CHIRALCEL OJ(250mm∗30mm,10mm); Begin B: 30%; End B: 30%; Flow Rate(mL/min): 80) to provide the two diastereoisomers:
BIO-2007817: Yield: 19.1 mg. UPLC-MS (ESI)+m/z: 553 [M + H]+. Rt = 2.84 min (SFC), 100% ee. 1H NMR (500MHz, METHANOL-d4) δ = 7.80 (m, 1H), 7.70–7.36 (s, 1H), 7.55 (m, 1H), 7.47–7.40 (m, 3H), 7.32 (m, 1H), 7.23 (m, 1H), 7.05 (m, 1H), 6.91 (m, 1H), 6.76 (brm, 1H), 5.65–5.27 (m, 2H), 4.86–4.76 (m, 1H), 4.51–4.34 (m, 2H), 4.20–4.08 (m, 1H), 4.01–3.76 (m, 5H), 3.41 (m, 3H), 2.91 (m, 2H), 2.17–2.03 (m, 2H), 1.88–1.71 (m, 1H), 1.09–0.81 (m, 6H).
BIO-2007818: Yield: 30.9 mg. UPLC-MS (ESI)+m/z: 553 [M + H]+. Rt = 3.49 min (SFC), 100% ee.1H NMR (500MHz, METHANOL-d4) δ = 7.85–7.29 (m, 7H), 7.26–7.20 (m, 1H), 7.07–7.01 (m, 1H), 6.91 (m, 1H), 6.75 (br, 1H), 5.71–5.12 (m, 2H), 4.79 (m, 1H), 4.68–4.04 (m, 3H), 3.98–3.70 (m, 5H), 3.34–3.33 (m, 3H), 2.93–2.85 (m, 2H), 2.16–2.02 (m, 2H), 1.91–1.70 (m, 1H), 1.11–0.53 (m, 6H)
The stereochemistry of the products was not determined, but was assigned based on the biological activities observed for BIO-1967173 and BIO-1967174.

(S)-1-((R)-6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-methoxy-2-phenylethan-1-one (BIO-2008550):
To a solution of (R)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone (100 mg, 249.68 mmol) in EtOAc (3 mL) was added triethylamine (75.79 mg, 749.04 mmol, 104.40 uL), T3P (158.89 mg, 249.68 umol, 1 mL, 50% purity) and rac-2-methoxy-2-phenyl-acetic acid (41.49 mg, 249.68 umol). The mixture was stirred at 20°C for 2 h. The solution was poured into water (10 mL) and the aqueous phase was extracted with ethyl acetate (3×10 mL). The combined organic layers were washed with water (3×10 mL), brine (2×20 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by prep-TLC (petroleum ether/EtOAc = 1/1, 2 elutions) to give P1 (Rf = 0.40, petroleum ether/EtOAc = 1/1) and P2 (Rf = 0.35, petroleum ether/EtOAc = 1/1). P2 was further purified by prep-HPLC (column: Phenomenex Synergi C18 150∗30mm∗4mm; condition: water(0.05%HCl)-ACN; from: 52%–82%) to provide BIO-2008550 (8.5 mg, 6% yield) as a white solid. The stereochemistry of the products was not determined, but was assigned based on the biological activities observed for BIO-1967173 and BIO-1967174.
BIO-2008550: Yield: 8.5 mg. UPLC-MS (ESI)+m/z: 549 [M + H]+. Rt = 1.40 min (SFC), 98% ee. 1H NMR (500MHz, METHANOL-d4) δ = 7.80–7.73 (m, 1H), 7.51–7.19 (m, 10H), 7.10–7.01 (m, 1H), 6.95–6.88 (m, 1H), 6.76 (m, 1H), 5.59–5.25 (m, 2H), 4.83–4.76 (m, 1H), 4.46–3.85 (m, 5H), 3.45 (m, 3H), 2.90 (m, 2H), 2.17–2.02 (m, 2H), 1.82–1.58 (m, 1H), 1.02–0.71 (m, 6H)
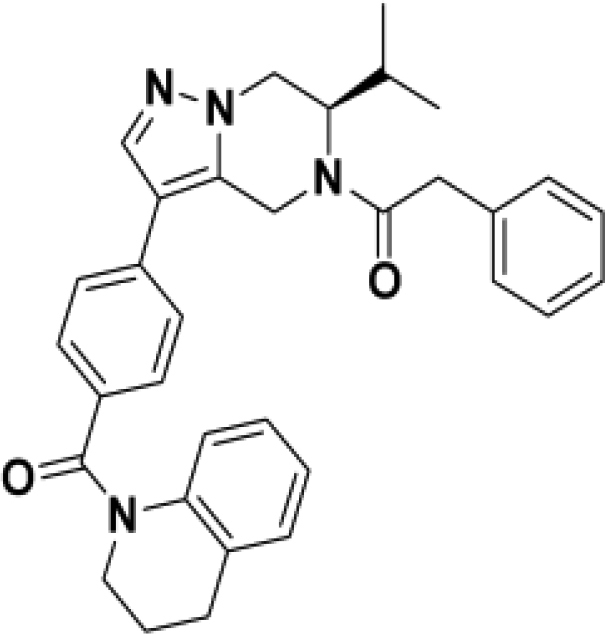
(R)-1-(6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one (BIO-1966561):
To a solution of (R)-1-(6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one (40.00 mg, 99.87 mmol) and N-ethyl-N-isopropyl-propan-2-amine (51.63 mg, 399.48 mmol, 69.77 uL) in DCM (3 mL) was added 2-phenylacetyl chloride (30.88 mg, 199.74 mmol, 26.39 mL) at room temperature. The solution was stirred for 3h, then was quenched with water followed by extraction with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (40–100% AcOEt in heptane) to afford (R)-1-(6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one. Yield: 44 mg (85%). UPLC-MS (ESI)+m/z: 519 [M + H]+. 1H NMR (400MHz, METHANOL-d4) δ = 7.78 (m, 1H), 7.43–7.17 (m, 10H), 7.06–6.98 (m, 1H), 6.92–6.84 (m, 1H), 6.73 (m, 1H), 5.65–5.14 (m, 1H), 4.82–3.80 (m, 8H), 2.90–2.84 (m, 2H), 2.11–1.99 (m, 2H), 1.83–1.60 (m, 1H), 1.06–0.75 (m, 6H).
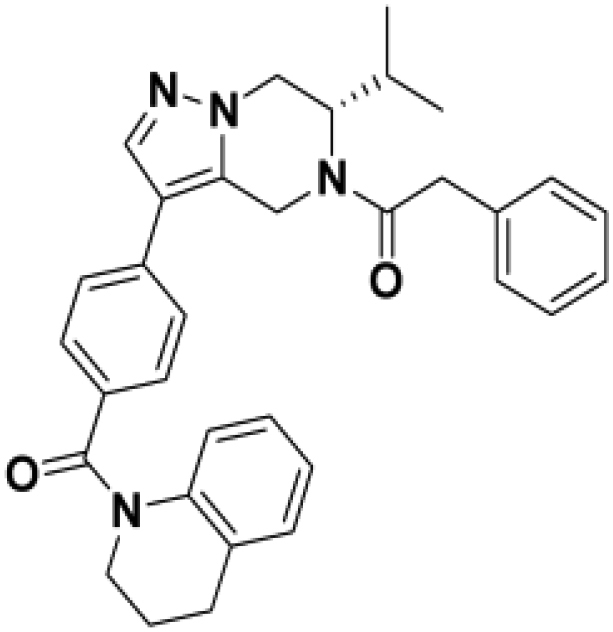
(S)-1-(6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one (BIO-1966560):
(S)-1-(6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one was prepared with the same procedure as described for (R)-1-(6-isopropyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one using (S)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone.

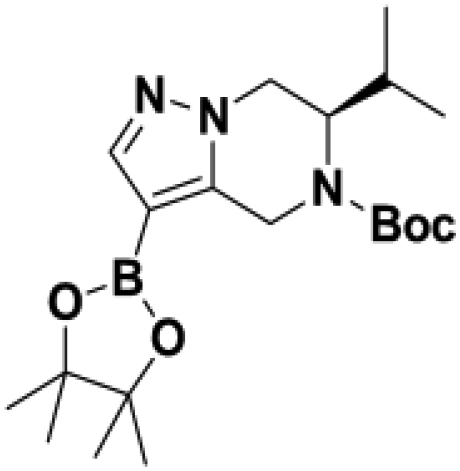
tert-butyl (R)-6-isopropyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate:
To a solution of tert-butyl (6R)-3-bromo-6-isopropyl-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazine-5-carboxylate (800 mg, 2.32 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (708.15 mg, 2.79 mmol) and potassium acetate (700 mg, 7.13 mmol) in Dioxane (10 mL) was added dichloropalladium;tricyclohexylphosphane (170 mg, 230.30 mmol) under inert atmosphere and the mixture was stirred at 90°C for 12 h. The mixture was diluted in water and extracted with AcOEt. The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered and concentrated to give tert-butyl (6R)-6-isopropyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazine-5-carboxylate (980 mg, crude) as a yellow oil which was used for next step without purification. Yield: 980 mg (crude). UPLC-MS (ESI)+m/z: 392 [M + H]+.
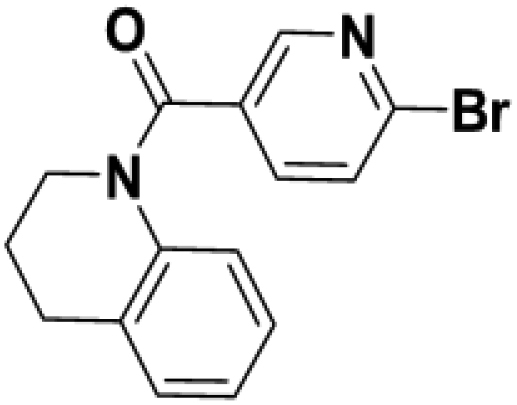
(6-bromopyridin-3-yl)(3,4-dihydroquinolin-1(2H)-yl)methanone:
A mixture of 6-bromopyridine-3-carboxylic acid (2 g, 9.90 mmol), triethylamine (2.00 g, 19.80 mmol, 2.76 mL) and HATU (7.55 g, 19.80 mmol) in DCM (40 mL) was stirred at 25°C for 30 min, then the 1,2,3,4-tetrahydroquinoline (1.32 g, 9.90 mmol, 1.24 mL) was added and the mixture was stirred at 25°C for 12 h under inert atmsophere. The mixture was diluted in water and extracted with DCM (30 mL × 3). The combined organic layers were washed with brine (30 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc= 9/1 to 4/1) to give (6-bromo-3-pyridyl)-(3,4-dihydro-2H-quinolin-1-yl)methanone (2.1 g, 6.62 mmol, 67% yield) as a yellow solid. Yield: 2.1g (67%). UPLC-MS (ESI)+m/z: 317 [M + H]+. 1H NMR (500MHz, METHANOL-d4) δ = 8.25 (s, 1H), 7.73–7.50 (m, 2H), 7.26 (d, J = 7.5 Hz, 1H), 7.11 (dt, J = 1.0, 7.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 6.69 (br s, 1H), 3.92 (t, J = 6.6 Hz, 2H), 2.89 (t, J = 6.6 Hz, 2H), 2.21–2.04 (m, 2H)
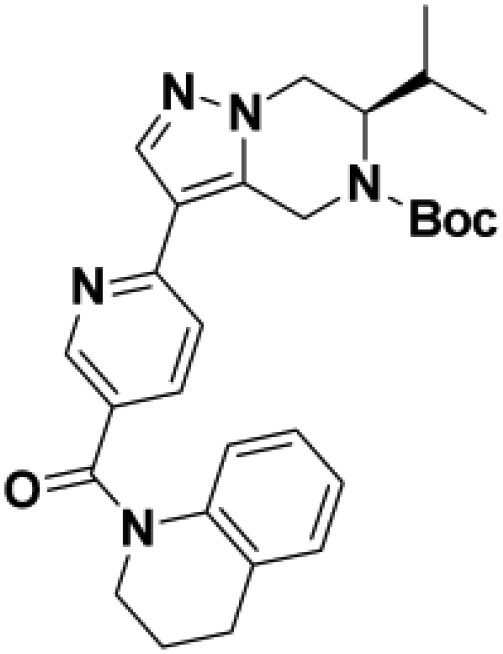
tert-butyl (R)-6-isopropyl-3-(5-(1,2,3,4-tetrahydroquinoline-1-carbonyl)pyridin-2-yl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate:
To a solution of tert-butyl (6R)-6-isopropyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazine-5-carboxylate (800.00 mg, 2.04 mmol), (6-bromo-3-pyridyl)-(3,4-dihydro-2H-quinolin-1-yl)methanone (648.45 mg, 2.04 mmol) and K2CO3 (847.65 mg, 6.13 mmol) in Dioxane (18 mL) and H20 (2 mL) was added ditert-butyl(cyclopentyl)phosphane; dichloropalladium;iron (133.24 mg, 204.44 mmol) under inert atmsophere and the mixture was stirred at 80°C for 3 h. The mixture was concentrated under reduced pressure to give a residue which was extracted with EtOAc (20 mL × 3). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (petroleum ether/EtOAc= 19/1 to 4/1) to give tert-butyl (6R)-3-[5-(3,4-dihydro-2H-quinoline-1-carbonyl)-2-pyridyl]-6-isopropyl-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazine-5-carboxylate as a yellow solid containing some impurities. Yield: 440 mg. UPLC-MS (ESI)+m/z: 502 [M + H]+.
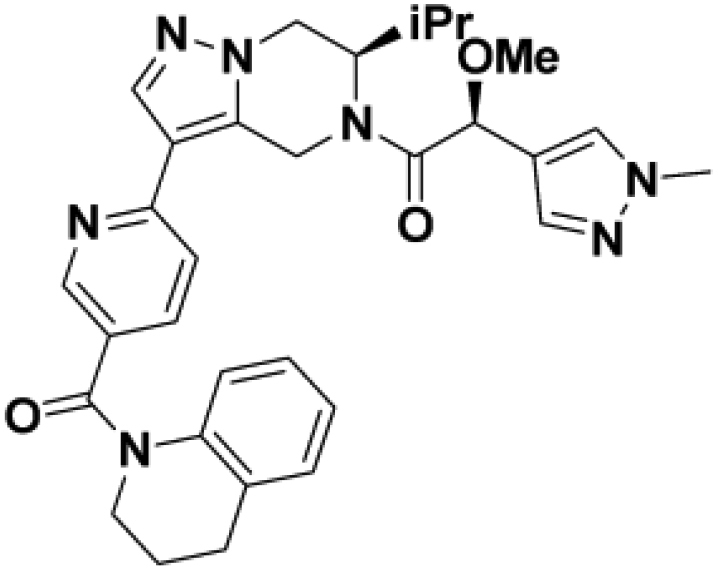
(S)-1-((R)-6-isopropyl-3-(5-(1,2,3,4-tetrahydroquinoline-1-carbonyl)pyridin-2-yl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-methoxy-2-(1-methyl-1H-pyrazol-4-yl)ethan-1-one (BIO-2007967):
To a solution of tert-butyl (6R)-3-[5-(3,4-dihydro-2H-quinoline-1-carbonyl)-2-pyridyl]-6-isopropyl-6,7-dihydro-4H-pyrazolo[1,5-a]pyrazine-5-carboxylate (140 mg, 279.10 mmol) in Dioxane (3 mL) was added HCl/dioxane (4 M, 1 mL) and the mixture was stirred at 25°C for 5 h. The mixture was concentrated under reduced pressure to give 3,4-dihydro-2H-quinolin-1-yl-[6-[(6R)-6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]-3-pyridyl]methanone (98 mg, crude) as a yellow solid which was used for next step without purification.
To a solution of 3,4-dihydro-2H-quinolin-1-yl-[6-[(6R)-6-isopropyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]-3-pyridyl]methanone (100 mg, 249.06 mmol), T3P (158.49 mg, 249.06 mmol, 50% purity) and Triethylamine (25.20 mg, 249.06 mmol, 34.71 mL) in EtOAc (2 mL) was added (rac)-2-(1-methylpyrazol-4-yl)propanoic acid (38.40 mg, 249.06 mmol) and the mixture was stirred at 25°C for 2 h. The mixture was concentrated under reduced pressure and purified by pre-HPLC (Column: YMC-Actus Triart C18 100∗30mm∗5mm; Condition: water (0.0%HCl)-ACN; 41%–71%) to provide a mixture of diastereomers which was further purified by by chiral SFC: (Column: DAICEL CHIRALPAK AD(250mm∗30mm,10mm); Condition: 0.1%NH3H2O IPA; 40%) to provide the two desired diastereomers. The stereochemistry of the purified diastereomers was not determined.
BIO-2007967: Yield: 12.6 mg. UPLC-MS (ESI)+m/z: 554 [M + H]+. Rt = 3.72 min (SFC), 100% ee. (500 MHz, METHANOL-d4) ppm: 8.42–8.61 (m, 1H), 7.97–8.08 (m, 1H), 7.37–7.76 (m, 4H), 7.20–7.31 (m, 1H), 7.02–7.12 (m, 1H), 6.88–6.98 (m, 1H), 6.64–6.82 (m, 1H), 5.50–5.85 (m, 1H), 5.22–5.45 (m, 1H), 4.74–4.80 (m, 1H), 3.90–4.55 (m, 5H), 3.78–3.90 (m, 3H), 3.32–3.44 (m, 3H), 2.84–2.97 (m, 2H), 2.01–2.17 (m, 2H), 1.62–1.80 (m, 1H), 0.73–1.03 (m, 6H).
Other Diastereomer: Yield: 12.9 mg. UPLC-MS (ESI)+m/z: 554 [M + H]+. Rt = 3.09 min (SFC), 100% ee. 1H NMR (500MHz, METHANOL-d4) δ = 8.45–8.53 (m, 1H), 7.98–8.08 (m, 1H), 7.34–7.74 (m, 4H), 7.23–7.29 (m, 1H), 7.05–7.11 (m, 1H), 6.89–6.98 (m, 1H), 6.68–6.78 (m, 1H), 5.48–5.94 (m, 2H), 5.24–5.38 (m, 1H), 4.62–4.81 (m, 2H), 3.91–4.51 (m, 4H), 3.68–3.91 (m, 3H), 3.32–3.38 (m, 3H), 2.86–2.96 (m, 2H), 2.04–2.16 (m, 2H), 1.66–1.85 (m, 1H), 0.52–1.06 (m, 6H).

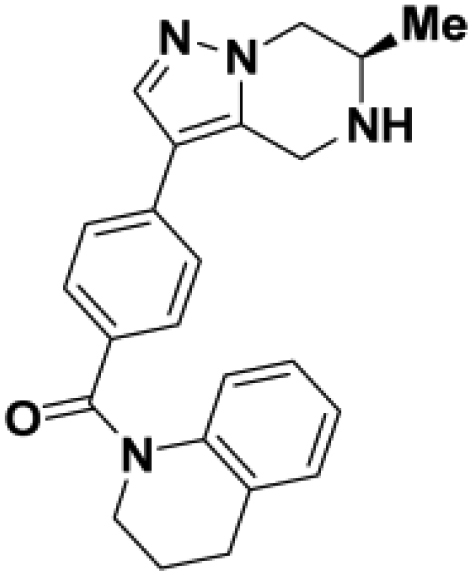
tert-butyl(R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate:
tert-butyl(R)-3-bromo-6-methyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate was prepared following General procedure A starting with tert-butyl (R)-(1-hydroxypropan-2-yl)carbamate.
A solution of tert-butyl(R)-3-bromo-6-methyl-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate, 3,4-dihydro-2H-quinolin-1-yl-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanone (584.12 mg, 1.61 mmol) and K2CO3 (555.60 mg, 4.02 mmol, 2.01 mL) in EtOH:water (10:1, 10 mL) was stirred at 75°C for 5 min. SiliaCat DPP-Pd (0.05 eq, 0.30mmol/g) was added and the reacting mixture was stirred at 75°C for 2h. The solution was cooled down, filtered over celite and washed with DCM/water. The aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by column chromatography on silica gel (0–70% AcOEt in DCM) to afford tert-butyl (R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate. Yield: 535 mg (84%). UPLC-MS (ESI)+m/z: 473 [M + H]+.
tert-butyl (R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazine-5(4H)-carboxylate (535 mg) was stirred in a 4N HCl solution in dioxane (15 mL) for 5h. The solution was concentrated to provide (R)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone Hydrochloride. Yield: 395 mg (72%). UPLC-MS (ESI)+m/z: 373 [M + H]+. 1H NMR (500MHz, DMSO-d6) δ = 8.00 (s, 1H), 7.46–7.38 (m, 4H), 7.22 (d, J = 7.3 Hz, 1H), 7.01 (dt, J = 1.2, 7.3 Hz, 1H), 6.96–6.89 (m, 1H), 6.83 (br d, J = 7.3 Hz, 1H), 4.79–4.61 (m, 2H), 4.53 (dd, J = 4.3, 13.4 Hz, 1H), 4.04 (dd, J = 10.4, 13.4 Hz, 1H), 3.97–3.87 (m, 1H), 3.78 (m, 2H), 3.48 (br, 1H), 2.84 (t, J = 6.4 Hz, 2H), 1.97 (quin, J = 6.9 Hz, 2H), 1.41 (d, J = 6.1 Hz, 3H)
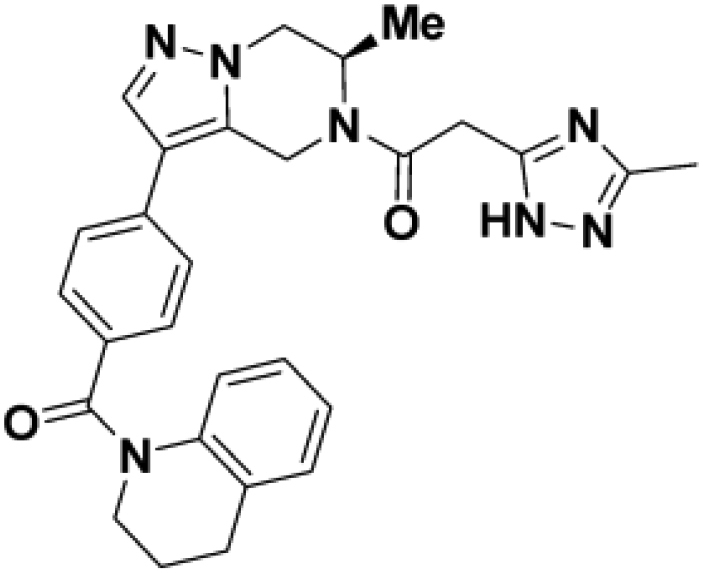
(R)-2-(3-methyl-1H-1,2,4-triazol-5-yl)-1-(6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)ethan-1-one (BIO-1984542):
To a solution (R)-(3,4-dihydroquinolin-1(2H)-yl)(4-(6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl)phenyl)methanone (15.0 mg, 40.3 mmol), 2-(3-methyl-1H-1,2,4-triazol-5-yl)acetic acid (11.4 mg, 80.6 mmol), and DIPEA (21.1 ul, 120.8 mmol) in DMF (1 mL) was added HATU (23.1 mg, 60.4 mmol) at room temperature and stirred for 16h. The mixture was concentrated, suspended in 5% MeOH in AcOEt and washed with water. The organic layer was dried over Na2SO4, filtered and concentrated. The reside was purified by prep HPLC (Waters XSelect CSH Prep C18 5mm OBD 19 × 100mm, 5 to 50% CH3CN in water, Ammonium buffer) to provide (R)-2-(3-methyl-1H-1,2,4-triazol-5-yl)-1-(6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)ethan-1-one (BIO-1984542). Yield: 10.5 mg (52%). UPLC-MS (ESI)+m/z: 495 [M + H]+.
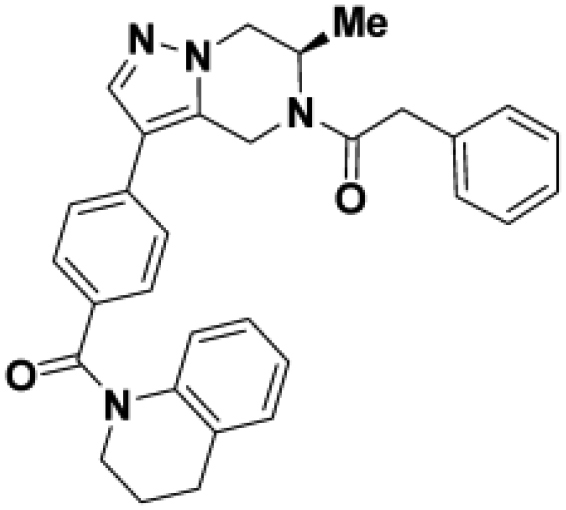
(R)-1-(6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one (BIO-1953719):
To a solution of 3,4-dihydro-2H-quinolin-1-yl-[4-[(6R)-6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]phenyl]methanone (25.00 mg, 61.14 mmol, Hydrochloride) and N-ethyl-N-isopropyl-propan-2-amine (23.70 mg, 183.41 mmol, 32.03 mL) in DCM (2 mL) was added 2-phenylacetyl chloride (14.18 mg, 91.70 mmol, 12.12 mL) at room temperature and stirred overnight. The solution was diluted in DCM and washed with water (2x). The combined organic layers were filtered and concentrated. The residue was purified by prep HPLC (Waters SunFire Prep C18 5mm OBD 19 × 100mm, 5 to 65% CH3CN in water, TFA buffer) to afford (R)-1-(6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylethan-1-one as a white solid. Yield: 15.2 mg (50%). UPLC-MS (ESI)+m/z: 491 [M + H]+. 1H NMR (500MHz, DMSO-d6) δ = 7.92 (m, 1H), 7.50–7.17 (m, 9H), 7.01 (m, 1H), 6.92 (m, 1H), 6.82 (m, 1H), 5.47–4.71 (m, 2H), 4.69–4.36 (m, 1H), 4.17–4.09 (m, 2H), 3.98–3.77 (m, 4H), 2.83 (m, 2H), 1.97 (m, 2H), 1.13–0.98 (m, 3H)
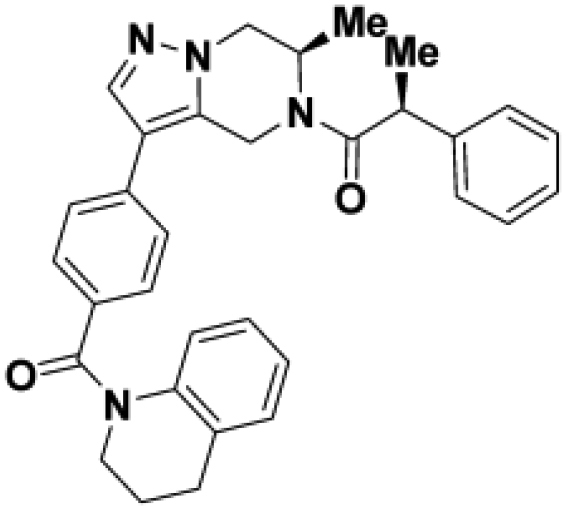
(S)-1-((R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one (BIO-1967173):
To a solution of 3,4-dihydro-2H-quinolin-1-yl-[4-[(6R)-6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]phenyl]methanone (28.00 mg, 68.47 mmol, Hydrochloride), (2S)-2-phenylpropanoic acid (20.56 mg, 136.94 mmol, 18.69 mL) and N-ethyl-N-isopropyl-propan-2-amine (35.40 mg, 273.88 mmol, 47.84 mL) in DCM (3 mL) was added HATU (39.16 mg, 102.71 umol) at room temperature and stirred overnight. The solution was diluted in water followed by extraction with DCM. The combined organic layers were dried over Na2SO4, filtered and concentrated. The residue was purified by prep HPLC (Waters SunFire Prep C18 5mm OBD 19 × 100mm, 5 to 65% CH3CN in water, TFA buffer) to afford (S)-1-((R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one (24.40 mg, 48.35 mmol, 71% yield) as a white solid. Yield: 24.4 mg (71%). UPLC-MS (ESI)+m/z: 505 [M + H]+. 1H NMR (500MHz, METHANOL-d4) δ = 7.78 (m, 1H), 7.44–7.11 (m, 10H), 7.10–6.97 (m, 1H), 6.89 (m, 1H), 6.74 (m, 1H), 5.59–5.07 (m, 1H), 5.34–4.69 (m, 1H), 4.47–3.12 (m, 6H), 2.93–2.85 (m, 2H), 2.16–2.01 (m, 2H), 1.42 (m, 3H), 1.31–0.95 (m, 3H).
(R)-1-((R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one (BIO-1967174):
To a solution of 3,4-dihydro-2H-quinolin-1-yl-[4-[(6R)-6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]phenyl]methanone (28.00 mg, 68.47 mmol, Hydrochloride), (2R)-2-phenylpropanoic acid (20.56 mg, 136.94 mmol, 18.69 mL), N-ethyl-N-isopropyl-propan-2-amine (35.40 mg, 273.89 mmol, 47.83 mL) in DCM (3 mL) was added HATU (39.16 mg, 102.71 mmol) at room temperature and stirred overnight. The solution was diluted in water followed by extraction with DCM. The combined organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by prep HPLC (Waters SunFire Prep C18 5um OBD 19 × 100mm, 5 to 65% CH3CN in water, TFA buffer) to afford (R)-1-((R)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one.
Yield: 24.8 mg (72%). UPLC-MS (ESI)+m/z: 505 [M + H]+. 1H NMR (500MHz, METHANOL-d4) δ = 7.85–7.60 (m, 1H), 7.45–6.99 (m, 11H), 6.95–6.84 (m, 1H), 6.75 (m, 1H), 5.57–4.95 (m, 1H), 5.44–4.77 (1H, m), 4.74–3.85 (m, 6H), 2.94–2.82 (m, 2H), 2.11–2.00 (m, 2H), 1.49–1.35 (m, 3H), 1.31–0.56 (m, 3H).
(S)-1-((S)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one (BIO-1968010):
To a solution of 3,4-dihydro-2H-quinolin-1-yl-[4-[(6S)-6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]phenyl]methanone (28.00 mg, 68.47 mmol, Hydrochloride), (2S)-2-phenylpropanoic acid (20.56 mg, 136.94 mmol, 18.69 mL) and N-ethyl-N-isopropyl-propan-2-amine (35.40 mg, 273.89 mmol, 47.83 mL) in DCM (3 mL) was added (39.16 mg, 102.71 mmol) at room temperature and stirred overnight. The solution was diluted in water followed by extraction with DCM. The combined organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by prep HPLC (Waters SunFire Prep C18 5mm OBD 19 × 100mm, 5 to 65% CH3CN in water, TFA buffer) to afford (S)-1-((S)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one. Yield: 31.2 mg (90%). UPLC-MS (ESI)+m/z: 505 [M + H]+.
(R)-1-((S)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one (BIO-1968011):
To a solution of 3,4-dihydro-2H-quinolin-1-yl-[4-[(6S)-6-methyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrazin-3-yl]phenyl]methanone (28.00 mg, 68.47 mmol, Hydrochloride), (2R)-2-phenylpropanoic acid (20.56 mg, 136.94 mmol, 18.69 uL) and N-ethyl-N-isopropyl-propan-2-amine (35.40 mg, 273.89 mmol, 47.83 mL) in DCM (3 mL) was added (39.16 mg, 102.71 mmol) at room temperature and stirred overnight. The solution was diluted in water followed by extraction with DCM. The combined organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by prep HPLC (Waters SunFire Prep C18 5mm OBD 19 × 100mm, 5 to 65% CH3CN in water, TFA buffer) to afford (R)-1-((S)-6-methyl-3-(4-(1,2,3,4-tetrahydroquinoline-1-carbonyl)phenyl)-6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)-2-phenylpropan-1-one.
Yield: 26.9 mg (78%). UPLC-MS (ESI)+m/z: 505 [M + H]+.
Quantificationandstatisticalanalysis
Statistical analysis
Statistical analyses were calculated in Prism 8.3 (Graphpad Software). According to the experimental set-up, we used ANOVA followed by Dunnett’s multiple comparisons test (MCT). The specific statistical tests used are indicated in the figure legends.
Parkin translocation assay
Image analysis was performed using Harmony High-Content Analysis software. Nuclei were detected, and a cell mask was defined based on diffuse GFP-Parkin signal. Punctate GFP-Parkin was masked based on contrast against diffuse signal within the cell mask. A negative mask was generated using the punctate mask to include only non-punctate signal, and the integrated intensity of each mask was determined per cell. When the integrated punctate signal was equal to the integrated residual signal, the masked cell was marked as having Parkin translocation. To determine the time to reach 50% of cells with Parkin translocation (T50), the percentage of cells with translocated Parkin was plotted as a function of time in Prism. A non-linear regression with the following equation was used to determine T50: Y= top−bottom/1 + 10(LogT50−X) × Hill Slope.
MitoQC
Image analysis was performed using Harmony High-Content Analysis software. Images from the GFP and mCherry channels were digitally sharpened, and masks drawn to create a set of green or red objects, respectively. Red objects with no corresponding green object were identified as mitochondria that had been trafficked to lysosomes: “mitolysosomes”. Mitolysosome abundance was normalized to the area of total masked red objects, and expressed as “mitolysosomes per mitochondria area.”
Acknowledgments
The authors would like to thank Kevin Guckian for a critical reading of the manuscript. The authors would like to thank Rebecca Haddad and Joanne Shearer at Cambridge Research Labs in the UK as well as Richard Nelson for their intellectual contributions towards troubleshooting and optimizing the miniaturization of the screen and executing the high-throughput screen.
Author contributions
A.W. and W.D.H. conceived of the presented idea, P.M., E.S., A.H, R.K., N.B, S.B., L.Y.W. & D.M carried out the experiments and performed the analysis. E.S., J.H., W.D.H, A.W., and L.S. supervised the findings of this work. All authors provided critical feedback and helped write the final version of the manuscript.
Declaration of interests
All authors are employees and shareholders at Biogen.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103650.
Supplemental information
Data and code availability
This study did not generate code or datasets that have been deposited in a repository.
References
- Allen G.F., Toth R., James J., Ganley I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14:1127–1135. doi: 10.1038/embor.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne A.N., Trempe J.F. Mechanisms of PINK1, ubiquitin and Parkin interactions in mitochondrial quality control and beyond. Cell Mol. Life Sci. 2019;76:4589–4611. doi: 10.1007/s00018-019-03203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky A., Kessler B.M., Casagrande R., Overkleeft H.S., Wilkinson K.D., Ploegh H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–5196. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchell L., Chaugule V.K., Walden H. Small, N-terminal tags activate Parkin E3 ubiquitin ligase activity by disrupting its autoinhibited conformation. PLoS One. 2012;7:e34748. doi: 10.1371/journal.pone.0034748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule V.K., Burchell L., Barber K.R., Sidhu A., Leslie S.J., Shaw G.S., Walden H. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011;30:2853–2867. doi: 10.1038/emboj.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M., Chan P., Chen R., Tan L., Lincoln S., Hernandez D., Forno L., Gwinn-Hardy K., Petrucelli L., Hussey J., et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann. Neurol. 2001;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- Garofalo A.W., Johnston J., Fatheree P.R. 2017. Triazole Benzamide Derivatives and the Compositions and Methods of Treatment Regarding the Same. [Google Scholar]
- Gladkova C., Maslen S.L., Skehel J.M., Komander D. Mechanism of parkin activation by PINK1. Nature. 2018;559:410–414. doi: 10.1038/s41586-018-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J.W., Ordureau A., Heo J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol.Cell Biol. 2018;19:93–108. doi: 10.1038/nrm.2017.129. [DOI] [PubMed] [Google Scholar]
- Hou X., Fiesel F.C., Truban D., Castanedes Casey M., Lin W.L., Soto A.I., Tacik P., Rousseau L.G., Diehl N.N., Heckman M.G., et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy. 2018;14:1404–1418. doi: 10.1080/15548627.2018.1461294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.M., Lazarou M., Wang C., Kane L.A., Narendra D.P., Youle R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191:933–942. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J., Garofalo A.W. 2017. Pyradazinonederivatives and the Compositions and Methods of Treatment Regarding the Same. [Google Scholar]
- Kane L.A., Lazarou M., Fogel A.I., Li Y., Yamano K., Sarraf S.A., Banerjee S., Youle R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J.Cell Biol. 2014;205:143–153. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D.G., Ritorto M.S., Hofmann K., Alessi D.R., Knebel A., Trost M., Muqit M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014;460:127–139. doi: 10.1042/BJ20140334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Academic Press; 2016. Pharmacology in Drug Discovery and Development: Understanding Drug Response. [Google Scholar]
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klosowiak J.L., Park S., Smith K.P., French M.E., Focia P.J., Freymann D.M., Rice S.E. Structural insights into Parkin substrate lysine targeting from minimal Miro substrates. Sci. Rep. 2016;6:33019. doi: 10.1038/srep33019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H.I., Campbell D.G., Gourlay R., Burchell L., Walden H., Maccartney T.J., Deak M., et al. Muqit, M.M. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012;2:120080. doi: 10.1098/rsob.120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Kumar A., Aguirre J.D., Condos T.E., Martinez-Torres R.J., Chaugule V.K., Toth R., Sundaramoorthy R., Mercier P., Knebel A., Spratt D.E., et al. Walden, H. Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J. 2015;34:2506–2521. doi: 10.15252/embj.201592337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie M.J., Ostaszewski B.L., Weihofen A., Schlossmacher M.G., Selkoe D.J. Dopamine covalently modifies and functionally inactivates parkin. Nat. Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lubbe S.J., Bustos B.I., Hu J., Krainc D., Joseph T., Hehir J., Tan M., Zhang W., Escott-Price V., Williams N.M., et al. Assessing the relationship between monoallelic PRKN mutations and Parkinson's risk. Hum. Mol. Genet. 2021;30:78–86. doi: 10.1093/hmg/ddaa273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpartida A.B., Williamson M., Narendra D.P., Wade-Martins R., Ryan B.J. Mitochondrial dysfunction and mitophagy in Parkinson's disease: from mechanism to therapy. Trends Biochem. Sci. 2021;46:329–343. doi: 10.1016/j.tibs.2020.11.007. [DOI] [PubMed] [Google Scholar]
- Matsuda N., Kitami T., Suzuki T., Mizuno Y., Hattori N., Tanaka K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 2006;281:3204–3209. doi: 10.1074/jbc.M510393200. [DOI] [PubMed] [Google Scholar]
- Miller S., Muqit M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson's disease. Neurosci.Lett. 2019;705:7–13. doi: 10.1016/j.neulet.2019.04.029. [DOI] [PubMed] [Google Scholar]
- Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J.Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offensperger F., Muller F., Jansen J., Hammler D., Gotz K.H., Marx A., Sirois C.L., Chamberlain S.J., Stengel F., Scheffner M. Identification of small-molecule activators of the ubiquitin ligase E6AP/UBE3A and angelman syndrome-derived E6AP/UBE3Avariants. Cell Chem. Biol. 2020;27:1510–1520 e6. doi: 10.1016/j.chembiol.2020.08.017. [DOI] [PubMed] [Google Scholar]
- Okatsu K., Oka T., Iguchi M., Kosako H., Tani N., Kimura M., Go E., Koyano F., Funayama M., Shiba-Fukushima K., et al. Matsuda, N. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun. 2012;3:1016. doi: 10.1038/ncomms2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A., Sarraf S.A., Duda D.M., Heo J.M., Jedrychowski M.P., Sviderskiy V.O., Olszewski J.L., Koerber J.T., Xie T., Beausoleil S.A., et al. Harper, J.W. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56:360–375. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao K.C., Stanley M., Han C., Lai Y.C., Murphy P., Balk K., Wood N.T., Corti O., Corvol J.C., Muqit M.M., Virdee S. Probes of ubiquitin E3 ligases enable systematic dissection of parkin activation. Nat. Chem. Biol. 2016;12:324–331. doi: 10.1038/nchembio.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S., Foote P.K., Krist D.T., Rice S.E., Statsyuk A.V. UbMES and UbFluor: novel probes for ring-between-ring (RBR) E3 ubiquitin ligase PARKIN. J. Biol. Chem. 2017;292:16539–16553. doi: 10.1074/jbc.M116.773200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S., Soya N., Truong L., Croteau N., Lukacs G., L, Trempe J.F. PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018;19 doi: 10.15252/embr.201744981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf S.A., Raman M., Guarani-Pereira V., Sowa M.E., Huttlin E.L., Gygi S.P., Harper J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V., Lilov A., Seirafi M., Vranas M., Rasool S., Kozlov G., Sprules T., Wang J., Trempe J.F., Gehring, K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015;34:2492–2505. doi: 10.15252/embj.201592237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V., Sung G., Soya N., Kozlov G., Blaimschein N., Miotto L.S., Trempe J.F., Lukacs G.L., Gehring K. Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol. 2018;25:623–630. doi: 10.1038/s41594-018-0088-7. [DOI] [PubMed] [Google Scholar]
- Seirafi M., Kozlov G., Gehring K. Parkin structure and function. FEBS J. 2015;282:2076–2088. doi: 10.1111/febs.13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukishima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., Hattori, N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2:1002. doi: 10.1038/srep01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K., Arano T., Matsumoto G., Inoshita T., Yoshida S., Ischihama Y., Ryu K.Y., Nukina N., Hattori N., Imai, Y. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet. 2014;10:e1004861. doi: 10.1371/journal.pgen.1004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K., Inoshita T., Sano O., Iwata H., Ishikawa K.I., Okano H., Akamatsu W., Imai Y., Hattori N. A cell-based high-throughput screening identified two compounds that enhance PINK1-parkin signaling. iScience. 2020;23:101048. doi: 10.1016/j.isci.2020.101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K., Ishikawa K.I., Inoshita T., Izawa N., Takanashi M., Sato S., Onodera O., Akamatsu W., Okano H., Imai Y., Hattori N. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson's disease. Hum. Mol. Genet. 2017;26:3172–3185. doi: 10.1093/hmg/ddx201. [DOI] [PubMed] [Google Scholar]
- Shlevkov E., Kramer T., Schapansky J., Lavoie M.J., Schwarz T.L. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. U S A. 2016;113:E6097–E6106. doi: 10.1073/pnas.1612283113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.A., Hartley R.C., Cocheme H.M., Murphy M.P. Mitochondrial pharmacology. Trends Pharmacol. Sci. 2012;33:341–352. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Springer W., Fiesel F.C., Caulfield T.R. 2018. Small Molecule Activators of Parkin Enzyme Function. [Google Scholar]
- Surmeier D.J., Guzman J.N., Sanchez-Padilla J., Goldberg J.A. What causes the death of dopaminergic neurons in Parkinson's disease? Prog. Brain Res. 2010;183:59–77. doi: 10.1016/S0079-6123(10)83004-3. [DOI] [PubMed] [Google Scholar]
- Tang M.Y., Vranas M., Krahn A.I., Pundlik S., Trempe J.F., Fon E.A. Structure-guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat. Commun. 2017;8:14697. doi: 10.1038/ncomms14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokarew J.M., El-Kodsi D.N., Lengacher N.A., Fehr T.K., Nguyen A.P., Shutinoski B., O'nuallain B., Jin M., Khan J.M., Ng A.C.H., et al. Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites. Acta Neuropathol. 2021;141:725–754. doi: 10.1007/s00401-021-02285-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe J.F., Sauve V., Grenier K., Seirafi M., Tang M.Y., Menade M., Al-Abdul-Wahid S., Krett J., Wong K., Kozlov G., et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Valente E.M., Abou-Sleiman P.M., Caputo V., Muqit M.M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A.R., Healy D.G., et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Wang C., Tan J.M., Ho M.W., Zaiden N., Wong S.H., Chew C.L., Eng P.W., Lim T.M., Dawson T.M., Lim K.L. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson’s disease-linked point mutations. J. Neurochem. 2005;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y.L., Selkoe D., Rice S., Steen J., Lavoie M.J., Schwarz T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T., Komander D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013;32:2099–2112. doi: 10.1038/emboj.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K., Youle R.J. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9:1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W., Macdougall E.J., Tang M.Y., Krahn A.I., Gan-Or Z., Trempe J.F., Fon E.A. The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson's disease. Hum. Mol. Genet. 2019;28:2811–2825. doi: 10.1093/hmg/ddz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M., Cortese G.P., Waites C.L. Parkinson's disease-linked Parkin mutations impair glutamatergic signaling in hippocampal neurons. BMC Biol. 2018;16:100. doi: 10.1186/s12915-018-0567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Huang X., Yoon E., Bandres-Ciga S., Blauwendraat C., Cade J.H., Wu B.P., Williams V.H., Schindler A.B., Brooks J., et al. Heterozygous PRKN mutations are common but do not increase the risk of Parkinson's disease. medRxiv. 2021 doi: 10.1101/2021.08.11.21261928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code or datasets that have been deposited in a repository.